 Ippei Kawano
Ippei Kawano Michaela Adamcova
Michaela Adamcova- Department of Physiology, Faculty of Medicine in Hradec Kralove, Charles University in Prague, Hradec Kralove, Czechia
Doxorubicin (DOX) is a chemotherapeutic drug widely used for cancer treatment, but its use is limited by cardiotoxicity. Although free radicals from redox cycling and free cellular iron have been predominant as the suggested primary pathogenic mechanism, novel evidence has pointed to topoisomerase II inhibition and resultant genotoxic stress as the more fundamental mechanism. Recently, a growing list of microRNAs (miRNAs) has been implicated in DOX-induced cardiotoxicity (DIC). This review summarizes miRNAs reported in the recent literature in the context of DIC. A particular focus is given to miRNAs that regulate cellular responses downstream to DOX-induced DNA damage, especially p53 activation, pro-survival signaling pathway inhibition (e.g., AMPK, AKT, GATA-4, and sirtuin pathways), mitochondrial dysfunction, and ferroptosis. Since these pathways are potential targets for cardioprotection against DOX, an understanding of how miRNAs participate is necessary for developing future therapies.
Introduction
Doxorubicin (DOX) is an anthracycline (ANT) antibiotic that is widely used as a chemotherapeutic agent against a variety of malignancies, namely breast cancer, prostate cancer, small cell lung carcinoma, Hodgkin’s lymphoma, and others. Despite the high efficacy, its use is significantly limited by its chronic cardiotoxicity. DOX-induced cardiotoxicity (DIC) risk increases as the cumulative dose exceeds 400–700 mg/m2 for adults and 300 mg/m2 for children (Renu et al., 2018). Patients with DIC could show symptoms ranging from subclinical left ventricular (LV) dysfunction to congestive heart failure (CHF) and death (Bansal et al., 2019). DOX-induced CHF could present acutely (weeks after the onset of treatment) or chronically (decades later), with most cases seen in the first year of therapy (Lipshultz et al., 2008; Cardinale et al., 2015). As DIC is irreversible, dose monitoring and early detection are critical to minimize morbidity and mortality. LV function can be monitored with speckle tracking echocardiography (STE) combined with serum biomarkers such as cardiac troponins and natriuretic peptides to predict DIC risk (Kang et al., 2013; Pudil et al., 2020).
Despite many decades of investigation, the mechanism of DIC has not been completely understood (Mitry and Edwards, 2016). Free radical reactive oxygen species (ROS) generation due to one-electron redox cycling by the quinone moiety of DOX and increased free cellular iron are the most favored hypotheses in the literature (Horenstein et al., 2000; Minotti et al., 2004; Šimůnek et al., 2009). However, other mechanisms have also been suggested, such as DNA damage due to topoisomerase II inhibition and double-stranded breaks (DSBs) (Pommier et al., 2010; Marinello et al., 2018), mitochondrial dysfunction (Štěrba et al., 2011; Osataphan et al., 2020; Wallace et al., 2020; Huang et al., 2022), altered calcium homeostasis (Wallace, 2007; Shinlapawittayatorn et al., 2022), and others. Recently, the ROS hypothesis has been questioned as the primary pathogenic mechanism (Pointon et al., 2010; Zhang et al., 2012; Ghigo et al., 2016; Kalyanaraman, 2020). Instead, inhibition of topoisomerase IIβ (Top2B) in cardiomyocytes and the downstream DNA damage response seems to be the predominant mechanism for mitochondrial dysfunction and ROS generation leading to DIC (Zhang et al., 2012; Vejpongsa and Yeh, 2014). Dexrazoxane, the only drug used in clinics to prevent DIC, has long been thought to achieve its effect through iron chelation (Štěrba et al., 2013). However, more recent evidence points to its interaction with Top2B, not its iron-chelating function, as the primary mechanism for its cardioprotective effect (Kaiserová et al., 2006; Lyu et al., 2007; Deng et al., 2014; Kollárová-Brázdová et al., 2020; Jirkovská et al., 2021; Jirkovský et al., 2021).
Although the understanding that connects DNA damage and DIC is incomplete, certain pathways have been implicated to play a role. Upon DNA damage such as DSBs, cells activate a group of serine/threonine kinases sensitive to DSBs, known as ataxia telangiectasia mutated (ATM), ATM- and RAD3-related (ATR), and DNA-dependent protein kinase (DNA-PK). ATM/ATR can stabilize p53, the master regulator of cell fate after DNA damage (Jackson and Bartek, 2009). In DOX-treated cardiomyocytes, both ATM and p53 are activated (L’Ecuyer et al., 2006; Yoshida et al., 2009; Piegari et al., 2013). Heterozygous p53 loss attenuated DOX-induced cardiomyocyte apoptosis in vivo, suggesting p53 activation by DOX contributes to DIC (Yoshida et al., 2009). Meanwhile, DNA-PK is required for the non-homologous end joining (NHEJ) of DSBs. DNA-PK can also activate the pro-survival AKT via the mammalian target of rapamycin (mTOR) complex 2 (mTORC2) (Dragoi et al., 2005; Bozulic et al., 2008; Liu et al., 2022). This DNA-PK-AKT axis activation has also been reported in DOX-treated cardiomyocytes (Gratia et al., 2012). While AKT activation may be beneficial for cardiomyocyte survival, in the context of DIC, it is later followed by AKT inhibition (Lee et al., 2006; Das et al., 2011; Sahu et al., 2019). Initial transient AKT activation may rather exacerbate cardiomyocyte energy deficiency and death in DIC by inhibiting AMP-activated protein kinase (AMPK) signaling (Gratia et al., 2012). In addition, the DNA-PK-AKT axis can stabilize p53 following DNA damage, independent of ATM/ATR (Boehme et al., 2008). In summary, DNA damage response in DOX-treated cardiomyocytes likely converges on p53 and AKT/AMPK signaling pathways that, in turn, regulate cell death. One of the major downstream targets of these pathways is the mitochondrion. Specifically, DOX-induced DNA damage and p53 activation may trigger mitochondrial membrane potential loss (L’Ecuyer et al., 2006; Sardão et al., 2009), mitochondrial fragmentation (Samant et al., 2014; Tang et al., 2017; Catanzaro et al., 2019), and impaired mitophagy (Hoshino et al., 2013; Li et al., 2022) in cardiomyocytes, potentiating cell death.
In recent years, microRNA (miRNA) has garnered attention as a novel therapeutic target for DIC (Holmgren et al., 2016; Ruggeri et al., 2018; Skála et al., 2019; Pereira et al., 2020). In the last few years, several experimental and clinical studies have described changes in miRNAs induced by the cardiotoxic effect of ANT with very controversial results (Ruggeri et al., 2018; Pereira et al., 2020; Chen et al., 2021). However, the role miRNAs play in the molecular pathways responsible for DIC is not wholly understood.
This paper aims to give an integrated overview of the impact of miRNAs on the early pathogenetic mechanism of DIC development. Particular attention is paid to the role of the DNA damage response, as it triggers various cellular changes. When the DNA damage exceeds the extent of cellular repair, cells commit themselves to regulated cell death (RCD), including apoptosis and ferroptosis, likely contributing to cardiac functional compromise (Christidi and Brunham, 2021). As a part of RCD, pro-survival signals (e.g., AMPK, AKT, GATA-4, and the sirtuin pathways) are inhibited, and mitochondria become fragmented and accumulate iron within their matrix. Multiple molecular pathways of this process are regulated by miRNAs, raising the possibility of attenuating DOX-induced RCD through targeting miRNAs.
miRNAs involved in p53 regulation
One of the primary pathways that leads to DOX-induced apoptosis is the oxidative DNA damage-ATM-p53 pathway (Kurz et al., 2004; Yoshida et al., 2009). DNA damage such as DSB activates ATM. One of the most critical downstream targets of ATM is p53, which is activated into a form with improved DNA binding affinity and rapidly accumulates inside of the cell to induce cell cycle arrest, DNA repair, and apoptosis (Lakin and Jackson, 1999; Cheng and Chen, 2010). By causing DSBs and increased ROS generation, DOX can activate this ATM-p53 pathway of DNA damage response (Kurz et al., 2004; Yoshida et al., 2009). p53 was also identified as the critical transcriptome regulator of DIC in a study using transcriptomic profiling, with p53-induced upregulation in death receptors potentially playing a role in increased cardiomyocyte apoptosis (McSweeney et al., 2019). However, the role p53 plays in DIC has also been reported to be cardioprotective through its impact on mitochondrial DNA (mtDNA) rather than apoptosis-inducing activity. Loss of p53 function led to the exacerbation of DIC that can be rescued by recovering mtDNA health (Li et al., 2019b).
The ATM-p53 axis induces the miR-34 family. Even without DNA damage, a stagnant pool of mature miR-34 is present, quickly activated upon DNA damage by ATM/Clp1-dependent 5′-phosphorylation and Ago2-loading (Salzman et al., 2016). Later, p53 also initiates the transcription of miR-34 (Chang et al., 2007; He et al., 2007; Raver-Shapira et al., 2007; Hermeking, 2012). In humans, three members of miR-34 exist, namely, miR-34a from chromosome 1p36 and miR-36b/c from chromosome 11q23. Among these three, miR-34a has been particularly implicated in the p53 response, as it directly targets TP53 and its inhibitors MDM4, YY1, MTA2, HDAC1, and SIRT1 (Yamakuchi et al., 2008; Kaller et al., 2011; Navarro and Lieberman, 2015). This double-negative regulation of p53 by miR-34a effectively serves as a positive feedback loop. Recently, a mathematical bifurcation analysis has proposed an exciting function for such a circuit regulating p53 dynamics (Gao et al., 2020), producing limit cycle oscillations needed for biochemical oscillators (Novák and Tyson, 2008; Gao et al., 2020). Therefore, miR-34a can trigger p53 fluctuation via its action on sirtuin 1 (SIRT1) mRNA. The amplitude of this activated p53 oscillation is determined by the intensity of DNA damage coded by the activated ATM level and the power of SIRT1 inhibition by miR-34a. A moderate increase in the intensity produces a sustained oscillation of p53 that activates genes involved in cell cycle arrest and DNA repair. However, a significant increase in intensity leads to a prolonged high-level steady state that triggers pro-apoptotic genes (Gao et al., 2020). This agrees with the current experimental data describing how p53 dynamics differentially regulates cell decision-making, between survival (cell cycle arrest, DNA repair) and death (apoptosis), likely through the modulation of its binding affinity to target genes. Pro-apoptotic genes have a lower affinity for p53 than pro-arrest genes (Purvis et al., 2012; Wu et al., 2017). Notably, p53 is not solely regulated by miR-34a to generate an oscillatory pattern, as miR-34 loss did not impair p53 function (Concepcion et al., 2012). Other mechanisms have also been proposed to produce p53 pulses, such as the well-studied p53-mouse double minute 2 homolog (MDM2) feedback loop (Lev Bar-Or et al., 2000; Mihalas et al., 2000) and p53-wild-type p53-induced phosphatase 1 (Wip1)-ATM/checkpoint kinase 2 (Chk2) negative feedback loop (Fiscella et al., 1997; Fujimoto et al., 2006; Shreeram et al., 2006; Boehme et al., 2008). Interestingly, the p53 pulse regulator Wip1 is targeted by miR-16, whose expression is under the regulation of p53 (Zhang et al., 2010; Lezina et al., 2013). This p53-miR16-Wip1 positive feedback loop may also impact cell fate decision (Issler and Mombach, 2017).
Besides the modulation of p53, miR-34a also directly inhibits G1/S transition by targeting c-Myc, n-Myc, CCND1, E2F, CDK4, CDK6, and MET and induces apoptosis through anti-apoptotic Bcl-2 and SIRT1 (Bommer et al., 2007; Yamakuchi et al., 2008; Kaller et al., 2011; Chen and Hu, 2012). Therefore, in addition to regulating cellular p53 dynamics, miR-34a directly impacts cell fate and regulation to aid in p53 function.
Unsurprisingly, after DOX exposure, miR-34a levels increase in rat cardiomyocytes, which is reversed by dexrazoxane (Zhu et al., 2017). In a rat model, antimiR-34a suppressed cardiomyocyte apoptosis via upregulating Bcl-2 and SIRT1 and reduced DIC (Piegari et al., 2016, 2020). SIRT1 prevents cardiac apoptosis by inhibiting the pro-apoptotic p66shc, increasing mitochondrial ROS generation (Zhu et al., 2017). Therefore, SIRT1 inhibition by miR-34a can exacerbate DOX-induced cardiac oxidative stress via the action of p66shc. Taken together, the miR-34 family, mainly miR-34a, is induced in cardiomyocytes upon DOX-induced DNA damage via the ATM-p53 axis. It acts synergistically with p53 to induce cardiac cell cycle arrest and apoptosis, contributing to DIC.
MiR-23a and miR-128 are also associated with p53 in DOX-induced cardiomyocyte apoptosis (Adlakha and Saini, 2013; Li et al., 2015). MiR-23a is upregulated by DOX treatment and exposure to hydrogen peroxide, and it promotes apoptosis by binding to p53 (Li et al., 2015). Upon binding, the complex associates with the miR-128 promoter region, increasing miR-128 expression (Li et al., 2015). An increased miR-128 level was reported in DOX-treated hearts (Zhang et al., 2021b). miR-128 promotes apoptosis by inhibiting SIRT1, resulting in increased p53 acetylation and activity (Adlakha and Saini, 2013; Li et al., 2015). Additionally, miR-128 also contributes to DIC by targeting peroxisome proliferator-activated receptor γ (PPAR-γ), a protective agent against cardiomyocyte oxidative stress and apoptosis (Ren et al., 2009; Zhang et al., 2021b).
