 Salma Nasser
Salma Nasser Dalaal M. Abdallah
Dalaal M. Abdallah Kawkab A. Ahmed
Kawkab A. Ahmed Yousra Abdel-Mottaleb
Yousra Abdel-Mottaleb Hanan S. El-Abhar
Hanan S. El-Abhar
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol., 25 October 2022
Sec. Gastrointestinal and Hepatic Pharmacology
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.1008085
Although dysautonomia was documented in inflammatory bowel disease, with activation of the stress-related sympathetic system, the role of agonists/antagonists of the adrenergic receptors is not conclusive. Moreover, ulcerative colitis was recently linked to dementia, but the potential role of the presenilin 1(PS1)/BACE-1/beta-amyloid (Aβ) axis has not been evaluated. Hence, we investigated the impact of mirabegron (β3-agonist) and/or carvedilol (β1/β2 antagonist) on iodoacetamide-induced ulcerative colitis with emphasis on the novel pathomechanism of the PS1/BACE-1/Aβ axis in ulcerative colitis, and its relation to the inflammatory cascade, fibrotic processes, and the gut barrier dysfunction. Ulcerated rats were either left untreated or treated for 8 days with mirabegron and/or carvedilol. Besides minimizing colon edema and weight loss, and improving colon structure, mirabegron and/or carvedilol abated colonic PS1/BACE-1/Aβ axis and the NOTCH1/NICD/HES1 hub besides the inflammatory cascade GSK3-β/NF-κΒ/TNF-α, and the oxidative stress marker malondialdehyde. The anti-fibrotic effect was verified by boosting SMAD-7 and inhibiting TGF-β1, α-SMA immunoexpression, and MTC staining. Moreover, the drugs improved the gut barrier function, attested by the increased goblet cells and expression of E-cadherin, and the inhibited expression of p(Y654)-β-catenin to preserve the E-cadherin/β-catenin adherens junction (AJ). These signaling pathways may be orchestrated by the replenished PPAR-γ, a transcription factor known for its anti-colitic effect.
Conclusion: Besides maintaining the gut barrier, mirabegron and/or carvedilol mediated their anti-colitic effect by their anti-oxidant, anti-inflammatory, and anti-fibrotic capacities. The therapeutic effect of these drugs depends partly on suppressing the harmful signaling pathways PS1/BACE-1/Aβ, NOTCH1/NICD/HES1, GSK3-β/NF-κΒ/TNF-α, and TGF-1β/α-SMA while enhancing PPAR-γ, SMAD-7, mucus, and AJ.
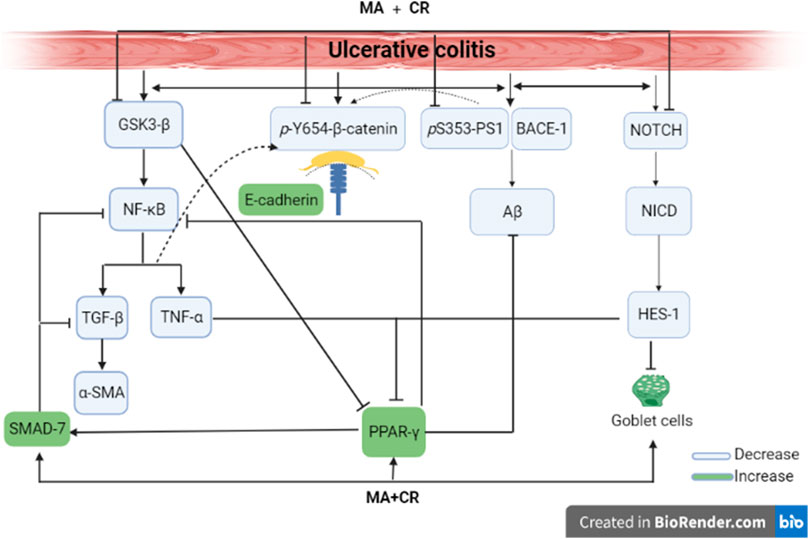
Graphical AbstractMirabegron and carvedilol anti-colitic effect. Both act by inhibiting PS1/BACE-1/Aβ, NOTCH1/NICD/HES1, GSK-3β/NF-κB/TNF-α, MDA, & TGF-β1/α-SMA, while boosting PPAR-γ, SMAD-7 to reduce amyloidosis, osidative stress, inflammation, and fibrosis, while amending E-cadherin/β-catenin adherens junction to enhance gut barrier integrity and replenishing goblet cells to protect the colon.
Ulcerative colitis is an inflammatory disease known to show a relapsing-remitting course pattern. The disease is highly prevalent affecting around 200–250 individuals per 100,000 and its widespread is not limited to gender, age, or ethnicity (Kobayashi et al., 2020; Bagalagel et al., 2022). Ulcerative colitis is caused by multiple factors that collectively compromise gut integrity, including genetic susceptibility, environmental factors, and immune dysregulation (Choi et al., 2017). The jeopardization of the intestinal barrier plays a crucial role in the pathophysiology of ulcerative colitis; this can be driven by epithelial cell dysfunction (Gitter et al., 2001; Mehta et al., 2015) or an inflammatory storm that compromises cells in the lamina propria, which leads to chronicity (Strober and Fuss, 2011). Moreover, colon fibrosis can ensue upon exposure to prolonged inflammation (Wynn, 2008; Binabaj et al., 2019).
Despite the extensively studied pathogenesis of this disease, the full mechanism responsible has not yet been fully elucidated. For instance, the possible deposition of amyloid and its role in the pathogenesis of this disease has not been extensively studied. For instance, the intestine of patients with Alzheimer’s disease (AD) revealed the expression of amyloid precursor protein (APP) (Shankle et al., 1993), and a plaque-like deposition (Arai et al., 1991) in the enteric nervous system (ENS). Additionally, it was reported that transgenic mice expressing mutant presenilin-1 (PS1), a critical component for the γ-secretase complex, or its precursor APP had a progressive accumulation of amyloid beta (Aβ) in the ENS (Puig et al., 2015). Indeed, the phosphorylation of PS1 causes a pathogenic conformational change in PS1/γ-secretase that increases the Aβ42/40 ratio (Maesako et al., 2017). These depositions decrease the number of enteric neurons and increase vulnerability to intestinal inflammation. Recent evidence also indicates that irritable bowel disease (IBD) has a positive correlation with the development of dementia (Zhang et al., 2021) and more specifically, the elevated proinflammatory cytokines and the increase in oxidative stress, cause blood-brain barrier dysfunction and accelerate the amyloid-β accumulation in the brain (Kesika et al., 2021). This notion is strengthened by evidence from studies of ADLPAPT mice, which carry three human transgenes: APP, PS1, and tau. These mice were found to have altered gut microbiota composition, as well as gut barrier dysfunction and chronic gut and systemic inflammation (El-Hakim et al., 2022).
Apart from its role in the cleavage of APP, PS1 also aids in the cleavage of NOTCH (De Strooper and Annaert, 2001). The impact of the NOTCH signaling pathway on ulcerative colitis has attracted more attention in the recent past (Lin et al., 2019; Li et al., 2020; Christopoulos et al., 2021; Roy et al., 2021). Previously, the overexpression of the NOTCH1 gene was recorded in the colon of dextran sulfate sodium-induced colitic mice (Noah and Shroyer, 2013) and its downstream target gene hairy and enhancer of split 1 (HES1) was reported to deplete the mucus-producing goblet cells (GCs) (Zheng et al., 2011). Indeed, this hub is a dominant regulator of cell fate during intestinal homeostasis, which indicates its crucial role in the pathogenesis of intestinal diseases partly by activating the inflammatory transcription factor nuclear factor kappa Β (NF-κB) (Zhang et al., 2010) and the pro-inflammatory surrogate marker tumor necrosis factor (TNF-α) (Ghorbaninejad et al., 2019). One of the upstream molecules of NF-κB is the glycogen synthase kinase-3 (GSK-3) β, a serine-threonine protein kinase that plays a pivotal role in ulcerative colitis. Aside from its positive correlation with PS1 and Aβ42 (Zhang et al., 2020), previous studies have reported the abundance of this kinase in an ulcerative colitis model (Uddin et al., 2013; Di Gregorio et al., 2017). Furthermore, its inhibition by selective inhibitors has been found to inhibit inflammation and tissue injury in acutely colitic rats by causing downregulation of NF-κΒ (Tantray et al., 2017).
In addition, GSK-3β can regulate peroxisome-proliferator activated receptor gamma (PPAR-γ), another transcription factor that plays an important role in anti-inflammatory reactions possibly by inhibiting the release of TNF-α (Xu et al., 2017). Moreover, in different inflammatory disease conditions, its agonists have been shown to offer protective effects via downregulating pro-inflammatory cytokines and inhibiting apoptosis (Vetuschi et al., 2018). It has also been recounted that the anti-inflammatory activity of 5 amino-salicylic acid (5-ASA), a first-line medication for the treatment of ulcerative colitis, requires the activation of epithelial PPAR-γ signaling, which makes it a potential target for therapeutic intervention in ulcerative colitis (Cevallos et al., 2021).
Regarding ulcerative colitis disease, no definitive treatments are present, which makes it attractive to repurpose available molecules. Despite the scarce data available on the role of adrenergic receptors (ARs) in this ailment, some studies correlate between gastrointestinal tract (GIT) and these ARs. Although in IBD patients the balance of autonomic nervous function is disturbed with sympathetic over-activity (Bai et al., 2009), the role of β-AR antagonism in protecting against ulcerative colitis is not conclusive. Previous studies reported that acetic acid-induced ulcerative colitis was alleviated using carvedilol, a β-and α1- AR blocker (Fatani et al., 2015), and nebivolol, a highly selective β1-AR antagonist with a weak β2-AR antagonistic effect and a β3-AR agonism (Aly Labib et al., 2016). These treatments were thought to be effective in their anti-inflammatory, antioxidant, and/or antiapoptotic properties. On the other hand, trinitrobenzene sulfonic acid (TNBS)-induced colitis was found to reduce the expression of β2-AR (Fan et al., 2013), and the administration of salbutamol, a β2-AR agonist, has also been found to ameliorate ulcerative colitis (Deng et al., 2022).
Concerning β3-AR, earlier studies reported that agonists of this receptor possess anti-motility effects on the colon (Lipworth, 1996). This was thought to occur by inhibiting pacemaker potentials (Wu et al., 2015), which suggests a possible role in the treatment of irritable bowel syndrome. Moreover, another study recounted that colitis-induced loss of the β3-AR is one reason behind diarrhea associated with this disease (Zhao et al., 2001). Moreover, the selective β3-agonist, SR58611, was found to reduce the severity of colitis via its anti-inflammatory potential and the upregulation of the β3 AR expression in inflamed tissues (Vasina et al., 2008). These findings were supported by the presence of the β3-AR on myenteric neurons to suggest a potential therapeutic target for gut inflammatory disease. The first β3-AR agonist is mirabegron (Bragg et al., 2014), which besides its approved use in the treatment of overactive urinary bladder syndrome (Mo et al., 2017), has other off-target effects by acting on muscarinic receptors, α1-AR, dopamine/noradrenaline transporters, and the sodium channel site 2 (Dehvari et al., 2018).
Based on the role of the dysregulated ARs in IBD (Jacobson et al., 2021), we aimed at studying the potential anti-colitic efficacy of mirabegron and/or carvedilol against the iodoacetamide (IAA)-induced ulcerative colitis model in rats. Our analyses emphasized the colonic PS1/BACE-1/Aβ hub, the NOTCH1 axis, physical barrier dysfunction, PPAR-γ/NF-κB, and the associated fibrotic process.
