 Chun Li1
Chun Li1 Yuan Jiang
Yuan Jiang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 21 December 2022
Sec. Pharmacogenetics and Pharmacogenomics
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.1007723
Triplex-forming oligonucleotides (TFOs) can bind to the major groove of double-stranded DNA with high specificity and affinity and inhibit gene expression. Triplex-forming oligonucleotides have gained prominence because of their potential applications in antigene therapy. In particular, the target specificity of triplex-forming oligonucleotides combined with their ability to suppress oncogene expression has driven their development as anti-cancer agents. So far, triplex-forming oligonucleotides have not been used for clinical treatment and seem to be gradually snubbed in recent years. But triplex-forming oligonucleotides still represent an approach to down-regulate the expression of the target gene and a carrier of active substances. Therefore, in the present review, we will introduce the characteristics of triplex-forming oligonucleotides and their anti-cancer research progress. Then, we will discuss the challenges in their application.
Numerous gene diseases are related to genome mutations, so the therapeutic strategies that enable targeted editing in genetic disease treatment have attracted more attention (Piotrowski-Daspit et al., 2018). Triplex-forming oligonucleotides (TFOs) can be composed of DNA, RNA, or synthetic base analogs linked by phosphodiester backbones (Chin and Glazer, 2009). TFOs can bind to the major groove of homopurine-homopyrimidine stretches of double-stranded DNA in a sequence-specific manner through Hoogsteen hydrogen bonding to form DNA triplexes (Moser and Dervan, 1987; Häner and Dervan, 1990; Reza and Glazer, 2014). Based on this property, TFOs can be used as tools to study gene regulation, DNA reparation, recombination, and mutagenesis (Giovannangeli and Hélène, 1997). In addition, TFOs can target specific genes to form the DNA triplex structures, so they have been investigated in the therapeutic strategies for gene diseases. Studies have predicted that almost 98% of annotated human genes have at least one TFOs target sequence (TTS) within the promoter/transcribed region, and 87% of TTSs are unique to one gene (Reza and Glazer, 2014; Lauria et al., 2020). A total of 519,971 TTSs can serve as potential target sites (Kaushik Tiwari et al., 2022). The high frequency of TTSs in the human genome provides targets for the design of TFOs as a therapeutic targeting approach in gene diseases. At least in principle, TFOs can serve as gene-targeting agents for the transient control of gene activity and alter gene expression through directed mutagenesis (Moasser, 2007). In fact, besides promoter/transcribed region, triplex sites can be located downstream of the transcription start site, including exons and introns, indicating an increased number of potential target genes for triplex-based strategies (Barre et al., 2000).
RNA interference (RNAi) is an effective mRNA-level intervention technology, but it does not change the genetic code, and gene silencing is transient. In comparison, the advantage of TFOs lies in their ability to target and permanently modify disease-specific genes (Zuin Fantoni et al., 2020). Paquet and colleagues suggested that the quantity of TFOs demanded to obtain a significant inhibition was very lower than that in siRNA, and the inhibition effects of the target gene induced by TFOs can last longer (Paquet et al., 2011). Strobel and colleagues chose the entire candidate region for the Huntington’s disease gene at the tip of human chromosome 4 as a target for testing the recognition and enzymatic cleavage effect of a 16-base TFO, then results showed that sequence-specific TFO recognition of the motif within the human genome, and may facilitate the regulation of human genetics (Strobel et al., 1991). So far, the study and application of TFOs are related to the treatment of Duchenne muscular dystrophy, beta-thalassemia mutation, arthritis, HIV infection, and cancer (Reza and Glazer, 2014). In particular, the target specificity of TFOs combined with their ability to suppress oncogene expression has driven their development as anti-cancer agents (Ricciardi et al., 2014; Pyne et al., 2021). Therefore, in the present review, we will introduce the characteristics of TFOs and their anti-cancer research progress. Then, we will discuss the challenges of TFOs in their application.
TFOs typically are 12–28 long oligonucleotides that bind to specific regions in duplex DNA as a third strand to form a triple helix formation (Beal and Dervan, 1991). The hydrogen bonds involved in triple-helix formation are referred to as Hoogsteen hydrogen bonds and are different from the hydrogen-bonding pattern of the Watson-Crick base pairs (Häner and Dervan, 1990). The Hoogsteen hydrogen bonds can afford the specificity and affinity of the TFOs for their target DNA duplex (Duca et al., 2008). These Hoogsteen bonds are generally weaker than Watson-Crick base pairing but can be stabilized in the presence of divalent cations (e.g., Mg2+, Ca2+, and Zn2+) to enhance triplex stability (Chin et al., 2007; Chin and Glazer, 2009). Moreover, the third-strand binding is kinetically slow compared to Watson-Crick base pairing, but triplexes are stable once formed, exhibiting half-lives on the order of days (Chin et al., 2007; Jain et al., 2008). The formation of triplexes depends on several factors, such as length, divalent cations, temperature, base composition, and the sequence context of the TFO-binding site. In particular, TFO sequence specificity relies on binding to oligopurine-oligopyrimidine target sequences. Two TFO motifs are possible: pyrimidine-rich TFOs bind in a parallel manner, while purine-rich TFOs bind in an anti-parallel orientation. The pyrimidine-rich TFOs consisting primarily of thymines (T) or protonated cytosines (C+) bind to DNA with AT and GC base pairs to form the canonical base triads T:AT and C+:GC with duplex DNA (Beal and Dervan, 1991; Hennessy et al., 2022). Typically, pyrimidine-rich TFOs can only form the triple-helix formation under acidic conditions. It's due to the N3-protonated cytosines and thymines of TFOs can recognize guanine and adenine to form Hoogsteen-type hydrogen bonds, respectively (Lee et al., 1979; Seio et al., 2020). The purine-rich TFOs consisting primarily of guanines (G) or adenines (A) bind to DNA with AT and GC base pairs in an anti-parallel direction via reverse Hoogsteen bonds to form the base triads G:GC and A:AT with duplex DNA (Praseuth et al., 1999; Mukherjee and Vasquez, 2011). Compared to the pyrimidine-rich TFOs, the purine-rich TFOs bind to DNA at neutral pH and require no protonation. However, the guanines in purine-rich TFOs contribute to forming G-quartets in physiologic concentrations of K+, and G-quartets presumably reduce the extent of bioavailable TFO. The schematic representation from Figure 1 illustrates the triplex binding code in the purine-rich and pyrimidine-rich TFO motifs. Generally, the oligopurine-oligopyrimidine target sequences are usually located in gene promoter regions, so the TFOs directed against these regulatory sites can selectively reduce transcription of the targeted genes.
