 Andrea Catte
Andrea Catte Venkata K. Ramaswamy
Venkata K. Ramaswamy Attilio Vittorio Vargiu
Attilio Vittorio Vargiu Giuliano Malloci
Giuliano Malloci Andrea Bosin
Andrea Bosin Paolo Ruggerone
Paolo Ruggerone
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol., 11 November 2022
Sec. Pharmacology of Infectious Diseases
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.1021916
This article is part of the Research TopicInsights in Pharmacology of Infectious Diseases: 2021View all 5 articles
The secondary transporters of the resistance-nodulation-cell division (RND) superfamily mediate multidrug resistance in Gram-negative bacteria like Pseudomonas aeruginosa. Among these RND transporters, MexB, MexF, and MexY, with partly overlapping specificities, have been implicated in pathogenicity. Only the structure of the former has been resolved experimentally, which together with the lack of data about the functional dynamics of the full set of transporters, limited a systematic investigation of the molecular determinants defining their peculiar and shared features. In a previous work (Ramaswamy et al., Front. Microbiol., 2018, 9, 1144), we compared at an atomistic level the two main putative recognition sites (named access and deep binding pockets) of MexB and MexY. In this work, we expand the comparison by performing extended molecular dynamics (MD) simulations of these transporters and the pathologically relevant transporter MexF. We employed a more realistic model of the inner phospholipid membrane of P. aeruginosa and more accurate force-fields. To elucidate structure/dynamics-activity relationships we performed physico-chemical analyses and mapped the binding propensities of several organic probes on all transporters. Our data revealed the presence, also in MexF, of a few multifunctional sites at locations equivalent to the access and deep binding pockets detected in MexB. Furthermore, we report for the first time about the multidrug binding abilities of two out of five gates of the channels deputed to peripheral (early) recognition of substrates. Overall, our findings help to define a common “recognition topology” characterizing Mex transporters, which can be exploited to optimize transport and inhibition propensities of antimicrobial compounds.
Pseudomonas aeruginosa is the leading cause of hospital-acquired infections worldwide due to the emergence and spread of multidrug-resistant strains susceptible to very few antimicrobial agents (Fischbach and Walsh, 2009; Poole, 2011). P. aeruginosa resistance to multiple antibiotics arises from the synergy between its low permeable outer membrane and the action of multidrug efflux systems, among which the resistance-nodulation-cell division (RND) superfamily of secondary transporters plays a major role (Hancock, 1998; Li et al., 2015).
RND transporters responsible for the export of multiple drugs in Gram-negative bacteria belong to the substrate-based subfamily known as the hydrophobe/amphiphile efflux (HAE) family (Nikaido, 2018), which contribute to both intrinsic and acquired resistance (Poole et al., 1993; Poole, 2001, Poole, 2005; Poole and Srikumar, 2001; Dreier and Ruggerone, 2015). They function as tripartite efflux systems constituted by an inner membrane protein (IMP) recognizing the substrates, a periplasmic membrane fusion protein (MFP, aka PAP), involved in the assembly and/or substrate transfer, and an outer membrane channel protein (OMP) shuttling the substrates to the extracellular environment of the bacterial cell (Alav et al., 2021; Klenotic et al., 2021). The extrusion of substrates is powered by the proton motive force, which provides the energy required for the transport of different antibiotics and toxic compounds.
The first member of the RND family in P. aeruginosa, namely the MexAB-OprM transporter, was identified about 30 years ago (Poole et al., 1993). Since the first structural study of AcrB [the homologue of MexB and the main RND transporter in Escherichia coli and other bacteria—(Kobylka et al., 2020)] published in 2002 (Murakami et al., 2002), many experimental structures of individual components as well as the assemblies of MexAB-OprM and AcrAB-TolC have been reported (Sennhauser et al., 2009; Nakashima et al., 2013; Eicher et al., 2014; Tsutsumi et al., 2019; Glavier et al., 2020) (see for instance (Alav et al., 2021) and (Klenotic et al., 2021) for a full list of references).
MexB shows a characteristic jellyfish-like morphology constituted by an asymmetric trimer with each protomer comprising three domains (Figure 1A) (Ruggerone et al., 2013): 1) A transmembrane domain (TMD) of 12 α-helices inserted into the inner membrane (IM), where the chemical energy of protonated aspartates is converted into motion; 2) a pore (porter) domain (PD) located in the periplasm, where the recruitment and transport of substrates occur; and 3) a periplasmic funnel domain (FD), which connects the RND transporter to the OMP via the assembly of MFPs (Symmons et al., 2015; Wang et al., 2017; Tsutsumi et al., 2019; Glavier et al., 2020; Chen et al., 2022). The transport of substrates occurs through a “functional rotation mechanism” in which protomers cycle in a concerted (not necessarily synchronous) fashion across three asymmetric states: Loose (L or access) in which a substrate binds to a peripheral site termed access (or proximal) pocket (APL); Tight (T or binding) in which the substrate binds to a deep (aka distal) pocket (DPT); and Open (O or extrusion) in which the substrate is released into the central funnel leading toward the extracellular space via different OMP proteins (Murakami et al., 2006; Seeger et al., 2006; Pos, 2009). The protein pockets APL and DPT (Figure 1B) were previously identified in AcrB as the binding sites responsible for the recognition and selectivity of different types of substrates based on their molecular weight or chemical type (Nakashima et al., 2011; Kobayashi et al., 2014; Schuster et al., 2016; Iyer et al., 2022). These binding pockets are separated by a flexible G-rich switch loop, which facilitates the transport of high-molecular-mass compounds from the APL (Nakashima et al., 2011; Eicher et al., 2012; Cha et al., 2014). A stretch of phenylalanine residues (namely F136, F178, F610, F615, F617, and F628), also known as the hydrophobic trap (HP-trap) and located in the DPT, were found to interact with substrates and moreover to stabilize inhibitors binding to the periplasmic region of the transporter (Vargiu and Nikaido, 2012; Sjuts et al., 2016).
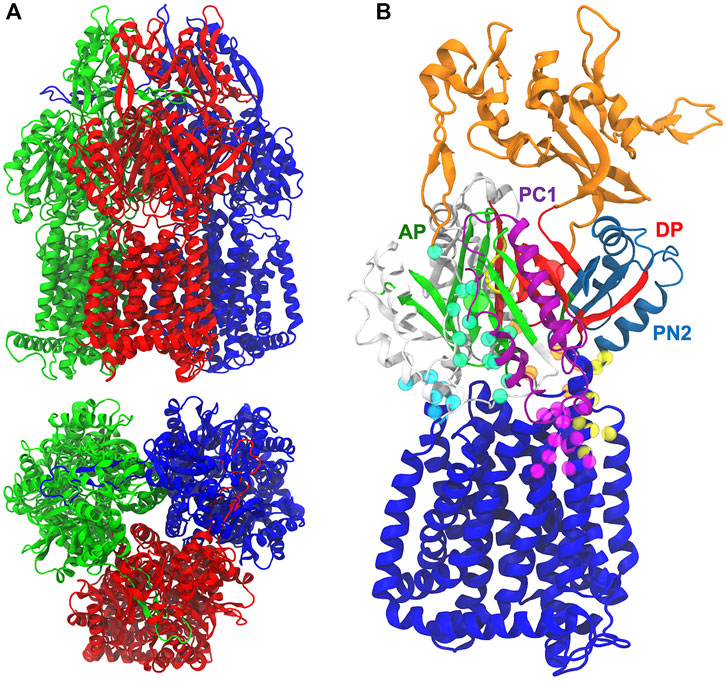
FIGURE 1. (A) Side and top views of MexB X-ray crystal structure (PDB ID 3W9I) Loose (L; red), Tight (T; blue), and Open (O; green) protomers are shown in different colors. (B) The pre-MD structure of the MexF Open protomer. The funnel and inner transmembrane domains are shown in orange and blue, respectively. AP and DP binding sites are in green and red, respectively, and their geometrical centers are represented by spheres. PC1 and PN2 subdomains are in purple and skyblue, respectively. The G-loop is in yellow. All other periplasmic residues are shown in white. CH1, CH2, CH3, CH4, and CH5 domains are highlighted with cyan, seagreen, orange, yellow, and magenta, respectively, spheres having as centers the position of alpha carbons.
Importantly, the DPT binding site was also observed experimentally in MexB (Nakashima et al., 2013), and both the APL and DPT were involved in the binding of substrates to the AdeB transporter of Acinetobacter baumannii (Morgan et al., 2021; Ornik-Cha et al., 2021). Thus, both the APL and the DPT seem to be shared binding spots across HAE-1 transporters that are deputed to the recognition of substrates and inhibitors (Nakashima et al., 2013; Sjuts et al., 2016).
Structural studies on AcrB also identified up to five channels leading from the surface of the protein to the APL and/or DPT: they are located at the membrane/periplasm interface (CH1), in the periplasm (CH2), at the interface between the periplasm and central cavity of the T protomer (CH3), within the interface between PN2 and PC1 domains (CH4) and at the membrane/periplasm interface adjacent to CH4 (hereafter CH5); Figure 1B). These channels appear to be crucial for the early recognition of substrates and for polyspecificity (Zwama et al., 2018; Alav et al., 2021): CH1 is likely mediating the transport of detergents and small drugs, such as β-lactams (Murakami et al., 2006; Sennhauser et al., 2007; Pos, 2009; Eicher et al., 2012); CH2 leading from the periplasm to the AP has been suggested to transport high molecular weight drugs such as macrolides (Seeger et al., 2006; Sennhauser et al., 2007, Sennhauser et al., 2009; Zwama et al., 2018); CH3 is possibly involved in sequestering planar aromatic cations, such as ethidium bromide or rhodamine 6G (Zwama et al., 2018); CH4 has been proposed to transport carboxylated drugs, such as fusidic acid and β-lactam antibiotics (Oswald et al., 2016). CH5, earlier named S1’, was also proposed by a recent in silico investigation as a putative binding site for fusidic acid and carboxylated β-lactams in AcrB (Tam et al., 2020), and later shown to catalyze allosteric binding of multiple molecules of fusidic acid to the transmembrane domain binding pocket (Tam et al., 2021). The substrate specificity of P. Aeruginosa MexB is very similar to that of AcrB from E. coli, as evidenced by its ability to transport macrolides such as erythromycin, β-lactams, chloramphenicol, ethidium bromide, fluoroquinolones, detergents, tetracyclines, etc. (Li et al., 1995). Previous experimental studies of MexB and its homologues (in particular, MexF and MexY transporters) were devoted to the identification of the amino acids (Middlemiss and Poole, 2004; Wehmeier et al., 2009) and domains responsible for substrate recognition (Tikhonova et al., 2002; Eda et al., 2003b), the substrates specificities of the different Mex efflux pumps (Masuda et al., 2000; Collu et al., 2012), and the structural basis of the binding of inhibitors to MexB and MexY (Nakashima et al., 2013). MexY, which is expected to be structurally similar to MexB and AcrB (Murata et al., 2002; Eda et al., 2003a), seems to transport aminoglycosides, such as gentamicin, tobramycin, amikacin, and isepamicin much more efficiently than MexB (Krahn et al., 2012; Lau et al., 2014; Dreier and Ruggerone, 2015). MexF, the third most relevant transporter in P. aeruginosa, is highly expressed in nfxC mutants (Llanes et al., 2011) to confer increased resistance to quinolones/fluoroquinolones, chloramphenicol, trimethoprim, and tetracycline (Vaccaro et al., 2008; Koch et al., 2013; Dreier and Ruggerone, 2015), as well as imipenem and some β-lactamase inhibitors (Kohler et al., 1997a; Li et al., 1998; Köhler et al., 1999). Systems overexpressing the MexEF-OprN efflux pump have been suggested to be among the first-step mutants induced by exposure to the aforementioned compounds, which may promote the acquisition of additional resistance mechanisms (Poole, 2011).
