 Yiwei Zeng
Yiwei Zeng Yunhui Chen
Yunhui Chen Su Zhang1
Su Zhang1 Mengnan Liu
Mengnan Liu- 1College of Acupuncture-moxibustion and Tuina, College of Basic Medicine, College of Nursing, College of Chinese Classics, Chengdu University of Traditional Chinese Medicine, Chengdu, China
- 2Traditional Chinese Medicine Hospital Affiliated to Southwest Medical University, Luzhou, China
- 3School of Humanities, Jiangxi University of Traditional Chinese Medicine, Nanchang, China
Methamphetamine (METH), an amphetamine-type psychostimulant, is highly abused worldwide. Chronic abuse of METH causes neurodegenerative changes in central dopaminergic neurons with numerous neuropsychiatric consequences. Neuronal apoptosis plays a critical role in METH-induced neurotoxicity and may provide promising pharmacological targets for preventing and treating METH addiction. In recent years, accumulating evidence has revealed that natural products may possess significant potentials to inhibit METH-evoked neuronal apoptosis. In this review, we summarized and analyzed the improvement effect of natural products on METH-induced neuronal apoptosis and their potential molecular mechanisms on modulating dopamine release, oxidative stress, mitochondrial-dependent apoptotic pathway, endoplasmic reticulum stress-mediated apoptotic pathway, and neuroinflammation. Hopefully, this review may highlight the potential value of natural products in modulating METH-caused neuronal apoptosis and provide useful information for future research and developments of novel and efficacious pharmacotherapies in this field.
Introduction
Methamphetamine (METH), also known as “ice” or “crystal,” is one of the most widely used and abused illicit amphetamine-type stimulants (Courtney and Ray, 2014; Jones et al., 2020). Statistics reveal that in 2019, approximately 27 million people (0.5% of the global population) reported using METH (UNODC, 2020). The number of METH abusers in China and the United States reached an estimated 1.18 million (out of 2.14 million drug users) and 1.7 million (more than half of them suffering METH use disorder), respectively (CNNCC, 2019; Elinore, 2020). Studies also demonstrate that METH administration is highly prevalent in Australia, Canada, and South Africa (Australian Institute of Health and Welfare, 2020; Bach et al., 2020; Sorsdahl et al., 2021). This highly addictive stimulant drug causes serious societal and public health concerns globally, and considerable attention and effort need to be devoted to researching and developing effective intervention strategies against METH-evoked neurotoxic consequences.
Epidemiological and clinical studies have demonstrated that METH is a potent central nervous system stimulant and can significantly impact the human body physically, behaviorally, cognitively, and psychiatrically (Schep et al., 2010; Kevil et al., 2019). Acute METH intake may cause euphoria, delusions, agitations, hypersexuality, or even hyperthermia, cerebrovascular hemorrhages, arrhythmias, heart attacks, and renal failure in high doses (Courtney and Ray, 2014; Glasner-Edwards and Mooney, 2014; Baradhi et al., 2019; Kevil et al., 2019). Long-term METH abuse elicits neuropsychiatric impairments, including cognitive decline, altered decision-making, psychosis, and psychomotor dysfunction (Krasnova and Cadet, 2009; Dean et al., 2013; Lappin et al., 2018; Mizoguchi and Yamada, 2019). Its withdrawal symptoms in abstinence include depression, agitation, fatigue, and intense craving (Krasnova and Cadet, 2009; Feng et al., 2020). These neuropsychiatric complications are mostly attributed to METH-induced neurotoxicity. Animal-based studies and human neuroimaging analysis have revealed that repeated use of METH can induce neurodegeneration of dopaminergic and serotoninergic terminals in the striatum, cortex, and hippocampus and primarily affects the nigrostriatal dopaminergic pathway (Ernst et al., 2000; Friend and Keefe, 2013; McFadden et al., 2018; Golsorkhdan et al., 2020). METH-evoked neurotoxicity may include direct damage to terminals, oxidative stress, mitochondria dysfunction, neuronal excitotoxicity, endoplasmic reticulum stress, and neuroinflammation, leading to dopaminergic neuron apoptosis (Jayanthi et al., 2004; Yang et al., 2018; Kim et al., 2020). Although substantial progress has been made in deciphering the mechanisms of METH-induced neuronal apoptosis, currently effective pharmacotherapies for METH addiction remain scarce (Yang et al., 2018). Hence, exploration of natural products as potential interventions against METH-elicited complications has become a research hotspot in recent years (Moszczynska and Callan, 2017; Chiang et al., 2019).
Natural products are bioactive components from natural organisms, including plants, animals, insects, marine organisms, microorganisms (Xie et al., 2021). These agents have been widely used by human societies for thousands of years (Baker et al., 2007), and their bioactivities are of great interest to enormous research fields (Ekiert and Szopa, 2020). In recent decades, a growing body of research has demonstrated that natural products exert anti-apoptotic effects on METH-induced neurotoxicity and may yield promising targets for further investigation of effective pharmacological interventions. Converging lines of evidence suggest that natural products can attenuate symptoms of addiction and drug use disorders and modulate the complex mechanisms underlying METH-evoked apoptosis in various ways (Lu et al., 2009). Moreover, METH users have been reported to have an increased risk of developing Parkinson’s disease. Natural products may garner considerable neuroprotective properties and effectively mitigate neurodegeneration of Parkinson’s disease that partially shares pathogenesis with METH-induced apoptosis, including dopamine release, oxidative stress, and mitochondria damage (Rios et al., 2016; Solayman et al., 2017). Their application may merit further research and development as a promising strategy for pharmacological intervention against METH-induced neuronal apoptosis. Hence, to provide useful insights for future research and development of novel and efficacious pharmacotherapies in this field, a systematic and comprehensive review is of necessity.
In this review, electronic databases, including PubMed, EMBASE, Web of Science, Cochrane Library, China National Knowledge Infrastructure, China Biomedical Literature Database, Wanfang Database, and Chinese Scientific Journals Database, were searched from the inception to August 2021 to identify eligible studies. All the included individual studies were assessed according to the Best practice in research—Overcoming common challenges in phytopharmacological research (Heinrich et al., 2020). The anti-apoptotic effects of natural products on METH-evoked neurotoxic consequences through mediating multiple pathways were summarized and analyzed, including dopamine release, oxidative stress, mitochondrial-dependent apoptotic pathway, endoplasmic reticulum stress-mediated apoptotic pathway, and neuroinflammation. The detailed information of natural products and their potential effects with mechanisms on modulating METH-evoked neuronal apoptosis are illustrated in Tables 1, 2.
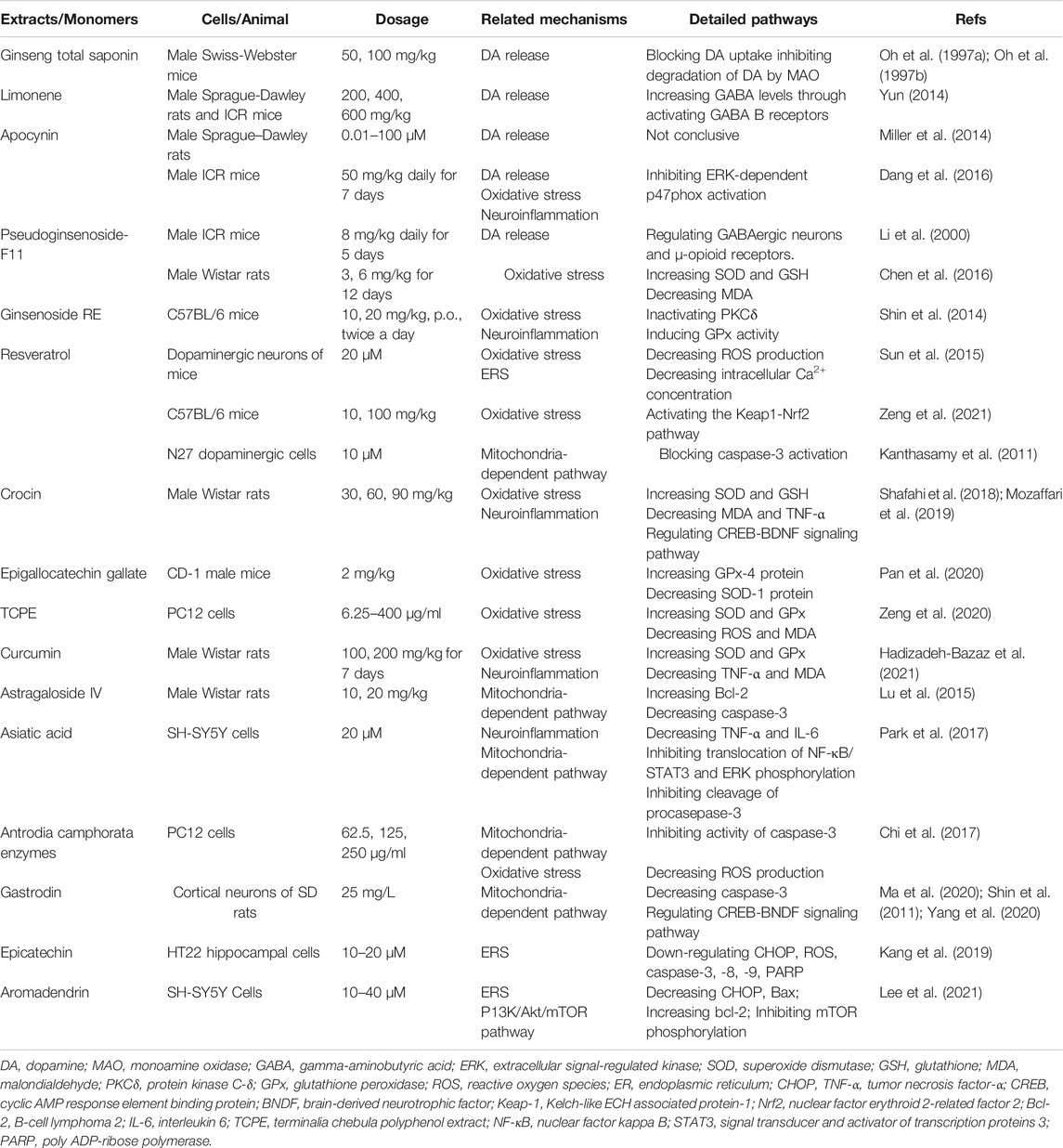
TABLE 1. Anti-apoptotic activities of natural products in METH-induced neuronal apoptosis.

TABLE 2. Detailed information and chemical structures of natural products.