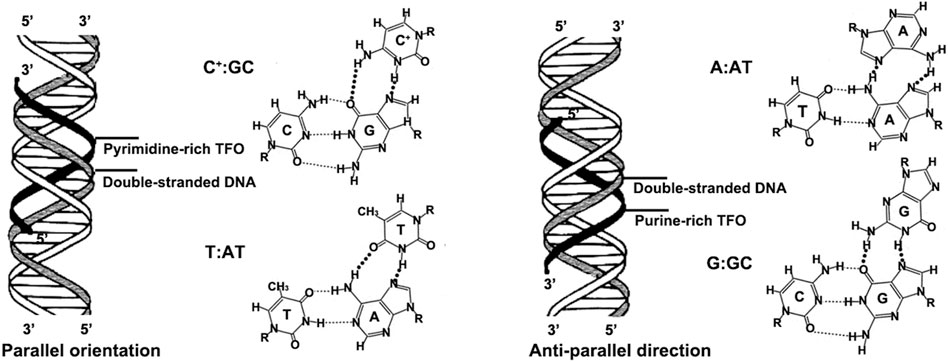
FIGURE 1. Triplex binding code in the pyrimidine-rich and purine-rich TFO motifs.
TFOs were designed primarily for binding the oncogenes and suppressing transcription to reduce cancer cell growth in anti-cancer research. For example, c-myc and bcl-2 are important therapeutic targets. In addition, some of the genes involved in epithelial-mesenchymal transition (EMT) or multidrug resistance (MDR) also can be the target gene.
The c-myc is a small family of proto-oncogenes and is located on human chromosome 8q24.12-q24.13 (Mai and Mushinski, 2003; Mastronikolis et al., 2019). c-myc acts as an essential transcription factor that regulates the expression of many genes involved in cell growth, proliferation, and apoptosis pathways (Liao and Dickson, 2000). c-myc promotes tumor transformation by the induction of genomic instability in critical genes, and amplification and (or) overexpression of the c-myc gene are associated with poor prognosis or decreased survival of patients with cancer (Garte, 1993; Wade and Wahl, 2006). Therefore, seeking therapies targeting the c-myc gene will be key to reversing cancerous growth in patients with cancer. Transcription of the human c-myc initiates from two major start sites designated P1 and P2 (Kim et al., 1998). P2 promoter is the major promoter for c-myc transcription and generates more than 75% of c-myc mRNA (Kim and Miller, 1995). Meanwhile, multiple protein-binding sites are within the P2 promoter. P2 promoter contains a 23 base pair purine-pyrimidine-rich motif (−62 to −40) and is a potential TTS for purine-purine-pyrimidine triplex formation. Kim and colleagues designed a purine-rich TFO targeted to the region of the c-myc P2 promoter and determined the TFO was oriented antiparallel to TTS in a sequence-specific manner and inhibits nuclear protein binding and in vitro transcription of the c-myc gene (Kim and Miller, 1995). A previous study first demonstrated that a TFO targeted to a nuclease-sensitive region upstream of the P1 promoter can inhibit c-myc gene transcription in a cell-free system (Cooney et al., 1988). Therefore, Kim and colleagues suggested that a TFO targeted to either P1 or P2 promoter is unlikely to inhibit c-myc expression completely. In the subsequent study, they used both P1- and P2-targeted TFOs to inhibit c-myc expression in the HeLa human cervical carcinoma cell line at the same time. To investigate the effects of phosphorothioate TFOs on c-myc transcription in vivo, they employed a luciferase reporter plasmid containing the cDNA encoding a firefly luciferase gene under the control of an 862-bp human c-myc promoter. After HeLa cells were cotransfected with both P1- and P2-targeted TFOs, the luciferase activity was inhibited by 90%. They suggested that TFOs may represent a gene-specific means of inhibiting c-myc gene expression (Kim et al., 1998). Some scholars found that P2-targeted TFOs can enhance the anti-cancer efficacy of gemcitabine (GEM) via TFO-targeted DNA damage-induced unscheduled DNA repair synthesis (UDS) in breast cancer cells (Christensen et al., 2006). They also exhibited superior antitumor activity in a mouse model of human colon cancer after being combined with GEM, suggesting that combining gene-specific TFOs with DNA-targeting drugs represents a more effective treatment strategy for solid tumors in humans (Boulware et al., 2014). Interestingly, certain anti-cancer drugs can be used as triplex stabilizing to improve the stability and biological activity of the P2-targeted TFOs, such as daunomycin (DNM). The attachment of DNM can increase the triplex stability and biological activity of the TFOs without compromising their specificity to c-myc gene. The DNM-conjugated TFOs can inhibit the c-myc gene transcription in vitro and reduce c-myc promoter activity in prostate and breast cancer cells while less effective against normal cells (Carbone et al., 2004; Napoli et al., 2006). In recent years, the booming field of nanotechnology provides a more effective strategy for TFOs delivery. Huo and colleagues first used ultrasmall 2 nm tiopronin-covered gold nanoparticles (Au-TIOP NPs) as carriers for P2-targeted TFOs delivery. 5′ amino-modified P2-targeted TFOs were linked to 2 nm Au-TIOP NPs through the carboxyl group of tiopronin. Based on the Au-TIOP NPs smaller than 10 nm that could enter the nucleus, the nanoparticle-conjugated TFOs were more effective at reducing c-myc RNA and c-myc protein in breast cancer cells than free TFOs (Huo et al., 2014). In the subsequent study, Huo and colleagues designed another single-stranded sequence to complementarily hybridize with the tail part of the nanoparticle-conjugated TFOs to block the ability of the TFOs to bind with the c-myc gene and then self-assemble into large-sized sunflower-like structures. The sunflower-like structures possess strong near-infrared (NIR) absorption and photothermal conversion ability. After breast cancer cells have taken up the sunflower-like nanostructures, they stand by in the cell cytoplasm until NIR irradiation. Upon NIR irradiation, the sunflower-like nanostructures could disassemble at the melting point (Tm) and release Au-TIOP NPs to enter the nucleus to help TFOs to achieve the c-myc oncogene silencing. This study provides a new approach for controlling c-myc gene silencing in time and space (Huo et al., 2019).
