 Andrew K. Evans
Andrew K. Evans Erwin Defensor
Erwin Defensor Mehrdad Shamloo
Mehrdad Shamloo
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Pharmacol., 30 November 2022
Sec. Neuropharmacology
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.1030609
This article is part of the Research TopicNeuroinflammatory and Oxidative/Nitrosative Pathways in Neuropsychiatric and Neurological Diseases and Their Possible Neuropharmacological Regulation, Volume IIView all 7 articles
Locus coeruleus (LC) noradrenergic (NE) neurons supply the main adrenergic input to the forebrain. NE is a dual modulator of cognition and neuroinflammation. NE neurons of the LC are particularly vulnerable to degeneration both with normal aging and in neurodegenerative disorders. Consequences of this vulnerability can be observed in both cognitive impairment and dysregulation of neuroinflammation. LC NE neurons are pacemaker neurons that are active during waking and arousal and are responsive to stressors in the environment. Chronic overactivation is thought to be a major contributor to the vulnerability of these neurons. Here we review what is known about the mechanisms underlying this neuronal vulnerability and combinations of environmental and genetic factors that contribute to confer risk to these important brainstem neuromodulatory and immunomodulatory neurons. Finally, we discuss proposed and potential interventions that may reduce the overall risk for LC NE neuronal degeneration.
Neurons of the locus coeruleus (LC) are the primary source of forebrain norepinephrine (NE). LC NE neurons are activated with waking and have a wide range of firing rates during various behavioral states related to attention, novelty, arousal, and in response to stressors, and their function is essential for adaptation, survival, and multiple aspects of cognitive function (Borodovitsyna et al., 2022; Chandler et al., 2019; Poe et al., 2020; Sara, 2009). The NE system is also essential for the regulation of neuroinflammation (Galea et al., 2003; Heneka et al., 2010; Feinstein et al., 2016; Weinshenker, 2018; Evans et al., 2020). Due to putative mechanisms that we will review here, LC NE neurons are one of the first sites of pathology in the aging brain and are highly susceptible to degeneration (Braak and Del, 2011; Mather and Harley, 2016; Kelly et al., 2017; Weinshenker, 2018). Given the role of the NE system in modulating neuroinflammation, it has been proposed that degeneration of the LC could be a common underlying etiological factor in progressive neuroinflammation observed widely in association with neurodegenerative disorders. An early loss of NE neurons and NE tone may contribute to and accelerate the deterioration of cognitive function and an exacerbation of neuroinflammation, contributing to the worsening of both symptoms and pathology in neurodegenerative disorders. This has opened the possibility that “selective neuronal vulnerability and degeneration” of the LC neurons may be an initiation point for the cascade of pathological events leading to a more widespread pathology and neuronal cell death in the aging and diseased brain (Mather and Harley, 2016). Why do LC neurons display this selective early-onset neuronal vulnerability? What factors determine the aberrant signaling leading to the initiation, temporal profile, and extent of this selective neuronal cell loss? Many possible scenarios have been suggested (see Figure 1), including chronic high metabolic demand, sensitivity to systemic inflammation, viral infection, or toxins in the environment, the persistent activity of oxidizing enzymes such as monoamine oxidase, mitochondrial dysfunction, oxidative and nitrosative stress, and cellular stress in response to misfolded proteins (Kang et al., 2020; Kang et al., 2022) (Zhang et al., 2014a; Wang et al., 2020). Here we review environmental and genetic factors contributing to the vulnerability of the LC and discuss common mechanisms and possible therapeutic interventions to attenuate this vulnerability. The aim of this mini-review is to increase awareness of the multiple risk factors that contribute to the vulnerability of the LC, to identify some common mechanisms of vulnerability, and to initiate further investigation into potential therapeutic interventions and approaches.
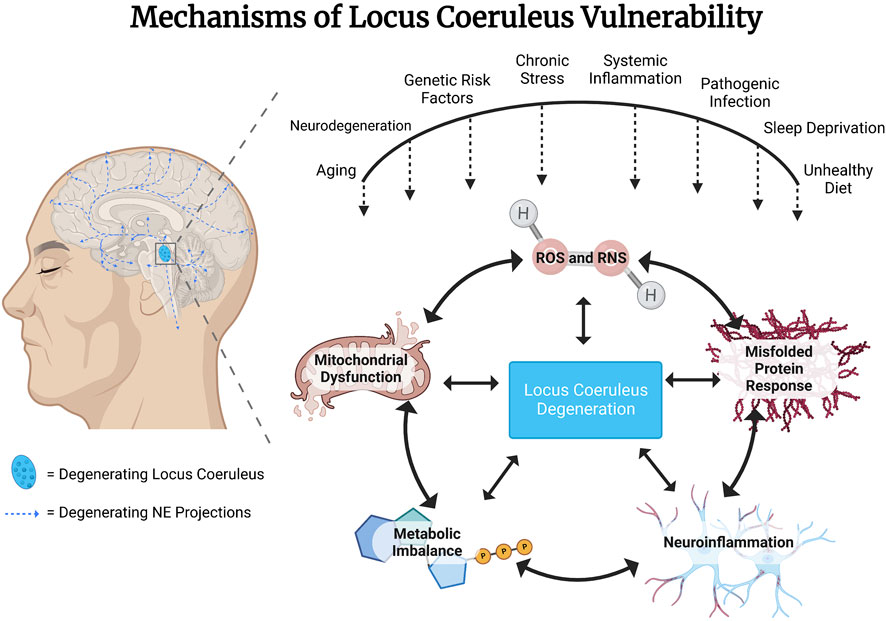
FIGURE 1. Genetic and environmental risk factors converge to confer vulnerability to locus coeruleus (LC) noradrenergic (NE) neurons. Proximate mechanisms of vulnerability that have been suggested to contribute to LC NE degeneration include actions of reactive oxygen and nitrogen species (ROS/RNS), response to misfolded proteins, neuroinflammatory mechanisms, metabolic imbalance and mitochondrial dysfunction due to high baseline rates of neuronal activity.
The LC NE system regulates diverse aspects of cognitive function, including arousal, attention, salience, learning and memory, and pain and stress responses; this role in cognitive function has been reviewed extensively (Aston-Jones et al., 2000; Valentino and Van Bockstaele, 2008; Sara, 2009; Roozendaal and McGaugh, 2011; Poe et al., 2020) (Rohampour et al., 2017; Farahani et al., 2021). The LC is a small nucleus comprising between 30,000 and 60,000 neurons in the human brain (German et al., 1988), and it has long been considered a homogeneous and broadly projecting nucleus with diffuse ascending and descending projections. Historically, functional specificity within the NE system has mainly been attributed to differences in excitatory and inhibitory postsynaptic receptor subtypes, receptor distribution patterns, and patterns of neuronal firing rates. However, anatomical subdivisions within the LC and the existence of some functional subsets of NE neurons with specific projection patterns have been described. For example, hippocampal projecting LC NE neurons were shown to be located in the dorsal core of the LC (Mason and Fibiger, 1979). Our understanding of LC function has further evolved in the last 10 years, in particular, as it relates to connectivity to other brain regions and tonic and phasic regulation of neuronal circuits, (Chandler et al., 2019; Poe et al., 2020). Recent investigation has demonstrated a robust functional specificity within subtypes of LC NE neurons. (Chandler et al., 2019). There is a functional topography with subpopulations of neurons receiving distinct inputs and having precise projections related to specific functions (Borodovitsyna et al., 2020). The concept of heterogeneity of the LC NE system complements nicely emerging evidence from other monoaminergic brainstem neuromodulatory systems, such as dopaminergic (Lammel et al., 2012; Beier et al., 2015; Watabe-Uchida and Uchida, 2018) and serotonergic (Lowry et al., 2007; Lowry et al., 2008; Hale and Lowry, 2011) systems. This functional heterogeneity within the LC may confer vulnerability to different subsets of LC NE neurons, which supports evidence for heterogeneity of vulnerability of LC neurons to degeneration in Alzheimer’s disease (AD) (German et al., 1992). Given this heterogeneity within the LC, it becomes less useful to talk about LC activation versus, for example, activation of hippocampal or prefrontal cortical projecting LC neurons as functionally defined collections of related neurons. The significance of this is illustrated in studies demonstrating that following bout of stress, LC NE neurons innervating the central nucleus of the amygdala become hyperactive and hyperexcitable. In contrast, those projecting to the prefrontal cortex have suppressed activity and excitability (Chandler et al., 2019). In this case, “activation of the LC” depends on which subpopulation of LC NE neurons is engaged in the specific task. This is especially relevant in the context of vulnerability of LC neurons in relation to chronic activation states.
Recent advances in optogenetic and chemogenetic techniques have enabled precise targeting of NE neuronal firing rates and have advanced our understanding of LC function. Chemogenetic selective activation of the LC has been shown to alter anxiety behavior and to induce large scale functional changes in neural connectivity associated with changes in pupil dilation (Zerbi et al., 2019). In a transgenic rat model (TgF344-AD) in which rats express mutant amyloid precursor protein and presenilin-1, rats show early hyperphosphorylation of tau in the LC, loss of hippocampal and cortical NE fibers, impaired reversal learning in the Morris Water Maze, and behavioral deficits were reversed with chemogenetic activation of the LC (Rorabaugh et al., 2017). In a Ts65Dn mouse model of down syndrome, in which LC NE neurons have been shown to degenerate, chemogenetic activation of LC NE neurons reversed deficits in novel object recognition (Fortress et al., 2015), while chemogenetic inhibition of LC NE neurons resulted in impaired novel object recognition and reversal learning and a potentiation of hippocampal microglia activation (Hamlett et al., 2020).
