
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 14 January 2022
Sec. Pharmacology of Ion Channels and Channelopathies
Volume 12 - 2021 | https://doi.org/10.3389/fphar.2021.814066
This article is part of the Research Topic Novel Pathophysiological and Pharmacological insight into the Kir/KATP Channelopathies of Cardiovascular & Musculoskeletal Apparatus View all 4 articles
 Marien J. C. Houtman1†
Marien J. C. Houtman1† Theres Friesacher2†
Theres Friesacher2† Xingyu Chen2
Xingyu Chen2 Eva-Maria Zangerl-Plessl2
Eva-Maria Zangerl-Plessl2 Marcel A. G. van der Heyden1
Marcel A. G. van der Heyden1 Anna Stary-Weinzinger2*
Anna Stary-Weinzinger2*Introduction: DEND syndrome is a rare channelopathy characterized by a combination of developmental delay, epilepsy and severe neonatal diabetes. Gain of function mutations in the KCNJ11 gene, encoding the KIR6.2 subunit of the IKATP potassium channel, stand at the basis of most forms of DEND syndrome. In a previous search for existing drugs with the potential of targeting Cantú Syndrome, also resulting from increased IKATP, we found a set of candidate drugs that may also possess the potential to target DEND syndrome. In the current work, we combined Molecular Modelling including Molecular Dynamics simulations, with single cell patch clamp electrophysiology, in order to test the effect of selected drug candidates on the KIR6.2 WT and DEND mutant channels.
Methods: Molecular dynamics simulations were performed to investigate potential drug binding sites. To conduct in vitro studies, KIR6.2 Q52R and L164P mutants were constructed. Inside/out patch clamp electrophysiology on transiently transfected HEK293T cells was performed for establishing drug-channel inhibition relationships.
Results: Molecular Dynamics simulations provided insight in potential channel interaction and shed light on possible mechanisms of action of the tested drug candidates. Effective IKIR6.2/SUR2a inhibition was obtained with the pore-blocker betaxolol (IC50 values 27–37 μM). Levobetaxolol effectively inhibited WT and L164P (IC50 values 22 μM) and Q52R (IC50 55 μM) channels. Of the SUR binding prostaglandin series, travoprost was found to be the best blocker of WT and L164P channels (IC50 2–3 μM), while Q52R inhibition was 15–20% at 10 μM.
Conclusion: Our combination of MD and inside-out electrophysiology provides the rationale for drug mediated IKATP inhibition, and will be the basis for 1) screening of additional existing drugs for repurposing to address DEND syndrome, and 2) rationalized medicinal chemistry to improve IKATP inhibitor efficacy and specificity.
Potassium carrying KATP channels transduce the cellular metabolic status to electrophysiological properties in many cell types. In pancreatic beta-cells, high intracellular ATP levels, and low MgADP levels, inhibit KATP channel activity which slightly depolarizes the cells resulting in insulin release (Ashcroft, 2005). KATP channels are octameric protein complexes consisting of a tetrameric pore-forming KIR6.1 or KIR6.2 protein assembly, encoded by KCNJ8 and KCNJ11, respectively, surrounded by a tetramer of SUR1 or SUR2A/B proteins, encoded by ABCC8 and ABCC9, respectively (Hibino et al., 2010). Gain of function mutations in the underlying genes associate with diverse disease phenotypes affecting one or more tissues and organs, like transient or permanent neonatal diabetes (ND) and Cantú disease (Gloyn et al., 2004; Harakalova et al., 2012). Binding of ATP to the KIR6.2 subunit has an inhibitory effect, whilst binding of nucleotides to the SUR subunit activates the channel (Sikimic et al., 2019). Gain of function mutations decrease the ability of ATP to inhibit the channel, either directly by affecting the ATP binding site, or indirectly by increasing channel open probability, thereby decreasing ATP affinity (Ashcroft, 2005). There is a clear relation between the extent of diabetes causing KCNJ11 gain of function strength and clinical phenotype. Whereas KIR6.2 channels harboring mild gain of function mutations result in transient or permanent ND, strong gain of function mutant KIR6.2 channels on the other hand associate with a complex phenotype of developmental delay, muscle weakness, dysmorphic features, epilepsy and neonatal diabetes, known as the DEND syndrome (Hattersley and Ashcroft, 2005). These strong gain of function mutations are generally of the indirect class.
Sulfonylurea type drugs, like tolbutamine and gliclazide, stimulate insulin secretion from beta-cells (Proks et al., 2002). Successful sulfonylurea therapy results in KATP channel inhibition, by direct and indirect mechanisms in which the pharmakon interacts with the SUR subunits, and thereby enhances ATP sensitivity of the KIR6.2 subunits and thus stimulate channel closure at lower intracellular ATP concentrations (De Wet and Proks, 2015). From a therapeutic perspective, the stronger gain of function mutations pose a pharmacological challenge since they are relatively resistant to sulfonylureas, and even in cases when the ND part of the phenotype is relieved, neurological symptoms are mostly less-, or non-sensitive to such treatment (Koster et al., 2005; Pearson et al., 2006). This is even worsened by difficulties of many existing and newly developed compounds to efficiently cross the blood-brain-barrier and reach therapeutic concentrations in the neural tissues (Ashcroft et al., 2017). Apart from this, some mutants associated with ND only, are also relatively resistant to sulfonylureas (Tammaro et al., 2008).
The KCNJ11 mutation Q52R associates with DEND syndrome, although clinical phenotype and therapeutic options vary between patients (Gloyn et al., 2004; Shaw and Majzoub, 2009; Ioacara et al., 2017; Helmi and Hussain, 2020). The L164P mutation however associates with the “milder” phenotype of permanent ND (Flanagan et al., 2006). Both mutations alter channel kinetics in a similar fashion, that is an increased open probability in the absence of ligand, which at least for the Q52R mutation was independent of the SUR isoform (Tammaro et al., 2006, 2008). The position of the mutations is shown in Figure 1A. Importantly, both mutations render the resulting KATP channel rather insensitive to sulfonylureas like tolbutamine (Pearson, et al., 2006; Tammaro et al., 2008; Babiker et al., 2016; Hashimoto et al., 2017). Therefore, there is a need for new compounds with desired pharmacokinetic/pharmacodynamic properties that effectively inhibit DEND syndrome associated mutant KATP channel and additional sulfonylurea resistant KATP channel variants.
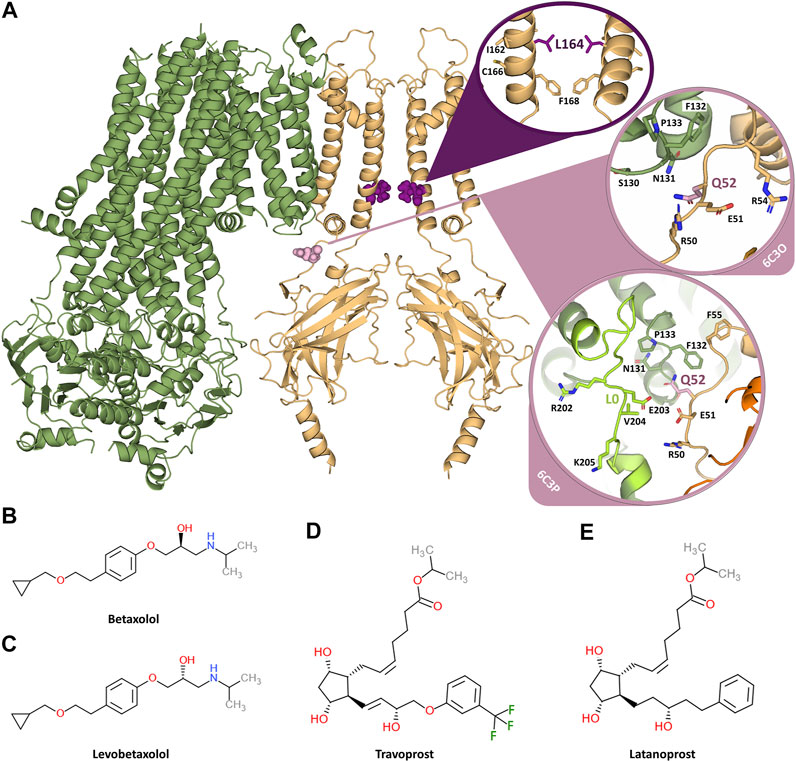
FIGURE 1. Overview of the KATP channel and the investigated inhibiting compounds. (A) Two opposing subunits of the KIR6.2 channel pore are shown in orange, one SUR1 subunit is shown in green. The two mutation sites L164 and Q52 are shown in purple and pink, respectively. The circles provide close-ups of the mutation sites. Since Q52 is located on the surface of KIR6.2 close to SUR1 and the orientation of the SUR1 subunits is different in the 6C3O and 6C3P structure, the close-ups of Q52 are shown for both structures. The loop L0, which comprises residues important for KIR-SUR coupling, is shown in light green for the 6C3P structure, while it is not modelled in the 6C3O structure. (B) Chemical structure of betaxolol. (C) Chemical structure of levobetaxolol. (D) Chemical structure of travoprost. (E) Chemical structure of latanoprost.
Previously, in a successful attempt to find potential inhibitors of Cantú Syndrome associated gain of function mutants, we set out to screen for potential KIR6.1 inhibiting compounds by applying Molecular Dynamics and pharmacophore modelling (Chen et al., 2019). This work resulted in a hit-list from DrugBank entries. Here, we expanded this in silico and in vitro work to KIR6.2 inhibiting compounds betaxolol, levobetaxolol, latanoprost and travoprost (for chemical structure, see Figures 1B–E) as well as glibenclamide, ethosuximide, fluvastatin, pazopanib and dinoprostone (for chemical structure see Supplementary Figure S1) focusing on Q52R and L164P gain of function mutations.
Nucleotide mutations resulting in Q52R and L164P were made in pCMV6-KIR6.2 (mouse) expression construct (a generous gift from J.A. Sánchez-Chapula (Centro Universitario de Investigaciones Biomédicas de la Universidad de Colima) using the Quickchange Site-Directed Mutagenesis kit (Agilent Technologies), by the use of custom made primers (Merck): (Q52R forward primer: caagaacatccgagagcggggccgcttcctgcaag, reverse primer: cttgcaggaagcggccccgctctcggatgttcttg; L164P forward primer: gatcaacgccatcatgccgggctgcatcttcatg, reverse primer: catgaagatgcagcccggcatgatggcgttgatc). Mutations were verified using Sanger based sequencing (Macrogen Europe B.V.) and SnapGene analysis software (GSL Biotech LLC).
