
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol., 03 January 2022
Sec. Experimental Pharmacology and Drug Discovery
Volume 12 - 2021 | https://doi.org/10.3389/fphar.2021.799561
This article is part of the Research TopicInsights in Experimental Pharmacology and Drug Discovery: 2021View all 15 articles
 Maria Grazia Morgese1*†
Maria Grazia Morgese1*† Maria Bove1†
Maria Bove1† Lorenzo Di Cesare Mannelli2
Lorenzo Di Cesare Mannelli2 Stefania Schiavone1
Stefania Schiavone1 Anna Laura Colia1
Anna Laura Colia1 Stefania Dimonte1
Stefania Dimonte1 Emanuela Mhillaj1
Emanuela Mhillaj1 Vladyslav Sikora1,3
Vladyslav Sikora1,3 Paolo Tucci1
Paolo Tucci1 Carla Ghelardini2
Carla Ghelardini2 Luigia Trabace1
Luigia Trabace1Alzheimer’s disease (AD), one of the most widespread neurodegenerative disorder, is a fatal global burden for the elder population. Although many efforts have been made, the search of a curative therapy is still ongoing. Individuating phenotypic traits that might help in investigating treatment response is of growing interest in AD research. AD is a complex pathology characterized by many comorbidities, such as depression and increased susceptibility to pain perception, leading to postulate that these conditions may rely on common biological substrates yet to be determined. In order to investigate those biological determinants to be associable with phenotypic traits, we used the rat model of amyloid beta-induced toxicity. This established model of early phase of AD is obtained by the intracerebroventricular injection of soluble amyloid beta1-42 (Aβ) peptide 7 days before performing experiments. In this model, we have previously reported increased immobility in the forced swimming test, reduced cortical serotonin levels and subtle alterations in the cognitive domain a depressive-like phenotype associated with subtle alteration in memory processes. In light of evaluating pain perception in this animal model, we performed two different behavioral tests commonly used, such as the paw pressure test and the cold plate test, to analyze mechanical hyperalgesia and thermal allodynia, respectively. Behavioural outcomes confirmed the memory impairment in the social recognition test and, compared to sham, Aβ-injected rats showed an increased selective susceptibility to mechanical but not to thermal stimulus. Behavioural data were then corroborated by neurochemical and biochemical biomarker analyses either at central or peripheral level. Data showed that the peptide injection evoked a significant increase in hypothalamic glutamate, kynurenine and dopamine content, while serotonin levels were reduced. Plasma Cystatin-C, a cysteine protease, was increased while serotonin and melatonin levels were decreased in Aβ-injected rats. Urinary levels paralleled plasma quantifications, indicating that Aβ-induced deficits in pain perception, mood and cognitive domain may also depend on these biomarkers. In conclusion, in the present study, we demonstrated that this animal model can mimic several comorbid conditions typical of the early phase of AD. Therefore, in the perspective of generating novel therapeutic strategies relevant to precision medicine in AD, this animal model and the biomarkers evaluated herein may represent an advantageous approach.
