
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol., 10 January 2023
Sec. Gastrointestinal and Hepatic Pharmacology
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.1071844
This article is part of the Research TopicLiver Diseases and Programmed Cell DeathView all 9 articles
 Le Li1,2
Le Li1,2 Zhijun Zhu1*
Zhijun Zhu1*Liver fibrosis, which is characterized by the excessive deposition of extracellular matrix (ECM) materials (primarily fibrillar collagen-I), is an abnormal repair reaction and pathological outcome of chronic liver diseases caused by alcohol abuse, non-alcoholic fatty liver disease, and chronic hepatitis B and C virus infections. Liver fibrosis often progresses to liver cirrhosis and hepatocellular carcinoma. Ferroptosis, characterized by lipid peroxidation, is a form of iron-dependent non-apoptotic cell death, and recent studies have reported that ferroptosis contribute to the development of liver fibrosis. Moreover, several agents have demonstrated therapeutic effects in experimental liver fibrosis models by inducing hepatic stellate cell (HSCs) ferroptosis. This review delineates the specific mechanism by which ferroptosis contributes to the development of liver fibrosis. Specifically, we focused on the different types of therapeutic agents that can induce HSCs ferroptosis and summarize their pharmacological effectiveness for liver fibrosis treatment. We suggest that HSCs ferroptosis may be a potential useful target of novel therapies for preventing and treating liver fibrosis.
Liver fibrosis, characterized by diffuse excessive and progressive deposition of the extracellular matrix (ECM) in liver, is a pathological state and abnormal repair reaction to chronic liver injury that is typically caused by long-standing liver damage resulting from infectious (hepatitis B and C viruses, HBV and HCV), metabolic (non-alcoholic fatty liver disease, NAFLD), toxin- or drug-induced, cholestatic, or autoimmune insults (Friedman and Pinzani, 2022). Continued liver fibrosis exponentially increases the risk of liver-related mortality regardless of the cause of liver disease. Liver fibrosis is one of the most important manifestations of chronic liver diseases and is typically implicated in the progression of disease to cirrhosis, which is associated with severe morbidity and mortality as well as higher incidence of hepatocellular carcinoma (Roehlen et al., 2020). Liver fibrosis has become one of the leading global causes of liver disease and is poised to become a leading indication for liver transplantation. What’s more, liver fibrosis can negatively affect the outcomes and prognosis of both adult and pediatric patients with liver transplantation and lead to retransplantation (Berumen et al., 2021). The molecular pathogenetic mechanism of liver fibrosis has not yet been fully elucidated, and to date, no effective anti-fibrotic drugs have become available to treat liver fibrosis (Gilgenkrantz et al., 2021). Therefore, it is of fundamental importance to delineate the molecular mechanisms underlying the development of liver fibrosis to develop effective management and treatment strategies.
In recent years, ferroptosis, iron-dependent non-apoptotic cell death characterized by lipid peroxidation, has been quickly gaining attention in chronic liver disease research due to the predisposition of the liver to oxidative stress injury and excessive iron accumulation (Wu et al., 2021). Accumulating evidence has demonstrated that ferroptosis plays an important role in the pathogenesis of liver fibrosis (Kim et al., 2020; Wu et al., 2021; Pan et al., 2021; Zhou et al., 2022; Chen et al., 2022). Some bioactive compounds have been demonstrated to exert therapeutic effects in experimental liver fibrosis models via pharmacological targeting by inducing HSCs ferroptosis or inhibiting hepatocytes ferroptosis. In the current review, we first delineated the specific mechanism by which ferroptosis contributes to the development of liver fibrosis. Specifically, we focused on the different types of therapeutic agents that can target HSCs or hepatocyte ferroptosis and summarize their pharmacological effectiveness for liver fibrosis treatment. This review is expected to improve our knowledge of the molecular mechanisms of ferroptosis in liver fibrosis and highlight strategies for targeting HSCs or hepatocyte ferroptosis as potential novel therapeutic targets.
Liver fibrosis leads to the accumulation of ECM proteins, mainly Type I and Type III collagens, to form fibrous scar tissues that replace damaged normal tissue and ultimately compromise normal liver function (Friedman, 2003; Kisseleva and Brenner, 2021). Generally, two types of chronic liver injuries, i.e., hepatotoxic and cholestatic injury, cause liver fibrosis (Kisseleva and Brenner, 2021). Hepatotoxic injury is triggered by chronic cellular injury of hepatocytes from external factors, including HBV and HCV infections, alcoholic steatohepatitis (ASH), NAFLD, and non-alcoholic steatohepatitis (NASH) (Bataller and Brenner, 2005). Primary and/or secondary diseases such as primary biliary cholangitis, primary sclerosing cholangitis, and biliary atresia obstruct or reduce bile flow in the liver, thereby cause cholestatic injury (Bataller and Brenner, 2005; Kisseleva and Brenner, 2021; Friedman and Pinzani, 2022). Regardless of cause, liver fibrosis cases share a common molecular mechanism involving epithelial or endothelial barrier disruption, hepatocyte injury and death, chronic inflammation, and HSCs activation (Dhar et al., 2020) (Figure 1).
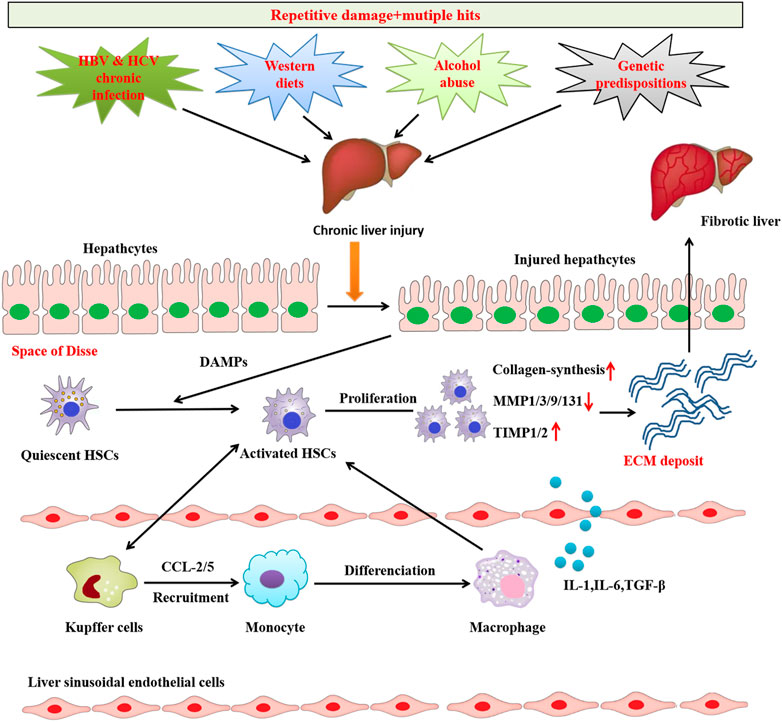
FIGURE 1. Common mechanisms of liver fibrosis. Repetitive damage and mutiple hits causes chronic hepatocyte injury, which causes release of damage-associated patterns (DAMPs) that activate Hepatic stellate cells (HSCs) and recruit immune cells. Activated HSCs are continuously activated and proliferated to secret abundant fibrogenic cytokines and produce excessive ECM, which causes the imbalance of pro-fibrosis/anti-fibrosismechanism.The pro-fibrosis mechanism leads to the abnormal formation of scar and eventually induces liver fibrosis.
