 Rui Wang
Rui Wang Yong Fen Qi
Yong Fen Qi Chun Hua Ding
Chun Hua Ding Da Zhang
Da Zhang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Pharmacol. , 04 November 2022
Sec. Cardiovascular and Smooth Muscle Pharmacology
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.1063241
This article is part of the Research Topic Gaseous Signalling Molecules: Cardiovascular Significance and Molecular Mechanisms View all 5 articles
Atrial fibrosis, a symbol of atrial structural remodelling, is a complex process involved in the occurrence and maintenance of atrial fibrillation (AF). Atrial fibrosis is regulated by multiple factors. Sulfur containing amino acids and their metabolites, such as hydrogen sulfide (H2S) and taurine, can inhibit the process of atrial fibrosis and alleviate atrial remodeling. However, homocysteine can promote the activation of atrial fibroblasts and further promote atrial fibrosis. In this review, we will focus on the recent progress in atrial structural changes and molecular mechanisms of atrial fibrosis, as well as the regulatory roles and possible mechanisms of sulfur containing amino acids and their metabolites in atrial fibrosis. It is expected to provide new ideas for clarifying the mechanism of atrial fibrosis and finding targets to inhibit the progress of atrial fibrosis.
It is estimated that the prevalence of atrial fibrillation (AF) in adults is between 2% and 4%, and a 2.3-fold rise is expected (Hindricks et al., 2021). As a major risk factor of cardiovascular events, stroke and sudden death, AF brings heavy economic and medical burden to individuals and society (Christophersen et al., 2017). Atrial fibrosis is the main pathophysiological factor for the occurrence and maintenance of AF (Sohns and Marrouche, 2020). Clinical studies indicate that AF is mainly due to atrial structural remodeling (van den Berg et al., 2021), atrial structural remodeling have been found even before the occurrence of AF in patients. Inhibition of atrial structural remodeling caused by atrial fibrosis is the key direction to inhibit the occurrence and further development of AF. Exploring the mechanisms and intervention strategies of atrial fibrosis can delay the progress of fibrosis, and then reduce the structural and electrical remodeling of the atrium, playing a pivotal role in the treatment of AF.
Many kinds of sulfur-containing amino acids and their metabolites are responsible for cardiovascular disease. Changed plasma sulfur-amino acid level are found in cardiovascular diseases, indicating the importance of proper regulation of thio metabolism for cardiovascular homeostasis (Mistry and Brewer, 2019). For example, H2S, the sulf-metabolite of methionine, plays a biological protective role such as reducing extravascular collagen deposition, inhibiting cardiovascular inflammatory reaction, relaxing blood vessels, attenuating cardiomyocyte apoptosis and inhibiting vascular smooth muscle cell proliferation (Zhang et al., 2019). In recent years, there is growing focus on the role of sulfur-containing amino acids and their metabolites in anti-atrial fibrosis. In this review, we will discuss the formation of atrial fibrosis, the metabolism of sulfur-containing amino acids and their role in the formation and progression of atrial fibrosis, and provided new insights into potential clinical applications and interventions.
Atrial fibroblasts are the major effector cells of atrial fibrosis, which would be activated, proliferated and transdifferentiated into myofibroblast during fibrosis (Krishnan et al., 2021; Li et al., 2021). The myofibroblasts showed greater mobile and contractile and produced a large amount of extracellular matrix (Frangogiannis, 2019, 2021). Under physiological conditions, atrial resident fibroblasts would rest and produce less extracellular matrix (Souders et al., 2009). However, when atrium was stimulated by mechanical injury, pressure overload, electrical stimulation and other stimulators, extracellular matrix would be abnormally deposited in tissues, leading to atrial fibrosis. The extracellular matrix is mainly composed of collagen I and collagen III (Li et al., 2018). Collagen I is the main structural component of cardiac interstitium, which is similar to fibers and mainly forms thick and hard rod-shaped structures around the outer membrane and muscle. In contrast, collagen III forms thinner and more flexible reticular fibers, which is more abundant in endocardium, thus increasing the elasticity of heart tissue (Frangogiannis, 2019). When atrial fibrosis was activated, the synthesis/degradation of atrial collagen I and collagen III proteins changed seriously, which lead to atrial remodeling. This pathophysiological process, which characterized by abnormal collagen metabolism, is related to the disorder of matrix metalloproteinase/tissue metalloproteinase inhibitor (MMP/TIMP) system and the increase of Kazal motif (RECK) level. In addition, previous studies showed that the expression of non fibrous collagen VI was up-regulated in patients with AF during the process (Polyakova et al., 2008). Although non fibrous collagen did not form large fiber bundles, they could still interact with collagen I and collagen III, which promoted the process of atrial fibrosis (Polyakova et al., 2008) (Figure 1).
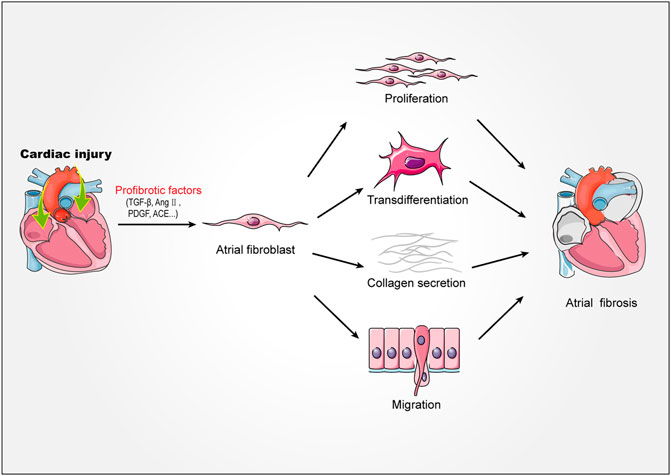
FIGURE 1. Mechanism of atrial fibrosis. Stimulated by injury factors TGF-β, AngⅡ, PDGF, ACE, etc. Atrial fibroblasts proliferate, migrate, transdifferentiate, secrete collagen fibers, and ultimately promote the formation of atrial fibrosis. TGF-β, transforming growth factor-β; AngⅡ, angiotensin II; PDGF, platelet derived growth factor; ACE, angiotensin-converting enzyme.