Alzheimer’s disease (AD) is the most common form of dementia in the elderly. It is nowadays becoming more evident that AD is heterogeneous in many aspects, ranging from biomolecular or clinical manifestations, thus indicating that AD cannot be explained with a single pathological process (Ferrari and Sorbi, 2021). In this light, studying differences in disease symptomatology, treatment responses may represent an innovative field of study for supporting and implementing precision medicine in AD. Indeed, this complex neurological disorder is often accompanied by a variety of comorbidities. Among these, depression is highly frequent in the way that some Authors have hypothesized that depressive symptoms may represent an early manifestation of this neurodegenerative disease (Pomara and Sidtis, 2010). Interestingly, the prevalence of chronic pain in dementia is quite elevated and it is esteemed to be between 30 and 80% (Defrin et al., 2015). On the other hand, several lines of evidence have reported alterations of pain perception in different psychiatric disorders (Lautenbacher and Krieg, 1994; Malejko et al., 2020). In particular, the pain experience has been frequently associated with emotional and cognitive dysfunctions, as well as to depressive states (Moriarty et al., 2011; Bushnell et al., 2013). Indeed, the comorbidity of pain and depression has been described as the “pain-depression syndrome or pain-depression dyad” (Thompson et al., 2016). More specifically, depression may induce a reduction of the pain threshold (Torta and Munari, 2010). This suggests a possible overlapping of neurobiological and molecular mechanisms underlying this comorbidity (Li, 2015), whose understanding may be crucially provided by preclinical investigations using animal models of depressive-like state. In this regard, we have previously reported that rats receiving an intracerebroventricular (icv) injection of soluble amyloid beta1-42 (Aβ) peptide showed increased immobility frequency in the forced swimming test, mimicking a behavioral despair state, also accompanied by neurochemical alterations, as well as changes in neurotrophin levels (Colaianna et al., 2010; Schiavone et al., 2017). Furthermore, in this animal model, depressive like symptoms are accompanied by subtle changes in spatial memory, mimicking the early manifestation of AD onset, featured by higher Aβ levels and alterations in emotional and cognitive domains. In this regard, we have previously fully validated this model. Immunohistochemical analyses were also previously carried out after the peptide injection (Trabace et al., 2007), and the peptide solution was fully evaluated by atomic force and transmission electron microscopy (Morgese et al., 2015). In particular, systemic soluble Aβ levels were still highier in treated injected rats 7 days after central administration of the peptide, while immunohistochemical analyses reveiled no gross signs of neurodegeneration within the area of Aβ diffusion in the periventricular parenchyma (Trabace et al., 2007; Morgese et al., 2015; Schiavone et al., 2017; Morgese et al., 2018). Therefore, in this model we generally assume that the supraphysiological levels of the peptide lead to profound unbalance in several brain circuitries (see (Trabace et al., 2007; Preda et al., 2008; Mura et al., 2010) for reference). Thus, from a behavioral and neurobiological point of view, this model can be considered as a valid animal model of early AD phase.
A key brain region involved in pain perception, particularly in its descending modulation, is the hypothalamus (Puopolo, 2019), which has also been reported to play key functions in the development of different depressive symptoms, acting as a crucial hub in a network of neurocircuits modulating these two comorbidities (Bao and Swaab, 2019). Interestingly, alteration in the hypothalamic area has also been reported in the early AD phase (Vercruysse et al., 2018). Of note, neurotransmitter systems implicated in pain perception, transmission and control physiologically overlap with those underlying the development of depressive disorders and cognition (Yang and Chang, 2019). Among them, dopaminergic pathways have been implicated in the central regulation and modulation of pain (Li C. et al., 2019). Conversely, nociceptive stimuli have been described to disrupt dopamine homeostasis in the central nervous system (Wood, 2008; Kim et al., 2015). Moreover, it has been reported that soluble Aβ could determine alterations in dopamine content in the rodent brain (Wu et al., 2007; Morgese and Trabace, 2019). Together with its crucial role in pain perception (Paredes et al., 2019), decreased central levels of serotonin (5-HT) have been widely described in depressive-like states induced by soluble Aβ (Colaianna et al., 2010; Schiavone et al., 2017; Morgese et al., 2018). In addition, drugs able to increase 5-HT levels can improve cognitive function in AD patients (Xie et al., 2019) and in rodent AD models (Ma et al., 2017). Increased levels of glutamate (GLU), a key player in the perception of pain and pain transmission from peripheral to central districts (Pereira and Goudet, 2018), were found in rodent brain following icv injection of soluble Aβ able to cause spatial memory impairment (Tucci et al., 2014), and whose accumulation might also lead to synaptic failure through disruption of the glutamatergic pathways (Findley et al., 2019). Of note, ketamine, an antagonist of the glutamatergic NMDA receptor, is able to reduce pain perception as well as depressive symptoms (Dowben et al., 2013; Hayley and Litteljohn, 2013), and in this model we have demonstrated that ketamine is able to prevent depressive-like phenotype in Aβ-treated rats (Schiavone et al., 2017).