Adult male Wistar rats (180–250 g) were housed under standard conditions (i.e., at 25°C ± 2°C, humidity 60%–70%, 12 h dark/light cycles) with free access to food and water. Before the experiment, animals were acclimated in cages for 1 week at the Future University in Egypt (FUE; Cairo, Egypt). The study protocol was approved by the Ethics Committee of the Faculty of Pharmacy, Cairo University (Cairo, Egypt) (PT 2288, 25/3/2019) and complies with the National Institutes of Health Guide for the care and use of laboratory animals (NIH, revised 2011). All experiments comply with ARRIVE guidelines.
Rats were fasted for 24 h with free access to water then anesthetized using thiopental (30 mg/kg; i.p). A single dose of 0.15 ml of 4% IAA (Sigma-Aldrich, MO, USA; in 1% methylcellulose) was instilled rectally via a catheter placed 8 cm proximal to the anus (Satoh et al., 1997) and animals were kept upside-down to prevent leakage of solution. Animals were then left for 7 days.
Animals were allocated into 6 groups (n = 6 each) comprising normal and ulcerated rats. Animals in the first group served as the normal control and rats in group 2 were assigned as the untreated ulcerated group. Rats in both groups were administered saline orally for 7 days. Ulcerated animals in groups 3 and 4 received carvedilol (CR; Selleckchem, TX, USA) at doses of 15 and 30 mg/kg (Fatani et al., 2015), whereas rats in group 5 were treated with mirabegron (MA; 30 mg/kg; Selleckchem, TX, USA) based on a pilot study testing 3 (Hatanaka et al., 2013), 10 (Sawada et al., 2013), and 30 mg/kg (de Oliveira et al., 2019; Supplementary Figures S1, S2). In the last group, ulcerated rats received a combination of mirabegron with the low dose of carvedilol (MA+CR15). All treatments were gavaged orally for 7 days.
Rats were weighed before induction of colitis and on the day of scarification to calculate the difference in body weight. The colon edema index was calculated according to the following formula (Ahmedy et al., 2020):
Following, the distal 10 cm of the colon was opened longitudinally and divided into two halves; the first half (n = 3) was fixed in 10% neutral buffered formalin for histopathological/immunological examinations, whereas the 2nd was further subdivided into 2 parts. The first portion (n = 6) was homogenized in normal saline for biochemical and ELISA assessments and the 2nd portion (n = 4) was kept in RIPA buffer with a protease inhibitor to assess the Western blot parameters. All samples were divided into aliquots and stored at −80°C.
The lipid peroxide content was measured colorimetrically at a wavelength of 534 nm using an MDA kit (Bio-diagnostic, Cairo, Egypt; Cat #: MD2529) and processed according to the manufacturer’s instructions.
The colon contents of NOTCH1 (MyBioSource, CA, USA, Cat#: MBS004226), E-cadherin (MyBioSource, Cat#: MBS761750), Aβ(1–42) peptide (MyBioSource, Cat#: MBS726579), SMAD7 (MyBioSource, Cat#: MBS2501417), PPAR-γ (MyBioSource, Cat#: MBS2508012), TNF-α (CUSABIO, TX, USA; Cat#: CSB-E11987r), and HES1 (LifeSpan Biosciences, WA, USA; Cat #: LS-F33052) were assessed using rat ELISA kits according to the manufacturer’s directions, and the produced color was measured spectrophotometrically at a wavelength 450 nm.
The protein concentration in the lysed samples was determined using the Bradford Protein Assay kit (BIO BASIC, ON, Canada, Cat# SK3041) according to the manufacturer’s instructions. Equal sample protein concentrations were added to 2x Laemmli sample buffer and heated at 95°C for 5 min, then loaded on the SDS-PAGE gel. The blots were transferred to a nitrocellulose membrane that was blocked with TEST buffer and 3% BSA at room temperature for 1 h, then incubated overnight at 4°C with the primary antibodies (ThermoFisher Scientific, MA, USA) for p(S353)-PS1 (Cat #: PA5-64705), Total-PSI (Cat #: PA5-96088), phosphorylated glycogen synthase kinase (p(S9) -GSK3β; Cat #: MA5-14873), p(Y654)-β-catenin (Cat #: PA5-36748), BACE-1 (Cat #: PA1-757), NICD (Cat #: MA5-11961), or β-actin (Cat #: AM4302) for protein normalization diluted in TEST. The blots were then rinsed 5 times with TEST each 5 min then incubated with the horseradish peroxidase (HRP)-conjugated 2ry antibody for 1 h at room temperature, and the blots were rinsed again. The chemiluminescent substrate (Clarity™ Western ECL substrate-Bio-Rad, CA, USA; Cat#170–5,060) was applied to the blot according to the manufacturer’s recommendation and the signals of target protein bands were captured using a CCD camera-based imager and analyzed using the Chemi Doc MP Bio-Rad imager (CA, USA #170–5,060) software.
The fixed colon specimen was processed in paraffin blocks by regular method and sectioned at 4 μm thickness and stained by H&E then examined by a BX43 light microscope (Olympus; Tokyo, Japan) in a blinded fashion. Quantitative histological assessment of colon lesions was carried out and scored from (0 ̶ 3) in five randomly examined microscopic fields per animal as follows: (0) no changes, 1) mild, 2) moderate, and 3) severe changes for mucosal necrosis, inflammatory cells infiltration, edema, hemorrhage, fibrosis, and depletion of mucous secreting glands (Riley et al., 1988; Salem and Wadie, 2017). The deparaffinized blocks were stained with Alcian blue for the determination of goblet cells (GCs) and Masson’s Trichrome (MTC) for collagen fibers (Bancroft J. and Layton C., 2019). The area percentage of fibrosis, as well as the number of positive GCs, were blindly visualized and quantified using CellSens dimensions (Olympus Software).
The deparaffinized colon slices were immunostained with primary antibodies (ThermoFisher Scientific) of NF-κB (Cat # 14–6,731-81), TGF-β1 (Cat # MA5-15065), or α-SMA (Cat #: 14–9,760-82), then incubated with goat anti-rabbit IgG (H+L) Superclonal™ Recombinant Secondary Antibody, HRP (Cat # A27036). Positive immune expression was expressed as area % from 5 randomly chosen fields per section using Image J Software 1.46a (NIH, MD, USA).
The equation of coefficient drug interaction
For the parametric data, values are presented as mean ± SD that were analyzed using one-ANOVA followed by Tukey’s posthoc test for multiple comparisons, p < 0.05. The non-parametric data are presented as median (min-max) that were analyzed using Kruskal-Wallis followed by Dunn’s posthoc test, p < 0.05. Graph pad prism® software package, version 7 (CA, USA) was used for calculation and drawing.
As compared to the control group, Figure 1 depicted a 39 g loss of (A) body weight in ulcerated rats relative to the initial weight along with a 3.7-fold increment in (B) colon weight. Nonetheless, all treatment regimens, except the low dose of carvedilol (CR15), restored body weight as compared to the control group, an effect that was extended to abate the colon index.
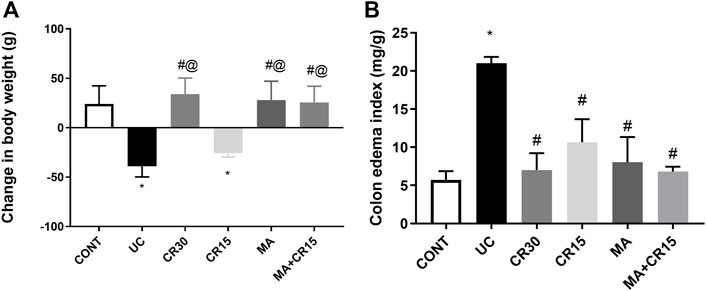
FIGURE 1. Effect of mirabegron and/or carvedilol on (A) body weight, and (B) colon edema index in rats with IAA-induced ulcerative colitis. Data are presented as mean ± SD (n = 6 rats/group). Statistical analysis was carried out using one-way ANOVA followed by Tukey’s posthoc test; p < 0.05, as compared to the (*) CONT, (#) UC, and (@) CR15 groups. CONT, control; CR, carvedilol; IAA, iodoacetamide; MA, mirabegron; UC, ulcerative colitis.
As presented in Figure 2, colon sections of (B-D) ulcerated rats reveal a marked increment in injury markers (mucosal necrosis, inflammatory cells infiltration, edema, hemorrhage, fibrosis, and depletion of mucous secreting glands), as compared to the normal section of the (A) control group. Contrariwise, the section of rats treated with (F) the low dose of carvedilol (CR15) divulges a moderate improvement in the injury markers but a slight upturn in the GCs. Meanwhile, treatment with the (E) high dose of carvedilol (CR30) shows an overall improvement in the colon structure, whereas a better recovery is detected in sections of rats treated with (H) mirabegron and (I) the combination (MA+CR15), with slight hyperplasia of mucous secreting glands and submucosal edema. The collective score of histopathological lesions is shown in panel J.
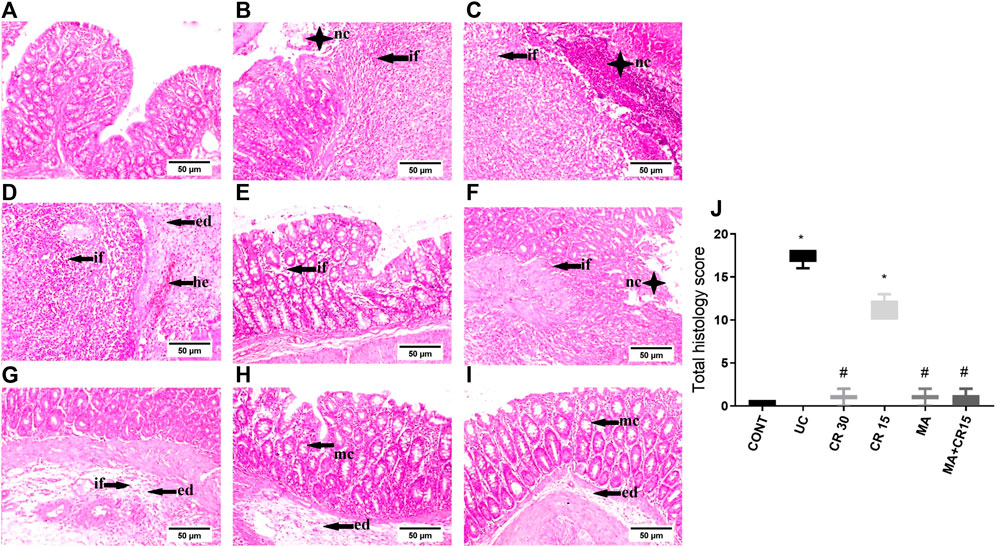
FIGURE 2. Effect of mirabegron and/or carvedilol on colon histopathological changes and total lesions score in rats with IAA-induced ulcerative colitis. Representative photomicrographs (H&E; scale bar, 50 μm) of the (B–D) UC model show marked mucosal necrosis (nc), massive inflammatory cells infiltration (if), marked edema (ed), and focal hemorrhage (he), compared to the section of the (A) CONT group that reveals normal morphology. Section of (E) CR30 shows few inflammatory cells infiltration in the lamina propria, whereas, sections of (F,G) CR15 depict mucosal/submucosal focal necrosis associated with inflammatory cells infiltration, fibroblasts proliferation, and submucosal edema. Treatment with (H) MA and (I) MA+CR15 reveals hyperplasia of mucous-secreting glands (mc) and slight submucosal edema. (J) represents total lesion scoring. Data are presented as median (min-max) (5 fields in each section, n = 3 rats/group); the data were analyzed using Kruskal-Wallis followed by Dunn’s posthoc test; p < 0.05. As compared to the (*) CONT and (#) ulcerated groups. CONT, control; CR, carvedilol; ed, edema; he, hemorrhage; if, inflammatory cells infiltration; IAA, iodoacetamide; nc, necrosis; MA, mirabegron; mc, mucous secreting glands; UC, ulcerative colitis.