The activation of HSCs is recognized a core event in liver fibrosis. Under normal conditions, HSCs containing retinoid lipid droplets are located at the space of Disse and show a quiescent phenotype. Quiescent HSCs function as major vitamin A-storing pericytes (Geerts, 2001; Iredale, 2007; Senoo et al., 2007), which are essential for maintaining the quiescent HSCs phenotype (Hazra et al., 2004; She et al., 2005). Chronic liver injury destroys the hepatocyte membrane and leads to hepatocyte necrosis and apoptosis, thereby causing hepatocyte injury. The injured hepatocyte releases damage-associated molecular patterns (DAMPs), which directly stimulate the transformation from quiescent HSCs to activated HSCs. During activation, quiescent HSCs decrease the expression of glial fibrillar acidic protein, vitamin A, and peroxisome proliferator-activated receptor gamma (PPARγ) and lose lipid droplets. Activated HSCs are equivalent to myofibroblasts that express fibrogenic genes such collagen Type I and alpha-smooth muscle actin (α-SMA) (Fallowfield et al., 2007), eventually leading to liver fibrosis (Gandhi, 2017; Schuppan et al., 2018). The imbalance between ECM synthesis and degradation causes excessive ECM accumulation and deposition in the space of Disse and the formation of hepatic scar tissue (Lackner and Tiniakos, 2019). ECM synthesis and degradation is regulated by the balance between matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs). Changes to the MMP/TIMP balance between ECM protein synthesis and degradation can lead to ECM deposition, resulting in the development of hepatic fibrosis. TIMPs inhibit MMPs that promote ECM degradation (Tacke and Weiskirchen, 2012). The activated HSCs acquire contractile capability, upregulate α-SMA expression, and secrete cytokines including transforming growth factor beta 1 (TGF-β1), platelet derived growth factor (PDGF), and connective tissue growth factor (CTGF) (Tan et al., 2021). The chemokines secreted by activated HSCs accumulate chemotactically, thereby perpetuating and aggravating inflammation and fibrogenesis in the inflammatory compartment. DAMPs are also involved in stimulating the activation of Kupffer cells and other immune cells. These cells further stimulate the activation of HSCs and maintain their survival by secreting pro-inflammatory and pro-fibrotic factors such as TGF-β1, PDGF, interleukin-1 beta (IL-1β), and tumor necrosis factor alpha (TNF-α) to induce inflammation (Tan et al., 2021).
Injury and death to hepatocytes that comprise 80% of the total liver cell population are important initial events in all liver disease etiologies. Dead hepatocytes release DAMPs, which play a vital role in the development of fibrosis and inflammation by transducing danger signals to HSCs and Kupffer cells (Berumen et al., 2021; Wang et al., 2022). In response to injury, hepatocytes alter their gene expression profiles and produce several newly expressed fibrogenic factors, such as osteopontin, TGF-β, NADPH oxidase 4 (NOX4), transcription regulator TAZ (WWTR1), and Indian Hedgehog and Notch (Xie et al., 2013; Lan et al., 2015; Wang et al., 2016; Zhu et al., 2018). Moreover, injured hepatocytes can secrete exosomes containing non-coding RNAs (ncRNAs) that regulate HSCs activation (Lee et al., 2017; Liu et al., 2019; Luo et al., 2021a; Luo et al., 2021b; Luan et al., 2022; Ma et al., 2022). Therefore, developing therapies to protect hepatocytes against damage is considered crucial in treating liver fibrosis.
Studies have increasingly shown that chronic inflammation is a detrimental and critical element of liver fibrosis, as hepatocyte-derived fibrogenic factors alone cannot directly activate HSCs. Many studies have suggested that inflammatory cells such as neutrophils (Gao and Bataller, 2011; Moles et al., 2014; Koyama et al., 2017; Mridha et al., 2017; Gehrke et al., 2018), resident macrophages of liver Kupffer cells (Hellerbrand et al., 1999; Bonniaud et al., 2005; de Gouville et al., 2005), Th17 cells, and bone marrow-derived monocytes (Meng et al., 2012) can promote HSCs activation by releasing cytokines and growth factors. For more detail regarding the role of inflammatory cells and cytokines in liver fibrosis pathogenesis, see previous reviews (Roehlen et al., 2020; Berumen et al., 2021; Kisseleva and Brenner, 2021).
The concept of ferroptosis was first reported by the Stockwell lab in 2012 based on three major research areas, which proposed a foundational understanding of ferroptosis: the mechanisms of lipid and amino acid metabolism (EAGLE, 1955; Mitchell et al., 1973; Bannai et al., 1977), the control of reactive oxygen species (ROS) (Ursini et al., 1982; Flohé, 2020), and the regulation of iron (Stockwell, 2022) (Figure 2). Ferroptosis, which is characterized by the iron-dependent oxidative modification of phospholipid membranes, is a non-apoptotic mode of regulated cell death (RCD) (Dixon et al., 2012). Ferroptosis reflects a delicate imbalance between inducers of ferroptosis and ferroptosis defense systems. When ferroptosis-promoting factors, including iron-dependent ROS and lipid peroxitation (LPO), significantly override the antioxidant defense systems, lipid peroxides lethally accumulate on cellular membranes, which induces membrane rupture and ultimately results in ferroptotic cell death (Lei et al., 2022). Ferroptosis is mainly driven by iron accumulation and LPO causing subsequent plasma membrane rupture (Tang and Kroemer, 2020). The induction of ferroptosis requires two key signals: the accumulation of free iron and the inhibition of the antioxidant SLC7A11-GSH-GPX4 system (Chen et al., 2021). Ferroptosis defense systems include the SLC7A11-reduced glutathione (GSH)-glutathione peroxidase 4 (GPX4), dihydroorotate dehydrogenase -dihydroubiquione (DHODH-CoQH2), ferroptosis suppressor protein 1-ubiquinol (FSP1-CoQH2), and GTP cyclohydrolase 1-tetrahydrobiopterin (GCH1-BH4) systems (Lei et al., 2022). Primary substrates for LPO are phospholipid polyunsaturated fatty acids (PL-PUFAs) due to their intrinsic susceptibility to peroxidation chemistry (Hadian and Stockwell, 2020). PL-PUFAs are generated by different enzymes such as acyl-coenzyme A [CoA] synthetase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase (LPCATs), which incorporate free PUFAs into phospholipids (PL). In non-enzymatic LPO, ACSL4 ligate PUFAs with coenzyme A (CoA) to produce acyl-CoA, which can be re-esterifed in phospholipids through LPCATs to produce PL-PUFAs. Acetyl CoA serve as building blocks for PUFAs synthesis, through the action of acetyl CoA carboxylase (ACC) (Hadian and Stockwell, 2020). Once PL-PUFAs are incorporated into membrane environments, arachidonate lipoxygenase (ALOXs) and cytochrome P450 oxidoreductase (POR), and labile iron use molecular oxygen (O2) to do a peroxidation reaction, resulting in the generation of peroxidated PL-PUFAs(PL-PUFA-OOH) (Hadian and Stockwell, 2020; Zou et al., 2020). This process requires hydrogen peroxide (H2O2) derived from an iron-dependent Fenton reaction or POR and NADPH oxidase (NOX) activation, or mitochondria electron transport chain pathways. During the last step of ferroptosis, LPO or its secondary products such as 4-HNE and MDA cause pore formation in plasma or organelle membranes, which triggers cell death. In recent years, ferroptosis has garnered substantial attention in liver fibrosis research, and there has been an increasing focus on targeting ferroptosis in potential treatment strategies (Wu et al., 2021; Pan et al., 2021).