Together with neuropathological dysfunctions at central levels, depression and AD have been described to be also associated with peripheral alterations. In particular, decreased plasma 5-HT levels have been detected in patients with major depression (Paul-Savoie et al., 2011). The 5-HT content in plasma has also been considered as a predictor of antidepressant treatment outcomes (Blardi et al., 2002; Holck et al., 2019; Sun et al., 2020). In addition, it has been suggested that the peripheral serotonergic system is negatively implicated in AD (Kumar et al., 1995). Moreover, a negative association has been found between peripheral 5-HT amount and pain perception after different noxious stimuli (Ernberg et al., 2000; Farhanchi et al., 2018). On the other hand, melatonin (MEL), a circadian rhythm–regulated hormone, has been shown to be protective in neurodegeneration associated with AD (Savaskan et al., 2005). Indeed, anti-amyloidogenic as well as free radical–scavenging properties have been reported for MEL leading to hypothesize that this molecule might represent a therapeutic candidate to inhibit AD progression (Rosales-Corral et al., 2012; Lin et al., 2013) or a valid biomarker for defining disease progression or therapeutic success (Nous et al., 2021).
Depressive symptoms have also been associated with increased peripheral levels of Cystatin-C (Cys-C), a low-molecular-weight protein, synthesized at a constant rate in all nucleated cells (Li H. et al., 2019; Huang et al., 2021). Cys-C has been thought to be crucially implicated in the development of soluble Aβ neuronal dysfunctions, also relating to cognitive impairment (Zeng et al., 2019). Of note, central and peripheral enhanced amount of this protein has been described as a pain biomarker (Mannes et al., 2003). Hence, in our animal model of early phase AD, we investigated possible alterations of pain threshold as possible comorbidity in the early AD phase and in the depressive-like state.
In addition, involvement of noradrenergic, dopaminergic, serotonergic, including the tryptophan derivate kynurenine (KYN), glutamatergic and gamma-aminobutyric acid (GABA)-ergic systems was assessed in the hypothalamus, an area whose dysfunction has been considered a putative driver of AD pathology (Ishii and Iadecola, 2015). Furthermore, possible alterations in peripheral, such as in plasma and in urine levels, of 5-HT, KYN, MEL and Cys-C amounts were also evaluated.
In this light, the establishment of an animal model that can allow to study different aspects of this neuropathology could represent a valid advantage in the perspective of building tailored treatment for AD patients, thus pursuing the goal of precision medicine for this complex neurologic and neuropsychiatric disorder.
Experiments were performed on a total number of 47 seven week-old male Wistar rats (Envigo, San Pietro al Natisone, Italy). Animals were constantly maintained under controlled conditions, with a room temperature of 22 ± 1°C, relative humidity of 55 ± 5% and a light/dark cycle of 12 h (light on from 7:00 a.m. to 7:00 p.m.). Rats were group-housed and food and water were provided ad libitum. We carried out all experimental procedures involving animals in conformity with the institutional guidelines of the Italian Ministry of Health (D.L. 26/2014), the Guide for the Care and Use of Mammals in Neuroscience and Behavioral Research (National Research Council 2004), the Directive 2010/63/EU of the European Parliament and of the Council of September 22, 2010, on the protection of animals used for scientific purposes, as well as the ARRIVE guidelines. The Italian Ministry of Health approved our experimental protocol (protocol number: B2EF8.15-aut.737-2017-PR). Animal welfare was daily monitored in order to detect any signs of animal suffering or distress during the entire experimental procedure. All efforts to reduce the number of animals used and their suffering were conducted.