Stock solutions were prepared as follows: Barium (Merck, cat. nr. 1.01719) at 1 M in H2O, sterile filtered; betaxolol (Merck, cat. nr. 1069903) at 0.1 M in H2O; dinoprostone (Merck, cat. nr. D2250000) at 0.1 M in EtOH; ethosuximide (Merck, cat. nr. E7138) at 0.1 M in EtOH; fluvastatin (Merck, cat. nr. SML0038) at 10 mM in H2O; glibenclamide (Merck, cat. nr. G0639) at 0.1 M in DMSO; latanoprost (Merck, cat. nr. PHR 1884) at 1 mM in EtOH; levobetaxolol (Cayman Chemical, cat. nr. 33435-250) at 0.1 M in H2O; travoprost (Toronto Research Chemicals, cat. nr. T715600) at 10 mM in DMSO. Barium was stored at 4°C and all other compounds were stored at −20°C until use.
HEK293T cells were cultured in DMEM medium (Lonza, cat. nr. 12–733F) and seeded on glass coverslips, coated with gelatin (Merck, cat. nr. G1890). After cell adherence, polyethylenimine (Polysciences, cat. nr. 23966) transfection was performed using either WT or mutant pCMV6-KIR6.2 in combination with pCMV6-SUR2A and pEGFP1 (0.16, 0.16 and 0.08 μg, respectively). IKATP measurements were performed essentially as described before (Houtman et al., 2019). Briefly, glass capillary pipettes (Harvard Apparatus, cat. nr. 30–0,040) filled with pipette solution (145 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 5 mM HEPES, pH 7.40 KOH) were introduced in a temperature controlled perfusion-chamber (CellMicroControls) filled with bath solution (131 mM KCl, 1 mM EGTA, 7.2 mM K2HPO4, 2.8 mM KH2PO4, 1 mM MgCl2, pH 7.20 KOH). After giga-seal formation, the pipette was pulled up and after brief air exposure inside-out patches were formed. Using an AxoPatch 200B amplifier and Clampex 10 software (Molecular Devices) a ramp protocol was run with holding potentials ranging from −100 mV to +100 mV. Inward and outward IKATP current levels at −80 mV and +50 mV were determined using Clampfit 10 software (Molecular Devices). All measurements were performed at room temperature (22°C). Data and statistical analysis (one-way ANOVA and t test) were performed using GraphPad Prism 8 software (GraphPad Software LLC). Data are presented as mean ± s.e.m.
Molecular modelling of drug interactions with the KATP channel were carried out using coarse-grained and classical MD simulations. An overview of the simulation runs is given in Supplementary Table S1.
The quatrefoil (PDB: 6C3O) and propeller (PDB:6C3P) structures (Lee et al., 2017) were set up with the Martini bilayer maker of the CHARMM-GUI web service (Jo et al., 2008; Qi et al., 2015; Hsu et al., 2017). For the coarse-grained MD simulations without SUR, the KIR6.2 pore was extracted from the KATP channel (PDB: 6C3O) and used as starting structure. Simulations were carried out based on the Martini2.2 coarse-grained model (Marrink et al., 2007; Monticelli et al., 2008), implementing the global elastic network (Periole et al., 2009). The channels were embedded in a palmitoyloleoylphosphatidylcholine (POPC) membrane and solvated with the non-polarizable water model in a 150 mM KCl solution. Simulations were carried out with Gromacs 2018 and 2020 (Abraham et al., 2015). Temperature was coupled to 310 K using the v-rescale thermostat (Bussi et al., 2007) with a coupling constant of 0.1 ps. The pressure was kept constant semi-isotropically at 1 bar with the Parrinello-Rahman barostat (τ = 2 ps) (Parrinello and Rahman, 1981). Simulations with betaxolol were computed with an integration step of 2 fs, whereas simulations with the prostaglandins required an integration step of 1 fs in order to increase the stability of the system. Latanoprost, travoprost and betaxolol were parametrized as described on the Martini homepage (Martini, 2021). The topologies of the ligands can be found in Supplementary Table S2.
All-atom MD simulations with KIR6.2 and betaxolol were performed as described previously (Chen et al., 2019, PDB:6C3O). For this purpose, betaxolol was docked into the binding sites, which were identified in the coarse-grained MD simulations, using the GOLD software (Verdonk et al., 2003) with ChemPLP scoring function (Korb et al., 2009). We used the Gromacs 2018 (Abraham et al., 2015) software with the Amber99sb force field (Hornak et al., 2006) and embedded the KIR6.2 channel pores in a palmitoyloleoylphosphatidylcholine (POPC) lipid bilayer [Berger lipids parameters (Berger et al., 1997)] with four PIP2 molecules bound to the channel. Ligand parameters for PIP2 and betaxolol were obtained using the Hartree-Fock geometry optimization with the 6-31G* basis set (Frisch et al., 2013) and the antechamber tools (Wang et al., 2004; Wang et al., 2006). The SPCE water model (Berendsen et al., 1987; Kusalik and Svishchev, 1994) was used and 150 mM KCl were added to the solvent. Five K+ ions were placed in the selectivity filter at sites S0 to S4. 2 times 1 μs MD simulations and 9 times 350 ns were run, including the four main identified betaxolol binding sites, obtained from coarse-grained simulations. The topology of betaxolol can be found in Supplementary Table S2.
The simulations were analyzed using the analysis tools of Gromacs (Abraham et al., 2015). Furthermore, the binding sites observed in the coarse-grained MD simulations were analyzed with the Python package MDAnalysis (Michaud-Agrawal et al., 2011; Gowers et al., 2016). In-house Python 3 scripts were used for plotting. The molecular structures were visualized with PyMol (PyMol, 2016) and VMD (Humphrey et al., 1996).
We first created the KIR6.2 Q52R and L164P mutant channels and subsequently tested these using inside-out recordings on membrane patches from transiently transfected HEK293T cells in the absence of MgATP/ADP. Barium, as a positive control, dose-dependently inhibited outward current (at +50 mV) equally well for mutant channels as for WT channels (IC50 37.4 ± 4.9 μM, Hill coefficient −0.87 (WT) vs. 65.2 ± 3.5 μM, Hill coefficient −1.1 (Q52R) (n.s.) and 36.5 ± 1.2 μM, Hill coefficient −1.1 (L164P) (n.s.), whereas inward current (at −80 mV) was less potently inhibited compared to the outward current for WT and not inhibited for Q52R and L164P channels (Figure 2A). The sulfonylurea drug glibenclamide potently inhibited inward and outward currents of the WT channels (IC50 0.9 ± 0.3 μM, Hill coefficient −0.41 and 0.6 ± 0.2 μM, Hill coefficient −0.40 for inward and outward currents respectively), whereas Q52R channels were insensitive for glibenclamide mediated block (<10% block at 10 μM) (Figure 2B). The L164P channels were less potently inhibited compared to WT channels (approx. 20–25% block at 10 μM). These data demonstrate the expected behavior of the mutant channels in relation to drug sensitivity of the WT channels.
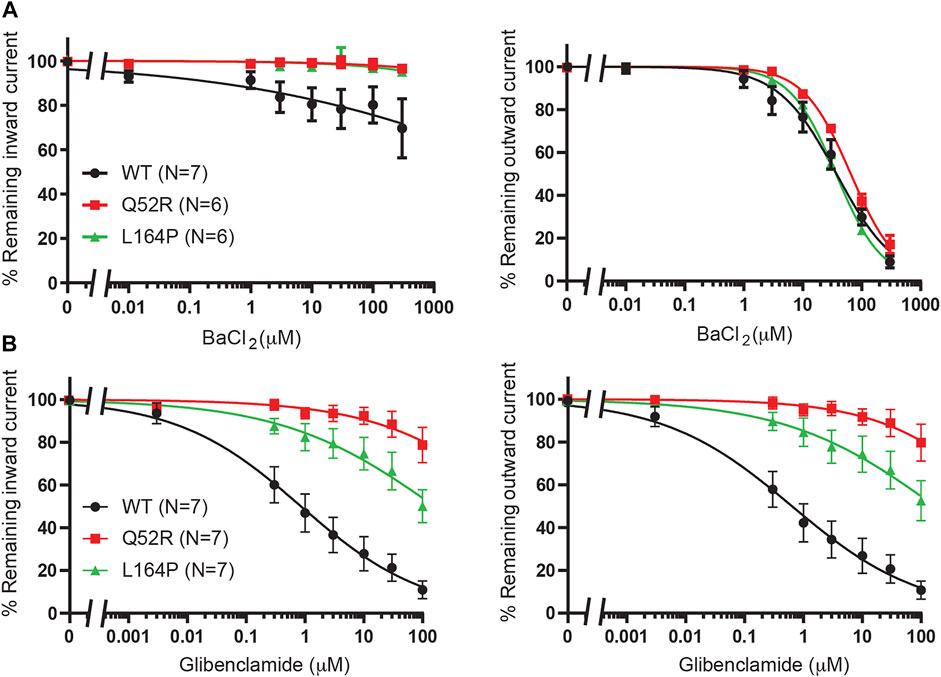
FIGURE 2. Pharmacological validation of Q52R and L164P KIR6.2 mutations. IC50 curves of KIR6.2/SUR2a inward (at −80 mV), left panels, and outward (at +50 mV), right panels, current inhibition in response to increasing concentrations of (A) BaCl2 and (B) glibenclamide. Wildtype KIR6.2/SUR2a serves as control. Data were fitted with Hill equation to estimate the IC50 values. Data are shown as mean ± SEM. N depicts number of measurements on independent inside-out patches.
We next expanded our previous findings, in which we demonstrated that betaxolol (no 5 on Chen et al., 2019 hit list) and Travoprost (no 7) were able to inhibit IKIR6.2/SUR2a outward currents.