Main Mechanisms Underlying METH-Induce Neuronal Apoptosis
METH-Induced Dopamine Release
Dopamine (DA) is a key neurotransmitter associated with the reinforcement effects of drug addiction. METH is a cationic molecule and chiral compound based around a phenylethylamine core (Homer et al., 2008) and can modulate the DA system with a myriad of influences on DA release. It has a high lipid solubility, penetrates the blood-brain barrier readily (Nordahl et al., 2003; Schep et al., 2010), and can be taken up by dopaminergic cells through binding to the dopamine transporter (DAT) due to its chemical similarity with DA (Iversen, 2006). Physiologically, DAT removes DA from the synapse to terminate its neurotransmitter effects (Wang et al., 2015). The binding of METH to DAT causes the blockage of the extracellular DA reuptake and the reverse transportation of DA outside the cell, leading to the increase of DA concentration in the synaptic cleft (Panenka et al., 2013). METH in high concentrations can cross the axons through lipophilic diffusion, exacerbating the accumulation of extracellular DA (Shin et al., 2018). Another primary substrate molecule involved in METH-induced DA release is the vesicular monoamine transporter-2 (VMAT-2), an integral membrane protein transporting DA from the cytosol to synaptic vesicles (Fleckenstein et al., 2009). This transportation is coupled to a vacuolar-type H+ pump ATPase. Studies show that METH, as a “weak base,” can disrupt the proton gradient of inner and outer sides of the vesicle required for DA sequestration, eliciting the DA leakage from the vesicle to the cytosol (Schwartz et al., 2006; Panenka et al., 2013). Moreover, METH may induce a decrease of VMAT-2 function and its density on the membrane (Eyerman and Yamamoto, 2005; McFadden et al., 2012). These interactions of METH with DAT and VMAT-2 result in excessive DA accumulation in the intracellular and extracellular surroundings.
METH-Induced Oxidative Stress and Mitochondria-Dependent Cell Apoptosis
Multiple pathways are involved in the complicated mechanism of METH-caused neurotoxicity, and oxidative stress has been considered an essential contributor to neuronal apoptosis. METH-induced cytosolic and synaptic DA accumulation results in the production of quinone and semi-quinone through autoxidation to generate large amounts of reactive oxygen species (ROS), such as hydrogen peroxide, hydroxyl radicals, and superoxide radicals. Furthermore, a small portion of DA produces H2O2 with the mediation of monoamine oxidase (MAO) (Hermida-Ameijeiras et al., 2004; McDonnell-Dowling and Kelly, 2017; Yang et al., 2018). METH also inhibits the production of antioxidants, including glutathione, superoxide dismutase (SOD), and catalase (Tata and Yamamoto, 2007; Huang et al., 2013). Thus, the imbalance between ROS and free radical scavengers contributes to the oxidative stress in dopaminergic terminals. Excessive ROS may result in lipids and protein metabolism disruption, mitochondrial dysfunction, and nuclear DNA damage, increasing susceptibility to neuronal damage and death (Potashkin and Meredith, 2006).
Mitochondria is an important site of METH-evoked ROS generation in neural cells. Several investigations have suggested that mitochondria dysfunction plays a crucial role in METH-induced dopaminergic apoptosis. ROS can inhibit key enzymes in the electron transport chain (ETC) of mitochondria, including complex II-III and IV (Wu et al., 2007; Halpin et al., 2014a; Dawson and Dawson, 2017; Moratalla et al., 2017). METH diffuses into mitochondria, and its accumulation disrupts oxidative phosphorylation and suppresses ATP synthesis (Krasnova and Cadet, 2009). In addition, METH inhibits the tricarboxylic acid cycle and ETC, which are essential to ATP production (Barbosa et al., 2015). In turn, the inhibition of ETC enhances ROS production, which may further restrain ETC components through positive feedback, aggravating the dysfunction of mitochondrial metabolism (Yang et al., 2018). Another critical mechanism underlying mitochondria dysfunction triggered by METH is the alteration of mitochondrial dynamics (biogenesis, fusion, fission, and mitophagy) (Michel et al., 2012; Anne Stetler et al., 2013). METH can decrease the expression of certain key molecules related to mitochondrial biogenesis (Valian et al., 2017; Beirami et al., 2018). Mitochondrial dysfunction causes the mitochondrial membrane permeabilization (Kroemer et al., 2007), and subsequently the release of cytochrome-c (Cyt-c), apoptosis-inducing factor (AIF), and Smac/Diablo into the cytosol (Cadet et al., 2003; Jayanthi et al., 2004). METH administration has been shown to up-regulate the expression of pro-apoptotic proteins (Bax, Bak, and Bid) and down-regulate the expression of anti-apoptotic proteins (Bcl-2, Bcl-XL, and BclW) (Jayanthi et al., 2001; Cadet et al., 2003). The increase in the Bax, Bak, and Bid can promote the release of Cyt-c from mitochondria to the cytosol (Roth and D'Sa, 2001). Subsequently, apoptosis protease activating factor 1 (Apaf-1) is activated, initiating the mitochondria-dependent cell apoptosis pathway (the intrinsic pathway) (Cain et al., 2002). Apaf-1 binding to Cyto-c constitutes the “apoptosome,” which serves as a molecular platform for the caspase-9 activation (Jiang and Wang, 2000; Bratton and Salvesen, 2010) and induces sequential caspase-3, -6, and -7 activations (Roth and D'Sa, 2001; Shin et al., 2018). Additionally, several studies have reported that protein kinase C-δ (PKCδ), a member of the PKC family, plays a critical role in the mitochondrial-dependent apoptotic cascade. PKCδ can be proteolytically activated by caspase-3, then translocate to the nucleus mediating DNA fragmentation and apoptosis. In turn, the activated PKCδ enhances caspase-3 production through a positive-feedback loop (Kanthasamy et al., 2003; Kaul et al., 2003).
METH-Induced Excitotoxicity and Endoplasmic Reticulum Stress
Excitotoxicity refers to the overloaded intracellular calcium levels initiated by excessive glutamate (Glu) release, activating apoptotic pathways, eventually resulting in cellular damage (Halpin et al., 2014b). Glu is the major excitatory neurotransmitter in the brain, and METH has been shown to evoke the excessive release of Glu in the striatum (Stephans and Yamamoto, 1994; Mark et al., 2004). Accumulated Glu binds to amino-5hydroxy-3-methyl-4-isoxazole propionic acid receptors and N-methyl-D-aspartate receptors to initiate downstream signaling pathways, leading to a surge of Ca2+ influx (Riddle et al., 2006; Halpin et al., 2014b; Kim et al., 2020). Ca2+ is an important intracellular second manager responsible for multiple signal transductions, which can be regulated by the endoplasmic reticulum (ER) through the sequestration of Ca2+. METH-induced excess intracellular Ca2+ causes the disturbance of intracellular Ca2+ concentrations, activates the protein kinases, phosphatase, and nitric oxide synthase (NOS) and eventually results in endoplasmic reticulum stress (ERS) (Bahar et al., 2016; Moratalla et al., 2017). In stress conditions, a decrease in the protein-folding capacity of ER may induce the accumulation of unfolded and misfolded proteins in ER, which causes unfold protein response (UPR) to remove those proteins and maintain ER homeostasis (Go et al., 2017). UPR is mediated by IRE1α, PERK, and ATF6 signaling proteins, triggering UPR-dependent apoptotic signaling by activating C/EBP homologous protein (CHOP) (Sano and Reed, 2013; Bahar et al., 2016). As a transcription factor, CHOP can up-regulate the expressions of Bax and Bak and down-regulate the expressions of Bcl-2 and Bcl-XL to promote apoptosis (Hu et al., 2018). The elevated Ca2+ activates the calpain, a cytosolic calcium-activated neutral cysteine endopeptidase. The activated calpain translocates from the cytosol to the membrane, where it cleaves procaspase-12 (Nakagawa and Yuan, 2000), then the active caspase-12 initiates positive feedback to activate caspase-9 and -3 to potentiate apoptosis (Martinez et al., 2010).
METH-Induced Neuroinflammation
The neuroinflammation induced by METH is closely related to the microglial activation and the production of pro-inflammatory cytokines (Goncalves et al., 2008). Microglial cells monitor neuronal homeostasis, serve as the immune defense against neuronal damage, and can be activated upon neuronal injury and neurotoxic agents (Lull and Block, 2010; Shaerzadeh et al., 2018). METH stimulation may elicit the morphological alteration of microglial cells (Xu et al., 2017). Neuroimaging has demonstrated that chronic METH administration induces microgliosis reaction in humans (Sekine et al., 2008). Activated microglial cells secrete enormous pro-inflammatory cytokines, including interleukin6 (IL-6) and tumor necrosis factor-α (TNF-α), which induce prolonged neuroinflammation (Goncalves et al., 2010). This inflammatory response may be associated with certain signaling pathways, such as nuclear factor kappa B (NF-κB) and signal transducer and transcription (STAT) pathway activator. Research has shown that METH can activate the NF-κB pathway, inducing its transfer from the cytoplasm to the nucleus and promoting the transcription of pro-inflammatory cytokines in microglia (Shah et al., 2012). METH also causes the increase of signal transducer and activator of transcription proteins 3 (STAT3) and its translocation to the nucleus (Zhao et al., 2019), which carries out the IL-6 signaling (Coelho-Santos et al., 2012). Additionally, studies have demonstrated that mitogen-activated protein kinase (MAPK) may play a crucial role in the METH-induced release of inflammatory cytokines (Yan et al., 2014; Park et al., 2017). The chronic neuroinflammation leads to overexpression of PUMA (p53-up-regulated modulator of apoptosis), a pro-apoptotic protein of the Bcl-2 family that drives Cyt-c release from the mitochondria to initiate caspase cascades (Yang et al., 2018).
Effects and Mechanisms of Natural Products on METH-Induced Apoptosis
Natural Products Targeting DA Release to Modulate METH-Induced Apoptosis
As described above, METH interferes with DA reuptake and causes DA oxidation. Elevated levels of DA readily diffuse in cells and cause oxidative damage, which is closely related to METH-initiated neurotoxic effects. Ginseng total saponin (GTS), an active biological component derived from Panax ginseng C. A. Mey., has been reported to be an effective intervention for the central dopaminergic system more than 2 decades ago. Studies conducted in 1994 and 1996 by Kim et al. first identified that GTS could attenuate conditioned place preference in mice, and this effect might be associated with inhibition of METH-induced dopaminergic activation (Kim et al., 1995; Kim et al., 1996). Their follow-up studies demonstrated that GTS pretreatment could partially protect striatal dopaminergic neurons from METH-induced depletions and reduce DA release and its degradation by MAO (Oh et al., 1997a; Oh et al., 1997b). The property of reducing DA release in GTS may alleviate DA autoxidation and ensuing oxidative stress and apoptosis activation. Limonene is a major compound in the essential oils of citrus fruit peels. In 2014, Lin et al. demonstrated that Limonene reduced extracellular DA levels elevated by METH in the nucleus accumbens of rats, which might be resulted from the increase of gamma-aminobutyric acid (GABA) levels (Yun, 2014). In the same year, Miller et al. proposed that subchronic use of Apocynin, a phenolic compound isolated from Picrorhiza kurroa Royle ex Benth., could significantly reduce the METH-stimulated DA release in rat striatum (Miller et al., 2014). In 2016, Dang et al. further studied the neuroprotective effects of Apocynin and found that this compound could attenuate mitochondrial dysfunction, microglial activation, and causal apoptotic signaling process through inhibiting the extracellular signal-regulated kinase (ERK)-dependent activation of p47phox (Dang et al., 2016). The major mechanisms of how natural products target DA release to attenuate METH-induced apoptosis are summarized in Figure 1.