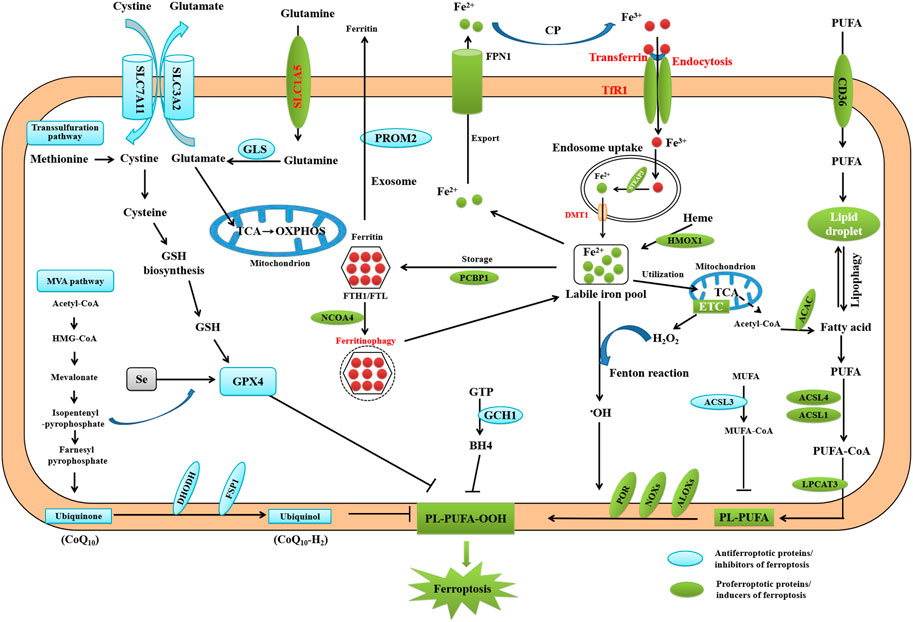
FIGURE 2. Core mechanisms of ferroptosis. Ferroptosis is mainly caused by iron-dependent lipid peroxidation. The initiation of ferroptosis requires two key signals, namely the accumulation of free iron and the inhibition of antioxidant SLC7A11/GPX4 system. The generation of polyunsaturated phospholipid (by ACSL4 and LPCAT3) and subsequent activation of ALOX have a main role in promoting lipid peroxidation. This process requires HO from an iron-mediated Fenton reaction or the activation of POR, NOX, or mitochondria electron transport chain pathways. The lipid peroxidation or its secondary products (e.g., 4-HNE and MDA) induce pore formation in plasma or organelle membrane, which eventually triggers cell death at the final step of ferroptosis. Alternatively, CoQ10 or tetrahydrobiopterin (BH4) inhibits ferroptosis independently of GSH. Importantly, all aspects of iron metabolism, including iron absorption, storage, export, and utilization, have an important regulatory effect on ferroptosis.
Since the first study reporting that the role of ferroptosis in liver fibrosis (Sui et al., 2018), accumulating evidence has shown that ferroptosis is involved in the pathogenesis of liver fibrosis. Numerous studies have demonstrated that ferroptosis exerts an inhibitory effect on liver fibrosis by inactivating HSCs and inducing HSCs death. Moreover, specific regulators of ferroptosis, including ELAV-like RNA binding protein 1 (ELAVL1) (Zhang et al., 2018), bromodomain-containing protein 7 (BRD7) (Zhang et al., 2020a), ZFP36 ring finger protein (ZFP36) (Zhang et al., 2020b), and tripartite motif-containing protein 26 (TRIM26) (Zhu et al., 2021), have been reported to play crucial roles in regulating ferroptosis in HSCs and function as promising targets in preventing liver fibrosis. ELAVL1, a ubiquitous RNA-binding protein, promoted autophagy activation by binding to BECN1/Beclin1 mRNA, thereby enhancing ferroptosis. Ablation of autophagy completely impaired ELAVL1-mediated ferroptosis, whereas induction of autophagy induced a synergistic effect with ELAVL1. In mice, ELAVL1 expression was upregulated by the ferroptosis inducers sorafenib and erastin by inhibiting the ubiquitin–proteasome pathway. In this process, upregulated ELAVL1 binds to BECN1 mRNA and promotes BECN1/Beclin1 generation, resulting in autophagy-dependent degradation of ferritin, HSCs ferroptosis, and attenuated liver fibrosis. Specific knockdown of ELAVL1 in HSCs impaired sorafenib-induced ferroptosis of HSCs in murine liver fibrosis (Zhang et al., 2018). ZFP36,a RNA-binding protein, can destabilize autophagy related 16 like 1 (ATG16L1) mRNA, thus inhibiting macroautophagy/autophagy activation and mediating ferroptosis resistance. The ferroptosis inducer sorafenib was shown to downregulate ZFP36 protein expression by the ubiquitin ligase F-box and WD repeat domain containing 7 (FBXW7/CDC4). Therefore, erastin and sorafenib treatment can ameliorate liver fibrosis by downregulating ZFP36, activating ferritinophagy, and ferroptosis in HSCs (Zhang et al., 2020a). Ferritinophagy is a type of autophagy mediated by nuclear receptor activator 4 (NCOA4), which plays a role in inducing ferroptosis by regulating iron homeostasis and producing ROS in cells (Gao et al., 2016; Hou et al., 2016; Liu et al., 2022). NCOA4 acts as a selective autophagy receptor and binds to ferritin heavy chain-1 (FTH1) of ferritin to mediate the transport of intracellular ferritin to autophagy lysosomes and finally release free iron (Mancias et al., 2014). NCOA4-mediated ferritinophagy degrade ferritin resulting in a feedback regulation mechanism of available iron in cells, and its activation will increase the content of available iron in cells (He et al., 2022). In mice, treatment with erastin and sorafenib alleviated liver fibrosis by inducing HSCs ferroptosis. HSCs-specific overexpression of Zfp36 impaired erastin- or sorafenib-induced HSCs ferroptosis, suggesting that ZFP36 may function as a ferroptosis inhibitor. Another study reported the ability of BRD7 to function as ferroptosis inducer targeting HSCs (Zhang et al., 2020b). BRD7 directly binds to promote p53 mitochondrial translocation, which subsequently forms complexes with and elevates the activity of solute carrier family 25 member 28 (SLC25A28), leading to excessive deposition of redox-active iron and HSCs ferroptosis. Genetically blocking the binding of BRD7 to p53 inhibited mitochondrial translocation of p53 and ferroptosis in HSCs. Silencing SLC25A2 impaired BRD7-or p53-induced HSCs ferroptosis. In vivo, the ferroptosis inducer erastin abrogated liver fibrosis by inducing HSCs ferroptosis. Moreover, HSCs-specific blockade of BRD7/P53/SLC25A28 axis could ameliorate erastin-induced ferroptosis in HSCs. It was shown that erastin and sorafenib can suppress murine liver fibrosis by inducing HSCs ferroptosis via the BRD7/P53/SLC25A28 axis (Zhang et al., 2020a). Moreover, sorafenib was shown to attenuate liver fibrosis by triggering HSCs ferroptosis via hypoxia-inducible factor (HIF)-1α/SLC7A11 signaling in a carbon tetrachloride (CCl4)-induced mouse liver fibrosis model (Yuan et al., 2022). A recent study also demonstrated that TRIM26, an E3 ubiquitin ligament, was downregulated in fibrotic liver tissues. TRIM26 overexpression promoted HSCs ferroptosis by mediating the ubiquitination and degradation of SLC7A11, thereby inhibiting HSCs proliferation (Zhu et al., 2021). Another recent study demonstrated that N6-methyladenosine (m6A) modification enhances HSCs ferroptosis while methylase METTL4 and demethylase FTO competitively stabilized YTHDF1-dependant BECN1 mRNA, thereby activating autophagy and eventually resulting in HSCs ferroptosis (Shen et al., 2021). Taken together, these findings suggest that the pharmacological induction of HSCs ferroptosis may represent a therapeutic target for novel liver fibrosis treatment strategies.
Few studies have reported on the role of hepatocyte ferroptosis in the development of liver fibrosis. The evidence suggests that hepatocyte and HSCs ferroptosis exerts opposite effects. High iron overload, serving as a driving factor in ferroptosis induction, increased the risk of developing liver fibrosis and cirrhosis. Such excessive iron accumulation enhanced ferroptosis in hepatocytes by inducing heme oxygenase 1 (HO-1) expression, which contributed to the progression of liver fibrosis, accompanied by the upregulation of the FGF21 protein level in vitro and in vivo (Wu et al., 2021). During ferroptosis, HO-1 may play a pro-death role in enhancing iron release, thereby being implicated in the initiation of ferroptosis (Ryter, 2021).
In a db/db mouse model of T2DM, increased TGF-β, collagen I, and collagen III levels in hepatic cells indicated greater liver fibrogenesis (Song et al., 2022). Ferroptosis activation was observed in hepatic cells, showing that increased ROS production; SOD, GSH-PX, and GSH downregulation; and MDA, 4-HNE, and NOX4 upregulation increased TfR1 expression, decreased FPN1 expression, and downregulated SLC7A11 and the Nrf2/HO-1/GPX4 signaling pathway (Song et al., 2022). The anti-obesity and diabetes drug liraglutide was also shown to alleviate liver fibrosis by inhibiting ferroptosis (Song et al., 2022). Taken together, these findings suggest that the induction of hepatocyte ferroptosis may be involved in the pathogenesis of T2DM-related liver fibrosis. However, the precise function of hepatocyte ferroptosis in the development and pathogenesis of liver fibrosis remains poorly understood. The molecular mechanism by which hepatocyte ferroptosis is regulated remains largely unknown and warrants further investigation.