The B cell lymphoma-2 (bcl-2) is one of the most important endogenous anti-apoptotic factors and is located on human chromosome 18q21 (Tomita, 2011). Bcl-2 overexpression will increase the mutation opportunities of other genes, resulting in the deterioration of normal cells into tumor cells. Meanwhile, the Bcl-2 gene increases the number of tumor cells and leads to the further development and proliferation of tumors by inhibiting apoptosis (Goldar et al., 2015). Shen and colleagues found two potential TTSs can bind the corresponding TFOs with high specificity and affinity downstream in the bcl-2 promoter region: from −666 to −647 and from −1113 to −1094 relative to the translation start site. Then they designed the corresponding purine-rich TFOs to inhibit the bcl-2 gene transcription in HeLa human cervical carcinoma cell line. In addition, they attempted to label the conversion-electron-emitter (99mtechnetium) to the 3′ end of TFOs via histidine to induce DNA double-strand breaks (DSB) when brought close to the DNA strand by TFOs. The formed 99mtechnetium-coupled TFOs also bind to the TTS to form stable triplexes and inhibit bcl-2 gene transcription in vitro, indicating that TFOs are a promising carrier for gene-radiotherapy and may provide an additional therapeutic method for cancers with bcl-2 overexpression (Shen et al., 2003). In addition to the major transcriptional promoter of bcl-2 gene, Shen and colleagues found that the 3′-untranslated region (3′UTR) of the human bcl-2 gene contains a potential TTS located +1946 to +1963 from the translation start site. 3′UTR also plays an important role in the regulation of gene expression. They demonstrated that the TFOs targeted to the 3′UTR of the bcl-2 gene can inhibit the gene expression in HeLa cells. The mechanism is to form a triple helix in 3′UTR to inhibit transcription elongation and affect the progression of RNA polymerase in its synthesis of the mRNA resulting in mRNA lacking a polyA tail is very unstable and rapidly degraded in Hela cells. They also found that TFO with an amino linkage at the 3′-end alone was particularly resistant to nuclease degradation, which may contribute to maintaining the effect of TFO on bcl-2 expression last for 72 h after transfection (Shen et al., 2001). Taniguchi and Sasaki labeled the non-natural nucleoside analogue, the W-shaped nucleoside analogue (WNA-βT), at the 3′-end of the bcl-2-targeted TFOs to increase the triplex stability. The TFOs with WNA- βT exhibited a higher antiproliferative effect than the corresponding natural TFOs in human A549 lung cancer cells. They suggested that the TFOs incorporating artificial nucleoside analogues may be powerful antigene agents for inhibition of the bcl-2 gene expression (Taniguchi and Sasaki, 2012).
The human EGF-like receptor 2 (HER2) is functionally implicated in the pathogenesis of human breast cancer and is an important prognostic and predictive marker in breast cancer (Moasser, 2007; Tapia et al., 2007). HER2 is located on human chromosome 17q12-21.3, encoding a transmembrane growth factor receptor with tyrosine kinase activity. The receptor protein interacts with HER family members and their ligands to regulate cell growth, differentiation, and proliferation through signal transduction (van de Vijver, 2001). HER2 overexpression promotes tumorigenesis that making HER2 is suitability as a drug target in cancer drug development. Kaushik and colleagues reported a TFO-based therapeutic strategy for targeting HER2 gene amplifications (Kaushik Tiwari et al., 2022). They designed two purine-rich TFOs (HER2-1 and HER2-205) to target the polypurine sequence in the promoter region of the HER2 gene at positions −218 to −245 relative to the transcription start site and the coding region beginning at position 205, respectively. When TFOs bind to the target polypurine sequence within the major groove of the double helix in an anti-parallel direction to form the DNA triplex structures will induce DNA perturbation that can impede replication fork progression, resulting in fork collapse and double-strand break (DSB) formation. The formation of DNA triplex structures makes sufficient DNA damage to activate apoptosis in breast cancer. Compared with the control group, TFOs (HER2-205) treatment of HER2-positive breast cancer xenografts resulted in a 52% reduction in tumor volume, equivalent to the 58% reduction observed with trastuzumab. It indicates that activating the DNA damage response with TFOs treatment is as effective as the administration of trastuzumab. They also tried to use biodegradable nanoparticles (NPs) to serve as a delivery platform. For example, PLA-HPG was screened from a copolymer of poly (lactic acid) (PLA) and hyperbranched polyglycerol (HPG). The TFOs encapsulated PLA-HPG NP delivery system can significantly improve the efficacy of TFO treatment. These findings offer a feasible therapeutic strategy for targeting tumors with HER-positive.
E26 oncogene homolog 2 (Ets-2) is a member of the ETS transcription factor family and is located on human chromosome 21q22. The abnormal expression of Ets2 exists in various cancer types, including prostate, breast, and thyroid cancers. It may contribute to the occurrence and progression of tumors by activating the expression of multiple genes that promote cell proliferation and invasion or prevent apoptotic cell death. Carbone and colleagues found a potential TTS located −40 to −65 relative to the transcription start site in the Ets2 promoter, which contains a region that can be bound to transcription factors of the Sp family. Sp1 sites may be critical for promoter activity of the Ets2 gene. They designed a pyrimidine-rich Ets2-targeting TFO, which has high affinity and specificity for the TTS, for inhibiting the bind of Sp1/Sp3 transcription factors and Sp1 site in vitro. They found that Ets2-targeting TFO can act as a selective transcriptional repressor of Ets2 transcription in human prostate cancer cells, suggesting that the triplex formation was the functional mechanism of the anti-transcriptional activity of the Ets2-TFO (Carbone et al., 2003). The pre-initiation complex (PIC) is formed by the assembles of RNA polymerase II (Pol II) and general transcription factors, like the TFIID complex, which contains the TATA-binding protein (TBP) and TBP-associated factors (TAFs), and is essential for the initiation of transcription. Carbone and colleagues suggested that Sp1 can interact with the TAFs proteins, such as TAFII130 and TAFII250, and then promote their assembly into PIC as a tethering moiety. Therefore, they further demonstrated that Ets2-targeting TFOs could prevent the binding of Sp1, TAFII130, and TAFII250 to the Ets2 promoter and interfere with the assembly of PIC. Those results further explain the potential mechanism of triplex DNA-mediated transcriptional repression in cancers with Ets-2 overexpression (Jain et al., 2009).