We previously demonstrated betaxolol mediated inhibition of KIR6.2/SUR2a current (Chen et al., 2019). To test the inhibitory capacity of betaxolol and its closely related homologue levobetaxolol (no 3 on Chen et al. (2019) hit-list) on Q52R and L164P mutants, we performed inside-out recordings on HEK293T cell membrane patches expressing either WT, Q52R or L164P KIR6.2/SUR2a channels. Both compounds did not significantly inhibit the inward component at −80 mV for concentrations up to 300 μM (Figures 3A,B). In contrast, outward current at +50 mV became inhibited by betaxolol with almost identical capacity for WT and mutants (IC50 27.5 ± 2.1 μM, Hill coefficient −0.98 vs. 37.1 ± 2.1 μM, Hill coefficient −1.17 (n.s.) and 30.6 ± 2.3 μM, Hill coefficient −0.87 (n.s.), for WT, Q52R and L164P, respectively) (Figure 3A). Levobetaxolol inhibited the outward current of Q52R channels less potently than for WT and L164P channels (IC50 22.7 ± 2.0 μM, Hill coefficient −0.91 vs. 55.3 ± 3.1 μM, Hill coefficient −1.19 (p < 0.01) and 21.8 ± 2.1 μM, Hill coefficient -0.88 (n.s.), for WT, Q52R and L164P, respectively) (Figure 3B). Interestingly, betaxolol also inhibited the classical inward rectifier KIR2.1 current with similar IC50 values (IC50 29.8 ± 3.7 μM, Hill coefficient −0.79) (Supplementary Figure S2). Sequential mutation of the classical polyamine binding sites within the cytosolic (E224A, D259A, F254A, E299A) and transmembrane (D172A) pore domains resulted in a mild increase in IC50 values, except for E224A (approximately 35% inhibition at 100 μM) (Supplementary Figure S2).
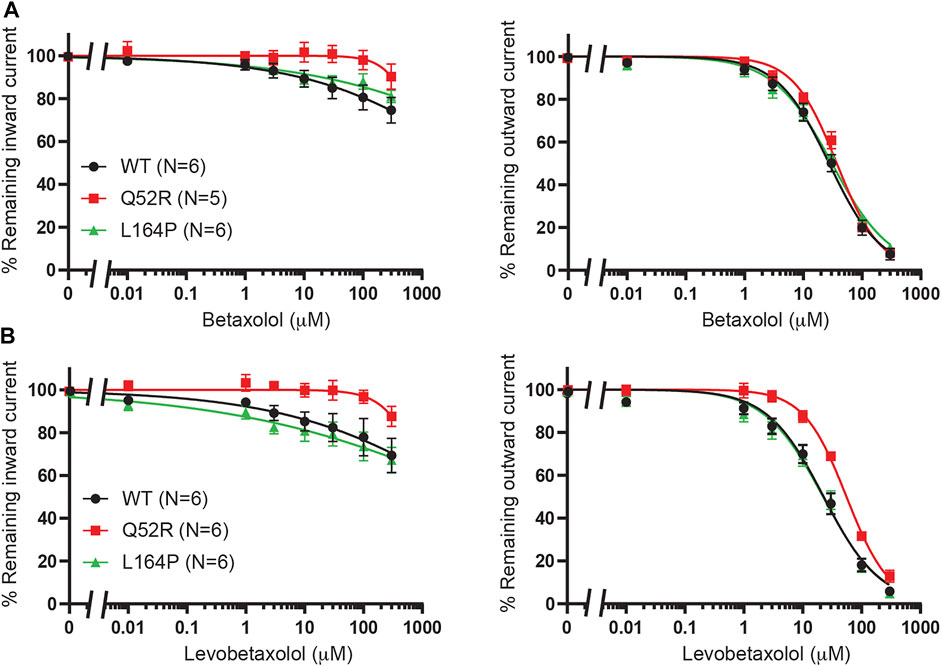
FIGURE 3. Inhibition of Q52R and L164P KIR6.2/SUR2a dependent current by betaxolol and levobetaxolol. IC50 curves of KIR6.2/SUR2a inward (at −80 mV), left panels, and outward (at +50 mV), right panels, current inhibition in response to increasing concentrations of (A) betaxolol and (B) levobetaxolol. Wildtype KIR6.2/SUR2a serves as control. Data were fitted with Hill equation to estimate the IC50 values. Data are shown as mean ± SEM. N depicts number of measurements on independent inside-out patches.
Aiming to sample putative binding sites of betaxolol at reasonable computational costs, we carried out coarse-grained molecular dynamics (MD) simulations with the WT KIR6.2 channel pore in presence of ten betaxolol drug molecules randomly placed in the solvent. The reasons not to include the SUR subunit in these simulations were: 1) electrophysiology measurements show voltage dependent inhibition, 2) the Hill coefficient was around 1, and 3) our experimental data shows comparable inhibition of the different KATP mutants by betaxolol. These arguments indicate that the drug acts on the channel pore rather than on the SUR subunit.
Ten replicate runs of 1 µs-long MD simulations unraveled three main binding sites for the drug, which are located close to the PIP2 binding site at the surface of helix M1, in the transmembrane cavity, and in the cytoplasmic domain at the G-loop region (Figure 4).
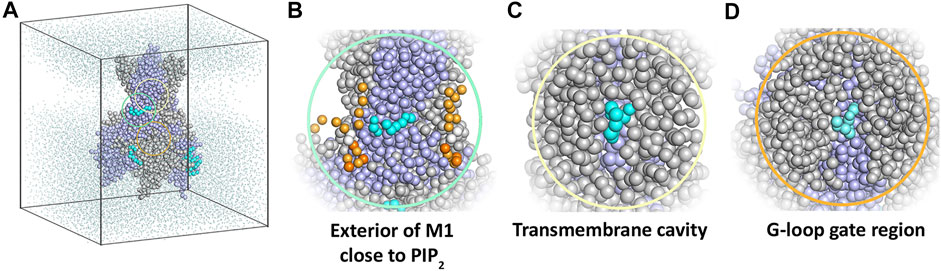
FIGURE 4. Coarse-grained MD simulation with the KIR6.2 channel pore and betaxolol. (A) Overview of a typical simulation system after 1 µs simulation. Different subunits of the KIR6.2 channel are shown as grey and lilac spheres, respectively. Bound betaxolol molecules are shown as cyan spheres. Water molecules are illustrated as dots, while lipid molecules are omitted for clarity. (B) Close-up of the exterior of M1, where a betaxolol molecule is bound near the PIP2 binding site. PIP2 molecules are shown as orange spheres. (C) Close-up of the transmembrane cavity. (D) Close-up of the G-loop region.
In order to analyze the binding sites in more detail, we performed atomistic MD simulations of the KIR6.2 channel pore in the presence of betaxolol docked at the identified interaction sites Figure 5A (Supplementary Table S1). Figures 5E–G shows typical snapshots of the respective betaxolol binding poses observed in the atomistic MD simulations. The binding site near PIP2 is formed by a cleft between the lower halves of transmembrane helix 1 (TM1) and 2 (TM2) of one subunit. Betaxolol predominantly engages in hydrophobic interactions with residues L56, L63 and M163. This binding pose is reminiscent of the binding site of rosiglitazone to the KIR6.1 channel pore identified in a previous study (Chen et al., 2019).
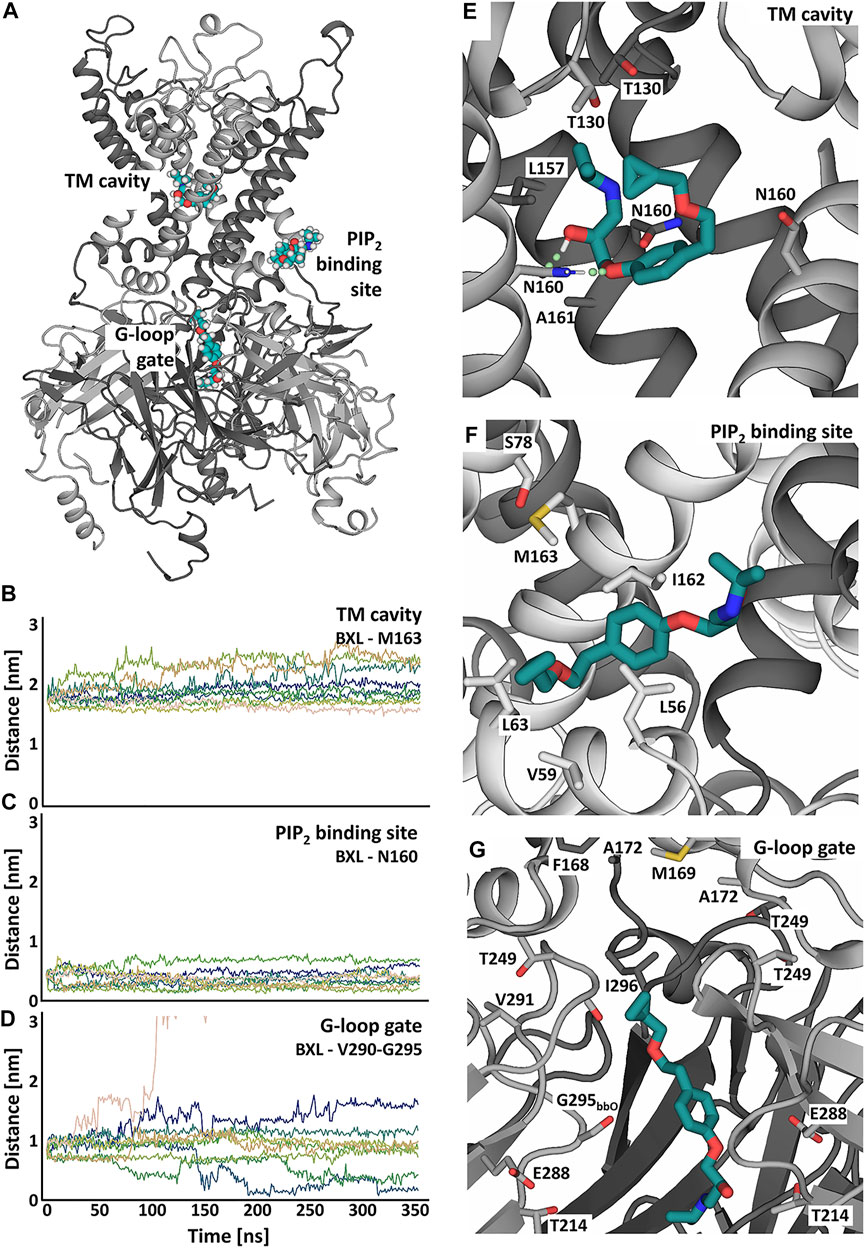
FIGURE 5. Putative betaxolol binding sites and stability investigated in atomistic MD simulations with the KIR6.2 channel pore. (A) Overview of the binding sites of betaxolol to the KIR channel pore. Different KIR subunits are shown in cartoon representation and colored in dark and light grey. The different betaxolol molecules are shown in teal. (B–D) Distances between betaxolol and the KIR6.2 channel in 9 replica of 350ns-long MD simulations. The differently colored lines represent the distances of the docked betaxolol molecules to the respective binding sites in the different runs. (E–G) Close-ups of the betaxolol binding sites at TM-cavity (E), PIP2 binding site (F), G-loop gate (G) seen at the end of run1 of the all-atom MD simulations. The drug is shown as teal sticks, with hydrogen bonds shown as green dotted lines.