The IAA-induced colon amyloidosis and the effect of the different treatment regimens were confirmed biochemically (Figures 3A–C). The ulcerated rats showed increments in both the APP degrading enzymes (A) PS1 (2.9 folds) and (B) BACE-1 (4.1 folds) to augment the formation of (C) Aβ (3.2 folds) in the colon, as compared to the control group. Nevertheless, post-treatment with the low dose of carvedilol (CR15) lessened PS1 by only 39%, whereas treatment with the high dose of carvedilol (CR30), mirabegron, and the combination regimen markedly reduced PS1 to reach a non-significant level as compared to the control group. Once more, treatment with MA alone and in combination returned the protein expression of BACE-1 to a comparable level of the control group. However, the effect of high dose of carvedilol (CR30) was less pronounced and the least effect was mediated by the low dose of carvedilol (CR15). Finally, in a comparable manner, the different treatments leveled off the Aβ content, with a better effect mediated by the combination regimen to surpass that of low dose of carvedilol (CR15) alone.
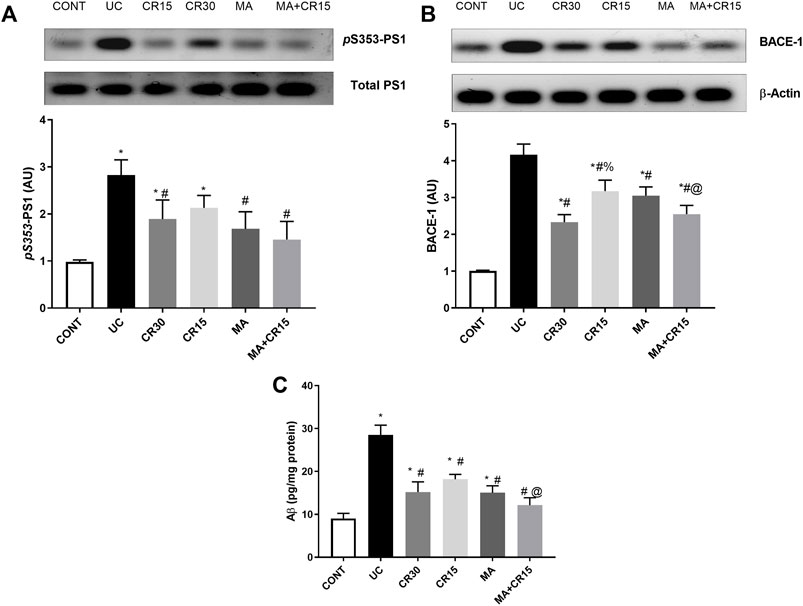
FIGURE 3. Effect of mirabegron and/or carvedilol on colon protein expression/content of (A) p(S353)-PS1, (B) BACE-1, and (C) Aβ(1–42) in rats with IAA-induced ulcerative colitis. Data are presented as mean ± SD (n = 4–6). Statistical analysis was carried out using one-way ANOVA followed by Tukey’s posthoc test; p < 0.05. As compared to the (*) CONT, (#) UC, and (@) CR15 groups. Aβ, beta-amyloid; BACE-1, beta-site APP-cleaving enzyme; CONT, control; CR, carvedilol; IAA, iodoacetamide; MA, mirabegron; PS1, presenilin; UC, ulcerative colitis.
The enhanced PS1 also cleaves NOTCH1 to activate its downstream pathway, a verity that was depicted in Figure 4, where IAA boosted the NOTCH1/NICD/HES1 signaling pathway to reach 2.3 (NOTCH1), 6.4 (NICD), and 3.2 (HES1) folds, respectively relative to the normal counterparts. These increments were almost equally reduced by all single regimens to achieve a significant level as compared to either ulcerated or control groups. However, treatment with the combination regimen reduced NICD to a non-significant level relative to the control group, an effect that was better than that of the low dose of carvedilol (CR15) alone. The impact of HES-1 on GC count was also evident from the results of our Alcian blue stained GC count (Figure 5), which revealed a sharp GC reduction in (B) colitic rats relative to the (A) control group. We also observed a strong increase in GC count in the (E) mirabegron-treated group, which exceeded that of the control group, and an even stronger effect in the group receiving the (F) combination regimen. Panel G summarizes these data.
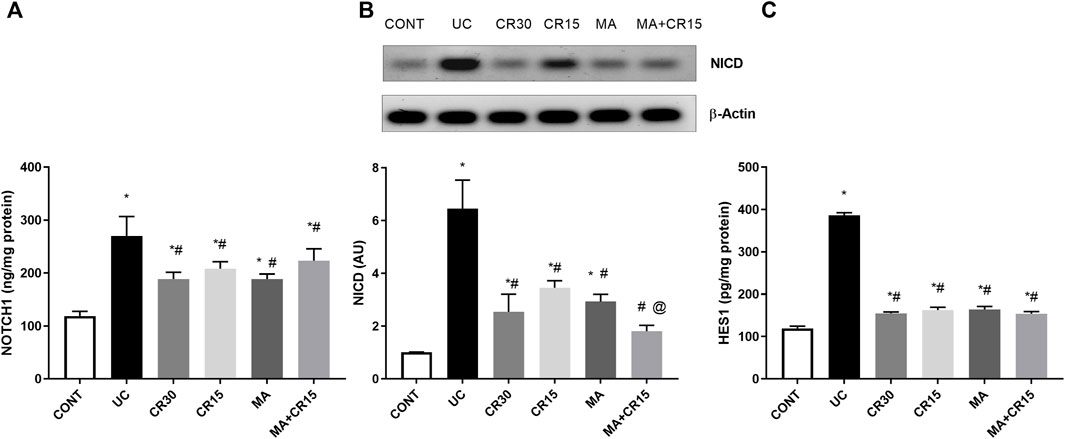
FIGURE 4. Effect of mirabegron and/or carvedilol on colon protein expression/content of (A) NOTCH1, (B) NICD, and (C) HES1 in rats with IAA-induced ulcerative colitis. Data are presented as mean ± SD (n = 4–6). Statistical analysis was carried out using one-way ANOVA followed by Tukey’s posthoc test; p < 0.05, as compared to the (*) CONT, (#) UC, and (@) CR15 groups. CONT, control; CR, carvedilol; IA, iodoacetamide; MA, mirabegron; NICD, nuclear intracellular domain; UC, ulcerative colitis.
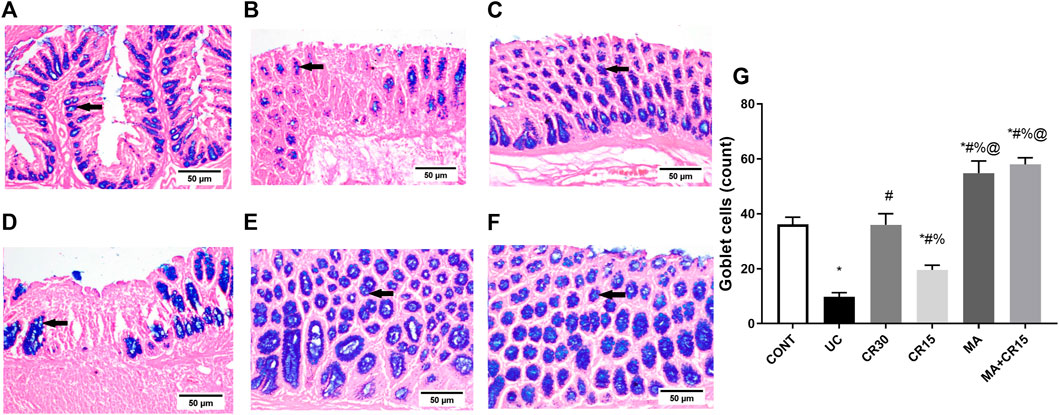
FIGURE 5. Effect of mirabegron and/or carvedilol on the number of goblet cells in the colon of rats with IAA-induced ulcerative colitis. Data are presented as mean ± SD (n = 3). Statistical analysis was carried out using one-way ANOVA followed by Tukey’s posthoc test; p < 0.05, as compared to the (*) CONT, (#) UC, (%) CR30, and (@) CR15 groups. Panels (A–F) represent colon photomicrographs stained with Alcian blue (scale bar, 50 μm). Section of (B) IAA model shows a sharp decrease in the number of GCs, compared to that of (A) CONT group. Section of the ulcerated rats treated with (D) CR15 shows an increase in GCs, whereas that of the (C) CR30 treated group reaches a normal level. However, treatment with (E) MA and (F) MA+CR15 shows a marked increase in the number of GCs that exceeded that in panel (A) (arrows). Panel (G) summarizes the above results (5 fields in each section). CONT, control; CR, carvedilol; IAA, iodoacetamide; MA, mirabegron; UC, ulcerative colitis.
In Figure 6, the insult revealed its inflammatory effect by reducing (A) PPAR-γ (32%) and (B) p(S9)-GSK-3β by 76%, whereas it accelerated the pro-inflammatory mediator (C) TNF-α to 5 folds and the lipid peroxidation marker (D) MDA to 3.9 folds when compared to the control group. Relative to the ulcerated group, all post-treatments normalized PPAR-γ and enhanced p-GSK3-β indicating its deactivation to different levels. Treatment with the high dose of carvedilol (CR30) or mirabegron alone elevated inactive p-GSK-3β significantly, whereas the addition of mirabegron to the low dose of carvedilol (CR15) enhanced its effect. Moreover, all treatments showed a comparable reduction in colon contents of TNF-α and MDA, except for the combination regimen, which normalized these contents and showed an additive interaction between the two drugs. The inflammatory condition was further strengthened by the strong protein expression of the transcription factor NF-κB (Figure 7B), compared to the section of (A) control normal rats. This expression was inhibited significantly with the best effect seen in sections of (E) mirabegron and (F) the combination (MA+CR15) that reveal a weak immune expression to mimic that of panel A. While the worst effect was seen in sections of (D) CR15 and a moderate decrement was seen in section of (C) CR30. These results are summarized in panel G.
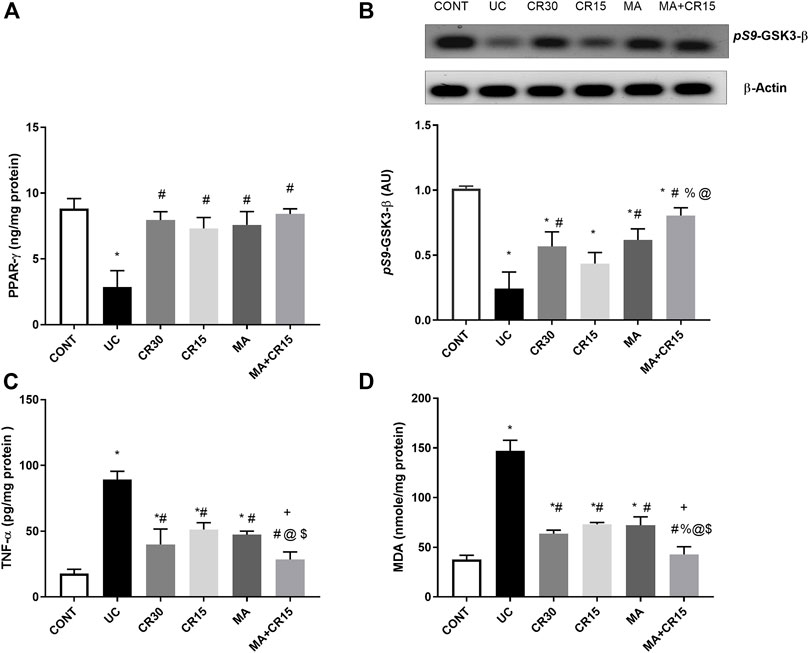
FIGURE 6. Effect of mirabegron and/or carvedilol on colon protein expression/content of (A) PPAR-γ, (B) p(S9)-GSK3-β, (C) TNF-α, and (D) MDA in rats with IAA-induced ulcerative colitis. Data are presented as mean ± SD (n = 4–6). Statistical analysis was carried out using one-way ANOVA followed by Tukey’s post-hoc test; p < 0.05. As compared to the (*) CONT, (#) UC, (%) CR30, (@) CR15, and ($) MA groups. (+) indicate additive interaction using CDI. CDI, coefficient of drug interaction; CONT, control; CR, carvedilol; GSK3-β, glycogen synthase kinase 3 beta; IAA, iodoacetamide; MA, mirabegron; MDA, malondialdehyde; PPAR-γ, peroxisome proliferator-activated receptor gamma; TNF-α, tumor necrosis factor-alpha; UC, ulcerative colitis.