Another p53 target that seems to play a role in DIC is miR-22. Upon DOX exposure, miR-22 is activated to target the cell cycle regulator p21 (Tsuchiya et al., 2011), which is also activated by p53 during stress conditions. It induces cell cycle arrest by inhibiting various cyclin-dependent kinases, eventually leading to cell senescence rather than apoptosis. However, under high stress, p53 induces the transcription of both miR-22 and p21, thereby suppressing p21 action and favoring apoptosis rather than senescence (Tsuchiya et al., 2011). DOX also induces miR-22 upregulation in murine cardiomyocytes (Xu et al., 2020). miR-22 inhibition has recently been reported to counteract DIC via directly targeting SIRT1 (Xu et al., 2020; Wang J et al., 2021). SIRT1 deacetylates the peroxisome proliferator-activated receptor gamma coactivator-1α (PGC-1α), a vital regulator of mitochondrial mass and function (Tang, 2016). It has been reported that when miR-22 is knocked out, SIRT1 suppression is removed, and the expression of PGC-1α and other mitochondrial biogenesis or mitophagy regulators are enhanced, alleviating mitochondrial dysfunction in DIC (Wang R et al., 2021).
In addition to the miRNAs mentioned above, p53 activates other miRNAs, such as miR-200, miR-15/16 family, miR-192/194/215 family, miR-145, and miR-107, although whether they play a role in DIC remains to be largely elusive (Hermeking, 2012). Another form of non-coding RNA, known as large intergenic non-coding RNA-p21 (lincRNA-p21), is also activated by p53 (Huarte et al., 2010). LincRNA-p21 might compete with mRNA for miRNA binding at the miRNA binding sites (MREs), functioning as competing endogenous RNAs (ceRNAs) (Amirinejad et al., 2020). For example, lincRNA-p21 can bind to the miR-181 family, miR-17-5p, miR-1277-5p, and other miRNAs to regulate their levels (Amirinejad et al., 2020). These downstream miRNA targets of p53 might also be altered by DOX treatment in cardiomyocytes.
Lastly, p53 can be therapeutically targeted by miR-146a, which is upregulated by DOX treatment (Horie et al., 2010; Pan et al., 2019). miR-146a directly inhibits TATA-binding protein-associated factor 9b (TAF9b), a p53 coactivator and stabilizer (Pan et al., 2019). Reduced p53 stability improves autophagy and reduces apoptosis in DOX-treated cardiomyocytes (Pan et al., 2019). MiR-146a also inhibits cardiomyocyte death by targeting cyclophilin D, which can otherwise trigger mitochondrial permeability transition pore (mPTP) opening and necrosis (Su et al., 2021). Although this mechanism of miR-146a action is not studied in DIC, since cyclophilin D does seem to participate in DIC, the connection is speculated (Dhingra et al., 2020). On the other hand, miR-146a has also been reported to exacerbate acute DIC, especially in patients simultaneously receiving trastuzumab (an anti-ErbB2 monoclonal antibody) through targeting ErbB4 (Horie et al., 2010). This is due to ErbB4 participating in the neuregulin-1 (NRG-1)-ErbB2/ErbB4-phosphoinositide 3-kinase (PI3K)-AKT-mTOR axis that is protective against DIC (Fukazawa et al., 2003; Bian et al., 2009).
miRNAs involved in the inhibition of pro-survival signaling pathways
AMPK pathway
How DOX treatment affects cardiac AMPK activity has been controversial. DOX treatment was reported to show activation, no difference, and inhibition of AMPK and autophagy (Koleini and Kardami, 2017; Christidi and Brunham, 2021). One potential hypothesis explaining conflicting observations might be that DOX initially activates AMPK and thus autophagy but later inhibits them. Nevertheless, it is generally accepted that DOX-induced AMPK inhibition plays a role in DIC, and therefore AMPK activation can be cardioprotective against DOX (Gratia et al., 2012; Wang et al., 2012; Timm and Tyler, 2020; Russo et al., 2021). Dysregulated AMPK activity and autophagy can lead to the accumulation of undegraded autophagosomes/autolysosomes, further potentiating cardiomyocyte deaths (Christidi and Brunham, 2021).
The mechanism connecting DOX treatment and AMPK modulation remains elusive, but oxidative and genotoxic stress are likely essential players. In response to DNA damage, DNA-PK can transiently activate the pro-survival AKT pathway, which can at least partially inhibit AMPK (Lee et al., 2006; Gratia et al., 2012). DOX-induced AMPK inhibition may further exacerbate genotoxic stress and cardiomyocyte apoptosis by p53 accumulation and reduced SIRT1 activity due to reduced NAD+/NADH ratio (Wang et al., 2012). Through AMPK inhibition, DOX may also attenuate mitochondrial biogenesis and oxidative metabolism via PGC-1α, reduce autophagy/mitophagy via ULK1, and increase fibrosis via TGF-β signaling, i.e., all factors that could contribute to DIC (Timm and Tyler, 2020). AMPK inhibition can at least partially explain metabolic alterations in DOX-treated cardiomyocytes, namely reduced fatty acid oxidation and oxidative phosphorylation, accompanied by persistent glycolysis, potentially culminating in energetic failure (Russo et al., 2021).
miR-451 might be involved in DOX-induced AMPK inhibition, potentially contributing to DIC (Li et al., 2019a). In DOX-treated mouse cardiomyocytes, the level of miR-451 was significantly enhanced, and the inhibition of miR-451 suppressed cardiomyocyte death and functional compromise (Li et al., 2019a). miR-451 attenuates AMPK signaling, and AMPK inhibition abolishes benefits obtained by miR-451 (Li et al., 2019a). These results indicate that DOX-induced miR-451 upregulation contributes to AMPK inhibition and DIC. Mechanistically, miR-451 might inhibit AMPK signaling by targeting MO25, a component of the upstream kinase of AMPK (Chen et al., 2012).
Another miRNA significantly upregulated by DOX that may alter AMPK signaling is miR-25. Recently, it was reported that miR-25 could exacerbate DOX-induced cardiac cell apoptosis and ROS production via targeting phosphatase and tensin homolog deleted on chromosome 10 (PTEN) (Li et al., 2020d). PTEN is a negative regulator of AKT signaling, and its loss in the heart leads to impaired AMPK signaling (Roe et al., 2015). Therefore, DOX-induced miR-25 upregulation might attenuate AMPK signaling via targeting PTEN. However, as mentioned in the next section, PTEN inhibition and AKT activation have also been reported to be cardioprotective against DIC (Yu et al., 2020; Meng and Xu, 2022). Further testing is required to determine whether the harmful effect of miR-25 depends on the alteration of AMPK or AKT pathways.
AKT pathway
DOX treatment can initially (2–8 h after treatment) transiently activate and later inhibit the pro-survival PI3K/AKT pathway (Lee et al., 2006; Das et al., 2011; Sahu et al., 2019). There is abundant evidence that suggests upregulating AKT signaling can ameliorate cardiomyocyte apoptosis in DIC (Taniyama and Walsh, 2002; Das et al., 2011; Maruyama et al., 2011; Guo et al., 2013; Cao et al., 2014; Sahu et al., 2019; Zhang et al., 2019; Li et al., 2020a; Zhang et al., 2020; Liao et al., 2022). AKT inhibition can be achieved by p53, which can transcriptionally activate PTEN, the AKT inhibitor (Feng et al., 2007). p53 has also been reported to inhibit mTOR signaling, a primary downstream target of AKT, which could lead to the loss of myocardial mass in DOX-treated hearts (Zhu et al., 2009). Interestingly, DOX-induced AKT inhibition can also be mediated by two miRNAs upregulated by DOX, miR-143 and miR-375 (Li et al., 2020b; Liu et al., 2020b). miR-143 upregulation exacerbates oxidative stress and cardiomyocyte apoptosis by inhibiting AKT (Li et al., 2020c). miR-375 upregulation was similarly associated with oxidative stress and cardiomyocyte apoptosis by directly targeting 3-phosphoinositide-dependent protein kinase 1 (PDK1), an AKT activator (Liu et al., 2020b). Inhibition of miR-143 or miR-375 attenuates cardiomyocyte apoptosis in an AKT-dependent fashion (Liu et al., 2020b; Li et al., 2020c).
Meanwhile, some miRNAs have been reported as AKT activators and may counteract DIC. These are, for example, miR-495-3p, miR-17-5p, and miR-21 (Tong et al., 2015; Yu et al., 2020; Meng and Xu, 2022). MiR-495-3p expression is downregulated in DOX-treated hearts, and it targets PTEN to activate AKT signaling (Meng and Xu, 2022). Unlike miR-25, this PTEN inhibition attenuated DOX-induced oxidative stress and DIC. Similarly, miR-17-5p also targets PTEN (Yu et al., 2020). MiR-17-5p may partially be responsible for the cardioprotective effect of dexrazoxane against DOX-induced apoptosis, as miR-17-5p is upregulated by dexrazoxane treatment (Yu et al., 2020). Meanwhile, the expression of miR-21 is increased in cardiomyocytes after DOX treatment. It targets PTEN and B cell translocation gene 2 (BTG2), another negative regulator of the AKT pathway (Yang et al., 2014; Tong et al., 2015).
GATA-4 pathway
DOX treatment inhibits GATA-4 activity, another critical survival factor that resists DIC (Kim et al., 2003; Kobayashi et al., 2010; Lenčová-Popelová et al., 2014). GATA-4 enhances anti-apoptotic Bcl2 expression, leading to reduced apoptosis and suppressing deleterious DOX-induced autophagy. (Aries et al., 2004; Kobayashi et al., 2010). GATA-4 also plays a role in cellular senescence, activated by ATM and Rad3-related (ATR) upon DNA damage (Kang et al., 2015). This can indicate that GATA-4 action might lead to cardiac senescence and functional impairment. Whether GATA-4 activation is a viable therapeutic strategy remains elusive.
DOX-induced GATA-4 suppression can be mediated by p53, which suppresses CBF/NF-Y binding to the CCAAT box of the promotor region of GATA-4 (Park et al., 2011). It could also be given by miR-208a upregulation, directly targeting GATA-4 (Tony et al., 2015). Hence, miR-208a silencing counteracts such DOX-induced functional compromise (Tony et al., 2015). However, contradicting reports exist regarding miR-208a expression after DOX treatment. Vacchi-Suzzi et al. described downregulation in rat hearts rather than upregulation (Vacchi-Suzzi et al., 2012). Meanwhile, miR-199a-3p is another miRNA that directly targets GATA-4, but its level was attenuated in cardiomyocytes by DOX treatment rather than increased (Xia et al., 2021). miR-199a-3p attenuated cardiomyocytes’ senescence and promoted cell proliferation, indicating a potential therapeutic effect in DIC (Xia et al., 2021).
Sirtuin pathways
Sirtuins are a family of highly conserved NAD+-dependent deacetylases and ADP-ribosyltransferases that play an essential role in metabolic regulation and DNA damage repair. There are currently seven members in this family, denoted SIRT1-7, localized in the cytoplasm (SIRT1, 2) or mitochondria (SIRT3, 4, 5) or the nucleus (SIRT1, 2, 6, 7) (Matsushima and Sadoshima, 2015; Mei et al., 2016). Sirtuins could serve a pro-survival role in cardiomyocytes, and at least the involvement of SIRT1, 2, 3, and 6 have been implicated in counteracting DIC (Christidi and Brunham, 2021; Li et al., 2021).
SIRT1 expression is downregulated by DOX exposure in neonatal rat cardiomyocytes, a change associated with increased oxidative stress and apoptosis (Ruan et al., 2015). SIRT1 overexpression or pharmacological activation attenuates DIC through various downstream targets. Specifically, pro-apoptotic p53, as well as p38MAPK and p66shc, are inhibited by SIRT1, while the cardioprotective AMPK is activated by SIRT1 via liver kinase B1 (LKB1) and sestrin 2 action (Zhang et al., 2011; Ruan et al., 2015; Wang et al., 2017, 2022; Wu et al., 2019). In addition to the AMPK pathway, SIRT1 activates cardioprotective PGC-1α and nuclear factor erythroid 2-related factor 2 (NRF2). It inhibits NOD-like receptor family pyrin domain-containing protein 3 (NLRP3) inflammasome and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) (Wang(a) et al., 2021). PGC-1α increases mitochondrial biogenesis, while NRF2 transcriptionally activates antioxidant enzymes, thereby contributing to the attenuation of oxidative stress and the alleviation of DIC (Li et al., 2014b; Guo et al., 2014; Zhang et al., 2021a). Meanwhile, NLRP3 inflammasome induces pyroptosis, exacerbating DIC (Meng et al., 2019; Sun et al., 2020).
DOX may inhibit this pro-survival SIRT1 pathway downstream to DNA damage. For example, p53 may inhibit SIRT1 via activating hypermethylation in cancer 1 (HIC1) (Wales et al., 1995; Chen et al., 2005). DNA damage-induced poly(ADP-ribose) polymerases (PARPs) activation can deplete NAD+, which compromises SIRT1 function (Pillai et al., 2005; Khadka et al., 2018). In addition to these pathways, at least a part of the mechanism involves miRNAs activated by p53 to target SIRT1, namely miR-34a, miR-22, and miR-128 (Adlakha and Saini, 2013; Zhu et al., 2017; Xu et al., 2020; Wang J et al., 2021). As previously mentioned, DNA damage by p53 can activate these miRNAs, which regulate p53 activity via SIRT1, or target other apoptosis genes. Interestingly, genotoxic stress has been reported to induce ATM-dependent activation of SIRT1, which, by deacetylating histones H1 and H4, promotes the recruitment of DNA repair machinery (Mei et al., 2016). Therefore, SIRT1 inhibition in the DOX-treated hearts may impair DNA repair and potentiate the cell towards apoptotic death.
SIRT2 might also serve a cardioprotective function via attenuating oxidative stress by deacetylation/activation of forkhead box O3a (FOXO3a), which upregulates manganese superoxide dismutase (MnSOD) (Matsushima and Sadoshima, 2015). As reported by Zhao et al., the SIRT2 expression is attenuated by DOX treatment and Nrf2 by the action of miR-140-5p in cardiomyocytes, thereby leading to increased oxidative stress (Zhao et al., 2018).