Some of the genes involved in EMT or MDR have also drawn the concerns of many researchers. EMT is a process that epithelial cells lose the apical-basal polarity and cell-cell adhesion and then transit to invasive mesenchymal cells. It is well-known that EMT plays an important role in cancer progression, metastasis, and even drug resistance (Du and Shim, 2016). The c-met gene encodes a receptor tyrosine kinase receptor (RTK), also commonly referred to as mesenchymal-epithelial transition factor (MET), and its ligand is the hepatocyte growth factor (HGF) (Faiella et al., 2022). The binding of MET to HGF contributes to the regulation of EMT. Singhal and colleagues designed a purine-rich TFO to bind the TTS located −144 to −119 relative to the transcription start site in the c-met promoter. The TTS contains the binding site of the transcription factor Sp1, and any mutation in this Sp1 binding site will inhibit the c-met transcription. They found that the c-met targeted TFOs lead to cell death and tumor regression in hepatoma, representing a promising tool for inhibiting cancer cell proliferation (Singhal et al., 2011). P-glycoprotein (P-gp/MDR1) is a multidrug efflux transporter that protects cells by exporting xenobiotics out from the plasma membrane to the extracellular space. The overexpression of P-gp as a cause of multidrug resistance is a key player in the multidrug-resistant phenotype in cancer (Callaghan et al., 2014; Pokharel et al., 2017). Based on the MDR1 gene (ABCB1) encoding P-gp, Stierlé and colleagues synthesized a DNM-conjugated TFO to pass the cell membrane, remain in DNM-resistant cell lines, and then inhibit the expression of the MDR1 gene and P-gp (Stierlé et al., 2008).
The experiments described in the previous section have shown that TFOs can be used as antigene agents to control the activity of the tumor-associated gene. But many challenges remain, particularly efficient uptake of TFOs into the nucleus, stability once inside the cell, and specific, high-affinity binding to TTS in a cellular environment.
Pyrimidine-rich TFOs require a protonated cytosine to form triplexes under acidic conditions, while guanine-rich TFOs tend to form G-quartets under physiological K+ concentrations, which has restricted their use as gene-targeting agents. Moreover, a quick degradation of TFOs also is a big drawback. Therefore, modifications are necessary for the TFOs design. Rusling introduced the nucleobase 6-amino-5-nitropyridin-2-one (Z) to take the place of cytosine in pyrimidine-rich TFOs to overcome the pH dependence of triplex formation (Rusling, 2021). In guanine-rich TFOs, the presence of N, N-diethylethylene-diamine-modified bases may also reduce G-quartet formation (Reza and Glazer, 2014). Torigoe and colleagues reported 2′-O,4′-C-aminomethylene-bridged nucleic acid (2′,4′-BNA(NC)) modification of TFOs significantly increased the nuclease resistance of TFOs and existing good stability in human serum (Torigoe et al., 2011; Torigoe et al., 2012). The TFO backbone can also be modified to improve intracellular resistance to nucleases, such as N,N-diethylethylenediamine (DEED), phosphoramidate, phosphorothioate, methylphosphonate or a peptide-like backbone to form so-called peptide-nucleic acids (PNAs) (Chin and Glazer, 2009; Reza and Glazer, 2014; Chandrasekaran and Rusling, 2018). In addition, the low stability of the triplex and limitations of the target DNA sequence are the major drawback in triplex formation by a natural TFO (Obika, 2004). TFOs inhibit transcription initiation through competition with transcription binding to DNA, but unmodified oligonucleotides may not form stable enough triplex complexes with their DNA target for the arrest of transcription elongation. Mayer and colleagues suggested that introducing the pyrrolidino isocytidine pseudonucleoside and the corresponding phosphoramidite building block into TFOs enhanced the DNA affinity due to the positive charge at the pyrrolidino subunit (Mayer et al., 2005). Faria and colleagues reportped that covalent attachment of an intercalating acridine derivative at the 5′ end of the TFOs inhibited transcriptional elongation in cells, indicating that improvement of triplex stability by modifications increases the intracellular efficacy of triplex-induced effects (Faria et al., 2000). The above modifications are smart and appropriate, but the influence on the biophysical characteristics and activity of TFO in vitro and in vivo needs further research.
TFOs must be effectively delivered to and taken up by cancer cells to significantly regulate the target oncogene phenotype and reduce off-target and toxicity effects. Given the characteristics of cell membrane potential, the addition of positively charged lysine residues can enhance the cellular uptake of TFOs (Ricciardi et al., 2014). Previous studies have reported some strategies for the delivery of TFOs into cells, including nucleofection, cell-penetrating peptides (CPPs), and nanoparticles (Ricciardi et al., 2014). The advantage of nucleofection is the commercially available kits are readily available, but can only be used in vitro and may lead to high levels of cell death compared to nanoparticle delivery or other methods. Conjugation of TFOs to CPPs or delivery in NPs contributes to effective transfection with lower cell death and direct in vivo delivery, representing a feasible delivery strategy (Ricciardi et al., 2014). But, until now, the Efficient delivery of TFOs into cells remains a challenge.
Once delivered into the cytoplasm, TFOs can be affected by cellular conditions, such as potassium and magnesium concentrations. Within the nucleus, the charge repulsion between the three polyanionic strands and accessibility of binding sites in a chromosomal context may affect the stable triplex formation (Reza and Glazer, 2014). Moreover, the stable triplex formation also requires contiguous homopurine bases to avoid pyrimidine interruptions within the purine stretch can inhibit triplex formation. Of course, modification can enhance the stability of TFOs but may affect the biological activity of TFOs. For instance, extensive sugar modification can decrease the biological activity of TFOs in vivo (Mukherjee and Vasquez., 2011).