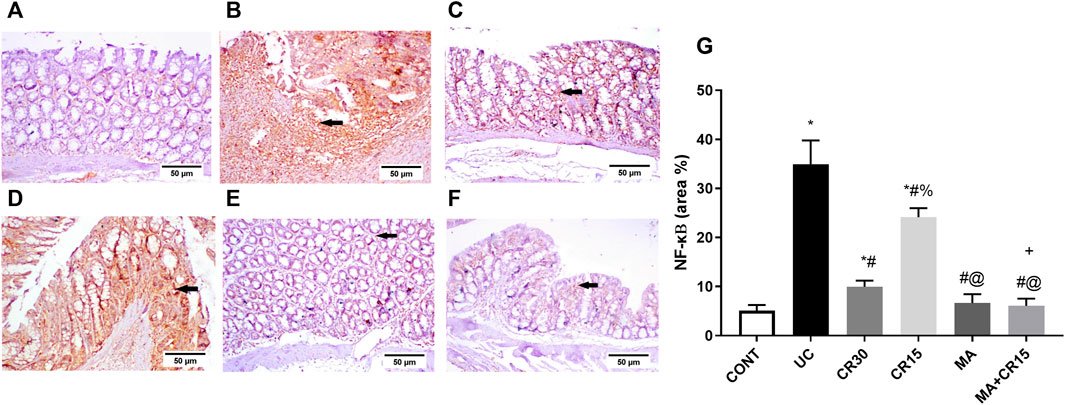
FIGURE 7. Effect of mirabegron and/or carvedilol on colon NF-κB immunoreactivity in rats with IAA-induced ulcerative colitis. Panels (A–F) represent colon photomicrographs of NF-κB immunoreactivity (scale bar, 50 μm). Section of (B) UC model shows strong positive immunostained cells, relative to the (A) CONT group that shows no immune expression. Treatment with (D) CR15 reduced the number of immunostained cells, whereas the section of (C) CR30 lessened it markedly. Sections of (E) MA and (F) MA+CR15 treated groups show a weak immune expression (arrows). (G) shows the % area of NF-κB staining (5 fields in each section). Data are presented as mean ± SD (n = 3). Statistical analysis was carried out using one-way ANOVA followed by Tukey’s posthoc test; p < 0.05. As compared to the (*) CONT, (#) UC, and (@) CR15. (+) additive interaction using CDI. CDI: coefficient of drug interaction; CONT, control; CR, carvedilol; IAA, iodoacetamide; MA, mirabegron; NF-κB, nuclear factor kappa B; UC, ulcerative colitis.
As depicted in Figure 8 and compared to the photomicrograph of the (A) control group, the (B) IAA model triggers fibrosis manifested by the increased expression of TGF-β1 (3.6 folds), an effect that was minimized after treatment with the (D) low dose of carvedilol (CR15) and reached a normal expression level in the groups treated with the (C) high dose of carvedilol (CR30), (E) mirabegron and (F) the combination (MA+CR15). These results are summarized in panel G. In an opposite pattern, the IAA-induced ulcerative colitis sharply reduced the anti-fibrotic marker (H) SMAD-7 by 76% vs the control group. However, all treatments partially increased it with the highest increment noted in the combination regimen (2.8 folds), as compared to the insult. The IAA-induced elevation in TGF-β1 protein expression entailed its downstream target α-SMA (Figure 9), as depicted in panel (B), and compared to the (A) control group to further confirm the fibrotic signal. The best effect was noticed in the (F) combination-treated group, which showed no significant difference when compared to the control group as depicted in panel (G).
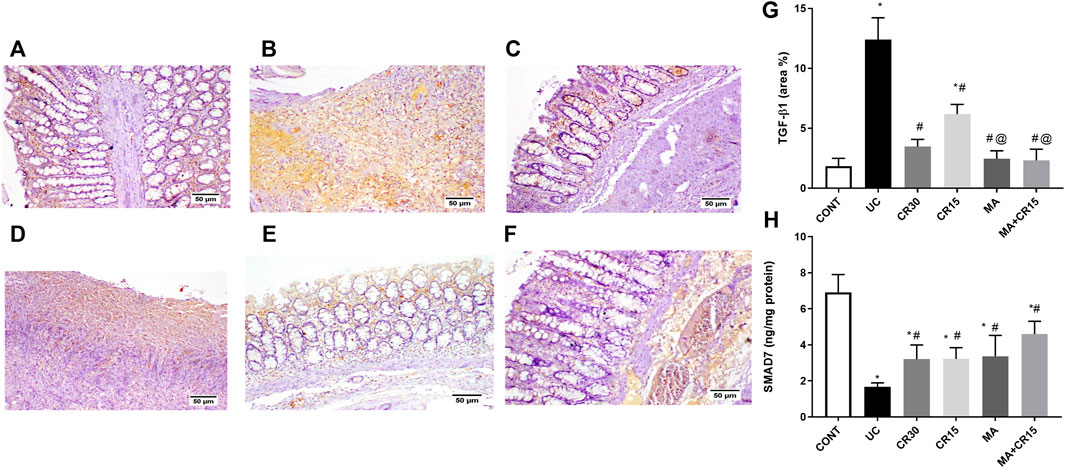
FIGURE 8. Effect of mirabegron and/or carvedilol on colon TGF-β1 immunoreactivity and SMAD7 content in rats with IAA-induced ulcerative colitis. Section of (A) CONT group, shows a normal expression of TGF-β1, while that of (B) UC group shows increased expression of TGF-β in the ulcerative mucosa. Treatment with (C) CR30 reveals moderate expression of TGF-β1, whereas treatment with (D) CR15 only lessened the expression of TGF-β1. Sections of (E) MA and (F) MA+CR15 treated groups show few expressions of TGF-β1 in the mucosa. (G) presents the % area of TGF-β1 staining (5 fields in each section) and (H) represents the effect on SMAD-7. Data are presented as mean ± SD (n = 3–6). Statistical analysis was carried out using one-way ANOVA followed by Tukey’s posthoc test; p < 0.05, as compared to the (*) CONT, (#) UC, and (@) CR15 groups. CONT, control; CR, carvedilol; IAA, iodoacetamide; MA, mirabegron; TGF-β1, transforming growth factor beta; UC, ulcerative colitis.
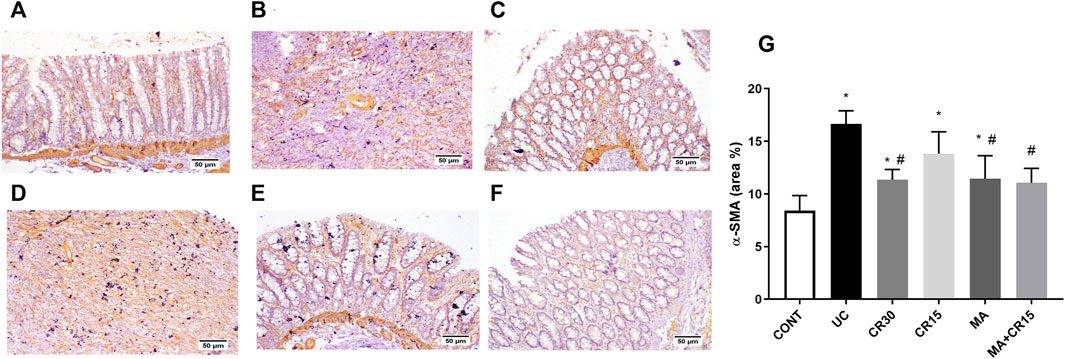
FIGURE 9. Effect of mirabegron and/or carvedilol on the colonic α-SMA immunoreactivity in rats with IAA-induced ulcerative colitis. Section of (A) CONT group shows a normal expression of α-SMA in the lamina epithelia, lamina propria, and muscularis mucosa, while that of (B) UC group and (D) CR15 treated group show higher expression of α-SMA in the injured intestinal mucosa. Treatment with (C) CR30, (E) MA, and (F) MA+CR15 reveals few expressions of α-SMA in the mucosa. (G) shows the % area of α-SMA staining (6 fields in each section). Data are presented as mean ± SD (n = 3 rats/group). Statistical analysis was carried out using one-way ANOVA followed by Tukey’s posthoc test; p < 0.05, as compared to the (*) CONT and (#) UC groups. CONT, control; CR, carvedilol; IAA, iodoacetamide; MA, mirabegron; α-SMA, alpha-smooth muscle actin; UC, ulcerative colitis.
Figure 10 further validates the fibrotic effect of the ulcerative colitis inducer, which causes a marked increase in collagen fibers deposition as shown in section (B), compared to the weak deposition of MTC-stained collagen fibers in the lamina propria of the (A) control group. However, the colon of rats treated with the (D) low dose of carvedilol (CR15) exhibited moderate deposition of collagen fibers to be further reduced in the (C) high dose (CR30)-treated group, but with a slight increase in the lamina propria and submucosa. A near normal deposition was visualized in the colon of rats treated with either (E) mirabegron or (F) the combination (MA+CR15).
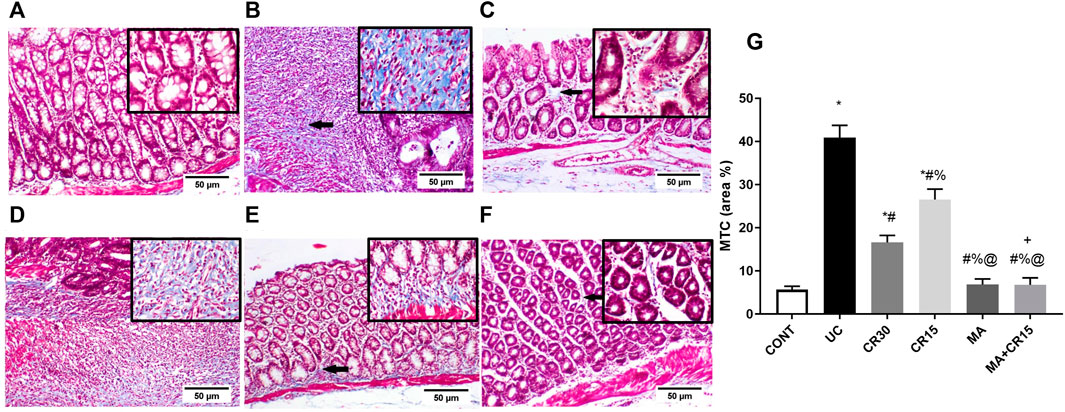
FIGURE 10. Effect of mirabegron and/or carvedilol on MTC stained collagen fibers in the colon of rats with IAA-induced ulcerative colitis. Panels of A-F represent colon photomicrographs stained with MTC (scale bar, 50 μm). Section of (A) CONT shows normal weak MTC stained collagen fibers, whereas the section of (B) ulcerated group reveals a marked increase in collagen fibers deposition. Treatment with (C) CR30 and (D) CR15 show mild and moderate increases in collagen fibers deposition, respectively. However, treatment with (E) MA and (F) MA+CR15 reveals weak collagen fibers deposition (arrows). (G) shows the % area of MTC staining (5 fields in each section). Data are presented as mean ± SD (n = 3 rats/group). Statistical analysis was carried out using one-way ANOVA followed by Tukey’s posthoc test; p < 0.05, as compared to the (*) CONT, (#) UC, (@) CR15, and (%) CR30 groups. (+) Indicates additive interaction using CDI. CDI, coefficient of drug interaction; CONT, control; CR, carvedilol; IAA, iodoacetamide; MA, mirabegron; MTC, Masson’s Trichrome; UC, ulcerative colitis.