Atrial fibrosis was divided into reactive fibrosis and repair fibrosis (Krishnan et al., 2021; S. Nattel, 2017). Atrial reactive fibrosis was considered a response fibrous tissue to cardiac inflammation or pressure overload. When the atrium initiated reactive fibrosis, the thin-fibrous tissue which layer around the myocardial bundle changed into thick sheath. By isolating muscle bundles, myocardial interstitial fibrosis could accelerate conduction. The presence of thick interstitial collagen strands could be associated with more rapid longitudinal conduction and persistent AF in patients (Krul et al., 2015). When the damage or death of myocardial caused by various factors occured, the repair fibrosis would be initiated, such as myocardial infarction (MI) scar. This type of fibrosis might be irreversible to a large extent. Different types of fibrosis would not necessarily mutually exclusive, and patients with atrial remodeling were likely to combine reactive fibrosis and repair fibrosis (Nattel et al., 2017).
Atrial remodeling associated with atrial fibrosis which could be divided into structural remodeling and electrophysiological remodeling (Pellman et al., 2010). When the atrial fibrosis process get started, the structural and electrical remodeling will be activated, such as inducing left atrial dysfunction (LADS) and increasing susceptibility to AF (Hu et al., 2021). Clinical studies indicated that the persistence AF was mainly attributed to structural remodeling (Yang et al., 2017; Beyer et al., 2021). In fact, cardiac fibroblasts are non excitable cells essentially, but they can transfer electric current between cardiac fibroblasts and myocardial cells through connexin (Andrade et al., 2014). The continuous accumulation of fibrin would lead to permanent tissue remodeling and organ damage. In the atrium, the continuous accumulation of fibrin will directly affect the mechanical function and electrical activity of the myocardium, leading to abnormal cardiac systolic and diastolic functions. These pathological processes change the electrophysiological characteristics of the original myocardium, leading to abnormal atrial electrical conduction and reentry of the atrial loop, thus directly or indirectly leading to arrhythmia, and thus leading to the occurrence and progress of AF (Pellman et al., 2010; Afzal et al., 2017). Therefore, prevention of atrial remodeling which related to atrial fibrosis is the key to prevent AF.
At present, several common mechanisms of promoting fibrosis have been identified. The classic Smad dependent pathway and non classic Smad independent pathway activated downstream of transforming growth factor (TGF-β) (Biernacka et al., 2011; Saadat et al., 2020), the activation of renin angiotensin aldosterone system (RAAS) (AlQudah et al., 2020), oxidative stress, and the activation of PDGF-JAK-STAT pathway mediated by platelet derived growth factor (PDGF) have been shown to be closely related to cardiac fibrosis (Everett and Olgin, 2007; Li et al., 2021). By reviewing the literature, we found that under different pathophysiological conditions, atrial and ventricular might exhibit different levels of fibrosis (Nakajima et al., 2000). Transgenic mice with TGF-β1 exhibited higher TGF-β1 levels in the atria than in the ventricles under the control of an α-MHC promoter. In this model, as shown by RNA microarray analysis, 80 fibrogenic genes are overexpressed in atrium, while only 2 genes are differentially expressed in ventricle (Rahmutula et al., 2013). Similarly, transgenic mice overexpressing angiotensin-converting enzyme (ACE) showed atrial hypertrophy and dilation with focal atrial fibrosis, but the ventricles were normal (Xiao et al., 2004). This specific fibrotic response to ACE overexpression in different parts of the heart could be partly explained by the different expression of AT1 receptor in atria and ventricles (Bauer et al., 1997). Studies have shown that atrial fibroblasts are stronger than ventricular fibroblasts in terms of TGF-β1 mediated fibrosis and oxidative response (Yeh et al., 2013). TGF-β1 through TGF-β1/Smad pathway phosphorylates Smad 2 to promote atrial fibrosis. These processes eventually lead to the accumulation of fibrous and non fibrous collagen, which in turn leads to excessive atrial fibrosis and AF (Polyakova et al., 2008). This indicates that the atrium has a stronger fibrotic response to various stimuli (Burstein et al., 2008). The results above indicate that the mechanisms of fibrosis in atrial and ventricular are different. Compared with ventricles, further studies are needed to determine whether other special signal pathways or mechanisms contribute to the development of atrial selective fibrosis.
Methionine (Met), as the starting amino acid for protein synthesis in eukaryotes, is one of the common sulfur containing amino acids. Met metabolism includes transmethylation, demethylation and transvulcanization.
In the process of methylation, Met is activated by methionine adenosyltransferase (MAT) to S-adenosylmethionine, and then S-adenosylmethionine transfers S-adenosylmethionine methyl through methyltransferase (MT) to S-adenosylhomocysteine. Finally, S-adenosylhomocysteine is hydrolyzed into adenosine and homocysteine (Hcy) by S-adenosylhomocysteine hydrolase (SAHH) to complete transmethylation (Das et al., 2019; Elwan et al., 2021). The demethylation of Hcy to Met is called demethylation. Under the catalysis of methionine synthetase (MTR), with vitamin B12 as the coenzyme, 5-methyltetrahydrofolate as the methyl donor, Hcy obtains methyl to generate Met (Portillo et al., 2020).
The catabolism of Met is mainly through transvulcanization. After Met is converted into Hcy, Hcy and serine (Ser) are condensed to form cystathionine under the catalysis of cystathionine β-synthase (CBS) (Hage et al., 2021). Next, cystathionide is catalyzed by cystathionine γ-lyase (CSE) to produce cysteine. Then, with cysteine as the central link, cysteine generates thiometabolic products through multiple metabolic pathways, which is called transvulcanization (Townsend et al., 2004).