Since short oligonucleotides were found that can bind in the major groove of the DNA duplex to form a triple helix formation and play a role in the control of gene expression, many scholars were devoted to exploring the mechanism of TFOs forming triple helix formation and the specific applications of TFOs as an anti-gene strategy during the past decades, especially the identification and manipulation of oncogenes that play a key role in the progression and maintenance of cancers. The application potential of TFOs in therapeutical and biotechnological aspects has been widely investigated. But up to now, they have not been utilized therapeutically. On the one hand, the inherent restrictions of TFOs still exist. Modifications are necessary but can’t cover everything. For instance, modifications can enhance the stability of TFOs but may affect biophysical characteristics and activity. Modification methods may also be complex, and economic costs need to be taken into account. On the other hand, several other gene-targeting methodologies designed to edit DNA are flourishing and aroused more research interest from scholars in recent years, such as zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and CRISPR/Cas9 platform (Chandrasekaran and Rusling, 2018). In short, TFO-related research seems to be gradually snubbed.
Important to note is that some bioactive substances, such as Auger-electron-emitting radionuclide 125I, pyrene, and psoralen, can be conjugated or labeled with TFOs to target specific sites in the genome based on the sequence-specific action of TFOs (Panyutin and Neumann, 1994; Sedelnikova et al., 2000; Li et al., 2007; Benfield et al., 2008; Majumdar et al., 2008; Dahmen et al., 2016). It indicates that TFOs can act as a carrier of active substances. In particular, the conjugation of TFOs to psoralen provides a powerful approach to modifying DNA in vitro and in cells (Mukherjee and Vasquez, 2011). Psoralen is a planar triheterocyclic compound containing furan and pyrone rings and can intercalate in DNA, and following exposure to ultraviolet (UVA) irradiation can form interstrand cross-links (ICLs) in double-stranded DNA (Vasquez, 2010). The psoralen-labeled TFOs (pso-TFOs) can be used to induce site-specific DNA damage by directly inactivating the interest gene under the control of UVA irradiation at 365 nm. Thus, pso-TFOs have the potential to irreversibly modify target genes (Barre et al., 2000; Diviacco et al., 2001). The use of pso-TFOs may be limited by UVA does not penetrate internal organs and psoralen potentiates carcinogenesis (Sargent et al., 2011), but it indicates that the covalently linking photoactivatable derivatives to TFOs contribute to achieving the timing of gene damage can be controlled by light. The photoactivation of photoactivatable derivatives-labeled TFOs could have several advantages. First, genetic manipulation could be controlled at the target location and specific time; Second, light irradiation is a clean and safe intervention; Third, the photoinduced targeted gene damage would be highly specific with insignificant toxic side effects (Kolevzon and Yavin, 2010). Photosensitizers (PS) with strong photosensitivity and low toxin may be the potential candidates to replace psoralen for conjugate with TFOs. PS labeled on TFOs may improve the stability of TFOs and achieve the photoinduced targeted gene damage by releasing reactive oxygen species (ROS). Interestingly, some PS also are sonosensitizer (e.g., hematoporphyrin, photofrin, and chlorin e6), which can be activated to generate ROS under ultrasound (Kuroki et al., 2007; Hong et al., 2020; Mai et al., 2020; Zhang et al., 2020). PS-labeled TFOs can be activated by visible light or ultrasound, that may provide advanced technology for clinical applications.
Moreover, the continuous research and innovation of drug delivery systems will provide an appropriate vehicle for the delivery of TFOs, such as nanoparticles, exosomes, and metal-organic frameworks (MOFs), which could be used as an ideal delivery system and would allow systemic injections of TFOs. Therefore, TFOs as an anti-gene technique for cancer therapy still have great attraction and application prospects and need further research and exploration.
CL, HZ, and YJ had the idea for the review. CL, ZZ, and CR wrote the first draft of the manuscript. QW, FP, and YD wrote sections of the manuscript. HZ and YJ proofread the manuscript.
This work was supported by the grants from Research project of the Science and Technology Department of Sichuan province (2021YJ0217), Research Project of Sichuan Applied Psychology Research Center of Chengdu Medical College (CSXL-202A13), Research project of Health Commission of Sichuan Province (19PJ189) and Special Nursing Project of Special Scientific Research Fund of First Affiliated Hospital of Chengdu Medical College (CYFY2020HL03).
The authors also express their sincere thanks to Fengbo Wang, Ke Wang, Xiaofei Wei, Xiaogang Shen, Yu Li, and Yamin Yue for their helpful assistance.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Barre, F. X., Ait-Si-Ali, S., Giovannangeli, C., Luis, R., Robin, P., Pritchard, L. L., et al. (2000). Unambiguous demonstration of triple-helix-directed gene modification. Proc. Natl. Acad. Sci. U. S. A. 97, 3084–3088. doi:10.1073/pnas.050368997
Beal, P. A., and Dervan, P. B. (1991). Second structural motif for recognition of DNA by oligonucleotide-directed triple-helix formation. Science 251, 1360–1363. doi:10.1126/science.2003222
Benfield, A. P., Macleod, M, C., Liu, Y., Wu, Q., Wensel, T. G., and Vasquez, K. M. (2008). Targeted generation of DNA strand breaks using pyrene-conjugated triplex-forming oligonucleotides. Biochemistry 47, 6279–6288. doi:10.1021/bi7024029
Boulware, S. B., Christensen, L. A., Thames, H., Coghlan, L., Vasquez, K. M., and Finch, R. A. (2014). Triplex-forming oligonucleotides targeting c-MYC potentiate the anti-tumor activity of gemcitabine in a mouse model of human cancer. Mol. Carcinog. 53, 744–752. doi:10.1002/mc.22026