The detailed information provided by structural studies enabled, with the aid of computer simulations, to shed some light into the molecular mechanisms of substrate recognition and transport by, as well as inhibition of RND transporters (Schulz et al., 2010; Vargiu et al., 2011, Vargiu et al., 2014; Collu et al., 2012; Ramaswamy et al., 2016, Ramaswamy et al., 2017b, Ramaswamy et al., 2017a, Ramaswamy et al., 2018; López et al., 2017; Atzori et al., 2019; Tam et al., 2020). However, while all Gram-negative bacteria are endowed with several such pumps, only the structure of one protein per bacterium has been resolved so far [AcrB in E. coli and Salmonella enterica (Johnson et al., 2020), MexB in P. aeruginosa (Nakashima et al., 2013; Tsutsumi et al., 2019), AdeB in Acinetobacter baumannii (Morgan et al., 2021; Ornik-Cha et al., 2021)]. This limits the understanding of which general molecular determinants are responsible for the specificity of each transporter, as well as of the shared properties that enable recognition of the same class of substrates by different pumps. This is particularly true for the accurate mapping of substrate preferences (and overlap) between the different channels in the different transporters, as a characterization of these channels has been partially done only for AcrB.
We recently exploited MD simulations and fragment-based protein mapping to identify chemotype-specific and multi-functional sites (MFSs) (Imai et al., 2011) in the APL and DPT of AcrB/AcrD from E. coli and of MexB/MexY from P. aeruginosa (Ramaswamy et al., 2017b, Ramaswamy et al., 2018). Focusing on the latter pathogen, in this study we present the first in silico comparative study of its main three transporters MexB, MexF, and MexY. State-of-the-art MD simulations were performed in multiple copies for each transporter, followed by analyses of their structural, dynamical, physico-chemical features, and the mapping of their putative hot-spots and MFSs. Importantly, this analysis covered not only the APL and the DPT, but also highlighted the preferential binding of different fragments to all the channel gates of each transporter.
Since there is no experimental structure of MexF, we built up an all-atom model of its asymmetric trimer structure by homology modeling using Modeler 9.13 (Sali and Blundell, 1993). The amino acid sequence of full length MexF transporter from P. aeruginosa PAO1 was retrieved from the UniProtKB database [(The Uniprot Consortium, 2015); https://www.uniprot.org] (UniProt ID Q9I0Y8), and subsequently searched for the best available template structures bearing homologous relationship to the query sequence using the NCBI-BLAST tool (Madden, 2013) against the Protein Data Bank (PDB, www.rcsb.org). The X-ray crystal structure of MexB at 2.7 Å resolution [PDB ID 3W9I (Nakashima et al., 2013)] was chosen as a template for the homology modeling of MexF. The protein sequences were optimally aligned by ClustalOmega (Sievers et al., 2011; Sievers and Higgins, 2014) and the results were visually inspected to ensure the absence of gaps in important secondary structure regions. Multiple sequence alignments were analyzed using EMBOSS Stretcher to calculate sequence identity and similarity (Rice et al., 2000). Modeler 9.13 was used to generate a total of 100 asymmetric models of MexF using an optimization method combining slow MD with a very thorough variable target function method through 300 iterations. This whole cycle was repeated twice unless the objective function (MOLPDF) was greater than 10. The resulting models were ranked using discrete optimized protein energy (DOPE) (Shen and Sali, 2006) score values, and the top 5 models (with the lowest DOPE score) were selected for individual structure quality checks. Each model was further subjected to loop refinement using Modeler, and to overall structure relaxation by energy minimizations using AMBER18 (Case et al., 2018). The most reliable model was then selected based on various geometric and stereochemical quality factors evaluated for backbone angles, side chains flips, rotamers, steric clashes etc. using PROCHECK (Laskowski et al., 1993), ERRAT (Colovos and Yeates, 1993), ProSA (Wiederstein and Sippl, 2007), Verify3D (Eisenberg et al., 1997) programs available in MolProbity (Williams et al., 2018) and Structure Analysis and Verification Server. We performed comparative structural evaluation by superimposition of the modeled MexF structures over the experimentally determined X-ray crystal structure of MexB used as a template. Likewise, the template structure was also evaluated with the same programs to serve as reference for the results obtained for the MexF models (data not shown). Visual inspections were performed with VMD1.9.3 (Humphrey et al., 1996) and PyMOL (Schrödinger, 2015).
All-atom MD simulations of MexF, MexB, and MexY were carried out at 310 K and 1 atm using the MD software package AMBER18 (Case et al., 2018). The X-ray crystal structure of MexB (PDB ID 3W9I) (Nakashima et al., 2013) and the homology models of MexF and MexY will be hereafter referred to as pre-MD. The most reliable homology model of MexF obtained as described above was used as the starting structure. The details of the validation of the pre-MD model of MexF are reported in the Supplementary Table S1. We considered the highest resolution X-ray crystal structure released by Nakashima et al. (2013) (PDB ID 3W9I) and the validated homology model published by Ramaswamy et al., in 2018 as initial models for MexB (Figure 1A) and MexY, respectively. The protonation states of aspartate, glutamate and histidine sidechains were assigned by comparing the pKa values calculated by PROPKA3 (Olsson et al., 2011) with the physiological pH experienced by each amino acid and considering its exposure to the solvent. As previously reported by Eicher et al., in 2014, we considered that periplasmic (i.e., H318 and H749 of MexF) and cytoplasmic (i.e., H525 and H534 of MexB and MexF, respectively) side chains exchanged protons with aqueous solutions at pH values of 5.5 and 7.5, respectively, while transmembrane domain residues (i.e., D407, D410 and D406 of MexB, MexF, and MexY, respectively) experienced intermediate pH values between these lower and upper limits (Eicher et al., 2014). Moreover, the ionizable residues exhibiting buried sidechains were left unprotonated. The protonation states of specific amino acids in each protomer (Eicher et al., 2014) were adopted with E346 (E349 and E345) and D923 (N935 and D919) protonated in both Loose (L) and Tight (T) protomers while deprotonated in the Open (O) protomer of MexB (MexF and MexY). The residues D407 (D410, D406), D408 (D411, D407), D566 (Q575, E563) and H338 (E341, R337) were protonated only in the Open protomer of MexB (MexF, MexY). MexF N935, Q575, and E341 and MexY R337 residues were not protonated. The periplasmic residues H318 and H749 were protonated only in all protomers of MexF, as a result of a pKas analysis performed on MexB and its homologues using PROPKA3 version 3.4.0 (Olsson et al., 2011). The same protonation employed by Ramaswamy et al. (2018) for MexY was used. Each protein was successively embedded in 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylethanolamine (POPE): 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylglycerol (POPG) (POPE:POPG = 4:3) lipid bilayer patches, solvated with explicit OPC (Optimal Point Charges) water molecules (Tian et al., 2020). The ratios of POPE and POPG molecules in lipid membranes containing MexB, MexF, and MexY were 380:285, 356:267, and 388:292, respectively. The total number of solvation waters for lipid membranes containing MexB, MexF, and MexY was 96,936, 99,309, and 98,527, respectively. The residual charge of the systems was neutralized by appropriate numbers of randomly placed K+ and Cl− ions to reach a physiological ionic strength of 0.15 M KCl (Schulz et al., 2010, Schulz et al., 2011, Schulz et al., 2015; Vargiu et al., 2011). The total number of atoms of each Mex transporter embedded in a hydrated lipid bilayer was about 520,000.
The ions count was suitably adjusted to set a physiological ionic strength of 0.15 M KCl. The insertion of the protein into a POPE:POPG lipid membrane was performed using the Packmol-Memgen software (Schott-Verdugo and Gohlke, 2019). The POPE:POPG molar ratio of 4:3 was chosen to mimic the planktonic lipid composition of the inner membrane of P. aeruginosa (Benamara et al., 2014). The topology and the initial coordinate files were created using the LEaP module of AmberTools20 (Case et al., 2021). Periodic boundary conditions were used and the distance between the protein and the edge of the box was set to be at least 30 Å in each direction. Multi-step energy minimization with a combination of steepest descent and conjugate gradient methods was carried out using the pmemd program implemented in AMBER20 to relax internal constraints of the systems by gradually releasing positional restraints. Following this, the systems were heated from 0 to 310 K by a 1 ns heating (0–100 K) under constant volume (NVT) followed by 5 ns of constant pressure heating (NPT) (100–310 K) with the phosphorous heads of lipids restrained along the z-axis to allow membrane merging and to bring the atmospheric pressure of the system to 1 bar. Langevin thermostat (collision frequency of 1 ps−1) was used to maintain a constant temperature, and multiple short equilibration steps of 500 ps under anisotropic pressure scaling (Berendsen barostat) in NPT conditions were performed to equilibrate the box dimensions. A time step of 2 fs was used during all these runs, while post-equilibrium MD simulations were performed with a time step of 4 fs under constant volume conditions after hydrogen mass repartitioning (Hopkins et al., 2015). The particle-mesh Ewald (PME) algorithm was used to evaluate long-range electrostatic forces with a non-bonded cutoff of 9 Å. During the MD simulations, the length of all R-H bonds was constrained with SHAKE algorithm. Coordinates were saved every 100 ps With respect to our previous study (Ramaswamy et al., 2017b), we employed updated atomistic models and more physiologically relevant lipid bilayers with a chemical composition similar to that of the inner membrane of P. aeruginosa (Benamara et al., 2014). The ff19SB (Tian et al., 2020) version of the all-atom Amber force field was used to represent the protein systems while Lipid17 (Gould et al., 2018) [an improved version of Lipid14 (Dickson et al., 2014)] parameters were used for the POPE:POPG lipid bilayer. After equilibration, multi-copy μs-long MD simulations were performed for each system, namely five ∼1 μs-long production simulations for each transporter (for a total simulation time of ∼15 µs). Trajectory analysis was done using cpptraj module of AmberTools20 and VMD1.9.3, and graphs were plotted using the xmgrace tool.
After pre-processing MexB, MexF, and MexY structures to assign charges and atomic radii using the PDB2PQR 3.5.0 (Dolinsky et al., 2004), the electrostatic potential surface maps were calculated using APBS 1.5 (Baker et al., 2001). All electrostatic potential calculations were performed at 0.15 M physiological salt concentration, with a solvent probe of radius 1.4 Å, a solvent dielectric constant of 78.5, a biomolecular dielectric constant of 2.0, a temperature of 310.15 K, a minimum grid spacing of 0.5 Å and keeping the other Poisson–Boltzmann parameters at default.
The radial distribution function (rdf) indicates the probability of finding water molecules at a certain distance from a region or residue of interest and is commonly used to analyze the solution structure revealed from either experimental or computer simulations data. The rdf analysis of water oxygen atoms was performed using cpptraj module of AMBER20, in which the rdf is computed from the histogram of the number of solvent particles found as a function of the distance R from an (ensemble of) atom(s), normalized by the expected number of solvent particles at that distance in bulk. Bin spacing of 0.1 and a maximum bin value of 4.0 was used in this case to calculate the rdf of all water oxygen atoms to each atom of APL and DPT over the entire length of the simulation.