Binding of betaxolol in the transmembrane cavity is mediated by hydrogen bonds to N160 (Figure 5E, Supplementary Figure S3B), which can be observed in approximately 70% of the simulation time, as well as hydrophobic interactions to L157, A161 and T130. The third binding site is located in the cytoplasmic domain near the G-loop gate, which constitutes another important regulative barrier for ion conduction. Betaxolol forms hydrophobic interactions with residues V290, I296, A172, M169 and the HBC gate composing residue F168. Additional hydrogen bonds can be observed between the drug and T214, E288 and the backbone oxygens of S212 and G295 (Supplementary Figures S3C–F).
Figures 5B–D shows distance measurements of the docked betaxolol molecules to the respective binding sites observed in 9 runs of 350 ns long. The analysis reveals a stable drug interaction near the PIP2 binding site and the TM cavity (Figures 5B,C), since dissociation of betaxolol is not observed in any of the simulations. For the interaction near the G-loop gate, a greater deviation of the drug from its original binding pose can be observed. Figure 5D shows that the distance between betaxolol and the G-loop gate composing residues V290-G295 decreases in two runs, which is caused by betaxolol detaching from its initial binding site and moving towards the narrowest parts of the G-loop gate, where it remains stable for the rest of the simulation time. Similar observations could be made in the two additional 1 μs-long MD simulations (Supplementary Figure S4).
Taken together, our simulations suggest that betaxolol plugs the conduction pathway by interacting with pore lining residues. This also provides an explanation for its voltage dependent block (outward >> inward) observed in electrophysiology measurements. In addition to this pore block, betaxolol binding near the PIP2 binding site might also influence the conductivity, a phenomenon which has already been described for other KIR channel blockers (López-Izquierdo et al., 2011; Ponce-Balbuena et al., 2012; Koepple et al., 2017; Scherer et al., 2017).
Given the relatively low potency (mid-micromolar) and state dependency of betaxolol and levobetaxolol, we also tested more lipophilic compounds with higher affinity towards KIR6.2/SUR2a channels. Travoprost, a synthetic prostaglandin derivative, was identified as a drug that inhibits both outward and inward currents (Chen et al., 2019). In addition, we decided to test a naturally occurring prostaglandin [dinoprostone, no 4 on Chen et al. (2019) hit-list] and an additional synthetic prostaglanding (latanoprost).
In order to investigate the reputative binding sites of travoprost, latanoprost and dinoprostone, we compared their inhibiting capacity on WT KIR6.2/SUR2a channel excised membrane patches, similar as described in Betaxolol and Levobetaxolol. Whereas, travoprost mediated potent inhibition (see below), considerably less inhibition was observed with latanoprost (approximately 35–40% inhibition at 30 μM), and no inhibition was observed with dinoprostone (approximately 10–20% inhibition at 100 μM) (Figure 6A). Subsequently, travoprost was tested on WT, Q52R and L164P channel patches (Figure 6B). Both inward and outward currents of WT channel were inhibited (IC50 2.1 ± 0.4 μM, Hill coefficient −0.90 and 2.0 ± 0.3 μM, Hill coefficient −0.86, respectively), while inhibition of L164P channels showed similar IC50 values, (IC50 3.4 ± 0.8 μM, Hill coefficient −0.67 (n.s.) and 4.0 ± 1.2 μM, Hill coefficient −0.64 (n.s.) for inward and outward L164P, respectively). No significant inhibition of Q52R channels was observed (approximately 15–20% inhibition at 10 μM), We next carried out coarse-grained molecular dynamics (MD) simulations with travoprost and latanoprost to identify their putative binding sites. In contrast to betaxolol, the experimental data for these two drugs does not allow the exclusion of binding sites on SUR. Hence, the coarse-grained MD simulations were conducted with the whole KATP channel complex, including the four SUR subunits. As a starting point for the simulations, we chose the highest resolution Cryo-EM structure pair available [PBD: 6C3O/6C3P (Lee et al., 2017)], which shows two very different orientations of the SUR subunits with regard to the KIR6.2 channel pore. For both Cryo-EM structures, a total of 30 μs coarse-grained MD simulations were conducted in the presence of travoprost and latanoprost, respectively (Supplementary Table S1).
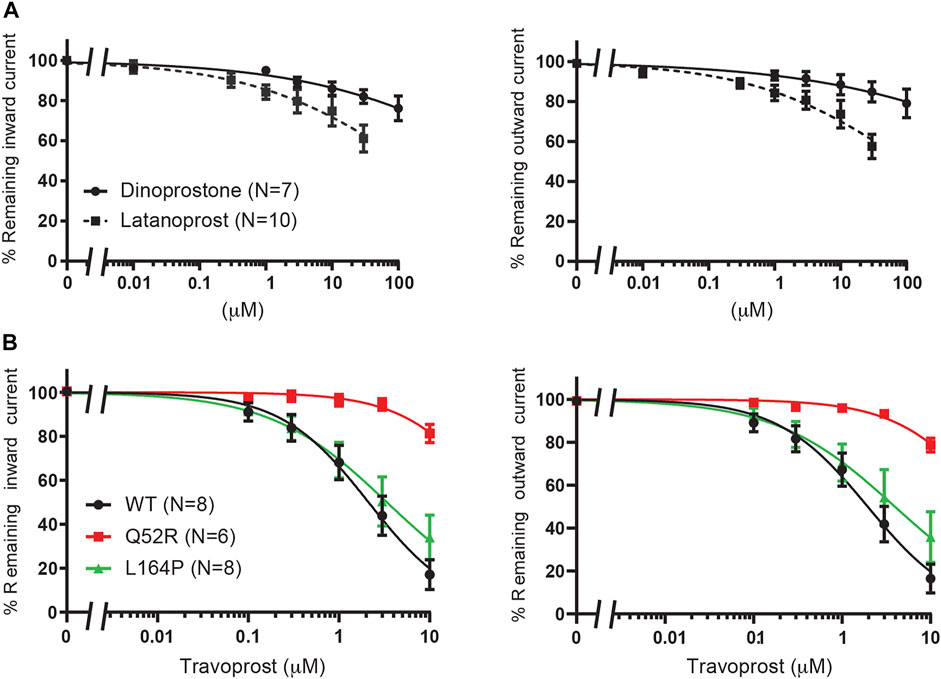
FIGURE 6. Evaluation of KIR6.2/SUR2a dependent currents in response to dinoprostone, latanoprost and travoprost. (A) IC50 curves of WT KIR6.2/SUR2a inward (at −80 mV), left panel, and outward (at +50 mV), right panel, current inhibition in response to increasing concentrations of dinoprostone or latanoprost. (B) IC50 curves of WT, Q52R and L164P KIR6.2/SUR2a inward (at −80 mV), left panel, and outward (at +50 mV), right panel, current inhibition in response to increasing concentrations of travoprost. Data were fitted with Hill equation to estimate the IC50 values. Data are shown as mean ± SEM. N depicts number of measurements on independent inside-out patches.
In general, the coarse-grained MD simulations unraveled a strong affinity of travoprost and latanoprost to the SUR subunits, where a broad range of binding sites could be identified. All of the drugs in the simulation systems bind to the protein complex (Supplementary Figure S5), whereby the interaction sites are almost exclusively located on SUR. A summary of the binding sites found in the simulations with the 6C3O and 6C3P structures is shown in Figures 7, 8, respectively. Since most of the binding sites were observed more than once, either by being detected in different simulations or in the same simulations on different subunits, similar binding poses were collected in interaction clusters in order to give a comprehensive overview of the affinities.
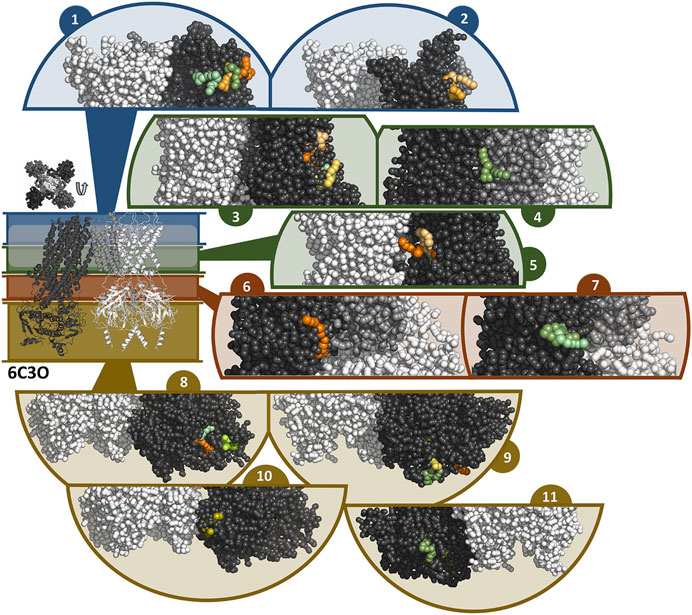
FIGURE 7. Binding sites of travoprost and latanoprost found in a total of 60 μs coarse-grained MD simulations with the 6C3O structure. The smaller figure on the left shows the overall structure of the 6C3O channel, the location of the lipid bilayer membrane is indicated with a white bar. For the sake of clarity, the structure is divided in different zones, which are shown in different colors (blue = extracellular part as well as the outer lipid layer of membrane, green = inner lipid layer of membrane, red = cytoplasmic domain near cell membrane, yellow = cytoplasmic domain). The figure above the overall structure shows the orientation of the SUR subunits in a top-down view of the channel. The different sub figures show close-ups of different interaction sites of the drugs to the channel. Travoprost is shown in shades of green, latanoprost in shades of orange and yellow. In each of the close-ups, all four subunits of the KIR channel are shown colored in white. For SUR, only one subunit is shown. The TM0 of SUR colored in middle grey, while the rest of SUR is colored in dark grey.