Recent studies have demonstrated that the pharmacological scavenging of HSCs by activating ferroptosis has potential as a novel therapeutic strategy for liver fibrosis (Figure 3). Mounting evidence from in vitro and in vivo models indicates that pharmacological induction of ferroptosis may mitigate liver fibrosis development (Table 1). Moreover, there is an increasing body of evidence showing that inducing HSCs ferroptosis using Curcumol (Zheng et al., 2022), ellagic acid (Li et al., 2022), phloridzin (Shi et al., 2022), liraglutide (Song et al., 2022), berberine (Yi et al., 2021), wogonoside (Liu et al., 2022), decursin (Que et al., 2022), celastrol (Luo et al., 2022),isoliquiritigenin (Huang et al., 2022), DHA (Zhang et al., 2021; Shen et al., 2022), wild bitter melon extract (WBME) (Ho et al., 2021),chrysophanol (Kuo et al., 2020), magnesium isoglycyrrhizinate (Sui et al., 2018), artesunate (Kong et al., 2019), and artemether (Wang et al., 2019) has potential as a therapeutic strategy to prevent or inhibit liver fibrosis. Curcumol (Zheng et al., 2022), ellagic acid (Li et al., 2022), wogonoside (Liu et al., 2022), decursin, sorafenib, berberine, celastrol (Luo et al., 2022), isoliquiritigenin (Huang et al., 2022), DHA (Zhang et al., 2021), WBME (Ho et al., 2021), and chrysophanol (Kuo et al., 2020) alleviate liver fibrosis through inducing ferroptosis by inhibiting SLC7A11. Curcumol inhibits GPX4, thereby inducing HSCs ferroptosis to alleviate liver fibrosis (Zheng et al., 2022). Isoliquiritigenin inhibits DHODH and upregulates POR, thereby inducing ferroptosis to alleviate liver fibrosis (Sui et al., 2018). Artesunate (Kong et al., 2019) and DHA (Zhang et al., 2021) induces NCOA4-mediated ferritinophagy to induce ferroptosis and alleviate liver fibrosis. Curcumol (Zheng et al., 2022) and DHA (Zhang et al., 2021) induced ACSL4-dependant ferroptosis to alleviate liver fibrosis. Berberine (Yi et al., 2021) and curcumol (Zheng et al., 2022) alleviate liver fibrosis through inhibiting FTH1/FTL. Ellagic acid (Li et al., 2022) and MgIG (Sui et al., 2018) downregulates FPN1 to induce ferroptosis and alleviate liver fibrosis. MgIG (Sui et al., 2018) upregulates TfR1 and HO-1 to induce ferroptosis and alleviate liver fibrosis (Sui et al., 2018). Celastrol also upregulates HO-1 to induce ferroptosis and alleviate liver fibrosis (Luo et al., 2022).
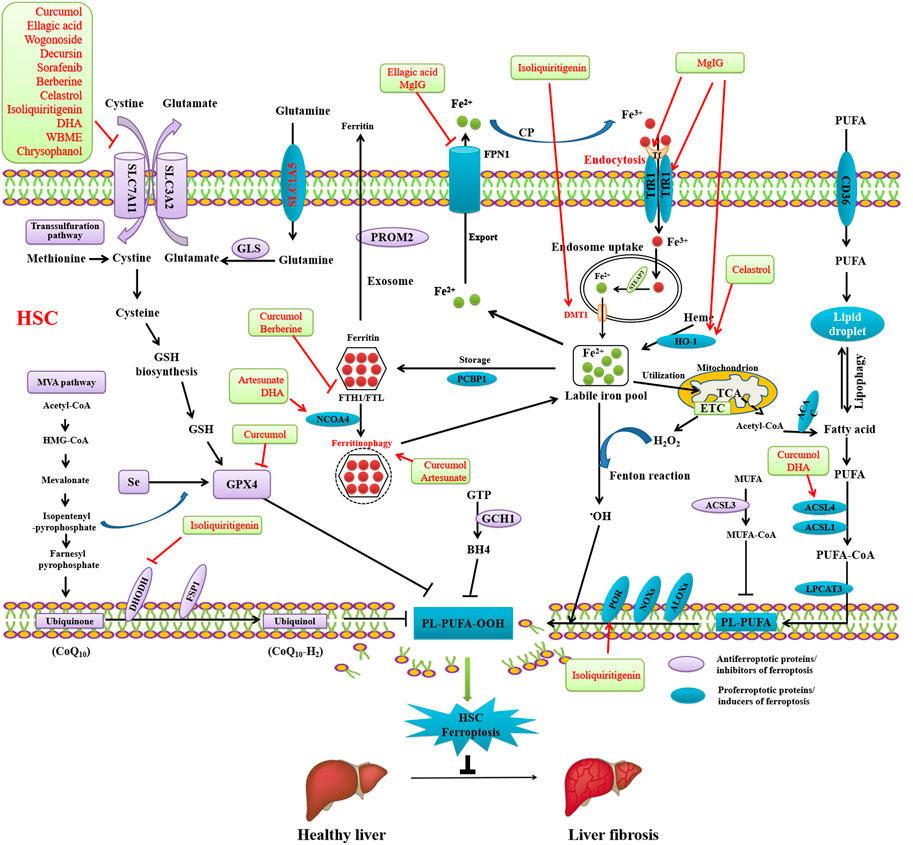
FIGURE 3. Reversing liver fibrosis through inducing ferroptosis by bioactivative compounds in HSCs. Curcumol, ellagic acid, wogonoside, decursin, sorafenib, berberine, celastrol, isoliquiritigenin, DHA, WBME, and chrysophanol alleviate liver fibrosis through inducing ferroptosis by inhibiting SLC7A11. Curcumol inhibits GPX4, thereby inducing HSCs ferroptosis to alleviate liver fibrosis. Isoliquiritigenin inhibits DHODH and upregulates POR, thereby inducing ferroptosis to alleviate liver fibrosis. Artesunate and DHA induces NCOA4-mediated ferritinophagy to induce ferroptosis and alleviate liver fibrosis. Curcumol and DHA induced ACSL4-dependant ferroptosis to alleviate liver fibrosis. Berberine and curcumol alleviate liver fibrosis through inhibiting FTH1/FTL. Ellagic acid and MgIG downregulates FPN1 to induce ferroptosis and alleviate liver fibrosis. MgIG upregulates TfR1 and HO-1 to induce ferroptosis and alleviate liver fibrosis. Celastrol also upregulates HO-1 to induce ferroptosis and alleviate liver fibrosis.