Since ulcerative colitis disturbs the membrane integrity, Figure 11 shows the impact of the IAA model on the AJ by studying its effect on the β-catenin/E-cadherin complex. In this figure, IAA caused an upsurge in the protein expression of (A) p(Y654)-β-catenin to reach 5.6 folds relative to the control group. On the contrary and to confirm the interrupted colon cell membrane, the insult leveled off the content of (B) E-cadherin by 59%, as compared to the normal rats. However, this picture was inverted in the treated groups, where all treatments decreased the protein expression of p(Y654)-β-catenin to a comparable level. Unexpectedly, carvedilol at its two doses failed to affect E-cadherin, while mirabegron alone or in combination succeeded to increase its colon content to reach almost the normal level.
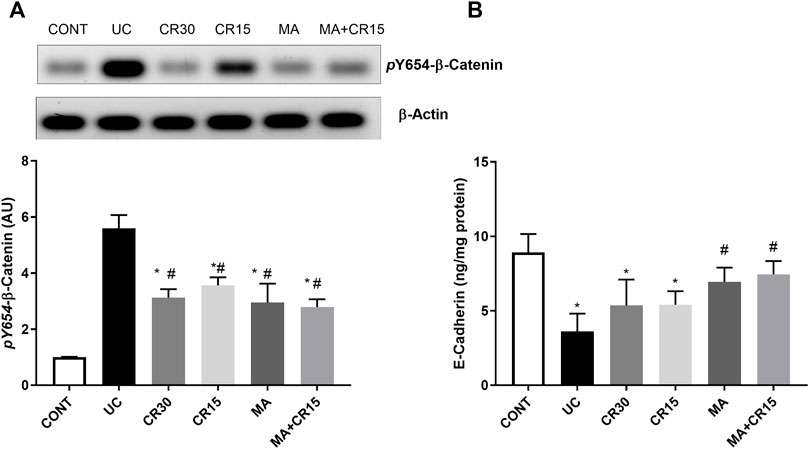
FIGURE 11. Effect of mirabegron and/or carvedilol on colon protein expression/content of (A) p(Y654)-β-catenin and (B) E-cadherin in rats with IAA-induced ulcerative colitis. Data are presented as mean ± SD (n = 4–6). Statistical analysis was carried out using one-way ANOVA followed by Tukey’s posthoc test; p < 0.05, as compared to the (*) CONT and (#) UC groups. CONT, control; CR, carvedilol; IAA, iodoacetamide; MA, mirabegron; UC, ulcerative colitis.
Our study is the first to highlight that the modulation of β-ARs using the β3-AR agonist mirabegron and the β1and β2 antagonist carvedilol offer a therapeutic potential against an ulcerative colitis model. These drugs prove their anti-inflammatory, antioxidant, and anti-fibrotic capacities besides maintaining gut barrier integrity. The anti-colitic mechanisms by which these drugs and their combination act reside partially on shutting down the PS1/BACE-1/Aβ axis, the NOTCH1/NICD/HES1 signaling, and the GSK-3β/NF-κB/TNF-α, as well as the lipid peroxide marker MDA. Besides, they modulate the TGF-β1/α-SMA/ SMAD-7 axis to highlight their anti-fibrotic role. Furthermore, by regulating the β-catenin/E-cadherin hub and the heightening of the GCs these drugs maintain cell integrity. Additionally, the β-AR modulators enhanced PPAR-γ, a transcription factor with an anti-colitic role. Indeed, our results showed that mirabegron has augmented the effect of the low dose of carvedilol to mimic approximately that of the high dose, where the low dose alone exhibited the least beneficial effect.
The impact of the β-ARs on colonic amyloidosis has not been studied before. Indeed, our study is the first to document the presence of amyloidosis-related proteins in a model of ulcerative colitis. Here, the IAA model resulted in an upsurge in the protein expression/content of PS1, BACE-1, and Aβ, whereas post-administration of mirabegron— with or without carvedilol— succeeded to turn this axis off. Amyloidosis is known to accompany AD centrally, but no direct role in ulcerative colitis has yet been reported. Nevertheless, the association between dementia and patients with IBD (Zhang et al., 2021) and the beneficial role of β3 agonism for AD (Tournissac et al., 2021) has been recently highlighted. In addition, earlier studies reported that Aβ or its precursor was present in the enteric neurons of patients with AD (Shankle et al., 1993; Puig et al., 2015; Yi et al., 2017). Although no studies have reported a relation between β3-AR agonists and PS1 or BACE-1, the beneficial role of a β3-AR agonist against Aβ was reported in a model of AD, where it helped to avert the Aβ-induced memory loss, whereas the effect of a β2 agonist was more limited (Gibbs, 2015). Previous studies, both these co-authors and others, also reached the same conclusion, where activation of β3-AR, but not β2, reduced Aβ and its production and prevented the associated memory loss in an AD model (Gibbs et al., 2010; Tournissac et al., 2021); these facts support our findings. The failure of β2-AR agonists to mediate similar effects may be owed to the binding of β2-AR to Aβ leading to its dysfunction and degeneration (Wang et al., 2011), a finding that can explain the effect of the β2-AR blocker carvedilol. This notion was reviewed earlier in 1999 when it was found that carvedilol was able to bind Aβ and prevent it from forming oligomeric fibrils (Howlett et al., 1999).
The tested drugs also exerted an anti-inflammatory effect by inhibiting the IAA-mediated increase of the transcription factor NF-κB, the inflammatory surrogate TNF-α, and their upstream molecule GSK-3β in addition to the oxidative stress marker MDA; this offers another mechanism for their anti-colitic effect since inflammation plays a pivotal role in the pathomechanism of ulcerative colitis (Uddin et al., 2013; Kwiecien et al., 2014). The anti-inflammatory effect of these drugs gives another explanation for the inhibited PS1/BACE-1/Aβ axis where the inflammatory cascade was recounted to activate the PS1/BACE-1/Aβ hub (Paouri et al., 2017) and to increase the formation of ROS, which in turn elevates also the AD-related markers (Kummer and Heneka, 2008; He et al., 2017). In line with the present work, GSK-3β was reported to be activated in ulcerative colitis, where it positively regulated NF-κB (Uddin et al., 2013), which then controls pro-inflammatory mediators like TNF-α (Demarchi et al., 2003). Moreover, the inhibition of GSK-3β in an AD model robustly decreased the oligomeric Aβ load in the mouse brain (DaRocha-Souto et al., 2012) and its activation augments PS1 to enhance the formation of Aβ42 (Zhang et al., 2020).
Besides the above mechanisms, the AR modulators tested herein succeeded to augment PPAR-γ, another transcription factor known for its anti-colitic mechanism. The correlation between β3-AR and PPAR-γ has been highlighted previously by several models, but not in ulcerative colitis. Previously, the activation of the β3-AR, in a PPAR-γ-dependent manner, was able to ameliorate liver steatosis and inflammation in nonalcoholic fatty liver disease (Wang et al., 2020), to cause the browning of white adipocytes to cure obesity (Fan et al., 2019), to offer anti-atherosclerotic effects (Shi et al., 2014), and to hinder preterm labor onset (Hadi et al., 2017). Moreover, the inverse correlation between PPAR-γ and NF-κB can explain its anti-inflammatory character and its inhibition in the present ulcerative colitis model can partake in the exaggerated inflammatory cascade as reported earlier (Latella et al., 2014; Noori et al., 2020). Indeed, the link between PPAR-γ and the PS1/BACE-1/Aβ trajectory has been highlighted in different AD models, where the increased PPAR-γ decreases Aβ (Camacho et al., 2004) and silences BACE-1 (Kummer and Heneka, 2008). Moreover, the PPAR-γ agonist 15d-PGJ2 was able to inhibit the expression of PS1 in a PPARγ-dependent manner in a model of AD (Lu et al., 2018). Moreover, this receptor inhibits inflammation (Viladomiu et al., 2013) and fibrosis (Vetuschi et al., 2018) to represent a hinge upon which the tested drugs mediate partly their effects.
Our study also showed that the treatment regimens succeeded in suppressing the NOTCH1/NICD/HES-1 trajectory that was activated in the colitic group. Although several studies have reported the activation of this pathway in ulcerative colitis models (Zheng et al., 2011; Ghorbaninejad et al., 2019; Christopoulos et al., 2021), scarce data exists that can link ARs with this signaling pathway. A previous study noted that stimulation of β2-AR receptor activates NOTCH1, a fact that can explicate the lowering effect of the β-blocker carvedilol to suppress the catecholamine-induced NOTCH1 activation (Chen et al., 2014). Since to date, no data is reporting the effect of β-3 AR or mirabegron on the NOTCH pathway, one can postulate that activation of PPAR-γ along with the inhibition of the inflammatory parameters and the PS1/BACE-1/Aβ hub can be the tether. In this regard, the increased PS1 was reported to activate the NOTCH trajectory (Selkoe, 2001) and to cleave NOTCH1 to its active form and accordingly HES1(Soriano et al., 2001; Lu et al., 2018; Jfri et al., 2019). Mutually, the activation of NOTCH1 enhances Aβ (Ahn and June, 2007) and BACE-1 (Leal et al., 2012). In addition, the interplay between inflammation and the NOTCH signaling has been amply reviewed (Quillard and Charreau, 2013; Fazio and Ricciardiello, 2016) and it has been reported that NOTCH1 and HES1 in co-operation with TNF-α can suppress PPAR-γ (Maniati et al., 2011) to play a crucial role in the pathogenesis of ulcerative colitis (Ghorbaninejad et al., 2019). On the other hand, another study stated that stimulated PPAR inhibits NOTCH1 (Liu et al., 2022). These data, hence, prove the crosstalk between the assessed parameters to pin down the anti-colitic effect of the tested drugs.
Furthermore, the ability of the used drugs to shut down the NOTCH1 trajectory explains the increased GCs, where previous data showed that hypo-activated NOTCH/HES1 enhances the formation of GCs (Crosnier et al., 2006; Zheng et al., 2011), which could give a novel insight into the present anti-ulcerogenic effect of both mirabegron and carvedilol. The tested drugs were also able to preserve cell integrity by inhibiting the protein expression of p(Y654)-β-catenin to maintain its link to E-cadherin, since the phosphorylation of β-catenin at this site interrupts the E-cadherin/β-catenin complex and cell adhesion in turn (Behrens et al., 1993; Taddei et al., 2002). The role of carvedilol to uphold the E-cadherin/β-catenin hub has been reported earlier in a liver fibrosis model (El-Wakeel et al., 2018). Moreover, the aptitude of mirabegron and carvedilol to reduce PS1 and the inflammatory markers facilitate the stabilization of the E-cadherin/β-catenin complex, since each of them can interrupt this adhesion (Nelson and Nusse, 2004; Twomey and McCarthy, 2006; Saito et al., 2014); however, the augmentation of PPAR-γ cannot be ruled out (Bocca et al., 2007).
The final mechanism by which the tested drugs alleviate IAA-induced ulcerative colitis was by their anti-fibrotic effect manifested by the reduction of the TGF-β1/α-SMA axis and the enhancement of SMAD-7, effects that were reverted by the ulcerogenic inducer. Indeed, TGF-β1 has a controversial role in the pathogenesis of ulcerative colitis (Feagins, 2010; Ihara et al., 2017), yet it was here part of the pathomechanism. The anti-fibrotic potential of the tested drugs has been reported earlier in non-ulcerative colitis models; first, carvedilol was able to decrease TGF-β1/α-SMA and increase SMAD-7 in renal (Wong et al., 2001) and hepatic (El-Wakeel et al., 2018) injury models, whereas mirabegron proved its anti-fibrotic potential against doxorubicin-induced cardiotoxicity via the inhibition of TGF-β/SMAD2/3 pathway (Freiwan et al., 2022). However, the aptitude of mirabegron to release noradrenaline (Mo et al., 2017) may also be one possible reason behind the augmentation of SMAD-7 (Kanamaru et al., 2001).