The NE system is also an important endogenous modulator of neuroinflammation and is involved in the recruitment of peripheral immune cells to the brain (Elenkov et al., 2000; Feinstein et al., 2002; Galea et al., 2003; Heneka et al., 2010; Madrigal et al., 2010; Hinojosa et al., 2011; Gyoneva and Traynelis, 2013). The anti-inflammatory effects of NE have been attributed mainly to the activation of beta-adrenergic receptors (ADRB1 and ADRB2) and modulation of both microglia and astrocyte function. Both ADRB1 and ADRB2 adrenergic receptors are expressed in microglia and astrocytes, although ADRB2 receptors are more highly expressed in both cell types (Zhang et al., 2014b). NE and beta-adrenergic agonists, via agonism of beta-adrenergic receptors, reduce the expression of proinflammatory cytokines in the brain (Feinstein et al., 2002; Zapater et al., 2012; Weber et al., 2015; Ardestani et al., 2017; Evans et al., 2020). An ADRB2 agonist, salmeterol, inhibits inflammasome activation and induction of tumor necrosis factor alpha (TNFa) and interleukin 1-beta (IL1b) in a lipopolysaccharide model of systemic inflammation (Song et al., 2018a). Another beta-adrenergic agonist, mabuterol, potentiates IL10 and attenuates macrophage inflammatory protein 1-alpha (MIP1a) in a lipopolysaccharide model of inflammation. Conversely, the beta-adrenergic antagonist propanol attenuates IL10 and potentiates MIP1a, demonstrating the bidirectional effects of noradrenergic pharmacology on the regulation of neuroimmune homeostasis in the brain (Evans et al., 2020). Given this role in the modulation of inflammation, degeneration of LC NE neurons and loss of adrenergic tone may result in dysregulation of neuroimmune homeostasis and potentiation of pathological neuroinflammation in neurodegenerative disorders (Feinstein et al., 2016) and may potentiate both inflammation and neuronal loss in neurodegenerative disorders such as Parkinson’s disease (PD) and AD (Weinshenker, 2008; Heneka et al., 2010; Weinshenker, 2018). Furthermore, the clinical use of beta-blockers for hypertension can further dysregulate the neuroimmune homeostasis in the vulnerable aging population and initiate a cascade of events leading to cognitive decline.
Loss of LC NE neurons occurs with normal aging and is accelerated in neurodegenerative disorders such as AD and PD, suggesting these neurons are fundamentally vulnerable to degeneration (Braak and Del, 2011; Kelly et al., 2017; Liu et al., 2019). The LC is one of the first brain regions to develop tau pathology in AD (Braak and Del, 2011) and to show significant early signs of neurodegeneration (German et al., 1992). Early-onset LC degeneration has been reported in patients with mild cognitive impairment before the full-blown manifestation of AD symptoms (Kelly et al., 2017). It has been suggested that degeneration of the LC and the extended LC network may actually precede and be a risk factor for more widespread degeneration in AD patients (Ross et al., 2015; Mather and Harley, 2016; Weinshenker, 2018; Jacobs et al., 2021). Under experimental conditions, loss of NE tone resulting from pharmacological antagonism or lesion of LC NE neurons exacerbates the behavioral deficits, neuroinflammation, and pathology observed in animal models of AD (Heneka et al., 2006; Kalinin et al., 2007; Heneka et al., 2010; Evans et al., 2020). In humans, pathology in the LC also precedes more widespread pathological cell death in neurodegenerative disorders such as AD and PD (Braak and Del, 2011; Betts et al., 2019). In an experimental model of AD, transfection of rat LC neurons with the gene for human mutant tau leads to spreading of pathological tau and behavioral changes related to odor discrimination learning in rats (Ghosh et al., 2019). Consequences of age-related degeneration of the LC NE system may be further exacerbated by the clinical use of brain-permeable beta-adrenergic antagonists, commonly prescribed beta-blockers for controlling hypertension and anxiety, which have been shown to impair cognitive function and potentiate inflammation (Gliebus and Lippa, 2007; Paran et al., 2010; Evans et al., 2020). Epidemiological studies have identified beta-blocker use as a risk factor for chronic neurodegenerative disorders such as PD and AD (Mittal et al., 2017; Cepeda et al., 2019).
Degeneration of the LC can be readily observed and quantified using imaging techniques such as structural MRI, but can also be detected early through more indirect measurement of physiological responses known to be controlled by the LC, such as pupillary dilation reflex, as well as in more complex indicators such as attention, and arousal (Marien et al., 2004; Grudzien et al., 2007; Jacobs et al., 2021; Mather and Harley, 2016; Weinshenker, 2018; Liu et al., 2019; Liu et al., 2020; Matchett et al., 2021). Notably, altered adrenergic signaling with an impact on cognitive function may precede the loss of noradrenergic neurons as the LC becomes dysregulated prior to degeneration (Goodman et al., 2021). This dysregulation may contribute to and precede degeneration as hyperactivity can lead to vulnerability factors such as metabolic and oxidative stress. Why is the LC so susceptible to dysregulation and degeneration? Both environmental and genetic factors may contribute to this vulnerability of LC NE neurons. Environmental risk factors for the development of AD coincide with factors that have been implicated in LC vulnerability, such as chronic stress, poor sleep hygiene, diet, and chronic inflammation (Bredesen, 2015; Ross et al., 2018; Zamore and Veasey, 2022). In terms of biological risk factors, functional studies have identified AD risk-related genes (e.g., TREM2, CD33) linked to the neuroimmune system (Griciuc and Tanzi, 2021) and proteomic changes implicating an altered metabolome (Johnson et al., 2022).
It is important to note that degeneration within the LC is not uniform (German et al., 1992), and biological features of subsets of neurons may contribute to differential vulnerability. Functional subsets of NE neurons, as defined by efferent projections, share morphological features, suggesting fundamental differences in subpopulations of LC NE neurons within the nucleus. New developments in targeting and identifying subpopulations of NE neurons in the LC using genetic, developmental, anatomical, and neurophysiological evidence have further characterized functional subsets of LC neurons with targeted projection sites and involved in specific cognitive functions (Totah et al., 2018; Chandler et al., 2019). Characterizing features of these neuronal subpopulations may help elucidate new mechanisms contributing to LC vulnerability.
Environmental factors that have been shown to impact LC vulnerability include systemic inflammation (Song et al., 2018b; Wang et al., 2020), viral infection, sleep (Zhang et al., 2014a; Zhu et al., 2015; Zamore and Veasey, 2022), diet (Chou et al., 2022) and stress (Ross et al., 2015) (see Figure 1). Many of these environmental factors have been modeled in rodents, enabling mechanistic studies of neurodegeneration and, specifically, LC degeneration. Underlying mechanistic factors contributing to neuronal vulnerability across these models include both oxidative and nitrosative stress, as discussed below.
Systemic inflammation can lead to neuroinflammation (Barrientos et al., 2005; Perry, 2010; Perry and Holmes, 2014) and presents a risk factor for the degeneration of LC NE neurons. Systemic inflammation induced by lipopolysaccharide in animal models has been shown to lead to progressive neurodegeneration with loss of neurons first seen in the locus coeruleus. Neuroinflammation and LC degeneration in this model were reduced in mice with the NADPH oxidase-2 (NOX2) gene deletion or mice treated with a NOX2 inhibitor, implicating NOX2 and production of reactive oxygen and nitrogen species in LC degeneration induced by systemic inflammation (Wang et al., 2020). Systemic inflammation in the presence of primed microglia in the context of aging or chronic neurodegenerative disease triggers a cascade of inflammatory signaling in the brain involving IL1b, neutrophil infiltration, and inducible nitric oxide synthase expression and is associated with increased neuronal death (Barrientos et al., 2005; Cunningham et al., 2005; Cunningham et al., 2009). In patients with Alzheimer’s disease, bouts of systemic inflammation and concentrations of circulating plasma TNFa is predictive of rates of cognitive decline (Holmes et al., 2009). We have observed that a subpopulation of LC NE neurons that are activated in response to systemic inflammation, labeled and chronically tracked with the FosTrap technique, are more susceptible to degeneration over time relative to those LC neurons not activated by systemic inflammation.
