 Cédric Boudou
Cédric Boudou Luce Mattio
Luce Mattio Alexey Koval
Alexey Koval Valentin Soulard1
Valentin Soulard1 Vladimir L. Katanaev
Vladimir L. Katanaev
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 28 October 2022
Sec. Pharmacology of Anti-Cancer Drugs
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.1045102
The Wnt-pathway has a critical role in development and tissue homeostasis and has attracted increased attention to develop anticancer drugs due to its aberrant activation in many cancers. In this study, we identified a novel small molecule series with a thienopyrimidine scaffold acting as a downstream inhibitor of the β-catenin-dependent Wnt-pathway. This novel chemotype was investigated using Wnt-dependent triple-negative breast cancer (TNBC) cell lines. Structure activity relationship (SAR) exploration led to identification of low micromolar compounds such as 5a, 5d, 5e and a novel series with quinazoline scaffold such as 9d. Further investigation showed translation of activity to inhibit cancer survival of HCC1395 and MDA-MB-468 TNBC cell lines without affecting a non-cancerous breast epithelial cell line MCF10a. This anti-proliferative effect was synergistic to docetaxel treatment. Collectively, we identified novel chemotypes acting as a downstream inhibitor of β-catenin-dependent Wnt-pathway that could expand therapeutic options to manage TNBC.
The β-catenin-dependent Wnt-pathway regulates various physiological processes, from embryonic development to tissue homeostasis, and regeneration in adults (Parsons et al., 2021). However, aberrant activation of the Wnt-pathway plays a critical role in cancer cell proliferation, survival, and metastasis as well as in maintenance of cancer stem cells. This overactivation is predominantly driven by mutations, such as loss-of-function mutations for negative regulators like APC, AXIN1 or AXIN2, gain-of-function mutations for β-catenin (CTNNB1) or TCF, and eventually dysregulation of Wnt receptor abundance through mutations of RNF43, ZNRF3 or RSPO (Bugter et al., 2021; Parsons et al., 2021). In addition, several cancers have been linked to overexpression of the Wnt-pathway components, such as Wnts, their receptors (FZDs and LRP5/6), or Porcupine—the enzyme regulating Wnt secretion and activity (Stewart, 2014; Zhan et al., 2017; Shaw et al., 2019a; Wang et al., 2021). As a result of such aberrations, β-catenin accumulates in cell nucleus and promotes transcription of oncogenic target genes.
Since the discovery of the first WNT family member in 1982, Wnt signaling has attracted increased attention in the drug discovery field. Targeting the β-catenin-dependent pathway has been the focus of multiple drug discovery programs both in academia and industry, but only a few have reached clinical trials (Blagodatski et al., 2014; Liu et al., 2021). Among them, there are five small molecules acting as Porcupine inhibitors (e.g., 1 and 2, in Figure 1), which are profiled in phase 1/2 clinical trials for different cancer indications (Flanagan et al., 2022), while CWP232291, PRI-724, and SM08502 target downstream components of the Wnt-pathway (Shaw et al., 2019a). CWP232291 is a peptidomimetic targeting Sam68, an RNA-binding protein, and is currently profiled on acute myeloid leukaemia in phase 1/2. PRI-724 (3, in Figure 1) affecting interaction between β-catenin and the transcription co-activator CBP, has been evaluated in clinical trials against multiple cancers and cirrhosis. Finally, SM08502, acting through CDC-like kinase, reduces the Wnt-pathway-related gene expression and is profiled in phase 1 (Tam et al., 2020).
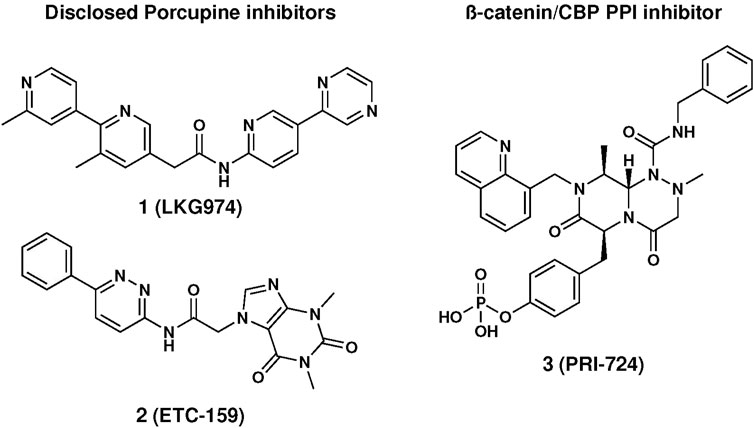
FIGURE 1. Structure of disclosed Wnt-inhibitors.
Our research has been aimed at discovering novel Wnt-pathway inhibitors (Koval and Katanaev, 2012; Katanaev et al., 2021; Larasati et al., 2022). We have previously developed a High-Throughput Screening (HTS) assay based on a triple-negative breast cancer (TNBC) cell line BT-20 that could be adapted to any β-catenin-dependent cancer cell line. In this assay, BT-20 cells are stably transfected with the Wnt-responsive TopFlash reporter, and activated by the in-house purified Wnt3a (Shaw et al., 2019b). A pilot screening of a commercial library of 1,000 compounds led to the identification of FSA as selective inhibitor of the Wnt signaling and cancer cell proliferation, in vitro and in vivo (Katanaev and Koval, 2021). This assay has also been used to drive the drug design of clofazimine derivatives acting as downstream inhibitors of the Wnt-pathway (Koval et al., 2021). In continuation of our efforts to identify novel chemotypes acting as β-catenin-dependent Wnt inhibitors and potential anticancer drugs, we here report a novel chemical series leading to compounds endowed with submicromolar activity in cancer cell lines, inhibiting the β-catenin-dependent Wnt signaling at downstream levels of the pathway.
The advantage of our platform lies in its ability to discriminate between inhibitors of the β-catenin-dependent Wnt signaling acting at upstream vs. downstream levels of the pathway, by stimulating the pathway at the different levels either with Wnt3a or with a GSK3β inhibitor, like CHIR99021 (Shaw et al., 2019b). Both stimulations abolish formation of the destruction complex, a multiprotein complex consisting of Axin, adenomatous polyposis coli (APC), glycogen synthase kinase three beta (GSK3β), and casein kinase one alpha (CK1α). Upon inactivation of the complex, β-catenin accumulates in the cytoplasm and translocates into the nucleus where it interacts with transcription factors. These downstream events are quantified by a Luciferase transcription-based readout assay (TopFlash assay).
During our investigation toward novel Wnt-inhibitors, we identified a thienopyrimidine 4a as a β-catenin-dependent Wnt-pathway inhibitor with micromolar potency in the TNBC cell line HCC1395. Both means of the pathway activation—at the upstream levels with Wnt3a and at the downstream levels with a GSK3β inhibitor CHIR99021—revealed a similar potency of 4a to inhibit the signaling, respectively 8.31 µM and 8.47 µM (Figure 2). This result led to the conclusion that 4a acts downstream of GSK3β in the Wnt-pathway.