Callaghan, R., Luk, F., and Bebawy, M. (2014). Inhibition of the multidrug resistance P-glycoprotein: Time for a change of strategy? Drug Metab. Dispos. 42, 623–631. doi:10.1124/dmd.113.056176
Carbone, G. M., McGuffie, E. M., Collier, A., and Catapano, C. V. (2003). Selective inhibition of transcription of the Ets2 gene in prostate cancer cells by a triplex-forming oligonucleotide. Nucleic Acids Res. 31, 833–843. doi:10.1093/nar/gkg198
Carbone, G. M., McGuffie, E., Napoli, S., Flanagan, C. E., Dembech, C., Negri, U., et al. (2004). DNA binding and antigene activity of a daunomycin-conjugated triplex-forming oligonucleotide targeting the P2 promoter of the human c-myc gene. Nucleic Acids Res. 32, 2396–2410. doi:10.1093/nar/gkh527
Chandrasekaran, A. R., and Rusling, D. A. (2018). Triplex-forming oligonucleotides: A third strand for DNA nanotechnology. Nucleic Acids Res. 46, 1021–1037. doi:10.1093/nar/gkx1230
Chin, J. Y., and Glazer, P. M. (2009). Repair of DNA lesions associated with triplex-forming oligonucleotides. Mol. Carcinog. 48, 389–399. doi:10.1002/mc.20501
Chin, J. Y., Schleifman, E. B., and Glazer, P. M. (2007). Repair and recombination induced by triple helix DNA. Front. Biosci. 12, 4288–4297. doi:10.2741/2388
Christensen, L. A., Finch, R. A., Booker, A. J., and Vasquez, K. M. (2006). Targeting oncogenes to improve breast cancer chemotherapy. Cancer Res. 66, 4089–4094. doi:10.1158/0008-5472.CAN-05-4288
Cooney, M., Czernuszewicz, G., Postel, E. H., Flint, S. J., and Hogan, M. E. (1988). Site-specific oligonucleotide binding represses transcription of the human c-myc gene in vitro. Science 241, 456–459. doi:10.1126/science.3293213
Dahmen, V., Pomplun, E., and Kriehuber, R. (2016). Iodine-125-labeled DNA-Triplex-forming oligonucleotides reveal increased cyto- and genotoxic effectiveness compared to Phosphorus-32. Int. J. Radiat. Biol. 92, 679–685. doi:10.3109/09553002.2016.1160157
Diviacco, S., Rapozzi, V., Xodo, L., Helene, C., Quadrifoglio, F., and Giovannangeli, C. (2001). Site-directed inhibition of DNA replication by triple helix formation. FASEB J. 15, 2660–2668. doi:10.1096/fj.01-0440com
Du, B., and Shim, J. S. (2016). Targeting epithelial-mesenchymal transition (EMT) to overcome drug resistance in cancer. Molecules 21, 965. doi:10.3390/molecules21070965
Duca, M., Vekhoff, P., Oussedik, K., Halby, L., and Arimondo, P. B. (2008). The triple helix: 50 years later, the outcome. Nucleic Acids Res. 36, 5123–5138. doi:10.1093/nar/gkn493
Faiella, A., Riccardi, F., Cartenì, G., Chiurazzi, M., and Onofrio, L. (2022). The emerging role of c-met in carcinogenesis and clinical implications as a possible therapeutic target. J. Oncol. 2022, 5179182. doi:10.1155/2022/5179182
Faria, M., Wood, C. D., Perrouault, L., Nelson, J. S., Winter, A., White, M. R., et al. (2000). Targeted inhibition of transcription elongation in cells mediated by triplex-forming oligonucleotides. Proc. Natl. Acad. Sci. U. S. A. 97, 3862–3867. doi:10.1073/pnas.97.8.3862
Giovannangeli, C., and Hélène, C. (1997). Progress in developments of triplex-based strategies. Antisense Nucleic Acid. Drug Dev. 7, 413–421. doi:10.1089/oli.1.1997.7.413
Goldar, S., Khaniani, M. S., Derakhshan, S. M., and Baradaran, B. (2015). Molecular mechanisms of apoptosis and roles in cancer development and treatment. Asian Pac J. Cancer Prev. 16, 2129–2144. doi:10.7314/apjcp.2015.16.6.2129
Häner, R., and Dervan, P. B. (1990). Single-strand DNA triple-helix formation. Biochemistry 29, 9761–9765. doi:10.1021/bi00494a001
Hennessy, J., McGorman, B., Molphy, Z., Farrell, N. P., Singleton, D., Brown, T., et al. (2022). A click chemistry approach to targeted DNA crosslinking with cis-platinum(II)-Modified triplex-forming oligonucleotides. Angew. Chem. Int. Ed. Engl. 61, e202110455. doi:10.1002/anie.202110455
Hong, L., Pliss, A. M., Zhan, Y., Zheng, W., Xia, J., Liu, L., et al. (2020). Perfluoropolyether Nanoemulsion Encapsulating Chlorin e6 for Sonodynamic and Photodynamic Therapy of Hypoxic Tumor. Nanomater. (Basel) 10, 2058. doi:10.3390/nano10102058
Huo, S., Gong, N., Jiang, Y., Chen, F., Guo, H., Gan, Y., et al. (2019). Gold-DNA nanosunflowers for efficient gene silencing with controllable transformation. Sci. Adv. 5, eaaw6264. doi:10.1126/sciadv.aaw6264
Huo, S., Jin, S., Ma, X., Xue, X., Yang, K., Kumar, A., et al. (2014). Ultrasmall gold nanoparticles as carriers for nucleus-based gene therapy due to size-dependent nuclear entry. ACS Nano 8, 5852–5862. doi:10.1021/nn5008572
Jain, A., Magistri, M., Napoli, S., Carbone, G. M., and Catapano, C. V. (2009). Mechanisms of triplex DNA-mediated inhibition of transcription initiation in cells. Biochimie 92, 317–320. doi:10.1016/j.biochi.2009.12.012
Jain, A., Wang, G., and Vasquez, K. M. (2008). DNA triple helices: Biological consequences and therapeutic potential. Biochimie 90, 1117–1130. doi:10.1016/j.biochi.2008.02.011
Kaushik Tiwari, M., Colon-Rios, D. A., Tumu, H. C. R., Liu, Y., Quijano, E., Krysztofiak, A., et al. (2022). Direct targeting of amplified gene loci for proapoptotic anticancer therapy. Nat. Biotechnol. 40, 325–334. doi:10.1038/s41587-021-01057-5
Kim, H. G., and Miller, D. M. (1995). Inhibition of in vitro transcription by a triplex-forming oligonucleotide targeted to human c-myc P2 promoter. Biochemistry 34, 8165–8171. doi:10.1021/bi00025a023
Kim, H. G., Reddoch, J. F., Mayfield, C., Ebbinghaus, S., Vigneswaran, N., Thomas, S., et al. (1998). Inhibition of transcription of the human c-myc protooncogene by intermolecular triplex. Biochemistry 37 (8), 2299–2304. doi:10.1021/bi9718191
Kolevzon, N., and Yavin, E. (2010). Site-specific DNA photocleavage and photomodulation by oligonucleotide conjugates. Oligonucleotides 20, 263–275. doi:10.1089/oli.2010.0247
Kuroki, M., Hachimine, K., Abe, H., Shibaguchi, H., Kuroki, M., Maekawa, S., et al. (2007). Sonodynamic therapy of cancer using novel sonosensitizers. Anticancer Res. 27, 3673–3677.