The inhibition of the fibrotic marker also participates in the anti-oxidant and anti-inflammatory potentials of the tested drugs, since TGF-β1 triggers redox imbalance, which in turn heightens NF-κB (Morry et al., 2017), to increase TGF-β1 (Suryavanshi and Kulkarni, 2017) in a feedforward pattern. Moreover, the stimulation of PPAR-γ increases SMAD7 to inhibit the TGF-β1 signaling pathway (Vallée et al., 2017), an effect that can be linked to its anti-oxidant character (Morel and Singer, 2014; Morry et al., 2017). In addition, TGF-β1 has been reported to crosstalk with PS1 in normal and pathological conditions (Ren et al., 1999), with GSK-3β (Baarsma et al., 2011), and with NOTCH signaling (Yi et al., 2017). Taken together, the current study sheds more light on the possible anti-colitic mechanisms of mirabegron/carvedilol by turning off the injurious effect of these trails.
Here we found that exogenous administration of mirabegron and/or carvedilol caused an upturn in the colon structure and GCs count in a model of ulcerative colitis. This anti-colitic effect may be attributed to the inhibition of oxidative stress, inflammation, fibrosis, and barrier dysfunction. These effects were partly achieved by inhibiting several harmful intersecting signaling pathways; viz., PS1/BACE-1/Aβ, NOTCH1/NICD/HES1, GSK-3β/NF-κB/TNF-α, MDA, and TGF-β1/α-SMA, and enhancing PPAR-γ, SMAD-7, as well as cell integrity (E-cadherin/β-catenin axis).
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
The animal study was reviewed and approved by Research Ethics Committee (Faculty Pharmacy, Cairo University, Cairo, Egypt) that complies with the National Institutes of Health guide for care and use of laboratory animals (NIH, revised 2011).
HS, DMA, and SN conceived the conception and design, SN carried out the experimental part, HS and YA supervised the practical part, HS, DMA, and SN helped in the acquisition of data, HS, DMA, and SN analyzed the results, KAA analyzed and interpreted the histopathological and immunohistochemical data, SN wrote the 1st draft of the article, KAA and YA revised the 1st draft of the article, HS and DMA major revision of the article, all authors participated revising the final manuscript.
We thank Prof. Nabila El-Maraghy (Professor of Pharmacology and Toxicology at Future University in Egypt) for her continuous support and constructive technical advice.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2022.1008085/full#supplementary-material
Aβ, amyloid beta; AJ, adherens junction; APP, amyloid precursor protein; BACE-1, beta-site APP cleaving enzyme-1; CR, carvedilol; GSK-3β, glycogen synthase kinase-3 beta; HES1, hairy and enhancer of split-1; MA, mirabegron; MDA, malondialdehyde; MTC, Masson’s Trichrome for collagen fibers; NF-κB, nuclear factor-kappa B; NICD, notch intracellular domain; PPAR-γ, peroxisome proliferator-activated receptor gamma; PS1, presenilin; α-SMA, α-smooth muscle actin; TGF-β1, transforming growth factor-beta 1; TNF-α, tumor necrosis factor-alpha.
Ahmedy, O. A., Ibrahim, S. M., Salem, H. H., and Kandil, E. A. (2020). Antiulcerogenic effect of melittin via mitigating TLR4/TRAF6 mediated NF-κB and p38MAPK pathways in acetic acid-induced ulcerative colitis in mice. Chem. Biol. Interact. 331, 109276. doi:10.1016/j.cbi.2020.109276
Ahn, Y., and Jun, Y. (2007). Measurement of pain-like response to various NICU stimulants for high-risk infants. Early Hum. Dev. 83, 255–262. doi:10.1016/j.earlhumdev.2006.05.022
Aly Labib, D., Shaker, O., and Elfarouk, L. (2016). Protective effects of nebivolol on acetic acid-induced ulcerative colitis in rats. Kasr Al Ainy Med. J. 22, 99. doi:10.4103/1687-4625.195889
Arai, H., Lee, V. M. ‐., Messinger, M. L., Greenberg, B. D., Lowery, D. E., and Trojanowski, J. Q. (1991). Expression patterns of ?-amyloid precursor protein (?-APP) in neural and nonneural human tissues from alzheimer's disease and control subjects. Ann. Neurol. 30, 686–693. doi:10.1002/ana.410300509
Baarsma, H. A., Spanjer, A. I. R., Haitsma, G., Engelbertink, L. H. J. M., Meurs, H., and Jonker, M. R. (2011). Activation of WNT/ β-catenin signaling in pulmonary fibroblasts by TGF-β 1 is increased in chronic obstructive pulmonary disease. PLoS One 6, e25450. doi:10.1371/journal.pone.0025450
Bagalagel, A., Diri, R., Noor, A., Almasri, D., Bakhsh, H. T., Kutbi, H. I., et al. (2022). The therapeutic effects of cycloastragenol in ulcerative colitis by modulating SphK/MIP-1α/miR-143 signalling. Basic Clin. Pharmacol. Toxicol. 27, 406–419. doi:10.1111/bcpt.13788
Bai, A., Lu, N., Guo, Y., Chen, J., and Liu, Z. (2009). Modulation of inflammatory response via α2-adrenoceptor blockade in acute murine colitis. Clin. Exp. Immunol. 156, 353–362. doi:10.1111/j.1365-2249.2009.03894.x
Bancroft, J., and Layton, C. (2019). Bancroft’s theory and practice of histological techniques. Bancroft’s Theory Pract. Histol. Tech. 1, 126–176. doi:10.1016/c2015-0-00143-510.1016/b978-0-7020-6864-5.00010-4
Behrens, J., Vakaet, L., Friis, R., Winterhager, E., Van Roy, F., Mareel, M. M., et al. (1993). Loss of epithelial differentiation and gain of invasiveness correlates with tyrosine phosphorylation of the E-cadherin/β-catenin complex in cells transformed with a temperature-sensitive v-SRC gene. J. Cell Biol. 120, 757–766. doi:10.1083/jcb.120.3.757
Binabaj, M. M., Asgharzadeh, F., Avan, A., Rahmani, F., Soleimani, A., Parizadeh, M. R., et al. (2019). EW-7197 prevents ulcerative colitis-associated fibrosis and inflammation. J. Cell. Physiol. 234, 11654–11661. doi:10.1002/jcp.27823
Bocca, C., Bozzo, F., Francica, S., Colombatto, S., and Miglietta, A. (2007). Involvement of PPARγ and E-cadherin/β-catenin pathway in the antiproliferative effect of conjugated linoleic acid in MCF-7 cells. Int. J. Cancer 121, 248–256. doi:10.1002/ijc.22646
Bragg, R., Hebel, D., Vouri, S. M., and Pitlick, J. M. (2014). Mirabegron: A beta-3 agonist for overactive bladder. Consult. Pharm. 29, 823–837. doi:10.4140/TCP.n.2014.823
Camacho, I. E., Serneels, L., Spittaels, K., Merchiers, P., Dominguez, D., and De Strooper, B. (2004). Peroxisome proliferator-activated receptor γ induces a clearance mechanism for the amyloid-β peptide. J. Neurosci. 24, 10908–10917. doi:10.1523/JNEUROSCI.3987-04.2004
Cevallos, S. A., Lee, J.-Y., Velazquez, E. M., Foegeding, N. J., Shelton, C. D., Tiffany, C. R., et al. (2021). 5-Aminosalicylic acid ameliorates colitis and checks dysbiotic Escherichia coli expansion by activating PPAR-γ signaling in the intestinal epithelium. MBio 12, 1–10. doi:10.1128/mbio.03227-20
Chen, H., Liu, D., Yang, Z., Sun, L., Deng, Q., Yang, S., et al. (2014). Adrenergic signaling promotes angiogenesis through endothelial cell-tumor cell crosstalk. Endocr. Relat. Cancer 21, 783–795. doi:10.1530/ERC-14-0236
Choi, C. H., Moon, W., Kim, Y. S., Kim, E. S., Lee, B. I., Jung, Y., et al. (2017). Second Korean guideline for the management of ulcerative colitis. Korean J. Gastroenterol. 69, 1–28. doi:10.4166/kjg.2017.69.1.1
Christopoulos, P. F., Gjølberg, T. T., Krüger, S., Haraldsen, G., Andersen, J. T., and Sundlisæter, E. (2021). Targeting the notch signaling pathway in chronic inflammatory diseases. Front. Immunol. 12, 668207. doi:10.3389/fimmu.2021.668207
Crosnier, C., Stamataki, D., and Lewis, J. (2006). Organizing cell renewal in the intestine: Stem cells, signals and combinatorial control. Nat. Rev. Genet. 75, 349–359. doi:10.1038/nrg1840
DaRocha-Souto, B., Coma, M., Pérez-Nievas, B. G., Scotton, T. C., Siao, M., Sánchez-Ferrer, P., et al. (2012). Activation of glycogen synthase kinase-3 beta mediates β-amyloid induced neuritic damage in Alzheimer’s disease. Neurobiol. Dis. 45, 425–437. doi:10.1016/j.nbd.2011.09.002
de Oliveira, M. G., Rojas-Moscoso, J. A., Bertollotto, G. M., Candido, T. Z., Kiguti, L. R. de A., Pupo, A. S., et al. (2019). Mirabegron elicits rat corpus cavernosum relaxation and increases in vivo erectile response. Eur. J. Pharmacol. 858, 172447. doi:10.1016/j.ejphar.2019.172447
De Strooper, B., and Annaert, W. (2001). Where Notch and Wnt signaling meet: The presenilin hub. J. Cell Biol. 152, 17–20. doi:10.1083/jcb.152.4.f17
Dehvari, N., da Silva Junior, E. D., Bengtsson, T., and Hutchinson, D. S. (2018). Mirabegron: Potential off target effects and uses beyond the bladder. Br. J. Pharmacol. 175, 4072–4082. doi:10.1111/bph.14121
Demarchi, F., Bertoli, C., Sandy, P., and Schneider, C. (2003). Glycogen synthase kinase-3β regulates NF-κB1/p105 stability. J. Biol. Chem. 278, 39583–39590. doi:10.1074/jbc.M305676200
Deng, L., Guo, H., Wang, S., Liu, X., Lin, Y., Zhang, R., et al. (2022). The attenuation of chronic ulcerative colitis by (R)-salbutamol in repeated DSS-induced mice. Oxid. Med. Cell. Longev. 2022, 1. doi:10.1155/2022/9318721
Di Gregorio, J., Sferra, R., Speca, S., Vetuschi, A., Dubuquoy, C., Desreumaux, P., et al. (2017). Role of glycogen synthase kinase-3β and PPAR-γ on epithelial-to-mesenchymal transition in DSS-induced colorectal fibrosis. PloS One 12, e0171093. doi:10.1371/journal.pone.0171093
El-Hakim, Y., Bake, S., Mani, K. K., and Sohrabji, F. (2022). Impact of intestinal disorders on central and peripheral nervous system diseases. Neurobiol. Dis. 165, 105627. doi:10.1016/J.NBD.2022.105627
El-Wakeel, S. A., Rahmo, R. M., and El-Abhar, H. S. (2018). Anti-fibrotic impact of Carvedilol in a CCl-4 model of liver fibrosis via serum microRNA-200a/SMAD7 enhancement to bridle TGF-β1/EMT track. Sci. Rep. 8, 1–13. doi:10.1038/s41598-018-32309-1
Fan, H., Liao, Y., Tang, Q., Chen, X. Y., Zhang, L. J., Liu, X. X., et al. (2013). Role of β2-adrenoceptor-β-arrestin2-nuclear factor-κB signal transduction pathway and intervention effects of oxymatrine in ulcerative colitis. Chin. J. Integr. Med. 187, 514–521. doi:10.1007/S11655-012-1146-3
Fan, L., Xu, H., Yang, R., Zang, Y., Chen, J., and Qin, H. (2019). Combination of capsaicin and capsiate induces browning in 3T3-L1 white adipocytes via activation of the peroxisome proliferator-activated receptor γ/β 3-adrenergic receptor signaling pathways. J. Agric. Food Chem. 67, 6232–6240. doi:10.1021/ACS.JAFC.9B02191
Fatani, A. J., Al-Hosaini, K. A., Ahmed, M. M., Abuohashish, H. M., Parmar, M. Y., and Al-Rejaie, S. S. (2015). Carvedilol attenuates inflammatory biomarkers and oxidative stress in a rat model of ulcerative colitis. Drug Dev. Res. 76, 204–214. doi:10.1002/ddr.21256
Fazio, C., and Ricciardiello, L. (2016). Inflammation and notch signaling: A crosstalk with opposite effects on tumorigenesis. Cell Death Dis. 7, e2515. doi:10.1038/cddis.2016.408
Feagins, L. A. (2010). Role of transforming growth factor-β in inflammatory bowel disease and colitis-associated colon cancer. Inflamm. Bowel Dis. 16, 1963–1968. doi:10.1002/ibd.21281