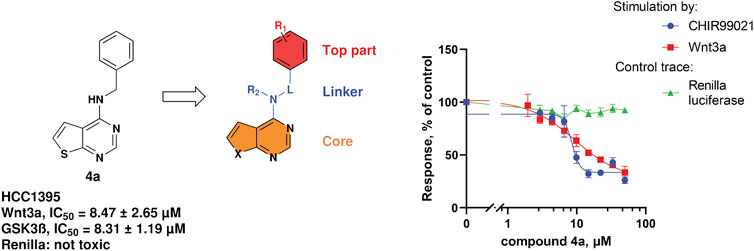
FIGURE 2. Structure of compound 4a, area of exploration for library synthesis, and concentration-dependent inhibition of the TopFlash readout.
We considered 4a as a good starting point for a drug discovery exploration. The compound has no cytotoxic activity (measured by Renilla luciferase expressed under the Wnt-independent CMV promoter, Figure 2) and has a good potency with respect to its low molecular weight (241 g/mol). The simplicity of the scaffold combined with synthetic accessibility allows building a library of compounds and performing structure activity relationship (SAR) studies of the three main parts of the molecule: the top part, the linker and the core (Figure 2).
The synthesis of thieno [2,3-d]pyrimidine derivatives is illustrated in Scheme 1. Commercially available 4-chlorothieno [2,3-d]pyrimidine was subjected to aromatic substitution with different primary and secondary amines to afford the benzyl derivatives (4a-e, 4g, 4i-l and 4o-q) and some of the linker replacement 5a-n in the 29%–81% yield. In some cases, formation of side products did not permit purification of the final compounds, or the corresponding amines were not available. Therefore, as an alternative approach, 4-chlorothieno [2,3-d]pyrimidine was converted into 4-aminothieno [2,3-d]pyrimidine (compound 6) with ammonium hydroxide in the 75% yield without purification. This second building block was reacted with differently substituted benzyl bromides or benzoyl chloride to afford compounds 4f, 4k, 4m, and 7 in moderate yields. With these synthetic approaches, it was possible to obtain a series of analogues of 4a modified in the top part and in the linker portions (Figure 2).
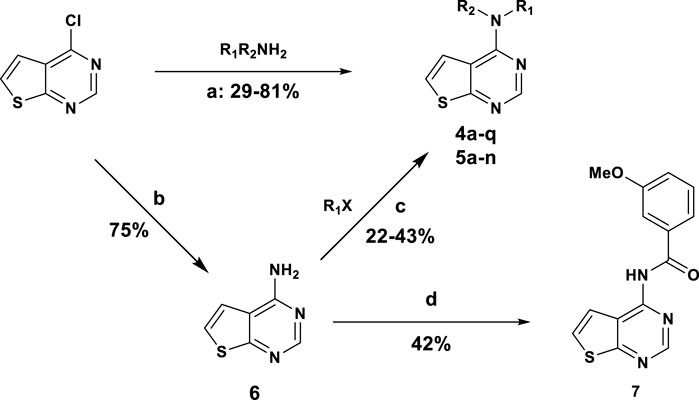
SCHEME 1. General synthesis of Thieno [2,3-d]pyrimidine derivatives: (A) DIPEA, DMF, 100°C, 4 h; (B) NH4OH, 90°C 12 h; (C) NaH 60%, THF, 0°C to rt, 12 h; (D) 3-methoxybenzenesulfonyl chloride, Et3N, DCM, rt, 24 h.
To perform core modifications, the thienopyrimidine nucleus was replaced with a series of heterocycles. For the quinazoline core, the synthetic approach used was similar to the one used for the thienopyrimidine derivatives. Commercially available 2,4-dichloroquinazoline was subjected to the nucleophilic aromatic substitution with different primary amines to afford the intermediates 8a-i in moderate to good yields (Scheme 2). Interestingly, although both C2 and C4 positions are electron-deficient, the reaction is regioselective toward the C4 position (Mohamed and Rao, 2015). In the second step, chlorine in position C2 was removed using reductive conditions. For intermediates 8a-f, 8f and 8h-i, ammonium formate in presence of palladium on charcoal at reflux was used to afford the expected compounds in the 11%–74% yield. For compounds 8e and 8g, bearing chlorine on the aromatic ring in the top part, a regioselective reduction of the chlorine in C2 of the quinazoline derivatives had to be performed. Therefore, compounds 9e and 9g were obtained in milder conditions with zinc, N,N,N′,N′-tetramethyl ethylenediamine, acetic acid in methanol at 45°C overnight.

SCHEME 2. General synthesis of Quinazoline core: (A) DIPEA, CH3CN, rt, 12 h; (B) 10% Pd/C, ammonium formate, EtOH, reflux, 12 h; (C) 1) Zn, TMEDA, AcOH, MeOH, 40°C–45°C, 12 h, 2) 2-mercaptonicotinic acid, rt, 30 min.
Preparation of pyrimidines (11 and 12), isoquinoline, 13 and quinoline 16 derivatives followed similar synthetic strategies and are described in the Supplementary Material.
The synthesized compounds were used to explore SAR of three defined areas of investigation (Figure 2). All compounds were tested in the HCC1395 TNBC cell line using the GSK3β inhibitor to induce β-catenin signaling since 4a acted downstream of GSK3β (Figure 2). Both inhibition of β-catenin signaling measured by the TopFlash reporter and cytotoxicity measured by the Renilla reporter were assessed.
SAR of benzyl substitution is depicted in Table 1. Overall, ortho substituents were not tolerated or resulted in toxicity, like the 2-fluoro derivative 4e. The only exception is the 2-methoxy analogue 4b, which showed a 3-fold improvement in the activity (IC50 = 2.79 µM) compared to the unsubstituted benzyl derivative 4a (IC50 = 8.31 µM), although with a partial inhibition of 40.7% compared to 72% of the parental compound. In the meta position, electron withdrawing groups were not tolerated (compounds 4f, 4i), except for compound 4i bearing chlorine, which resulted in the activity similar to that of 4a. However, potency was modestly improved by introducing electron donating groups, such as methoxy (4c) or methyl (4n) groups. In the para position, electron donating (methoxy, 4d) or withdrawing (cyano, 4m) groups abrogated the activity, whereas fluorine 4g, chlorine 4j and methyl 4o groups showed a 2-fold improvement in the activity compared to 4a. Therefore, derivatives combining the substituents which gave the best results in the inhibitory activity were designed and synthesized. Pleasingly, compounds 4p and 4q, bearing a methoxy group in the ortho position and fluoro or chloro in the para position, respectively, resulted in the IC50 of 2.59 µM and 1.33 µM.