There are several further catabolic pathways of cysteine generated in the process of sulfurization. First of all, cysteine can form cystathionine and H2S with Hcy under the catalysis of CSE. Cysteine transvulcanization is further cracked into cysteine (Cys), α-Ketobutyrate and ammonia under the catalysis of CSE, which are involved in metabolism, such as entering the tricarboxylic acid cycle or excreted by urine (Portillo et al., 2020). Secondly, Cys can be further decomposed into cystine, which is catalyzed by CSE to produce thiocysteine and pyruvate. Thiocysteine and pyruvate are further decomposed to produce cysteine and H2S. At the same time, cysteine can also generate 3-mercaptopyruvate under the action of 2-cysteine aminotransferase (CAT), and 3-mercaptopyruvate can generate H2S and pyruvate under the further catalysis of 3-mercaptopyruvate sulfurtransfer (3-MST), which is carried out in mitochondria (Kolluru et al., 2022).
In addition, the catabolism of cysteine can also produce sulfur dioxide and taurine. Cys is first oxidized to form cysteine sulfinate under the action of cysteine dioxygenase (CDO). Cysteine sulfinate generates β-sulfinylpyruvate through transamination under the catalysis of aspartate aminotransferase (AAT). Then β-sulfinylpyruvate decomposes spontaneously into pyruvate and sulfur dioxide. Cysteine sulfinate can also generate sub taurine and carbon dioxide under the action of cysteine sulfite decarboxylase (CSAD), and then generate taurine (Huang et al., 2016) (Figure 2).
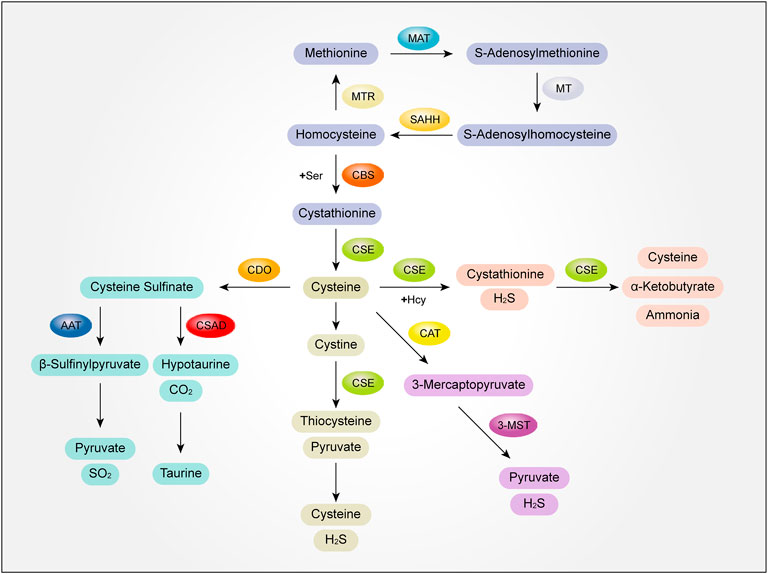
FIGURE 2. The mutual transformation mechanism of sulfur-containing amino acids and their metabolites. MAT, methionine adenosyltransferase; MT, methyltransferase; SAHH, S-adenosylhomocysteine hydrolase; MTR, methionine synthetase; Ser,serine; CBS, cystathionine β-synthase; CSE, cystathionine γ-lyase; Hcy, homocysteine; H2S, hydrogen sulfide; CAT, 2-cysteine aminotransferase; 3-MST, 3-mercaptopyruvate sulfurtransfer; AAT, aspartate aminotransferase; CSAD, cysteine sulfite decarboxylase; SO2, sulfur dioxide.
Homocysteine (Hcy) is a non essential amino acid containing sulfur. As an independent risk factor of cardiovascular disease, Hcy plays a complex role in the pathophysiology of cardiac remodeling (Yao et al., 2017). Cardiovascular diseases with hyperhomocysteinemia (hHcy) are significantly related to cardiac fibrosis (Kar et al., 2019; Deng et al., 2022). Clinical studies have found that hHcy can promote cardiac hypertrophy in patients with hypertension. In addition, Hcy can promote cardiac inflammation and fibrosis by exacerbating AngII-stimulated expression of Calcineurin and nuclear factor of activated T cells (NFAT) (Deng et al., 2022).
Atrial fibrosis is an inevitable pathological determinent of several different heart diseases, especially AF. High plasma Hcy is considered to be a risk factor for AF (Nasso et al., 2013; Yao et al., 2017) and is closely related to the formation of atrial fibrosis (Svenningsson et al., 2020). Atrial structural remodeling can be detected in patients with both paroxysmal or permanent AF. HHcy is an key pathological feature in the etiology of atrial fibrosis (Iguchi et al., 2018). The activation level of transient receptor potential 3 (TRPC3) channel is closely related to the degree of atrial fibrosis and the incidence rate of AF. It was found that Hcy mediates atrial fibrosis by triggering the activation of TRPC3 signaling pathway (Han et al., 2020). TRPC3 is an indispensable factor in regulating the development mechanism of fibrosis and promoting the transformation of fibroblasts into myofibroblasts, which has adverse effects on the regulation of collagen (Numaga-Tomita et al., 2017). Hcy can combine with G protein coupled receptor (GPCR) and activate phospholipase C (PLC) to produce intracellular messenger 1,2-diacylglycerol (DAG). DAG recruits protein kinase C (PKC) and subsequently affects the downstream signaling pathway of cardiac fibrosis (Vacek et al., 2009). TRPC3 is directly activated by PKC phosphorylation. Higher Hcy level can combine with GPCRs, thus activating the downstream PKC/TRPC3 signal pathway, promoting the formation of atrial fibrosis. On the other hand, studies have found that atrial fibrosis induced by hHcy is also mediated by the interaction between TRPC3 channel and sirtuin-1 (SIRT1). Patients with hHcy showed elevated TRPC3 protein levels, decreased SIRT1 levels, and activation of TGF-β signal pathway. In addition, the experiment showed that TAC mice fed with hHcy developed heart failure which the type of preserved ejection fraction (HFpEF) accompanied with atrial fibrosis. Over-expression of SIRT1 and knockdown of TRPC3 could effectively control Hcy mediated atrial fibrosis in mice. The results showed that TRPC3 is required for the activation and translocation of TGF-β into the nucleus of fibroblasts by mediating the trafficking and activity of SIRT1. The clinical analysis also showed that compared with the sinus rhythm group, the level of TRPC3 RNA in patients with AF increased, while the level of SIRT1 RNA decreased.