Viruses are capable of triggering inflammatory processes in the CNS by direct infiltration or by systemic activation of the immune system (Dahm et al., 2016; Agner and Klein, 2018; Xie et al., 2021). Neurological symptoms of viral infection may include fever, vomiting, chronic fatigue, impaired consciousness, and cognitive deficits (Wasay et al., 2008; Bathoorn et al., 2011; Chen et al., 2021; Wouk et al., 2021). While the acute immune response can be beneficial in promoting CNS repair and slowing down viral replication, chronic and/or irreversible complications that may persist long after viral clearance could be detrimental to brain function (Berth et al., 2009; Amor et al., 2014; Walker, 2018). The LC appears to be particularly vulnerable to toxins and infections, possibly due to extensive innervation of CNS vasculature and close proximity to the fourth ventricle, exposing it to potential pathogens in circulating blood and cerebrospinal fluid (Pamphlett, 2014; Mather and Harley, 2016). Although the causal link between viral infections and neurodegeneration is undetermined, many studies have investigated the association between the two factors and provided hypotheses describing how different viruses may impact neurodegeneration. For instance, herpes simplex virus 1 (HSV-1), which is widely implicated in neurodegenerative disorders (Itzhaki, 2014; Harris and Harris, 2015; Tzeng et al., 2018), can enter a dormant state in the trigeminal ganglion, which projects to the LC (Baringer and Swoveland, 1973). When reactivated, it has been hypothesized that HSV-1 may spread to the LC (Mather and Harley, 2016), where it can induce Aβ accumulation which can contribute to neurodegeneration (Santana et al., 2012; Gupta and Goyal, 2016). The H5N1 influenza virus has also been implicated in neurodegenerative disease. H5N1 infection in a mouse model induced parkinsonian-like symptoms and activated microglia. Alpha-synuclein aggregation, a hallmark of neurodegenerative disorders, was also induced and persisted after the infection was resolved. Behavioral impairments and brain dysfunction have been reported with infection by Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) (Niazkar et al., 2020), the virus that causes COVID-19, with some patients experiencing post-COVID symptoms that may last for years. Symptoms may be related to elevated proinflammatory cytokines, prolonged neuroinflammation, and potentially hypoxia (de Erausquin et al., 2021). The direct viral infection of the brain has also been proposed as a cause for these symptoms, as SARS-CoV-2 viral proteins were detected in the brain tissue of patients who died from COVID-19 (Matschke et al., 2020). It has been suggested that increased COVID-19 susceptibility and severity could increase the risk for neurodegenerative disorders (Li et al., 2022). While further investigations are required to clarify a causal link, these collective findings demonstrate the importance of understanding the neurodegenerative sequelae of viral infections.
Sleep deprivation or poor sleep hygiene (e.g., frequent waking, apnea) is a risk factor for Alzheimer’s disease (Ross et al., 2015; Bredesen et al., 2016; Feinstein et al., 2016) and in animal models has been shown to be causally linked to degeneration of LC NE neurons (Zhu et al., 2016). LC NE neurons are most active during wakefulness, with enhanced activity during times of novelty and unpredictability, and are quiet during sleep. LC NE neurons are vulnerable to neurodegeneration following prolonged wakefulness (Zamore and Veasey, 2022). Increased metabolic demand from short bouts of sleep deprivation (i.e., NE neurons remaining active for extended periods of wakefulness) is accompanied by a neuroprotective upregulation of mitochondrial sirtuin 3 (SirT3) signaling (Zhang et al., 2014a). SirT3 has been proposed to have many neuroprotective functions, such as deacetylation of mitochondrial ETC proteins and upregulation of molecules such as glutathione dehydrogenase and activation of isocitrate dehydrogenase, both of which increase levels of reduced glutathione, serving as an important antioxidant. It has been proposed that the extended periods of increased wakefulness and chronic activation of LC NE neurons may also lead to prolonging monoamine production in LC, leading to oxidative stress and shutting off the transient neuroprotective upregulation of mitochondrial SirT3 signaling (Zhang et al., 2014a). Other mechanisms linking extended wakefulness with neuronal vulnerability include NOS activation, mitochondrial stress, and activation of cleaved caspase-3 (Zhang et al., 2014a).
Insulin resistance and neuroinflammation resulting from a high-fat, high-carbohydrate, Western-style diet have both been identified as risk factors for the development of Alzheimer’s disease (Liu et al., 2011; Bredesen, 2015). Postmortem brain from patients with AD show deficits in the insulin–PI3K–AKT signaling pathway, which could contribute to tau hyperphosphorylation in the LC through activation of glycogen synthase kinase-3β (Liu et al., 2011). Rats sustained on a high-fat carbohydrate-rich diet for 30 weeks show loss of NE neurons in the LC and the substantia nigra (as measured by tyrosine hydroxylase-positive neurons). This cell loss is associated with a deficit in astrocytes (Chou et al., 2022). Astrocytes contribute to a neuroprotective environment by preventing nitrosative damage, as discussed below (Madrigal et al., 2007; Madrigal et al., 2017).
Stress and elevated glucocorticoid levels engage LC NE neurons (Curtis et al., 2002; Valentino and Van Bockstaele, 2008) and may cause long-term aberrant signaling changes leading to neuronal vulnerability (Ross et al., 2015; Borodovitsyna et al., 2018). Chronic social stress increases mu opioid receptor expression in the LC and results in chronic suppression of LC activity that resembles opiate dependence, an effect that endures long after the stress has ended (Chaijale et al., 2013). In light of recent work identifying differential effects of stress on subpopulations of LC NE neurons, this opioid-dependent suppression of LC activity could be within a subpopulation of functionally relevant NE neurons, but this remains to be seen (Borodovitsyna et al., 2020). These changes in response to increased endogenous opioid tone have been linked to corticotrophin-releasing hormone signaling from the amygdala. Social stress also upregulates proinflammatory gene expression in the LC, including IL1β (Wood et al., 2015). Stress increases nitric oxide synthesis in the brain, which may lead to degeneration (Madrigal et al., 2006). In experimental rodent models of stress, chronic unpredictable mild stress results in the generation of reactive oxygen species in the brain coupled with neuroinflammation. The effects of stress on neuroinflammation can be exacerbated in transgenic mice in which the mitochondrial uncoupling protein 2 (UCP2) has been knocked out, as reviewed by (Anderson and Maes, 2014). In humans, psychological stress is associated with increased markers of oxidative stress in students (Sivonova et al., 2004).
Genome Wide Association Studies and large-scale proteomic analyses have identified genetic risk factors and biological pathways related to susceptibility to developing AD, several of which may contribute directly to neuronal vulnerability (Zhang et al., 2013; Griciuc and Tanzi, 2021; Johnson et al., 2022). Proteomic analyses have identified biological pathways underlying AD risks, such as cellular metabolism and mitochondrial oxidative stress, as a clear source of vulnerability for the development of AD (Johnson et al., 2022). Genes and proteins that have been identified include Triggering receptors expressed on myeloid cells 2 (TREM2), sialic acid binding Ig-like lectin 3 (Siglec-3; CD33), apolipoprotein E version 4 (APOE4), and mitogen-activated protein kinase (MAPK) signaling (Johnson et al., 2022). Other proteins such as glutamate pyruvate transaminase 2 (GPT2), a metabolic enzyme related to cellular metabolism and the Krebs cycle, implicate mitochondrial metabolism in LC NE neuronal health as KO of gpt2 in mice results in LC degeneration and are also linked to a neurological developmental disorder in children (Baytas et al., 2022). CD33 mutation impairs phagocytosis of beta-amyloid by microglia (Griciuc et al., 2013; Griciuc et al., 2020). In a newly developed mouse model of genetic risk for sporadic or late-onset Alzheimer’s disease (LOAD mice) transgenic mice express humanized APOE4 and TREM2 (Oblak et al., 2021; Oblak et al., 2022) alongside a humanized beta-amyloid precursor protein. This is an important advancement in modeling disease mechanisms in the laboratory as, up until this point, transgenic models have focused mainly on genes related to early onset AD, such as APP and presenilin mutations, which represent only a small fraction of human AD patients. Broadening these models to include genetic risk factors for sporadic or late-onset AD may lead to more scaleable results. Studies combining environmental risk factors with LOAD genetic susceptibility mark a new path forward (Kotredes et al., 2021; Oblak et al., 2022).
NE neurons of the LC are pacemaker neurons with a high metabolic rate during waking, arousal, and stress. The increased metabolic demand on LC NE neurons, with the extended activity of monoamine oxidase and related accumulation of free oxygen radicals, may contribute to their vulnerability (Zhang et al., 2014a; Wang et al., 2020). Overactivation of NE neurons may also lead to the accumulation of neurotoxic metabolites shown to induce tau pathology (Kang et al., 2020; Kang et al., 2022). NE neurons produce a toxic metabolite, DOPEGAL, when overactivated, which contributes to tau hyperphosphorylation (Kang et al., 2022). Hyperactivation of LC NE neurons may also contribute to mitochondrial oxidative stress through activity-dependent increases in nitric oxide production (Sanchez-Padilla et al., 2014), triggering aberrant cellular signaling and leading to neuronal cell death. LC neurons are also susceptible to tau hyperphosphorylation through alpha-2a adrenergic receptor signaling on LC neurons which, through an autoreceptor mechanism, has been shown to potentiate tau hyperphosphorylation (Zhang et al., 2020). Conversely, antagonism of alpha-2a adrenergic receptors, which blocks autoreceptors on LC neurons and increases NE release, reduces NOS expression and prevents beta-amyloid-induced nitrosative damage (Kalinin et al., 2006). Even under normal physiological conditions, LC NE neurons show higher levels of mitochondrial oxidative stress, which is then exacerbated with further cellular stress inducers such as systemic inflammation (Wang et al., 2020). Aged mice have evidence of oxidative stress and changes in key mitochondrial proteins involved in energy homeostasis in the LC at an early stage before any evident sign of neuronal degeneration (Evans et al., 2021).
Molecular damage attributed to reactive oxygen species (ROS) is common in postmortem brains of Alzheimer’s disease patients, including lipid oxidation and oxidative DNA damage. Beta-amyloid aggregation induces the production of ROS and may mediate some of the cellular damage associated with plaque pathology (Ono et al., 2006). Damage to membrane-bound transporters and ion channels through oxidation has been proposed to underlie compromised mitochondrial function, excitotoxicity, apoptosis, and synaptic degeneration (Mattson, 2004). Beta-amyloid induces astrocytic activation of NADPH oxidase and generation of reactive oxygen species leading to failure of mitochondrial respiration and neuronal death (Abramov et al., 2004). Intracellular beta-amyloid aggregation is also associated with decreased intracellular concentrations of the important endogenous antioxidant glutathione (Madrigal et al., 2007). Understanding the longitudinal progression of the cellular injury and mechanisms underlying selective neuronal vulnerability in the LC is essential for understanding AD pathophysiology and will advance the development of therapeutic interventions.