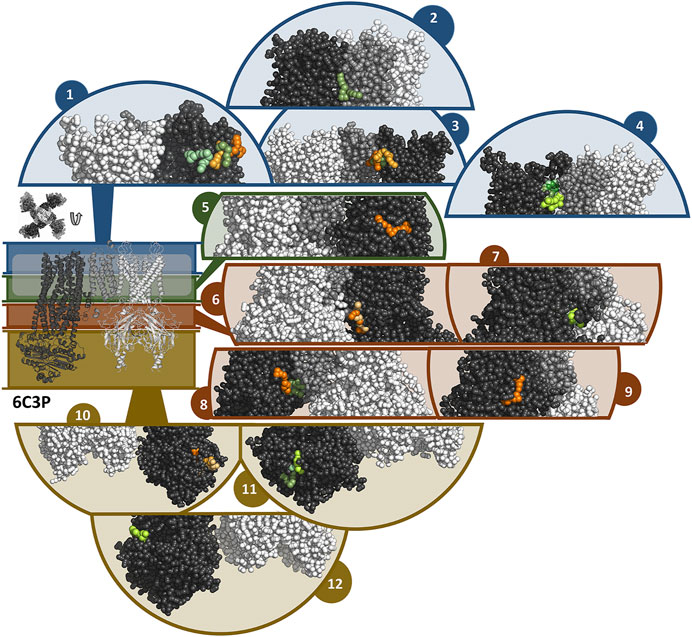
FIGURE 8. Binding sites of travoprost and latanoprost found in a total of 60 μs coarse-grained MD simulations with the 6C3P structure. The smaller figure on the left shows the overall structure of the 6C3P channel, the location of the lipid bilayer membrane is indicated with a white bar. For the sake of clarity, the structure is divided in different zones, which are shown in different colors (blue = extracellular part as well as the outer lipid layer of membrane, green = inner lipid layer of membrane, red = cytoplasmic domain near cell membrane, yellow = cytoplasmic domain). The figure above the overall structure shows the orientation of the SUR subunits in a top-down view of the channel. The different sub figures show close-ups of different interaction sites of the drugs to the channel. Travoprost is shown in shades of green, latanoprost in shades of orange and yellow. In each of the close-ups, all four subunits of the KIR channel are shown colored in white. For SUR, only one subunit is shown. The TM0 of SUR colored in middle grey, while the rest of SUR is colored in dark grey.
In the simulations with 6C3O and the 6C3P, 11 and 12 interaction clusters can be identified, respectively. Importantly, clusters 1, 4, 5, and 10 are identical between the structures and hence are detected independently of the starting configuration of the simulation. Cluster 1 is located between the POPC membrane and TMD1 as well as TMD2, whereby the observed binding poses deviate from each other to a greater extent than in other clusters. This site is quite frequently occupied, as it is found by 2 travoprost molecules and 3 latanoprost molecules, as well as 1 travoprost and 4 latanoprost molecules in simulations with 6C3O and 6C3P, respectively. Clusters 4 and 5 are located approximately at the level of the intracellular boundary of the membrane. Cluster 4 is located at the interface of TMD0 and TMD1 and is only occupied by travoprost molecules (1 in 6C3O, 4 in 6C3P), while both travoprost and latanoprost are seen in cluster 5 at the surface of TMD2 (1 travoprost and 2 latanoprosts molecules in 6C3O, 1 latanoprost molecule in 6C3P). In simulations with the 6C3O structure, the latter interaction site is framed by TMD2 and the KIR channel, whereas the more extended, “propeller”-like conformation of 6C3P places this binding site between SUR and the membrane. Another interaction site observed for both 6C3O and 6C3P is cluster 10, which is located at the cytoplasmic part of SUR on the TMD2.
The rest of the interaction clusters are only seen for either 6C3O or 6C3P. For 6C3O, interaction cluster 3 is well occupied (2 travoprost molecules, 3 latanoprost molecules) and contains a range of different binding poses at the surface of TMD1. Another interaction, shown in cluster 2, is located at TMD1 approximately at the same level as cluster 1 and is only observed for latanoprost. The binding pose shown in cluster 6 is found by one travoprost molecule and is in direct vicinity of cluster 4, while interaction cluster 7 is located below the well-populated cluster 3 at the surface of TMD2. Furthermore, a variety of affinity sites (clusters 8, 9 and 11) were identified at the surface of the cytoplasmic domain.
In the simulations with 6C3P, interaction cluster 2 stands out since it is the only cluster in direct contact with the KIR6.2 channel pore. This site is occupied by one travoprost molecule in all simulations with 6C3P and is sandwiched between the membrane and the KATP channel, where it makes contact with both the TM2 of KIR and the TMD0 of SUR. Cluster 3 is approximately on the same level as cluster 4, interacting with TMD1 in vicinity of TMD0. The interaction cluster 8 is positioned at the surface of TMD0 and TMD1 near the cytoplasmic boundary of the membrane, and separated by the lower ends of these two interacting helices. Cluster 6 is close to the ATP binding site on the KIR subunit. Two latanoprost as well as one travoprost molecule and one latanoprost molecule are occupying the interaction sites in cluster 6 and 8, respectively. Cluster 9 is below cluster 3 found in the simulations with 6C3O, but is only detected by one travoprost molecule, whereas two additional binding poses are seen at the lower cytoplasmic end of SUR, represented as clusters 11 and 12. Interestingly, cluster 11 is close to the ATP binding site at the degenerate nucleotide binding domain of the SUR subunit (cite Lee et al., 2017). Further information about the number of drug molecules in each interaction cluster as well as distance measurements and contacting residues are summarized in Supplementary Table S3.
Finally, fluvastatin [no 2 on Chen et al. (2019) hit list], ethosuximide and pazopanib were tested. Ethosuximide is a KIR2.1 and KIR3 inhibitor with favorable properties for crossing the blood brain barrier (Colombo et al., 2008; Kobayashi et al., 2009; Huang and Kuo 2015). Clinically, pazopanib induces hypoglycemia as an adverse effect in 0.1–1% of patients. All three compounds did not significantly inhibit KIR6.2/SUR2a current (approximately 20% inhibition for fluvastatin and ethosuximide at 100 μM, and approximately 40% inhibition at 30 μM for pazopanib) (Figure 9).
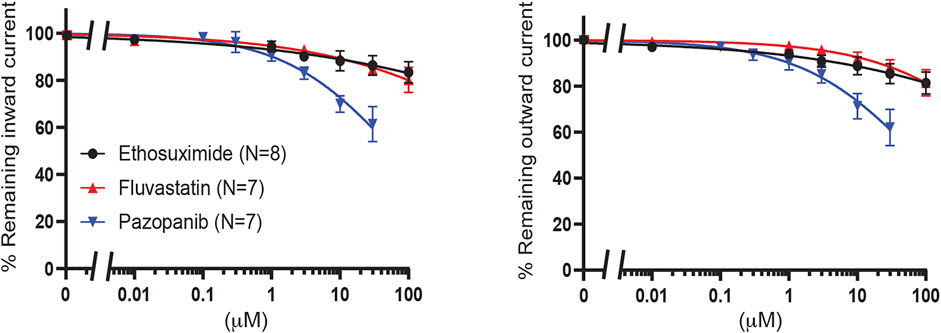
FIGURE 9. Evaluation of KIR6.2/SUR2a dependent currents in response to ethosuximide, fluvastatin and pazopanib. IC50 curves of WT KIR6.2/SUR2a inward (at −80 mV), left panel, and outward (at +50 mV), right panel, current inhibition in response to increasing concentrations drugs. Data were fitted with Hill equation to estimate the IC50 values. Data are shown as mean ± SEM. N depicts number of measurements on independent inside-out patches.
In this work, we use a combination of electrophysiology and molecular modelling in order to investigate putative binding sites of betaxolol, travoprost and latanoprost as well as the molecular mechanism underlying KATP channel inhibition by these compounds. Inside-out recordings presented here were performed in the absence of Mg-ATP and ADP in the bath solution, to best mimic the MD conditions. This most likely resulted in 6–11 fold higher glibenclamide IC50 values than in the presence of 0.15 mM MgATP (Houtman et al., 2019), as seen also in measurements of tolbutamide block in presence and absence of ATP (Miyamura et al., 2000). Nevertheless, Q52R and L164P displayed reduced glibenclamide sensitivity as expected, which therefore is a MgATP independent characteristic. This latter finding also applies to the reduced travoprost mediated inhibition sensitivity as observed for the Q52R mutant.
We established the effects of each compound on both the inward and outward component of the IKIR6.2/SUR2a current. From a physiological perspective, the outward component is most relevant, but when comparing the voltage-dependence of block (inward vs. outward) insights can be gained on the nature of block, e.g., direct pore block vs. inference in PIP2-channel interaction.
MD simulations with betaxolol were carried out in presence of the KIR6.2 channel pore without SUR1. The motivation for this setup is twofold: firstly, electrophysiological experiments show that betaxolol inhibits the WT as well as many of the tested mutations, including L164P and Q52R, in a similar fashion (Figure 3A). Secondly, the mutations L164P and Q52R were reported to impair the coupling of the KIR channel to SUR (Tammaro et al., 2008; Tammaro et al., 2006, Pratt et al., 2012) and to be partially resistant to inhibition by SUR-binding sulfonylureas (Pearson, et al., 2006; Tammaro et al., 2008). Since an allosteric inhibition would require drug binding to overcome the disruptive effect of both mutations in a similar fashion, we deem it likely that betaxolol blocks the open channel pore instead of inhibiting the channel allosterically. In addition, the Hill coefficient of -0.98 for WT channels is in line with pore block. Assuming a pore block, it is sensible to simulate the KATP complex without the SUR subunits, since this reduction of the system size drastically decreases the computational cost of simulating the system.
Coarse-grained and atomistic MD simulations show binding of betaxolol at the transmembrane cavity and near the G-loop gate, where it plugs the ion conduction pathway of the KATP channel (Figures 4C,D, 5E,G). A resembling blockage can be commonly encountered in the KIR2.x channel family, where a range of compounds, including pentamidine (De Boer et al., 2010), quinidine (Koepple et al., 2017) and chloroquine (Rodriguez-Menchaca et al., 2008), have been shown to prevent ion flux by blocking the cytoplasmic pore.
The betaxolol binding sites at the transmembrane cavity and near the G-loop gate are situated in vicinity to the disease mutation L164P and could generally explain why betaxolol effectively inhibits this mutant. In a previous study, we predicted that the inserted proline mainly leads to a widening of the pore and increased solvation of the gates, thereby preventing proper closure of the channel (Bründl et al., 2021). Thus, binding of the drug to the inner cavity and/or the G-loop gate would prevent ion flux by blocking the permeation path through the channel.