A cluster analysis of MD trajectories was performed using the average-linkage hierarchical agglomerative clustering method implemented in the cpptraj module of AMBER20 (Case et al., 2021). Such clustering helps to reduce the number of structures for analysis while retaining the large conformational space sampled during MD simulations. In this approach, we clustered in thirteen separate instances the trajectory based on root mean square deviation (RMSD) (cutoff set to 3 Å) of APL, DPT, CH1, CH2, CH3, CH4, and CH5 in both L and T protomers. The amino acids constituting each binding pocket and channel of investigated Mex transporters are reported in Supplementary Tables S2, S3, respectively. For each protein, the representative structures from each of the top 5 clusters generated in each of the thirteen different cases considered (APL, DPT, CH1, CH2, CH3, CH4, and CH5 in L and T) were used to perform the fragment-based mapping (see below). All non-protein molecules were stripped from the trajectory during post-processing to reduce additional memory usage and to speed up file processing.
Fragment-based methods exploit small organic ligands characterized by different physicochemical properties (Supplementary Figure S1) to identify putative hotspots for drug binding on a protein (Ciulli et al., 2006; Kozakov et al., 2015; Radoux et al., 2016; Wakefield et al., 2020; Hadfield et al., 2022; Smilova et al., 2022). These methods can be used to refine the choice of compounds to be screened in more accurate drug design investigations at both experimental and computational levels. The FTMap server (Kozakov et al., 2015) employed in this work implements the FTSite algorithm to discover druggable binding sites on proteins and to identify specific fragments that could serve as scaffolds for drug design attempts. FTMap identifies the important hot spots based on the consensus clusters of 16 standard probes, which include molecules varying in size, shape, and polarity (Supplementary Figure S1). Such a diverse library of probes captures a range of interaction types that include hydrophilic, namely via hydrogen bond donor (HBD) and acceptor (HBA) molecules, hydrophobic (aliphatic), and aromatic interactions. The regions where clusters of different probes of the same or different types overlap together are marked as CSs and MFSs, respectively, and are ranked based on the number of their clusters. Clusters near a top-ranked one are merged together and the protein residues within this region become the top-ranked putative ligand binding site. We identify as MFS a cluster of organic ligands displaying all types of interactions within 5 Å of any residue of the binding site of interest (Imai et al., 2011). The same cutoff of 5 Å has also been used to estimate the number of various types of organic probes interacting with different channels of each transporter.
In pharmaceutical science, the logarithm of the partition coefficient (P) between octanol and water phases (log Po/w) is one of the most used descriptors to adequately model the distribution between lipophilic cell membranes (octanol) and hydrophilic blood plasma (water) (Leo et al., 1971; Sangster, 1997). The three-dimensional distribution of lipophilicity on a molecular surface can be described using the molecular lipophilicity potential (MLP), which represents the lipophilic contributions of all polar and hydrophobic fragments of a molecule (Broto et al., 1984; Ghose and Crippen, 1986; Gaillard et al., 1994; Testa et al., 1996). To characterize the lipophilicity of APL and DPT binding pockets in MexB, MexF, and MexY, we calculated the Lipophilic Index (LI) of pre-MD and the ten most populated cluster structures using the MLP-Pocket sub-program implemented in the MLP Tools plugin (Oberhauser et al., 2014) of PyMOL version 2.4.1 (Schrödinger, 2015). The location of each binding site was defined by its Cartesian coordinates and center. The cutoff was 4.0 Å for each calculation.
The volume of APL and DPT binding pockets was monitored during MD simulations using the two-probe sphere method of rbcavity subprogram of the rDock suite (Ruiz-Carmona et al., 2014). This analysis provided detailed information on the pocket volume and plasticity of the site. The binding site volume was identified by a fast grid-based cavity detection algorithm (Morley and Afshar, 2004) within a sphere of radius 13 Å, centered over the pockets, using large and small probe radii of 6.0 and 1.5 Å, respectively.
Overall, the aminoacid sequences are well conserved across the strains of MexB, MexF, and MexY deposited in the UniProtKB database (accessed April 2021; Supplementary Figure S2). MexF displays the smallest sequence identity with both MexB and MexY (Supplementary Table S4). When analyzing in more detail the sequence conservation at key sites, we noticed that both the AP and DP of MexF and MexY are less conserved than the whole proteins, sharing respectively a percentage of identical (similar) residues of 37 (55) and 33 (54) % and 35 (48) and 31 (53) % with the corresponding site on MexB (Supplementary Figure S3). The AP of MexB and both the AP and the DP of MexY exhibit a more predominant distribution of negatively charged residues, while the balance is almost neutral in MexF (Supplementary Table S5). The AP of MexY also contains the lowest number of hydrophobic residues, while the AP and DP of MexF contain more polar residues than MexB and MexY. The HP-trap, lined by 6 phenylalanines in MexB, conserves its hydrophobic character in both MexF and MexY, although only 3 and 2 phenylalanines line the site in these proteins (Supplementary Figure S3). A similar comparison for the CHs sites reveals that (Supplementary Figure S3): CH1 is well conserved between MexB and MexF, while MexY displays several differences with respect to MexF and, moreover, MexB; CH2 turns out to be the less conserved entrance gate, with each transporter featuring a very dissimilar amino acid composition; at CH3, MexF displays a large similarity with MexY, and to a minor extent with MexB; CH4 and CH5 are overall the most conserved sites.
All-atom MD simulations of MexB, MexF, and MexY embedded in a POPE: POPG lipid bilayer were performed using as starting conformation the corresponding pre-MD structure, respectively the X-ray crystal structure of MexB (PDB ID 3W9I) (Nakashima et al., 2013) and the homology models of MexF and MexY. The stability of both MexF and MexY models and their suitability for subsequent quantitative analyses were further validated by performing five independent μs-long MD simulations (which were performed also for MexB). The analysis of Cα-RMSDs of the whole protein and individual protomers of each RND transporter, using as a reference their initial structure (Supplementary Figure S4), highlighted that a plateau was reached within at most ∼0.4 μs in all MD simulations. The results of several analyses performed on these equilibrated trajectories are reported below.
MexF features an APL with the largest percentage of polar residues but the number of positively charged residues is very low compared to MexB and MexY (Supplementary Table S5), leading to a wider distribution of negatively charged patches of electrostatic potential in the pre-MD structure of the former transporter (Figure 2A). This difference in the distribution of charges at the APL persists, although less pronounced when comparing the most populated structure extracted from the cluster analysis of the MD simulations. In agreement with previous findings (Ramaswamy et al., 2018), we also observed a difference in the electrostatic potential surface at the APL between MexB and MexY, the latter exposing more positively charged patches (Figure 2A). The DPT in pre-MD structures of MexF and MexB showed electrostatic potential surfaces more positive than those observed for the APL, while MexY displayed an almost equal distribution of positive and negative charges (Figure 2B). Similarly to the APL results, the projection of the electrostatic potential onto the surface of the DPT of most populated cluster structures resulted in a mainly positively charged environment in MexB and MexF as compared to MexY (Figure 2B), which is consistent with the relative percentages of positively vs. negatively charged residues in this site (Supplementary Table S5).
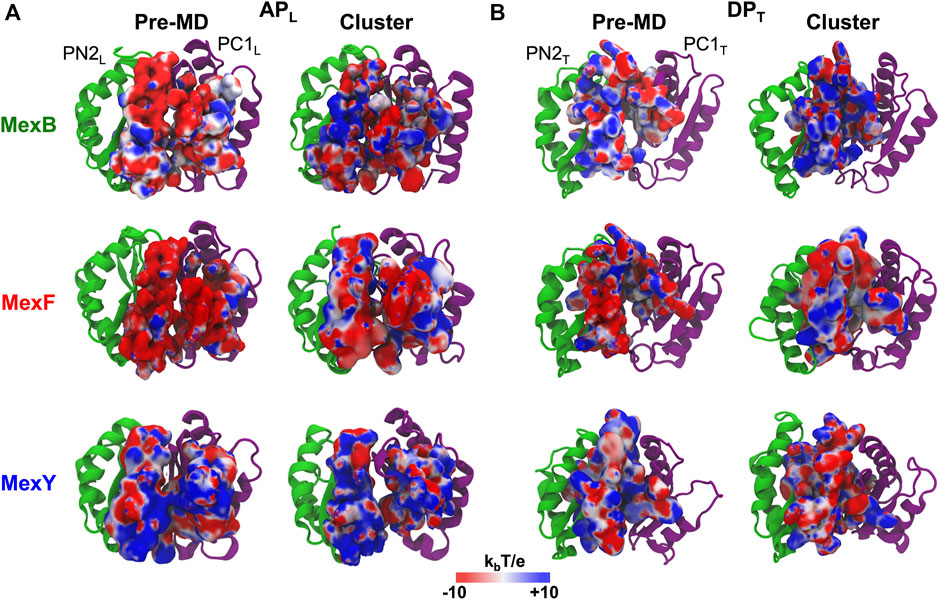
FIGURE 2. Electrostatic potential of (A) APL and (B) DPT of MexB, MexF, and MexY. The electrostatic potential is plotted on the molecular surface representation of each binding pocket in the Pre-MD (left) and the most populated cluster (right) structures of MexB and its isoforms as seen from a periplasm. The color code is red to blue from negative (−10 kbT/e) to positive (+10 kbT/e) potentials, where kb is the Boltzmann constant, T is the absolute temperature and e is the electron charge. PN2L, PC1L, PN2T, and PC1T domains of each RND transporter are in green and purple cartoons, respectively.
The hydration levels in binding sites of MexB and its homologs were evaluated by calculating the rdf of water oxygen atoms around APL, DPT, and HP-trapT residues (Supplementary Figure S5). As previously reported in (Ramaswamy et al., 2018), we observed a first solvation shell at around 2 Å for each binding site of MexB, MexF, and MexY. In particular, MexF has a slightly less hydrated APL than MexB and MexY. MexF, MexB, and MexY show comparable hydration profiles at the DPT. Interestingly, the HP-trapT has similar hydration levels in the first shell of all transporters, while it is more hydrated in the second shell of MexF and MexY as compared to MexB. These results were confirmed by hydration analyses performed on MD simulations of the three RND transporters (Supplementary Figure S5).
In this work, we employed the FTMap server to probe AP and DP hot spots in different protomers of MexB, MexF, and MexY (Kozakov et al., 2015; Ramaswamy et al., 2017b, Ramaswamy et al., 2018). Among the putative binding sites identified with this methodology, the so-called consensus sites (CS; that is, regions on the protein where clusters of more than one probe of the same type do overlap, see Materials and Methods) and moreover the MFSs (that is, sites including all four kinds of probe functionalities: H-bond acceptor, H-bond donor, aliphatic, and aromatic) sites where probes of different kind cluster together) are particularly relevant for polyspecific transporters such as MexB and homologs (Kozakov et al., 2015; Ramaswamy et al., 2017b, Ramaswamy et al., 2018).
In the following, we will focus on the comparison between results obtained for MexF and the other two transporters. Two and one MFSs were detected respectively in the DPT and in the APL of the pre-MD structure of MexF, increasing on average by 0.5 and 1 across the equilibrium MD trajectory (Table 1; Figures 3A,B). While these values are overall comparable with those obtained for MexB and MexY (Table 1; Supplementary Figures S6, S7, the number of MFS sites in the DPT is the largest for MexF, which is in line with our findings on the average number of organic probes of any kind that can bind to this site in the different transporters (Supplementary Table S6). Both this site and the AP feature, in all transporters, a few hotspots for each type of organic probe: HBD, HBA, ALI, and ARO (Figure 4; Supplementary Table S6); a finding that is consistent with their polyspecificity. MexY features the largest (lowest) number of MFSs in the APL (DPT) among all the transporters, a result that is also consistent with the average number of hotspots found in these pockets (Table S6).