Multiple strategies combining lifestyle changes such as improved sleep, diet, stress reduction, and physical activity with natural pharmacological therapeutics targeting mechanisms (e.g., reactive oxygen and nitrogen species, inflammation, autophagy, NAD deficiencies, mitochondrial stress) may be effective at preventing or slowing the course of LC or neuronal degeneration (see Figure 2). These hypotheses, while still needing much further validation and confirmation, have been built on preclinical animal studies and epidemiological studies in humans.
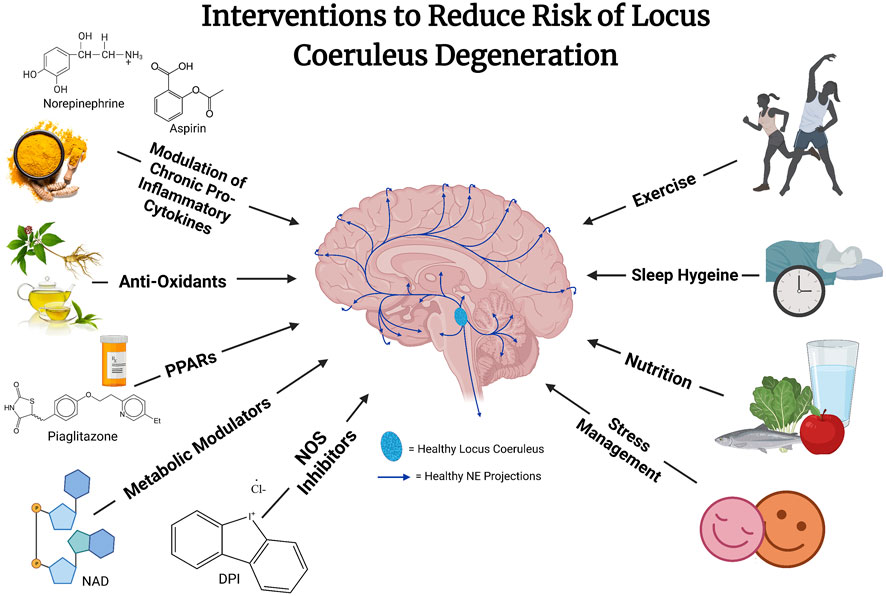
FIGURE 2. The risk of locus coeruleus degeneration may be reduced through a combination of different interventions. These interventions remain hypothetical and likely will work in concert to reduce vulnerability to what is likely a multi-dimensional disease etiology. Future well designed and controlled studies are needed to explore the efficacy of these approaches.
Fasting and dietary restriction have been shown to reduce cellular stress and may be neuroprotective (Mattson et al., 2004). Whereas an unhealthy diet may contribute to systemic inflammation and neuroinflammation, healthy nutrition can contribute to a healthy and responsive immune system that can help control systemic inflammation. Balanced nutrition and access to vitamins are essential for a healthy immune system and for replenishing precursors for important biochemical metabolic pathways and antioxidants. Exercise has been shown to have long-term anti-inflammatory effects (De Miguel et al., 2021), and exercise/environmental enrichment is protective against beta-amyloid-induced hippocampal impairment and inflammation in mice (Li et al., 2013; Xu et al., 2018). Whereas short-term effects of exercise have been shown to activate the immune system, this is thought to “train” the immune system and lead to the development of a more responsive immune system capable of being controlled more tightly and reducing the risk of chronic inflammation. Good sleep hygiene allows NE neurons to replenish antioxidant glutathione levels and protective sirtuin pathways, as described above (Zamore and Veasey, 2022). Finally, stress reduction, through a multitude of practices including but not limited to practices such as therapy sessions, meditation, and establishing a strong social support system, has been shown to reduce systemic markers of inflammation, increase cognitive performance, and is thought to reduce risk for psychiatric disease, including neurodegenerative disorders. It should also be noted that these lifestyle factors are also highly interrelated and can be difficult to dissect as exercise is a form of stress reduction, and stress reduction is linked with improved sleep and improved nutrition. The idea that the prevention of neurodegenerative disease may require a multifactorial approach using combined therapies, including lifestyle factors, is gaining momentum in the field (Bredesen et al., 2016).
Anti-inflammatory agents, such as non-steroidal anti-inflammatory drugs, have been proposed to reduce the risk for neurodegenerative diseases such as AD, and epidemiological studies have shown mixed results. In mouse models of AD, increasing adrenergic tone results in cognitive improvement and attenuation of beta-amyloid load, neuroinflammation, and tau pathology (Heneka et al., 2010; Coutellier et al., 2014; Ardestani et al., 2017). Chronic inhibition of NOS with a NOS inhibitor, Diphenyleneiodonium (DPI), can be neuroprotective and reverse mitochondrial stress and oxidative damage in animal models of LC degeneration induced by systemic inflammation (Wang et al., 2020). NE inhibits nitric oxide synthase by neuronal production of the neuroprotective CX3CL1 (Feinstein et al., 1993), which reduces nitric oxide production in microglia (Madrigal et al., 2017). NE has been shown to increase the production of the endogenous antioxidant glutathione in a cascade thought to involve the activation of peroxisome proliferator-activated receptors (PPARs) (Madrigal et al., 2007). NE has been suggested to be neuroprotective through its effects on neuroplasticity, inflammation, energy metabolism, and oxidative stress, for review, see (Marien et al., 2004). Antioxidant properties of NE may underlie the neuroprotective effects of NE on dopaminergic neurons in culture (Troadec et al., 2001). A study including four million Norwegians showed a reduced risk for neurodegenerative disease using the commonly prescribed beta-adrenergic agonist, salbutamol. In contrast, the use of the beta-adrenergic antagonist propranolol correlated with increased risk and worsening clinical outcomes (Mittal et al., 2017). This was later confirmed in a follow study by Janssen Pharmaceutical on 117 million people demonstrating adverse effects of the commonly prescribed beta-blocker propranolol (Cepeda et al., 2019).
mTOR (mechanistic target of rapamycin) is an important serine/threonine protein kinase with a critical regulatory function in cellular and neuronal homeostasis in the regulation of mitochondrial function, energy metabolism, and autophagy. mTOR plays a key pathological role in blocking insulin receptor signaling, autophagy, inflammation, and removal of the misfolded protein aggregates such beta-amyloid. Inhibition of the mTOR has been shown to be neuroprotective in neurodegenerative disorders, as reviewed (Querfurth and Lee, 2021). It has also been suggested and shown that mTOR inhibition can lead to the prolongation of life in mice. This has now been investigated in multiple ongoing human studies (Harrison et al., 2009). Finally, NAD depletion has been suggested to be one of the major factors associated with neurocognitive disorders and associated with neuronal cell death, as reviewed (Guerreiro et al., 2020). Therefore a systemic supplementation of NAD or pharmacological inhibition of the CD38 enzyme responsible for the degradation of the NAD has been explored as an additional potential therapeutic approach (Guerreiro et al., 2020).
Locus coeruleus noradrenergic neurons are one of the first sites of pathology in neurodegenerative disorders. Their early degeneration, coupled with a loss of noradrenergic tone, can contribute to both cognitive impairment and enhanced pathology and unchecked neuroinflammation associated with these disorders. Risk factors associated with a higher incidence of brain disorders, such as systemic inflammation, have been shown to lead to regional neuroinflammation and chronic degeneration of locus coeruleus noradrenergic neurons in experimental models. These findings warrant further investigation to understand why LC neurons display this selective early-onset neuronal vulnerability in aging and neurodegenerative diseases. What factors determine the aberrant cellular cascades leading to the initiation, temporal profile, and extent of this selective neuronal cell loss? In addition, experimental and epidemiological findings have identified the long-term use of CNS active blood-brain barrier permeable beta-blockers as a risk factor for the deterioration of cognitive function in neurodegenerative disorders such as Parkinson’s and Alzheimer’s. While beta-blockers are important for cardiovascular and blood pressure maintenance, a risk-to-benefit assessment and alternative treatment with a different mode of action should be explored. Our future effort should be focused on the elucidation of mechanistic pathways triggered by risk factors such as systemic inflammation, sleep disruption, chronic stress, and viral infections leading to selective neuronal cell loss in LC and other key brain regions. Identifying underlying mechanisms responsible for the particular vulnerability of LC and examining factors contributing to its resiliency through which protection or prevention may be possible will be essential for understanding the pathophysiology of neurodegenerative disorders and the development of effective therapeutic approaches for its prevention.
AE, ED, and MS wrote and revised the manuscript.
This work was partially supported by the National Institutes of Health (NIH R21 NS097945) (NIH R01 AG054533) awarded to MS.
We would like to thank Sarah Blumenfeld for her artistic and conceptual contribution to Figures 1 and Figure 2. Figures were created with BioRender.com.