SIRT3 suppresses Bcl-2-like 19 kDa-interacting protein 3 (Bnip3), a critical contributor to DIC, by promoting mitophagy and apoptosis (Dhingra et al., 2014; Saito and Sadoshima, 2015; Du et al., 2017). Although no studies report miRNAs targeting SIRT3 in the context of DIC, to our knowledge, miR-195 may be a candidate. MiR-195 is upregulated in human cardiomyocyte cell lines by DOX treatment, and it might promote cardiomyocyte apoptosis targeting Bcl-2 and SIRT3 (Zhang et al., 2018; Chen et al., 2019a).
SIRT6 attenuates DIC by upregulating endogenous antioxidant levels and attenuating apoptosis (Wu et al., 2021). The latter is achieved by SIRT6 interaction with p53, which leads to the repression of p53 transcription and binding to the Fas/FasL promotor region to reduce apoptotic signaling (Wu et al., 2021). SIRT6 also enhances GATA-4 chromatin binding capacity via TIP60 recruitment, counteracting DOX-induced apoptosis (Peng et al., 2020). SIRT6 is targeted by miR-330-5p, upregulated in DOX-treated cardiomyocytes (Han et al., 2020). MiR-330-5p exacerbates DIC by inhibiting SIRT6 and the anti-apoptotic survivin and sarcoplasmic reticulum Ca2+-ATPase 2a (SERCA2a) (Han et al., 2020). The miR-330-5p sponge circular RNA ITCH (CircITCH) prevents DIC (Han et al., 2020).
miRNAs involved in DOX-induced mitochondrial fission and mitophagy
DIC is also caused by altered mitochondrial dynamics (Osataphan et al., 2020). Specifically, DOX treatment leads to dynamin-related protein 1 (Drp1)-dependent mitochondrial fission, resulting in enhanced apoptosis propensity (Catanzaro et al., 2019). Indeed, when mitochondrial fission is inhibited through Drp1 knockdown or by the mitochondrial division inhibitor (mdivi-1), it attenuates cardiac cell death and DOX-induced functional compromise (Gharanei et al., 2013; Catanzaro et al., 2019).
Mitochondrial fission can be induced by p53, which can transcriptionally activate Drp1 (Li et al., 2010). This process is counteracted by miR-30 and miR-499-5p, two cardioprotective miRNAs downregulated in the heart after DOX treatment (Li et al., 2010; Roca-Alonso et al., 2015; Wan et al., 2019). miR-30 family downregulates p53 expression, reducing Drp1-mediated mitochondrial fission and apoptosis (Li et al., 2010). Of note, miR-30 can also counteract DIC by targeting β1-and β2-adrenoceptors (β1AR, β2AR) and Giα-2 of the β-Adrenergic signaling pathway, as well as the pro-apoptotic BNIP3L/NIX (Roca-Alonso et al., 2015). Furthermore, miR-30a and miR-30e target Beclin-1 to preserve cardiomyocyte autophagy and avoid DOX-induced apoptosis (Lai et al., 2017; Zhang et al., 2021c). Meanwhile, miR-499-5p directly targets p21 and both α- and β-Isoforms of the calcineurin subunits, which favor mitochondrial fission by increasing Drp1 dephosphorylation and accumulation in mitochondria (Wang et al., 2010; Wan et al., 2019). Interestingly, miR-499 expression is transcriptionally suppressed by p53 (Wang et al., 2010). To summarize, DOX treatment downregulates miR-30 while also causing DNA damage, leading to p53 activation. p53, in turn, suppresses miR-499-5p as well as transcriptionally activating Drp1, resulting in increased mitochondrial fission and apoptosis.
DOX-induced mitochondrial fragmentation is also mediated by miR-532-3p, miR-23a, and miR-140 (Li et al., 2014a; Wang et al., 2015; Du et al., 2019). miR-532-3p is upregulated by DOX and targets apoptosis repressor with caspase recruitment domain (ARC), a critical negative regulator of mitochondrial fission and apoptosis (Wang et al., 2015). This miR-532-3p-ARC axis of DOX-induced apoptosis was reported to be present only in cardiomyocytes but not in cancer cells. In addition to the pro-apoptotic role via binding to p53, DOX-induced miR-23a expression favors mitochondrial fission and cardiomyocyte apoptosis by directly targeting PGC-1α, an inhibitor of Drp1 phosphorylation and activation (Du et al., 2019). Meanwhile, miR-140, another miRNA upregulated by DOX in cardiomyocytes, targets and downregulates the pro-fusion protein mitofusin 1 (Mfn1) rather than manipulating Drp1 activity, aiding in mitochondrial fission and cardiomyocyte apoptosis (Li et al., 2014a).
Another aspect of mitochondrial dysfunction in DIC is dysregulated mitophagy. Mitophagy refers to mitochondrial autophagy, or the selective engulfment of damaged mitochondria into autophagosomes for lysosomal degradation (Bravo-San Pedro et al., 2017). Impairing mitophagy leads to the accumulation of damaged mitochondria and compromises cellular bioenergetics. In cardiomyocytes, two significant pathways induce mitophagy: PTEN-induced kinase 1 (PINK1)/Parkin pathway and Bnip3/NIX pathway (Koleini and Kardami, 2017). While Bnip3/NIX-dependent mitophagy’s role in DIC is elusive, contradictory findings have been reported for the PINK1/Parkin pathway in DIC. Some of them described that DIC is due to DOX attenuating Parkin-dependent mitophagy (Hoshino et al., 2013), while others the opposite (Yin et al., 2018; Catanzaro et al., 2019). Hoshino et al. (2013) showed that DOX upregulates cytosolic p53, which directly binds to Parkin and prevents its translocation to mitochondria. They, therefore, suggest that increased mitophagy may be therapeutic for DIC. In contrast, Yin et al. (2018) and Catanzaro et al. (2019) reported DOX activates Parkin-mediated mitophagy, and inhibition of mitophagy by mdivi-1 or Parkin knockdown mitigated DIC. The discrepancies may be due to differences in experimental conditions.
Reports on the role of miRNA in DOX-induced mitophagy are scarce. One recent study showed that miR-147-y may counter DOX-induced cardiomyocyte death by augmenting mitophagy (Gao et al., 2022). miR-147-y achieves its effect by targeting Raptor, a subunit of mTOR complex 1 (mTORC1) and a potential negative regulator of mitophagy (Gao et al., 2022). Another recent article found that DOX suppresses mitophagy by reducing miR-152-3p expression via DNA methyltransferase 1 (DNMT1) (Deng et al., 2022). When DNMT1 is inactive, miR-153-3p enhances mitophagy by targeting E26 transformation specific-1 (ETS1) (Deng et al., 2022). Both studies seem to support the idea that DOX suppresses cardiac mitophagy.
miRNAs involved in DOX-induced ferroptosis
The close relationship between cardiac iron accumulation and DIC has been widely discussed in the literature. DOX quinone moiety redox cycling and inactivation of iron regulatory proteins (IRP1/2) have been reported as the primary mechanistic connection (Minotti et al., 2001; Xu et al., 2005; Šimůnek et al., 2009; Gammella et al., 2014). More recently, DIC has been mainly linked to mitochondrial iron accumulation (Ichikawa et al., 2014). Mechanistically, the dysfunction of the mitochondrial iron exporter ABCB8 correlates with mitochondrial iron level, cellular ROS, and DIC severity (Ichikawa et al., 2014; Menon and Kim, 2022). Impaired ABCB8 function could be induced by iron overload, resulting in increased DOX accumulation in the mitochondria (Menon and Kim, 2022). Mitochondrial iron exacerbated DIC independently on Top2B.
Iron overload is associated with cardiomyocyte ferroptosis (Sumneang et al., 2020). Ferroptosis is a relatively recently discovered mechanism of RCD, and it involves intracellular iron-dependent accumulation of lipid peroxides and impaired antioxidant activity of glutathione peroxidase 4 (GPx4) (Dixon et al., 2012; Li et al., 2020a). It was recently reported that DOX downregulates GPx4 in addition to forming a complex with iron in the mitochondria to promote lipid peroxidation, culminating in mitochondria-dependent ferroptosis (Tadokoro et al., 2020). Significantly, the inhibition of ferroptosis by ferrostatin-1 combined with the inhibition of apoptosis by zVAD-FMK prevented DOX-induced cardiomyocyte death, while using either one only achieved partial prevention (Tadokoro et al., 2020). Since ferrostatin-1 alone or zVAD-FMK alone both seem to account for a significant fraction of DOX-induced cardiomyocyte death, both ferroptosis and apoptosis appear to be the primary forms of cell death in DIC (Tadokoro et al., 2020).
Ferroptosis could also be potentiated by p53, although the interaction is complex, with some p53 downstream targets favoring and others inhibiting ferroptosis (Liu et al., 2020a; Liu and Gu, 2022). Part of the pro-ferroptosis mechanism concerns p53’s impact on cysteine metabolism. Cysteine is an essential component of glutathione (GSH), the main antioxidant required for GPx4 activity. Therefore, a lack of intracellular cysteine leads to the potentiation of ferroptosis (Badgley et al., 2020; Poltorack and Dixon, 2022). Specifically, p53 represses the expression of SLC7A11, a cystine/glutamate transporter, leading to reduced cystine (Jiang et al., 2015). p53 also represses ELAVL1 expression, stabilizing the posttranscriptional level of LINC00336, an endogenous sponge for miR-6852 (Wang et al., 2019). miR-6852 targets cystathionine-β-synthase (CBS) mRNA, which codes the enzyme converting homocysteine to cystathionine reducing intracellular cysteine. Furthermore, p53 also induces the long noncoding RNA (lncRNA) PVT1, an endogenous sponge for miR-214 (Lu et al., 2020; Olivero et al., 2020). miR-214 attenuates ferroptosis via directly targeting transferrin receptor 1 (TfR1), PVT1, and p53 (Lu et al., 2020). However, others report that miR-214-3p enhances ferroptosis by targeting activating transcription factor 4 (ATF4) (Bai et al., 2020). AFT4 is a transcriptional activator that counteracts ferroptosis through its downstream targets, namely SLC7A11 and heat shock protein family A member 5 (HSPA5), which is a GPx4 activator (Chen et al., 2017b; Chen et al., 2017a; Chen et al., 2019b). The role of these miRNAs is yet to be reported in the context of DOX-induced ferroptosis, a topic that awaits future studies.
NRF2 might be another target of regulation by miRNAs in DOX-induced cardiomyocyte ferroptosis. NRF2 is a transcriptional activator of antioxidant proteins, and its action counteracts ferroptosis by preventing lipid peroxidation and free iron accumulation (Dodson et al., 2019). In addition to this anti-ferroptotic effect, NRF2 can also ameliorate DIC by maintaining autophagy (Li et al., 2014b). NRF2 action is enhanced by miR-152, which directly targets Keap1, the inhibitor of NRF2 (Zhang et al., 2021a). miR-152 ameliorates DIC by attenuating oxidative stress and apoptosis, potentially preventing ferroptosis (Zhang et al., 2021a). Furthermore, miR-152 may suppress ferroptosis by targeting TfR1 (Kindrat et al., 2016). Interestingly, NRF2 also transcriptionally represses miR-214, disinhibiting AFT4 and preventing ferroptosis (Gao et al., 2016).
The miRNA regulation of DOX-induced ferroptosis is a poorly understood topic, and the significance of the miRNAs mentioned above is speculative. One miRNA that has been reported to participate in DOX-induced ferroptosis is miR-7-5p. DOX treatment upregulates methyltransferase-like 14 (METTL14), which stabilizes lncRNA KCNQ1OT1, a miR-7-5p sponge (Zhuang et al., 2021). Since miR-7-5p directly targets TfR1, KCNQ1OT1 action disinhibits TfR1, contributing to iron accumulation and ferroptosis. Indeed, miR-7-5p mimic prevented DOX-induced iron accumulation and lipid ROS generation (Zhuang et al., 2021).
MiRNA-based therapeutics for cancer
Increasing evidence shows that altered miRNA expression profiles and unique miRNA signaling pathways are present in different types of cancers. Some miRNAs can function as oncogenes (known as oncomiRs) or tumor suppressors (TS-miRs) during tumor development and progression (Garzon et al., 2009; Lee and Dutta, 2009; Ventura and Jacks, 2009; Wang and Wu, 2012; di Leva et al., 2014). OncomiRs such as the miR-17–92 cluster, miR-21, miR-106a, miR-155, miR-372, and miR-373 target and inhibit various tumor suppressor genes, thereby enhancing cancer survival and proliferation (Zhang et al., 2007; Wang and Wu, 2012). It is not surprising that some of them, such as miR-17-5p and miR-21, also favor cell survival and mitigate DIC in the heart (Tong et al., 2015; Yu et al., 2020). On the other hand, TS-miRs such as miR-15a/16-1, Let-7, and miR-34a counter cell proliferation and favor apoptosis (Zhang et al., 2007; Lee and Dutta, 2009; Ventura and Jacks, 2009; Wang and Wu, 2012). Some of them, such as miR-34a, may also exacerbate cardiomyocyte apoptosis (Piegari et al., 2016, 2020). In the recent decade, anti-oncomiRs and TS-miR mimics have emerged as novel therapeutic strategies to normalize the gene regulatory network and signaling pathways and reverse the phenotype in cancer cells (Forterre et al., 2020). For example, the TS-miR miR-34a is considered to be a potential tumor marker and a promising cancer therapeutic candidate (Kalfert et al., 2020). Although the phase 1 study of MRX34, a liposomal miR-34a mimic, in patients with advanced solid tumors was closed early, miRNA-based gene therapy provides an attractive anti-tumor approach for integrated cancer therapy (Hong et al., 2020). If these therapeutics were to be combined with DOX, specific anti-cancer miRNAs might simultaneously exacerbate DIC. On the other hand, miRNAs that may mitigate DIC can also enhance cancer cell survival. Therefore, it is crucial to develop a highly specific delivery system that minimizes unintentional miRNA effects.