In conclusion, the increase of Hcy level promotes the proliferation and differentiation of atrial fibroblasts, which shows that Hcy can combine with GPCRs and increase the protein level of TPRC3. TRPC3 is a negative regulator of SIRT1. Under the stimulation of homocysteine, TRPC3 can directly interact with SIRT1, leading to activation of TGF signal pathway, thereby inducing collagen accumulation and forming atrial fibrosis (Han et al., 2020). But the molecular interaction between Hcy and atrial myocytes and atrial fibroblasts remains to be clarified.
Taurine is a conditionally essential amino acid, which is expressed in most animal tissues. In recent years, research has found that taurine, with its characteristics of anti-oxidative stress (Baliou et al., 2021) and anti-inflammation (Qaradakhi et al., 2020), plays a protective role in cardiovascular diseases such as reducing atherosclerosis, improving myocardial function and reducing cardiovascular risk factors (Zulli, 2011). Taurine can reduce myocyte hypertrophy and fibrosis by activating SIRT1-p53 pathway, significantly relieving cardiac dysfunction after TAC (Liu et al., 2020). Futher more, taurine can reduce iron mediated cardiac fibrosis by playing the role of anti-oxidative stress (Oudit et al., 2004). The lack of taurine can cause atrophic cardiac remodeling in wistar rats (Pansani et al., 2012). It is also found that the anti-atrial structural remodeling effect of taurine is related to its anti-inflammatory effect (Yang et al., 2017). In the rats with atrial fibrosis, taurine significantly decreased the serum or plasma levels of tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), AngII, high-sensitive C-reactive protein (Hs-CRP) and matrix metallaproteinase-9 (MMP-9). Meanwhile, it reduced the degree of interstitial fibrosis significantly. Electron microscopy showed that taurine could significantly reduce the ultrastructure damage of atrial cells, and could reverse the change of gap junction and maintain the integrity of myocardial ultrastructure. The results indicated that taurine could reduce the degree of atrial fibrosis partly by reducing the levels of inflammatory factors and fibrogenic molecules. Taurine can inhibit atrial structural remodeling caused by AF through by protecting the integrity of myocardial ultrastructure (Yang et al., 2017). However, the potential mechanism of taurine regulating the activation of inflammatory signaling pathway and inhibiting atrial fibrosis still needs to be further explored.
At present, hydrogen sulfide (H2S) has been defined as the third gas signal molecule after nitric oxide (NO) and carbon monoxide (CO) (Zhao et al., 2022). In vivo, endogenous H2S is mainly produced by CBS, CSE and (3-mercaptosulfurtransferase) 3-MST, among which 3-MST is the key enzyme in mitochondria to produce H2S (Bełtowski, 2015). In the cardiovascular system, H2S is mainly generated endogenously through the catalysis of CSE and participates in a variety of physiological and pathophysiological processes such as inhibition of oxidative stress, inflammation, cell death, cell proliferation and transformation (Zhang et al., 2019). Clinical studies have found that the expression of CSE and 3-MST in the left atrial appendage of patients with AF is decreased, and the expression of CBS is increased, accompanied by significant atrial fibrosis. Patients with AF showed that the significantly reduced level of H2S in plasma (Hu et al., 2021). Further experiments showed that these changes were related to the increased warburg effect and the occurrence of endoplasmic reticulum stress (ERS). Supplementation of H2S can reduce atrial fibrosis by inhibiting warburg effect and ERS induced by AngII (Hu et al., 2021). Another study confirmed that during the occurrence of AF, stable sulfide stored in mitochondria was mobilized to produce free sulfide in the blood to offset myocardial damage caused by oxidative stress (Watts et al., 2021). Therefore, the longer the duration of AF, the long-term stable sulfide storage reserves are consumed more, which leads to the decreased plasma H2S content and the intensification of oxidative stress reaction, thus aggravating the occurrence of atrial fibrosis (Watts et al., 2021). Compared with the atrial tissues of patients with sinus rhythm, the expression of CSE in atrial tissues of AF was lower and atrial fibrosis was significant. The level of CBS in patients with AF is significantly higher than that in patients with sinus rhythm, which may be related to feedback regulation of CSE and CBS (Nandi and Mishra, 2017).
As an endogenous gas transmitter, H2S also plays an pivotal role in many cell systems by regulating the activity of ion channels. H2S was the first opener of (ATP Sensitive K+) KATP channel identified in vascular smooth muscle cells (Tang et al., 2005). Through activation of KATP channels, H2S lowers blood pressure, protects heart from ischaemia and reperfusion injury (Givvimani et al., 2011; J. Huang et al., 2012). Interestingly, other studies have found that the mechanism of H2S inhibiting atrial fibrosis is independent of KATP channel. H2S can potentially regulate cardiac fibrosis by inhibiting the activation of large conductance Ca2+-activated K+current (BKCa), transient outward K+current (IKto) and inward rectifying K+current (IKir), leading to decreased proliferation and suppression of transforming growth factor-β1 (TGF-β1)–induced myofibroblast transformation of atrial fibroblasts (Sheng et al., 2013). In addition to the ion channel pathway, H2S can also reduce the incidence rate of AF and alleviate atrial fibrosis by activating PI3K/Akt/eNOS pathway (Xue et al., 2020). Su et al. (2021). found in the model of fibrosis in human atrial fibroblasts (HAFs) induced by Ang II that the proliferation and migration of HAFs were enhanced, the expression of microRNA-133a (miR-133a) was reduced, and connective tissue growth factor (CTGF) and fibrosis markers such as collagen I, collagen III and the level of α- SMA increased. After H2S intervention, the fibrosis, proliferation and migration of HAFs induced by Ang II were reduced. Further research found that CTGF was the direct target of miR-133a to regulate atrial fibrosis. H2S improved HAFs fibrosis by significantly increasing the expression of miR-133a.