The disease mutation Q52R is located in the loop connecting the slide helix with the C-terminal domain (CTD) of the of KIR channel, in vicinity to the PIP2 binding site (Puljung, 2018) (Figure 1A). Furthermore, the 6C3P structure shows that Q52 extends towards the cytoplasmic loop L0 of SUR1, where it resides within 5 Å of residue E203 (Figure 1A). The role of the interaction between Q52- KIR6.2 and E203-SUR1 in mediating KIR6.2 gating by SUR was confirmed by a mutation study, which demonstrated that the crosslinking of E203C in SUR1 and Q52C in KIR6.2 via a Cys-Cys bridge locks the channel in a closed state (Pratt et al., 2012). However, the effect of the Q52R mutation on the channel structure is currently unclear and thus, needs to be elucidated in future investigations. Nevertheless, putative pore blockers such as betaxolol and levobetaxolol can inhibit this mutant as well, and therefore might provide a basis for drug development for sulfonyl-urea resistant DEND mutations.
Nowadays, the most successful therapy for the DEND syndrome is based on KATP channel inhibition by sulfonylureas, which bind to the SUR subunits and exert inhibition in an allosteric manner. The inability of sulfonylureas to cross the blood-brain-barrier and alleviate the neurological problems of the disease, as well as the fact that some mutations are resistant to these compounds call for the development of a more diverse set of KATP inhibitors (Kharade et al., 2016). Betaxolol is different from the sulfonylureas in that it blocks the ion conduction pathway, and thus can be counted among a small set of KATP channel inhibitors targeting the channel pore. Other compounds with an affinity to the KIR channel are verapamil (Ninomiya et al., 2003), tamoxifen (Ponce-Balbuena et al., 2010) and rosiglitazone (Yu et al., 2012), whereby the latter two have been reported to interfere with the channel activation by PIP2. In addition to the development of chemically based inward rectifier channel inhibitors and activators (Van der Schoor et al., 2020; Walsh, 2020), progress is made also in the field of short peptide toxins as channel inhibitors (Doupnik, 2017). The small protein SpTx-1 was shown to inhibit a range of permanent ND causing mutations, including Q52R, by binding to KIR6.2 (Ramu et al., 2018). Furthermore, the recently studied centipede toxin, SsTx-4, displayed low nanomolar IC50 values for KIR6.2/SUR1 channels, including the Q52R mutant channel (Tang et al., 2021).
Upon oral dosing (40 mg, 15 days p.o.), betaxolol plasmalevels of approximately 0.54 μM were found (Lipworth et al., 1991). Whereas IC50 values around 30 μM were observed for KIR6.2/SUR2a outward current, approximately 20% reduction of outward current is already found at 5 μM. Drugs with an affinity to the KIR6.2 channel pore might therefore provide a powerful tool to inhibit sulfonylurea-resistant DEND mutations, which makes betaxolol a promising starting point for the development of a novel medication against this syndrome.
However, it should be mentioned that betaxolol is not able to cross the blood-brain-barrier (BBB) to a meaningful extent and thus, is unlikely to suppress the neurological and developmental symptoms associated with the DEND syndrome.
In addition to pore blockage, the simulations unraveled another interaction site for betaxolol, which is located near the binding site of PIP2. In support of this finding, certain drugs were reported affecting KIR channels by blocking the pore and simultaneously exerting an allosteric inhibition by interacting with to the PIP2 binding site (López-Izquierdo et al., 2011; Koepple et al., 2017; Scherer et al., 2017). One of these compounds is chloroquine, which has been shown to inhibit KIR6.2 containing KATP channels in this two-fold manner (Ponce-Balbuena et al., 2012). A fast voltage-dependent inhibition observed at positive potentials was suggested to originate in a blockage of the cytoplasmic channel pore, while a slower inhibition might stem from an impaired channel activation by PIP2 caused by drug interference with this site. The experimentally observed voltage dependence of KATP inhibition by betaxolol (Chen et al., 2019, and current data) could be explained by a channel block in the central cavity region or close to the G-loop, and therefore supports the hypothesis of betaxolol being a KATP pore blocker. However, drug binding near the PIP2 binding site might additionally influence the conductivity, as it is seen for chloroquine, and we therefore cannot exclude an allosteric effect involved in KATP channel inhibition by betaxolol.
Travoprost and latanoprost are both synthetic analogues of prostaglandin, which belongs to the group of eicosanoids. In coarse-grained MD simulations with the KATP channel, travoprost and latanoprost exhibited a high affinity to the SUR subunits. Our functional measurements demonstrate that travoprost, and to a lesser extent latanoprost, inhibits both inward and outward components of the WT IKATP channels, in contrast to betaxolol which displays voltage dependent block (outward >> inward). Based on these observations, we consider it more likely that travoprost and latanoprost act by binding to SUR1 rather than to the KIR6.2 channel pore. The variety of interactions observed for both drugs could be interpreted as the presence of multiple binding sites on the SUR subunits. Although the data obtained from the MD simulations indicates the presence of multiple prostaglandin interaction sites with similar affinity, we cannot exclude the existence of one or more high affinity binding sites.
Interestingly, in the simulations of 6C3P, we found two binding clusters near sites important for KATP regulation. Cluster 6 shows binding of travoprost and latanoprost in proximity to the inhibitory ATP binding site at the KIR subunit. Cluster 11 shows binding of travoprost to the degenerate site of the SUR subunit. Since ATP binding to either KIR or SUR exhibits regulatory effects, potential binding of compounds to or close to these binding sites might compete with or change the ability of ATP regulation of this channel. However, since the details of KATP regulation are still unknown, the mechanism by which these compounds modulate the channel remains obscure.
In the context of the DEND syndrome, a therapeutic approach based on a drug with binding affinity to SUR might not be the best choice. The reason for this is that some of the DEND syndrome causing mutations, including Q52R and L164P, are known to impair the coupling of SUR to the KIR channel pore (Tammaro et al., 2006; Tammaro et al., 2008, Pratt et al., 2012). This could undermine the effect of a SUR-bound KATP channel inhibitor and adds an unwanted factor of uncertainty in predicting the efficiency of the inhibition. A further characterization of the binding behavior of this drug class to KATP channels might be interesting for the treatment of other KATP channelopathies.
Fluvastatin is on the Chen hitlist (no 2), but did not result in KIR6.2/SUR2a inhibition. Pharmacophore based database searches provides enrichment of hit rates compared to random sampling, but does comes with false positives (Seidel et al., 2019), like in this case fluvastatin.
A 2010 case report described improved glycemic control in a prostate cancer patient, who also presented glibenclamide resistant Type 2 diabetes, upon treatment with pazopanib (400 mg/p.d.) (Böhm et al., 2010). The authors suggested that pazopanib mediated inhibition of PDGF-receptor signaling is the key factor in this finding. Our result of a non-significant change in IKIR6.2/SUR2a upon application of 30 μM pazopanib does not disagree with their suggestion. However, at a daily dosing of 800 mg, peak plasma concentrations were found as high as 132 μM (NIH Daily Med 2014). We did not perform functional measurements at such concentrations.
Our study comes with several limitations. A shortcoming of our study is that we exclusively simulate channels with a closed pore module due to a lack of open KATP structures. Two studies, which modelled the opening of KIR2 channels either by forcing the channel into a conductive state (Li et al., 2015) or by introducing channel-opening mutations (Zangerl-Plessl et al., 2020), identified motions in the CTD, bending of the inner transmembrane helix (TM2), and a widening at the Helix Bundle Crossing (HBC) gate to be associated with the acquisition of an activated state. However, a rearrangement of the residues lining the pore of the channel was not reported. Based on these findings, one can expect that the residues in the TM cavity remain available for drug interaction independently of the conductive state of the channel and thus, the betaxolol block at this site to be relatively unchanged upon channel opening. Nevertheless, channel opening was accompanied by structural changes at the G-loop region in targeted MD simulations (Li et al., 2015). Hence, the possibility of an altered binding behavior of betaxolol near the G-loop gate in the open channel should not be neglected. A further limitation to the closed crystal structure is that betaxolol is not able to interact with the HBC gate, and we therefore cannot assess the interplay of the compound with the gate comprising residue F168 in the simulations. Secondly, a number of symptoms, including muscle weakness, of the DEND syndrome is generally considered to involve the KIR6.2/SUR1 channels (Clark et al., 2010). Our experimental evaluation uses KIR6.2/SUR2a channels instead. It was shown that the SUR isoform is important to confer the cellular metabolic status to IKATP activity of a mutant channel (Clark et al., 2010). In our inside-out measurements we did not took the metabolic status into account, as Mg-ATP and ADP were not included in the bath solution. Therefore, we reason that the SUR isoform is not essential in our current experimental evaluations. Moreover, changes in Q52R single channel kinetics are independent of SUR isoform (Tammaro et al., 2006). Finally, we did not confirm potential binding sites as suggested by MD by mutational analysis in the current study, which could be considered as a limitation.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
MJH, TF, and XC performed research. MJH, TF, E-MZ-P, MAH, and AS-W analysed the results. MAH and AS-W designed the study. MAH and TF wrote the paper. All the authors reviewed and approved the final manuscript.