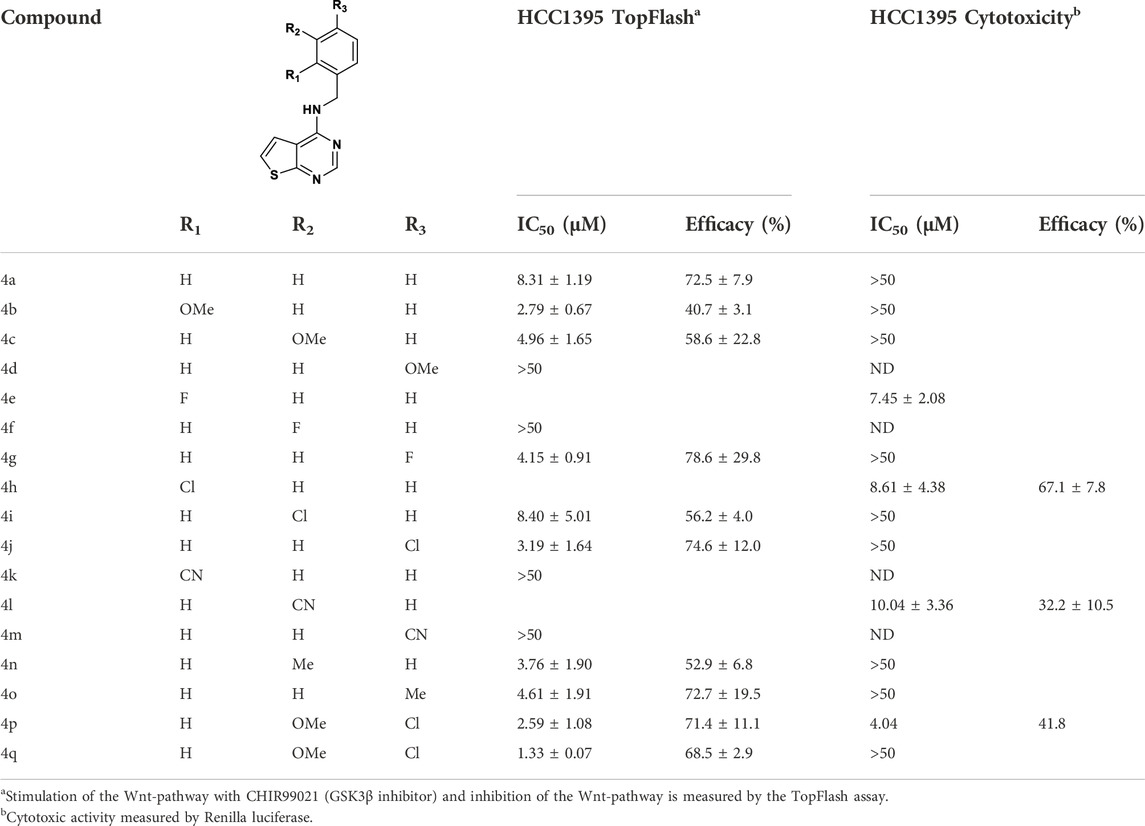
TABLE 1. SAR exploration of the benzyl top part of thieno[2,3-d]pyrimidine.
To investigate contribution of the linker between the thienopyrimidine core and the aromatic rings, we initially probed the influence of an additional methyl group, preparing a series of 1-phenylethylamine analogues (Table 2). Except for the 3-methoxy analogue 5b, which was inactive, the other derivatives resulted in 2 to 7-fold more potent compounds. In particular, 5d (4-chloro), 5e (4-methyl) and 5a (2-methoxy) showed submicromolar activities of 0.6, 0.63, and 0.77 µM, respectively. Unfortunately, this modification of the linker resulted in partial inhibition (<50% efficacy). Polar groups such as an amide linker (Liu et al., 2021) were not tolerated (Table 3). Alkylation of the amino group provided different results with benzyl or 2-phenylethan-1-amine top parts. Addition of ethyl group on compound 4c resulted in toxicity at 8.30 µM with compound 5i, whereas addition of a methyl group on compound 5g resulted in the similar potency with compound 5j (IC50 = 2.04 µM, 100% efficacy) (Table 3). This loss in potency could derive from different SAR between benzyl and 2-phenylethan-1-amine. Then, we investigated the impact of the linker length (Table 3) and found that compounds without a linker between the core and the top part, like 5f, or bearing a three-carbon chain (5h) were inactive. On the other side, a two-carbons linker (5g) was three times more potent than 4a with 89% of inhibition (IC50 = 2.64 and 8.31 µM respectively). Attracted by the potency of 5g, we decided to introduce several in-house available substituted 2-phenylethan-1-amines. As for the benzyl derivatives, chlorine substitution was not tolerated in the ortho position (5k, IC50 = 42.87 µM), but resulted in good potency in the meta (5l, IC50 = 3.07 µM) and para (5l, IC50 = 1.79 µM) positions. Unlike the benzyl analogue 4c, the methoxy group in the meta position was not tolerated (5n).
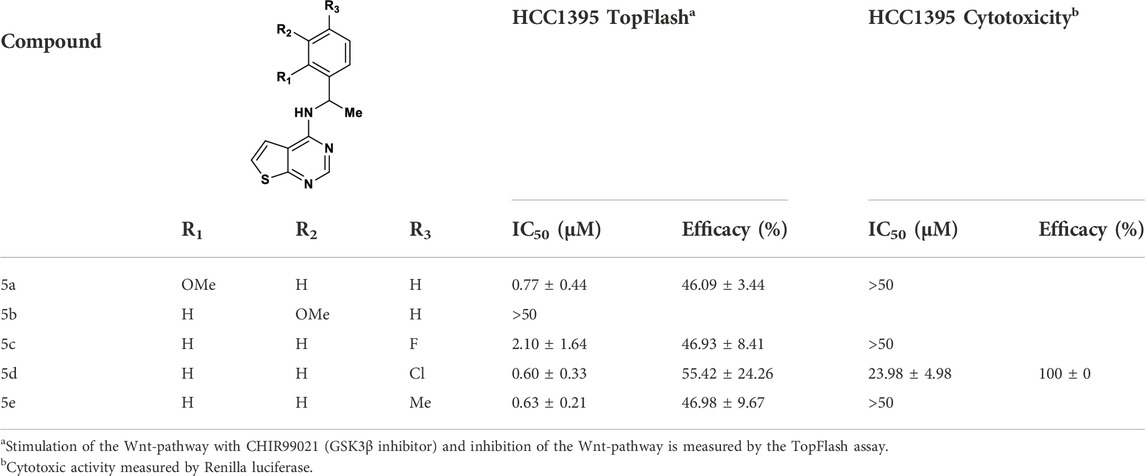
TABLE 2. SAR exploration of the 1-phenylethylamine top part of thieno[2,3-d]pyrimidine.

TABLE 3. SAR exploration of the linker part of thieno[2,3-d]pyrimidine.