Conclusion
DNA damage response is increasingly recognized as the essential pathogenic mechanism of DIC, which should be reflected in our current understanding of the role of miRNAs. Increasing evidence points to the hypothesis that excess genotoxic stress in the heart due to DOX treatment may alter miRNA expression to promote RCDs, mainly apoptosis and ferroptosis. We have reviewed evidence suggesting miRNAs may impact RCDs via directly modulating p53 dynamics and various pro-survival signaling pathways (Figure 1; Table 1). Counteracting RCD-promoting miRNA changes, therefore, may prevent DIC. Certain aspects of this hypothesis, such as the mutual regulation between p53 and miRNAs in DIC, still require further experimental confirmation.
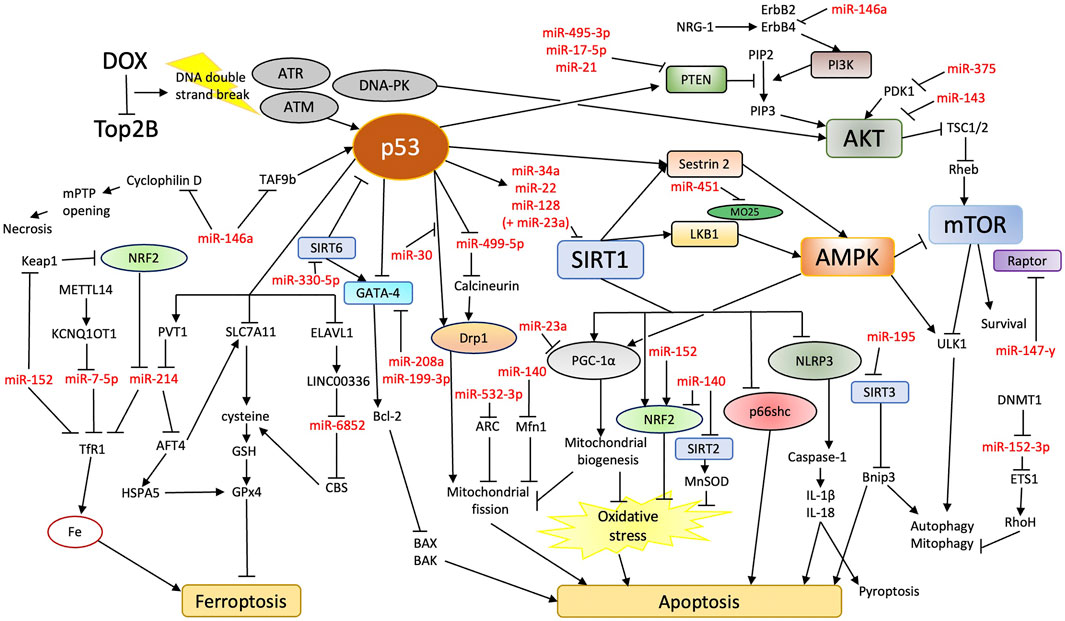
FIGURE 1. Overview of potential miRNA involvement in DIC. DOX treatment increases apoptosis and ferroptosis in cardiomyocytes. Rather than oxidative stress, DOX inhibition of Top2B and subsequent DNA damage response may be the primary contributor to cardiomyocyte death. miRNAs may aid in the regulation of molecular pathways that connect DNA damage response and apoptosis/ferroptosis.
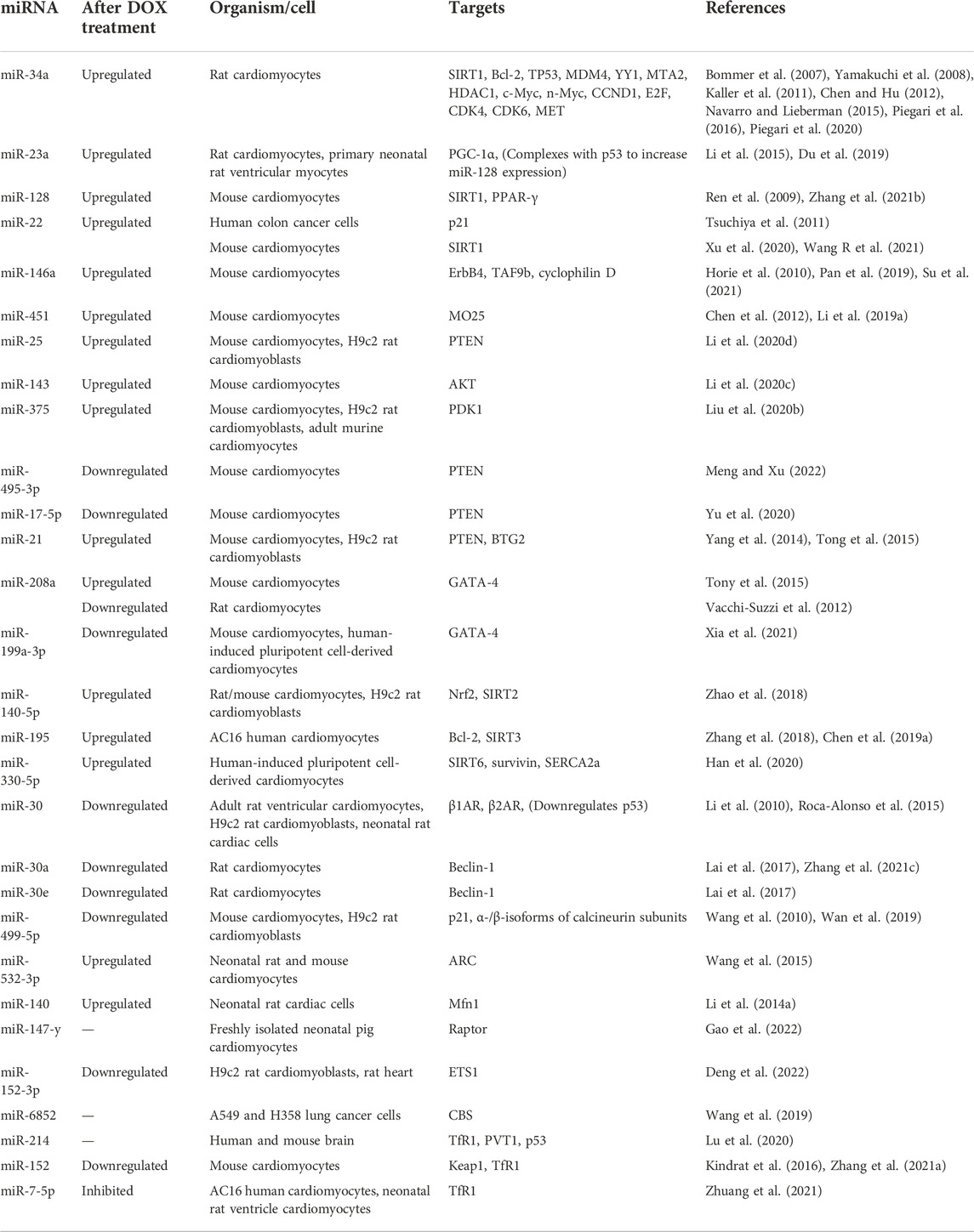
TABLE 1. List of miRNAs potentially involved in DIC.
It is also necessary to highlight the effects of the same miRNA in proliferating cancer cells and non-proliferating cardiomyocytes. Although the role of miRNAs in carcinogenesis is relatively well understood, the data about the DIC are inconsistent. Furthermore, mice and rat models are currently predominant in the literature, so confirming the findings in human patients would be ultimately required for clinical translation.
Author contributions
Conceptualization, MA; Writing of the original draft, IK; Preparation of the figures and the table, IK; Review and editing, MA and IK; Funding acquisition, MA. Both authors have read and agreed to the final version of the manuscript.
Funding
This work was supported by the Project InoMed CZ.02.1.01/0.0/0.0/18_069/0010046 co-funded by the ERDF.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adlakha, Y. K., and Saini, N. (2013). miR-128 exerts pro-apoptotic effect in a p53 transcription-dependent and -independent manner via PUMA-Bak axis. Cell. Death Dis. 4, e542. doi:10.1038/cddis.2013.46
Amirinejad, R., Rezaei, M., and Shirvani-Farsani, Z. (2020). An update on long intergenic noncoding RNA p21: A regulatory molecule with various significant functions in cancer. Cell. Biosci. 10, 82–89. doi:10.1186/s13578-020-00445-9
Aries, A., Paradis, P., Lefebvre, C., Schwartz, R. J., and Nemer, M. (2004). Essential role of GATA-4 in cell survival and drug-induced cardiotoxicity. Proc. Natl. Acad. Sci. U. S. A. 101, 6975–6980. doi:10.1073/PNAS.0401833101
Badgley, M. A., Kremer, D. M., Carlo Maurer, H., DelGiorno, K. E., Lee, H. J., Purohit, V., et al. (2020). Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 368, 85–89. doi:10.1126/science.aaw9872
Bai, T., Liang, R., Zhu, R., Wang, W., Zhou, L., and Sun, Y. (2020). MicroRNA-214-3p enhances erastin-induced ferroptosis by targeting ATF4 in hepatoma cells. J. Cell. Physiol. 235, 5637–5648. doi:10.1002/JCP.29496
Bansal, N., Adams, M. J., Ganatra, S., Colan, S. D., Aggarwal, S., Steiner, R., et al. (2019). Strategies to prevent anthracycline-induced cardiotoxicity in cancer survivors. Cardiooncology. 5, 18–22. doi:10.1186/S40959-019-0054-5
Bian, Y., Sun, M., Silver, M., Ho, K. K. L., Marchionni, M. A., Caggiano, A. O., et al. (2009). Neuregulin-1 attenuated doxorubicin-induced decrease in cardiac troponins. Am. J. Physiol. Heart Circ. Physiol. 297, H1974–H1983. doi:10.1152/ajpheart.01010.2008
Boehme, K. A., Kulikov, R., and Blattner, C. (2008). p53 stabilization in response to DNA damage requires Akt/PKB and DNA-PK. Proc. Natl. Acad. Sci. U. S. A. 105, 7785–7790. doi:10.1073/pnas.0703423105
Bommer, G. T., Gerin, I., Feng, Y., Kaczorowski, A. J., Kuick, R., Love, R. E., et al. (2007). p53-Mediated activation of miRNA34 candidate tumor-suppressor genes. Curr. Biol. 17, 1298–1307. doi:10.1016/j.cub.2007.06.068
Bozulic, L., Surucu, B., Hynx, D., and Hemmings, B. A. (2008). PKBalpha/Akt1 acts downstream of DNA-PK in the DNA double-strand break response and promotes survival. Mol. Cell. 30, 203–213. doi:10.1016/j.molcel.2008.02.024
Bravo-San Pedro, J. M., Kroemer, G., and Galluzzi, L. (2017). Autophagy and mitophagy in cardiovascular disease. Circ. Res. 120, 1812–1824. doi:10.1161/CIRCRESAHA.117.311082
Cao, Y., Ruan, Y., Shen, T., Huang, X., Li, M., Yu, W., et al. (2014). Astragalus polysaccharide suppresses doxorubicin-induced cardiotoxicity by regulating the PI3k/Akt and p38MAPK pathways. Oxid. Med. Cell. Longev. 2014, 674219. doi:10.1155/2014/674219
Cardinale, D., Colombo, A., Bacchiani, G., Tedeschi, I., Meroni, C. A., Veglia, F., et al. (2015). Early detection of anthracycline cardiotoxicity and improvement with heart failure therapy. Circulation 131, 1981–1988. doi:10.1161/CIRCULATIONAHA.114.013777
Catanzaro, M. P., Weiner, A., Kaminaris, A., Li, C., Cai, F., Zhao, F., et al. (2019). Doxorubicin-induced cardiomyocyte death is mediated by unchecked mitochondrial fission and mitophagy. FASEB J. 33, 11096–11108. doi:10.1096/FJ.201802663R
Chang, T. C., Wentzel, E. A., Kent, O. A., Ramachandran, K., Mullendore, M., Lee, K. H., et al. (2007). Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol. Cell. 26, 745–752. doi:10.1016/J.MOLCEL.2007.05.010
Chen, D., Fan, Z., Rauh, M., Buchfelder, M., Eyupoglu, I. Y., and Savaskan, N. (2017a). ATF4 promotes angiogenesis and neuronal cell death and confers ferroptosis in a xCT-dependent manner. Oncogene 36, 5593–5608. doi:10.1038/ONC.2017.146
Chen, D., Rauh, M., Buchfelder, M., Eyupoglu, I. Y., and Savaskan, N. (2017b). The oxido-metabolic driver ATF4 enhances temozolamide chemo-resistance in human gliomas. Oncotarget 8, 51164–51176. doi:10.18632/ONCOTARGET.17737
Chen, F., and Hu, S. J. (2012). Effect of microRNA-34a in cell cycle, differentiation, and apoptosis: A review. J. Biochem. Mol. Toxicol. 26, 79–86. doi:10.1002/JBT.20412
Chen, H., Untiveros, G. M., McKee, L. A. K., Perez, J., Li, J., Antin, P. B., et al. (2012). Micro-RNA-195 and -451 regulate the LKB1/AMPK signaling Axis by targeting MO25. PLoS One 7, e41574. doi:10.1371/JOURNAL.PONE.0041574
Chen, S., Wang, J., and Zhou, Y. (2019a). Long non-coding RNA SNHG1 protects human AC16 cardiomyocytes from doxorubicin toxicity by regulating miR-195/Bcl-2 axis. Biosci. Rep. 39, 20191050. doi:10.1042/BSR20191050
Chen, W. Y., Wang, D. H., Yen, R. C., Luo, J., Gu, W., and Baylin, S. B. (2005). Tumor suppressor HIC1 directly regulates SIRT1 to modulate p53-dependent DNA-damage responses. Cell. 123, 437–448. doi:10.1016/j.cell.2005.08.011
Chen, Y., Mi, Y., Zhang, X., Ma, Q., Song, Y., Zhang, L., et al. (2019b). Dihydroartemisinin-induced unfolded protein response feedback attenuates ferroptosis via PERK/ATF4/HSPA5 pathway in glioma cells. J. Exp. Clin. Cancer Res. 38, 402. doi:10.1186/s13046-019-1413-7
Chen, Y., Yingjie, X. U., Deng, Z., Wang, Y., Zheng, Y., Jiang, W., et al. (2021). MicroRNA expression profiling involved in doxorubicin-induced cardiotoxicity using high-throughput deep-sequencing analysis. Oncol. Lett. 22, 560–569. doi:10.3892/ol.2021.12821
Cheng, Q., and Chen, J. (2010). Mechanism of p53 stabilization by ATM after DNA damage. Cell. Cycle 9, 472–478. doi:10.4161/CC.9.3.10556
Christidi, E., and Brunham, L. R. (2021). Regulated cell death pathways in doxorubicin-induced cardiotoxicity. Cell. Death Dis. 12 (4 12), 339–415. doi:10.1038/s41419-021-03614-x
Concepcion, C. P., Han, Y. C., Mu, P., Bonetti, C., Yao, E., D’Andrea, A., et al. (2012). Intact p53-dependent responses in miR-34–deficient mice. PLoS Genet. 8, e1002797. doi:10.1371/JOURNAL.PGEN.1002797
Das, J., Ghosh, J., Manna, P., and Sil, P. C. (2011). Taurine suppresses doxorubicin-triggered oxidative stress and cardiac apoptosis in rat via up-regulation of PI3-K/Akt and inhibition of p53, p38-JNK. Biochem. Pharmacol. 81, 891–909. doi:10.1016/J.BCP.2011.01.008
Deng, S., Yan, T., Jendrny, C., Nemecek, A., Vincetic, M., Gödtel-Armbrust, U., et al. (2014). Dexrazoxane may prevent doxorubicin-induced DNA damage via depleting both Topoisomerase II isoforms. BMC Cancer 14, 842. doi:10.1186/1471-2407-14-842
Deng, Z., Yao, J., Xiao, N., Han, Y., Wu, X., Ci, C., et al. (2022). DNA methyltransferase 1 (DNMT1) suppresses mitophagy and aggravates heart failure via the microRNA-152-3p/ETS1/RhoH axis. Lab. Invest. 102, 782–793. doi:10.1038/s41374-022-00740-8
Dhingra, R., Guberman, M., Rabinovich-Nikitin, I., Gerstein, J., Margulets, V., Gang, H., et al. (2020). Impaired NF-κB signalling underlies cyclophilin D-mediated mitochondrial permeability transition pore opening in doxorubicin cardiomyopathy. Cardiovasc. Res. 116, 1161–1174. doi:10.1093/CVR/CVZ240
Dhingra, R., Margulets, V., Chowdhury, S. R., Thliveris, J., Jassal, D., Fernyhough, P., et al. (2014). MicroRNAs in cancer. Annu. Rev. Pathol. Mech. Dis. U. S. A. 111, E5537–E5544. doi:10.1073/PNAS.1414665111
di Leva, G., Garofalo, M., and Croce, C. M. (2014). microRNAs in cancer. Annu. Rev. Pathol. 9, 287–314. doi:10.1146/ANNUREV-PATHOL-012513-104715
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., et al. (2012). Ferroptosis: An iron-dependent form of non-apoptotic cell death. Cell. 149, 1060–1072. doi:10.1016/J.CELL.2012.03.042
Dodson, M., Castro-Portuguez, R., and Zhang, D. D. (2019). NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 23, 101107. doi:10.1016/J.REDOX.2019.101107
Dragoi, A.-M., Fu, X., Ivanov, S., Zhang, P., Sheng, L., Wu, D., et al. (2005). DNA-PKcs, but not TLR9, is required for activation of Akt by CpG-DNA. EMBO J. 24, 779–789. doi:10.1038/sj.emboj.7600539