Lauria, T., Slator, C., McKee, V., Müller, M., Stazzoni, S., Crisp, A. L., et al. (2020). A click chemistry approach to developing molecularly targeted DNA scissors. Chemistry 26, 16782–16792. doi:10.1002/chem.202002860
Lee, J. S., Johnson, D. A., and Morgan, A. R. (1979). Complexes formed by (pyrimidine)n . (purine)n DNAs on lowering the pH are three-stranded. Nucleic Acids Res. 6, 3073–3091. doi:10.1093/nar/6.9.3073
Li, H., Broughton-Head, V. J., Fox, K. R., and Brown, T. (2007). Photoinduced crosslinking of double-helical DNA by psoralen covalently linked to a triple helix-forming oligonucleotide under near-physiological conditions. Nucleosides Nucleotides Nucleic Acids 26, 1005–1009. doi:10.1080/15257770701508554
Liao, D. J., and Dickson, R. B. (2000). c-Myc in breast cancer. Endocr. Relat. Cancer 7, 143–164. doi:10.1677/erc.0.0070143
Mai, B., Wang, X., Liu, Q., Zhang, K., and Wang, P. (2020). The application of DVDMS as a sensitizing agent for sono-/photo-therapy. Front. Pharmacol. 11, 19. doi:10.3389/fphar.2020.00019
Mai, S., and Mushinski, J. F. (2003). c-Myc-induced genomic instability. J. Environ. Pathol. Toxicol. Oncol. 22, 179–199. doi:10.1615/jenvpathtoxoncol.v22.i3.30
Majumdar, A., Muniandy, P. A., Liu, J., Liu, J. L., Liu, S. T., Cuenoud, B., et al. (2008). Targeted gene knock in and sequence modulation mediated by a psoralen-linked triplex-forming oligonucleotide. J. Biol. Chem. 283, 11244–11252. doi:10.1074/jbc.M800607200
Mastronikolis, N., Ragos, V., Kyrodimos, E., Chrysovergis, A., Papanikolaou, V., Mastronikolis, S., et al. (2019). Mechanisms of C-myc oncogenic activity in head and neck squamous cell carcinoma. J. BUON 24, 2242–2244.
Mayer, A., Häberli, A., and Leumann, C. J. (2005). Synthesis and triplex forming properties of pyrrolidino pseudoisocytidine containing oligodeoxynucleotides. Org. Biomol. Chem. 3, 1653–1658. doi:10.1039/b502799c
Moasser, M. M. (2007). The oncogene HER2: Its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene 26, 6469–6487. doi:10.1038/sj.onc.1210477
Moser, H. E., and Dervan, P. B. (1987). Sequence-specific cleavage of double helical DNA by triple helix formation. Science 238, 645–650. doi:10.1126/science.3118463
Mukherjee, A., and Vasquez, K. M. (2011). Triplex technology in studies of DNA damage, DNA repair, and mutagenesis. Biochimie 93, 1197–1208. doi:10.1016/j.biochi.2011.04.001
Napoli, S., Negri, U., Arcamone, F., Capobianco, M. L., Carbone, G. M., and Catapano, C. V. (2006). Growth inhibition and apoptosis induced by daunomycin-conjugated triplex-forming oligonucleotides targeting the c-myc gene in prostate cancer cells. Nucleic Acids Res. 34, 734–744. doi:10.1093/nar/gkj473
Obika, S. (2004). Development of bridged nucleic acid analogues for antigene technology. Chem. Pharm. Bull. (Tokyo) 52, 1399–1404. doi:10.1248/cpb.52.1399
Panyutin, I. G., and Neumann, R. D. (1994). Sequence-specific DNA double-strand breaks induced by triplex forming 125I labeled oligonucleotides. Nucleic Acids Res. 22, 4979–4982. doi:10.1093/nar/22.23.4979
Paquet, J., Henrionnet, C., Pinzano, A., Vincourt, J. B., Gillet, P., Netter, P., et al. (2011). Alternative for anti-TNF antibodies for arthritis treatment. Mol. Ther. 19, 1887–1895. doi:10.1038/mt.2011.156
Piotrowski-Daspit, A. S., Glaze, P. M., and Saltzman, W. M. (2018). Debugging the genetic code: Non-viral in vivo delivery of therapeutic genome editing technologies. Curr. Opin. Biomed. Eng. 7, 24–32. doi:10.1016/j.cobme.2018.08.002
Pokharel, D., Roseblade, A., Oenarto, V., Lu, J. F., and Bebawy, M. (2017). Proteins regulating the intercellular transfer and function of P-glycoprotein in multidrug-resistant cancer. Ecancermedicalscience 11, 768. doi:10.3332/ecancer.2017.768
Praseuth, D., Guieysse, A. L., and Hélène, C. (1999). Triple helix formation and the antigene strategy for sequence-specific control of gene expression. Biochim. Biophys. Acta 1489, 181–206. doi:10.1016/s0167-4781(99)00149-9
Pyne, A. L. B., Noy, A., Main, K. H. S., Velasco-Berrelleza, V., Piperakis, M. M., Mitchenall, L. A., et al. (2021). Base-pair resolution analysis of the effect of supercoiling on DNA flexibility and major groove recognition by triplex-forming oligonucleotides. Nat. Commun. 12, 1053. doi:10.1038/s41467-021-21243-y
Reza, F., and Glazer, P. M. (2014). Triplex-mediated genome targeting and editing. Methods Mol. Biol. 1114, 115–142. doi:10.1007/978-1-62703-761-7_8
Ricciardi, A. S., McNeer, N. A., Anandalingam, K. K., Saltzman, W. M., and Glazer, P. M. (2014). Targeted genome modification via triple helix formation. Methods Mol. Biol. 1176, 89–106. doi:10.1007/978-1-4939-0992-6_8
Rusling, D, A. (2021). Triplex-forming properties and enzymatic incorporation of a base-modified nucleotide capable of duplex DNA recognition at neutral pH. Nucleic Acids Res. 49, 7256–7266. doi:10.1093/nar/gkab572