MS is a co-founder of CuraSen Therapeutics. MS and AE own shares of stock in CuraSen Therapeutics.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abramov, A. Y., Canevari, L., and Duchen, M. R. (2004). Beta-amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J. Neurosci. 24, 565–575. doi:10.1523/JNEUROSCI.4042-03.2004
Agner, S. C., and Klein, R. S. (2018). Viruses have multiple paths to central nervous system pathology. Curr. Opin. Neurol. 31, 313–317. doi:10.1097/WCO.0000000000000556
Amor, S., Peferoen, L. A. N., Vogel, D. Y. S., Breur, M., van der Valk, P., Baker, D., et al. (2014). Inflammation in neurodegenerative diseases-an update. Immunology 142, 151–166. doi:10.1111/imm.12233
Anderson, G., and Maes, M. (2014). Oxidative/nitrosative stress and immuno-inflammatory pathways in depression: Treatment implications. Curr. Pharm. Des. 20, 3812–3847. doi:10.2174/13816128113196660738
Ardestani, P. M., Evans, A. K., Yi, B., Nguyen, T., Coutellier, L., and Shamloo, M. (2017). Modulation of neuroinflammation and pathology in the 5XFAD mouse model of Alzheimer's disease using a biased and selective beta-1 adrenergic receptor partial agonist. Neuropharmacology 116, 371–386. doi:10.1016/j.neuropharm.2017.01.010
Aston-Jones, G., RaJkowski, J., and Cohen, J. (2000). Locus coeruleus and regulation of behavioral flexibility and attention. Prog. Brain Res. 126, 165–182. doi:10.1016/S0079-6123(00)26013-5
Baringer, J. R., and Swoveland, P. (1973). Recovery of herpes-simplex virus from human trigeminal ganglions. N. Engl. J. Med. 288, 648–650. doi:10.1056/NEJM197303292881303
Barrientos, R. M., Higgins, E. A., Biedenkapp, J. C., Sprunger, D. B., Wright-Hardesty, K. J., Watkins, L. R., et al. (2005). Peripheral infection and aging interact to impair hippocampal memory consolidation. Neurobiol. Aging 27, 723–732. doi:10.1016/j.neurobiolaging.2005.03.010
Bathoorn, E., Vlaminckx, B. J. M., Schoondermark-Stolk, S., Donders, R., van der Meulen, M., and Thijsen, S. F. T. (2011). Primary Epstein-Barr virus infection with neurological complications. Scand. J. Infect. Dis. 43, 136–144. doi:10.3109/00365548.2010.531760
Baytas, O., Davidson, S. M., DeBerardinis, R. J., and Morrow, E. M. (2022). Mitochondrial enzyme GPT2 regulates metabolic mechanisms required for neuron growth and motor function in vivo. Hum. Mol. Genet. 31, 587–603. doi:10.1093/hmg/ddab269
Beier, K. T., Steinberg, E. E., DeLoach, K. E., Xie, S., Miyamichi, K., Schwarz, L., et al. (2015). Circuit architecture of VTA dopamine neurons revealed by systematic input-output mapping. Cell 162, 622–634. doi:10.1016/j.cell.2015.07.015
Berth, S. H., Leopold, P. L., and Morfini, G. N. (2009). Virus-induced neuronal dysfunction and degeneration. Front. Biosci. 14, 5239–5259. doi:10.2741/3595
Betts, M. J., Kirilina, E., Otaduy, M. C. G., Ivanov, D., Acosta-Cabronero, J., Callaghan, M. F., et al. (2019). Locus coeruleus imaging as a biomarker for noradrenergic dysfunction in neurodegenerative diseases. Brain 142, 2558–2571. doi:10.1093/brain/awz193
Borodovitsyna, O., Duffy, B. C., Pickering, A. E., and Chandler, D. J. (2020). Anatomically and functionally distinct locus coeruleus efferents mediate opposing effects on anxiety-like behavior. Neurobiol. Stress 13, 100284. doi:10.1016/j.ynstr.2020.100284
Borodovitsyna, O., Joshi, N., and Chandler, D. (2018). Persistent stress-induced neuroplastic changes in the locus coeruleus/norepinephrine system. Neural Plast. 2018, 1892570. doi:10.1155/2018/1892570
Borodovitsyna, O., Tkaczynski, J. A., Corbett, C. M., Loweth, J. A., and Chandler, D. J. (2022). Age- and sex-dependent changes in locus coeruleus physiology and anxiety-like behavior following acute stressor exposure. Front. Behav. Neurosci. 16, 808590. doi:10.3389/fnbeh.2022.808590
Braak, H., and Del, T. K. (2011). The pathological process underlying Alzheimer's disease in individuals under thirty. Acta Neuropathol. 121, 171–181. doi:10.1007/s00401-010-0789-4
Bredesen, D. E., Amos, E. C., Canick, J., Ackerley, M., Raji, C., Fiala, M., et al. (2016). Reversal of cognitive decline in Alzheimer's disease. Aging (Albany NY) 8, 1250–1258. doi:10.18632/aging.100981
Bredesen, D. E. (2015). Metabolic profiling distinguishes three subtypes of Alzheimer's disease. Aging (Albany NY) 7, 595–600. doi:10.18632/aging.100801
Cepeda, M. S., Kern, D. M., Seabrook, G. R., and Lovestone, S. (2019). Comprehensive real-world assessment of marketed medications to guide Parkinson's drug discovery. Clin. Drug Investig. 39, 1067–1075. doi:10.1007/s40261-019-00830-4
Chaijale, N. N., Curtis, A. L., Wood, S. K., Zhang, X. Y., Bhatnagar, S., Reyes, B. A., et al. (2013). Social stress engages opioid regulation of locus coeruleus norepinephrine neurons and induces a state of cellular and physical opiate dependence. Neuropsychopharmacology 38, 1833–1843. doi:10.1038/npp.2013.117
Chandler, D. J., Jensen, P., McCall, J. G., Pickering, A. E., Schwarz, L. A., and Totah, N. K. (2019). Redefining noradrenergic neuromodulation of behavior: Impacts of a modular locus coeruleus architecture. J. Neurosci. 39, 8239–8249. doi:10.1523/JNEUROSCI.1164-19.2019
Chen, X., Laurent, S., Onur, O. A., Kleineberg, N. N., Fink, G. R., Schweitzer, F., et al. (2021). A systematic review of neurological symptoms and complications of COVID-19. J. Neurol. 268, 392–402. doi:10.1007/s00415-020-10067-3
Chou, M. C., Lee, H. C., Liu, Y. C., Yen, P. S. Y., Liu, C. K., Chen, C. H., et al. (2022). Long-term high-fat diet consumption depletes glial cells and tyrosine hydroxylase-containing neurons in the brain of middle-aged rats. Cells 11, 295. doi:10.3390/cells11020295
Coutellier, L., Ardestani, P. M., and Shamloo, M. (2014). β1-adrenergic receptor activation enhances memory in Alzheimer's disease model. Ann. Clin. Transl. Neurol. 1, 348–360. doi:10.1002/acn3.57
Cunningham, C., Campion, S., Lunnon, K., Murray, C. L., Woods, J. F. C., Deacon, R. M. J., et al. (2009). Systemic inflammation induces acute behavioral and cognitive changes and accelerates neurodegenerative disease. Biol. Psychiatry 65, 304–312. doi:10.1016/j.biopsych.2008.07.024
Cunningham, C., Wilcockson, D. C., Campion, S., Lunnon, K., and Perry, V. H. (2005). Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J. Neurosci. 25, 9275–9284. doi:10.1523/JNEUROSCI.2614-05.2005
Curtis, A. L., Bello, N. T., Connolly, K. R., and Valentino, R. J. (2002). Corticotropin-releasing factor neurones of the central nucleus of the amygdala mediate locus coeruleus activation by cardiovascular stress. J. Neuroendocrinol. 14, 667–682. doi:10.1046/j.1365-2826.2002.00821.x
Dahm, T., Rudolph, H., Schwerk, C., Schroten, H., and Tenenbaum, T. (2016). Neuroinvasion and inflammation in viral central nervous system infections. Mediat. Inflamm. 2016, 8562805. doi:10.1155/2016/8562805
de Erausquin, G. A., Snyder, H., Carrillo, M., Hosseini, A. A., Brugha, T. S., Seshadri, S., et al. (2021). The chronic neuropsychiatric sequelae of COVID-19: The need for a prospective study of viral impact on brain functioning. Alzheimers Dement. 17, 1056–1065. doi:10.1002/alz.12255
De Miguel, Z., Khoury, N., Betley, M. J., Lehallier, B., Willoughby, D., Olsson, N., et al. (2021). Exercise plasma boosts memory and dampens brain inflammation via clusterin. Nature 600, 494–499. doi:10.1038/s41586-021-04183-x
Elenkov, I. J., Wilder, R. L., Chrousos, G. P., and Vizi, E. S. (2000). The sympathetic nerve-an integrative interface between two supersystems: The brain and the immune system. Pharmacol. Rev. 52, 595–638.