We finally evaluated replacement of the core and, without surprise, the bioisosteric replacement of thieno [2,3-d]pyrimidine by quinazoline was well tolerated (Table 4). Other modifications of the bottom part led to drastic loss of potency (Table S1). Substituted quinazolines with 2-chloroquinazoline (8a), 7-dimethylthieno [3,2-d]pyrimidine (10) and single heteroaromatics 2-chloropyrimidine (11) or pyrimidine (12) were no longer active. Moreover, we observed that both nitrogens of the pyrimidine ring from quinazoline were necessary for the activity, since the isoquinoline 13 and quinoline 16 derivatives were inactive or toxic at 18.67 µM. Attracted by the potency of quinazoline 9b with IC50 of 3.58 µM compared to 4.96 µM for thieno [2,3-d]pyrimidine 4c, we evaluated SAR transfer synthesizing quinazoline derivatives bearing the best modifications obtained from the thieno [2,3-d]pyrimidine analogues previously described. With few exceptions, the resulting compounds had similar or better potencies and better efficacies (Table 4). For substituents of the phenyl ring, compounds bearing a substituent in the para position showed a good activity: 4-methoxy was active for the quinazoline version (9c, with IC50 = 4.76 µM, 81.5%) and 4-fluoro (9b), 4-chloro (9e) and 4-methyl (9f) were more potent with IC50 of 0.67, 2.18, and 1.22 µM, respectively. Also, with the quinazoline core, a longer linker did not lead to activity improvement, as we observed with the 4-chlorophenylethyl derivative 9g, found to be inactive. On the other hand, addition of methyl to provide 1-phenylethylamine derivatives resulted either in a drastic reduction of activity (9h), or in compounds with similar activities, if we compare 9f with 9i with 1.22 µM and 1.88 µM, respectively.
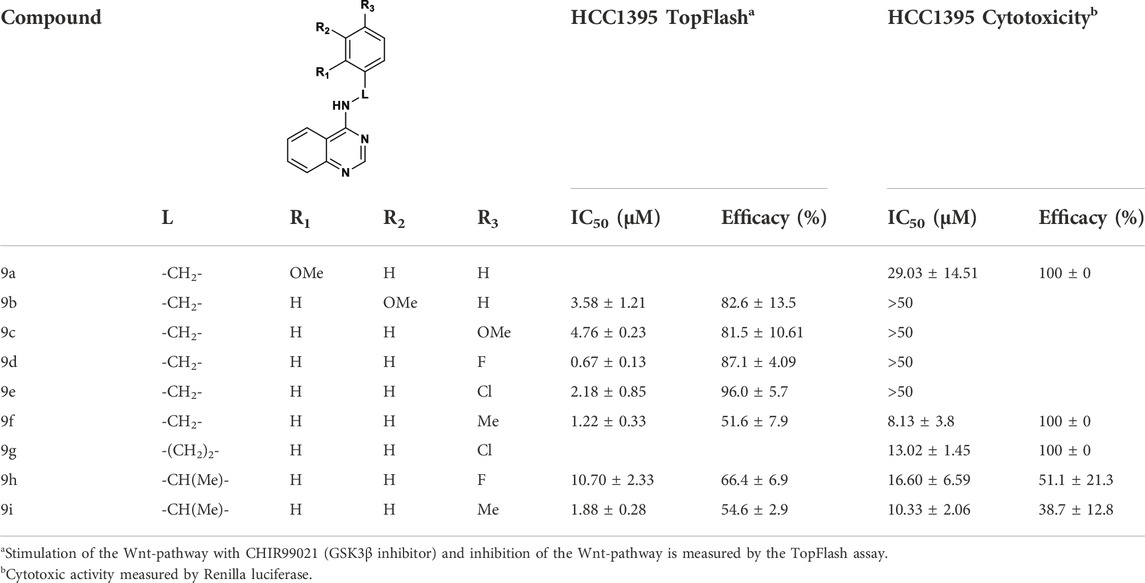
TABLE 4. SAR exploration of quinazoline.
In general, this investigation of SAR of the three areas of the hit compound led to up to 10-fold improvements from the initial compound 4a (IC50 = 8.31 µM) to compound 9d with IC50 of 0.67 µM.
To assess the translation of the compounds’ anti-Wnt potency—ranging from two-digits micromolar to submicromolar values—into inhibition of cell proliferation, we applied the classical MTT assay. As illustrated in Figure 3A, there was a good correlation for HCC1395 cells between the TopFlash and MTT assays with r = 0.648. Interestingly, the partial efficacy observed for some compounds in the TopFlash assay appeared not to influence the potency in the MTT survival assay. We further evaluated whether our compounds could inhibit proliferation of a large panel of TNBC cell lines and of the non-cancerous breast fibrocystic MCF10a cells (Figure 3B and Supplementary Table S2). We analyzed the obtained set of IC50’s by independent clustering that resulted in a clear separation of the cell lines tested into 2 groups: highly sensitive ones (HCC1395 and MDA-MB-468 lines) and poorly sensitive ones (BT20, HCC1806 and MDA-MB-231). Cell lines with high susceptibility to the compounds’ treatment systematically demonstrated lower IC50s as compared to the others. Those with the low antiproliferative sensitivity also clustered with the control non-cancerous MCF10a cells, which indicates strong selectivity of the compounds towards a subset cancer cells. In this regard, only few compounds such as 5j, 5h and 8a (Figure 3A in red, see Supplementary Table S2) were considered to have promiscuous effects with no selectivity over the control cells (less than 3-fold difference in IC50), although 5h should rather be considered as inactive (IC50 > 50 µM).
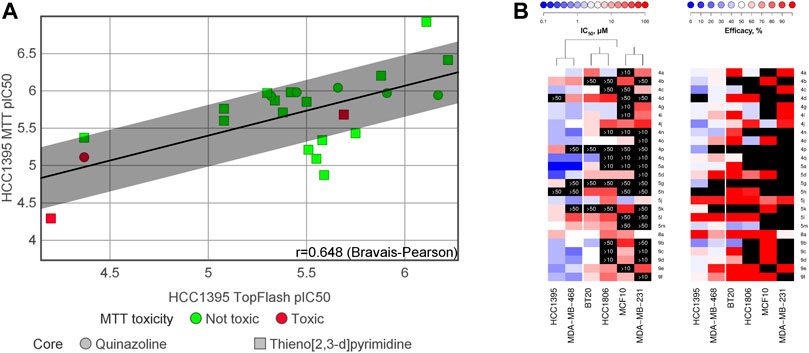
FIGURE 3. (A) Correlation between inhibition of the Wnt-pathway (measured by the TopFlash assay) and inhibition of TNBC cell proliferation (measured by the MTT assay). “MTT toxicity” refers the ratio between the cell proliferation IC50 measured on MCF10a (non-cancerous breast cells) by the IC50 measured on HCC1395 (TNBC cells); “Not toxic” refers to this ratio being less than 3-fold. (B) Heatmaps summarizing IC50 and efficacy of the compounds against diverse TNBC cell lines and the control non-cancerous MCF10a cell line. Only compounds that had previously demonstrated a specific anti-Wnt activity in HCC1395 cells were tested; black blocks indicate no activity within the tested concentration range, with the maximal tested concentration indicated on the block. HCC1395 and MDA-MB-468 cell lines compose a distinct cluster with overall higher susceptibility to the compounds, whereas the other tested TNBC cell lines cluster together with MCF10a cells, demonstrating lower responses.