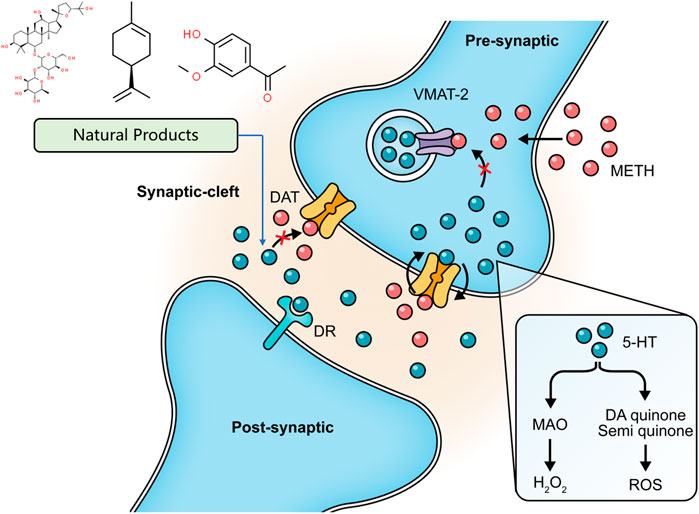
FIGURE 1. Natural products targeting DA release to attenuate METH-induced apoptosis.
Natural Products Targeting Oxidative Stress and Mitochondria-Dependent Pathway to Modulate METH-Induced Apoptosis
Oxidative stress and mitochondria dysfunction have been considered major mechanisms underlying METH-induced apoptosis. Regulating the generation of ROS and antioxidants may be efficacious strategies to ameliorate mitochondria dysfunction and neural damage. Pseudoginsenoside-F11 (PF11), an ocotillol-type saponin derived from American Ginseng (Panax quinquefolius L.) (Li et al., 2000), has been demonstrated to mediate neuroinflammation through decreasing the activation of microglial and the subsequent expression of pro-inflammatory factors and oxidative stress through increasing antioxidant activity (Wang et al., 2014; Zhang et al., 2019). In 2003, a study by Wu et al. showed that PF11 could antagonize the depletion of dopaminergic contents in mice induced by chronic METH administration (Wu et al., 2003). In 2016, Chen et al. further revealed that PF11 could attenuate the METH-evoked oxidative stress in the hippocampus of rats through increasing SOD and GSH-Px activities and decreasing malondialdehyde (MDA) level (Chen. et al., 2016). PF11 also regulated GABAergic neurons and μ-opioid receptors, which are considered to be involved in METH-induced DA release (Fu et al., 2016). Gastrodia elata Blume is a well-known herb in traditional Chinese medicine and has often been used to treat various neurological disorders. In early 2011, Shin et al. reported that its methanol extract could significantly attenuate METH-induced ROS in striatal dopaminergic neurons of mice (Shin et al., 2011). Ginsenoside RE, a validated constituent extracted from Panax ginseng C. A. Mey. (Liu et al., 2019), showed an antioxidative effect against METH-induced apoptosis. In 2014, Shin et al. proved the mediation effect of Ginsenoside RE for oxidative stress in mitochondria through inhibiting PKCδ (Shin et al., 2014). In 2015, in vitro studies by Nam et al. also suggested that Ginsenoside RE could induce the cytosolic and mitochondrial GPx activity and the mitochondrial translocation of cleaved PKCδ (Nam et al., 2015). Resveratrol, a polyphenol found in red wine, has considerable neuroprotective effects, including antioxidation, anti-inflammation, and anti-apoptosis (Howitz et al., 2003). In 2015, Sun et al. reported that it could significantly decrease METH-induced ROS production in vitro and the compensatory SOD generation. They also observed that resveratrol might reduce the intracellular Ca2+ concentration, suggesting it exerts neuroprotective effects on METH-caused apoptosis through targeting oxidative stress and ER stress (Sun et al., 2015). Another study by Zeng et al. also suggested that the antioxidative effect of resveratrol might attribute to the activation of the keap1-Nrf2 pathway (Zeng et al., 2021). In 2018, Shafahi et al. reported that intervention with crocin, a bioactive constituent of saffron (Crocus sativus L.), could regulate the increased SOD and GSH and the ensuing decreased caspase-3 activation in rats stimulated by METH (Shafahi et al., 2018). In 2019, an in vivo study by Mozaffari et al. demonstrated that the neuroprotective effects of crocin might be associated with the Cyclic AMP response element-binding protein (CREB) and the Brain-derived neurotrophic factor (BDNF) signaling pathway (Mozaffari et al., 2019). Epigallocatechin gallate (EGCG), a polyphenol compound derived from green tea, is a potent antioxidant. In 2020, Pan et al. observed that EGCG pretreatment could reverse the reduced GPx-4 protein level in the striatum of mice and the overexpression of compensatory SOD-1 protein induced by METH, indicating its effects in preventing METH-caused apoptosis (Pan et al., 2020). Terminalia chebula polyphenol extract (TCPE) is obtained from Terminalia chebula Retz. In 2020, Zeng et al. reported that TCPE could protect PC12 cells from METH-elicited DNA damage and apoptosis, which might be related to the inhibition of intracellular ROS and MDA production and the enhancement of SOD and GPx activities (Zeng et al., 2020). Curcumin (CUR) is the main component of turmeric (Curcuma longa L.). In 2021, a study by Hadizadeh-Bazaz et al. reported that CUR could increase SOD and GSH and decrease MDA in the hippocampal neurons of mice injected with METH. They also found that CUR could reduce the TNF-α levels to attenuate the neuroinflammation. These effects of CUR contributed to a significant reduction in METH-induced apoptosis (Hadizadeh-Bazaz et al., 2021).
Several natural products targeting mitochondria-related apoptotic proteins have been explored in recent years. In 2011, Kanthas et al. reported that resveratrol pretreatment could protect dopaminergic cells from MTTH-induced cell death through blocking caspase-3 activation and the subsequent DNA fragmentation (Kanthasamy et al., 2011). Astragaloside IV is the main bioactive component in Astragalus mongholicus bunge. In 2015, a study by Sui et al. revealed that the Bcl-2 and caspase-3 were overexpressed in METH-dependent mice, and the former might be a compensatory reaction against apoptosis. Astragaloside IV could enhance the Bcl-2 expression and inhibit the caspase-3 expression and activation (Lu et al., 2015). Asiatic acid is a pentacyclic triterpene extracted from Centella asiatica (L.) Urb. and possesses multiple bioactivities, including antioxidation, anti-inflammation, and anti-apoptosis. An in vitro study conducted in 2017 by Park et al. revealed that Asiatic acid inhibited the cleavage of caspase-3 to reduce the subsequent cleavage of PARP (Park et al., 2017). Antrodia camphorata (M. Zang et C. H. Su) Sheng. H. Wu, Ryvarden et T. T. Chang is a medicinal mushroom endemic to Southeast China. In 2017, a study by Chi et al. showed that its enzymes could suppress the METH-evoked mitochondrial apoptosis pathway in PC12 cells by reducing the intracellular ROS levels and inhibiting procaspase-3 cleavage (Chi et al., 2017). In 2020, Yan et al. investigated the anti-apoptotic effect of Gastrodin (GAS), the major bioactive component of Gastrodia elata Blume, and reported that GAS intervention could decline the METH-stimulated caspase-3 overexpression in cortical neurons of rats (Yan et al., 2020). An in vitro experiment by Ma et al. also observed that GAS could decrease TUNEL-positive cells in the cortical neurons stimulated by METH through modulating CREB/BNDF signaling pathway (Ma et al., 2020). The major mechanisms of how natural products target oxidative stress and mitochondria-dependent apoptotic pathway to attenuate METH-induced apoptosis are summarized in Figure 2.
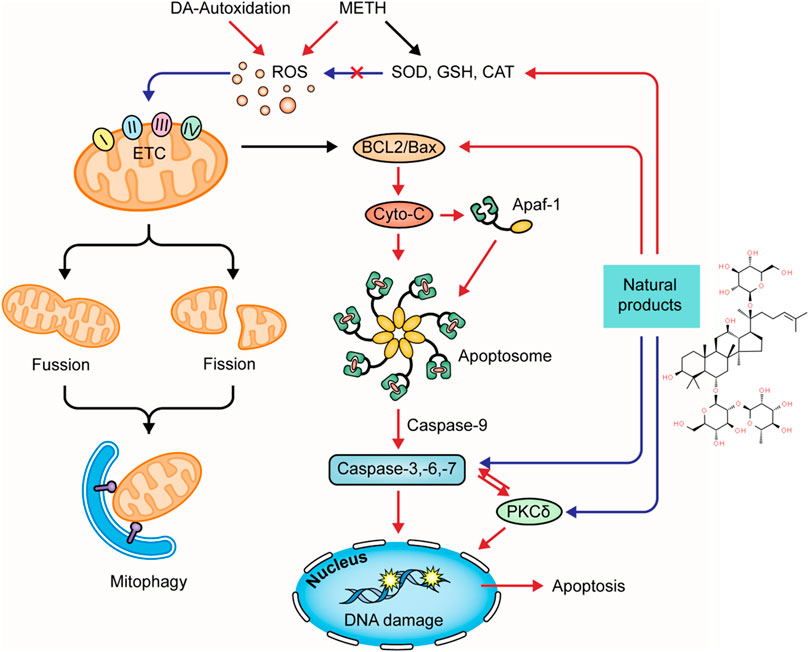
FIGURE 2. Natural products targeting on oxidative stress and mitochondria-dependent apoptotic pathway to attenuate METH-induced apoptosis.