Sargent, R. G., Kim, S., and Gruenert, D. C. (2011). Oligo/polynucleotide-based gene modification: Strategies and therapeutic potential. Oligonucleotides 21, 55–75. doi:10.1089/oli.2010.0273
Sedelnikova, O. A., Panyutin, I. G., Luu, A. N., Reed, M. W., Licht, T., Gottesman, M. M., et al. (2000). Targeting the human mdr1 gene by 125I-labeled triplex-forming oligonucleotides. Antisense Nucleic Acid. Drug Dev. 10 (6), 443–452. doi:10.1089/oli.1.2000.10.443
Seio, K., Yamaguchi, K., Yamazaki, A., Kanamori, T., and Masaki, Y. (2020). Transcription of DNA duplex containing deoxypseudouridine and deoxypseudoisocytidine, and inhibition of transcription by triplex forming oligonucleotide that recognizes the modified duplex. Nucleosides Nucleotides Nucleic Acids 39, 892–904. doi:10.1080/15257770.2020.1714652
Shen, C., Buck, A., Mehrke, G., Polat, B., Gross, H., Bachem, M., et al. (2001). Triplex forming oligonucleotide targeted to 3'UTR downregulates the expression of the bcl-2 proto-oncogene in HeLa cells. Nucleic Acids Res. 29, 622–628. doi:10.1093/nar/29.3.622
Shen, C., Rattat, D., Buck, A., Mehrke, G., Polat, B., Ribbert, H., et al. (2003). Targeting bcl-2 by triplex-forming oligonucleotide--a promising carrier for gene-radiotherapy. Cancer Biother Radiopharm. 18, 17–26. doi:10.1089/108497803321269296
Singhal, G., Akhter, M. Z., Stern, D. F., Gupta, S. D., Ahuja, A., Sharma, U., et al. (2011). DNA triplex-mediated inhibition of MET leads to cell death and tumor regression in hepatoma. Cancer Gene Ther. 18, 520–530. doi:10.1038/cgt.2011.21
Stierlé, V., Duca, M., Halby, L., Senamaud-Beaufort, C., Capobianco, M. L., Laigle, A., et al. (2008). Targeting MDR1 gene: Synthesis and cellular study of modified daunomycin-triplex-forming oligonucleotide conjugates able to inhibit gene expression in resistant cell lines. Mol. Pharmacol. 73, 1568–1577. doi:10.1124/mol.107.042010
Strobel, S. A., Doucette-Stamm, L. A., Riba, L., Housman, D. E., and Dervan, P. B. (1991). Site-specific cleavage of human chromosome 4 mediated by triple-helix formation. Science 254, 1639–1642. doi:10.1126/science.1836279
Taniguchi, Y., and Sasaki, S. (2012). An efficient antigene activity and antiproliferative effect by targeting the Bcl-2 or survivin gene with triplex forming oligonucleotides containing a W-shaped nucleoside analogue (WNA-βT). Org. Biomol. Chem. 10, 8336–8341. doi:10.1039/c2ob26431e
Tapia, C., Savic, S., Wagner, U., Schönegg, R., Novotny, H., Grilli, B., et al. (2007). HER2 gene status in primary breast cancers and matched distant metastases. Breast Cancer Res. 9, R31. doi:10.1186/bcr1676
Tomita, N. (2011). BCL2 and MYC dual-hit lymphoma/leukemia. J. Clin. Exp. Hematop 51, 7–12. doi:10.3960/jslrt.51.7
Torigoe, H., Nakagawa, O., Imanishi, T., Obika, S., and Sasaki, K. (2012). Chemical modification of triplex-forming oligonucleotide to promote pyrimidine motif triplex formation at physiological pH. Biochimie 94, 1032–1040. doi:10.1016/j.biochi.2012.01.003
Torigoe, H., Rahman, S. M., Takuma, H., Sato, N., Imanishi, T., Obika, S., et al. (2011). 2'-O, 4'-C-aminomethylene-bridged nucleic acid modification with enhancement of nuclease resistance promotes pyrimidine motif triplex nucleic acid formation at physiological pH. Chemistry 17, 2742–2751. doi:10.1002/chem.201002745
van de Vijver, M. J. (2001). Assessment of the need and appropriate method for testing for the human epidermal growth factor receptor-2 (HER2). Eur. J. Cancer 37, S11–S17. doi:10.1016/S0959-8049(00)00403-2
Vasquez, K. M. (2010). Targeting and processing of site-specific DNA interstrand crosslinks. Environ. Mol. Mutagen 51, 527–539. doi:10.1002/em.20557
Wade, M., and Wahl, G. M. (2006). c-Myc, genome instability, and tumorigenesis: the devil is in the details. Curr. Top. Microbiol. Immunol. 302, 169–203. doi:10.1007/3-540-32952-8_7
Zhang, Y., Bi, L., Hu, Z., Cao, W., and Zhuang, D. (2020). Hematoporphyrin monomethyl ether-mediated sonodynamic therapy induces A-253 cell apoptosis. Oncol. Lett. 19, 3223–3228. doi:10.3892/ol.2020.11419
Keywords: triplex-forming oligonucleotides (TFOs), triple helix formation, oncogenes, anti-gene technique, cancer therapy
Citation: Li C, Zhou Z, Ren C, Deng Y, Peng F, Wang Q, Zhang H and Jiang Y (2022) Triplex-forming oligonucleotides as an anti-gene technique for cancer therapy. Front. Pharmacol. 13:1007723. doi: 10.3389/fphar.2022.1007723
Received: 30 July 2022; Accepted: 12 December 2022;
Published: 21 December 2022.
Edited by:
Gao Yong-Guang, Northwestern Polytechnical University, ChinaReviewed by:
David Rusling, University of Portsmouth, United KingdomCopyright © 2022 Li, Zhou, Ren, Deng, Peng, Wang, Zhang and Jiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hong Zhang, MjAwMDAxMkB0b25namkuZWR1LmNu; Yuan Jiang, ODU3NDE5MjBAcXEuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.