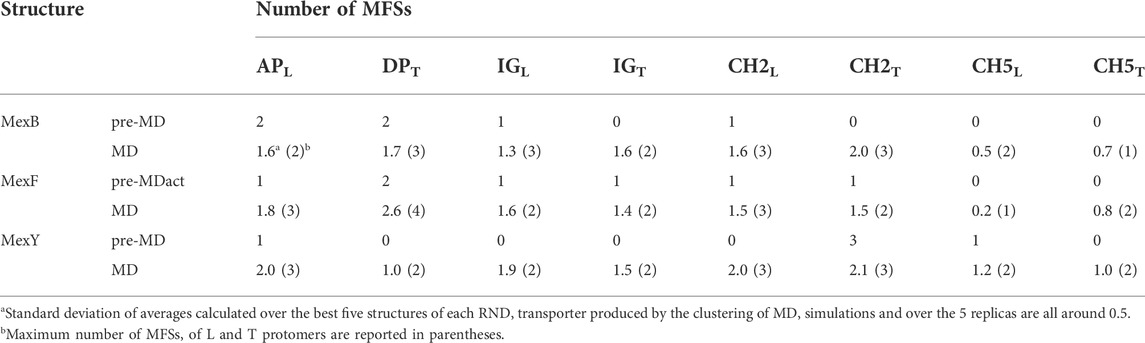
TABLE 1. MFSs identified in MexB, MexF, and MexY L and T protomers before (pre-MD) and along the MD simulation (average and maximum values are calculated over all replicas).
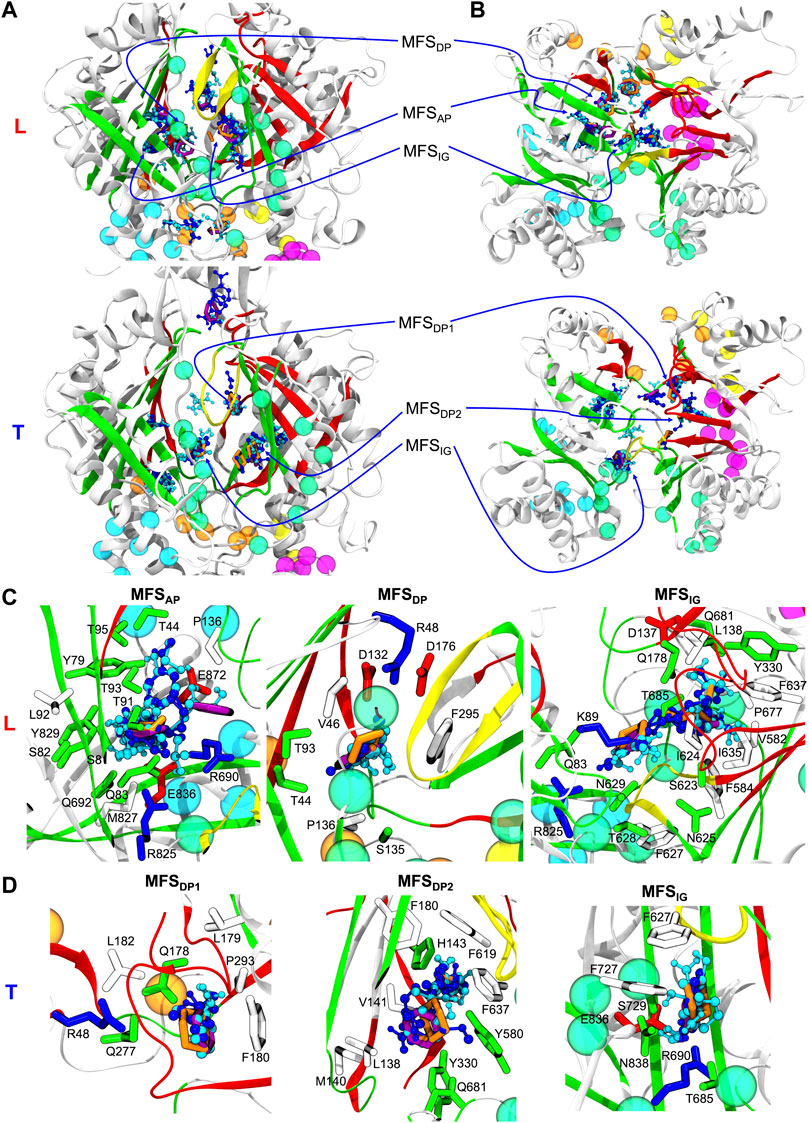
FIGURE 3. (A) Side and (B) top views of Loose (L) and Tight (T) protomers of the MexF pre-MD structure highlighting MFSs obtained with the FTMap server fragment-based mapping. Hydrogen bond donors, hydrogen bond acceptors, aliphatic, and aromatic organic probes are shown in celestial blue, blue, orange, and purple, respectively. The same color code of Figure 1 is applied to the protein and its channels. For the sake of clarity, only MFSs and CSs are shown. MFSIG (IG: Interface/G-loop) is in proximity of the G-loop at the interface between AP and DP binding pockets. MFSDP1 and MFSDP2 denote the two different MFSs of the T protomer’s DP. (C) and (D) Zoomed views of MFSs located in L and T protomers, respectively, show sidechains of MexF residues interacting with each MFS, defined as those amino acids having at least one atom within 3.5 Å of any atom of the MFS small organic probes. Polar, negatively charged, positively charged, and hydrophobic sidechains are colored in green, red, blue, and white, respectively. For clarity, only MFSs of interest are shown in each panel, other adjacent MFSs and CSs are omitted.
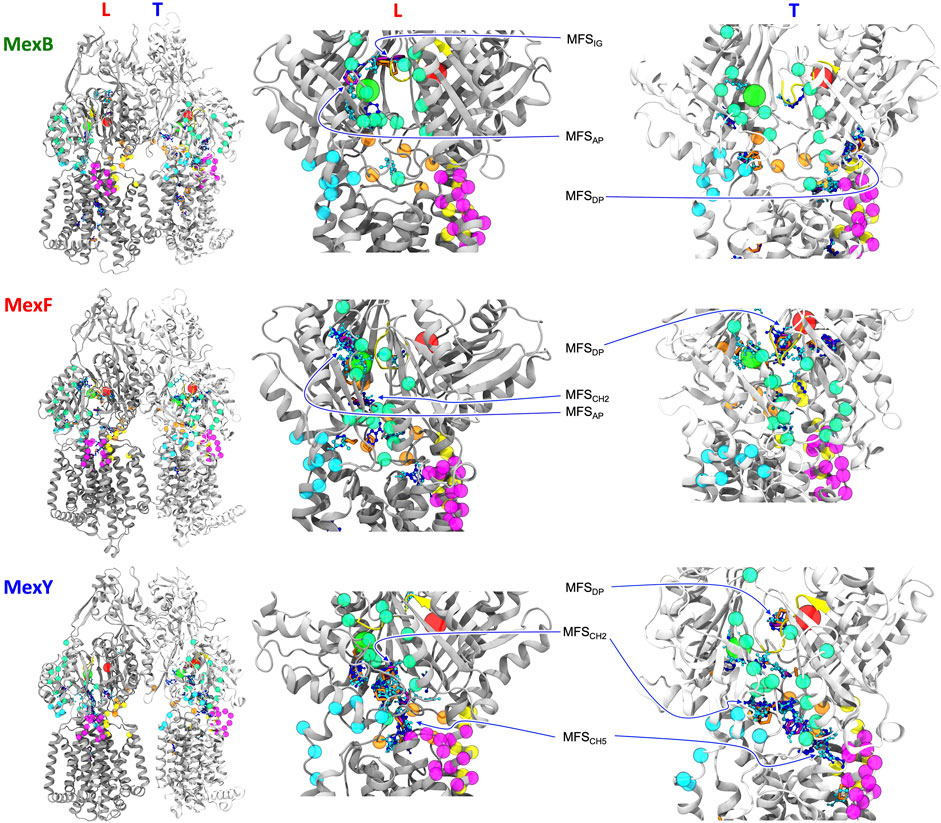
FIGURE 4. L and T protomers of MexB, MexF, and MexY best representative structures highlighting MFSs obtained with the FTMap server fragment-based mapping. Hydrogen bond donors, hydrogen bond acceptors, aliphatic and aromatic organic probes are shown with the same color code in Figure 3. For clarity, AP and DP binding sites are represented by green and red spheres, respectively, centered at the geometric center of each domain. The same color code of Figure 1 is applied to the protein binding pockets, switch loop and channels. All the other protein residues of L and T protomers are shown in gray and white, respectively. The definition of MFSAP, MFSDP and MFSIG is the same of Figure 3. MFSCH2 and MFSCH5 indicate MFSs in proximity of CH2 and CH5 channels, respectively. The sites not labeled as MFS are all CSs.
Our data confirm the trend reported previously for MexB and MexY and show a more marked difference in the number of MFSs at DPT, whose average values also increased in both proteins with respect to ref. (Ramaswamy et al., 2018). These differences could be ascribed to the different MD simulations protocol employed (new water model and protein force field, see Materials and Methods) and/or the more complex lipid bilayer environment (i.e., the protein is inserted into a POPE:POPG (4:3) lipid membrane, in which neutral and, especially, negatively charged lipids can interact with the protein and affect its conformation in a way different from in a pure POPE lipid bilayer (Bruzzese et al., 2018; Liko et al., 2018). Moreover, our observation of MFSs in the transmembrane domain of each transporter (Supplementary Figure S8) could be affected by the chemical composition of the surrounding lipid environment, confirming the presence of cytoplasmic binding sites in MexB (Ohene-Agyei et al., 2012) and, also, in its homologs MexF and MexY.
For each Mex transporter, we also investigated the composition of the amino acids lining the MFSs (defined as the residues having at least one atom within 3.5 Å from any probe). In MexF, the MFSs within the APL (Figure 3C; Supplementary Figure S9) displayed more interactions with polar and charged residues than the MFSs in DPT (Figure 3D; Supplementary Figure S9). Of note, the MFSAP was in contact with several highly conserved residues between MexF (S81, S82, T91, T93, and E836) and MexB (S79, S80, T89, T91, and E825; see Supplementary Figure S6). In contrast, only one residue was shared between MexF and MexY at this site (E836—Q820 in MexY—see Supplementary Figure S7). During the MD simulations, despite the specific residues near the MFSs within the AP varied with respect to the initial conformation, the location of these MFSs did not change significantly (Supplementary Figure S9).
The MFSDP in the T protomer of MexF is surrounded by several hydrophobic residues and, notably, by three aspartic acid residues (i.e., D132, D137, and D176; Supplementary Figure S9). This excess of negative charges, which is even larger than that observed in MexY and MexB, could be related to a more favorable interaction with positively charged drugs, such as aminoglycosides. Conserved residues D132, A134 in MexF and E129, A131 in MexY were interacting with MFSs in the DP of their corresponding proteins (Supplementary Figure S9).
We also monitored the occurrence of MFSs near all channel gates (Figure 1B). Among the five channels, we found that CH2 and CH5 showed a more significant population of MFSs (considering either the values from pre-MD or the averages from MD simulations in Table 1), pointing to the possibility of establishing relatively strong interactions with different drugs even at peripheral sites on these transporters. Consistently, these two entrance gates also were able to accommodate, on average, the largest number of different types of small organic probes (Supplementary Table S6; Supplementary Figure S10). Notably, in all transporters, the average number of MFSs at CH2 was nearly comparable with that found at APL. CH5 displayed the highest average number of MFSs in MexY; no MFSs were found in the pre-MD structures of the other two transporters. As for CH2, when estimating the average number of organic probes at CH5T, we noticed the same trend as for the MFS (Supplementary Table S6). This is interesting as both MexY and MexB have been proposed to recognize β-lactam antibiotics (Alav et al., 2021), and CH5T was proposed to be a putative entry gate for carboxylated drugs from this family (Tam et al., 2020). Conserved residues L683 and T685 in MexF (L669 and S671 in MexY) were found in proximity to MFSCH2. Regarding CH5, we observed a non-vanishing number of MFSs only in the L and T protomers of MexY (Table 1 and Figure 4; Supplementary Figure S9). Residues E564, P665, L666, and G99 were found in the proximity of the MFSs in both protomers. CH3 and CH4 also feature some interaction with all probes, although to a reduced amount as compared to CH2 and CH5 (Supplementary Table S6). Finally, CH1 seems virtually unable to bind any probe.