Natural Products Targeting Endoplasmic Reticulum Stress-Mediated Pathway to Modulate METH-Induced Apoptosis
Targeting the ERS-mediated pathway is also a feasible way for natural products to modulate METH-induced neuronal apoptosis. In 2015, Sun et al. observed that resveratrol might reduce the intracellular Ca2+ overload and exert neuroprotective effects on METH-caused apoptosis of dopaminergic neurons through attenuating ERS (Sun et al., 2015). In 2019, Kang et al. reported that Epicatechin, a bioactive flavonoid in green tea, could protect hippocampal neurons from METH-induced cell apoptosis via reducing ERS, and its potential mechanisms might associate with the down-regulation of CHOP, ROS, caspase-3, -8, -9, and PARP (Kang et al., 2019). In 2021, Lee et al. revealed that pretreatment with aromadendrin, a flavonoid obtained from Abies sibirica Ledeb. (Syn. Pinus sibirica Ledeb), suppressed METH-induced cell death in SH-SY5Y cells by decreasing CHOP and Bax and increasing Bcl-2. It also exhibited a preventative effect on cell apoptosis via restoring the phosphorylation of mTOR to inhibit the PI3K/Akt/mTOR pathway (Lee et al., 2021). The major mechanisms of how natural products target ERS to attenuate METH-induced apoptosis are summarized in Figure 3.
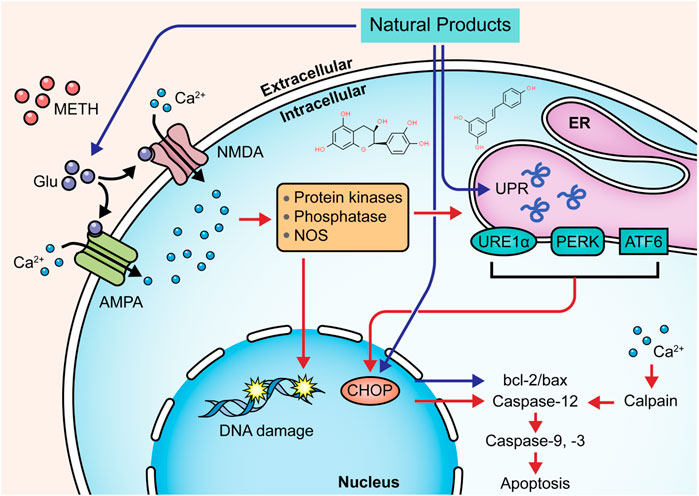
FIGURE 3. Natural products targeting ERS-mediated pathway to attenuate METH-induced apoptosis.
Natural Products Targeting Neuroinflammation to Modulate METH-Induced Apoptosis
As stated before, the neuroinflammation induced by activated microglia is crucial in METH-caused neurotoxicity. Accordingly, ameliorating neuroinflammation may be a promising approach to control METH-induced neurotoxicity. In 2014, Shin et al. found that Ginsenoside RE could attenuate the METH-induced activation of microglial in PKCδ (+/+) mice, suggesting that Ginsenoside RE could mediate apoptosis via inactivation of PKCδ (Shin et al., 2014). In 2017, an investigation by Park et al. demonstrated that Asiatic acid could relieve METH-mediated dopaminergic neuroinflammation of SH-SY5Y cells by reducing pro-inflammatory cytokines secretion, such as TNFα and IL-6, and down-regulated the expression of TNF receptor via inhibiting NF-κB, STAT3, and MAPK-ERK signaling pathway. Moreover, Asiatic acid also exerted the inhibitory effects on caspase-3 cleavage and the subsequent PARP cleavage and the mediative effects on Bcl-2 and Bax (Park et al., 2017). In addition, in 2018, Shafahi et al. reported that crocin treatment could reduce the level of TNF-α in the hippocampus of rats stimulated by METH, suggesting that crocin could also exert an antiapoptotic effect by mediating METH-induced neuroinflammation (Shafahi et al., 2018). The major mechanisms of how natural products target neuroinflammation to attenuate METH-induced apoptosis are summarized in Figure 4.
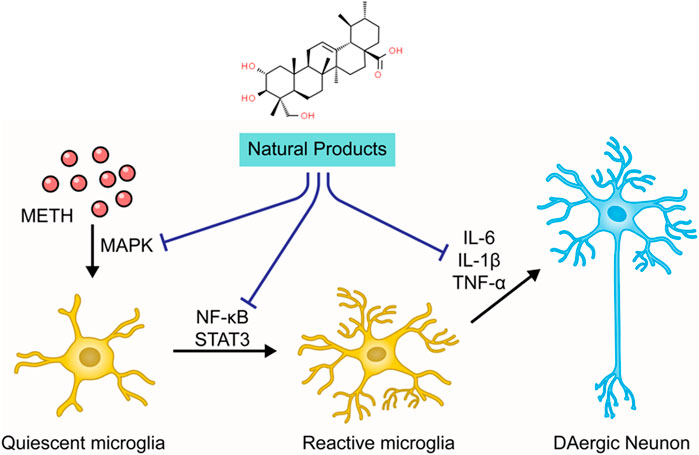
FIGURE 4. Natural products targeting neuroinflammation to attenuate METH-induced apoptosis.
Conclusion and Perspectives
Most of the METH-elicited neuropsychiatric consequences are attributable to its neurotoxicity, including direct damage to central dopaminergic and serotonergic terminals and the subsequent neurodegenerative changes upon the activation of neuronal autophagy, apoptosis, and necrosis (Davidson et al., 2001; Yu et al., 2015; Sanchez et al., 2018). In recent years, important progress has been achieved in interpreting the complex mechanism underlying METH-produced neurotoxicity. Neuronal apoptosis has been considered the main pathogenic factor in METH-induced neurodegeneration. Animal studies have confirmed that pro-apoptotic changes in the brain are highly associated with METH-induced neuropsychiatric symptoms, such as locomotor sensitization, psychosis, memory impairments, and cognitive impairments (Shin et al., 2017). With the decrease of central monoaminergic terminals and the depletion of dopamine and serotonin, these psychiatric complications are sustained (Rusyniak, 2013), leading to failed anti-addiction treatments, relapse to drug use, and increased risk of various neurodegenerative diseases (Shukla and Vincent, 2020). In view of the devastating societal and public health burdens caused by METH abuse, further in-depth explorations in pharmacotherapies are highly anticipated. In this review, we summarized and analyzed these natural products that have been proven to possess significantly in vitro and in vivo protective effects against METH-induced neuronal apoptosis through mediating multiple pro-apoptotic factors, such as DA release, oxidative stress, mitochondria-dependent apoptotic pathway, ERS-mediated apoptotic pathway, and neuroinflammation.
Notwithstanding the numerous studies conducted to excavate the effects of natural agents in the prevention and treatment of METH-caused neuronal apoptosis, the discovery and development of novel and effective novel pharmacotherapies still require more sophisticated works in both preclinical and clinical investigations. First, METH abuse involves the activation of multiple apoptotic pathways. However, current research has primarily focused on using a single natural agent to block a single pathway. Given the complex nature of METH-initiated neurotoxicity, strategies that combine multiple natural compounds and target multiple pathways may yield more encouraging medications. Interestingly, the protective effect of certain natural products, such as Crocin, Resveratrol, Curcumin, and Apocynin, on METH-evoked apoptosis have been attributed to the regulation of multiple targets simultaneously. From the perspective of optimal “cocktail” drug design, they may yield a promising future in novel anti-METH strategies (Radhakrishnan and Tidor, 2008). Approaches combining systems biology with traditional computational-aided drug design are applicable for developing drug “cocktail” regimens to modulate the core target network of METH. Second, the available studies have focused on the effects of natural products on oxidative stress, and the interpretation of their actions on other pathogenic factors is still insufficient. Several natural agents have demonstrated promising potential to regulate neuronal apoptosis in other fields, including neurodegenerative diseases (Kim and Kim, 2018), which indicate that discovery and exploration of more potential natural candidates can be anticipated.
Third, the specific molecular and genetic mechanisms underlying the anti-apoptotic effects of natural products are still elusive. The safety of natural products and their interaction with conventional drugs have become major concerns. Therefore, further studies on their pharmacokinetics, pharmacodynamics, and toxicological profiles are of great necessity (Jha and Rathi, 2008; Nauffal and Gabardi, 2016; Gaston et al., 2020). Follow-up studies need to be performed to evaluate the potential toxicity of the natural agents. In addition, no clinical trial regarding the anti-apoptotic effects of natural products for METH-induced neuronal apoptosis has been documented, and future studies may devote more efforts to designing high-quality and well-designed clinical trials to verify their therapeutic effects and explore the relative mechanism. Lastly, new and emerging insightful scientific findings on METH-induced neuronal apoptosis have continually updated our understanding of its biomechanisms. For example, recent studies confirmed that CCAAT/enhancer-binding protein (C/EBP-β), Lipocalin2 (lcn2), and Sigma-1 receptor (σ-1R) might play an important role in METH-induced microglial apoptosis (Shen et al., 2016; Chen et al., 2019). These new findings may provide new directions for future studies of natural agents and yield more refined and robust evidence. In addition, it is also worth noting that except for the natural products stated above, other agents, such as Vitamin C (L-ascorbate) and Melatonin, also have the potentials to be developed as candidate anti-apoptotic drugs in clinical management (Huang et al., 2012; Nguyen et al., 2015).
In conclusion, this review summarized and analyzed these natural products with anti-apoptotic properties to prevent and treat METH-induced neurotoxicity by modulating DA release, oxidative stress, mitochondria dysfunction, caspase cascades, and neuroinflammation. Hopefully, this review may provide helpful information for their further exploration preclinically and clinically.
Author Contributions
All authors contributed to the article. YR, YC, and BS conceived and designed this paper. YZ, YC, SZ, HR, JX, ML, and YR summarized and analyzed the data. YZ and YC drafted the paper. YC, BS, and YR revised and edited the paper.
Funding
This research was supported by the Major Research and Development Project and the International Cooperation and Exchange Project of Sichuan Provincial Science and Technology Department (No. 2018SZ071, No. 2017HH0004), the National Natural Science Foundation of China (No. 81873239, No. 81603537), the Sichuan Provincial Administration of Traditional Chinese Medicine (No. 2021MS464), and the Youth Scholarship of Chengdu University of Traditional Chinese Medicine (No. QNXZ2019043).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Anne Stetler, R., Leak, R. K., Gao, Y., and Chen, J. (2013). The Dynamics of the Mitochondrial Organelle as a Potential Therapeutic Target. J. Cereb. Blood Flow Metab. 33 (1), 22–32. doi:10.1038/jcbfm.2012.158
Australian Institute of Health and Welfare (2020). National Drug Strategy Household Survey 2019. Available at: https://www.aihw.gov.au/reports/illicit-use-of-drugs/national-drug-strategy-household-survey-2019/contents/summary (Accessed July 10, 2021).