We continued by analysis of the compounds’ capacity to suppress Wnt signaling in TNBC cells independently of an external Wnt ligand and beyond the artificial TopFlash reporter system. To this end, we assessed the expression levels of the transcription factor c-Myc, a well-proven key target gene of the pathway (Rennoll and Yochum, 2015; Koval et al., 2018; Ahmed et al., 2019). Effects of the compounds on c-Myc levels in HCC1395 cells upon treatment by the indicated compounds, chosen based on their higher potency (IC50 between 0.6 µM and 2.59 µM in the TopFlash assay), are summarized in Figure 4. As expected, Wnt inhibition by the compounds resulted in profoundly decreased endogenous c-Myc levels.
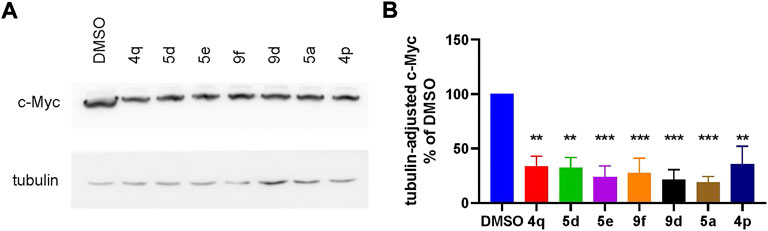
FIGURE 4. (A) Representative Western blot and (B) quantification of suppression of the Wnt target gene c-Myc by high-potent compounds (taken at 5 µM). Statistical significance was assessed by one-way ANOVA and is shown as **p < 0.01, ***p < 0.001.
Compound 5d (IC50 = 0.6 µM, Table 2) was used as a representative to analyze the selectivity of the chemotype towards the Wnt-pathway, by profiling it against a panel of different transcriptional reporters with significant basal activities in HCC1395 cells (Figure 5). These results demonstrate that none other signaling pathways are affected by the compound. We thus conclude that the anti-proliferative effect of the compound is related exclusively to the suppressed Wnt signaling.
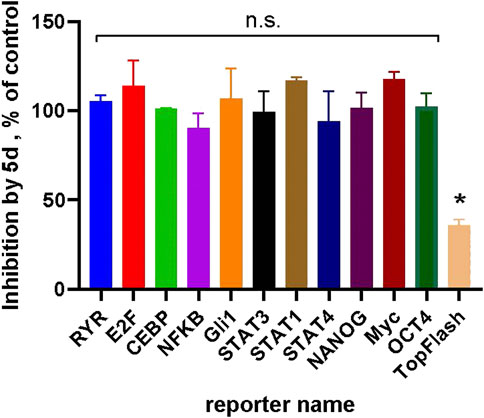
FIGURE 5. Compound 5d (at 5 µM) demonstrates selectivity towards the Wnt-pathway: no inhibition in a panel of luciferase reporters with measurable basal activities in HCC1395 cells could be observed by the compound. Inhibition of the Wnt3a-stimulated TopFlash signal is shown as a positive control. Statistical significance was assessed by one-sample t-test and is shown as *p < 0.05; “n.s.”—not significant.
The fact that some TNBC cell lines were highly sensitive to the compounds, while others were much less so (see Figure 3), brought us to hypothesize that different gene expression determinants existed between these two sets of cancer lines. We took advantage of the RNAseq-derived gene expression datasets in CCLE (Cancer Cell Line Encyclopedia (Barretina et al., 2012)) available for the five TNBC lines used in our studies. In order to pinpoint the genes most likely responsible for the differential response among the two groups of cell lines, we extracted 419 genes that had at least 4-fold higher or lower average expression levels in the HCC1395 and MDA-MB-468 lines as compared to the other cell lines. Pathway enrichment analysis of this list performed with the DAVID online tool (Huang et al., 2009; Sherman et al., 2022) identified the Wnt-pathway among the top over-represented pathways in this list, with 10 genes (Figure 6A and Supplementary Table S3). Indeed, expression levels of these 10 Wnt component and target genes cluster the 5 TNBC cell lines into the same two groups as by the potency to the compound treatment (Figure 6B). Plotting StringDB (Szklarczyk et al., 2021) interactions (physical and genetic) among these genes place them in a tightly interconnected network (Figure 6C). Taken together, these data indicate that the differential capacity of the compounds to inhibit proliferation of these lines is driven by peculiarities of the Wnt-pathway organization in them—the phenomenon previously predicted by us (Koval and Katanaev, 2018).
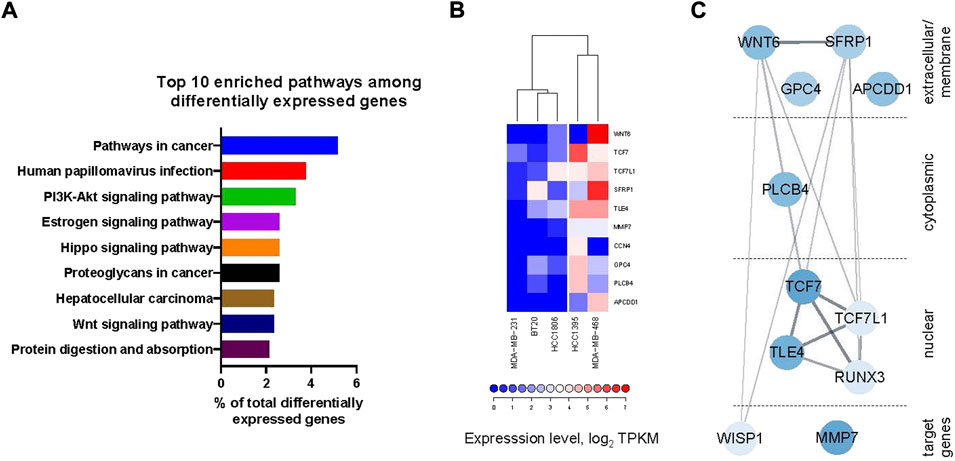
FIGURE 6. (A) Top 10 enriched pathways (by DAVID online tool) among the genes differentially expressed between the lines highly sensitive to the compound treatment (HCC1395 and MDA-MB-468) and those with poor sensitivity (HCC 1806, BT20 and MDA-MB-231) (see also Supplementary Table S3) includes Wnt-pathway. (B) Heatmap of the expression levels of differentially expressed Wnt-pathway component and target genes with clustering clearly delineates the cell lines sensitive to the compound treatment. (C) Representation of the differentially expressed Wnt-related genes as a network with physical and genetic interactions overlaid from StringDB. Intensity of coloration is proportional to the overall difference in the expression levels between the sensitive and insensitive lines.
The Wnt-pathway is implicated in chemoresistance to conventional chemotherapies and drives a poor chemotherapy response in a broad range of tumors (O’Reilly et al., 2015; Martin-Orozco et al., 2019; Zhong and Virshup, 2020). To assess how combination with our compounds affects the anti-proliferative capacity of docetaxel—one of the first-line treatments for TNBC (Mandapati and Lukong, 2022), we performed the Loewe synergism assessment over a complete dose-response range of both compounds followed by analysis in the COMBENEFIT software in Matlab (Figure 7 and Supplementary Figure S1) (Di Veroli et al., 2016).