The APL of MexF displayed the largest LI value in the pre-MD structures (Supplementary Table S7). Concerning MexB and MexY, despite different absolute values, the same trend reported in (Ramaswamy et al., 2018) is confirmed by our analyses. As in that work, the conformational changes underwent by the transporters during MD simulations have a drastic effect on the values of the LI. While the LI trend for MexB and MexY remains the same despite a large increase in both values, a significant reduction occurred in the APL of MexF (Supplementary Figure S11). This is likely to be associated with an increase and a decrease, along the MD simulations, of the APL volume respectively in MexB/Y and MexF (Supplementary Table S8 and Supplementary Figure S12), which together with the chemical composition of the site influence the evaluation of the LI (Ramaswamy et al., 2017b). As far as regards the DPT, in the pre-MD structures, the largest LI value was again found for MexF, followed by MexB and MexY, featuring a value almost half of that in the other transporters (Supplementary Table S7). For this pocket too, a remarkable smoothing of the differences among the transporters occurred during the MD simulation (although MexF still displayed the largest LI value). Indeed, both MexY and MexB exhibited a larger decrease in the DPT volume as compared to MexF (respectively ∼41% and ∼27% vs. ∼13%) as compared to their pre-MD structures (Supplementary Table S8; Supplementary Figure S12).
The overexpression of MexEF-OprN in P. aeruginosa (Köhler et al., 1997b, Köhler et al., 1999) is associated with increased resistance to fluoroquinolones, tetracyclines, and chloramphenicol in cystic fibrosis patients (Linares et al., 2005), and is found in various hospital strains together with MexAB-OprM and MexXY-OprM (Wolter et al., 2004; Morita et al., 2015). The study of these RND transporters at a molecular level could aid the development of drugs and inhibitors able to increase the susceptibility of this efflux pumps and, at the same time, to reduce their resistance to various classes of antibiotics.
Prompted by this consideration and with the goal of getting insights into the molecular determinants accounting for the different specificities of these three Mex transporters, we performed a systematic comparison of the physico-chemical, structural, dynamical, and thermodynamical features of the APL and DPT binding sites of MexB, MexY, and (for the first time) MexF. The number, strength, and spatial distribution of MFS at these sites determines the level of promiscuity of their interactions (Ciulli et al., 2006). As evidenced by Ramaswamy et al., in 2018, the MFSs in the DPT of MexB are placed exactly at the binding positions observed for several substrates like minocycline (Murakami et al., 2006; Nakashima et al., 2011; Eicher et al., 2012), doxorubicin (Murakami et al., 2006; Eicher et al., 2012), and inhibitors like D13-9001 (Nakashima et al., 2013) and MBX (Sjuts et al., 2016). Furthermore, we focused for the first time on comparatively addressing the role of the different gates of the channels leading to the APL and the DPT binding pockets (Alav et al., 2021). The importance of multiple gates on the surface of RND transporters for polyspecific transport has been highlighted by numerous investigations. For instance, in the AcrB protein from E. coli up to four channels have been implicated in the recognition and transport of different classes of substrates, including detergents and β-lactams (mediated by CH1 and CH4) (Murakami et al., 2006; Sennhauser et al., 2007; Pos, 2009; Eicher et al., 2012), macrolides (mediated by CH2) (Seeger et al., 2006; Sennhauser et al., 2007, Sennhauser et al., 2009; Zwama et al., 2018), ethidium bromide and rhodamine 6G (mediated by CH3) (Zwama et al., 2018), and fusidic acid and carboxylated drugs (mediated by CH4 and CH5) (Tam et al., 2020).
We performed state-of-the-art MD simulations of each transporter embedded in hydrated membranes composed of phospholipid mixtures resembling the true P. aeruginosa inner membrane (Benamara et al., 2014). These were followed by analyses of hydration, electrostatics, volumetric profiles, as well as a fragment-based mapping of each Mex protein to monitor the presence of possible MFSs using small organic probes featuring different physicochemical properties (Supplementary Figure S1) (Imai et al., 2011; Ramaswamy et al., 2018). Although CH4 and CH5 gates are overall the most conserved among all investigated Mex transporters, CH4 displays identity percentages between MexB and MexF, MexF and MexY, and MexB and MexY lower than those observed for CH5.
The electrostatic features of the APL and DPT are overall in agreement with the known specificity for charged substrates transported by these proteins. The greater negative charge patch on the surface of DPT in MexY is functional to attract positively charged aminoglycosides and disfavor negatively charged molecules; however, the presence of scattered positive charges may feebly favor the binding of β-lactams (especially zwitterionic). Likewise, MexF and especially MexB with a more positive electrostatic potential surface in the DPT tend to attract negatively charged as well as zwitterionic β-lactams and extrude them with greater efficiency along with weakly acidic quinolones, such as cinoxacin and nalidixic acid. In comparison, cationic antibiotics such as oleandomycin, erythromycin, and puromycin are pumped out with low efficiency (Tikhonova et al., 2002). The observed changes in electrostatic potential surfaces of APL and DPT among pre-MD and most populated cluster structures are related to (expected) conformational changes taking place in the transporters during the MD simulations (Simsir et al., 2021).
The analysis of the fragment-based mapping of MexF highlighted the presence of a few MFSs in locations equivalent to the APL and the DPT of MexB and MexY (Ramaswamy et al., 2018), suggesting that these pockets mediate polyspecific recognition also in this member of the Mex family of transporters. The predominance of MFSs and moreover HBD and HBA probes found in the DPT of MexF (Table 1; Supplementary Table S6), together with the balance between positively and negatively charged residues (Supplementary Table S5) and its higher LI compared to MexB and MexY (Supplementary Table S7), might be key for the recognition of amphipathic compounds such as fluoroquinolones, tetracycline, and small hydrophilic compounds such as chloramphenicol (Fukuda et al., 1995; Kohler et al., 1997a; Aires et al., 2002; Fetar et al., 2011). Recognition of low-molecular-mass compounds seems to be mainly mediated by the DP also for this transporter, as already suggested for other members. The APL features instead a comparable number of MFSs among all transporters, although the LI remains the highest for MexF when calculated on the pre-MD structures. This, together with the lower percentage of charged residues lining the AP in MexF compared to MexB and MexY (Supplementary Figure S3; Supplementary Table S5), might disfavor effective interactions with substrates rich in HBD and HBA groups (such as aminoglycosides and carbapenems). These differences in LI values could also be interpreted considering the relative percentages of polar and hydrophobic amino acids lining the MFSs. For instance, compared to MexF and MexB, in MexY the MFSAP within the L protomer was surrounded by a large number of negatively charged residues (i.e., K79, D615, and D811), which could rationalize the efficient recognition of positively charged substrates by the latter transporter (Supplementary Figures S5, S7).
Additionally, a relevant number of MFSs at the interface (switch- or G-loop) between APL and DPT of each transporter was detected (Table 1), confirming the general belief that this region is an intermediate step in the transport of substrates from the APL to the DPT sites. Furthermore, we have observed that two channels among all, namely CH2 and CH5, are endowed with MFS in all the transporters (the latter mostly in MexY), suggesting their possible involvement in the early recognition of several small organic probes (and thus substrates, such as antibiotics, functionalized with these chemical groups). This is relevant also from a pharmaceutical perspective, as it points to the possibility of targeting these sites in drug design efforts (Collu et al., 2012; Kumar Roy and Patra, 2020)—for instance to inhibit the translocation of substrates from the periplasm to the APL and DPT binding pockets—and protein engineering (Mao et al., 2002; Middlemiss and Poole, 2004; Krishnamoorthy et al., 2008; Ohene-Agyei et al., 2012; Wang et al., 2015; Kumar Roy and Patra, 2020; Marshall and Bavro, 2020; Zwama and Nishino, 2021; Cacciotto et al., 2022).
Overall, our results suggest that, despite the different specificities of the Mex transporters, recognition and transport in this family are mediated by a shared “topology” which accounts for redundancy but allows subtle sequence alterations at specific site to confer peculiar binding abilities to each member.
In perspective, we intend to expand this analysis to MexD, which is another important member of the Mex RND transporters family lacking detailed structural information (Gotoh et al., 1999), as well as to the transporters AcrB and AcrD from E. coli, in particular, to understand at the molecular level of detail how entry gates mediate polyspecific transport (Husain and Nikaido, 2010; Husain et al., 2011; Nakashima et al., 2011; Eicher et al., 2012; Oswald et al., 2016; Schuster et al., 2016; Zwama et al., 2018; Tam et al., 2020; Zwama and Nishino, 2021).
The data presented in the study are deposited in the Zenodo repository, with associated DOI: 10.5281/zenodo.7274367.
AC, AVV, GM, and PR designed research; AC and VR performed research; AC, AVV, GM, AB, and PR analyzed the data; AC and AVV wrote the manuscript with contributions from all authors; AVV, GM, and PR supervised research; PR provided fundings.
This work was supported by NIAID/NIH grant no. R01AI136799 (to AC, AV, GM, and PR).
We thank Giovanni Serra for his technical assistance with computational resources.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer IB declared a past co-editorship with one of the authors AVV to the handling editor.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2022.1021916/full#supplementary-material
Aires, J. R., Pechère, J. C., van Delden, C., and Köhler, T. (2002). Amino acid residues essential for function of the MexF efflux pump protein of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 46, 2169–2173. doi:10.1128/AAC.46.7.2169-2173.2002
Alav, I., Kobylka, J., Kuth, M. S., Pos, K. M., Picard, M., Blair, J. M. A., et al. (2021). Structure, assembly, and function of tripartite efflux and type 1 secretion systems in gram-negative bacteria. Chem. Rev. 121, 5479–5596. doi:10.1021/acs.chemrev.1c00055
Atzori, A., Malloci, G., Prajapati, J. D., Basciu, A., Bosin, A., Kleinekathöfer, U., et al. (2019). Molecular interactions of cephalosporins with the deep binding pocket of the RND transporter AcrB. J. Phys. Chem. B 123, 4625–4635. doi:10.1021/acs.jpcb.9b01351
Baker, N. A., Sept, D., Joseph, S., Holst, M. J., and McCammon, J. A. (2001). Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U. S. A. 98, 10037–10041. doi:10.1073/pnas.181342398
Benamara, H., Rihouey, C., Abbes, I., Mlouka, M. A., Hardouin, J., Jouenne, T., et al. (2014). Characterization of membrane lipidome changes in Pseudomonas aeruginosa during biofilm growth on glass wool. PLoS One 9, e108478. doi:10.1371/journal.pone.0108478
Broto, P., Moreau, G., and Vandycke, C. (1984). Molecular structures: Perception, autocorrelation descriptor and sar studies: System of atomic contributions for the calculation of the n-octanol/water partition coefficients. Eur. J. Med. Chem. 19 (1), 71–78.