Bach, P., Hayashi, K., Milloy, M. J., Nosova, E., Kerr, T., Wood, E., et al. (2020). Characterising the Increasing Prevalence of crystal Methamphetamine Use in Vancouver, Canada, from 2006-2017: A Gender-Based Analysis. Drug Alcohol. Rev. 39 (7), 932–940. doi:10.1111/dar.13126
Bahar, E., Kim, H., and Yoon, H. (2016). ER Stress-Mediated Signaling: Action Potential and Ca(2+) as Key Players. Int. J. Mol. Sci. 17 (9), 1558. doi:10.3390/ijms17091558
Baker, D. D., Chu, M., Oza, U., and Rajgarhia, V. (2007). The Value of Natural Products to Future Pharmaceutical Discovery. Nat. Prod. Rep. 24 (6), 1225–1244. doi:10.1039/b602241n
Baradhi, K. M., Pathireddy, S., Bose, S., and Aeddula, N. R. (2019). Methamphetamine (N-Methylamphetamine)-Induced Renal Disease: Underevaluated Cause of End-Stage Renal Disease (ESRD). BMJ Case Rep. 12 (9), e230288. doi:10.1136/bcr-2019-230288
Barbosa, D. J., Capela, J. P., Feio-Azevedo, R., Teixeira-Gomes, A., Bastos, M. D. L., and Carvalho, F. (2015). Mitochondria: Key Players in the Neurotoxic Effects of Amphetamines. Arch. Toxicol. 89 (10), 1695–1725. doi:10.1007/s00204-015-1478-9
Beirami, E., Oryan, S., Seyedhosseini Tamijani, S. M., Ahmadiani, A., and Dargahi, L. (2018). Intranasal Insulin Treatment Restores Cognitive Deficits and Insulin Signaling Impairment Induced by Repeated Methamphetamine Exposure. J. Cel Biochem. 119 (2), 2345–2355. doi:10.1002/jcb.26398
Bratton, S. B., and Salvesen, G. S. (2010). Regulation of the Apaf-1-Caspase-9 Apoptosome. J. Cel Sci. 123 (Pt 19), 3209–3214. doi:10.1242/jcs.073643
Cadet, J. L., Jayanthi, S., and Deng, X. (2003). Speed Kills: Cellular and Molecular Bases of Methamphetamine-Induced Nerve Terminal Degeneration and Neuronal Apoptosis. FASEB J. 17 (13), 1775–1788. doi:10.1096/fj.03-0073rev
Cain, K., Bratton, S. B., and Cohen, G. M. (2002). The Apaf-1 Apoptosome: a Large Caspase-Activating Complex. Biochimie 84 (2-3), 203–214. doi:10.1016/s0300-9084(02)01376-7
Chen, X., Yang, X., Wang, J., and Li, X. (2016). Effects of Psuedoginsenoside F11 on the Oxidation Material Levels in the Serum and Brain Tissue of Methamphetamine - Dependent Rats (China). West China J. Pharm. Sci. 31 (02), 152–153. doi:10.13375/j.cnki.wcjps.2016.02.013
Chen, X., Lu, J., Zhao, X., Chen, C., Qiao, D., Wang, H., et al. (2019). Role of C/EBP-β in Methamphetamine-Mediated Microglial Apoptosis. Front. Cel Neurosci. 13, 366. doi:10.3389/fncel.2019.00366
Chi, D., Sun, Y., and Pan, S. (2017). The Protective Effect and its Mechanism of Antrodia Camphorata Enzyme on the Cytotoxicity Injury of PC12 Cells Induced by Amphetamine (China). Chin. J. Clin. Anat. 35 (4), 397408–398401. doi:10.13418/j.issn.1001-165x.2017.04.009
Chiang, M., Lombardi, D., Du, J., Makrum, U., Sitthichai, R., Harrington, A., et al. (2019). Methamphetamine-associated Psychosis: Clinical Presentation, Biological Basis, and Treatment Options. Hum. Psychopharmacol. 34 (5), e2710. doi:10.1002/hup.2710
CNNCC (2019). National Drug Report. China National Narcotics Control Committee. Available at: http://www.gov.cn/xinwen/2020-06/28/content_5522443.htm (Accessed July 10, 2021).
Coelho-Santos, V., Gonçalves, J., Fontes-Ribeiro, C., and Silva, A. P. (2012). Prevention of Methamphetamine-Induced Microglial Cell Death by TNF-α and IL-6 through Activation of the JAK-STAT Pathway. J. Neuroinflammation 9, 103. doi:10.1186/1742-2094-9-103
Courtney, K. E., and Ray, L. A. (2014). Methamphetamine: an Update on Epidemiology, Pharmacology, Clinical Phenomenology, and Treatment Literature. Drug Alcohol Depend. 143, 11–21. doi:10.1016/j.drugalcdep.2014.08.003
Dang, D. K., Shin, E. J., Nam, Y., Ryoo, S., Jeong, J. H., Jang, C. G., et al. (2016). Apocynin Prevents Mitochondrial Burdens, Microglial Activation, and Pro-apoptosis Induced by a Toxic Dose of Methamphetamine in the Striatum of Mice via Inhibition of P47phox Activation by ERK. J. Neuroinflammation 13, 12. doi:10.1186/s12974-016-0478-x
Davidson, C., Gow, A. J., Lee, T. H., and Ellinwood, E. H. (2001). Methamphetamine Neurotoxicity: Necrotic and Apoptotic Mechanisms and Relevance to Human Abuse and Treatment. Brain Res. Brain Res. Rev. 36 (1), 1–22. doi:10.1016/s0165-0173(01)00054-6
Dawson, T. M., and Dawson, V. L. (2017). Mitochondrial Mechanisms of Neuronal Cell Death: Potential Therapeutics. Annu. Rev. Pharmacol. Toxicol. 57, 437–454. doi:10.1146/annurev-pharmtox-010716-105001
Dean, A. C., Groman, S. M., Morales, A. M., and London, E. D. (2013). An Evaluation of the Evidence that Methamphetamine Abuse Causes Cognitive Decline in Humans. Neuropsychopharmacology 38 (2), 259–274. doi:10.1038/npp.2012.179
Ekiert, H. M., and Szopa, A. (2020). Biological Activities of Natural Products. Molecules 25 (23), 5769. doi:10.3390/molecules25235769
Elinore, F. M (2020). National Survey on Drug Use and Health-2019. Substance Abuse and Mental Health Services Administration. Available at: https://www.samhsa.gov/data/data-we-collect/nsduh-national-survey-drug-use-and-health (Accessed July 10, 2021).
Ernst, T., Chang, L., Leonido-Yee, M., and Speck, O. (2000). Evidence for Long-Term Neurotoxicity Associated with Methamphetamine Abuse: A 1H MRS Study. Neurology 54 (6), 1344–1349. doi:10.1212/wnl.54.6.1344
Eyerman, D. J., and Yamamoto, B. K. (2005). Lobeline Attenuates Methamphetamine-Induced Changes in Vesicular Monoamine Transporter 2 Immunoreactivity and Monoamine Depletions in the Striatum. J. Pharmacol. Exp. Ther. 312 (1), 160–169. doi:10.1124/jpet.104.072264
Feng, L., He, W., Lin, S., Ruan, Y., Yuan, C., Qiu, H., et al. (2020). The Association between Interleukin-8 Levels and the Development of Withdrawal Symptoms during Methamphetamine Abstinence. Hum. Psychopharmacol. 35 (4), e2736. doi:10.1002/hup.2736
Fleckenstein, A. E., Volz, T. J., and Hanson, G. R. (2009). Psychostimulant-induced Alterations in Vesicular Monoamine Transporter-2 Function: Neurotoxic and Therapeutic Implications. Neuropharmacology 56 (Suppl. 1), 133–138. doi:10.1016/j.neuropharm.2008.07.002
Friend, D. M., and Keefe, K. A. (2013). A Role for D1 Dopamine Receptors in Striatal Methamphetamine-Induced Neurotoxicity. Neurosci. Lett. 555, 243–247. doi:10.1016/j.neulet.2013.08.039
Fu, K., Lin, H., Miyamoto, Y., Wu, C., Yang, J., Uno, K., et al. (2016). Pseudoginsenoside-F11 Inhibits Methamphetamine-Induced Behaviors by Regulating Dopaminergic and GABAergic Neurons in the Nucleus Accumbens. Psychopharmacology 233 (5), 831–840. doi:10.1007/s00213-015-4159-8
Gaston, T. E., Mendrick, D. L., Paine, M. F., Roe, A. L., and Yeung, C. K. (2020). "Natural" Is Not Synonymous with "Safe": Toxicity of Natural Products Alone and in Combination with Pharmaceutical Agents. Regul. Toxicol. Pharmacol. 113, 104642. doi:10.1016/j.yrtph.2020.104642
Glasner-Edwards, S., and Mooney, L. J. (2014). Methamphetamine Psychosis: Epidemiology and Management. CNS Drugs 28 (12), 1115–1126. doi:10.1007/s40263-014-0209-8
Go, B. S., Kim, J., Yang, J. H., and Choe, E. S. (2017). Psychostimulant-Induced Endoplasmic Reticulum Stress and Neurodegeneration. Mol. Neurobiol. 54 (6), 4041–4048. doi:10.1007/s12035-016-9969-0
Golsorkhdan, S. A., Boroujeni, M. E., Aliaghaei, A., Abdollahifar, M. A., Ramezanpour, A., Nejatbakhsh, R., et al. (2020). Methamphetamine Administration Impairs Behavior, Memory and Underlying Signaling Pathways in the hippocampus. Behav. Brain Res. 379, 112300. doi:10.1016/j.bbr.2019.112300
Gonçalves, J., Martins, T., Ferreira, R., Milhazes, N., Borges, F., Ribeiro, C. F., et al. (2008). Methamphetamine-induced Early Increase of IL-6 and TNF-Alpha mRNA Expression in the Mouse Brain. Ann. N. Y Acad. Sci. 1139, 103–111. doi:10.1196/annals.1432.043
Gonçalves, J., Baptista, S., Martins, T., Milhazes, N., Borges, F., Ribeiro, C. F., et al. (2010). Methamphetamine-induced Neuroinflammation and Neuronal Dysfunction in the Mice hippocampus: Preventive Effect of Indomethacin. Eur. J. Neurosci. 31 (2), 315–326. doi:10.1111/j.1460-9568.2009.07059.x