This work was supported by the doctoral program “Molecular drug targets” W1232 of the Austrian Science Fund (FWF; http://www.fwf.ac.at), the DOC Fellowship of the Austrian Academy of Sciences 26156 and from the Hochschuljubiläumsstiftung der Stadt Wien (H-269241/2020). E-MZ-P received funding from the Zukunftskolleg program, grant number ZK-81B, from the Austrian Science Fund.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The computational results presented have been achieved in part using the Vienna Scientific Cluster (VSC).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2021.814066/full#supplementary-material
Abraham, M. J., Murtola, T., Schulz, R., Páll, S., Smith, J. C., Hess, B., et al. (2015). Gromacs: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 1-2, 19–25. doi:10.1016/j.softx.2015.06.001
Ashcroft, F. M. (2005). ATP-sensitive Potassium Channelopathies: Focus on Insulin Secretion. J. Clin. Invest. 115, 2047–2058. doi:10.1172/JCI25495
Ashcroft, F. M., Puljung, M. C., and Vedovato, N. (2017). Neonatal Diabetes and the KATP Channel: From Mutation to Therapy. Trends Endocrinol. Metab. 28, 377–387. doi:10.1016/j.tem.2017.02.003
Babiker, T., Vedovato, N., Patel, K., Thomas, N., Finn, R., Männikkö, R., et al. (2016). Successful Transfer to Sulfonylureas in KCNJ11 Neonatal Diabetes Is Determined by the Mutation and Duration of Diabetes. Diabetologia 59, 1162–1166. doi:10.1007/s00125-016-3921-8
Berendsen, H. J. C., Grigera, J. R., and Straatsma, T. P. (1987). The Missing Term in Effective Pair Potentials. J. Phys. Chem. 91, 6269–6271. doi:10.1021/j100308a038
Berger, O., Edholm, O., and Jähnig, F. (1997). Molecular Dynamics Simulations of a Fluid Bilayer of Dipalmitoylphosphatidylcholine at Full Hydration, Constant Pressure, and Constant Temperature. Biophys. J. 72, 2002–2013. doi:10.1016/S0006-3495(97)78845-3
Böhm, S., Hess, D., Gillessen, S., and Brändle, M. (2010). Improved Glycemic Control with the Multi-Receptor Tyrosine Kinase Inhibitor Pazopanib. Diabetes Care 33, e82. doi:10.2337/dc10-0436
Bründl, M., Pellikan, S., and Stary-Weinzinger, A. (2021). Simulating PIP2-Induced Gating Transitions in Kir6.2 Channels. Front. Mol. Biosci. 8, 711975. doi:10.3389/fmolb.2021.711975
Bussi, G., Donadio, D., and Parrinello, M. (2007). Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 126, 014101. doi:10.1063/1.2408420
Chen, X., Garon, A., Wieder, M., Houtman, M. J. C., Zangerl-Plessl, E. M., Langer, T., et al. (2019). Computational Identification of Novel Kir6 Channel Inhibitors. Front. Pharmacol. 10, 549. doi:10.3389/fphar.2019.00549
Clark, R. H., McTaggart, J. S., Webster, R., Mannikko, R., Iberl, M., Sim, X. L., et al. (2010). Muscle Dysfunction Caused by a KATP Channel Mutation in Neonatal Diabetes Is Neuronal in Origin. Science 329, 458–461. doi:10.1126/science.1186146
Colombo, S., Petri, S., Shalomov, B., Reddy, H. P., Tabak, G., Dhindsa, R. S., et al. (2008). G Protein-Coupled Potassium Channels Implicated in Mouse and Cellular Models of GNB1 Encephalopathy. BioRxivPreprint. Available at: https://https://www.biorxiv.org/content/10.1101/697235v1 (Accessed August 30, 2021).
De Boer, T. P., Nalos, L., Stary, A., Kok, B., Houtman, M. J., Antoons, G., et al. (2010). The Anti-protozoal Drug Pentamidine Blocks KIR2.X-Mediated Inward Rectifier Current by Entering the Cytoplasmic Pore Region of the Channel. Br. J. Pharmacol. 159, 1532–1541. doi:10.1111/j.1476-5381.2010.00658.x
De Wet, H., and Proks, P. (2015). Molecular Action of Sulphonylureas on KATP Channels: a Real Partnership between Drugs and Nucleotides. Biochem. Soc. Trans. 43, 901–907. doi:10.1042/BST20150096
Doupnik, C. A. (2017). Venom-derived Peptides Inhibiting Kir Channels: Past, Present, and Future. Neuropharmacology 127, 161–172. doi:10.1016/j.neuropharm.2017.07.011
Flanagan, S. E., Edghill, E. L., Gloyn, A. L., Ellard, S., and Hattersley, A. T. (2006). Mutations in KCNJ11, Which Encodes Kir6.2, Are a Common Cause of Diabetes Diagnosed in the First 6 Months of Life, with the Phenotype Determined by Genotype. Diabetologia 49, 1190–1197. doi:10.1007/s00125-006-0246-z
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2013). Gaussian 09, Revision D.01. Wallingford CT: Gaussian. Inc.
Gloyn, A. L., Pearson, E. R., Antcliff, J. F., Proks, P., Bruining, G. J., Slingerland, A. S., et al. (2004). Activating Mutations in the Gene Encoding the ATP-Sensitive Potassium-Channel Subunit Kir6.2 and Permanent Neonatal Diabetes. N. Engl. J. Med. 350, 1838–1849. doi:10.1056/NEJMoa032922
Gowers, R., Linke, M., Barnoud, J., Reddy, T., Melo, M., Seyler, S., et al. (2016). “MDAnalysis: A Python Package for the Rapid Analysis of Molecular Dynamics Simulations,” in Proceedings of the 15th Python in Science Conference. Editors S. Benthall, and S. Rostrup (Austin, TX: SciPy), 98–105. doi:10.25080/Majora-629e541a-00e
Harakalova, M., van Harssel, J. J., Terhal, P. A., van Lieshout, S., Duran, K., Renkens, I., et al. (2012). Dominant Missense Mutations in ABCC9 Cause Cantú Syndrome. Nat. Genet. 44, 793–796. doi:10.1038/ng.2324
Hashimoto, Y., Dateki, S., Hirose, M., Satomura, K., Sawada, H., Mizuno, H., et al. (2017). Molecular and Clinical Features of KATP -channel Neonatal Diabetes Mellitus in Japan. Pediatr. Diabetes 18, 532–539. doi:10.1111/pedi.12447
Hattersley, A. T., and Ashcroft, F. M. (2005). Activating Mutations in Kir6.2 and Neonatal Diabetes: New Clinical Syndromes, New Scientific Insights, and New Therapy. Diabetes 54, 2503–2513. doi:10.2337/diabetes.54.9.2503
Helmi, M., and Hussain, S. (2020). Severe Developmental Delay, Epilepsy and Neonatal Diabetes (DEND) Syndrome: A Case Report. J. ASEAN Fed. Endocr. Soc. 35, 125–128. doi:10.15605/jafes.035.01.22
Hibino, H., Inanobe, A., Furutani, K., Murakami, S., Findlay, I., and Kurachi, Y. (2010). Inwardly Rectifying Potassium Channels: Their Structure, Function, and Physiological Roles. Physiol. Rev. 90, 291–366. doi:10.1152/physrev.00021.2009
Hornak, V., Abel, R., Okur, A., Strockbine, B., Roitberg, A., and Simmerling, C. (2006). Comparison of Multiple Amber Force fields and Development of Improved Protein Backbone Parameters. Proteins 65, 712–725. doi:10.1002/prot.21123
Houtman, M. J. C., Chen, X., Qile, M., Duran, K., van Haaften, G., Stary-Weinzinger, A., et al. (2019). Glibenclamide and HMR1098 Normalize Cantú Syndrome-Associated Gain-Of-Function Currents. J. Cel Mol Med 23, 4962–4969. doi:10.1111/jcmm.14329
Hsu, P. C., Bruininks, B. M. H., Jefferies, D., Cesar Telles de Souza, P., Lee, J., Patel, D. S., et al. (2017). CHARMM-GUI Martini Maker for Modeling and Simulation of Complex Bacterial Membranes with Lipopolysaccharides. J. Comput. Chem. 38, 2354–2363. doi:10.1002/jcc.24895
Huang, C. W., and Kuo, C. C. (2015). Flow- and Voltage-dependent Blocking Effect of Ethosuximide on the Inward Rectifier K⁺ (Kir2.1) Channel. Pflugers Arch. 467, 1733–1746. doi:10.1007/s00424-014-1611-2
Humphrey, W., Dalke, A., and Schulten, K. (1996). VMD: Visual Molecular Dynamics. J. Mol. Graph 14, 2728–338. doi:10.1016/0263-7855(96)00018-5
Ioacara, S., Flanagan, S., Fröhlich-Reiterer, E., Goland, R., and Fica, S. (2017). First Case of Neonatal Diabetes with KCNJ11 Q52R Mutation Successfully Switched from Insulin to Sulphonylurea Treatment. J. Diabetes Investig. 8, 716–719. doi:10.1111/jdi.12620
Jo, S., Kim, T., Iyer, V. G., and Im, W. (2008). CHARMM-GUI: a Web-Based Graphical User Interface for CHARMM. J. Comput. Chem. 29, 1859–1865. doi:10.1002/jcc.20945
Kharade, S. V., Nichols, C., and Denton, J. S. (2016). The Shifting Landscape of KATP Channelopathies and the Need for 'sharper' Therapeutics. Future Med. Chem. 8, 789–802. doi:10.4155/fmc-2016-0005
Kobayashi, T., Hirai, H., Iino, M., Fuse, I., Mitsumura, K., Washiyama, K., et al. (2009). Inhibitory Effects of the Antiepileptic Drug Ethosuximide on G Protein-Activated Inwardly Rectifying K+ Channels. Neuropharmacology 56, 499–506. doi:10.1016/j.neuropharm.2008.10.003
Koepple, C., Scherer, D., Seyler, C., Scholz, E., Thomas, D., Katus, H. A., et al. (2017). Dual Mechanism for Inhibition of Inwardly Rectifying Kir2.X Channels by Quinidine Involving Direct Pore Block and PIP2-Interference. J. Pharmacol. Exp. Ther. 361, 209–218. doi:10.1124/jpet.116.238287
Korb, O., Stützle, T., and Exner, T. E. (2009). Empirical Scoring Functions for Advanced Protein-Ligand Docking with PLANTS. J. Chem. Inf. Model. 49, 84–96. doi:10.1021/ci800298z
Koster, J. C., Remedi, M. S., Dao, C., and Nichols, C. G. (2005). ATP and Sulfonylurea Sensitivity of Mutant ATP-Sensitive K+ Channels in Neonatal Diabetes: Implications for Pharmacogenomic Therapy. Diabetes 54, 2645–2654. doi:10.2337/diabetes.54.9.2645
Kusalik, P. G., and Svishchev, I. M. (1994). The Spatial Structure in Liquid Water. Science 265, 1219–1221. doi:10.1126/science.265.5176.1219
Lee, K. P. K., Chen, J., and MacKinnon, R. (2017). Molecular Structure of Human KATP in Complex with ATP and ADP. Elife 6, e32481. doi:10.7554/eLife.32481
Li, J., Lü, S., Liu, Y., Pang, C., Chen, Y., Zhang, S., et al. (2015). Identification of the Conformational Transition Pathway in PIP2 Opening Kir Channels. Sci. Rep. 5, 11289. doi:10.1038/srep11289
Lipworth, B. J., Irvine, N. A., and McDevitt, D. G. (1991). The Effects of Chronic Dosing on the Beta 1 and Beta 2-adrenoceptor Antagonism of Betaxolol and Atenolol. Eur. J. Clin. Pharmacol. 40, 467–471. doi:10.1007/BF00315224
López-Izquierdo, A., Aréchiga-Figueroa, I. A., Moreno-Galindo, E. G., Ponce-Balbuena, D., Rodríguez-Martínez, M., Ferrer-Villada, T., et al. (2011). Mechanisms for Kir Channel Inhibition by Quinacrine: Acute Pore Block of Kir2.X Channels and Interference in PIP2 Interaction with Kir2.X and Kir6.2 Channels. Pflugers Arch. 462, 505–517. doi:10.1007/s00424-011-0995-5
Marrink, S. J., Risselada, H. J., Yefimov, S., Tieleman, D. P., and De Vries, A. H. (2007). The MARTINI Force Field: Coarse Grained Model for Biomolecular Simulations. J. Phys. Chem. B 111, 7812–7824. doi:10.1021/jp071097f
Martini, (2021). General Purpose Coarse-Grained Force Field. Available at: http://cgmartini.nl (Assessed on November 11, 2021).