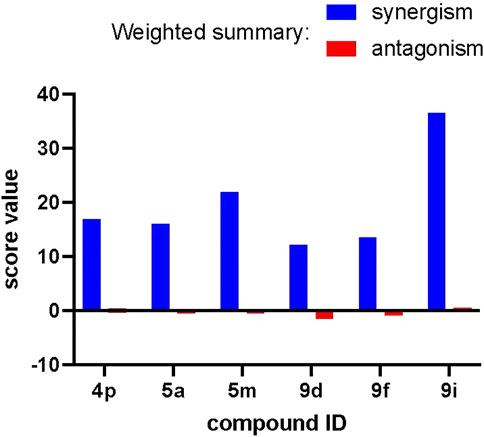
FIGURE 7. Strong synergism between our compounds and docetaxel in the anti-proliferative assays. Weighted summary synergism and antagonism scores obtained for different compounds after analysis with the COMBENEFIT software are presented.
The weighted summary synergism and antagonism scores indicate that compounds 4p, 5a, 5m, 9d, 9f, and 9i exert exclusively synergistic effects when used as a co-treatment with docetaxel. This effect is overall similar among the compounds (best for 9i) and can be characterized as mild since it scores 15–30 on a scale of 0–100. The dose-by-dose matrices calculated for each compound presented at Supplementary Figure S1 indicate that for most of the compounds, major zones of synergism can be seen at the highest concentrations of our compounds and intermediate concentrations of docetaxel. This serves as a strong indication that application of these compounds in vivo might be useful in combination with a conventional chemotherapy through reduction of the chemotherapy dose necessary to obtain a complete response.
From the initial thienopyrimidine hit with an IC50 of 8.31 µM, SAR exploration of three portions of the scaffold led to a 10-fold improvement with four compounds active below the µM range in HCC1395 cells. Although further SAR exploration would be necessary to fully understand the properties driving potency in the HCC1395 cell-based assay, elements of SAR were identified. From exploration of the top part (Table 1, Table 2 and Table 4), electron density of the phenyl does not seem to be critical as electron withdrawing or donating groups have little influence on the potency. Lipophilic or polar interactions are more important in this region. Also, for the thieno [2,3-d]pyrimidine derivatives, the most potent compounds are 1-phenylethylamine analogues (compounds 5a, 5d and 5e; Table 2), of which only racemic mixture were tested. It would be interesting to prepare and test each enantiomer. For the core modifications, a better activity was achieved with the quinazoline nucleus replacing the thienopyrimidine core. We also observed some differences between thieno [2,3-d]pyrimidine and quinazoline SAR (Table 1 and Table 4). Although we expected a good shape similarity for both cores, we could speculate that the subtle electron density difference resulting from the bioisosteric replacement of the thiophene part by phenyl affects the interaction with the biological target.
Based on the assay paradigm, we could conclude that these compounds act below the destruction complex within the Wnt-pathway, as they inhibited the β-catenin-dependent signaling induced by an inhibitor of GSK3β, CHIR99021. Moreover, our exploration of the genetic differences between the cancer lines with strong and weak proliferative response to the derivatives highlighted a set of the Wnt signaling component genes likely responsible for this effect, where TCF7 (a.k.a TCF-1) and TCF7L1 (a.k.a TCF-3) and their immediate regulators and co-factors TLE4 and RUNX3 occupy the central place. Taken together with a significant evidence of non-redundant relationships among TCF/LEF transcriptional factors (Arce et al., 2006; Mao and Byers, 2011), it beckons us that the molecular target of our compounds might be hidden among unique partners of these transcriptional factors. This proposition is also reinforced by the clear specificity of action for our compounds to Wnt signaling with no significant inhibition of other signal transduction pathway observed in the HCC1395 cell line.
The search for such factors—molecular targets of our compounds—is essential for further development of this promising series of compounds. The selective mechanism of action of our compounds might be a double-edged sword: while it is likely behind their capacity to avoid strong effects on healthy cells represented by MCF-10a in our study, on the other hand it might also limit the scope of therapeutic applications of the compounds. With proliferation of 2 out of 5 TNBC lines strongly affected by the compounds, deciphering of the molecular target and of the full mechanism of action will be a key for evaluation of their scope within TNBC and other cancers, as well as for eventual patient stratification for the application of this novel therapy. Immortalized cancer cell lines as well as 2D cultures have certain limitations in translation of the results obtained to a more complex 3D milieu of the tumor in the patients. However, our findings represent a promising step forward in the development of the long-desired Wnt-targeting agents. Future investigations will follow the logical steps of the preclinical development using animal models, and should further provide interesting insights into the roles of Wnt signaling in the tumor resistance, given the synergistic relationships between our compounds and the conventional chemotherapeutic docetaxel.
From the initial potency of the thienopyrimidine 4a with IC50 of 8.31 µM on the HCC1395 cell line, investigation of three portions of the molecule to generate a library of compounds led to identification of submicromolar inhibitors. Through orthogonal readouts, we confirmed that these thienopyrimidine derivative compounds act as downstream inhibitors of the β-catenin-dependent Wnt-pathway. Targeting downstream components would be more efficacious for the tumors where the pathway is induced by loss-of-function mutations in APC or Axin or by gain-of-function mutations in β-catenin or TCF, as compared to the upstream pathway inhibitors such as Porcupine inhibitors. Inhibition of the Wnt-pathway is translated into differential efficacy on inhibition of TNBC cell proliferation. Although we have not yet identified the exact molecular target, our analyses indicate the target to be among the partners of β-catenin-dependent transcriptional factors. Identification of the molecular target would unlock further development through the target-based assay to support further SAR exploration to identify compounds suitable for in vivo evaluation. Such findings will also be key to define the scope of application, determine potential biomarkers, and permit patient stratification.
The Wnt3a or CHIR99021-stimulated luciferase activity in HCC1395 TNBC cell line stably transfected with the TopFlash reporter was analyzed essentially as described for the BT-20 TNBC cell line (Shaw et al., 2019b; Koval et al., 2021). Reporter cells were seeded at 10,000 cells/well in a white opaque 384-well plate in the final volume of 20 μl of DMEM medium supplemented with 10% FCS. The cells were maintained incubated at 37°C, 5% CO2 overnight for attachment. Subsequently, they were transfected by a plasmid encoding constitutively expressed Renilla luciferase under the CMV promoter overall as described in the manufacturer’s protocol using 12 μg/ml of DNA and 40 μl/ml XtremeGENE nine reagent. The next day, the medium in each well was replaced with a 10 μl of fresh medium containing compound of interest, and, after 1 h of preincubation with the compounds, additional 10 μl of medium supplemented with Wnt3a [purified as described (Willert et al., 2003; Koval and Katanaev, 2011)] or CHIR99021 were added creating respective final concentrations of 2.5 μg/ml or 5 μM of each. In case of other signal transduction reporters, no additional stimulation was performed. Compound dilutions were prepared by serial dilution in DMSO and diluted with the amount of medium necessary to obtain their final concentrations indicated on the figures and tables and maintain concentration of DMSO of 0.5% in all assay points. After overnight incubation, the supernatant in each well was removed, and the luciferase activity was measured as described (Dyer et al., 2000; Huber et al., 2021; Pellissier et al., 2021). The culture medium was completely removed from all wells of the plate. Finally, the luciferase activity of the firefly and Renilla luciferases was detected sequentially in individual wells of a 384-well plate through injection of corresponding measurement solutions and immediate reading (400 ms integration time) in Infinite M Plex multifunctional plate reader with injection module.