Hadizadeh-Bazaz, M., Vaezi, G., Khaksari, M., and Hojati, V. (2021). Curcumin Attenuates Spatial Memory Impairment by Anti-oxidative, Anti-apoptosis, and Anti-inflammatory Mechanism against Methamphetamine Neurotoxicity in Male Wistar Rats: Histological and Biochemical Changes. Neurotoxicology 84, 208–217. doi:10.1016/j.neuro.2021.03.011
Halpin, L. E., Collins, S. A., and Yamamoto, B. K. (2014a). Neurotoxicity of Methamphetamine and 3,4-methylenedioxymethamphetamine. Life Sci. 97 (1), 37–44. doi:10.1016/j.lfs.2013.07.014
Halpin, L. E., Northrop, N. A., and Yamamoto, B. K. (2014b). Ammonia Mediates Methamphetamine-Induced Increases in Glutamate and Excitotoxicity. Neuropsychopharmacology 39 (4), 1031–1038. doi:10.1038/npp.2013.306
Heinrich, M., Appendino, G., Efferth, T., Fürst, R., Izzo, A. A., Kayser, O., et al. (2020). Best Practice in Research - Overcoming Common Challenges in Phytopharmacological Research. J. Ethnopharmacol. 246, 112230. doi:10.1016/j.jep.2019.112230
Hermida-Ameijeiras, A., Méndez-Alvarez, E., Sánchez-Iglesias, S., Sanmartín-Suárez, C., and Soto-Otero, R. (2004). Autoxidation and MAO-Mediated Metabolism of Dopamine as a Potential Cause of Oxidative Stress: Role of Ferrous and Ferric Ions. Neurochem. Int. 45 (1), 103–116. doi:10.1016/j.neuint.2003.11.018
Homer, B. D., Solomon, T. M., Moeller, R. W., Mascia, A., DeRaleau, L., and Halkitis, P. N. (2008). Methamphetamine Abuse and Impairment of Social Functioning: a Review of the Underlying Neurophysiological Causes and Behavioral Implications. Psychol. Bull. 134 (2), 301–310. doi:10.1037/0033-2909.134.2.301
Howitz, K. T., Bitterman, K. J., Cohen, H. Y., Lamming, D. W., Lavu, S., Wood, J. G., et al. (2003). Small Molecule Activators of Sirtuins Extend Saccharomyces cerevisiae Lifespan. Nature 425 (6954), 191–196. doi:10.1038/nature01960
Hu, H., Tian, M., Ding, C., and Yu, S. (2018). The C/EBP Homologous Protein (CHOP) Transcription Factor Functions in Endoplasmic Reticulum Stress-Induced Apoptosis and Microbial Infection. Front. Immunol. 9, 3083. doi:10.3389/fimmu.2018.03083
Huang, Y. N., Wang, J. Y., Lee, C. T., Lin, C. H., Lai, C. C., and Wang, J. Y. (2012). L-ascorbate Attenuates Methamphetamine Neurotoxicity through Enhancing the Induction of Endogenous Heme Oxygenase-1. Toxicol. Appl. Pharmacol. 265 (2), 241–252. doi:10.1016/j.taap.2012.08.036
Huang, M. C., Lin, S. K., Chen, C. H., Pan, C. H., Lee, C. H., and Liu, H. C. (2013). Oxidative Stress Status in Recently Abstinent Methamphetamine Abusers. Psychiatry Clin. Neurosci. 67 (2), 92–100. doi:10.1111/pcn.12025
Iversen, L. (2006). Neurotransmitter Transporters and Their Impact on the Development of Psychopharmacology. Br. J. Pharmacol. 147 (Suppl. 1), S82–S88. doi:10.1038/sj.bjp.0706428
Jayanthi, S., Deng, X., Bordelon, M., McCoy, M. T., and Cadet, J. L. (2001). Methamphetamine Causes Differential Regulation of Pro-death and Anti-death Bcl-2 Genes in the Mouse Neocortex. FASEB J. 15 (10), 1745–1752. doi:10.1096/fj.01-0025com
Jayanthi, S., Deng, X., Noailles, P. A., Ladenheim, B., and Cadet, J. L. (2004). Methamphetamine Induces Neuronal Apoptosis via Cross-Talks between Endoplasmic Reticulum and Mitochondria-dependent Death Cascades. FASEB J. 18 (2), 238–251. doi:10.1096/fj.03-0295com
Jha, V., and Rathi, M. (2008). Natural Medicines Causing Acute Kidney Injury. Semin. Nephrol. 28 (4), 416–428. doi:10.1016/j.semnephrol.2008.04.010
Jiang, X., and Wang, X. (2000). Cytochrome C Promotes Caspase-9 Activation by Inducing Nucleotide Binding to Apaf-1. J. Biol. Chem. 275 (40), 31199–31203. doi:10.1074/jbc.C000405200
Jones, C. M., Compton, W. M., and Mustaquim, D. (2020). Patterns and Characteristics of Methamphetamine Use Among Adults - United States, 2015-2018. Morb. Mortal. Wkly. Rep. 69 (12), 317–323. doi:10.15585/mmwr.mm6912a1
Kang, Y., Lee, J. H., Seo, Y. H., Jang, J. H., Jeong, C. H., Lee, S., et al. (2019). Epicatechin Prevents Methamphetamine-Induced Neuronal Cell Death via Inhibition of ER Stress. Biomol. Ther. (Seoul) 27 (2), 145–151. doi:10.4062/biomolther.2018.092
Kanthasamy, A. G., Kitazawa, M., Kanthasamy, A., and Anantharam, V. (2003). Role of Proteolytic Activation of Protein Kinase Cdelta in Oxidative Stress-Induced Apoptosis. Antioxid. Redox Signal. 5 (5), 609–620. doi:10.1089/152308603770310275
Kanthasamy, K., Gordon, R., Jin, H., Anantharam, V., Ali, S., Kanthasamy, A. G., et al. (2011). Neuroprotective Effect of Resveratrol against Methamphetamine-Induced Dopaminergic Apoptotic Cell Death in a Cell Culture Model of Neurotoxicity. Curr. Neuropharmacol. 9 (1), 49–53. doi:10.2174/157015911795017353
Kaul, S., Kanthasamy, A., Kitazawa, M., Anantharam, V., and Kanthasamy, A. G. (2003). Caspase-3 Dependent Proteolytic Activation of Protein Kinase C delta Mediates and Regulates 1-Methyl-4-Phenylpyridinium (MPP+)-induced Apoptotic Cell Death in Dopaminergic Cells: Relevance to Oxidative Stress in Dopaminergic Degeneration. Eur. J. Neurosci. 18 (6), 1387–1401. doi:10.1046/j.1460-9568.2003.02864.x
Kevil, C. G., Goeders, N. E., Woolard, M. D., Bhuiyan, M. S., Dominic, P., Kolluru, G. K., et al. (2019). Methamphetamine Use and Cardiovascular Disease. Arterioscler Thromb. Vasc. Biol. 39 (9), 1739–1746. doi:10.1161/ATVBAHA.119.312461
Kim, C., and Kim, B. (2018). Anti-Cancer Natural Products and Their Bioactive Compounds Inducing ER Stress-Mediated Apoptosis: A Review. Nutrients 10 (8), 1021. doi:10.3390/nu10081021
Kim, H. S., Kang, J. G., Rheu, H. M., Cho, D. H., and Oh, K. W. (1995). Blockade by Ginseng Total Saponin of the Development of Methamphetamine Reverse Tolerance and Dopamine Receptor Supersensitivity in Mice. Planta Med. 61 (1), 22–25. doi:10.1055/s-2006-957991
Kim, H. S., Jang, C. G., Park, W. K., Oh, K. W., Rheu, H. M., Cho, D. H., et al. (1996). Blockade by Ginseng Total Saponin of Methamphetamine-Induced Hyperactivity and Conditioned Place Preference in Mice. Gen. Pharmacol. 27 (2), 199–204. doi:10.1016/0306-3623(95)02023-3
Kim, B., Yun, J., and Park, B. (2020). Methamphetamine-Induced Neuronal Damage: Neurotoxicity and Neuroinflammation. Biomol. Ther. (Seoul) 28 (5), 381–388. doi:10.4062/biomolther.2020.044
Krasnova, I. N., and Cadet, J. L. (2009). Methamphetamine Toxicity and Messengers of Death. Brain Res. Rev. 60 (2), 379–407. doi:10.1016/j.brainresrev.2009.03.002
Kroemer, G., Galluzzi, L., and Brenner, C. (2007). Mitochondrial Membrane Permeabilization in Cell Death. Physiol. Rev. 87 (1), 99–163. doi:10.1152/physrev.00013.2006
Lappin, J. M., Darke, S., and Farrell, M. (2018). Methamphetamine Use and Future Risk for Parkinson's Disease: Evidence and Clinical Implications. Drug Alcohol Depend. 187, 134–140. doi:10.1016/j.drugalcdep.2018.02.032
Lee, H.-S., Kim, E.-N., and Jeong, G.-S. (2021). Aromadendrin Protects Neuronal Cells from Methamphetamine-Induced Neurotoxicity by Regulating Endoplasmic Reticulum Stress and PI3K/Akt/mTOR Signaling Pathway. Int. J. Mol. Sci. 22 (5), 2274. doi:10.3390/ijms22052274
Li, Z., Wu, C. F., Pei, G., Guo, Y. Y., and Li, X. (2000). Antagonistic Effect of Pseudoginsenoside-F11 on the Behavioral Actions of Morphine in Mice. Pharmacol. Biochem. Behav. 66 (3), 595–601. doi:10.1016/s0091-3057(00)00260-4
Liu, M., Bai, X., Yu, S., Zhao, W., Qiao, J., Liu, Y., et al. (2019). Ginsenoside Re Inhibits ROS/ASK-1 Dependent Mitochondrial Apoptosis Pathway and Activation of Nrf2-Antioxidant Response in Beta-Amyloid-Challenged SH-SY5Y Cells. Molecules 24 (15), 2687. doi:10.3390/molecules24152687
Lu, L., Liu, Y., Zhu, W., Shi, J., Liu, Y., Ling, W., et al. (2009). Traditional Medicine in the Treatment of Drug Addiction. Am. J. Drug Alcohol. Abuse 35 (1), 1–11. doi:10.1080/00952990802455469
Lu, S., Xinxin, L., and Kun, L. (2015). Caspase-3 and Bcl-2 Expression in Methamphetamine-dependent Rat Brain Interfered with Astragaloside IV (China). J. Apoplexy Nervous Dis. 32 (6), 505–507. doi:10.19845/j.cnki.zfysjjbzz.2015.06.007
Lull, M. E., and Block, M. L. (2010). Microglial Activation and Chronic Neurodegeneration. Neurotherapeutics 7 (4), 354–365. doi:10.1016/j.nurt.2010.05.014