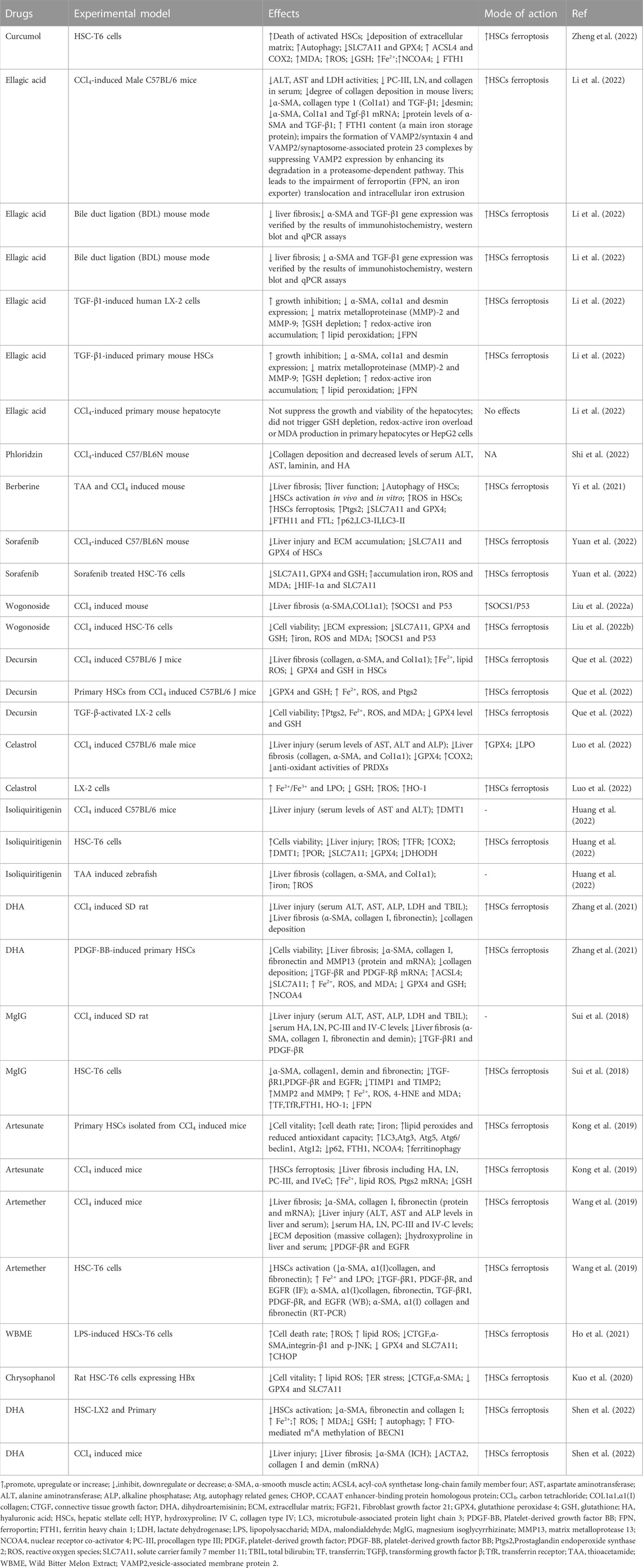
TABLE 1. Compounds alleviate liver fibrosis through inducing ferroptosis in HSCs.
To the best of our knowledge, there have been few reports on the pharmacological inhibition of hepatocyte ferroptosis for treating liver fibrosis (Wu et al., 2021) (Table 2). Recombinant FGF21 significantly protected against liver fibrosis by inhibiting hepatocyte ferroptosis (Wu et al., 2021). FGF21 ablation aggravated iron overload-induced hepatocyte ferroptosis, and FGF21 inhibited of HO-1 expression through promoting its ubiquitination and degradation. These findings indicate that FGF21 functions as a novel ferroptosis suppressor, and its activation in hepatocytes may serve as a potential therapeutic target for liver fibrosis. A recent study demonstrated that liraglutide can alleviate liver fibrosis by inhibiting hepatocyte ferroptosis through upregulating SLC7A11 expression and the Nrf2/HO-1/GPX4 pathway (Song et al., 2022). Liraglutide improved liver function and suppressed liver fibrosis in db/db mice; moreover, it inhibited ROS production and LPO by upregulating SOD, GSH-PX, and GSH activity and downregulating MDA, 4-HNE, and NOX4 expression (Song et al., 2022). Liraglutide was also found to attenuate iron accumulation by decreasing TfR1 expression and increasing FPN1 expression (Song et al., 2022). Together, these results show that liraglutide has potential for inhibiting T2DM-related liver fibrosis by limiting hepatocyte ferroptosis.

TABLE 2. Compounds alleviate liver fibrosis through inhibiting ferroptosis in hepatocytes.
This review summarizes the recent progress of research on the pathological pathways and regulation mechanisms of ferroptosis in liver fibrosis and reviews the application of ferroptosis modulators to mitigate liver fibrosis. Moreover, new targets were identified for future treatment and prevention of liver fibrosis through targeting ferroptosis. Emerging evidence have suggested that pharmacological induction of HSCs ferroptosis or inhibition of hepatocytes ferroptosis both have potential as therapeutic targets for managing or inhibiting liver fibrosis, which elucidates possibilities for the discovery of novel targets and drug strategies for liver fibrosis. However, the current literature on the role of ferroptosis in liver fibrosis remains somewhat limited, and more studies are required to clarify its role and functional mechanisms. Relatively little is known about how ferroptosis orchestrates diverse cellular events. Firstly, the regulatory mechanism underlying the ferroptosis of hepatocytes in liver fibrosis must be identified. Secondly, ferroptosis of different cell types may exert widely variable effects, and strategies aiming to induce ferroptosis in HSCs to inhibit liber fibrosis may aggravate fibrosis by inducing ferroptosis in hepatocytes owing to these differential effects. Therefore, ferroptosis-targeted therapies may have unpredictable effects in some patients with liver fibrosis and lead to side effects. Considering this, ferroptosis-targeted therapies require careful consideration. Possible side effects may be largely alleviated by the development of cell type–targeting drug delivery systems. At present, possible unwarranted toxic adverse events, poor drug targeting, and limited treatment effect among others are the major problems the anti-liver fibrosis drugs should to overcome (Poelstra and Schuppan, 2011; Peng et al., 2021). Current treatment strategies for liver fibrosis are directly targeting to eliminate related injury factors, inhibit the activation of HSCs, increase the degradation of ECM and decrease inflammatory reactions (Ebrahimi et al., 2018; Peng et al., 2021). HSCs are the primary effector cells in liver fibrosis, and most drugs are required to target HSC to treat liver fibrosis. And how to target HSCs has been the main focus of anti-liver fibrosis research in recent years. The nano-drug delivery system is a new and safe drug delivery system with many advantages are now widely used for the treatment of liver fibrosis (Zhou et al., 2022). Numerous nanocarriers have been designed to combat liver fibrosis through modifying the surface receptors of HSCs or specific ligands of highly expressed receptors on the surface of inorganic nanoparticles, liposomes, protein polymers, and other nanodrug delivery systems such as HSCs surface receptors or high expression of receptor specificity ligand (Peng et al., 2021). Targeting nanocarriers to specific liver cell types have the potential to suppress off-target and adverse effects, thereby increasing drugs efficacy (Kumar et al., 2021). Hepatocytes are the primary target of lipotoxicity and oxidative stress in NAFLD and NAFLD-associated liver fibrosis (Kumar et al., 2021). So targeting inhibition of ferroptosis through nanocarriers maybe a ideal strategies to treat NAFLD-associated liver fibrosis with few side effects. Together, cell-specificity reached by cell-specific drug carriers according to different causes that result in liver fibrosis is essential for most anti-liver fibrosis drugs. The future research should continues to develop smart nanocarriers to facilitate a personalized medicine for liver fibrosis. Furthermore, most data reported in the literature are derived from experimental studies that do not directly report clinical applications and implications. Hence, more clinical studies must be conducted to inform practical treatment and management strategies. Despite these considerations, the current evidence strongly indicates that targeting ferroptosis marks a significant new direction for treating liver fibrosis.