Du, J., Hang, P., Pan, Y., Feng, B., Zheng, Y., Chen, T., et al. (2019). Inhibition of miR-23a attenuates doxorubicin-induced mitochondria-dependent cardiomyocyte apoptosis by targeting the PGC-1α/Drp1 pathway. Toxicol. Appl. Pharmacol. 369, 73–81. doi:10.1016/J.TAAP.2019.02.016
Du, Q., Zhu, B., Zhai, Q., and Yu, B. (2017). Sirt3 attenuates doxorubicin-induced cardiac hypertrophy and mitochondrial dysfunction via suppression of Bnip3. Am. J. Transl. Res. 9, 3360–3373.
Feng, Z., Hu, W., de Stanchina, E., Teresky, A. K., Jin, S., Lowe, S., et al. (2007). The regulation of AMPK beta1, TSC2, and PTEN expression by p53: Stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 67, 3043–3053. doi:10.1158/0008-5472.CAN-06-4149
Fiscella, M., Zhang, H., Fan, S., Sakaguchi, K., Shen, S., Mercer, W. E., et al. (1997). Wip1, a novel human protein phosphatase that is induced in response to ionizing radiation in a p53-dependent manner. Proc. Natl. Acad. Sci. U. S. A. 94, 6048–6053. doi:10.1073/pnas.94.12.6048
Forterre, A., Komuro, H., Aminova, S., and Harada, M. (2020). A comprehensive review of cancer MicroRNA therapeutic delivery strategies. Cancers (Basel) 12, 18522–E1921. doi:10.3390/CANCERS12071852
Fujimoto, H., Onishi, N., Kato, N., Takekawa, M., Xu, X. Z., Kosugi, A., et al. (2006). Regulation of the antioncogenic Chk2 kinase by the oncogenic Wip1 phosphatase. Cell. Death Differ. 13, 1170–1180. doi:10.1038/sj.cdd.4401801
Fukazawa, R., Miller, T. A., Kuramochi, Y., Frantz, S., Kim, Y. D., Marchionni, M. A., et al. (2003). Neuregulin-1 protects ventricular myocytes from anthracycline-induced apoptosis via erbB4-dependent activation of PI3-kinase/Akt. J. Mol. Cell. Cardiol. 35, 1473–1479. doi:10.1016/J.YJMCC.2003.09.012
Gammella, E., Maccarinelli, F., Buratti, P., Recalcati, S., and Cairo, G. (2014). The role of iron in anthracycline cardiotoxicity. Front. Pharmacol. 5, 25. doi:10.3389/FPHAR.2014.00025
Gao, C., Liu, H., and Yan, F. (2020). Dynamic behavior of p53 driven by delay and a microrna-34a-mediated feedback loop. Int. J. Mol. Sci. 21, E1271. doi:10.3390/IJMS21041271
Gao, H., Xian, G., Zhong, G., Huang, B., Liang, S., Zeng, Q., et al. (2022). Alleviation of doxorubicin-induced cardiomyocyte death through miR-147-y-mediated mitophagy. Biochem. Biophys. Res. Commun. 609, 176–182. doi:10.1016/j.bbrc.2022.04.013
Gao, M., Liu, Y., Chen, Y., Yin, C., Chen, J. J., and Liu, S. (2016). miR-214 protects erythroid cells against oxidative stress by targeting ATF4 and EZH2. Free Radic. Biol. Med. 92, 39–49. doi:10.1016/J.FREERADBIOMED.2016.01.005
Garzon, R., Calin, G. A., and Croce, C. M. (2009). MicroRNAs in cancer. Annu. Rev. Med. 60, 167–179. doi:10.1146/annurev.med.59.053006.104707
Gharanei, M., Hussain, A., Janneh, O., and Maddock, H. (2013). Attenuation of doxorubicin-induced cardiotoxicity by mdivi-1: A mitochondrial division/mitophagy inhibitor. PLoS One 8, e77713. doi:10.1371/JOURNAL.PONE.0077713
Ghigo, A., Li, M., and Hirsch, E. (2016). New signal transduction paradigms in anthracycline-induced cardiotoxicity. Biochim. Biophys. Acta 1863, 1916–1925. doi:10.1016/J.BBAMCR.2016.01.021
Gratia, S., Kay, L., Potenza, L., Seffouh, A., Novel-Chate, V., Schnebelen, C., et al. (2012). Inhibition of AMPK signalling by doxorubicin: At the crossroads of the cardiac responses to energetic, oxidative, and genotoxic stress. Cardiovasc. Res. 95, 290–299. doi:10.1093/CVR/CVS134
Guo, J., Guo, Q., Fang, H., Lei, L., Zhang, T., Zhao, J., et al. (2014). Cardioprotection against doxorubicin by metallothionein Is associated with preservation of mitochondrial biogenesis involving PGC-1α pathway. Eur. J. Pharmacol. 737, 117–124. doi:10.1016/J.EJPHAR.2014.05.017
Guo, R., Lin, J., Xu, W., Shen, N., Mo, L., Zhang, C., et al. (2013). Hydrogen sulfide attenuates doxorubicin-induced cardiotoxicity by inhibition of the p38 MAPK pathway in H9c2 cells. Int. J. Mol. Med. 31, 644–650. doi:10.3892/ijmm.2013.1246
Han, D., Wang, Y., Wang, Y., Dai, X., Zhou, T., Chen, J., et al. (2020). The tumor-suppressive human circular RNA CircITCH sponges miR-330-5p to ameliorate doxorubicin-induced cardiotoxicity through upregulating SIRT6, survivin, and SERCA2a. Circ. Res. 127, E108–E125. doi:10.1161/CIRCRESAHA.119.316061
He, L., He, X., Lim, L. P., de Stanchina, E., Xuan, Z., Liang, Y., et al. (2007). A microRNA component of the p53 tumour suppressor network. Nature 447, 1130–1134. doi:10.1038/NATURE05939
Hermeking, H. (2012). MicroRNAs in the p53 network: Micromanagement of tumour suppression. Nat. Rev. Cancer 12 (9 12), 613–626. doi:10.1038/nrc3318
Holmgren, G., Synnergren, J., Andersson, C. X., Lindahl, A., and Sartipy, P. (2016). MicroRNAs as potential biomarkers for doxorubicin-induced cardiotoxicity. Toxicol. Vitro 34, 26–34. doi:10.1016/J.TIV.2016.03.009
Hong, D. S., Kang, Y. K., Borad, M., Sachdev, J., Ejadi, S., Lim, H. Y., et al. (2020). Phase 1 study of MRX34, a liposomal miR-34a mimic, in patients with advanced solid tumours. Br. J. Cancer 122, 1630–1637. doi:10.1038/S41416-020-0802-1
Horenstein, M. S., vander Heide, R. S., and L’Ecuyer, T. J. (2000). Molecular basis of anthracycline-induced cardiotoxicity and its prevention. Mol. Genet. Metab. 71, 436–444. doi:10.1006/MGME.2000.3043
Horie, T., Ono, K., Nishi, H., Nagao, K., Kinoshita, M., Watanabe, S., et al. (2010). Acute doxorubicin cardiotoxicity is associated with miR-146a-induced inhibition of the neuregulin-ErbB pathway. Cardiovasc. Res. 87, 656–664. doi:10.1093/CVR/CVQ148
Hoshino, A., Mita, Y., Okawa, Y., Ariyoshi, M., Iwai-Kanai, E., Ueyama, T., et al. (2013). Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat. Commun. 4, 2308. doi:10.1038/ncomms3308
Huang, J., Wu, R., Chen, L., Yang, Z., Yan, D., and Li, M. (2022). Understanding anthracycline cardiotoxicity from mitochondrial aspect. Front. Pharmacol. 13, 291. doi:10.3389/fphar.2022.811406
Huarte, M., Guttman, M., Feldser, D., Garber, M., Koziol, M. J., Kenzelmann-Broz, D., et al. (2010). A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell. 142, 409–419. doi:10.1016/J.CELL.2010.06.040
Ichikawa, Y., Ghanefar, M., Bayeva, M., Wu, R., Khechaduri, A., Naga Prasad, S. v., et al. (2014). Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J. Clin. Invest. 124, 617–630. doi:10.1172/JCI72931
Issler, M. V. C., and Mombach, J. C. M. (2017). MicroRNA-16 feedback loop with p53 and Wip1 can regulate cell fate determination between apoptosis and senescence in DNA damage response. PLoS One 12, e0185794. doi:10.1371/journal.pone.0185794
Jackson, S. P., and Bartek, J. (2009). The DNA-damage response in human biology and disease. Nature 461, 1071–1078. doi:10.1038/nature08467
Jiang, L., Kon, N., Li, T., Wang, S. J., Su, T., Hibshoosh, H., et al. (2015). Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520, 57–62. doi:10.1038/nature14344
Jirkovská, A., Karabanovich, G., Kubeš, J., Skalická, V., Melnikova, I., Korábečný, J., et al. (2021). Structure-activity relationship study of dexrazoxane analogues reveals ICRF-193 as the most potent bisdioxopiperazine against anthracycline toxicity to cardiomyocytes due to its strong topoisomerase IIβ interactions. J. Med. Chem. 64, 3997–4019. doi:10.1021/acs.jmedchem.0c02157
Jirkovský, E., Jirkovská, A., Bavlovič-Piskáčková, H., Skalická, V., Pokorná, Z., Karabanovich, G., et al. (2021). Clinically translatable prevention of anthracycline cardiotoxicity by dexrazoxane is mediated by topoisomerase II beta and not metal chelation. Circ. Heart Fail. 14, e008209. doi:10.1161/CIRCHEARTFAILURE.120.008209
Kaiserová, H., den Hartog, G. J. M., Šimůnek, T., Schröterová, L., Kvasničková, E., and Bast, A. (2006). Iron is not involved in oxidative stress-mediated cytotoxicity of doxorubicin and bleomycin. Br. J. Pharmacol. 149, 920–930. doi:10.1038/SJ.BJP.0706930
Kalfert, D., Ludvikova, M., Pesta, M., Ludvik, J., Dostalova, L., and Kholová, I. (2020). Multifunctional roles of miR-34a in cancer: A review with the emphasis on head and neck squamous cell carcinoma and thyroid cancer with clinical implications. Diagnostics 10, 563. doi:10.3390/diagnostics10080563
Kaller, M., Liffers, S. T., Oeljeklaus, S., Kuhlmann, K., Röh, S., Hoffmann, R., et al. (2011). Genome-wide characterization of miR-34a induced changes in protein and mRNA expression by a combined pulsed SILAC and microarray analysis. Mol. Cell. Proteomics 10, M111.010462. doi:10.1074/MCP.M111.010462
Kalyanaraman, B. (2020). Teaching the basics of the mechanism of doxorubicin-induced cardiotoxicity: Have we been barking up the wrong tree? Redox Biol. 29, 101394. doi:10.1016/J.REDOX.2019.101394
Kang, C., Xu, Q., Martin, T. D., Li, M. Z., Demaria, M., Aron, L., et al. (2015). The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 349, aaa5612. doi:10.1126/science.aaa5612
Kang, Y., Shu, X. H., Sun, M. M., Pan, C. Z., and Ge, J. B. (2013). Two-dimensional speckle tracking echocardiography in early detection and prediction of cardiotoxocity during epirubicine based chemotherapy. Eur. Heart J. 34, P2974. doi:10.1093/EURHEARTJ/EHT309.P2974
Khadka, D., Kim, H.-J., Oh, G.-S., Shen, A., Lee, S., Lee, S.-B., et al. (2018). Augmentation of NAD+ levels by enzymatic action of NAD(P)H quinone oxidoreductase 1 attenuates adriamycin-induced cardiac dysfunction in mice. J. Mol. Cell. Cardiol. 124, 45–57. doi:10.1016/j.yjmcc.2018.10.001
Kim, Y., Ma, A. G., Kitta, K., Fitch, S. N., Ikeda, T., Ihara, Y., et al. (2003). Anthracycline-induced suppression of GATA-4 transcription factor: Implication in the regulation of cardiac myocyte apoptosis. Mol. Pharmacol. 63, 368–377. doi:10.1124/MOL.63.2.368
Kindrat, I., Tryndyak, V., Conti, A., Shpyleva, S., Mudalige, T. K., Kobets, T., et al. (2016). MicroRNA-152-mediated dysregulation of hepatic transferrin receptor 1 in liver carcinogenesis. Oncotarget 7, 1276–1287. doi:10.18632/ONCOTARGET.6004