Indicated TNBC cell lines were detached and resuspended at 120,000 cells/ml and added into each well of a transparent 384-well plate in the final volume of 20 µl/well. The cells were maintained in DMEM containing 10% FBS at 37°C, 5% CO2 overnight. Next day, the medium in each well was replaced by 40 μl of the fresh one containing the indicated concentrations of compounds. In case of drug combination experiments, each compound dilution was delivered in corresponding well in 20 μl of medium. Compound dilutions were prepared by serial dilution in DMSO and diluted with the amount of medium necessary to obtain their final concentrations indicated on the figures and tables and maintain concentration of DMSO of 0.5% in all assay points. After incubation for 3–4 days, depending on the cell line, the medium in each well was replaced by 25 μl of 0.5 mg/ml Thiazolyl blue solution in 1xPBS following by incubation for 3 h at 37°C. Then the solution was removed and 25 μl DMSO was added into each well. Absorbance at 510 nm was measured in a Tecan Infinite M200 PRO plate reader.
HCC1395 TNBC cell line was seeded at 100,000 cells/well in 24 well plates. On the next day, the medium was replaced by the fresh one pre-warmed at 37°C containing the indicated compounds. After 24 h incubation, the medium was removed, followed by washing twice with 500 μl of 1x PBS. The cells were lysed in the well by addition of 30 μl of ice-cold RIPA buffer (1x TBS, 4 mM EDTA, 1% Triton, 0.1% SDS, 1x Protease inhibitor cocktail (Roche)) and incubated on ice for 10 min. The samples were resuspended and then centrifuged at 18,000 g at 4°C to remove debris. 15 μl of the supernatants each were further analyzed by Western blot with antibodies against c-Myc (Abcam) and α-Tubulin (Sigma) at 1:1,000 dilutions.
The data was visualized analyzed in either GraphPad Prism nine using indicated statistical analysis or in R using gplots package (Warnes et al., 2009). Dataset from CCLE (Barretina et al., 2012) were used in the study and pathway enrichment analysis was performed by DAVID online tool (Sherman et al., 2022). Drug combination data was treated in COMBENEFIT software (Di Veroli et al., 2016). Network was built using Cytoscape with StringDB plugin (Shannon et al., 2003; Szklarczyk et al., 2021).
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
LM, VS, and CB synthesized and characterized the compounds. AK tested the compounds in vitro biology assays. CB and LM analyzed the SAR based on the TopFlash assay in HCC1395 and designed the compounds from the different synthesis iterations. CB, AK, and VK analyzed the results and wrote the manuscript. All authors discussed the results and contributed to the manuscript. VK supervised the work.
This work was supported by grant from Innosuisse, 48262.1 IP-LS to VK. Open access funding was provided by the University of Geneva.
We are thankful to Léonardo Scapozza and his team who have generously provided access to the chemistry laboratory.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2022.1045102/full#supplementary-material
Ahmed, K., Koval, A., Xu, J., Bodmer, A., and Katanaev, V. L. (2019). Towards the first targeted therapy for triple-negative breast cancer: Repositioning of clofazimine as a chemotherapy-compatible selective Wnt pathway inhibitor. Cancer Lett. 449, 45–55. doi:10.1016/j.canlet.2019.02.018
Arce, L., Yokoyama, N. N., and Waterman, M. L. (2006). Diversity of LEF/TCF action in development and disease. Oncogene 25 (57), 7492–7504. doi:10.1038/sj.onc.1210056
Barretina, J., Caponigro, G., Stransky, N., Venkatesan, K., Margolin, A. A., Kim, S., et al. (2012). The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483 (7391), 603–607. doi:10.1038/nature11003
Blagodatski, A., Poteryaev, D., and Katanaev, V. L. (2014). Targeting the Wnt pathways for therapies. Mol. Cell. Ther. 2 (1), 28. doi:10.1186/2052-8426-2-28
Bugter, J. M., Fenderico, N., and Maurice, M. M. (2021). Mutations and mechanisms of Wnt-pathway tumour suppressors in cancer. Nat. Rev. Cancer 21 (1), 5–21. doi:10.1038/s41568-020-00307-z
Di Veroli, G. Y., Fornari, C., Wang, D., Mollard, S., Bramhall, J. L., Richards, F. M., et al. (2016). Combenefit: An interactive platform for the analysis and visualization of drug combinations. Bioinformatics 32 (18), 2866–2868. doi:10.1093/bioinformatics/btw230
Dyer, B. W., Ferrer, F. A., Klinedinst, D. K., and Rodriguez, R. (2000). A noncommercial dual luciferase enzyme assay system for reporter gene analysis. Anal. Biochem. 282 (1), 158–161. doi:10.1006/abio.2000.4605
Flanagan, D. J., Woodcock, S. A., Phillips, C., Eagle, C., and Sansom, O. J. (2022). Targeting ligand-dependent wnt pathway dysregulation in gastrointestinal cancers through porcupine inhibition. Pharmacol. Ther. 238, 108179. doi:10.1016/j.pharmthera.2022.108179
Huang, D. W., Sherman, B. T., and Lempicki, R. A. (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4 (1), 44–57. doi:10.1038/nprot.2008.211
Huber, R., Marcourt, L., Koval, A., Schnee, S., Righi, D., Michellod, E., et al. (2021). Chemoenzymatic synthesis of complex phenylpropanoid derivatives by the botrytis cinerea secretome and evaluation of their wnt inhibition activity. Front. Plant Sci. 12, 805610. doi:10.3389/fpls.2021.805610
Katanaev, V., and Koval, A. (2021). Pyrazole derivatives as nhibitors of the wnt signalling pathway.Wo2019166616a1
Katanaev, V. L., Blagodatski, A., Xu, J., Khotimchenko, Y., and Koval, A. (2021). Mining natural compounds to target WNT signaling: Land and sea tales. Handb. Exp. Pharmacol. 269, 215–248. doi:10.1007/164_2021_530
Koval, A., Bassanini, I., Xu, J., Tonelli, M., Boido, V., Sparatore, F., et al. (2021). Optimization of the clofazimine structure leads to a highly water-soluble C3-aminopyridinyl riminophenazine endowed with improved anti-Wnt and anti-cancer activity in vitro and in vivo. Eur. J. Med. Chem. 222, 113562. doi:10.1016/j.ejmech.2021.113562