Bruzzese, A., Gil, C., Dalton, J. A. R., and Giraldo, J. (2018). Structural insights into positive and negative allosteric regulation of a G protein-coupled receptor through protein-lipid interactions. Sci. Rep. 8, 4456. doi:10.1038/s41598-018-22735-6
Cacciotto, P., Basciu, A., Oliva, F., Malloci, G., Zacharias, M., Ruggerone, P., et al. (2022). Molecular rationale for the impairment of the MexAB-OprM efflux pump by a single mutation in MexA. Comput. Struct. Biotechnol. J. 20, 252–260. doi:10.1016/j.csbj.2021.11.042
Case, D. A., Aktulga, H. M., Belfon, K., Ben-Shalom, I. Y., Brozell, S. R., Cerutti, D. S., et al. (2021). Amber 2021 San Francisco: University of California.
Case, D. A., Ben-Shalom, I. Y., Brozell, S. R., Cerutti, D. S., Cheatham, T. E., Cruzeiro, V. W. D., et al. (2018). AMBER18 San Francisco: University of California.
Cha, H. -j., Muller, R. T., and Pos, K. M. (2014). Switch-loop flexibility affects transport of large drugs by the promiscuous AcrB multidrug efflux transporter. Antimicrob. Agents Chemother. 58, 4767–4772. doi:10.1128/AAC.02733-13
Chen, M., Shi, X., Yu, Z., Fan, G., Serysheva, I. I., Baker, M. L., et al. (2022). In situ structure of the AcrAB-TolC efflux pump at subnanometer resolution. Structure 30, 107–113. e3. doi:10.1016/j.str.2021.08.008
Ciulli, A., Williams, G., Smith, A. G., Blundell, T. L., and Abell, C. (2006). Probing hot spots at Protein−Ligand binding sites: A fragment-based approach using biophysical methods. J. Med. Chem. 49, 4992–5000. doi:10.1021/jm060490r
Collu, F., Vargiu, A. V., Dreier, J., Cascella, M., and Ruggerone, P. (2012). Recognition of imipenem and meropenem by the RND-transporter MexB studied by computer simulations. J. Am. Chem. Soc. 134, 19146–19158. doi:10.1021/ja307803m
Colovos, C., and Yeates, T. O. (1993). Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 2, 1511–1519. doi:10.1002/pro.5560020916
Dickson, C. J., Madej, B. D., Skjevik, Å. A., Betz, R. M., Teigen, K., Gould, I. R., et al. (2014). Lipid14: The amber lipid force field. J. Chem. Theory Comput. 10, 865–879. doi:10.1021/ct4010307
Dolinsky, T. J., Nielsen, J. E., McCammon, J. A., and Baker, N. A. (2004). PDB2PQR: An automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 32, W665–W667. doi:10.1093/nar/gkh381
Dreier, J., and Ruggerone, P. (2015). Interaction of antibacterial compounds with RND efflux pumps in Pseudomonas aeruginosa. Front. Microbiol. 6, 660. doi:10.3389/fmicb.2015.00660
Eda, S., Maseda, H., and Nakae, T. (2003a). An elegant means of self-protection in gram-negative bacteria by recognizing and extruding xenobiotics from the periplasmic space. J. Biol. Chem. 278, 2085–2088. doi:10.1074/jbc.C200661200
Eda, S., Yoneyama, H., and Nakae, T. (2003b). Function of the MexB efflux-transporter divided into two halves. Biochemistry 42, 7238–7244. doi:10.1021/bi0300074
Eicher, T., Cha, H., Seeger, M. A., Brandstätter, L., El-Delik, J., Bohnert, J. A., et al. (2012). Transport of drugs by the multidrug transporter AcrB involves an access and a deep binding pocket that are separated by a switch-loop. Proc. Natl. Acad. Sci. U. S. A. 109, 5687–5692. doi:10.1073/pnas.1114944109
Eicher, T., Seeger, M. A., Anselmi, C., Zhou, W., Brandstätter, L., Verrey, F., et al. (2014). Coupling of remote alternating-access transport mechanisms for protons and substrates in the multidrug efflux pump AcrB. Elife 3, e03145. doi:10.7554/eLife.03145
Eisenberg, D., Lüthy, R., and Bowie, J. U. (1997). “[20] VERIFY3D: Assessment of protein models with three-dimensional profiles,” in Methods in enzymology macromolecular crystallography Part B (United States: Academic Press), 396–404. Available at: https://www.sciencedirect.com/science/article/pii/S0076687997770228.
Fetar, H., Gilmour, C., Klinoski, R., Daigle, D. M., Dean, C. R., and Poole, K. (2011). mexEF-oprN multidrug efflux operon of Pseudomonas aeruginosa: Regulation by the MexT activator in response to nitrosative stress and chloramphenicol. Antimicrob. Agents Chemother. 55, 508–514. doi:10.1128/AAC.00830-10
Fischbach, M. A., and Walsh, C. T. (2009). Antibiotics for emerging pathogens. Science 325, 1089–1093. doi:10.1126/science.1176667
Fukuda, H., Hosaka, M., Iyobe, S., Gotoh, N., Nishino, T., and Hirai, K. (1995). nfxC-type quinolone resistance in a clinical isolate of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 39, 790–792. doi:10.1128/AAC.39.3.790
Gaillard, P., Carrupt, P. A., Testa, B., and Boudon, A. (1994). Molecular lipophilicity potential, a tool in 3D QSAR: Method and applications. J. Comput. Aided. Mol. Des. 8, 83–96. doi:10.1007/BF00119860
Ghose, A. K., and Crippen, G. M. (1986). Atomic physicochemical parameters for three-dimensional structure-directed quantitative structure-activity relationships I. Partition coefficients as a measure of hydrophobicity. J. Comput. Chem. 7, 565–577. doi:10.1002/jcc.540070419
Glavier, M., Puvanendran, D., Salvador, D., Decossas, M., Phan, G., Garnier, C., et al. (2020). Antibiotic export by MexB multidrug efflux transporter is allosterically controlled by a MexA-OprM chaperone-like complex. Nat. Commun. 11, 4948. doi:10.1038/s41467-020-18770-5
Gotoh, N., Kusumi, T., Tsujimoto, H., Wada, T., and Nishino, T. (1999). Topological analysis of an RND family transporter, MexD of Pseudomonas aeruginosa. FEBS Lett. 458, 32–36. doi:10.1016/S0014-5793(99)01116-3
Gould, I., Skjevik, A., Dickson, C., Madej, B., and Walker, R. (2018). Lipid17: A comprehensive AMBER force field for the simulation of zwitterionic and anionic lipids. in preparation.
Hadfield, T. E., Imrie, F., Merritt, A., Birchall, K., and Deane, C. M. (2022). Incorporating target-specific pharmacophoric information into deep generative models for fragment elaboration. J. Chem. Inf. Model. 62, 2280–2292. doi:10.1021/acs.jcim.1c01311
Hancock, R. E. W. (1998). Resistance mechanisms in Pseudomonas aeruginosa and other nonfermentative gram-negative bacteria. Clin. Infect. Dis. 27, S93–S99. doi:10.1086/514909
Hopkins, C. W., le Grand, S., Walker, R. C., and Roitberg, A. E. (2015). Long-time-step molecular dynamics through hydrogen mass repartitioning. J. Chem. Theory Comput. 11, 1864–1874. doi:10.1021/ct5010406
Humphrey, W., Dalke, A., and Schulten, K. (1996). Vmd: Visual molecular dynamics. J. Mol. Graph. 14, 33–38. doi:10.1016/0263-7855(96)00018-5
Husain, F., Bikhchandani, M., and Nikaido, H. (2011). Vestibules are part of the substrate path in the multidrug efflux transporter AcrB of Escherichia coli. J. Bacteriol. 193, 5847–5849. doi:10.1128/JB.05759-11
Husain, F., and Nikaido, H. (2010). Substrate path in the AcrB multidrug efflux pump of Escherichia coli. Mol. Microbiol. 78, 320–330. doi:10.1111/j.1365-2958.2010.07330.x
Imai, T., Miyashita, N., Sugita, Y., Kovalenko, A., Hirata, F., and Kidera, A. (2011). Functionality mapping on internal surfaces of multidrug transporter AcrB based on molecular theory of solvation: Implications for drug efflux pathway. J. Phys. Chem. B 115, 8288–8295. doi:10.1021/jp2015758
Iyer, R., Ferrari, A., Rijnbrand, R., and Erwin, A. L. (2022). A fluorescent microplate Assay quantifies bacterial efflux and demonstrates two distinct compound binding sites in AcrB. Antimicrob. Agents Chemother. 59, 2388–2397. doi:10.1128/AAC.05112-14
Johnson, R. M., Fais, C., Parmar, M., Cheruvara, H., Marshall, R. L., Hesketh, S. J., et al. (2020). Cryo-EM structure and molecular dynamics analysis of the fluoroquinolone resistant mutant of the acrb transporter from salmonella. Microorganisms 8, E943. doi:10.3390/microorganisms8060943
Klenotic, P. A., Moseng, M. A., Morgan, C. E., and Yu, E. W. (2021). Structural and functional diversity of resistance–nodulation–cell division transporters. Chem. Rev. 121, 5378–5416. doi:10.1021/acs.chemrev.0c00621
Kobayashi, N., Tamura, N., van Veen, H. W., Yamaguchi, A., and Murakami, S. (2014). β-Lactam selectivity of multidrug transporters AcrB and AcrD resides in the proximal binding pocket. J. Biol. Chem. 289, 10680–10690. doi:10.1074/jbc.M114.547794
Kobylka, J., Kuth, M. S., Müller, R. T., Geertsma, E. R., and Pos, K. M. (2020). AcrB: A mean, keen, drug efflux machine. Ann. N. Y. Acad. Sci. 1459, 38–68. doi:10.1111/nyas.14239
Koch, D. C., Raunest, M., Harder, T., and Kandt, C. (2013). Unilateral access regulation: Ground state dynamics of the Pseudomonas aeruginosa outer membrane efflux duct OprM. Biochemistry 52, 178–187. doi:10.1021/bi3014714
Köhler, T., Epp, S. F., Curty, L. K., and Pechère, J. C. (1999). Characterization of MexT, the regulator of the MexE-MexF-OprN multidrug efflux system of Pseudomonas aeruginosa. J. Bacteriol. 181, 6300–6305. doi:10.1128/jb.181.20.6300-6305.1999
Köhler, T., Michéa-Hamzehpour, M., Henze, U., Gotoh, N., Curty, L. K., and Pechère, J. C. (1997a). Characterization of MexE-MexF-OprN, a positively regulated multidrug efflux system of Pseudomonas aeruginosa. Mol. Microbiol. 23, 345–354. doi:10.1046/j.1365-2958.1997.2281594.x
Kohler, T., Michea-Hamzehpour, M., Plesiat, P., Kahr, A. L., and Pechere, J. C. (1997b). Differential selection of multidrug efflux systems by quinolones in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 41, 2540–2543. doi:10.1128/aac.41.11.2540
Kozakov, D., Grove, L. E., Hall, D. R., Bohnuud, T., Mottarella, S., Luo, L., et al. (2015). The FTMap family of web servers for determining and characterizing ligand binding hot spots of proteins. Nat. Protoc. 10, 733–755. doi:10.1038/nprot.2015.043
Krahn, T., Gilmour, C., Tilak, J., Fraud, S., Kerr, N., Lau, C. H.-F., et al. (2012). Determinants of intrinsic aminoglycoside resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 56, 5591–5602. doi:10.1128/AAC.01446-12
Krishnamoorthy, G., Tikhonova, E. B., and Zgurskaya, H. I. (2008). Fitting periplasmic membrane fusion proteins to inner membrane transporters: Mutations that enable Escherichia coli AcrA to function with Pseudomonas aeruginosa MexB. J. Bacteriol. 190, 691–698. doi:10.1128/JB.01276-07
Kumar Roy, R., and Patra, N. (2020). Configuration flipping in distal pocket of multidrug transporter MexB impacts the efflux inhibitory mechanism. Chemphyschem. 21, 2516–2524. doi:10.1002/cphc.202000759
Laskowski, R. A., MacArthur, M. W., Moss, D. S., and Thornton, J. M. (1993). Procheck: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291. doi:10.1107/S0021889892009944
Lau, C. H. F., Hughes, D., and Poole, K. (2014). MexY-promoted aminoglycoside resistance in Pseudomonas aeruginosa: Involvement of a putative proximal binding pocket in aminoglycoside recognition. mBio 5, e01068. doi:10.1128/mBio.01068-14
Leo, A., Hansch, C., and Elkins, D. (1971). Partition coefficients and their uses. Chem. Rev. 71, 525–616. doi:10.1021/cr60274a001
Li, X.-Z., Plésiat, P., and Nikaido, H. (2015). The challenge of efflux-mediated antibiotic resistance in gram-negative bacteria. Clin. Microbiol. Rev. 28, 337–418. doi:10.1128/CMR.00117-14
Li, X. Z., Nikaido, H., and Poole, K. (1995). Role of mexA-mexB-oprM in antibiotic efflux in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 39, 1948–1953. doi:10.1128/aac.39.9.1948
Li, X. Z., Zhang, L., Srikumar, R., and Poole, K. (1998). Beta-lactamase inhibitors are substrates for the multidrug efflux pumps of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 42, 399–403. doi:10.1128/aac.42.2.399
Liko, I., Degiacomi, M. T., Lee, S., Newport, T. D., Gault, J., Reading, E., et al. (2018). Lipid binding attenuates channel closure of the outer membrane protein OmpF. Proc. Natl. Acad. Sci. U. S. A. 115, 6691–6696. doi:10.1073/pnas.1721152115
Linares, J. F., López, J. A., Camafeita, E., Albar, J. P., Rojo, F., and Martínez, J. L. (2005). Overexpression of the multidrug efflux pumps MexCD-OprJ and MexEF-OprN is associated with a reduction of type III secretion in Pseudomonas aeruginosa. J. Bacteriol. 187, 1384–1391. doi:10.1128/JB.187.4.1384-1391.2005
Llanes, C., Köhler, T., Patry, I., Dehecq, B., van Delden, C., and Plésiat, P. (2011). Role of the MexEF-OprN efflux system in low-level resistance of Pseudomonas aeruginosa to ciprofloxacin. Antimicrob. Agents Chemother. 55, 5676–5684. doi:10.1128/AAC.00101-11
López, C. A., Travers, T., Pos, K. M., Zgurskaya, H. I., and Gnanakaran, S. (2017). Dynamics of intact MexAB-OprM efflux pump: Focusing on the MexA-OprM interface. Sci. Rep. 7, 16521. doi:10.1038/s41598-017-16497-w
Madden, T. (2013). “The BLAST sequence analysis tool,” in The NCBI handbook. Editors M. Jo, and O. Jim (Bethesda, MD: National Center for Biotechnology Information).