Evans, A. K., Ardestani, P. M., Yi, B., Park, H. H., Lam, R. K., and Shamloo, M. (2020). Beta-adrenergic receptor antagonism is proinflammatory and exacerbates neuroinflammation in a mouse model of Alzheimer's Disease. Neurobiol. Dis. 146, 105089. doi:10.1016/j.nbd.2020.105089
Evans, A. K., Park, H. H., Saw, N. L., Singhal, K., Ogawa, G., Leib, R. D., et al. (2021). Age-related neuroinflammation and pathology in the locus coeruleus and hippocampus: Beta-adrenergic antagonists exacerbate impairment of learning and memory in aged mice. Neurobiol. Aging 106, 241–256. doi:10.1016/j.neurobiolaging.2021.06.012
Farahani, F., Azizi, H., Janahmadi, M., Seutin, V., and Semnanian, S. (2021). Formalin-induced inflammatory pain increases excitability in locus coeruleus neurons. Brain Res. Bull. 172, 52–60. doi:10.1016/j.brainresbull.2021.04.002
Feinstein, D. L., GalEa, E., and Reis, D. J. (1993). Norepinephrine suppresses inducible nitric oxide synthase activity in rat astroglial cultures. J. Neurochem. 60, 1945–1948. doi:10.1111/j.1471-4159.1993.tb13425.x
Feinstein, D. L., Heneka, M. T., Gavrilyuk, V., Dello Russo, C., Weinberg, G., and Galea, E. (2002). Noradrenergic regulation of inflammatory gene expression in brain. Neurochem. Int. 41, 357–365. doi:10.1016/s0197-0186(02)00049-9
Feinstein, D. L., Kalinin, S., and Braun, D. (2016). Causes, consequences, and cures for neuroinflammation mediated via the locus coeruleus: Noradrenergic signaling system. J. Neurochem. 139 (2), 154–178. doi:10.1111/jnc.13447
Fortress, A. M., Hamlett, E. D., Vazey, E. M., Aston-Jones, G., Cass, W. A., Boger, H. A., et al. (2015). Designer receptors enhance memory in a mouse model of Down syndrome. J. Neurosci. 35, 1343–1353. doi:10.1523/JNEUROSCI.2658-14.2015
Galea, E., Heneka, M. T., Dello Russo, C., and Feinstein, D. L. (2003). Intrinsic regulation of brain inflammatory responses. Cell. Mol. Neurobiol. 23, 625–635. doi:10.1023/a:1025084415833
German, D. C., Manaye, K. F., White, C. L., Woodward, D. J., McIntire, D. D., Smith, W. K., et al. (1992). Disease-specific patterns of locus coeruleus cell loss. Ann. Neurol. 32, 667–676. doi:10.1002/ana.410320510
German, D. C., Walker, B. S., Manaye, K., Smith, W. K., Woodward, D. J., and North, A. J. (1988). The human locus coeruleus: Computer reconstruction of cellular distribution. J. Neurosci. 8, 1776–1788. doi:10.1523/jneurosci.08-05-01776.1988
Ghosh, A., Torraville, S. E., Mukherjee, B., Walling, S. G., Martin, G. M., Harley, C. W., et al. (2019). An experimental model of braak's pretangle proposal for the origin of Alzheimer's disease: The role of locus coeruleus in early symptom development. Alzheimers Res. Ther. 11, 59. doi:10.1186/s13195-019-0511-2
Gliebus, G., and Lippa, C. F. (2007). The influence of beta-blockers on delayed memory function in people with cognitive impairment. Am. J. Alzheimers Dis. Other Demen. 22, 57–61. doi:10.1177/1533317506295889
Goodman, A. M., Langner, B. M., Jackson, N., Alex, C., and McMahon, L. L. (2021). Heightened hippocampal beta-adrenergic receptor function drives synaptic potentiation and supports learning and memory in the TgF344-AD rat model during prodromal Alzheimer's disease. J. Neurosci. 41, 5747–5761. doi:10.1523/JNEUROSCI.0119-21.2021
Griciuc, A., Federico, A. N., Natasan, J., Forte, A. M., McGinty, D., Nguyen, H., et al. (2020). Gene therapy for Alzheimer's disease targeting CD33 reduces amyloid beta accumulation and neuroinflammation. Hum. Mol. Genet. 29, 2920–2935. doi:10.1093/hmg/ddaa179
Griciuc, A., Serrano-Pozo, A., Parrado, A. R., Lesinski, A. N., Asselin, C. N., Mullin, K., et al. (2013). Alzheimer's disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 78, 631–643. doi:10.1016/j.neuron.2013.04.014
Griciuc, A., and Tanzi, R. E. (2021). The role of innate immune genes in Alzheimer's disease. Curr. Opin. Neurol. 34, 228–236. doi:10.1097/WCO.0000000000000911
Grudzien, A., Shaw, P., Weintraub, S., Bigio, E., Mash, D. C., and Mesulam, M. M. (2007). Locus coeruleus neurofibrillary degeneration in aging, mild cognitive impairment and early Alzheimer's disease. Neurobiol. Aging 28, 327–335. doi:10.1016/j.neurobiolaging.2006.02.007
Guerreiro, S., Privat, A. L., Bressac, L., and Toulorge, D. (2020). CD38 in neurodegeneration and neuroinflammation. Cells 9, E471. doi:10.3390/cells9020471
Gupta, A., and Goyal, R. (2016). Amyloid beta plaque: A culprit for neurodegeneration. Acta Neurol. belg. 116, 445–450. doi:10.1007/s13760-016-0639-9
Gyoneva, S., and Traynelis, S. F. (2013). Norepinephrine modulates the motility of resting and activated microglia via different adrenergic receptors. J. Biol. Chem. 288, 15291–15302. doi:10.1074/jbc.M113.458901
Hale, M. W., and Lowry, C. A. (2011). Functional topography of midbrain and pontine serotonergic systems: Implications for synaptic regulation of serotonergic circuits. Psychopharmacol. Berl. 213, 243–264. doi:10.1007/s00213-010-2089-z
Hamlett, E. D., Ledreux, A., Gilmore, A., Vazey, E. M., Aston-Jones, G., Boger, H. A., et al. (2020). Inhibitory designer receptors aggravate memory loss in a mouse model of down syndrome. Neurobiol. Dis. 134, 104616. doi:10.1016/j.nbd.2019.104616
Harris, S. A., and Harris, E. A. (2015). Herpes simplex virus type 1 and other pathogens are key causative factors in sporadic Alzheimer's disease. J. Alzheimers Dis. 48, 319–353. doi:10.3233/JAD-142853
Harrison, D. E., Strong, R., Sharp, Z. D., Nelson, J. F., Astle, C. M., Flurkey, K., et al. (2009). Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460, 392–395. doi:10.1038/nature08221
Heneka, M. T., Nadrigny, F., Regen, T., Martinez-Hernandez, A., Dumitrescu-Ozimek, L., Terwel, D., et al. (2010). Locus ceruleus controls Alzheimer's disease pathology by modulating microglial functions through norepinephrine. Proc. Natl. Acad. Sci. U. S. A. 107, 6058–6063. doi:10.1073/pnas.0909586107
Heneka, M. T., Ramanathan, M., Jacobs, A. H., Dumitrescu-Ozimek, L., Bilkei-Gorzo, A., Debeir, T., et al. (2006). Locus ceruleus degeneration promotes Alzheimer pathogenesis in amyloid precursor protein 23 transgenic mice. J. Neurosci. 26, 1343–1354. doi:10.1523/JNEUROSCI.4236-05.2006
Hinojosa, A. E., Garcia-Bueno, B., Leza, J. C., and Madrigal, J. L. M. (2011). Regulation of CCL2/MCP-1 production in astrocytes by desipramine and atomoxetine: Involvement of α2 adrenergic receptors. Brain Res. Bull. 86, 326–333. doi:10.1016/j.brainresbull.2011.09.014
Holmes, C., Cunningham, C., Zotova, E., Woolford, J., Dean, C., Kerr, S., et al. (2009). Systemic inflammation and disease progression in Alzheimer disease. Neurology 73, 768–774. doi:10.1212/WNL.0b013e3181b6bb95
Itzhaki, R. F. (2014). Herpes simplex virus type 1 and Alzheimer's disease: Increasing evidence for a major role of the virus. Front. Aging Neurosci. 6, 202. doi:10.3389/fnagi.2014.00202
Jacobs, H. I. L., Riphagen, J. M., Ramakers, I. H. G. B., and Verhey, F. R. J. (2021). Alzheimer's disease pathology: Pathways between central norepinephrine activity, memory, and neuropsychiatric symptoms. Mol. Psychiatry 26, 897–906. doi:10.1038/s41380-019-0437-x
Johnson, E. C. B., Carter, E. K., Dammer, E. B., Duong, D. M., Gerasimov, E. S., Liu, Y., et al. (2022). Large-scale deep multi-layer analysis of Alzheimer's disease brain reveals strong proteomic disease-related changes not observed at the RNA level. Nat. Neurosci. 25, 213–225. doi:10.1038/s41593-021-00999-y
Kalinin, S., Gavrilyuk, V., Polak, P. E., Vasser, R., Zhao, J., Heneka, M. T., et al. (2007). Noradrenaline deficiency in brain increases beta-amyloid plaque burden in an animal model of Alzheimer's disease. Neurobiol. Aging 28, 1206–1214. doi:10.1016/j.neurobiolaging.2006.06.003
Kalinin, S., Polak, P. E., Madrigal, J. L. M., Gavrilyuk, V., Sharp, A., Chauhan, N., et al. (2006). Beta-amyloid-dependent expression of NOS2 in neurons: Prevention by an alpha2-adrenergic antagonist. Antioxid. Redox Signal. 8, 873–883. doi:10.1089/ars.2006.8.873
Kang, S. S., Liu, X., Ahn, E. H., Xiang, J., Manfredsson, F. P., Yang, X., et al. (2020). Norepinephrine metabolite DOPEGAL activates AEP and pathological Tau aggregation in locus coeruleus. J. Clin. Invest. 130, 422–437. doi:10.1172/JCI130513
Kang, S. S., Meng, L., Zhang, X., Wu, Z., Mancieri, A., Xie, B., et al. (2022). Tau modification by the norepinephrine metabolite DOPEGAL stimulates its pathology and propagation. Nat. Struct. Mol. Biol. 29, 292–305. doi:10.1038/s41594-022-00745-3
Kelly, S. C., He, B., Perez, S. E., Ginsberg, S. D., Mufson, E. J., and Counts, S. E. (2017). Locus coeruleus cellular and molecular pathology during the progression of Alzheimer's disease. Acta Neuropathol. Commun. 5, 8. doi:10.1186/s40478-017-0411-2
Kotredes, K. P., Oblak, A., Pandey, R. S., Lin, P. B. C., Garceau, D., Williams, H., et al. (2021). Uncovering disease mechanisms in a novel mouse model expressing humanized APOEε4 and Trem2*R47H. Front. Aging Neurosci. 13, 735524. doi:10.3389/fnagi.2021.735524
Lammel, S., Lim, B. K., Ran, C., Huang, K. W., Betley, M. J., Tye, K. M., et al. (2012). Input-specific control of reward and aversion in the ventral tegmental area. Nature 491, 212–217. doi:10.1038/nature11527
Li, C., Liu, J., Lin, J., and Shang, H. (2022). COVID-19 and risk of neurodegenerative disorders: A mendelian randomization study. Transl. Psychiatry 12, 283. doi:10.1038/s41398-022-02052-3
Li, S., Jin, M., Zhang, D., Yang, T., Koeglsperger, T., Fu, H., et al. (2013). Environmental novelty activates β2-adrenergic signaling to prevent the impairment of hippocampal LTP by Aβ oligomers. Neuron 77, 929–941. doi:10.1016/j.neuron.2012.12.040
Liu, K. Y., Acosta-Cabronero, J., Cardenas-Blanco, A., Loane, C., Berry, A. J., Betts, M. J., et al. (2019). In vivo visualization of age-related differences in the locus coeruleus. Neurobiol. Aging 74, 101–111. doi:10.1016/j.neurobiolaging.2018.10.014
Liu, K. Y., Kievit, R. A., Tsvetanov, K. A., Betts, M. J., Duzel, E., Rowe, J. B., et al. (2020). Noradrenergic-dependent functions are associated with age-related locus coeruleus signal intensity differences. Nat. Commun. 11, 1712. doi:10.1038/s41467-020-15410-w
Liu, Y., Liu, F., Grundke-Iqbal, I., Iqbal, K., and Gong, C. X. (2011). Deficient brain insulin signalling pathway in Alzheimer's disease and diabetes. J. Pathol. 225, 54–62. doi:10.1002/path.2912
Lowry, C. A., Evans, A. K., Gasser, P. J., Hale, M. W., Staub, D. R., Shekhar, A., et al. (2008). “Topographical organization and chemoarchitecture of the dorsal raphe nucleus and the median raphe nucleus,” in Serotonin and sleep: Molecular, functional and clinical aspects. Editor J. M. Monti. Editors (Basel: Birkhauser), 25–68.