Freiwan, M., Kovács, M. G., Kovács, Z. Z. A., Szűcs, G., Dinh, H., Losonczi, R., et al. (2022). Investigation of the antiremodeling effects of losartan, mirabegron and their combination on the development of doxorubicin-induced chronic cardiotoxicity in a rat model. Int. J. Mol. Sci. 23, 2201. doi:10.3390/IJMS23042201
Ghorbaninejad, M., Heydari, R., Mohammadi, P., Shahrokh, S., Haghazali, M., Khanabadi, B., et al. (2019). Contribution of NOTCH signaling pathway along with TNF-α in the intestinal inflammation of ulcerative colitis. Gastroenterol. Hepatol. Bed Bench 12, S80–S86. doi:10.22037/ghfbb.v12i0.1821
Gibbs, M. E., Maksel, D., Gibbs, Z., Hou, X., Summers, R. J., and Small, D. H. (2010). Memory loss caused by β-amyloid protein is rescued by a β3-adrenoceptor agonist. Neurobiol. Aging 31, 614–624. doi:10.1016/j.neurobiolaging.2008.05.018
Gibbs, M. (2015). Reflections on glycogen and β-amyloid: Why does glycogenolytic β2-adrenoceptor stimulation not rescue memory after β-amyloid? Metab. Brain Dis. 30, 345–352. doi:10.1007/S11011-014-9563-Y
Gitter, A. H., Wullstein, F., Fromm, M., and Schulzke, J. D. (2001). Epithelial barrier defects in ulcerative colitis: Characterization and quantification by electrophysiological imaging. Gastroenterology 121, 1320–1328. doi:10.1053/gast.2001.29694
Hadi, T., Douhard, R., Dias, A. M. M., Wendremaire, M., Pezzè, M., Bardou, M., et al. (2017). Beta3 adrenergic receptor stimulation in human macrophages inhibits NADPHoxidase activity and induces catalase expression via PPARγ activation. Biochim. Biophys. Acta - Mol. Cell Res. 1864, 1769–1784. doi:10.1016/j.bbamcr.2017.07.003
Hatanaka, T., Ukai, M., Watanabe, M., Someya, A., Ohtake, A., Suzuki, M., et al. (2013). Effect of mirabegron, a novel β3-adrenoceptor agonist, on bladder function during storage phase in rats. Naunyn. Schmiedeb. Arch. Pharmacol. 386, 71–78. doi:10.1007/s00210-012-0814-3
He, L., He, T., Farrar, S., Ji, L., Liu, T., and Ma, X. (2017). Antioxidants maintain cellular redox homeostasis by elimination of reactive oxygen species. Cell. Physiol. biochem. 44, 532–553. doi:10.1159/000485089
Howlett, D. R., George, A. R., Owen, D. E., Ward, R. V., and Markwell, R. E. (1999). Common structural features determine the effectiveness of carvedilol, daunomycin and rolitetracycline as inhibitors of Alzheimer β-amyloid fibril formation. Biochem. J. 343, 419–423. doi:10.1042/bj3430419
Ihara, S., Hirata, Y., and Koike, K. (2017). TGF-Β in inflammatory bowel disease: A key regulator of immune cells, epithelium, and the intestinal microbiota. J. Gastroenterol. 52, 777–787. doi:10.1007/s00535-017-1350-1
Jacobson, A., Yang, D., Vella, M., and Chiu, I. M. (2021). The intestinal neuro-immune axis: Crosstalk between neurons, immune cells, and microbes. Mucosal Immunol. 14, 555–565. doi:10.1038/s41385-020-00368-1
Jfri, A. H., O’Brien, E. A., Litvinov, I. V., Alavi, A., and Netchiporouk, E. (2019). Hidradenitis suppurativa: Comprehensive review of predisposing genetic mutations and changes. J. Cutan. Med. Surg. 23, 519–527. doi:10.1177/1203475419852049
Kanamaru, C., Yasuda, H., Takeda, M., Ueda, N., Suzuki, J., Tsuchida, T., et al. (2001). Smad7 is induced by norepinephrine and protects rat hepatocytes from activin A-induced growth inhibition. J. Biol. Chem. 276, 45636–45641. doi:10.1074/jbc.M105302200
Kesika, P., Suganthy, N., Sivamaruthi, B. S., and Chaiyasut, C. (2021). Role of gut-brain axis, gut microbial composition, and probiotic intervention in Alzheimer’s disease. Life Sci. 264, 118627. doi:10.1016/J.LFS.2020.118627
Kobayashi, T., Siegmund, B., Le Berre, C., Wei, S. C., Ferrante, M., Shen, B., et al. (2020). Ulcerative colitis. Nat. Rev. Dis. Prim. 6, 1. doi:10.1038/s41572-020-0205-x
Kummer, M. P., and Heneka, M. T. (2008). PPARs in Alzheimer’s disease. PPAR Res. 1, 403896. doi:10.1155/2008/403896
Kwiecien, S., Jasnos, K., Magierowski, M., Sliwowski, Z., Pajdo, R., Brzozowski, B., et al. (2014). Lipid peroxidation, reactive oxygen species and antioxidative factors in the pathogenesis of gastric mucosal lesions and mechanism of protection against oxidative stress - induced gastric injury. J. Physiol. Pharmacol. 65, 613–622.
Latella, G., Di Gregorio, J., Flati, V., Rieder, F., and Lawrance, I. C. (2014). Mechanisms of initiation and progression of intestinal fibrosis in IBD. Scand. J. Gastroenterol. 50, 53–65. doi:10.3109/00365521.2014.968863
Leal, M. C., Surace, E. I., Holgado, M. P., Ferrari, C. C., Tarelli, R., Pitossi, F., et al. (2012). Notch signaling proteins HES-1 and Hey-1 bind to insulin degrading enzyme (IDE) proximal promoter and repress its transcription and activity: Implications for cellular Aβ metabolism. Biochim. Biophys. Acta - Mol. Cell Res. 1823, 227–235. doi:10.1016/j.bbamcr.2011.09.014
Li, Y., Zhang, T., Guo, C., Geng, M., Gai, S., Qi, W., et al. (2020). Bacillus subtilis RZ001 improves intestinal integrity and alleviates colitis by inhibiting the Notch signalling pathway and activating ATOH-1. Pathog. Dis. 78, ftaa016. doi:10.1093/FEMSPD/FTAA016
Lin, J. C., Wu, J. Q., Wang, F., Tang, F. Y., Sun, J., Xu, B., et al. (2019). QingBai decoction regulates intestinal permeability of dextran sulphate sodium-induced colitis through the modulation of notch and NF-κB signalling. Cell Prolif. 52, e12547. doi:10.1111/CPR.12547
Lipworth, B. J. (1996). Clinical pharmacology of β3-adrenoceptors. Br. J. Clin. Pharmacol. 42, 291–300. doi:10.1046/j.1365-2125.1996.04222.x
Liu, B., Deng, C., and Tan, P. (2022). Ombuin ameliorates diabetic nephropathy in rats by anti-inflammation and antifibrosis involving Notch 1 and PPAR γ signaling pathways. Drug Dev. Res. 83 (6), 1270–1280. doi:10.1002/DDR.21956
Lu, C., Luo, Z. Y., Tai, Q. X., Wang, P., and Guan, P. P. (2018). Transferrin is responsible for mediating the effects of iron ions on the regulation of anterior pharynx-defective-1α/β and Presenilin 1 expression via PGE2 and PGD2 at the early stage of Alzheimer’s Disease. Aging (Albany. NY) 10, 3117–3135. doi:10.18632/aging.101615
Maesako, M., Horlacher, J., Zoltowska, K. M., Kastanenka, K. V., Kara, E., Svirsky, S., et al. (2017). Pathogenic PS1 phosphorylation at Ser367. Elife 6, e19720. doi:10.7554/ELIFE.19720
Maniati, E., Bossard, M., Cook, N., Candido, J. B., Emami-Shahri, N., Nedospasov, S. A., et al. (2011). Crosstalk between the canonical NF-κB and Notch signaling pathways inhibits Pparγ expression and promotes pancreatic cancer progression in mice. J. Clin. Invest. 121, 4685–4699. doi:10.1172/JCI45797
Mehta, S., Nijhuis, A., Kumagai, T., Lindsay, J., and Silver, A. (2015). Defects in the adherens junction complex (E-cadherin/ β-catenin) in inflammatory bowel disease. Cell Tissue Res. 360, 749–760. doi:10.1007/s00441-014-1994-6
Mo, W., Michel, M. C., Lee, X. W., Kaumann, A. J., and Molenaar, P. (2017). The β3-adrenoceptor agonist mirabegron increases human atrial force through β1-adrenoceptors: An indirect mechanism? Br. J. Pharmacol. 174, 2706–2715. doi:10.1111/bph.13897
Morel, J., and Singer, M. (2014). Statins, fibrates, thiazolidinediones and resveratrol as adjunctive therapies in sepsis: Could mitochondria be a common target? Intensive Care Med. Exp. 2, 9. doi:10.1186/2197-425x-2-9
Morry, J., Ngamcherdtrakul, W., and Yantasee, W. (2017). Oxidative stress in cancer and fibrosis: Opportunity for therapeutic intervention with antioxidant compounds, enzymes, and nanoparticles. Redox Biol. 11, 240–253. doi:10.1016/j.redox.2016.12.011
Nelson, W. J., and Nusse, R. (2004). Convergence of wnt, β-catenin, and cadherin pathways. Science. 303, 1483–1487. doi:10.1126/science.1094291
Noah, T. K., and Shroyer, N. F. (2013). Notch in the intestine: Regulation of homeostasis and pathogenesis. Annu. Rev. Physiol. 75, 263–288. doi:10.1146/annurev-physiol-030212-183741
Noori, T., Dehpour, A. R., Sureda, A., Fakhri, S., Sobarzo-Sanchez, E., Farzaei, M. H., et al. (2020). The role of glycogen synthase kinase 3 beta in multiple sclerosis. Biomed. Pharmacother. 132, 110874. doi:10.1016/j.biopha.2020.110874
Paouri, E., Tzara, O., Zenelak, S., and Georgopoulos, S. (2017). Genetic deletion of tumor necrosis factor-α attenuates amyloid-β production and decreases amyloid plaque formation and glial response in the 5xfad model of Alzheimer’s disease. J. Alzheimer’s Dis. 60, 165–181. doi:10.3233/JAD-170065
Puig, K. L., Lutz, B. M., Urquhart, S. A., Rebel, A. A., Zhou, X., Manocha, G. D., et al. (2015). Overexpression of mutant amyloid-β protein precursor and presenilin 1 modulates enteric nervous system. J. Alzheimers. Dis. 44, 1263–1278. doi:10.3233/JAD-142259
Quillard, T., and Charreau, B. (2013). Impact of Notch signaling on inflammatory responses in cardiovascular disorders. Int. J. Mol. Sci. 14, 6863–6888. doi:10.3390/ijms14046863
Ren, R. F., Lah, J. J., Diehlmann, A., Kim, E. S., Hawver, D. B., Levey, A. I., et al. (1999). Differential effects of transforming growth factor-βs and glial cell line-derived neurotrophic factor on gene expression of presenilin-1 in human post-mitotic neurons and astrocytes. Neuroscience 93, 1041–1049. doi:10.1016/S0306-4522(99)00215-8
Riley, S. A., Mani, V., Goodman, M. J., Herd, M. E., Dutt, S., and Turnberg, L. A. (1988). Comparison of delayed-release 5-aminosalicylic acid (mesalazine) and sulfasalazine as maintenance treatment for patients with ulcerative colitis. Gastroenterology 94, 1383–1389. doi:10.1016/0016-5085(88)90677-4
Roy, B. C., Ahmed, I., Stubbs, J., Zhang, J., Attard, T., Septer, S., et al. (2021). DCLK1 isoforms and aberrant Notch signaling in the regulation of human and murine colitis. Cell Death Discov. 71 (7), 1–14. doi:10.1038/s41420-021-00526-9
Saito, T., Yoshida, K., Matsumoto, K., Saeki, K., Tanaka, Y., Ong, S. M., et al. (2014). Inflammatory cytokines induce a reduction in E-cadherin expression and morphological changes in MDCK cells. Res. Vet. Sci. 96, 288–291. doi:10.1016/j.rvsc.2014.02.005
Salem, H. A., and Wadie, W. (2017). Effect of niacin on inflammation and angiogenesis in a murine model of ulcerative colitis. Sci. Rep. 7, 1–8. doi:10.1038/s41598-017-07280-y
Satoh, H., Sato, F., Takami, K., and Szabo, S. (1997). New ulcerative colitis model induced by sulfhydryl blockers in rats and the effects of antiinflammatory drugs on the colitis. Jpn. J. Pharmacol. 73, 299–309. doi:10.1254/jjp.73.299
Sawada, N., Nomiya, M., Hood, B., Koslov, D., Zarifpour, M., and Andersson, K. E. (2013). Protective effect of a β3-adrenoceptor agonist on bladder function in a rat model of chronic bladder ischemia. Eur. Urol. 64, 664–671. doi:10.1016/j.eururo.2013.06.043
Selkoe, D. J. (2001). Presenilin, notch, and the Genesis and treatment of Alzheimer’s disease. Proc. Natl. Acad. Sci. U. S. A. 98, 11039–11041. doi:10.1073/pnas.211352598
Shankle, W. R., Landing, B. H., Ang, S. M., Chui, H., Villarreal-Engelhardt, G., and Zarow, C. (1993). Studies of the enteric nervous system in alzheimer disease and other dementias of the elderly: Enteric neurons in alzheimer disease. Mod. Pathol. 6, 10–14.