Mao, W., Warren, M. S., Black, D. S., Satou, T., Murata, T., Nishino, T., et al. (2002). On the mechanism of substrate specificity by resistance nodulation division (RND)-type multidrug resistance pumps: The large periplasmic loops of MexD from Pseudomonas aeruginosa are involved in substrate recognition. Mol. Microbiol. 46, 889–901. doi:10.1046/j.1365-2958.2002.03223.x
Marshall, R. L., and Bavro, V. N. (2020). Mutations in the TolC periplasmic domain affect substrate specificity of the AcrAB-TolC pump. Front. Mol. Biosci. 7, 166. doi:10.3389/fmolb.2020.00166
Masuda, N., Sakagawa, E., Ohya, S., Gotoh, N., Tsujimoto, H., and Nishino, T. (2000). Substrate specificities of MexAB-OprM, MexCD-OprJ, and MexXY-OprM efflux pumps in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 44, 3322–3327. doi:10.1128/aac.44.12.3322-3327.2000
Middlemiss, J. K., and Poole, K. (2004). Differential impact of MexB mutations on substrate selectivity of the MexAB-OprM multidrug efflux pump of Pseudomonas aeruginosa. J. Bacteriol. 186, 1258–1269. doi:10.1128/JB.186.5.1258-1269.2004
Morgan, C. E., Glaza, P., Leus, I. V., Trinh, A., Su, C. C., Cui, M., et al. (2021). Cryoelectron microscopy structures of AdeB illuminate mechanisms of simultaneous binding and exporting of substrates. mBio 12 (1), e03690-20. doi:10.1128/mBio.03690-20
Morita, Y., Tomida, J., and Kawamura, Y. (2015). Efflux-mediated fluoroquinolone resistance in the multidrug-resistant Pseudomonas aeruginosa clinical isolate PA7: Identification of a novel MexS variant involved in upregulation of the mexEF-oprN multidrug efflux operon. Front. Microbiol. 6, 8. doi:10.3389/fmicb.2015.00008
Morley, S. D., and Afshar, M. (2004). Validation of an empirical RNA-ligand scoring function for fast flexible docking using Ribodock. J. Comput. Aided. Mol. Des. 18, 189–208. doi:10.1023/b:jcam.0000035199.48747.1e
Murakami, S., Nakashima, R., Yamashita, E., Matsumoto, T., and Yamaguchi, A. (2006). Crystal structures of a multidrug transporter reveal a functionally rotating mechanism. Nature 443, 173–179. doi:10.1038/nature05076
Murakami, S., Nakashima, R., Yamashita, E., and Yamaguchi, A. (2002). Crystal structure of bacterial multidrug efflux transporter AcrB. Nature 419, 587–593. doi:10.1038/nature01050
Murata, T., Kuwagaki, M., Shin, T., Gotoh, N., and Nishino, T. (2002). The substrate specificity of tripartite efflux systems of Pseudomonas aeruginosa is determined by the RND component. Biochem. Biophys. Res. Commun. 299, 247–251. doi:10.1016/S0006-291X(02)02626-8
Nakashima, R., Sakurai, K., Yamasaki, S., Hayashi, K., Nagata, C., Hoshino, K., et al. (2013). Structural basis for the inhibition of bacterial multidrug exporters. Nature 500, 102–106. doi:10.1038/nature12300
Nakashima, R., Sakurai, K., Yamasaki, S., Nishino, K., and Yamaguchi, A. (2011). Structures of the multidrug exporter AcrB reveal a proximal multisite drug-binding pocket. Nature 480, 565–569. doi:10.1038/nature10641
Nikaido, H. (2018). RND transporters in the living world. Res. Microbiol. 169, 363–371. doi:10.1016/j.resmic.2018.03.001
Oberhauser, N., Nurisso, A., and Carrupt, P.-A. (2014). MLP tools: A PyMOL plugin for using the molecular lipophilicity potential in computer-aided drug design. J. Comput. Aided. Mol. Des. 28, 587–596. doi:10.1007/s10822-014-9744-0
Ohene-Agyei, T., Lea, J. D., and Venter, H. (2012). Mutations in MexB that affect the efflux of antibiotics with cytoplasmic targets. FEMS Microbiol. Lett. 333, 20–27. doi:10.1111/j.1574-6968.2012.02594.x
Olsson, M. H. M., Søndergaard, C. R., Rostkowski, M., and Jensen, J. H. (2011). PROPKA3: Consistent treatment of internal and surface residues in empirical p Ka predictions. J. Chem. Theory Comput. 7, 525–537. doi:10.1021/ct100578z
Ornik-Cha, A., Wilhelm, J., Kobylka, J., Sjuts, H., Vargiu, A. v., Malloci, G., et al. (2021). Structural and functional analysis of the promiscuous AcrB and AdeB efflux pumps suggests different drug binding mechanisms. Nat. Commun. 12, 6919. doi:10.1038/s41467-021-27146-2
Oswald, C., Tam, H.-K., and Pos, K. M. (2016). Transport of lipophilic carboxylates is mediated by transmembrane helix 2 in multidrug transporter AcrB. Nat. Commun. 7, 13819. doi:10.1038/ncomms13819
Poole, K. (2005). Efflux-mediated antimicrobial resistance. J. Antimicrob. Chemother. 56, 20–51. doi:10.1093/jac/dki171
Poole, K., Krebes, K., McNally, C., and Neshat, S. (1993). Multiple antibiotic resistance in Pseudomonas aeruginosa: Evidence for involvement of an efflux operon. J. Bacteriol. 175, 7363–7372. doi:10.1128/jb.175.22.7363-7372.1993
Poole, K. (2001). Multidrug efflux pumps and antimicrobial resistance in Pseudomonas aeruginosa and related organisms. J. Mol. Microbiol. Biotechnol. 3, 255–264. Available at: http://www.ncbi.nlm.nih.gov/pubmed/11321581.
Poole, K. (2011). Pseudomonas aeruginosa: Resistance to the max. Front. Microbiol. 2, 65. doi:10.3389/fmicb.2011.00065
Poole, K., and Srikumar, R. (2001). Multidrug efflux in Pseudomonas aeruginosa: Components, mechanisms and clinical significance. Curr. Top. Med. Chem. 1, 59–71. doi:10.2174/1568026013395605
Pos, K. M. (2009). Drug transport mechanism of the AcrB efflux pump. Biochim. Biophys. Acta 1794, 782–793. doi:10.1016/j.bbapap.2008.12.015
Radoux, C. J., Olsson, T. S. G., Pitt, W. R., Groom, C. R., and Blundell, T. L. (2016). Identifying interactions that determine fragment binding at protein hotspots. J. Med. Chem. 59, 4314–4325. doi:10.1021/acs.jmedchem.5b01980
Ramaswamy, V. K., Cacciotto, P., Malloci, G., Ruggerone, P., and Vargiu, A. V. (2016). “Multidrug efflux pumps and their inhibitors characterized by computational modeling,” in Efflux-mediated antimicrobial resistance in bacteria: Mechanisms, regulation and clinical implications. Editors X.-Z. Li, C. A. Elkins, and H. I. Zgurskaya (Cham: Springer International Publishing). doi:10.1007/978-3-319-39658-3_30
Ramaswamy, V. K., Cacciotto, P., Malloci, G., Vargiu, A. V., and Ruggerone, P. (2017a). Computational modelling of efflux pumps and their inhibitors. Essays Biochem. 61, 141–156. doi:10.1042/EBC20160065
Ramaswamy, V. K., Vargiu, A. V., Malloci, G., Dreier, J., and Ruggerone, P. (2018). Molecular determinants of the promiscuity of MexB and MexY multidrug transporters of Pseudomonas aeruginosa. Front. Microbiol. 9, 1144. doi:10.3389/fmicb.2018.01144
Ramaswamy, V. K., Vargiu, A. V., Malloci, G., Dreier, J., and Ruggerone, P. (2017b). Molecular rationale behind the differential substrate specificity of bacterial RND multi-drug transporters. Sci. Rep. 7, 8075. doi:10.1038/s41598-017-08747-8
Rice, P., Longden, I., and Bleasby, A. (2000). Emboss: The European molecular biology open software suite. Trends Genet. 16, 276–277. doi:10.1016/S0168-9525(00)02024-2
Ruggerone, P., Satoshi, M., Klaas, M. P., and Attilio, V. V. (2013). RND efflux pumps: Structural information translated into function and inhibition mechanisms. Curr. Top. Med. Chem. 13, 3079–3100. doi:10.2174/15680266113136660220
Ruiz-Carmona, S., Alvarez-Garcia, D., Foloppe, N., Garmendia-Doval, A. B., Juhos, S., Schmidtke, P., et al. (2014). rDock: A fast, versatile and open source program for docking ligands to proteins and nucleic acids. PLoS Comput. Biol. 10, e1003571. doi:10.1371/journal.pcbi.1003571
Šali, A., Blundell, T. L., and Sali, A. (1993). Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815. doi:10.1006/jmbi.1993.1626
Sangster, J. (1997). Octanol-water partition coefficients: Fundamentals and physical chemistry. Chichester: Wiley.