Atrial fibrillation (AF) is the most common malignant arrhythmia in clinic. It is estimated that more than 33 million people worldwide are affected by AF (Christophersen et al., 2017). In recent years, China has become the country with the largest number of patients with AF in the world, and the prevalence of AF will further increase with the aging of the population in China (Christophersen et al., 2017; Hindricks et al., 2021). Currently, research shows that atrial fibrosis is an important reason for the occurrence and progress of AF, and its formation and progress are related to transforming growth factor1 (TGF-β1), renin angiotensin aldosterone system (RAAS), inflammation, rlatelet-derived growth factor (PDGF) and miRNAs (Li et al., 2021). However, the mechanisms have not been fully clarified. More and more studies have focused on the important role of sulfur-containing amino acids and their metabolites in the formation and development of atrial fibrosis. Taurine and H2S inhibit atrial fibrosis through their anti-inflammatory effects and ion channel regulation (Sheng et al., 2013; Yang et al., 2017), and homocysteine inhibits atrial fibrosis by promoting the interaction between TRPC3 and SIRT1 (Han et al., 2020). These findings provide new ideas for the prevention and treatment of atrial fibrosis and AF.
Although people have known some about the operations of sulfur-containing amino acids and their metabolites (Townsend et al., 2004; Kolluru et al., 2022), there are still a lot of specific mechanisms, mediators and conditions which might affect the progression of atrial fibrosis need to be explored. For example, how the metabolites of sulfur-containing amino acids affect the progression of atrial fibrosis?How the key enzymes of sulfide synthesis play a role in atrial structural remodeling and electrical remodeling?What are the direct targets of sulfur-containing amino acids and their metabolites in promoting or inhibiting the formation and progression of atrial fibrosis? The role of sulfur-containing amino acids and their metabolites in the occurrence and development of atrial fibrosis and the related molecular mechanisms have not been clarified, and a lot of in-depth research works are still needed in the future. Further elucidating the mechanism of sulfur-containing amino acids and their metabolites in the development of atrial fibrosis will provide a new basis for early diagnosis, prevention and targeted treatment of atrial fibrosis.
RW and DZ prepared the manuscript. DZ, YQ, and CD conceived the idea and reviewed the drafts and provided important information for the completion.
This work was supported by National Natural Science Foundation of China (Nos 82000289 and 32071113), and the Beijing Natural Science Foundation of China (No. 7212059).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Afzal, M. R., Samanta, A., Chatta, J., Ansari, B., Atherton, S., Sabzwari, S., et al. (2017). Adjunctive ablation strategies improve the efficacy of pulmonary vein isolation in non-paroxysmal atrial fibrillation: A systematic review and meta-analysis. Expert Rev. cardiovasc. Ther. 15 (3), 227–235. doi:10.1080/14779072.2017.1294064
AlQudah, M., Hale, T. M., and Czubryt, M. P. (2020). Targeting the renin-angiotensin-aldosterone system in fibrosis. Matrix Biol. 91-92, 92–108. doi:10.1016/j.matbio.2020.04.005
Andrade, J., Khairy, P., Dobrev, D., and Nattel, S. (2014). The clinical profile and pathophysiology of atrial fibrillation: Relationships among clinical features, epidemiology, and mechanisms. Circ. Res. 114 (9), 1453–1468. doi:10.1161/circresaha.114.303211
Baliou, S., Adamaki, M., Ioannou, P., Pappa, A., Panayiotidis, M. I., Spandidos, D. A., et al. (2021). Protective role of taurine against oxidative stress (Review). Mol. Med. Rep. 24 (2), 605. doi:10.3892/mmr.2021.12242
Bauer, P., Regitz-Zagrosek, V., Kallisch, H., Linz, W., Schoelkens, B., Hildebrandt, A. G., et al. (1997). Myocardial angiotensin receptor type 1 gene expression in a rat model of cardiac volume overload. Basic Res. Cardiol. 92 (3), 139–146. doi:10.1007/bf00788631
Bełtowski, J. (2015). Hydrogen sulfide in pharmacology and medicine--An update. Pharmacol. Rep. 67 (3), 647–658. doi:10.1016/j.pharep.2015.01.005
Beyer, C., Tokarska, L., Stühlinger, M., Feuchtner, G., Hintringer, F., Honold, S., et al. (2021). Structural cardiac remodeling in atrial fibrillation. JACC. Cardiovasc. Imaging 14 (11), 2199–2208. doi:10.1016/j.jcmg.2021.04.027
Biernacka, A., Dobaczewski, M., and Frangogiannis, N. G. (2011). TGF-β signaling in fibrosis. Growth factors 29 (5), 196–202. doi:10.3109/08977194.2011.595714