Kobayashi, S., Volden, P., Timm, D., Mao, K., Xu, X., and Liang, Q. (2010). Transcription factor GATA4 inhibits doxorubicin-induced autophagy and cardiomyocyte death. J. Biol. Chem. 285, 793–804. doi:10.1074/JBC.M109.070037
Koleini, N., and Kardami, E. (2017). Autophagy and mitophagy in the context of doxorubicin-induced cardiotoxicity. Oncotarget 8, 46663–46680. doi:10.18632/ONCOTARGET.16944
Kollárová-Brázdová, P., Jirkovská, A., Karabanovich, G., Pokorná, Z., Piskáčková, H. B., Jirkovský, E., et al. (2020). Investigation of structure-activity relationships of dexrazoxane analogs reveals topoisomerase IIβ interaction as a prerequisite for effective protection against anthracycline cardiotoxicity. J. Pharmacol. Exp. Ther. 373, 402–415. doi:10.1124/JPET.119.264580
Kurz, E. U., Douglas, P., and Lees-Miller, S. P. (2004). Doxorubicin activates ATM-dependent phosphorylation of multiple downstream targets in part through the generation of reactive oxygen species *. J. Biol. Chem. 279, 53272–53281. doi:10.1074/JBC.M406879200
Lai, L., Chen, J., Wang, N., Zhu, G., Duan, X., and Ling, F. (2017). MiRNA-30e mediated cardioprotection of ACE2 in rats with Doxorubicin-induced heart failure through inhibiting cardiomyocytes autophagy. Life Sci. 169, 69–75. doi:10.1016/J.LFS.2016.09.006
Lakin, N. D., and Jackson, S. P. (1999). Regulation of p53 in response to DNA damage. Oncogene 18, 7644–7655. doi:10.1038/sj.onc.1203015
L’Ecuyer, T., Sanjeev, S., Thomas, R., Novak, R., Das, L., Campbell, W., et al. (2006). DNA damage is an early event in doxorubicin-induced cardiac myocyte death. Am. J. Physiol. Heart Circ. Physiol. 291, H1273–H1280. doi:10.1152/ajpheart.00738.2005
Lee, E. R., Kim, J. Y., Kang, Y. J., Ahn, J. Y., Kim, J. H., Kim, B. W., et al. (2006). Interplay between PI3K/Akt and MAPK signaling pathways in DNA-damaging drug-induced apoptosis. Biochim. Biophys. Acta 1763, 958–968. doi:10.1016/J.BBAMCR.2006.06.006
Lee, Y. S., and Dutta, A. (2009). MicroRNAs in cancer. Annu. Rev. Pathol. 4, 199–227. doi:10.1146/ANNUREV.PATHOL.4.110807.092222
Lenčová-Popelová, O., Jirkovský, E., Mazurová, Y., Lenčo, J., Adamcová, M., Šimůnek, T., et al. (2014). Molecular remodeling of left and right ventricular myocardium in chronic anthracycline cardiotoxicity and post-treatment follow up. PLoS One 9, e96055. doi:10.1371/JOURNAL.PONE.0096055
Lev Bar-Or, R., Maya, R., Segel, L. A., Alon, U., Levine, A. J., and Oren, M. (2000). Generation of oscillations by the p53-mdm2 feedback loop: A theoretical and experimental study. Proc. Natl. Acad. Sci. U. S. A. 97, 11250–11255. doi:10.1073/pnas.210171597
Lezina, L., Purmessur, N., Antonov, A., Ivanova, T., Karpova, E., Krishan, K., et al. (2013). miR-16 and miR-26a target checkpoint kinases Wee1 and Chk1 in response to p53 activation by genotoxic stress. Cell. Death Dis. 4, e953. doi:10.1038/cddis.2013.483
Li, D., Yang, Y., Wang, S., He, X., Liu, M., Bai, B., et al. (2021). Role of acetylation in doxorubicin-induced cardiotoxicity. Redox Biol. 46, 102089. doi:10.1016/J.REDOX.2021.102089
Li, J., Aung, L. H. H., Long, B., Qin, D., An, S., and Li, P. (2015). miR-23a binds to p53 and enhances its association with miR-128 promoter. Sci. Rep. 5 (1 5), 16422–16513. doi:10.1038/srep16422
Li, J., Cao, F., Yin, H., Huang, Z., Lin, Z., Mao, N., et al. (2020a). Ferroptosis: Past, present and future. Cell. Death Dis. 11 (2 11), 88–13. doi:10.1038/s41419-020-2298-2
Li, J., Donath, S., Li, Y., Qin, D., Prabhakar, B. S., and Li, P. (2010). miR-30 regulates mitochondrial fission through targeting p53 and the dynamin-related protein-1 pathway. PLoS Genet. 6, e1000795. doi:10.1371/JOURNAL.PGEN.1000795
Li, J., Li, Y., Jiao, J., Wang, J., Li, Y., Qin, D., et al. (2014a). Mitofusin 1 is negatively regulated by MicroRNA 140 in cardiomyocyte apoptosis. Mol. Cell. Biol. 34, 1788–1799. doi:10.1128/MCB.00774-13
Li, J., Wan, W., Chen, T., Tong, S., Jiang, X., and Liu, W. (2019a). MiR-451 silencing inhibited doxorubicin exposure-induced cardiotoxicity in mice. Biomed. Res. Int. 2019, 1528278. doi:10.1155/2019/1528278
Li, J., Wang, P. Y., Long, N. A., Zhuang, J., Springer, D. A., Zou, J., et al. (2019b). P53 prevents doxorubicin cardiotoxicity independently of its prototypical tumor suppressor activities. Proc. Natl. Acad. Sci. U. S. A. 116, 19626–19634. doi:10.1073/pnas.1904979116
Li, L. L., Li, L. L., Wei, L., Wei, L., Wei, L., Zhang, N., et al. (2020b). Levosimendan protects against doxorubicin-induced cardiotoxicity by regulating the PTEN/akt pathway. Biomed. Res. Int. 2020, 8593617. doi:10.1155/2020/8593617
Li, S., Wang, W., Niu, T., Wang, H., Li, B., Shao, L., et al. (2014b). Nrf2 deficiency exaggerates doxorubicin-induced cardiotoxicity and cardiac dysfunction. Oxid. Med. Cell. Longev. 2014, 748524. doi:10.1155/2014/748524
Li, W., Wang, X., Liu, T., Zhang, Q., Cao, J., Jiang, Y., et al. (2022). Harpagoside protects against doxorubicin-induced cardiotoxicity via P53-parkin-mediated mitophagy. Front. Cell. Dev. Biol. 10, 813370. doi:10.3389/fcell.2022.813370
Li, X. Q., Liu, Y. K., Yi, J., Dong, J. S., Zhang, P. P., Wan, L., et al. (2020c). MicroRNA-143 increases oxidative stress and myocardial cell apoptosis in a mouse model of doxorubicin-induced cardiac toxicity. Med. Sci. Monit. 26, 9203944–e920401. doi:10.12659/MSM.920394
Li, Z., Li, H., Liu, B., Luo, J., Qin, X., Gong, M., et al. (2020d). Inhibition of MiR-25 attenuates doxorubicin-induced apoptosis, reactive oxygen species production and DNA damage by targeting pten. Int. J. Med. Sci. 17, 1415–1427. doi:10.7150/IJMS.41980
Liao, Z.-Q., Jiang, Y.-N., Su, Z.-L., Bi, H.-L., Li, J.-T., Li, C.-L., et al. (2022). Rutaecarpine inhibits doxorubicin-induced oxidative stress and apoptosis by activating AKT signaling pathway. Front. Cardiovasc. Med. 0, 809689. doi:10.3389/FCVM.2021.809689
Lipshultz, S. E., Alvarez, J. A., and Scully, R. E. (2008). Anthracycline associated cardiotoxicity in survivors of childhood cancer. Heart 94, 525–533. doi:10.1136/HRT.2007.136093
Liu, J., Zhang, C., Wang, J., Hu, W., and Feng, Z. (2020a). The regulation of ferroptosis by tumor suppressor p53 and its pathway. Int. J. Mol. Sci. 21, 83877–E8419. doi:10.3390/IJMS21218387
Liu, L., Dai, X., Yin, S., Liu, P., Hill, E. G., Wei, W., et al. (2022). DNA-PK promotes activation of the survival kinase AKT in response to DNA damage through an mTORC2-ECT2 pathway. Sci. Signal. 15, eabh2290. doi:10.1126/scisignal.abh2290
Liu, T. H., Chen, W. H., Chen, X. D., Liang, Q. E., Tao, W. C., Jin, Z., et al. (2020b). The effects of inhibition of MicroRNA-375 in a mouse model of doxorubicin-induced cardiac toxicity. Med. Sci. Monit. 26, 9205577–e920561. doi:10.12659/MSM.920557
Liu, Y., and Gu, W. (2022). p53 in ferroptosis regulation: the new weapon for the old guardian. Cell. Death Differ. 29 (5 29), 895–910. doi:10.1038/s41418-022-00943-y
Lu, J., Xu, F., and Lu, H. (2020). LncRNA PVT1 regulates ferroptosis through miR-214-mediated TFR1 and p53. Life Sci. 260, 118305. doi:10.1016/J.LFS.2020.118305
Lyu, Y. L., Kerrigan, J. E., Lin, C.-P., Azarova, A. M., Tsai, Y.-C., Ban, Y., et al. (2007). Topoisomerase IIbeta mediated DNA double-strand breaks: Implications in doxorubicin cardiotoxicity and prevention by dexrazoxane. Cancer Res. 67, 8839–8846. doi:10.1158/0008-5472.CAN-07-1649
Marinello, J., Delcuratolo, M., and Capranico, G. (2018). Anthracyclines as topoisomerase II poisons: From early studies to new perspectives. Int. J. Mol. Sci. 19, E3480. doi:10.3390/IJMS19113480
Maruyama, S., Shibata, R., Ohashi, K., Ohashi, T., Daida, H., Walsh, K., et al. (2011). Adiponectin ameliorates doxorubicin-induced cardiotoxicity through akt protein-dependent mechanism. J. Biol. Chem. 286, 32790–32800. doi:10.1074/JBC.M111.245985
Matsushima, S., and Sadoshima, J. (2015). The role of sirtuins in cardiac disease. Am. J. Physiol. Heart Circ. Physiol. 309, H1375–H1389. doi:10.1152/AJPHEART.00053.2015
McSweeney, K. M., Bozza, W. P., Alterovitz, W. L., and Zhang, B. (2019). Transcriptomic profiling reveals p53 as a key regulator of doxorubicin-induced cardiotoxicity. Cell. Death Discov. 5 (1 5), 102–111. doi:10.1038/s41420-019-0182-6
Mei, Z., Zhang, X., Yi, J., Huang, J., He, J., and Tao, Y. (2016). Sirtuins in metabolism, DNA repair and cancer. J. Exp. Clin. Cancer Res. 35, 182. doi:10.1186/S13046-016-0461-5
Meng, J., and Xu, C. (2022). MicroRNA-495-3p diminishes doxorubicin-induced cardiotoxicity through activating AKT. J. Cell. Mol. Med. 26, 2076–2088. doi:10.1111/JCMM.17230
Meng, L., Lin, H., Zhang, J., Lin, N., Sun, Z., Gao, F., et al. (2019). Doxorubicin induces cardiomyocyte pyroptosis via the TINCR-mediated posttranscriptional stabilization of NLR family pyrin domain containing 3. J. Mol. Cell. Cardiol. 136, 15–26. doi:10.1016/J.YJMCC.2019.08.009
Menon, A. V., and Kim, J. (2022). Iron promotes cardiac doxorubicin retention and toxicity through downregulation of the mitochondrial exporter ABCB8. Front. Pharmacol. 13, 322. doi:10.3389/fphar.2022.817951
Mihalas, G. I., Simon, Z., Balea, G., and Popa, E. (2000). Possible oscillatory behavior in P53–MDM2 interaction computer simulation. J. Biol. Syst. 08, 21–29. doi:10.1142/S0218339000000031
Minotti, G., Menna, P., Salvatorelli, E., Cairo, G., and Gianni, L. (2004). Anthracyclines: Molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol. Rev. 56, 185–229. doi:10.1124/PR.56.2.6
Minotti, G., Ronchi, R., Salvatorelli, E., Menna, P., and Cairo, G. (2001). Doxorubicin irreversibly inactivates iron regulatory proteins 1 and 2 in cardiomyocytes: Evidence for distinct metabolic pathways and implications for iron-mediated cardiotoxicity of antitumor therapy. Cancer Res. 61, 8422–8428.