Schott-Verdugo, S., and Gohlke, H. (2019). PACKMOL-memgen: A simple-to-use, generalized workflow for membrane-protein–lipid-bilayer system building. J. Chem. Inf. Model. 59, 2522–2528. doi:10.1021/acs.jcim.9b00269
Schulz, R., Vargiu, A., Collu, F., Kleinekathöfer, U., and Ruggerone, P. (2010). Functional rotation of the transporter AcrB: Insights into drug extrusion from simulations. PLoS Comput. Biol. 6, e1000806. doi:10.1371/journal.pcbi.1000806
Schulz, R., Vargiu, A., Ruggerone, P., and Kleinekathöfer, U. (2015). Computational study of correlated domain motions in the AcrB efflux transporter. Biomed. Res. Int. 2015, 487298. doi:10.1155/2015/487298
Schulz, R., Vargiu, A., Ruggerone, P., and Kleinekathöfer, U. (2011). Role of water during the extrusion of substrates by the efflux transporter AcrB. J. Phys. Chem. B 115, 8278–8287. doi:10.1021/jp200996x
Schuster, S., Vavra, M., and Kern, W. (2016). Evidence of a substrate-discriminating entrance channel in the lower porter domain of the multidrug resistance efflux pump AcrB. Antimicrob. Agents Chemother. 60, 4315–4323. doi:10.1128/AAC.00314-16
Seeger, M. A., Schiefner, A., Eicher, T., Verrey, F., Diederichs, K., and Pos, K. M. (2006). Structural asymmetry of AcrB trimer suggests a peristaltic pump mechanism. Science 313, 1295–1298. doi:10.1126/science.1131542
Sennhauser, G., Amstutz, P., Briand, C., Storchenegger, O., and Grütter, M. G. (2007). Drug export pathway of multidrug exporter AcrB revealed by DARPin inhibitors. PLoS Biol. 5, e7. doi:10.1371/journal.pbio.0050007
Sennhauser, G., Bukowska, M. A., Briand, C., and Grütter, M. G. (2009). Crystal structure of the multidrug exporter MexB from Pseudomonas aeruginosa. J. Mol. Biol. 389, 134–145. doi:10.1016/j.jmb.2009.04.001
Shen, M., and Sali, A. (2006). Statistical potential for assessment and prediction of protein structures. Protein Sci. 15, 2507–2524. doi:10.1110/ps.062416606
Sievers, F., and Higgins, D. G. (2014). “Clustal omega, accurate alignment of very large numbers of sequences,” in Multiple sequence alignment methods. Editor D. J. Russell (Totowa, NJ: Humana Press), 105–116. Available at: http://link.springer.com/10.1007/978-1-62703-646-7_6.
Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539. doi:10.1038/msb.2011.75
Simsir, M., Broutin, I., Mus-Veteau, I., and Cazals, F. (2021). Studying dynamics without explicit dynamics: A structure-based study of the export mechanism by AcrB. Proteins 89, 259–275. doi:10.1002/prot.26012
Sjuts, H., Vargiu, A., Kwasny, S. M., Nguyen, S. T., Kim, H.-S., Ding, X., et al. (2016). Molecular basis for inhibition of AcrB multidrug efflux pump by novel and powerful pyranopyridine derivatives. Proc. Natl. Acad. Sci. U. S. A. 113, 3509–3514. doi:10.1073/pnas.1602472113
Smilova, M. D., Curran, P. R., Radoux, C. J., von Delft, F., Cole, J. C., Bradley, A. R., et al. (2022). Hotspots api: A Python package for the detection of small molecule binding hotspots and application to structure-based drug design. J. Chem. Inf. Model. 62, 1911–1916. doi:10.1021/acs.jcim.9b00996
Symmons, M. F., Marshall, R. L., and Bavro, V. N. (2015). Architecture and roles of periplasmic adaptor proteins in tripartite efflux assemblies. Front. Microbiol. 6, 513. doi:10.3389/fmicb.2015.00513
Tam, H.-K., Malviya, V. N., Foong, W.-E., Herrmann, A., Malloci, G., Ruggerone, P., et al. (2020). Binding and transport of carboxylated drugs by the multidrug transporter AcrB. J. Mol. Biol. 432, 861–877. doi:10.1016/j.jmb.2019.12.025
Tam, H. K., Foong, W. E., Oswald, C., Herrmann, A., Zeng, H., and Pos, K. M. (2021). Allosteric drug transport mechanism of multidrug transporter AcrB. Nat. Commun. 12, 3889. doi:10.1038/s41467-021-24151-3
Testa, B., Carrupt, P.-A., Gaillard, P., Billois, F., and Weber, P. (1996). Lipophilicity in molecular modeling. Pharm. Res. 13, 335–343. doi:10.1023/A:1016024005429
The UniProt Consortium (2015). UniProt: A hub for protein information. Nucleic Acids Res. 43, D204–D212. doi:10.1093/nar/gku989
Tian, C., Kasavajhala, K., Belfon, K. A. A., Raguette, L., Huang, H., Migues, A. N., et al. (2020). ff19SB: Amino-Acid-Specific protein backbone parameters trained against quantum mechanics energy surfaces in solution. J. Chem. Theory Comput. 16, 528–552. doi:10.1021/acs.jctc.9b00591
Tikhonova, E. B., Wang, Q., and Zgurskaya, H. I. (2002). Chimeric analysis of the multicomponent multidrug efflux transporters from gram-negative bacteria. J. Bacteriol. 184, 6499–6507. doi:10.1128/JB.184.23.6499-6507.2002
Tsutsumi, K., Yonehara, R., Ishizaka-Ikeda, E., Miyazaki, N., Maeda, S., Iwasaki, K., et al. (2019). Structures of the wild-type MexAB–OprM tripartite pump reveal its complex formation and drug efflux mechanism. Nat. Commun. 10, 1520. doi:10.1038/s41467-019-09463-9
Vaccaro, L., Scott, K. A., and Sansom, M. S. P. (2008). Gating at both ends and breathing in the middle: Conformational dynamics of TolC. Biophys. J. 95, 5681–5691. doi:10.1529/biophysj.108.136028
Vargiu, A. V., Collu, F., Schulz, R., Pos, K. M., Zacharias, M., Kleinekathöfer, U., et al. (2011). Effect of the F610A mutation on substrate extrusion in the AcrB transporter: Explanation and rationale by molecular dynamics simulations. J. Am. Chem. Soc. 133, 10704–10707. doi:10.1021/ja202666x
Vargiu, A. V., and Nikaido, H. (2012). Multidrug binding properties of the AcrB efflux pump characterized by molecular dynamics simulations. Proc. Natl. Acad. Sci. U. S. A. 109, 20637–20642. doi:10.1073/pnas.1218348109
Vargiu, A. V., Ruggerone, P., Opperman, T. J., Nguyen, S. T., and Nikaido, H. (2014). Molecular mechanism of MBX2319 inhibition of Escherichia coli AcrB multidrug efflux pump and comparison with other inhibitors. Antimicrob. Agents Chemother. 58, 6224–6234. doi:10.1128/AAC.03283-14
Wakefield, A. E., Yueh, C., Beglov, D., Castilho, M. S., Kozakov, D., Keserü, G. M., et al. (2020). Benchmark sets for binding hot spot identification in fragment-based ligand discovery. J. Chem. Inf. Model. 60, 6612–6623. doi:10.1021/acs.jcim.0c00877
Wang, Z., Fan, G., Hryc, C. F., Blaza, J. N., Serysheva, I. I., Schmid, M. F., et al. (2017). An allosteric transport mechanism for the AcrAB-TolC multidrug efflux pumpe24905. Elife 6. doi:10.7554/eLife.24905
Wang, Z., Zhong, M., Lu, W., Chai, Q., and Wei, Y. (2015). Repressive mutations restore function-loss caused by the disruption of trimerization in Escherichia coli multidrug transporter AcrB. Front. Microbiol. 6, 4. doi:10.3389/fmicb.2015.00004
Wehmeier, C., Schuster, S., Fahnrich, E., Kern, W., and Bohnert, J. A. (2009). Site-directed mutagenesis reveals amino acid residues in the Escherichia coli RND efflux pump AcrB that confer macrolide resistance. Antimicrob. Agents Chemother. 53, 329–330. doi:10.1128/AAC.00921-08
Wiederstein, M., and Sippl, M. J. (2007). ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 35, W407–W410. doi:10.1093/nar/gkm290
Williams, C. J., Headd, J. J., Moriarty, N. W., Prisant, M. G., Videau, L. L., Deis, L. N., et al. (2018). MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 27, 293–315. doi:10.1002/pro.3330
Wolter, D. J., Smith-Moland, E., Goering, R. v., Hanson, N. D., and Lister, P. D. (2004). Multidrug resistance associated with mexXY expression in clinical isolates of Pseudomonas aeruginosa from a Texas hospital. Diagn. Microbiol. Infect. Dis. 50, 43–50. doi:10.1016/j.diagmicrobio.2004.05.004
Zwama, M., and Nishino, K. (2021). Ever-adapting rnd efflux pumps in gram-negative multidrug-resistant pathogens: A race against time. Antibiotics 10, 774. doi:10.3390/antibiotics10070774
Zwama, M., Yamasaki, S., Nakashima, R., Sakurai, K., Nishino, K., and Yamaguchi, A. (2018). Multiple entry pathways within the efflux transporter AcrB contribute to multidrug recognition. Nat. Commun. 9, 124. doi:10.1038/s41467-017-02493-1
AP Access (aka Proximal) Pocket
CS Consensus Site
DOPE Discrete Optimized Protein Energy
DP Deep (aka Distal) Pocket
FD Funnel Domain
HAE Hydrophobe/Amphiphile Efflux
HBA Hydrogen Bond Acceptor
HBD Hydrogen Bond Donor
HP-trap Hydrophobic Trap
IG Interface/G-loop
IM Inner Membrane
IMP Inner Membrane Protein
LI Lipophilic Index
MD Molecular Dynamics
MFP Membrane Fusion Protein
MFS Multi-Functional Site
MLP Molecular Lipophilicity Potential
OMP Outer Membrane Protein
OPC Optimal Point Charges
P Partition coefficient
PAP Periplasmic Adaptor Protein
PD Porter (aka Pore) Domain
PDB Protein Data Bank
PME Particle-Mesh Ewald
POPE 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylethanolamine
POPG 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylglycerol
rdf Radial Distribution Function
RMSD Root Mean Square Deviation
RND Resistance-Nodulation-cell Division
Keywords: RND efflux pumps, multidrug transporter, Pseudomonas aeruginosa, antibiotic resistance, molecular dynamics, molecular modeling
Citation: Catte A, K. Ramaswamy V, Vargiu AV, Malloci G, Bosin A and Ruggerone P (2022) Common recognition topology of mex transporters of Pseudomonas aeruginosa revealed by molecular modelling. Front. Pharmacol. 13:1021916. doi: 10.3389/fphar.2022.1021916
Received: 17 August 2022; Accepted: 24 October 2022;
Published: 11 November 2022.
Edited by:
Hendrik W. Van Veen, University of Cambridge, United KingdomReviewed by:
Isabelle Broutin, Centre National de la Recherche Scientifique (CNRS), FranceCopyright © 2022 Catte, K. Ramaswamy, Vargiu, Malloci, Bosin and Ruggerone. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Attilio Vittorio Vargiu, dmFyZ2l1QGRzZi51bmljYS5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.