Burstein, B., Libby, E., Calderone, A., and Nattel, S. (2008). Differential behaviors of atrial versus ventricular fibroblasts: A potential role for platelet-derived growth factor in atrial-ventricular remodeling differences. Circulation 117 (13), 1630–1641. doi:10.1161/circulationaha.107.748053
Christophersen, I. E., Rienstra, M., Roselli, C., Yin, X., Geelhoed, B., Barnard, J., et al. (2017). Large-scale analyses of common and rare variants identify 12 new loci associated with atrial fibrillation. Nat. Genet. 49 (6), 946–952. doi:10.1038/ng.3843
Das, T. K., Javadzadeh, A., Dey, A., Sabesan, P., Théberge, J., Radua, J., et al. (2019). Antioxidant defense in schizophrenia and bipolar disorder: A meta-analysis of mrs studies of anterior cingulate glutathione. Prog. Neuropsychopharmacol. Biol. Psychiatry 91, 94–102. doi:10.1016/j.pnpbp.2018.08.006
Deng, Y., Li, Z., An, X., Fan, R., Wang, Y., Li, J., et al. (2022). Hyperhomocysteinemia promotes cardiac hypertrophy in hypertension. Oxid. Med. Cell. Longev. 2022, 1486157. doi:10.1155/2022/1486157
Elwan, H., Xie, C., Miao, L. P., Dong, X., Zou, X. T., Mohany, M., et al. (2021). Methionine alleviates aflatoxinb1-induced broiler chicks embryotoxicity through inhibition of caspase-dependent apoptosis and enhancement of cellular antioxidant status. Poult. Sci. 100 (8), 101103. doi:10.1016/j.psj.2021.101103
Everett, T. H. T., and Olgin, J. E. (2007). Atrial fibrosis and the mechanisms of atrial fibrillation. Heart rhythm. 4 (3), S24–S27. doi:10.1016/j.hrthm.2006.12.040
Frangogiannis, N. G. (2021). Cardiac fibrosis. Cardiovasc. Res. 117 (6), 1450–1488. doi:10.1093/cvr/cvaa324
Frangogiannis, N. G. (2019). The extracellular matrix in ischemic and nonischemic heart failure. Circ. Res. 125 (1), 117–146. doi:10.1161/circresaha.119.311148
Givvimani, S., Munjal, C., Gargoum, R., Sen, U., Tyagi, N., Vacek, J. C., et al. (2011). Hydrogen sulfide mitigates transition from compensatory hypertrophy to heart failure. J. Appl. Physiol. 110 (4), 1093–1100. doi:10.1152/japplphysiol.01064.2010
Hage, H., Rosso, M. N., and Tarrago, L. (2021). Distribution of methionine sulfoxide reductases in fungi and conservation of the free-methionine-R-sulfoxide reductase in multicellular eukaryotes. Free Radic. Biol. Med. 169, 187–215. doi:10.1016/j.freeradbiomed.2021.04.013
Han, L., Tang, Y., Li, S., Wu, Y., Chen, X., Wu, Q., et al. (2020). Protective mechanism of SIRT1 on Hcy-induced atrial fibrosis mediated by TRPC3. J. Cell. Mol. Med. 24 (1), 488–510. doi:10.1111/jcmm.14757
Hindricks, G., Potpara, T., Dagres, N., Arbelo, E., Bax, J. J., Blomström-Lundqvist, C., et al. (2021). 2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association for Cardio-Thoracic Surgery (EACTS): The Task Force for the diagnosis and management of atrial fibrillation of the European Society of Cardiology (ESC) Developed with the special contribution of the European Heart Rhythm Association (EHRA) of the ESC. Eur. Heart J. 42 (5), 373–498. doi:10.1093/eurheartj/ehaa612
Hu, H. J., Wang, X. H., Liu, Y., Zhang, T. Q., Chen, Z. R., Zhang, C., et al. (2021). Hydrogen sulfide ameliorates angiotensin II-induced atrial fibrosis progression to atrial fibrillation through inhibition of the warburg effect and endoplasmic reticulum stress. Front. Pharmacol. 12, 690371. doi:10.3389/fphar.2021.690371
Huang, J., Wang, D., Zheng, J., Huang, X., and Jin, H. (2012). Hydrogen sulfide attenuates cardiac hypertrophy and fibrosis induced by abdominal aortic coarctation in rats. Mol. Med. Rep. 5 (4), 923–928. doi:10.3892/mmr.2012.748
Huang, Y., Tang, C., Du, J., and Jin, H. (2016). Endogenous sulfur dioxide: A new member of gasotransmitter family in the cardiovascular system. Oxid. Med. Cell. Longev. 2016, 8961951. doi:10.1155/2016/8961951
Iguchi, M., Tezuka, Y., Ogawa, H., Hamatani, Y., Takagi, D., An, Y., et al. (2018). Incidence and risk factors of stroke or systemic embolism in patients with atrial fibrillation and heart failure - the fushimi AF registry. Circ. J. 82 (5), 1327–1335. doi:10.1253/circj.CJ-17-1155
Kar, S., Shahshahan, H. R., Kambis, T. N., Yadav, S. K., Li, Z., Lefer, D. J., et al. (2019). Hydrogen sulfide ameliorates homocysteine-induced cardiac remodeling and dysfunction. Front. Physiol. 10, 598. doi:10.3389/fphys.2019.00598
Kolluru, G. K., Shackelford, R. E., Shen, X., Dominic, P., and Kevil, C. G. (2022). Sulfide regulation of cardiovascular function in health and disease. Nat. Rev. Cardiol., 1–17. doi:10.1038/s41569-022-00741-6
Krishnan, A., Chilton, E., Raman, J., Saxena, P., McFarlane, C., Trollope, A. F., et al. (2021). Are interactions between epicardial adipose tissue, cardiac fibroblasts and cardiac myocytes instrumental in atrial fibrosis and atrial fibrillation? Cells 10 (9), 2501. doi:10.3390/cells10092501