Ma, C. L., Li, L., Yang, G. M., Zhang, Z. B., Zhao, Y. N., Zeng, X. F., et al. (2020). Neuroprotective Effect of Gastrodin in Methamphetamine-Induced Apoptosis through Regulating cAMP/PKA/CREB Pathway in Cortical Neuron. Hum. Exp. Toxicol. 39 (8), 1118–1129. doi:10.1177/0960327120911438
Mark, K. A., Soghomonian, J. J., and Yamamoto, B. K. (2004). High-dose Methamphetamine Acutely Activates the Striatonigral Pathway to Increase Striatal Glutamate and Mediate Long-Term Dopamine Toxicity. J. Neurosci. 24 (50), 11449–11456. doi:10.1523/JNEUROSCI.3597-04.2004
Martinez, J. A., Zhang, Z., Svetlov, S. I., Hayes, R. L., Wang, K. K., and Larner, S. F. (2010). Calpain and Caspase Processing of Caspase-12 Contribute to the ER Stress-Induced Cell Death Pathway in Differentiated PC12 Cells. Apoptosis 15 (12), 1480–1493. doi:10.1007/s10495-010-0526-4
McDonnell-Dowling, K., and Kelly, J. P. (2017). The Role of Oxidative Stress in Methamphetamine-Induced Toxicity and Sources of Variation in the Design of Animal Studies. Curr. Neuropharmacol. 15 (2), 300–314. doi:10.2174/1570159x14666160428110329
McFadden, L. M., Stout, K. A., Vieira-Brock, P. L., Allen, S. C., Nielsen, S. M., Wilkins, D. G., et al. (2012). Methamphetamine Self-Administration Acutely Decreases Monoaminergic Transporter Function. Synapse 66 (3), 240–245. doi:10.1002/syn.21506
McFadden, L. M., Cordie, R., Livermont, T., and Johansen, A. (2018). Behavioral and Serotonergic Changes in the Frontal Cortex Following Methamphetamine Self-Administration. Int. J. Neuropsychopharmacol. 21 (8), 758–763. doi:10.1093/ijnp/pyy044
Michel, S., Wanet, A., De Pauw, A., Rommelaere, G., Arnould, T., and Renard, P. (2012). Crosstalk between Mitochondrial (Dys)function and Mitochondrial Abundance. J. Cel Physiol. 227 (6), 2297–2310. doi:10.1002/jcp.23021
Miller, D. K., Oelrichs, C. E., Sun, G. Y., and Simonyi, A. (2014). Subchronic Apocynin Treatment Attenuates Methamphetamine-Induced Dopamine Release and Hyperactivity in Rats. Life Sci. 98 (1), 6–11. doi:10.1016/j.lfs.2013.12.031
Mizoguchi, H., and Yamada, K. (2019). Methamphetamine Use Causes Cognitive Impairment and Altered Decision-Making. Neurochem. Int. 124, 106–113. doi:10.1016/j.neuint.2018.12.019
Moratalla, R., Khairnar, A., Simola, N., Granado, N., García-Montes, J. R., Porceddu, P. F., et al. (2017). Amphetamine-related Drugs Neurotoxicity in Humans and in Experimental Animals: Main Mechanisms. Prog. Neurobiol. 155, 149–170. doi:10.1016/j.pneurobio.2015.09.011
Moszczynska, A., and Callan, S. P. (2017). Molecular, Behavioral, and Physiological Consequences of Methamphetamine Neurotoxicity: Implications for Treatment. J. Pharmacol. Exp. Ther. 362 (3), 474–488. doi:10.1124/jpet.116.238501
Mozaffari, S., Ramezany Yasuj, S., Motaghinejad, M., Motevalian, M., and Kheiri, R. (2019). Crocin Acting as a Neuroprotective Agent against Methamphetamine-Induced Neurodegeneration via CREB-BDNF Signaling Pathway. Iran J. Pharm. Res. 18 (2), 745–758. doi:10.22037/ijpr.2019.2393
Nakagawa, T., and Yuan, J. (2000). Cross-talk between Two Cysteine Protease Families. Activation of Caspase-12 by Calpain in Apoptosis. J. Cel Biol. 150 (4), 887–894. doi:10.1083/jcb.150.4.887
Nam, Y., Wie, M. B., Shin, E. J., Nguyen, T. T., Nah, S. Y., Ko, S. K., et al. (2015). Ginsenoside Re Protects Methamphetamine-Induced Mitochondrial Burdens and Proapoptosis via Genetic Inhibition of Protein Kinase C δ in Human Neuroblastoma Dopaminergic SH-SY5Y Cell Lines. J. Appl. Toxicol. 35 (8), 927–944. doi:10.1002/jat.3093
Nauffal, M., and Gabardi, S. (2016). Nephrotoxicity of Natural Products. Blood Purif. 41 (1-3), 123–129. doi:10.1159/000441268
Nguyen, X. K., Lee, J., Shin, E. J., Dang, D. K., Jeong, J. H., Nguyen, T. T., et al. (2015). Liposomal Melatonin Rescues Methamphetamine-Elicited Mitochondrial Burdens, Pro-apoptosis, and Dopaminergic Degeneration through the Inhibition PKCδ Gene. J. Pineal Res. 58 (1), 86–106. doi:10.1111/jpi.12195
Nordahl, T. E., Salo, R., and Leamon, M. (2003). Neuropsychological Effects of Chronic Methamphetamine Use on Neurotransmitters and Cognition: a Review. J. Neuropsychiatry Clin. Neurosci. 15 (3), 317–325. doi:10.1176/jnp.15.3.317
Oh, K. W., Kim, H. S., and Wagner, G. C. (1997a). Inhibitory Effects of Ginseng Total Saponin on Methamphetamine-Induced Striatal Dopamine Increase in Mice. Arch. Pharm. Res. 20 (5), 516–518. doi:10.1007/BF02973952
Oh, K. W., Kim, H. S., and Wagner, G. C. (1997b). Ginseng Total Saponin Inhibits the Dopaminergic Depletions Induced by Methamphetamine. Planta Med. 63 (1), 80–81. doi:10.1055/s-2006-957610
Pan, A. L., Hasalliu, E., Hasalliu, M., and Angulo, J. A. (2020). Epigallocatechin Gallate Mitigates the Methamphetamine-Induced Striatal Dopamine Terminal Toxicity by Preventing Oxidative Stress in the Mouse Brain. Neurotox Res. 37 (4), 883–892. doi:10.1007/s12640-020-00177-1
Panenka, W. J., Procyshyn, R. M., Lecomte, T., MacEwan, G. W., Flynn, S. W., Honer, W. G., et al. (2013). Methamphetamine Use: a Comprehensive Review of Molecular, Preclinical and Clinical Findings. Drug Alcohol Depend. 129 (3), 167–179. doi:10.1016/j.drugalcdep.2012.11.016
Park, J. H., Seo, Y. H., Jang, J. H., Jeong, C. H., Lee, S., and Park, B. (2017). Asiatic Acid Attenuates Methamphetamine-Induced Neuroinflammation and Neurotoxicity through Blocking of NF-kB/STAT3/ERK and Mitochondria-Mediated Apoptosis Pathway. J. Neuroinflammation 14 (1), 240. doi:10.1186/s12974-017-1009-0
Potashkin, J. A., and Meredith, G. E. (2006). The Role of Oxidative Stress in the Dysregulation of Gene Expression and Protein Metabolism in Neurodegenerative Disease. Antioxid. Redox Signal. 8 (1-2), 144–151. doi:10.1089/ars.2006.8.144
Radhakrishnan, M. L., and Tidor, B. (2008). Optimal Drug Cocktail Design: Methods for Targeting Molecular Ensembles and Insights from Theoretical Model Systems. J. Chem. Inf. Model. 48 (5), 1055–1073. doi:10.1021/ci700452r
Riddle, E. L., Fleckenstein, A. E., and Hanson, G. R. (2006). Mechanisms of Methamphetamine-Induced Dopaminergic Neurotoxicity. AAPS J. 8 (2), E413–E418. doi:10.1007/BF02854914
Ríos, J. L., Onteniente, M., Picazo, D., and Montesinos, M. C. (2016). Medicinal Plants and Natural Products as Potential Sources for Antiparkinson Drugs. Planta Med. 82 (11-12), 942–951. doi:10.1055/s-0042-107081
Roth, K. A., and D'Sa, C. (2001). Apoptosis and Brain Development. Ment. Retard. Dev. Disabil. Res. Rev. 7 (4), 261–266. doi:10.1002/mrdd.1036
Rusyniak, D. E. (2013). Neurologic Manifestations of Chronic Methamphetamine Abuse. Psychiatr. Clin. North. Am. 36 (2), 261–275. doi:10.1016/j.psc.2013.02.005
Sanchez, A., Malaty, I. A., Khanna, A., Busl, K., Youn, T. S., Nagaraja, N., et al. (2018). Bilateral Basal Ganglia Necrosis Secondary to Methamphetamine. Mov Disord. Clin. Pract. 5 (5), 555–556. doi:10.1002/mdc3.12649
Sano, R., and Reed, J. C. (2013). ER Stress-Induced Cell Death Mechanisms. Biochim. Biophys. Acta 1833 (12), 3460–3470. doi:10.1016/j.bbamcr.2013.06.028
Schep, L. J., Slaughter, R. J., and Beasley, D. M. (2010). The Clinical Toxicology of Metamfetamine. Clin. Toxicol. (Phila) 48 (7), 675–694. doi:10.3109/15563650.2010.516752
Schwartz, K., Weizman, A., and Rehavi, M. (2006). The Effect of Psychostimulants on [3H]dopamine Uptake and Release in Rat Brain Synaptic Vesicles. J. Neural Transm. (Vienna) 113 (9), 1347–1352. doi:10.1007/s00702-005-0383-4
Sekine, Y., Ouchi, Y., Sugihara, G., Takei, N., Yoshikawa, E., Nakamura, K., et al. (2008). Methamphetamine Causes Microglial Activation in the Brains of Human Abusers. J. Neurosci. 28 (22), 5756–5761. doi:10.1523/JNEUROSCI.1179-08.2008
Shaerzadeh, F., Streit, W. J., Heysieattalab, S., and Khoshbouei, H. (2018). Methamphetamine Neurotoxicity, Microglia, and Neuroinflammation. J. Neuroinflammation 15 (1), 341. doi:10.1186/s12974-018-1385-0
Shafahi, M., Vaezi, G., Shajiee, H., Sharafi, S., and Khaksari, M. (2018). Crocin Inhibits Apoptosis and Astrogliosis of Hippocampus Neurons against Methamphetamine Neurotoxicity via Antioxidant and Anti-inflammatory Mechanisms. Neurochem. Res. 43 (12), 2252–2259. doi:10.1007/s11064-018-2644-2