Michaud-Agrawal, N., Denning, E. J., Woolf, T. B., and Beckstein, O. (2011). MDAnalysis: a Toolkit for the Analysis of Molecular Dynamics Simulations. J. Comput. Chem. 32, 2319–2327. doi:10.1002/jcc.21787
Miyamura, A., Kakei, M., Ichinari, K., Okamura, M., Oketani, N., and Tei, C. (2000). On the Mechanism of ADP-Induced Alteration of Sulphonylurea Sensitivity in Cardiac ATP-Sensitive K(+) Channels. Br. J. Pharmacol. 130, 1411–1417. doi:10.1038/sj.bjp.0703423
Monticelli, L., Kandasamy, S. K., Periole, X., Larson, R. G., Tieleman, D. P., and Marrink, S. J. (2008). The MARTINI Coarse-Grained Force Field: Extension to Proteins. J. Chem. Theor. Comput 4, 819–834. doi:10.1021/ct700324x
Nih DailyMed, (2014). Current Medication Information for Votrient (Pazopanib Hydrochloride) Tablet, Film Coated (Revised: June 2014). Availableat: https://dailymed.nlm.nih.gov/dailymed/lookup.cfm?setid=f5b7b3a4-c3a4-4722-8ca5-8f6a5c622553 (Assessed on August 30, 2021).
Ninomiya, T., Takano, M., Haruna, T., Kono, Y., and Horie, M. (2003). Verapamil, a Ca2+ Entry Blocker, Targets the Pore-Forming Subunit of Cardiac Type KATP Channel (Kir6.2). J. Cardiovasc. Pharmacol. 42, 161–168. doi:10.1097/00005344-200308000-00002
Parrinello, M., and Rahman, A. (1981). Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 52, 7182–7190. doi:10.1063/1.328693
Pearson, E. R., Flechtner, I., Njølstad, P. R., Malecki, M. T., Flanagan, S. E., Larkin, B., et al. Neonatal Diabetes International Collaborative Group (2006). Switching from Insulin to Oral Sulfonylureas in Patients with Diabetes Due to Kir6.2 Mutations. N. Engl. J. Med. 355, 467–477. doi:10.1056/NEJMoa061759
Periole, X., Cavalli, M., Marrink, S. J., and Ceruso, M. A. (2009). Combining an Elastic Network with a Coarse-Grained Molecular Force Field: Structure, Dynamics, and Intermolecular Recognition. J. Chem. Theor. Comput 5, 2531–2543. doi:10.1021/ct9002114
Ponce-Balbuena, D., Moreno-Galindo, E. G., López-Izquierdo, A., Ferrer, T., and Sánchez-Chapula, J. A. (2010). Tamoxifen Inhibits Cardiac ATP-Sensitive and Acetylcholine-Activated K+ Currents in Part by Interfering with Phosphatidylinositol 4,5-Bisphosphate-Channel Interaction. J. Pharmacol. Sci. 113, 66–75. doi:10.1254/jphs.10024fp
Ponce-Balbuena, D., Rodríguez-Menchaca, A. A., López-Izquierdo, A., Ferrer, T., Kurata, H. T., Nichols, C. G., et al. (2012). Molecular Mechanisms of Chloroquine Inhibition of Heterologously Expressed Kir6.2/SUR2A Channels. Mol. Pharmacol. 82, 803–813. doi:10.1124/mol.112.079152
Pratt, E. B., Zhou, Q., Gay, J. W., and Shyng, S. L. (2012). Engineered Interaction Between SUR1 and Kir6.2 That Enhances ATP Sensitivity in KATP Channels. J Gen Physiol. 140, 175–187. doi:10.1085/jgp.201210803
Proks, P., Reimann, F., Green, N., Gribble, F., and Ashcroft, F. (2002). Sulfonylurea Stimulation of Insulin Secretion. Diabetes 51 (Suppl. 3), S368–S376. doi:10.2337/diabetes.51.2007.s368
Puljung, M. C. (2018). Cryo-electron Microscopy Structures and Progress toward a Dynamic Understanding of KATP Channels. J. Gen. Physiol. 150, 653–669. doi:10.1085/jgp.201711978
Qi, Y., Ingólfsson, H. I., Cheng, X., Lee, J., Marrink, S. J., and Im, W. (2015). CHARMM-GUI Martini Maker for Coarse-Grained Simulations with the Martini Force Field. J. Chem. Theor. Comput 11, 4486–4494. doi:10.1021/acs.jctc.5b00513
Ramu, Y., Xu, Y., and Lu, Z. (2018). A Novel High-Affinity Inhibitor against the Human ATP-Sensitive Kir6.2 Channel. J. Gen. Physiol. 150, 969–976. doi:10.1085/jgp.201812017
Rodríguez-Menchaca, A. A., Navarro-Polanco, R. A., Ferrer-Villada, T., Rupp, J., Sachse, F. B., Tristani-Firouzi, M., et al. (2008). The Molecular Basis of Chloroquine Block of the Inward Rectifier Kir2.1 Channel. Proc. Natl. Acad. Sci. U S A. 105, 1364–1368. doi:10.1073/pnas.0708153105
Scherer, D., Schworm, B., Seyler, C., Xynogalos, P., Scholz, E. P., Thomas, D., et al. (2017). Inhibition of Inwardly Rectifying Kir2.X Channels by the Novel Anti-cancer Agent Gambogic Acid Depends on Both Pore Block and PIP2 Interference. Naunyn Schmiedebergs Arch. Pharmacol. 390, 701–710. doi:10.1007/s00210-017-1372-5
Seidel, T., Schuetz, D. A., Garon, A., and Langer, T. (2019). The Pharmacophore Concept and its Applications in Computer-Aided Drug Design. Prog. Chem. Org. Nat. Prod. 110, 99–141. doi:10.1007/978-3-030-14632-0_4
Shaw, N. D., and Majzoub, J. A. (2009). Permanent Neonatal Diabetes in a Patient with a KCNJ11/Q52R Mutation Accompanied by Intermittent Hypoglycemia and Liver Failure. Int. J. Pediatr. Endocrinol. 2009, 453240. doi:10.1155/2009/453240
Sikimic, J., McMillen, T. S., Bleile, C., Dastvan, F., Quast, U., Krippeit-Drews, P., et al. (2019). ATP Binding without Hydrolysis Switches Sulfonylurea Receptor 1 (SUR1) to Outward-Facing Conformations that Activate KATP Channels. J. Biol. Chem. 294, 3707–3719. doi:10.1074/jbc.RA118.005236
Tammaro, P., Flanagan, S. E., Zadek, B., Srinivasan, S., Woodhead, H., Hameed, S., et al. (2008). A Kir6.2 Mutation Causing Severe Functional Effects In Vitro Produces Neonatal Diabetes without the Expected Neurological Complications. Diabetologia 51, 802–810. doi:10.1007/s00125-008-0923-1
Tammaro, P., Proks, P., and Ashcroft, F. M. (2006). Functional Effects of Naturally Occurring KCNJ11 Mutations Causing Neonatal Diabetes on Cloned Cardiac KATP Channels. J. Physiol. 571, 3–14. doi:10.1113/jphysiol.2005.099168
Tang, D., Xu, J., Li, Y., Zhao, P., Kong, X., Hu, H., et al. (2021). Molecular Mechanisms of Centipede Toxin SsTx-4 Inhibition of Inwardly Rectifying Potassium Channels. J. Biol. Chem. 297, 101076. doi:10.1016/j.jbc.2021.101076
Van der Schoor, L., van Hattum, E. J., de Wilde, S. M., Harlianto, N. I., van Weert, A. J., Bloothooft, M., et al. (2020). Towards the Development of AgoKirs: New Pharmacological Activators to Study Kir2.X Channel and Target Cardiac Disease. Int. J. Mol. Sci. 21, 5746. doi:10.3390/ijms21165746
Verdonk, M. L., Cole, J. C., Hartshorn, M. J., Murray, C. W., and Taylor, R. D. (2003). Improved Protein-Ligand Docking Using GOLD. Proteins 52, 609–623. doi:10.1002/prot.10465
Walsh, K. B. (2020). Screening Technologies for Inward Rectifier Potassium Channels: Discovery of New Blockers and Activators. SLAS Discov. 25, 420–433. doi:10.1177/2472555220905558
Wang, J., Wang, W., Kollman, P. A., and Case, D. A. (2006). Automatic Atom Type and Bond Type Perception in Molecular Mechanical Calculations. J. Mol. Graph Model. 25, 247–260. doi:10.1016/j.jmgm.2005.12.005
Wang, J., Wolf, R. M., Caldwell, J. W., Kollman, P. A., and Case, D. A. (2004). Development and Testing of a General Amber Force Field. J. Comput. Chem. 25, 1157–1174. doi:10.1002/jcc.20035
Yu, L., Jin, X., Cui, N., Wu, Y., Shi, Z., Zhu, D., et al. (2012). Rosiglitazone Selectively Inhibits K(ATP) Channels by Acting on the K(IR) 6 Subunit. Br. J. Pharmacol. 167, 26–36. doi:10.1111/j.1476-5381.2012.01934.x
Keywords: SUR2, DEND syndrome, betaxolol, travoprost, patch clamp, KIR6.2, molecular dynamics
Citation: Houtman MJC, Friesacher T, Chen X, Zangerl-Plessl E-M, Heyden MAGvd and Stary-Weinzinger A (2022) Development of IKATP Ion Channel Blockers Targeting Sulfonylurea Resistant Mutant KIR6.2 Based Channels for Treating DEND Syndrome. Front. Pharmacol. 12:814066. doi: 10.3389/fphar.2021.814066
Received: 12 November 2021; Accepted: 23 December 2021;
Published: 14 January 2022.
Edited by:
Saïd Bendahhou, University of Nice Sophia Antipolis, FranceReviewed by:
Show-Ling Shyng, Oregon Health and Science University, United StatesCopyright © 2022 Houtman, Friesacher, Chen, Zangerl-Plessl, Heyden and Stary-Weinzinger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna Stary-Weinzinger, YW5uYS5zdGFyeUB1bml2aWUuYWMuYXQ=
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.