Koval, A., and Katanaev, V. L. (2018). Dramatic dysbalancing of the Wnt pathway in breast cancers. Sci. Rep. 8 (1), 7329. doi:10.1038/s41598-018-25672-6
Koval, A., and Katanaev, V. L. (2012). Platforms for high-throughput screening of Wnt/Frizzled antagonists. Drug Discov. Today 17 (23–24), 1316–1322. doi:10.1016/j.drudis.2012.07.007
Koval, A., and Katanaev, V. L. (2011). Wnt3a stimulation elicits G-protein-coupled receptor properties of mammalian Frizzled proteins. Biochem. J. 433 (3), 435–440. doi:10.1042/BJ20101878
Koval, A., Pieme, C. A., Queiroz, E. F., Ragusa, S., Ahmed, K., Blagodatski, A., et al. (2018). Tannins from Syzygium guineense suppress Wnt signaling and proliferation of Wnt-dependent tumors through a direct effect on secreted Wnts. Cancer Lett. 435, 110–120. doi:10.1016/j.canlet.2018.08.003
Larasati, Y., Boudou, C., Koval, A., and Katanaev, V. L. (2022). Unlocking the Wnt pathway: Therapeutic potential of selective targeting FZD7 in cancer. Drug Discov. Today 27 (3), 777–792. doi:10.1016/j.drudis.2021.12.008
Liu, Z., Wang, P., Wold, E. A., Song, Q., Zhao, C., Wang, C., et al. (2021). Small-molecule inhibitors targeting the canonical WNT signaling pathway for the treatment of cancer. J. Med. Chem. 64 (8), 4257–4288. doi:10.1021/acs.jmedchem.0c01799
Mandapati, A., and Lukong, K. E. (2022). Triple negative breast cancer: Approved treatment options and their mechanisms of action. J. Cancer Res. Clin. Oncol. doi:10.1007/s00432-022-04189-6
Mao, C. D., and Byers, S. W. (2011). Cell-context dependent TCF/LEF expression and function: Alternative tales of repression, de-repression and activation potentials. Crit. Rev. Eukaryot. Gene Expr. 21 (3), 207–236. doi:10.1615/critreveukargeneexpr.v21.i3.10
Martin-Orozco, E., Sanchez-Fernandez, A., Ortiz-Parra, I., and Ayala-San Nicolas, M. (2019). WNT signaling in tumors: The way to evade drugs and immunity. Front. Immunol. 10, 2854. doi:10.3389/fimmu.2019.02854
Mohamed, T., and Rao, P. P. N. (2015). Facile approaches toward the synthesis of N4-monosubstituted quinazolin-2, 4-diamines. Tetrahedron Lett. 56 (49), 6882–6885. doi:10.1016/j.tetlet.2015.10.096
O’Reilly, E. A., Gubbins, L., Sharma, S., Tully, R., Guang, M. H. Z., Weiner-Gorzel, K., et al. (2015). The fate of chemoresistance in triple negative breast cancer (TNBC). BBA Clin. 3, 257–275. doi:10.1016/j.bbacli.2015.03.003
Parsons, M. J., Tammela, T., and Dow, L. E. (2021). WNT as a driver and dependency in cancer. Cancer Discov. 11 (10), 2413–2429. doi:10.1158/2159-8290.CD-21-0190
Pellissier, L., Koval, A., Marcourt, L., Ferreira Queiroz, E., Lecoultre, N., Leoni, S., et al. (2021). Isolation and identification of isocoumarin derivatives with specific inhibitory activity against wnt pathway and metabolome characterization of lasiodiplodia venezuelensis. Front. Chem. 9, 664489. doi:10.3389/fchem.2021.664489
Rennoll, S., and Yochum, G. (2015). Regulation of MYC gene expression by aberrant Wnt/β-catenin signaling in colorectal cancer. World J. Biol. Chem. 6 (4), 290–300. doi:10.4331/wjbc.v6.i4.290
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13 (11), 2498–2504. doi:10.1101/gr.1239303
Shaw, H. V., Koval, A., and Katanaev, V. L. (2019b). A high-throughput assay pipeline for specific targeting of frizzled GPCRs in cancer. Methods Cell Biol. 149, 57–75. doi:10.1016/bs.mcb.2018.08.006
Shaw, H. V., Koval, A., and Katanaev, V. L. (2019a). Targeting the wnt signalling pathway in cancer: Prospects and perils. Swiss Med. Wkly. 149, w20129. doi:10.4414/smw.2019.20129
Sherman, B. T., Hao, M., Qiu, J., Jiao, X., Baseler, M. W., Lane, H. C., et al. (2022). DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 50, W216–W221. doi:10.1093/nar/gkac194[
Stewart, D. J. (2014). Wnt signaling pathway in non-small cell lung cancer. J. Natl. Cancer Inst. 106 (1), djt356. doi:10.1093/jnci/djt356
Szklarczyk, D., Gable, A. L., Nastou, K. C., Lyon, D., Kirsch, R., Pyysalo, S., et al. (2021). The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 49 (D1), D605–D612. doi:10.1093/nar/gkaa1074
Tam, B. Y., Chiu, K., Chung, H., Bossard, C., Nguyen, J. D., Creger, E., et al. (2020). The CLK inhibitor SM08502 induces anti-tumor activity and reduces Wnt pathway gene expression in gastrointestinal cancer models. Cancer Lett. 473, 186–197. doi:10.1016/j.canlet.2019.09.009
Wang, C., Chen, Q., and Xu, H. (2021). Wnt/β-catenin signal transduction pathway in prostate cancer and associated drug resistance. Discov. Oncol. 12 (1), 40. doi:10.1007/s12672-021-00433-6
Warnes, G. R., Bolker, B., Bonebakker, L., Gentleman, R., Huber, W., Liaw, A., et al. (2009). gplots: Various R programming tools for plotting data, 2, 1. R package version.4
Willert, K., Brown, J. D., Danenberg, E., Duncan, A. W., Weissman, I. L., Reya, T., et al. (2003). Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature 423 (6938), 448–452. doi:10.1038/nature01611
Zhan, T., Rindtorff, N., and Boutros, M. (2017). Wnt signaling in cancer. Oncogene 36 (11), 1461–1473. doi:10.1038/onc.2016.304
Keywords: Wnt signaling, triple-negative breast cancer, β-catenin, cancer survival, medicinal chemistry, structure activity relationship, thienopyrimidine, quinazoline
Citation: Boudou C, Mattio L, Koval A, Soulard V and Katanaev VL (2022) Wnt-pathway inhibitors with selective activity against triple-negative breast cancer: From thienopyrimidine to quinazoline inhibitors. Front. Pharmacol. 13:1045102. doi: 10.3389/fphar.2022.1045102
Received: 15 September 2022; Accepted: 17 October 2022;
Published: 28 October 2022.
Edited by:
Ming Ji, Institute of Materia Medica, ChinaReviewed by:
Songwen Lin, Chinese Academy of Medical Sciences and Peking Union Medical College, ChinaCopyright © 2022 Boudou, Mattio, Koval, Soulard and Katanaev. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vladimir L. Katanaev, VmxhZGltaXIuS2F0YW5hZXZAdW5pZ2UuY2g=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.