Shah, A., Silverstein, P. S., Singh, D. P., and Kumar, A. (2012). Involvement of Metabotropic Glutamate Receptor 5, AKT/PI3K Signaling and NF-κB Pathway in Methamphetamine-Mediated Increase in IL-6 and IL-8 Expression in Astrocytes. J. Neuroinflammation 9, 52. doi:10.1186/1742-2094-9-52
Shen, K., Zhang, Y., Lv, X., Chen, X., Zhou, R., Nguyen, L. K., et al. (2016). Molecular Mechanisms Involving Sigma-1 Receptor in Cell Apoptosis of BV-2 Microglial Cells Induced by Methamphetamine. CNS Neurol. Disord. Drug Targets 15 (7), 857–865. doi:10.2174/1871527315666160518122816
Shin, E. J., Bach, J. H., Nguyen, T. T., Nguyen, X. K., Jung, B. D., Oh, K. W., et al. (2011). Gastrodia Elata Bl Attenuates Methamphetamine-Induced Dopaminergic Toxicity via Inhibiting Oxidative Burdens. Curr. Neuropharmacol. 9 (1), 118–121. doi:10.2174/157015911795016967
Shin, E. J., Shin, S. W., Nguyen, T. T., Park, D. H., Wie, M. B., Jang, C. G., et al. (2014). Ginsenoside Re Rescues Methamphetamine-Induced Oxidative Damage, Mitochondrial Dysfunction, Microglial Activation, and Dopaminergic Degeneration by Inhibiting the Protein Kinase Cδ Gene. Mol. Neurobiol. 49 (3), 1400–1421. doi:10.1007/s12035-013-8617-1
Shin, E. J., Dang, D. K., Tran, T. V., Tran, H. Q., Jeong, J. H., Nah, S. Y., et al. (2017). Current Understanding of Methamphetamine-Associated Dopaminergic Neurodegeneration and Psychotoxic Behaviors. Arch. Pharm. Res. 40 (4), 403–428. doi:10.1007/s12272-017-0897-y
Shin, E. J., Tran, H. Q., Nguyen, P. T., Jeong, J. H., Nah, S. Y., Jang, C. G., et al. (2018). Role of Mitochondria in Methamphetamine-Induced Dopaminergic Neurotoxicity: Involvement in Oxidative Stress, Neuroinflammation, and Pro-apoptosis-A Review. Neurochem. Res. 43 (1), 66–78. doi:10.1007/s11064-017-2318-5
Shukla, M., and Vincent, B. (2020). The Multi-Faceted Impact of Methamphetamine on Alzheimer's Disease: From a Triggering Role to a Possible Therapeutic Use. Ageing Res. Rev. 60, 101062. doi:10.1016/j.arr.2020.101062
Solayman, M., Islam, M. A., Alam, F., Khalil, M. I., Kamal, M. A., and Gan, S. H. (2017). Natural Products Combating Neurodegeneration: Parkinson's Disease. Curr. Drug Metab. 18 (1), 50–61. doi:10.2174/1389200217666160709204826
Sorsdahl, K., Stein, D. J., Pasche, S., Jacobs, Y., Kader, R., Odlaug, B., et al. (2021). A Novel Brief Treatment for Methamphetamine Use Disorders in South Africa: a Randomised Feasibility Trial. Addict. Sci. Clin. Pract. 16 (1), 3. doi:10.1186/s13722-020-00209-3
Stephans, S. E., and Yamamoto, B. K. (1994). Methamphetamine-induced Neurotoxicity: Roles for Glutamate and Dopamine Efflux. Synapse 17 (3), 203–209. doi:10.1002/syn.890170310
Sun, D., Yue, Q., Guo, W., Li, T., Zhang, J., Li, G., et al. (2015). Neuroprotection of Resveratrol against Neurotoxicity Induced by Methamphetamine in Mouse Mesencephalic Dopaminergic Neurons. Biofactors 41 (4), 252–260. doi:10.1002/biof.1221
Tata, D. A., and Yamamoto, B. K. (2007). Interactions between Methamphetamine and Environmental Stress: Role of Oxidative Stress, Glutamate and Mitochondrial Dysfunction. Addiction 102 (Suppl. 1), 49–60. doi:10.1111/j.1360-0443.2007.01770.x
UNODC (2020). World Drug Report 2021. Available at: https://wdr.unodc.org/wdr2020/index.html (Accessed July 10, 2021).
Valian, N., Ahmadiani, A., and Dargahi, L. (2017). Escalating Methamphetamine Regimen Induces Compensatory Mechanisms, Mitochondrial Biogenesis, and GDNF Expression, in Substantia Nigra. J. Cel Biochem 118 (6), 1369–1378. doi:10.1002/jcb.25795
Wang, X., Wang, C., Wang, J., Zhao, S., Zhang, K., Wang, J., et al. (2014). Pseudoginsenoside-F11 (PF11) Exerts Anti-neuroinflammatory Effects on LPS-Activated Microglial Cells by Inhibiting TLR4-Mediated TAK1/IKK/NF-κB, MAPKs and Akt Signaling Pathways. Neuropharmacology 79, 642–656. doi:10.1016/j.neuropharm.2014.01.022
Wang, K. H., Penmatsa, A., and Gouaux, E. (2015). Neurotransmitter and Psychostimulant Recognition by the Dopamine Transporter. Nature 521 (7552), 322–327. doi:10.1038/nature14431
Wu, C. F., Liu, Y. L., Song, M., Liu, W., Wang, J. H., Li, X., et al. (2003). Protective Effects of Pseudoginsenoside-F11 on Methamphetamine-Induced Neurotoxicity in Mice. Pharmacol. Biochem. Behav. 76 (1), 103–109. doi:10.1016/s0091-3057(03)00215-6
Wu, C. W., Ping, Y. H., Yen, J. C., Chang, C. Y., Wang, S. F., Yeh, C. L., et al. (2007). Enhanced Oxidative Stress and Aberrant Mitochondrial Biogenesis in Human Neuroblastoma SH-SY5Y Cells during Methamphetamine Induced Apoptosis. Toxicol. Appl. Pharmacol. 220 (3), 243–251. doi:10.1016/j.taap.2007.01.011
Xie, Q., Li, H., Lu, D., Yuan, J., Ma, R., Li, J., et al. (2021). Neuroprotective Effect for Cerebral Ischemia by Natural Products: A Review. Front. Pharmacol. 12, 607412. doi:10.3389/fphar.2021.607412
Xu, E., Liu, J., Liu, H., Wang, X., and Xiong, H. (2017). Role of Microglia in Methamphetamine-Induced Neurotoxicity. Int. J. Physiol. Pathophysiol. Pharmacol. 9 (3), 84–100.
Yan, T., Li, L., Sun, B., Liu, F., Yang, P., Chen, T., et al. (2014). Luteolin Inhibits Behavioral Sensitization by Blocking Methamphetamine-Induced MAPK Pathway Activation in the Caudate Putamen in Mice. PLoS One 9 (6), e98981. doi:10.1371/journal.pone.0098981
Yang, X., Wang, Y., Li, Q., Zhong, Y., Chen, L., Du, Y., et al. (2018). The Main Molecular Mechanisms Underlying Methamphetamine- Induced Neurotoxicity and Implications for Pharmacological Treatment. Front. Mol. Neurosci. 11, 186. doi:10.3389/fnmol.2018.00186
Yang, G., Zeng, X., Li, J., Leung, C. K., Zhang, D., Hong, S., et al. (2020). Protective Effect of Gastrodin for Methamphetamine-Induced Neurotoxicity (China). Chinses Pharmacol. Bull. 36 (2), 238–245. doi:10.3969/j.issn.1001-1978.2020.02.017
Yu, S., Zhu, L., Shen, Q., Bai, X., and Di, X. (2015). Recent Advances in Methamphetamine Neurotoxicity Mechanisms and its Molecular Pathophysiology. Behav. Neurol. 2015, 103969. doi:10.1155/2015/103969
Yun, J. (2014). Limonene Inhibits Methamphetamine-Induced Locomotor Activity via Regulation of 5-HT Neuronal Function and Dopamine Release. Phytomedicine 21 (6), 883–887. doi:10.1016/j.phymed.2013.12.004
Zeng, Q., Lin, K., Liang, Z., Xiong, Q., Duan, J., Li, C., et al. (2020). Protective Effect and Mechanism of Terminalia Chebula Polyphenol Extract on Methamphetamine-Induced Injury in PC12 Cells. Sci. Techn. Food Industry 41 (05), 299–304. doi:10.13386/j.issn1002-0306.2020.05.049
Zeng, Q., Xiong, Q., Zhou, M., Tian, X., Yue, K., Li, Y., et al. (2021). Resveratrol Attenuates Methamphetamine-Induced Memory Impairment via Inhibition of Oxidative Stress and Apoptosis in Mice. J. Food Biochem. 45 (2), e13622. doi:10.1111/jfbc.13622
Zhang, Z., Yang, H., Yang, J., Xie, J., Xu, J., Liu, C., et al. (2019). Pseudoginsenoside-F11 Attenuates Cognitive Impairment by Ameliorating Oxidative Stress and Neuroinflammation in D-galactose-T-reated M-ice. Int. Immunopharmacol. 67, 78–86. doi:10.1016/j.intimp.2018.11.026
Keywords: methamphetamine, dopaminergic neurons, apoptosis, natural products, neurodegenaration
Citation: Zeng Y, Chen Y, Zhang S, Ren H, Xia J, Liu M, Shan B and Ren Y (2022) Natural Products in Modulating Methamphetamine-Induced Neuronal Apoptosis. Front. Pharmacol. 12:805991. doi: 10.3389/fphar.2021.805991
Received: 31 October 2021; Accepted: 09 December 2021;
Published: 04 January 2022.
Edited by:
Hong Zhang, Shanghai University of Traditional Chinese Medicine, ChinaReviewed by:
Jia Liu, China Academy of Chinese Medical Sciences, ChinaMailan Liu, Hunan University of Chinese Medicine, China
Copyright © 2022 Zeng, Chen, Zhang, Ren, Xia, Liu, Shan and Ren. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yunhui Chen, Y2hlbnl1bmh1aUBjZHV0Y20uZWR1LmNu; Baozhi Shan, c2hhbmJhb3poaUBqeHV0Y20uZWR1LmNu; Yulan Ren, cmVueXVsYW5AY2R1dGNtLmVkdS5jbg==
‡ORCID: Yiwei Zeng, https://orcid.org/0000-0002-7042-2503; Yunhui Chen, https://orcid.org/0000-0002-3555-8018
†These authors have contributed equally to this work