Mitry, M. A., and Edwards, J. G. (2016). Doxorubicin induced heart failure: Phenotype and molecular mechanisms. Int. J. Cardiol. Heart Vasc. 10, 17–24. doi:10.1016/J.IJCHA.2015.11.004
Navarro, F., and Lieberman, J. (2015). miR-34 and p53: New insights into a complex functional relationship. PLoS One 10, e0132767. doi:10.1371/JOURNAL.PONE.0132767
Novák, B., and Tyson, J. J. (2008). Design principles of biochemical oscillators. Nat. Rev. Mol. Cell. Biol. 9 (12 9), 981–991. doi:10.1038/nrm2530
Olivero, C. E., Martínez-Terroba, E., Zimmer, J., Liao, C., Tesfaye, E., Hooshdaran, N., et al. (2020). p53 activates the long noncoding RNA Pvt1b to inhibit myc and suppress tumorigenesis. Mol. Cell. 77, 761–774. e8. doi:10.1016/j.molcel.2019.12.014
Osataphan, N., Phrommintikul, A., Chattipakorn, S. C., and Chattipakorn, N. (2020). Effects of doxorubicin-induced cardiotoxicity on cardiac mitochondrial dynamics and mitochondrial function: Insights for future interventions. J. Cell. Mol. Med. 24, 6534–6557. doi:10.1111/JCMM.15305
Pan, J. A., Tang, Y., Yu, J. Y., Zhang, H., Zhang, J. F., Wang, C. Q., et al. (2019). miR-146a attenuates apoptosis and modulates autophagy by targeting TAF9b/P53 pathway in doxorubicin-induced cardiotoxicity. Cell. Death Dis. 10 (9 10), 668–715. doi:10.1038/s41419-019-1901-x
Park, A. M., Nagase, H., Liu, L., Vinod Kumar, S., Szwergold, N., Wong, C. M., et al. (2011). Mechanism of anthracycline-mediated down-regulation of GATA4 in the heart. Cardiovasc. Res. 90, 97–104. doi:10.1093/CVR/CVQ361
Peng, L., Qian, M., Liu, Z., Tang, X., Sun, J., Jiang, Y., et al. (2020). Deacetylase-independent function of SIRT6 couples GATA4 transcription factor and epigenetic activation against cardiomyocyte apoptosis. Nucleic Acids Res. 48, 4992–5005. doi:10.1093/NAR/GKAA214
Pereira, J. D., Tosatti, J. A. G., Simões, R., Luizon, M. R., Gomes, K. B., and Alves, M. T. (2020). microRNAs associated to anthracycline-induced cardiotoxicity in women with breast cancer: A systematic review and pathway analysis. Biomed. Pharmacother. 131, 110709. doi:10.1016/J.BIOPHA.2020.110709
Piegari, E., Angelis, A., Cappetta, D., Russo, R., Esposito, G., Costantino, S., et al. (2013). Doxorubicin induces senescence and impairs function of human cardiac progenitor cells. Basic Res. Cardiol. 108, 334. doi:10.1007/s00395-013-0334-4
Piegari, E., Cozzolino, A., Ciuffreda, L. P., Cappetta, D., de Angelis, A., Urbanek, K., et al. (2020). Cardioprotective effects of miR-34a silencing in a rat model of doxorubicin toxicity. Sci. Rep. 10 (1 10), 12250–12312. doi:10.1038/s41598-020-69038-3
Piegari, E., Russo, R., Cappetta, D., Esposito, G., Urbanek, K., Dell’Aversana, C., et al. (2016). MicroRNA-34a regulates doxorubicin-induced cardiotoxicity in rat. Oncotarget 7, 62312–62326. doi:10.18632/ONCOTARGET.11468
Pillai, J. B., Isbatan, A., Imai, S., and Gupta, M. P. (2005). Poly(ADP-ribose) polymerase-1-dependent cardiac myocyte cell death during heart failure is mediated by NAD+ depletion and reduced Sir2alpha deacetylase activity. J. Biol. Chem. 280, 43121–43130. doi:10.1074/jbc.M506162200
Pointon, A. v., Walker, T. M., Phillips, K. M., Luo, J., Riley, J., Zhang, S. D., et al. (2010). Doxorubicin in vivo rapidly alters expression and translation of myocardial electron transport chain genes, leads to ATP loss and caspase 3 activation. PLoS One 5, 127333–e12817. doi:10.1371/JOURNAL.PONE.0012733
Poltorack, C. D., and Dixon, S. J. (2022). Understanding the role of cysteine in ferroptosis: Progress & paradoxes. FEBS J. 289, 374–385. doi:10.1111/FEBS.15842
Pommier, Y., Leo, E., Zhang, H., and Marchand, C. (2010). DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol. 17, 421–433. doi:10.1016/J.CHEMBIOL.2010.04.012
Pudil, R., Mueller, C., Čelutkienė, J., Henriksen, P. A., Lenihan, D., Dent, S., et al. (2020). Role of serum biomarkers in cancer patients receiving cardiotoxic cancer therapies: A position statement from the cardio-oncology study group of the heart failure association and the cardio-oncology council of the European society of cardiology. Eur. J. Heart Fail. 22, 1966–1983. doi:10.1002/EJHF.2017
Purvis, J. E., Karhohs, K. W., Mock, C., Batchelor, E., Loewer, A., and Lahav, G. (2012). p53 dynamics control cell fate. Science 336, 1440–1444. doi:10.1126/science.1218351
Raver-Shapira, N., Marciano, E., Meiri, E., Spector, Y., Rosenfeld, N., Moskovits, N., et al. (2007). Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol. Cell. 26, 731–743. doi:10.1016/j.molcel.2007.05.017
Ren, Y., Sun, C., Sun, Y., Tan, H., Wu, Y., Cui, B., et al. (2009). PPAR gamma protects cardiomyocytes against oxidative stress and apoptosis via Bcl-2 upregulation. Vasc. Pharmacol. 51, 169–174. doi:10.1016/J.VPH.2009.06.004
Renu, K., Abilash, V. G., Tirupathi, T. P., and Arunachalam, S. (2018). Molecular mechanism of doxorubicin-induced cardiomyopathy – an update. Eur. J. Pharmacol. 818, 241–253. doi:10.1016/J.EJPHAR.2017.10.043
Roca-Alonso, L., Castellano, L., Mills, A., Dabrowska, A. F., Sikkel, M. B., Pellegrino, L., et al. (2015). Myocardial MiR-30 downregulation triggered by doxorubicin drives alterations in β-adrenergic signaling and enhances apoptosis. Cell. Death Dis. 6 (5 6), e1754. doi:10.1038/cddis.2015.89
Roe, N. D., Xu, X., Kandadi, M. R., Hu, N., Pang, J., Weiser-Evans, M. C. M., et al. (2015). Targeted deletion of PTEN in cardiomyocytes renders cardiac contractile dysfunction through interruption of Pink1–AMPK signaling and autophagy. Biochim. Biophys. Acta 1852, 290–298. doi:10.1016/J.BBADIS.2014.09.002
Ruan, Y., Dong, C., Patel, J., Duan, C., Wang, X., Wu, X., et al. (2015). SIRT1 suppresses doxorubicin-induced cardiotoxicity by regulating the oxidative stress and p38MAPK pathways. Cell. Physiol. biochem. 35, 1116–1124. doi:10.1159/000373937
Ruggeri, C., Gioffré, S., Achilli, F., Colombo, G. I., and D’Alessandra, Y. (2018). Role of microRNAs in doxorubicin-induced cardiotoxicity: An overview of preclinical models and cancer patients. Heart fail. Rev. 23, 109–122. doi:10.1007/s10741-017-9653-0
Russo, M., della Sala, A., Tocchetti, C. G., Porporato, P. E., and Ghigo, A. (2021). Metabolic aspects of anthracycline cardiotoxicity. Curr. Treat. Options Oncol. 22, 18. doi:10.1007/S11864-020-00812-1
Sahu, R., Dua, T. K., Das, S., de Feo, V., and Dewanjee, S. (2019). Wheat phenolics suppress doxorubicin-induced cardiotoxicity via inhibition of oxidative stress, MAP kinase activation, NF-κB pathway, PI3K/Akt/mTOR impairment, and cardiac apoptosis. Food Chem. Toxicol. 125, 503–519. doi:10.1016/J.FCT.2019.01.034
Saito, T., and Sadoshima, J. (2015). Molecular mechanisms of mitochondrial autophagy/mitophagy in the heart. Circ. Res. 116, 1477–1490. doi:10.1161/CIRCRESAHA.116.303790
Salzman, D. W., Nakamura, K., Nallur, S., Dookwah, M. T., Metheetrairut, C., Slack, F. J., et al. (2016). miR-34 activity is modulated through 5′-end phosphorylation in response to DNA damage. Nat. Commun. 7, 10954. doi:10.1038/NCOMMS10954
Samant, S. A., Zhang, H. J., Hong, Z., Pillai, V. B., Sundaresan, N. R., Wolfgeher, D., et al. (2014). SIRT3 deacetylates and activates OPA1 to regulate mitochondrial dynamics during stress. Mol. Cell. Biol. 34, 807–819. doi:10.1128/MCB.01483-13
Sardão, V. A., Oliveira, P. J., Holy, J., Oliveira, C. R., and Wallace, K. B. (2009). Doxorubicin-induced mitochondrial dysfunction is secondary to nuclear p53 activation in H9c2 cardiomyoblasts. Cancer Chemother. Pharmacol. 64, 811–827. doi:10.1007/s00280-009-0932-x
Shinlapawittayatorn, K., Chattipakorn, S. C., and Chattipakorn, N. (2022). The effects of doxorubicin on cardiac calcium homeostasis and contractile function. J. Cardiol. 80, 125–132. doi:10.1016/J.JJCC.2022.01.001
Shreeram, S., Demidov, O. N., Hee, W. K., Yamaguchi, H., Onishi, N., Kek, C., et al. (2006). Wip1 phosphatase modulates ATM-dependent signaling pathways. Mol. Cell. 23, 757–764. doi:10.1016/j.molcel.2006.07.010
Šimůnek, T., Štěrba, M., Popelová, O., Adamcová, M., Hrdina, R., and Gerši, V. (2009). Anthracycline-induced cardiotoxicity: Overview of studies examining the roles of oxidative stress and free cellular iron. Pharmacol. Rep. 61, 154–171. doi:10.1016/S1734-1140(09)70018-0
Skála, M., Hanousková, B., Skálová, L., and Matoušková, P. (2019). MicroRNAs in the diagnosis and prevention of drug-induced cardiotoxicity. Arch. Toxicol. 93, 1–9. doi:10.1007/s00204-018-2356-z
Štěrba, M., Popelová, O., Lenčo, J., Fučíková, A., Brčáková, E., Mazurová, Y., et al. (2011). Proteomic insights into chronic anthracycline cardiotoxicity. J. Mol. Cell. Cardiol. 50, 849–862. doi:10.1016/J.YJMCC.2011.01.018
Štěrba, M., Popelová, O., Vávrová, A., Jirkovský, E., Kovaříková, P., Geršl, V., et al. (2013). Oxidative stress, redox signaling, and metal chelation in anthracycline cardiotoxicity and pharmacological cardioprotection. Antioxid. Redox Signal. 18, 899–929. doi:10.1089/ARS.2012.4795
Su, Q., Xu, Y., Cai, R., Dai, R., Yang, X., Liu, Y., et al. (2021). miR-146a inhibits mitochondrial dysfunction and myocardial infarction by targeting cyclophilin D. Mol. Ther. Nucleic Acids 23, 1258–1271. doi:10.1016/J.OMTN.2021.01.034
Sumneang, N., Siri-Angkul, N., Kumfu, S., Chattipakorn, S. C., and Chattipakorn, N. (2020). The effects of iron overload on mitochondrial function, mitochondrial dynamics, and ferroptosis in cardiomyocytes. Arch. Biochem. Biophys. 680, 108241. doi:10.1016/J.ABB.2019.108241
Sun, Z., Lu, W., Lin, N., Lin, H., Zhang, J., Ni, T., et al. (2020). Dihydromyricetin alleviates doxorubicin-induced cardiotoxicity by inhibiting NLRP3 inflammasome through activation of SIRT1. Biochem. Pharmacol. 175, 113888. doi:10.1016/J.BCP.2020.113888
Tadokoro, T., Ikeda, M., Ide, T., Deguchi, H., Ikeda, S., Okabe, K., et al. (2020). Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight 5, 132747. doi:10.1172/JCI.INSIGHT.132747
Tang, B. L. (2016). Sirt1 and the mitochondria. Mol. Cells 39, 87–95. doi:10.14348/MOLCELLS.2016.2318
Tang, H., Tao, A., Song, J., Liu, Q., Wang, H., and Rui, T. (2017). Doxorubicin-induced cardiomyocyte apoptosis: Role of mitofusin 2. Int. J. Biochem. Cell. Biol. 88, 55–59. doi:10.1016/j.biocel.2017.05.006
Taniyama, Y., and Walsh, K. (2002). Elevated myocardial akt signaling ameliorates doxorubicin-induced congestive heart failure and promotes heart growth. J. Mol. Cell. Cardiol. 34, 1241–1247. doi:10.1006/JMCC.2002.2068
Timm, K. N., and Tyler, D. J. (2020). The role of AMPK activation for cardioprotection in doxorubicin-induced cardiotoxicity. Cardiovasc. Drugs Ther. 34, 255–269. doi:10.1007/S10557-020-06941-X