Lowry, C. A., Hollis, J. H., de Vries, A., Pan, B., Brunet, L. R., Hunt, J. R. F., et al. (2007). Identification of an immune-responsive mesolimbocortical serotonergic system: Potential role in regulation of emotional behavior. Neuroscience 146, 756–772. doi:10.1016/j.neuroscience.2007.01.067
Madrigal, J. L., Caso, J. R., Garcia-Bueno, B., Gutierrez, I. L., and Leza, J. C. (2017). Noradrenaline induces CX3CL1 production and release by neurons. Neuropharmacology 114, 146–155. doi:10.1016/j.neuropharm.2016.12.001
Madrigal, J. L., Garcia-Bueno, B., Caso, J. R., Perez-Nievas, B. G., and Leza, J. C. (2006). Stress-induced oxidative changes in brain. CNS Neurol. Disord. Drug Targets 5, 561–568. doi:10.2174/187152706778559327
Madrigal, J. L., Garcia-Bueno, B., Hinojosa, A. E., Polak, P., Feinstein, D. L., and Leza, J. C. (2010). Regulation of MCP-1 production in brain by stress and noradrenaline-modulating drugs. J. Neurochem. 113, 543–551. doi:10.1111/j.1471-4159.2010.06623.x
Madrigal, J. L., Kalinin, S., Richardson, J. C., and Feinstein, D. L. (2007). Neuroprotective actions of noradrenaline: Effects on glutathione synthesis and activation of peroxisome proliferator activated receptor delta. J. Neurochem. 103, 2092–2101. doi:10.1111/j.1471-4159.2007.04888.x
Marien, M. R., Colpaert, F. C., and Rosenquist, A. C. (2004). Noradrenergic mechanisms in neurodegenerative diseases: A theory. Brain Res. Brain Res. Rev. 45, 38–78. doi:10.1016/j.brainresrev.2004.02.002
Mason, S. T., and Fibiger, H. C. (1979). Regional topography within noradrenergic locus coeruleus as revealed by retrograde transport of horseradish peroxidase. J. Comp. Neurol. 187, 703–724. doi:10.1002/cne.901870405
Matchett, B. J., Grinberg, L. T., Theofilas, P., and Murray, M. E. (2021). The mechanistic link between selective vulnerability of the locus coeruleus and neurodegeneration in Alzheimer's disease. Acta Neuropathol. 141, 631–650. doi:10.1007/s00401-020-02248-1
Mather, M., and Harley, C. W. (2016). The locus coeruleus: Essential for maintaining cognitive function and the aging brain. Trends Cogn. Sci. 20, 214–226. doi:10.1016/j.tics.2016.01.001
Matschke, J., Lutgehetmann, M., Hagel, C., Sperhake, J. P., Schroder, A. S., Edler, C., et al. (2020). Neuropathology of patients with COVID-19 in Germany: A post-mortem case series. Lancet. Neurol. 19, 919–929. doi:10.1016/S1474-4422(20)30308-2
Mattson, M. P., Duan, W., Wan, R., and Guo, Z. (2004). Prophylactic activation of neuroprotective stress response pathways by dietary and behavioral manipulations. NeuroRx 1, 111–116. doi:10.1602/neurorx.1.1.111
Mattson, M. P. (2004). Pathways towards and away from Alzheimer's disease. Nature 430, 631–639. doi:10.1038/nature02621
Mittal, S., Bjornevik, K., Im, D. S., Flierl, A., Dong, X., Locascio, J. J., et al. (2017). β2-Adrenoreceptor is a regulator of the α-synuclein gene driving risk of Parkinson's disease. Science 357, 891–898. doi:10.1126/science.aaf3934
Niazkar, H. R., Zibaee, B., Nasimi, A., and Bahri, N. (2020). The neurological manifestations of COVID-19: A review article. Neurol. Sci. 41, 1667–1671. doi:10.1007/s10072-020-04486-3
Oblak, A. L., Kotredes, K. P., Pandey, R. S., Reagan, A. M., Ingraham, C., Perkins, B., et al. (2022). Plcg2M28L interacts with high fat/high sugar diet to accelerate Alzheimer's disease-relevant phenotypes in mice. Front. Aging Neurosci. 14, 886575. doi:10.3389/fnagi.2022.886575
Oblak, A. L., Lin, P. B., Kotredes, K. P., Pandey, R. S., Garceau, D., Williams, H. M., et al. (2021). Comprehensive evaluation of the 5XFAD mouse model for preclinical testing applications: A MODEL-AD study. Front. Aging Neurosci. 13, 713726. doi:10.3389/fnagi.2021.713726
Ono, K., Hamaguchi, T., Naiki, H., and Yamada, M. (2006). Anti-amyloidogenic effects of antioxidants: Implications for the prevention and therapeutics of Alzheimer's disease. Biochim. Biophys. Acta 1762, 575–586. doi:10.1016/j.bbadis.2006.03.002
Pamphlett, R. (2014). Uptake of environmental toxicants by the locus ceruleus: A potential trigger for neurodegenerative, demyelinating and psychiatric disorders. Med. Hypotheses 82, 97–104. doi:10.1016/j.mehy.2013.11.016
Paran, E., Anson, O., and Lowenthal, D. T. (2010). Cognitive function and antihypertensive treatment in the elderly: A 6-year follow-up study. Am. J. Ther. 17, 358–364. doi:10.1097/MJT.0b013e3181bf325c
Perry, V. H. (2010). Contribution of systemic inflammation to chronic neurodegeneration. Acta Neuropathol. 120, 277–286. doi:10.1007/s00401-010-0722-x
Perry, V. H., and Holmes, C. (2014). Microglial priming in neurodegenerative disease. Nat. Rev. Neurol. 10, 217–224. doi:10.1038/nrneurol.2014.38
Poe, G. R., Foote, S., Eschenko, O., Johansen, J. P., Bouret, S., Aston-Jones, G., et al. (2020). Locus coeruleus: A new look at the blue spot. Nat. Rev. Neurosci. 21, 644–659. doi:10.1038/s41583-020-0360-9
Querfurth, H., and Lee, H. K. (2021). Mammalian/mechanistic target of rapamycin (mTOR) complexes in neurodegeneration. Mol. Neurodegener. 16, 44. doi:10.1186/s13024-021-00428-5
Rohampour, K., Azizi, H., Fathollahi, Y., and Semnanian, S. (2017). Peripheral nerve injury potentiates excitatory synaptic transmission in locus coeruleus neurons. Brain Res. Bull. 130, 112–117. doi:10.1016/j.brainresbull.2017.01.012
Roozendaal, B., and McGaugh, J. L. (2011). Memory modulation. Behav. Neurosci. 125, 797–824. doi:10.1037/a0026187
Rorabaugh, J. M., Chalermpalanupap, T., Botz-Zapp, C. A., Fu, V. M., Lembeck, N. A., Cohen, R. M., et al. (2017). Chemogenetic locus coeruleus activation restores reversal learning in a rat model of Alzheimer's disease. Brain 140, 3023–3038. doi:10.1093/brain/awx232
Ross, J. A., Gliebus, G., and Van Bockstaele, E. J. (2018). Stress induced neural reorganization: A conceptual framework linking depression and Alzheimer's disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 85, 136–151. doi:10.1016/j.pnpbp.2017.08.004
Ross, J. A., McGonigle, P., and Van Bockstaele, E. J. (2015). Locus Coeruleus, norepinephrine and Aβ peptides in Alzheimer's disease. Neurobiol. Stress 2, 73–84. doi:10.1016/j.ynstr.2015.09.002
Sanchez-Padilla, J., Guzman, J. N., Ilijic, E., Kondapalli, J., Galtieri, D. J., Yang, B., et al. (2014). Mitochondrial oxidant stress in locus coeruleus is regulated by activity and nitric oxide synthase. Nat. Neurosci. 17, 832–840. doi:10.1038/nn.3717
Santana, S., Recuero, M., Bullido, M. J., Valdivieso, F., and Aldudo, J. (2012). Herpes simplex virus type I induces the accumulation of intracellular beta-amyloid in autophagic compartments and the inhibition of the non-amyloidogenic pathway in human neuroblastoma cells. Neurobiol. Aging 33, e19–e33. doi:10.1016/j.neurobiolaging.2010.12.010
Sara, S. J. (2009). The locus coeruleus and noradrenergic modulation of cognition. Nat. Rev. Neurosci. 10, 211–223. doi:10.1038/nrn2573