Conception and design: all authors; administrative support: all authors; collection and assembly of data: all authors; data analysis and interpretation: all authors; manuscript writing: LL; final approval of manuscript: all authors.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Bannai, S., Tsukeda, H., and Okumura, H. (1977). Effect of antioxidants on cultured human diploid fibroblasts exposed to cystine-free medium. Biochem. Biophys. Res. Commun. 74, 1582–1588. doi:10.1016/0006-291x(77)90623-4
Bataller, R., and Brenner, D. A. (2005). Liver fibrosis. J. Clin. Invest. 115, 209–218. doi:10.1172/JCI24282
Berumen, J., Baglieri, J., Kisseleva, T., and Mekeel, K. (2021). Liver fibrosis: Pathophysiology and clinical implications. WIREs Mech. Dis. 13, e1499. doi:10.1002/wsbm.1499
Bonniaud, P., Margetts, P. J., Ask, K., Flanders, K., Gauldie, J., and Kolb, M. (2005). TGF-beta and Smad3 signaling link inflammation to chronic fibrogenesis. J. Immunol. 175. 5390–5395. doi:10.4049/jimmunol.175.8.5390
Chen, J., Li, X., Ge, C., Min, J., and Wang, F. (2022). The multifaceted role of ferroptosis in liver disease. Cell. Death Differ. 29, 467–480. doi:10.1038/s41418-022-00941-0
Chen, X., Kang, R., Kroemer, G., and Tang, D. (2021). Ferroptosis in infection, inflammation, and immunity. J. Exp. Med. 218, e20210518. doi:10.1084/jem.20210518
de Gouville, A. C., Boullay, V., Krysa, G., Pilot, J., Brusq, J. M., Loriolle, F., et al. (2005). Inhibition of TGF-beta signaling by an ALK5 inhibitor protects rats from dimethylnitrosamine-induced liver fibrosis. Br. J. Pharmacol. 145, 166–177. doi:10.1038/sj.bjp.0706172
Dhar, D., Baglieri, J., Kisseleva, T., and Brenner, D. A. (2020). Mechanisms of liver fibrosis and its role in liver cancer. Exp. Biol. Med. (Maywood, N.J.) 245, 96–108. doi:10.1177/1535370219898141
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., et al. (2012). Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell. 149, 1060–1072. doi:10.1016/j.cell.2012.03.042
Eagle, H. (1955). Nutrition needs of mammalian cells in tissue culture. Science 122, 501–514. doi:10.1126/science.122.3168.501
Ebrahimi, H., Naderian, M., and Sohrabpour, A. A. (2018). New concepts on reversibility and targeting of liver fibrosis; A review article. Middle East J. Dig. Dis. 10 (3), 133–148. doi:10.15171/mejdd.2018.103
Fallowfield, J. A., Mizuno, M., Kendall, T. J., Constandinou, C. M., Benyon, R. C., Duffield, J. S., et al. (2007). Scar-associated macrophages are a major source of hepatic matrix metalloproteinase-13 and facilitate the resolution of murine hepatic fibrosis. J. Immunol. 178. 5288–5295. doi:10.4049/jimmunol.178.8.5288
Flohé, L. (2020). Looking back at the early stages of redox biology. Antioxidants (Basel) 9, 1254. doi:10.3390/antiox9121254
Friedman, S. L. (2003). Liver fibrosis - from bench to bedside. J. Hepatol. 38 (1), S38–S53. doi:10.1016/s0168-8278(02)00429-4
Friedman, S. L., and Pinzani, M. (2022). Hepatic fibrosis 2022: Unmet needs and a blueprint for the future. Hepatology 75, 473–488. doi:10.1002/hep.32285
Gandhi, C. R. (2017). Hepatic stellate cell activation and pro-fibrogenic signals. J. Hepatol. 67, 1104–1105. doi:10.1016/j.jhep.2017.06.001
Gao, B., and Bataller, R. (2011). Alcoholic liver disease: Pathogenesis and new therapeutic targets. Gastroenterology 141, 1572–1585. doi:10.1053/j.gastro.2011.09.002
Gao, M., Monian, P., Pan, Q., Zhang, W., Xiang, J., and Jiang, X. (2016). Ferroptosis is an autophagic cell death process. Cell. Res. 26, 1021–1032. doi:10.1038/cr.2016.95
Geerts, A. (2001). History, heterogeneity, developmental biology, and functions of quiescent hepatic stellate cells. Semin. Liver Dis. 21, 311–335. doi:10.1055/s-2001-17550
Gehrke, N., Nagel, M., Straub, B. K., Wörns, M. A., Schuchmann, M., Galle, P. R., et al. (2018). Loss of cellular FLICE-inhibitory protein promotes acute cholestatic liver injury and inflammation from bile duct ligation. Am. J. Physiol. Gastrointest. Liver Physiol. 314, G319–G333. doi:10.1152/ajpgi.00097.2017
Gilgenkrantz, H., Mallat, A., Moreau, R., and Lotersztajn, S. (2021). Targeting cell-intrinsic metabolism for antifibrotic therapy. J. Hepatol. 74, 1442–1454. doi:10.1016/j.jhep.2021.02.012
Hadian, K., and Stockwell, B. R. (2020). SnapShot: Ferroptosis. Cell. 181 (5), 1188–1188.e1. doi:10.1016/j.cell.2020.04.039
Hazra, S., Xiong, S., Wang, J., Rippe, R. A., Krishna, V., Chatterjee, K., et al. (2004). Peroxisome proliferator-activated receptor gamma induces a phenotypic switch from activated to quiescent hepatic stellate cells. J. Biol. Chem. 279, 11392–11401. doi:10.1074/jbc.M310284200
He, J., Li, Z., Xia, P., Shi, A., FuChen, X., Zhang, J., et al. (2022). Ferroptosis and ferritinophagy in diabetes complications. Mol. Metab. 60, 101470. doi:10.1016/j.molmet.2022.101470
Hellerbrand, C., Stefanovic, B., Giordano, F., Burchardt, E. R., and Brenner, D. A. (1999). The role of TGFbeta1 in initiating hepatic stellate cell activation in vivo. J. Hepatol. 30, 77–87. doi:10.1016/s0168-8278(99)80010-5
Ho, C. H., Huang, J. H., Sun, M. S., Tzeng, I. S., Hsu, Y. C., and Kuo, C. Y. (2021). Wild bitter melon extract regulates LPS-induced hepatic stellate cell activation, inflammation, endoplasmic reticulum stress, and ferroptosis. Evid. Based Complement. Altern. Med. 2021, 6671129. doi:10.1155/2021/6671129
Hou, W., Xie, Y., Song, X., Sun, X., Lotze, M. T., Zeh, H. J., et al. (2016). Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 12, 1425–1428. doi:10.1080/15548627.2016.1187366
Huang, S., Wang, Y., Xie, S., Lai, Y., Mo, C., Zeng, T., et al. (2022). Isoliquiritigenin alleviates liver fibrosis through caveolin-1-mediated hepatic stellate cells ferroptosis in zebrafish and mice. Phytomedicine 101, 154117. doi:10.1016/j.phymed.2022.154117
Iredale, J. P. (2007). Models of liver fibrosis: Exploring the dynamic nature of inflammation and repair in a solid organ. J. Clin. Invest. 117, 539–548. doi:10.1172/JCI30542
Kim, K. M., Cho, S. S., and Ki, S. H. (2020). Emerging roles of ferroptosis in liver pathophysiology. Arch. Pharm. Res. 43, 985–996. doi:10.1007/s12272-020-01273-8
Kisseleva, T., and Brenner, D. (2021). Molecular and cellular mechanisms of liver fibrosis and its regression. Nat. Rev. Gastroenterol. Hepatol. 18, 151–166. doi:10.1038/s41575-020-00372-7
Kong, Z., Liu, R., and Cheng, Y. (2019). Artesunate alleviates liver fibrosis by regulating ferroptosis signaling pathway. Biomed. Pharmacother. 109, 2043–2053. doi:10.1016/j.biopha.2018.11.030
Koyama, Y., Wang, P., Liang, S., Iwaisako, K., Liu, X., Xu, J., et al. (2017). Mesothelin/mucin 16 signaling in activated portal fibroblasts regulates cholestatic liver fibrosis. J. Clin. Invest. 127, 1254–1270. doi:10.1172/JCI88845