Krul, S. P., Berger, W. R., Smit, N. W., van Amersfoorth, S. C., Driessen, A. H., van Boven, W. J., et al. (2015). Atrial fibrosis and conduction slowing in the left atrial appendage of patients undergoing thoracoscopic surgical pulmonary vein isolation for atrial fibrillation. Circ. Arrhythm. Electrophysiol. 8 (2), 288–295. doi:10.1161/circep.114.001752
Li, L., Zhao, Q., and Kong, W. (2018). Extracellular matrix remodeling and cardiac fibrosis. Matrix Biol. 68-69, 490–506. doi:10.1016/j.matbio.2018.01.013
Li, C. Y., Zhang, J. R., Hu, W. N., and Li, S. N. (2021). Atrial fibrosis underlying atrial fibrillation (Review). Int. J. Mol. Med. 47 (3), 9. doi:10.3892/ijmm.2020.4842
Liu, J., Ai, Y., Niu, X., Shang, F., Li, Z., Liu, H., et al. (2020). Taurine protects against cardiac dysfunction induced by pressure overload through SIRT1-p53 activation. Chem. Biol. Interact. 317, 108972. doi:10.1016/j.cbi.2020.108972
Mistry, R. K., and Brewer, A. C. (2019). Redox-dependent regulation of sulfur metabolism in biomolecules: Implications for cardiovascular health. Antioxid. Redox Signal. 30 (7), 972–991. doi:10.1089/ars.2017.7224
Nakajima, H., Nakajima, H. O., Salcher, O., Dittiè, A. S., Dembowsky, K., Jing, S., et al. (2000). Atrial but not ventricular fibrosis in mice expressing a mutant transforming growth factor-beta(1) transgene in the heart. Circ. Res. 86 (5), 571–579. doi:10.1161/01.res.86.5.571
Nandi, S. S., and Mishra, P. K. (2017). H(2)S and homocysteine control a novel feedback regulation of cystathionine beta synthase and cystathionine gamma lyase in cardiomyocytes. Sci. Rep. 7 (1), 3639. doi:10.1038/s41598-017-03776-9
Nasso, G., Bonifazi, R., Romano, V., Brigiani, M. S., Fiore, F., Bartolomucci, F., et al. (2013). Increased plasma homocysteine predicts arrhythmia recurrence after minimally invasive epicardial ablation for nonvalvular atrial fibrillation. J. Thorac. Cardiovasc. Surg. 146 (4), 848–853. doi:10.1016/j.jtcvs.2012.07.099
Nattel, S. (2017). Molecular and cellular mechanisms of atrial fibrosis in atrial fibrillation. JACC. Clin. Electrophysiol. 3 (5), 425–435. doi:10.1016/j.jacep.2017.03.002
Numaga-Tomita, T., Oda, S., Shimauchi, T., Nishimura, A., Mangmool, S., and Nishida, M. (2017). TRPC3 channels in cardiac fibrosis. Front. Cardiovasc. Med. 4, 56. doi:10.3389/fcvm.2017.00056
Oudit, G. Y., Trivieri, M. G., Khaper, N., Husain, T., Wilson, G. J., Liu, P., et al. (2004). Taurine supplementation reduces oxidative stress and improves cardiovascular function in an iron-overload murine model. Circulation 109 (15), 1877–1885. doi:10.1161/01.cir.0000124229.40424.80
Pansani, M. C., Azevedo, P. S., Rafacho, B. P., Minicucci, M. F., Chiuso-Minicucci, F., Zorzella-Pezavento, S. G., et al. (2012). Atrophic cardiac remodeling induced by taurine deficiency in Wistar rats. PLoS One 7 (7), e41439. doi:10.1371/journal.pone.0041439
Pellman, J., Lyon, R. C., and Sheikh, F. (2010). Extracellular matrix remodeling in atrial fibrosis: Mechanisms and implications in atrial fibrillation. J. Mol. Cell. Cardiol. 48 (3), 461–467. doi:10.1016/j.yjmcc.2009.09.001
Polyakova, V., Miyagawa, S., Szalay, Z., Risteli, J., and Kostin, S. (2008). Atrial extracellular matrix remodelling in patients with atrial fibrillation. J. Cell. Mol. Med. 12 (1), 189–208. doi:10.1111/j.1582-4934.2008.00219.x
Portillo, F., Vázquez, J., and Pajares, M. A. (2020). Protein-protein interactions involving enzymes of the mammalian methionine and homocysteine metabolism. Biochimie 173, 33–47. doi:10.1016/j.biochi.2020.02.015
Qaradakhi, T., Gadanec, L. K., McSweeney, K. R., Abraham, J. R., Apostolopoulos, V., and Zulli, A. (2020). The anti-inflammatory effect of taurine on cardiovascular disease. Nutrients 12 (9), E2847. doi:10.3390/nu12092847
Rahmutula, D., Marcus, G. M., Wilson, E. E., Ding, C. H., Xiao, Y., Paquet, A. C., et al. (2013). Molecular basis of selective atrial fibrosis due to overexpression of transforming growth factor-β1. Cardiovasc. Res. 99 (4), 769–779. doi:10.1093/cvr/cvt074
Saadat, S., Noureddini, M., Mahjoubin-Tehran, M., Nazemi, S., Shojaie, L., Aschner, M., et al. (2020). Pivotal role of TGF-β/smad signaling in cardiac fibrosis: Non-coding RNAs as effectual players. Front. Cardiovasc. Med. 7, 588347. doi:10.3389/fcvm.2020.588347
Sheng, J., Shim, W., Wei, H., Lim, S. Y., Liew, R., Lim, T. S., et al. (2013). Hydrogen sulphide suppresses human atrial fibroblast proliferation and transformation to myofibroblasts. J. Cell. Mol. Med. 17 (10), 1345–1354. doi:10.1111/jcmm.12114
Sohns, C., and Marrouche, N. F. (2020). Atrial fibrillation and cardiac fibrosis. Eur. Heart J. 41 (10), 1123–1131. doi:10.1093/eurheartj/ehz786