Sivonova, M., Zitnanova, I., Hlincikova, L., Skodacek, I., Trebaticka, J., and Durackova, Z. (2004). Oxidative stress in University students during examinations. Stress 7, 183–188. doi:10.1080/10253890400012685
Song, N., Fang, Y., Sun, X., Jiang, Q., Song, C., Chen, M., et al. (2018a). Salmeterol, agonist of β2-aderenergic receptor, prevents systemic inflammation via inhibiting NLRP3 inflammasome. Biochem. Pharmacol. 150, 245–255. doi:10.1016/j.bcp.2018.02.009
Song, S., Jiang, L., Oyarzabal, E. A., Wilson, B., Li, Z., Shih, Y. Y. I., et al. (2018b). Loss of brain norepinephrine elicits neuroinflammation-mediated oxidative injury and selective caudo-rostral neurodegeneration. Mol. Neurobiol. 56, 2653–2669. doi:10.1007/s12035-018-1235-1
Totah, N. K., Neves, R. M., Panzeri, S., Logothetis, N. K., and Eschenko, O. (2018). The locus coeruleus is a complex and differentiated neuromodulatory system. Neuron 99, 1055–1068. doi:10.1016/j.neuron.2018.07.037
Troadec, J. D., MarienM., , DariosF.,, , HartmAnn, A., RubergM., , ColpaertF., , et al. (2001). Noradrenaline provides long-term protection to dopaminergic neurons by reducing oxidative stress. J. Neurochem. 79, 200–210. doi:10.1046/j.1471-4159.2001.00556.x
Tzeng, N. S., Chung, C. H., Lin, F. H., Chiang, C. P., Yeh, C. B., Huang, S. Y., et al. (2018). Anti-herpetic medications and reduced risk of dementia in patients with herpes simplex virus infections-a nationwide, population-based cohort study in taiwan. Neurotherapeutics 15, 417–429. doi:10.1007/s13311-018-0611-x
Valentino, R. J., and Van Bockstaele, E. (2008). Convergent regulation of locus coeruleus activity as an adaptive response to stress. Eur. J. Pharmacol. 583, 194–203. doi:10.1016/j.ejphar.2007.11.062
Walker, K. A. (2018). Inflammation and neurodegeneration: Chronicity matters. Aging (Albany NY) 11, 3–4. doi:10.18632/aging.101704
Wang, Q., Oyarzabal, E. A., Song, S., Wilson, B., Santos, J. H., and Hong, J. S. (2020). Locus coeruleus neurons are most sensitive to chronic neuroinflammation-induced neurodegeneration. Brain Behav. Immun. 87, 359–368. doi:10.1016/j.bbi.2020.01.003
Wasay, M., Channa, R., Jumani, M., Shabbir, G., Azeemuddin, M., and Zafar, A. (2008). Encephalitis and myelitis associated with dengue viral infection clinical and neuroimaging features. Clin. Neurol. Neurosurg. 110, 635–640. doi:10.1016/j.clineuro.2008.03.011
Watabe-Uchida, M., and Uchida, N. (2018). Multiple dopamine systems: Weal and woe of dopamine. Cold Spring Harb. Symp. Quant. Biol. 83, 83–95. doi:10.1101/sqb.2018.83.037648
Weber, M. D., Frank, M. G., Tracey, K. J., Watkins, L. R., and Maier, S. F. (2015). Stress induces the danger-associated molecular pattern HMGB-1 in the hippocampus of male sprague dawley rats: A priming stimulus of microglia and the NLRP3 inflammasome. J. Neurosci. 35, 316–324. doi:10.1523/JNEUROSCI.3561-14.2015
Weinshenker, D. (2008). Functional consequences of locus coeruleus degeneration in Alzheimer's disease. Curr. Alzheimer Res. 5, 342–345. doi:10.2174/156720508784533286
Weinshenker, D. (2018). Long road to ruin: Noradrenergic dysfunction in neurodegenerative disease. Trends Neurosci. 41, 211–223. doi:10.1016/j.tins.2018.01.010
Wood, S. K., Wood, C. S., Lombard, C. M., Lee, C. S., Zhang, X. Y., Finnell, J. E., et al. (2015). Inflammatory factors mediate vulnerability to a social stress-induced depressive-like phenotype in passive coping rats. Biol. Psychiatry 78, 38–48. doi:10.1016/j.biopsych.2014.10.026
Wouk, J., Rechenchoski, D. Z., Rodrigues, B. C. D., Ribelato, E. V., and Faccin-Galhardi, L. C. (2021). Viral infections and their relationship to neurological disorders. Arch. Virol. 166, 733–753. doi:10.1007/s00705-021-04959-6
Xie, J., Van Hoecke, L., and Vandenbroucke, R. E. (2021). The impact of systemic inflammation on Alzheimer's disease pathology. Front. Immunol. 12, 796867. doi:10.3389/fimmu.2021.796867
Xu, H., Rajsombath, M. M., Weikop, P., and Selkoe, D. J. (2018). Enriched environment enhances β-adrenergic signaling to prevent microglia inflammation by amyloid-β. EMBO Mol. Med. 10, e8931. doi:10.15252/emmm.201808931
Zamore, Z., and Veasey, S. C. (2022). Neural consequences of chronic sleep disruption. Trends Neurosci. 45, 678–691. doi:10.1016/j.tins.2022.05.007
Zapater, P., Gomez-Hurtado, I., Peiro, G., Gonzalez-Navajas, J. M., Garcia, I., Gimenez, P., et al. (2012). Beta-adrenergic receptor 1 selective antagonism inhibits norepinephrine-mediated TNF-alpha downregulation in experimental liver cirrhosis. PLoS One 7, e43371. doi:10.1371/journal.pone.0043371
Zerbi, V., Floriou-Servou, A., Markicevic, M., Vermeiren, Y., Sturman, O., Privitera, M., et al. (2019). Rapid reconfiguration of the functional connectome after chemogenetic locus coeruleus activation. Neuron 103, 702–718. doi:10.1016/j.neuron.2019.05.034
Zhang, B., Gaiteri, C., Bodea, L. G., Wang, Z., McElwee, J., Podtelezhnikov, A. A., et al. (2013). Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer's disease. Cell 153, 707–720. doi:10.1016/j.cell.2013.03.030
Zhang, J., Zhu, Y., Zhan, G., Fenik, P., Panossian, L., Wang, M. M., et al. (2014a). Extended wakefulness: Compromised metabolics in and degeneration of locus ceruleus neurons. J. Neurosci. 34, 4418–4431. doi:10.1523/JNEUROSCI.5025-12.2014
Zhang, Y., Chen, K., Sloan, S. A., Bennett, M. L., Scholze, A. R., O'Keeffe, S., et al. (2014b). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 34, 11929–11947. doi:10.1523/JNEUROSCI.1860-14.2014
Zhang, F., Gannon, M., Chen, Y., Yan, S., Zhang, S., Feng, W., et al. (2020). β-amyloid redirects norepinephrine signaling to activate the pathogenic GSK3β/tau cascade. Sci. Transl. Med. 12, eaay6931. doi:10.1126/scitranslmed.aay6931
Zhu, Y., Fenik, P., Zhan, G., Somach, R., and Veasey, S. (2016). Intermittent short sleep results in lasting sleep wake disturbances and degeneration of locus coeruleus and orexinergic neurons. Sleep 39, 1601–1611. doi:10.5665/sleep.6030
Keywords: locus coeruleus, noradrenaline, neuroinflammation, oxidative stress, neurodegeneration, beta-adrenergic, Alzheimer’s disease
Citation: Evans AK, Defensor E and Shamloo M (2022) Selective Vulnerability of the Locus Coeruleus Noradrenergic System and its Role in Modulation of Neuroinflammation, Cognition, and Neurodegeneration. Front. Pharmacol. 13:1030609. doi: 10.3389/fphar.2022.1030609
Received: 29 August 2022; Accepted: 14 November 2022;
Published: 30 November 2022.
Edited by:
Marta P. Pereira, Spanish National Research Council (CSIC), SpainReviewed by:
Jason Langley, University of California, Riverside, United StatesCopyright © 2022 Evans, Defensor and Shamloo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mehrdad Shamloo, c2hhbWxvb0BzdGFuZm9yZC5lZHU=; Andrew K. Evans, YWtldmFuc0BzdGFuZm9yZC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.