Shi, S., Li, Y., Guo, Y., and Wang, Z. (2014). Effect of beta-3 adrenoceptor stimulation on the levels of ApoA-I, PPARα, and PPARγ in apolipoprotein E–deficient mice. J. Cardiovasc. Pharmacol. 64, 407–411. doi:10.1097/FJC.0000000000000133
Soriano, S., Kang, D. E., Fu, M., Pestell, R., Chevallier, N., Zheng, H., et al. (2001). Presenilin 1 negatively regulates β-catenin/T cell factor/lymphoid enhancer factor-1 signaling independently of β-amyloid precursor protein and notch processing. J. Cell Biol. 152, 785–794. doi:10.1083/jcb.152.4.785
Strober, W., and Fuss, I. J. (2011). Proinflammatory cytokines in the pathogenesis of inflammatory bowel diseases. Gastroenterology 140, 1756–1767. doi:10.1053/j.gastro.2011.02.016
Suryavanshi, S. V., and Kulkarni, Y. A. (2017). NF-Κβ: A potential target in the management of vascular complications of diabetes. Front. Pharmacol. 8, 798. doi:10.3389/fphar.2017.00798
Taddei, M. L., Chiarugi, P., Cirri, P., Buricchi, F., Fiaschi, T., Giannoni, E., et al. (2002). β-catenin interacts with low-molecular-weight protein tyrosine phosphatase leading to cadherin-mediated cell-cell adhesion increase. Cancer Res. 62, 6489–6499.
Tantray, M. A., Khan, I., Hamid, H., Alam, M. S., Dhulap, A., and Ganai, A. A. (2017). Oxazolo[4,5-b]pyridine-Based piperazinamides as GSK-3β inhibitors with potential for attenuating inflammation and suppression of pro-inflammatory mediators. Weinheim: Arch. Pharm., 350. doi:10.1002/ARDP.201700022
Tournissac, M., Vu, T. M., Vrabic, N., Hozer, C., Tremblay, C., Mélançon, K., et al. (2021). Repurposing beta3-adrenergic receptor agonists for Alzheimer’s disease: Beneficial effects on recognition memory and amyloid pathology in a mouse model. Alzheimers. Res. Ther. 13, 103. doi:10.1101/2020.05.25.11445410.1186/s13195-021-00842-3
Twomey, C., and McCarthy, J. V. (2006). Presenilin-1 is an unprimed glycogen synthase kinase-3β substrate. FEBS Lett. 580, 4015–4020. doi:10.1016/j.febslet.2006.06.035
Uddin, M. J., Jeong, S. O., Zheng, M., Chen, Y., Cho, G. J., Chung, H. T., et al. (2013). Carbon monoxide attenuates dextran sulfate sodium-induced colitis via inhibition of GSK-3 andbeta; Signaling. Oxid. Med. Cell. Longev. 2013, 210563. doi:10.1155/2013/210563
Vallée, A., Lecarpentier, Y., Guillevin, R., and Vallée, J. N. (2017). Interactions between TGF-β1, canonical WNT/β-catenin pathway and PPAR γ in radiation-induced fibrosis. Oncotarget 8, 90579–90604. doi:10.18632/oncotarget.21234
Vasina, V., Abu-Gharbieh, E., Barbara, G., De Giorgio, R., Colucci, R., Blandizzi, C., et al. (2008). The β3-adrenoceptor agonist SR58611A ameliorates experimental colitis in rats. Neurogastroenterol. Motil. 20, 1030–1041. doi:10.1111/j.1365-2982.2008.01138.x
Vetuschi, A., Pompili, S., Gaudio, E., Latella, G., and Sferra, R. (2018). PPAR-γ with its anti-inflammatory and anti-fibrotic action could be an effective therapeutic target in IBD. Eur. Rev. Med. Pharmacol. Sci. 22, 8839–8848. doi:10.26355/eurrev_201812_16652
Viladomiu, M., Hontecillas, R., Yuan, L., Lu, P., and Bassaganya-Riera, J. (2013). Nutritional protective mechanisms against gut inflammation. J. Nutr. Biochem. 24, 929–939. doi:10.1016/j.jnutbio.2013.01.006
Wang, D., Yuen, E. Y., Zhou, Y., Yan, Z., and Xiang, Y. K. (2011). Amyloid β peptide-(1 - 42) induces internalization and degradation of β 2 adrenergic receptors in prefrontal cortical neurons. J. Biol. Chem. 286, 31852–31863. doi:10.1074/jbc.M111.244335
Wang, Z., Li, S., Wang, R., Guo, L., Xu, D., Zhang, T., et al. (2020). The protective effects of the β3 adrenergic receptor agonist BRL37344 against liver steatosis and inflammation in a rat model of high-fat diet-induced nonalcoholic fatty liver disease (NAFLD). Mol. Med. 26, 54. doi:10.1186/S10020-020-00164-4
Wong, V. Y., Laping, N. J., Nelson, A. H., Contino, L. C., Olson, B. A., Gygielko, E., et al. (2001). Renoprotective effects of carvedilol in hypertensive-stroke prone rats may involve inhibition of TGFβ expression. Br. J. Pharmacol. 134, 977–984. doi:10.1038/sj.bjp.0704329
Wu, M. J., Kee, K. H., Na, J., Kim, S. W., Bae, Y., Shin, D. H., et al. (2015). Pituitary adenylate cyclase-activating polypeptide inhibits pacemaker activity of colonic interstitial cells of cajal. Korean J. Physiol. Pharmacol. 19, 435. doi:10.4196/KJPP.2015.19.5.435
Wynn, T. A. (2008). Cellular and molecular mechanisms of fibrosis. J. Pathol. 214, 199–210. doi:10.1002/path.2277
Xu, B., Li, Y. L., Xu, M., Yu, C. C., Lian, M. Q., Tang, Z. Y., et al. (2017). Geniposide ameliorates TNBS-induced experimental colitis in rats via reducing inflammatory cytokine release and restoring impaired intestinal barrier function. Acta Pharmacol. Sin. 38, 688. doi:10.1038/APS.2016.168
Yan, F., Zhang, Q. Y., Jiao, L., Han, T., Zhang, H., Qin, L. P., et al. (2009). Synergistic hepatoprotective effect of Schisandrae lignans with Astragalus polysaccharides on chronic liver injury in rats. Phytomedicine 16, 805–813. doi:10.1016/j.phymed.2009.02.004
Yi, W., Shen, R. W., Han, B., Li, Z., Le, X., Zhang, F. Y., et al. (2017). Notch signaling mediated by TGF-β/Smad pathway in concanavalin A-induced liver fibrosis in rats. World J. Gastroenterol. 23, 2330–2336. doi:10.3748/wjg.v23.i13.2330
Zhang, B., Wang, H. E., Bai, Y. M., Tsai, S. J., Su, T. P., Chen, T. J., et al. (2021). Inflammatory bowel disease is associated with higher dementia risk: A nationwide longitudinal study. Gut 70, 85–91. doi:10.1136/gutjnl-2020-320789
Zhang, T., Chen, D., and Lee, T. H. (2020). Phosphorylation signaling in APP processing in Alzheimer’s disease. Int. J. Mol. Sci. 21, 209. doi:10.3390/ijms21010209
Zhang, W., Xu, W., and Xiong, S. (2010). Blockade of Notch1 signaling alleviates murine lupus via blunting macrophage activation and M2b polarization. J. Immunol. 184, 6465–6478. doi:10.4049/jimmunol.0904016
Zhao, A., Bossone, C., Piñeiro-Carrero, V., and Shea-Donohue, T. (2001). Colitis-induced alterations in adrenergic control of circular smooth muscle in vitro in rats. J. Pharmacol. Exp. Ther. 299 (2), 768–774.
Keywords: mirabegron, carvedilol, presenilin, PPAR-γ, notch, β-Amyloid, smad-7, adherens junction
Citation: Nasser S, Abdallah DM, Ahmed KA, Abdel-Mottaleb Y and El-Abhar HS (2022) The novel anti-colitic effect of β-adrenergic receptors via modulation of PS1/BACE-1/Aβ axis and NOTCH signaling in an ulcerative colitis model. Front. Pharmacol. 13:1008085. doi: 10.3389/fphar.2022.1008085
Received: 31 July 2022; Accepted: 12 October 2022;
Published: 25 October 2022.
Edited by:
Somasundaram Arumugam, National Institute of Pharmaceutical Education and Research, Kolkata, IndiaReviewed by:
María José Peral, Sevilla University, SpainCopyright © 2022 Nasser, Abdallah, Ahmed, Abdel-Mottaleb and El-Abhar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dalaal M. Abdallah, ZGFsYWFsLmFiZGFsbGFoQHBoYXJtYS5jdS5lZHUuZWc=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.