Tong, Z., Jiang, B., Wu, Y., Liu, Y., Li, Y., Gao, M., et al. (2015). MiR-21 protected cardiomyocytes against doxorubicin-induced apoptosis by targeting BTG2. Int. J. Mol. Sci. 16, 14511–14525. doi:10.3390/IJMS160714511
Tony, H., Yu, K., and Qiutang, Z. (2015). MicroRNA-208a silencing attenuates doxorubicin induced myocyte apoptosis and cardiac dysfunction. Oxid. Med. Cell. Longev. 2015, 597032. doi:10.1155/2015/597032
Tsuchiya, N., Izumiya, M., Ogata-Kawata, H., Okamoto, K., Fujiwara, Y., Nakai, M., et al. (2011). Tumor suppressor miR-22 determines p53-dependent cellular fate through post-transcriptional regulation of p21. Cancer Res. 71, 4628–4639. doi:10.1158/0008-5472.CAN-10-2475
Vacchi-Suzzi, C., Bauer, Y., Berridge, B. R., Bongiovanni, S., Gerrish, K., Hamadeh, H. K., et al. (2012). Perturbation of microRNAs in rat heart during chronic doxorubicin treatment. PLoS One 7, 40395. doi:10.1371/JOURNAL.PONE.0040395
Vejpongsa, P., and Yeh, E. T. H. (2014). Topoisomerase 2β: A promising molecular target for primary prevention of anthracycline-induced cardiotoxicity. Clin. Pharmacol. Ther. 95, 45–52. doi:10.1038/CLPT.2013.201
Ventura, A., and Jacks, T. (2009). MicroRNAs and cancer: Short RNAs go a long way. Cell. 136, 586–591. doi:10.1016/J.CELL.2009.02.005
Wales, M. M., Biel, M. A., Deiry, W. el, Nelkin, B. D., Issa, J.-P., Cavenee, W. K., et al. (1995). p53 activates expression of HIC-1, a new candidate tumour suppressor gene on 17p13.3. Nat. Med. 1, 570–577. doi:10.1038/nm0695-570
Wallace, K. B. (2007). Adriamycin-induced interference with cardiac mitochondrial calcium homeostasis. Cardiovasc. Toxicol. 7, 101–107. doi:10.1007/s12012-007-0008-2
Wallace, K. B., Sardão, V. A., and Oliveira, P. J. (2020). Mitochondrial determinants of doxorubicin-induced cardiomyopathy. Circ. Res. 126, 926–941. doi:10.1161/CIRCRESAHA.119.314681
Wan, Q., Xu, T., Ding, W., Zhang, X., Ji, X., Yu, T., et al. (2019). miR-499-5p attenuates mitochondrial fission and cell apoptosis via p21 in doxorubicin cardiotoxicity. Front. Genet. 10, 734. doi:10.3389/fgene.2018.00734
Wang, J., Zhang, J., Xiao, M., Wang, S., Wang, (b), J., Guo, Y., et al. (2021). Molecular mechanisms of doxorubicin-induced cardiotoxicity: Novel roles of sirtuin 1-mediated signaling pathways. Cell. Mol. Life Sci. 78 (7 78), 3105–3125. doi:10.1007/S00018-020-03729-Y
Wang, J., Tang, Y., Zhang, J., Wang, J., Xiao, M., Lu, G., et al. (2022). Cardiac SIRT1 ameliorates doxorubicin-induced cardiotoxicity by targeting sestrin 2. Redox Biol. 52, 102310. doi:10.1016/J.REDOX.2022.102310
Wang, J. X., Jiao, J. Q., Li, Q., Long, B., Wang, K., Liu, J. P., et al. (2010). miR-499 regulates mitochondrial dynamics by targeting calcineurin and dynamin-related protein-1. Nat. Med. 17 (1 17), 71–78. doi:10.1038/nm.2282
Wang, J. X., Zhang, X. J., Feng, C., Sun, T., Wang, K., Wang, Y., et al. (2015). MicroRNA-532-3p regulates mitochondrial fission through targeting apoptosis repressor with caspase recruitment domain in doxorubicin cardiotoxicity. Cell. Death Dis. 6, e1677. doi:10.1038/CDDIS.2015.41
Wang, M., Mao, C., Ouyang, L., Liu, Y., Lai, W., Liu, N., et al. (2019). Long noncoding RNA LINC00336 inhibits ferroptosis in lung cancer by functioning as a competing endogenous RNA. Cell. Death Differ. 26, 2329–2343. doi:10.1038/S41418-019-0304-Y
Wang, R., Xu, Y., Niu, X., Fang, Y., Guo, D., Chen, J., et al. (2021). MiR-22 inhibition alleviates cardiac dysfunction in doxorubicin-induced cardiomyopathy by targeting the sirt1/PGC-1α pathway. Front. Physiol. 12, 289. doi:10.3389/fphys.2021.646903
Wang, S., Song, P., and Zou, M. H. (2012). Inhibition of AMP-activated protein kinase α (AMPKα) by doxorubicin accentuates genotoxic stress and cell death in mouse embryonic fibroblasts and cardiomyocytes: Role of p53 and SIRT1. J. Biol. Chem. 287, 8001–8012. doi:10.1074/JBC.M111.315812
Wang, S., Wang, Y., Zhang, Z., Liu, Q., and Gu, J. (2017). Cardioprotective effects of fibroblast growth factor 21 against doxorubicin-induced toxicity via the SIRT1/LKB1/AMPK pathway. Cell. Death Dis. 8 (8 8), e3018. doi:10.1038/cddis.2017.410
Wang, V., and Wu, W. (2012). MicroRNA-based therapeutics for cancer. BioDrugs 23, 15–23. doi:10.2165/00063030-200923010-00002
Wu, M., Ye, H., Tang, Z., Shao, C., Lu, G., Chen, B., et al. (2017). p53 dynamics orchestrates with binding affinity to target genes for cell fate decision. Cell. Death Dis. 8 (10 8), e3130. doi:10.1038/cddis.2017.492
Wu, S., Lan, J., Li, L., Wang, X., Tong, M., Fu, L., et al. (2021). Sirt6 protects cardiomyocytes against doxorubicin-induced cardiotoxicity by inhibiting P53/Fas-dependent cell death and augmenting endogenous antioxidant defense mechanisms. Cell. Biol. Toxicol. 2021, 1–22. doi:10.1007/s10565-021-09649-2
Wu, Y. Z., Zhang, L., Wu, Z. X., Shan, T. T., and Xiong, C. (2019). Berberine ameliorates doxorubicin-induced cardiotoxicity via a SIRT1/p66Shc-mediated pathway. Oxid. Med. Cell. Longev. 2019, 2150394. doi:10.1155/2019/2150394
Xia, W., Chang, B., Li, L., Hu, T., Ye, J., Chen, H., et al. (2021). MicroRNA therapy confers anti-senescent effects on doxorubicin-related cardiotoxicity by intracellular and paracrine signaling. Aging (Albany NY) 13, 25256–25270. doi:10.18632/AGING.203743
Xu, C., Liu, C. H., and Zhang, D. L. (2020). MicroRNA-22 inhibition prevents doxorubicin-induced cardiotoxicity via upregulating SIRT1. Biochem. Biophys. Res. Commun. 521, 485–491. doi:10.1016/J.BBRC.2019.10.140
Xu, X., Persson, H. L., and Richardson, D. R. (2005). Molecular Pharmacology of the interaction of anthracyclines with iron. Mol. Pharmacol. 68, 261–271. doi:10.1124/MOL.105.013383
Yamakuchi, M., Ferlito, M., and Lowenstein, C. J. (2008). miR-34a repression of SIRT1 regulates apoptosis. Proc. Natl. Acad. Sci. U. S. A. 105, 13421–13426. doi:10.1073/PNAS.0801613105
Yang, Q., Yang, K., and Li, A. (2014). microRNA-21 protects against ischemia-reperfusion and hypoxia-reperfusion- induced cardiocyte apoptosis via the phosphatase and tensin homolog/Akt- dependent mechanism. Mol. Med. Rep. 9, 2213–2220. doi:10.3892/mmr.2014.2068
Yin, J., Guo, J., Zhang, Q., Cui, L., Zhang, L., Zhang, T., et al. (2018). Doxorubicin-induced mitophagy and mitochondrial damage is associated with dysregulation of the PINK1/parkin pathway. Toxicol. Vitro 51, 1–10. doi:10.1016/j.tiv.2018.05.001
Yoshida, M., Shiojima, I., Ikeda, H., and Komuro, I. (2009). Chronic doxorubicin cardiotoxicity is mediated by oxidative DNA damage-ATM-p53-apoptosis pathway and attenuated by pitavastatin through the inhibition of Rac1 activity. J. Mol. Cell. Cardiol. 47, 698–705. doi:10.1016/J.YJMCC.2009.07.024
Yu, X., Ruan, Y., Shen, T., Qiu, Q., Yan, M., Sun, S., et al. (2020). Dexrazoxane protects cardiomyocyte from doxorubicin-induced apoptosis by modulating miR-17-5p. Biomed. Res. Int. 2020, 5107193. doi:10.1155/2020/5107193
Zhang, B., Pan, X., Cobb, G. P., and Anderson, T. A. (2007). microRNAs as oncogenes and tumor suppressors. Dev. Biol. 302, 1–12. doi:10.1016/J.YDBIO.2006.08.028
Zhang, C., Feng, Y., Qu, S., Wei, X., Zhu, H., Luo, Q., et al. (2011). Resveratrol attenuates doxorubicin-induced cardiomyocyte apoptosis in mice through SIRT1-mediated deacetylation of p53. Cardiovasc. Res. 90, 538–545. doi:10.1093/CVR/CVR022
Zhang, S., Liu, X., Bawa-Khalfe, T., Lu, L. S., Lyu, Y. L., Liu, L. F., et al. (2012). Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med. 18 (11 18), 1639–1642. doi:10.1038/nm.2919
Zhang, W., Lai, X., and Guo, X. F. (2021a). Activation of Nrf2 by miR-152 inhibits doxorubicin-induced cardiotoxicity via attenuation of oxidative stress, inflammation, and apoptosis. Oxid. Med. Cell. Longev. 2021, 8860883. doi:10.1155/2021/8860883
Zhang, W. bin, Zheng, Y. F., and Wu, Y. G. (2021b). Inhibition of miR-128-3p attenuated doxorubicin-triggered acute cardiac injury in mice by the regulation of PPAR-γ. PPAR Res. 2021, 7595374. doi:10.1155/2021/7595374
Zhang, X., Hu, C., Kong, C. Y., Song, P., Wu, H. M., Xu, S. C., et al. (2019). FNDC5 alleviates oxidative stress and cardiomyocyte apoptosis in doxorubicin-induced cardiotoxicity via activating AKT. Cell. Death Differ. 27 (2 27), 540–555. doi:10.1038/s41418-019-0372-z
Zhang, X., Ji, R., Liao, X., Castillero, E., Kennel, P. J., Brunjes, D. L., et al. (2018). MicroRNA-195 regulates metabolism in failing myocardium via alterations in sirtuin 3 expression and mitochondrial protein acetylation. Circulation 137, 2052–2067. doi:10.1161/CIRCULATIONAHA.117.030486
Zhang, X., Lv, S., Zhang, W., Jia, Q., Wang, L., Ding, Y., et al. (2021c). Shenmai injection improves doxorubicin cardiotoxicity via miR-30a/Beclin 1. Biomed. Pharmacother. 139, 111582. doi:10.1016/J.BIOPHA.2021.111582
Zhang, X., Wan, G., Mlotshwa, S., Vance, V., Berger, F. G., Chen, H., et al. (2010). Oncogenic Wip1 phosphatase is inhibited by miR-16 in the DNA damage signaling pathway. Cancer Res. 70, 7176–7186. doi:10.1158/0008-5472.CAN-10-0697
Zhang, Y., Ma, C., Liu, C., and Wei, F. (2020). Luteolin attenuates doxorubicin-induced cardiotoxicity by modulating the PHLPP1/AKT/Bcl-2 signalling pathway. PeerJ 2020, e8845. doi:10.7717/peerj.8845
Zhao, L., Qi, Y., Xu, L., Tao, X., Han, X., Yin, L., et al. (2018). MicroRNA-140-5p aggravates doxorubicin-induced cardiotoxicity by promoting myocardial oxidative stress via targeting Nrf2 and Sirt2. Redox Biol. 15, 284–296. doi:10.1016/J.REDOX.2017.12.013
Zhu, J. N., Fu, Y. H., Hu, Z. Q., Li, W. Y., Tang, C. M., Fei, H. W., et al. (2017). Activation of miR-34a-5p/Sirt1/p66shc pathway contributes to doxorubicin-induced cardiotoxicity. Sci. Rep. 7 (1 7), 11879–11912. doi:10.1038/s41598-017-12192-y
Zhu, W., Soonpaa, M. H., Chen, H., Shen, W., Payne, R. M., Liechty, E. A., et al. (2009). Acute doxorubicin cardiotoxicity is associated with p53-induced inhibition of the mammalian target of rapamycin pathway. Circulation 119, 99–106. doi:10.1161/CIRCULATIONAHA.108.799700
Keywords: microRNA, doxorubicin, cardiotoxicity, genotoxic stress, p53
Citation: Kawano I and Adamcova M (2022) MicroRNAs in doxorubicin-induced cardiotoxicity: The DNA damage response. Front. Pharmacol. 13:1055911. doi: 10.3389/fphar.2022.1055911
Received: 28 September 2022; Accepted: 11 November 2022;
Published: 21 November 2022.
Edited by:
Jianqiang Xu, Dalian University of Technology, ChinaReviewed by:
Arun Samidurai, Virginia Commonwealth University, United StatesManikandan Panchatcharam, Louisiana State University Health Shreveport, United States
Jun Ren, Fudan University, China
Copyright © 2022 Kawano and Adamcova. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michaela Adamcova, YWRhbWNvdmFAbGZoay5jdW5pLmN6