Kumar, S., Duan, Q., Wu, R., Harris, E. N., and Su, Q. (2021a). Pathophysiological communication between hepatocytes and non-parenchymal cells in liver injury from NAFLD to liver fibrosis. Adv. Drug Deliv. Rev. 176, 113869. doi:10.1016/j.addr.2021.113869
Kumar, V., Xin, X., Ma, J., Tan, C., Osna, N., and Mahato, R. I. (2021b). Therapeutic targets, novel drugs, and delivery systems for diabetes associated NAFLD and liver fibrosis. Adv. Drug Deliv. Rev. 176, 113888. doi:10.1016/j.addr.2021.113888
Kuo, C. Y., Chiu, V., Hsieh, P. C., Huang, C. Y., Huang, S. J., Tzeng, I. S., et al. (2020). Chrysophanol attenuates Hepatitis B virus X protein-induced hepatic stellate cell fibrosis by regulating endoplasmic reticulum stress and ferroptosis. J. Pharmacol. Sci. 144 (3), 172–182. doi:10.1016/j.jphs.2020.07.014
Lackner, C., and Tiniakos, D. (2019). Fibrosis and alcohol-related liver disease. J. Hepatol. 70, 294–304. doi:10.1016/j.jhep.2018.12.003
Lan, T., Kisseleva, T., and Brenner, D. A. (2015). Deficiency of NOX1 or NOX4 prevents liver inflammation and fibrosis in mice through inhibition of hepatic stellate cell activation. PLoS ONE 10, e0129743. doi:10.1371/journal.pone.0129743
Lee, Y. S., Kim, S. Y., Ko, E., Lee, J. H., Yi, H. S., Yoo, Y. J., et al. (2017). Exosomes derived from palmitic acid-treated hepatocytes induce fibrotic activation of hepatic stellate cells. Sci. Rep. 7, 3710. doi:10.1038/s41598-017-03389-2
Lei, G., Zhuang, L., and Gan, B. (2022). Targeting ferroptosis as a vulnerability in cancer. Nat. Rev. Cancer 22, 381–396. doi:10.1038/s41568-022-00459-0
Li, L., Wang, K., Jia, R., Xie, J., Ma, L., Hao, Z., et al. (2022). Ferroportin-dependent ferroptosis induced by ellagic acid retards liver fibrosis by impairing the SNARE complexes formation. Redox Biol. 56, 102435. doi:10.1016/j.redox.2022.102435
Liu, G., Wei, C., Yuan, S., Zhang, Z., Li, J., Zhang, L., et al. (2022a). Wogonoside attenuates liver fibrosis by triggering hepatic stellate cell ferroptosis through SOCS1/P53/SLC7A11 pathway. Phytother. Res. 36, 4230–4243. doi:10.1002/ptr.7558
Liu, M. Z., Kong, N., Zhang, G. Y., Xu, Q., Xu, Y., Ke, P., et al. (2022b). The critical role of ferritinophagy in human disease. Front. Pharmacol. 13, 933732. doi:10.3389/fphar.2022.933732
Liu, R., Li, X., Zhu, W., Wang, Y., Zhao, D., Wang, X., et al. (2019). Cholangiocyte-derived exosomal long noncoding RNA H19 promotes hepatic stellate cell activation and cholestatic liver fibrosis. Hepatology 70, 1317–1335. doi:10.1002/hep.30662
Luan, S. H., Yang, Y. Q., Ye, M. P., Liu, H., Rao, Q. F., Kong, J. L., et al. (2022). ASIC1a promotes hepatic stellate cell activation through the exosomal miR-301a-3p/BTG1 pathway. Int. J. Biol. Macromol. 211, 128–139. doi:10.1016/j.ijbiomac.2022.05.041
Luo, P., Liu, D., Zhang, Q., Yang, F., Wong, Y. K., Xia, F., et al. (2022). Celastrol induces ferroptosis in activated HSCs to ameliorate hepatic fibrosis via targeting peroxiredoxins and HO-1. Acta Pharm. Sin. B 12 (5), 2300–2314. doi:10.1016/j.apsb.2021.12.007
Luo, X., Luo, S. Z., Xu, Z. X., Zhou, C., Li, Z. H., Zhou, X. Y., et al. (2021a). Lipotoxic hepatocyte-derived exosomal miR-1297 promotes hepatic stellate cell activation through the PTEN signaling pathway in metabolic-associated fatty liver disease. World J. Gastroenterol. 27, 1419–1434. doi:10.3748/wjg.v27.i14.1419
Luo, X., Xu, Z. X., Wu, J. C., Luo, S. Z., and Xu, M. Y. (2021b). Hepatocyte-derived exosomal miR-27a activateshepatic stellate cells through the inhibitionof PINK1-mediated mitophagy in MAFLD. Mol. Ther. Nucleic Acids 26, 1241–1254. doi:10.1016/j.omtn.2021.10.022
Ma, L., Wei, J., Zeng, Y., Liu, J., Xiao, E., Kang, Y., et al. (2022). Mesenchymal stem cell-originated exosomal circDIDO1 suppresses hepatic stellate cell activation by miR-141-3p/PTEN/AKT pathway in human liver fibrosis. Drug Deliv. 29, 440–453. doi:10.1080/10717544.2022.2030428
Mancias, J. D., Wang, X., Gygi, S. P., Harper, J. W., and Kimmelman, A. C. (2014). Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 509, 105–109. doi:10.1038/nature13148
Meng, F., Wang, K., Aoyama, T., Grivennikov, S. I., Paik, Y., Scholten, D., et al. (2012). Interleukin-17 signaling in inflammatory, Kupffer cells, and hepatic stellate cells exacerbates liver fibrosis in mice. Gastroenterology 143, 765–776. e3. doi:10.1053/j.gastro.2012.05.049
Mitchell, J. R., Jollow, D. J., Potter, W. Z., Gillette, J. R., and Brodie, B. B. (1973). Acetaminophen-induced hepatic necrosis. IV. Protective role of glutathione. J. Pharmacol. Exp. Ther. 187, 211–217.
Moles, A., Murphy, L., Wilson, C. L., Chakraborty, J. B., Fox, C., Park, E. J., et al. (2014). A TLR2/S100A9/CXCL-2 signaling network is necessary for neutrophil recruitment in acute and chronic liver injury in the mouse. J. Hepatol. 60, 782–791. doi:10.1016/j.jhep.2013.12.005
Mridha, A. R., Wree, A., Robertson, A., Yeh, M. M., Johnson, C. D., Van Rooyen, D. M., et al. (2017). NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 66, 1037–1046. doi:10.1016/j.jhep.2017.01.022
Pan, Q., Luo, Y., Xia, Q., and He, K. (2021). Ferroptosis and liver fibrosis. Int. J. Med. Sci. 18, 3361–3366. doi:10.7150/ijms.62903
Peng, W., Cheng, S., Bao, Z., Wang, Y., Zhou, W., Wang, J., et al. (2021). Advances in the research of nanodrug delivery system for targeted treatment of liver fibrosis. Biomed. Pharmacother. 137, 111342. doi:10.1016/j.biopha.2021.111342
Poelstra, K., and Schuppan, D. (2011). Targeted therapy of liver fibrosis/cirrhosis and its complications. J. Hepatol. 55, 726–728. doi:10.1016/j.jhep.2011.04.008
Que, R., Cao, M., Dai, Y., Zhou, Y., Chen, Y., and Lin, L. (2022). Decursin ameliorates carbon tetrachloride-induced liver fibrosis by facilitating ferroptosis of hepatic stellate cells. Biochem. Cell. Biol. 100, 378–386. doi:10.1139/bcb-2022-0027
Roehlen, N., Crouchet, E., and Baumert, T. F. (2020). Liver fibrosis: Mechanistic concepts and therapeutic perspectives. Cells 9, 875. doi:10.3390/cells9040875
Ryter, S. W. (2021). Heme oxgenase-1, a cardinal modulator of regulated cell death and inflammation. Cells 10, 515. doi:10.3390/cells10030515
Schuppan, D., Surabattula, R., and Wang, X. Y. (2018). Determinants of fibrosis progression and regression in NASH. J. Hepatol. 68, 238–250. doi:10.1016/j.jhep.2017.11.012
Senoo, H., Kojima, N., and Sato, M. (2007). Vitamin A-storing cells (stellate cells). Vitam. Horm. 75, 131–159. doi:10.1016/S0083-6729(06)75006-3
She, H., Xiong, S., Hazra, S., and Tsukamoto, H. (2005). Adipogenic transcriptional regulation of hepatic stellate cells. J. Biol. Chem. 280, 4959–4967. doi:10.1074/jbc.M410078200
Shen, M., Guo, M., Li, Y., Wang, Y., Qiu, Y., Shao, J., et al. (2022). m(6 A methylation is required for dihydroartemisinin to alleviate liver fibrosis by inducing ferroptosis in hepatic stellate cells. Free Radic. Biol. Med. 182, 246–259. doi:10.1016/j.freeradbiomed.2022.02.028