Souders, C. A., Bowers, S. L., and Baudino, T. A. (2009). Cardiac fibroblast: The renaissance cell. Circ. Res. 105 (12), 1164–1176. doi:10.1161/circresaha.109.209809
Su, H., Su, H., Liu, C. H., Hu, H. J., Zhao, J. B., Zou, T., et al. (2021). H(2)S inhibits atrial fibrillation-induced atrial fibrosis through miR-133a/CTGF axis. Cytokine 146, 155557. doi:10.1016/j.cyto.2021.155557
Svenningsson, M. M., Svingen, G. F. T., Lysne, V., Ueland, P. M., Tell, G. S., Pedersen, E. R., et al. (2020). Transsulfuration metabolites and the association with incident atrial fibrillation - an observational cohort study among Norwegian patients with stable angina pectoris. Int. J. Cardiol. 317, 75–80. doi:10.1016/j.ijcard.2020.05.010
Tang, G., Wu, L., Liang, W., and Wang, R. (2005). Direct stimulation of K(ATP) channels by exogenous and endogenous hydrogen sulfide in vascular smooth muscle cells. Mol. Pharmacol. 68 (6), 1757–1764. doi:10.1124/mol.105.017467
Townsend, D. M., Tew, K. D., and Tapiero, H. (2004). Sulfur containing amino acids and human disease. Biomed. Pharmacother. 58 (1), 47–55. doi:10.1016/j.biopha.2003.11.005
Vacek, T. P., Sen, U., Tyagi, N., Kumar, M., Moshal, K. S., Passmore, J. C., et al. (2009). Homocysteine effects classical pathway of GPCR down regulation: Galpha(q/11), Galpha(12/13), G(i/o). Mol. Cell. Biochem. 321 (1-2), 1–8. doi:10.1007/s11010-008-9904-4
van den Berg, N. W. E., Neefs, J., Kawasaki, M., Nariswari, F. A., Wesselink, R., Fabrizi, B., et al. (2021). Extracellular matrix remodeling precedes atrial fibrillation: Results of the PREDICT-AF trial. Heart rhythm. 18 (12), 2115–2125. doi:10.1016/j.hrthm.2021.07.059
Watts, M., Kolluru, G. K., Dherange, P., Pardue, S., Si, M., Shen, X., et al. (2021). Decreased bioavailability of hydrogen sulfide links vascular endothelium and atrial remodeling in atrial fibrillation. Redox Biol. 38, 101817. doi:10.1016/j.redox.2020.101817
Xiao, H. D., Fuchs, S., Campbell, D. J., Lewis, W., Dudley, S. C., Kasi, V. S., et al. (2004). Mice with cardiac-restricted angiotensin-converting enzyme (ACE) have atrial enlargement, cardiac arrhythmia, and sudden death. Am. J. Pathol. 165 (3), 1019–1032. doi:10.1016/s0002-9440(10)63363-9
Xue, X., Ling, X., Xi, W., Wang, P., Sun, J., Yang, Q., et al. (2020). Exogenous hydrogen sulfide reduces atrial remodeling and atrial fibrillation induced by diabetes mellitus via activation of the PI3K/Akt/eNOS pathway. Mol. Med. Rep. 22 (3), 1759–1766. doi:10.3892/mmr.2020.11291
Yang, Q., Wu, G., Han, L., Feng, Y., Lin, S., Lv, Q., et al. (2017). Taurine reverses atrial structural remodeling in ach-cacl(2) induced atrial fibrillation rats. Adv. Exp. Med. Biol. 975 (2), 831–841. doi:10.1007/978-94-024-1079-2_65
Yao, Y., Shang, M. S., Dong, J. Z., and Ma, C. S. (2017). Homocysteine in non-valvular atrial fibrillation: Role and clinical implications. Clin. Chim. Acta. 475, 85–90. doi:10.1016/j.cca.2017.10.012
Yao, Y., Yao, W., Bai, R., Lu, Z. H., Tang, R. B., Long, D. Y., et al. (2017). Plasma homocysteine levels predict early recurrence after catheter ablation of persistent atrial fibrillation. Europace 19 (1), 66–71. doi:10.1093/europace/euw081
Yeh, Y. H., Kuo, C. T., Chang, G. J., Qi, X. Y., Nattel, S., and Chen, W. J. (2013). Nicotinamide adenine dinucleotide phosphate oxidase 4 mediates the differential responsiveness of atrial versus ventricular fibroblasts to transforming growth factor-β. Circ. Arrhythm. Electrophysiol. 6 (4), 790–798. doi:10.1161/circep.113.000338
Zhang, D., Wang, X., Chen, S., Chen, S., Yu, W., Liu, X., et al. (2019). Endogenous hydrogen sulfide sulfhydrates IKKβ at cysteine 179 to control pulmonary artery endothelial cell inflammation. Clin. Sci. 133 (20), 2045–2059. doi:10.1042/cs20190514
Zhao, X., Cao, Y., Jin, H., Wang, X., Zhang, L., Zhang, Y., et al. (2022). Hydrogen sulfide promotes thyroid hormone synthesis and secretion by upregulating sirtuin-1. Front. Pharmacol. 13, 838248. doi:10.3389/fphar.2022.838248
Keywords: sulfur-containing amino acid, atrial fibrosis, hydrogen sulfide, taurine, homocysteine
Citation: Wang R, Qi YF, Ding CH and Zhang D (2022) Sulfur-containing amino acids and their metabolites in atrial fibrosis. Front. Pharmacol. 13:1063241. doi: 10.3389/fphar.2022.1063241
Received: 06 October 2022; Accepted: 24 October 2022;
Published: 04 November 2022.
Edited by:
Hongliang Dai, Jinzhou Medical University, ChinaReviewed by:
Zheng Rong Huang, First Affiliated Hospital of Xiamen University, ChinaCopyright © 2022 Wang, Qi, Ding and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Da Zhang, emhhbmdkYTIwMThAMTI2LmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.