Shen, M., Li, Y., Wang, Y., Shao, J., Zhang, F., Yin, G., et al. (2021). N6-methyladenosine modification regulates ferroptosis through autophagy signaling pathway in hepatic stellate cells. Redox Biol. 47, 102151. doi:10.1016/j.redox.2021.102151
Shi, Y., Yan, T., Lu, X., Li, K., Nie, Y., Jiao, C., et al. (2022). Phloridzin reveals new treatment strategies for liver fibrosis. Pharm. (Basel) 15 (7), 896. doi:10.3390/ph15070896
Song, J. X., An, J. R., Chen, Q., Yang, X. Y., Jia, C. L., Xu, S., et al. (2022). Liraglutide attenuates hepatic iron levels and ferroptosis in db/db mice. Bioengineered 13 (4), 8334–8348. doi:10.1080/21655979.2022.2051858
Stockwell, B. R. (2022). Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell. 185, 2401–2421. doi:10.1016/j.cell.2022.06.003
Sui, M., Jiang, X., Chen, J., Yang, H., and Zhu, Y. (2018). Magnesium isoglycyrrhizinate ameliorates liver fibrosis and hepatic stellate cell activation by regulating ferroptosis signaling pathway. Biomed. Pharmacother. = Biomedecine Pharmacother. 106, 125–133. doi:10.1016/j.biopha.2018.06.060
Tacke, F., and Weiskirchen, R. (2012). Update on hepatic stellate cells: Pathogenic role in liver fibrosis and novel isolation techniques. Expert Rev. Gastroenterol. Hepatol. 6, 67–80. doi:10.1586/egh.11.92
Tan, Z., Sun, H., Xue, T., Gan, C., Liu, H., Xie, Y., et al. (2021). Liver fibrosis: Therapeutic targets and advances in drug therapy. Front. Cell. Dev. Biol. 9, 730176. doi:10.3389/fcell.2021.730176
Tang, D., and Kroemer, G. (2020). Ferroptosis. Curr. Biol. 30, R1292–R1297. doi:10.1016/j.cub.2020.09.068
Ursini, F., Maiorino, M., Valente, M., Ferri, L., and Gregolin, C. (1982). Purification from pig liver of a protein which protects liposomes and biomembranes from peroxidative degradation and exhibits glutathione peroxidase activity on phosphatidylcholine hydroperoxides. Biochim. Biophys. Acta 710, 197–211. doi:10.1016/0005-2760(82)90150-3
Wang, F. D., Zhou, J., and Chen, E. Q. (2022). Molecular mechanisms and potential new therapeutic drugs for liver fibrosis. Front. Pharmacol. 13, 787748. doi:10.3389/fphar.2022.787748
Wang, L., Zhang, Z., Li, M., Wang, F., Jia, Y., Zhang, F., et al. (2019). P53-dependent induction of ferroptosis is required for artemether to alleviate carbon tetrachloride-induced liver fibrosis and hepatic stellate cell activation. IUBMB Life 71 (1), 45–56. doi:10.1002/iub.1895
Wang, X., Zheng, Z., Caviglia, J. M., Corey, K. E., Herfel, T. M., Cai, B., et al. (2016). Hepatocyte TAZ/WWTR1 promotes inflammation and fibrosis in nonalcoholic steatohepatitis. Cell. Metab. 24, 848–862. doi:10.1016/j.cmet.2016.09.016
Wu, A., Feng, B., Yu, J., Yan, L., Che, L., Zhuo, Y., et al. (2021a). Fibroblast growth factor 21 attenuates iron overload-induced liver injury and fibrosis by inhibiting ferroptosis. Redox Biol. 46, 102131. doi:10.1016/j.redox.2021.102131
Wu, J., Wang, Y., Jiang, R., Xue, R., Yin, X., Wu, M., et al. (2021b). Ferroptosis in liver disease: New insights into disease mechanisms. Cell. Death Discov. 7, 276. doi:10.1038/s41420-021-00660-4
Xie, G., Karaca, G., Swiderska-Syn, M., Michelotti, G. A., Krüger, L., Chen, Y., et al. (2013). Cross-talk between Notch and Hedgehog regulates hepatic stellate cell fate in mice. Hepatology 58, 1801–1813. doi:10.1002/hep.26511
Yi, J., Wu, S., Tan, S., Qin, Y., Wang, X., Jiang, J., et al. (2021). Berberine alleviates liver fibrosis through inducing ferrous redox to activate ROS-mediated hepatic stellate cells ferroptosis. Cell. Death Discov. 7 (1), 374. doi:10.1038/s41420-021-00768-7
Yuan, S., Wei, C., Liu, G., Zhang, L., Li, J., Li, L., et al. (2022). Sorafenib attenuates liver fibrosis by triggering hepatic stellate cell ferroptosis via HIF-1α/SLC7A11 pathway. Cell. Prolif. 55, e13158. doi:10.1111/cpr.13158
Zhang, Z., Wang, X., Wang, Z., Cao, Y., and Wei, Z. (2021). Dihydroartemisinin alleviates hepatic fibrosis through inducing ferroptosis in hepatic stellate cells. Biofactors 47 (5), 801–818. doi:10.1002/biof.1764
Zhang, Z., Guo, M., Li, Y., Shen, M., Kong, D., Shao, J., et al. (2020a). RNA-binding protein ZFP36/TTP protects against ferroptosis by regulating autophagy signaling pathway in hepatic stellate cells. Autophagy 16, 1482–1505. doi:10.1080/15548627.2019.1687985
Zhang, Z., Guo, M., Shen, M., Kong, D., Zhang, F., Shao, J., et al. (2020b). The BRD7-P53-SLC25A28 axis regulates ferroptosis in hepatic stellate cells. Redox Biol. 36, 101619. doi:10.1016/j.redox.2020.101619
Zhang, Z., Yao, Z., Wang, L., Ding, H., Shao, J., Chen, A., et al. (2018). Activation of ferritinophagy is required for the RNA-binding protein ELAVL1/HuR to regulate ferroptosis in hepatic stellate cells. Autophagy 14, 2083–2103. doi:10.1080/15548627.2018.1503146
Zheng, Y., Zhao, T., Wang, J., Jiang, R., Huang, J., Li, W., et al. (2022). Curcumol alleviates liver fibrosis through inducing autophagy and ferroptosis in hepatic stellate cells. FASEB J. 36, e22665. doi:10.1096/fj.202200933RR
Zhou, L., Li, Y., Liang, Q., Liu, J., and Liu, Y. (2022a). Combination therapy based on targeted nano drug co-delivery systems for liver fibrosis treatment: A review. J. Drug Target 30, 577–588. doi:10.1080/1061186X.2022.2044485
Zhou, X., Fu, Y., Liu, W., Mu, Y., Zhang, H., Chen, J., et al. (2022b). Ferroptosis in chronic liver diseases: Opportunities and challenges. Front. Mol. Biosci. 9, 928321. doi:10.3389/fmolb.2022.928321
Zhu, C., Kim, K., Wang, X., Bartolome, A., Salomao, M., Dongiovanni, P., et al. (2018). Hepatocyte Notch activation induces liver fibrosis in nonalcoholic steatohepatitis. Sci. Transl. Med. 10, eaat0344. doi:10.1126/scitranslmed.aat0344
Zhu, Y., Zhang, C., Huang, M., Lin, J., Fan, X., and Ni, T. (2021). TRIM26 induces ferroptosis to inhibit hepatic stellate cell activation and mitigate liver fibrosis through mediating SLC7A11 ubiquitination. Front. Cell. Dev. Biol. 9, 644901. doi:10.3389/fcell.2021.644901
Keywords: liver fibrosis, liver transplantation, ferroptosis, ferroptosis inducer, treatment, bioactive compounds
Citation: Li L and Zhu Z (2023) Pharmacological modulation of ferroptosis as a therapeutic target for liver fibrosis. Front. Pharmacol. 13:1071844. doi: 10.3389/fphar.2022.1071844
Received: 17 October 2022; Accepted: 29 December 2022;
Published: 10 January 2023.
Edited by:
Chao Mao, University of Texas MD Anderson Cancer Center, United StatesReviewed by:
Marta Bento Afonso, Research Institute for Medicines (iMed.ULisboa), PortugalCopyright © 2023 Li and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhijun Zhu, emh1LXpoaWp1bkBvdXRsb29rLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.