The human Aβ1–42 peptide (Tocris, Bristol, United Kingdom) was freshly prepared by using sterile double distilled water as vehicle (final concentration 4 μM), as previously described (Bove et al., 2018). An anesthetic cocktail solution (0.85 ml/kg) containing ketamine (100 mg/ml), xylazine (100 mg/ml) and acepromazine (10 mg/ml), dissolved in saline, was injected intraperitoneally in seven week-old rats. Rats were then secured in a stereotaxic frame (David Kopf Instruments, Tujunga, CA, United States) and underwent an icv infusion based on the following coordinates from bregma according to Paxinos and Watson rat brain atlas (Paxinos, 1986): AP = −0.5, ML = +1.2 and DV = −3.2. The incisor bar was set at −3.3 mm. A Hamilton micro-syringe was used to infuse 5 μl of soluble Aβ peptide, with an infusion rate of 2 μl/min over 2.5 min. Subsequently, the needle was left in place for 5 min in order to avoid elapsing. Vehicle was delivered to sham animals used as controls. Once dissecting the brain, it was verified whether the needle was placed in the correct way. Seven days after icv administration, experimental procedures were carried out.
The test was performed according to Di Cesare Mannelli et al. (Di Cesare Mannelli et al., 2015), by using an analgesimeter (Ugo Basile, Varese, Italy) to determine the nociceptive threshold in rats. This test is widely used as a test for hyperalgesia-like measurement (Deuis et al., 2017). Briefly, a blunt conical mechanical probe was used to apply an increasing pressure to the dorsal surface of the hind paw of slightly restrained rats. The constantly increasing pressure was applied until rats vocalized or showed a withdrawal reflex. Results were expressed in grams of tolerated mechanical pressure. The arbitrary cut-off value was set at 100 g and a blind observer collected the data.
The test was performed by using a cold plate kept at a constant temperature (4 ± 1°C) as floor in a stainless steel box (12 cm × 20 cm × 10 cm), as reported by Pacini et al. (Pacini et al., 2016). This test is widely used as a test for allodynia-like measurement (Deuis et al., 2017). Each animal was placed in the box and the latency to first lick the hind paw, expressed in seconds, was recorded as a measure of pain-related behaviour. A cut-off time of 60 s was adopted.
This test was used to analyze short-term memory by using a natural animal behaviour as social interacting with a similar (Mathiasen and Dicamillo, 2010) Briefly, rats were housed individually in plastic cages 3 days before the test. Juvenile rats used as stimulus animals were isolated in individual cages 30 min prior the beginning of the experiment. The social recognition test consisted in two trials of 5 min each, separated by an inter-trial of 30 min. During the first trial (T1) the stimulus animal is placed in the test animal home cage for 5 min. Then, an inter-trial of 30 min in which the stimulus returned to its cage was carried out. Subsequently, the trial 2 (T2) took place and the test animal was re-exposed to the stimulus animal. Time spent sniffing and social interacting, expressed in seconds, was scored by a blind observer. In order to exclude exploratory impairments, the time spent exploring the environment was also recorded.
At the end of the behavioural experiments, rats were euthanized, plasma was collected and brains were dissected by using a chilled rat brain matrix (World Precision Instruments, Inc. FL, United States) according to Paxinos and Watson rats brain atlas (Paxinos, 1986). The hypothalamus was weighed, frozen in liquid nitrogen and stored at − 80°C until ex vivo quantifications were performed.
Neurochemical analyses were performed by using HPLC. Hypothalamic samples were homogenized in 10 volumes (w/V) of 0.1 N perchloric acid, stored on ice for 30 min and then centrifuged at 10.000 × g for 10 min at 4°C, as previously described (Zotti et al., 2013). Plasma and urine samples were homogenized with 0.1 volumes (V/V) of 8% perchloric acid and centrifuged at 10.000 × g for 15 min at 4°C. Supernatants were then collected and HPLC analyses were carried out. Hypothalamic noradrenaline (NA), dopamine (DA) and 5-HT concentrations, plasma and urine 5-HT and 5-hydroxy-indole acetic acid (5-HIAA) levels were determined by using a LC18 reverse phase column (Kinetex, 150 mm × 4.2 mm, ODS 5 μm; Phenomenex, Castel Maggiore-Bologna, Italy). An electrochemical detector with a thin-layer amperometric cell (Ultimate 3000RS–ECD, Dionex, ThermoScientifics, Milan, Italy) at a working potential of 0.400 V was used. As previously described, KYN (Morgese et al., 2021b) and MEL were quantified with the same method by applying a working potential of 0.550 V. The flow rate was maintained at 0.7 ml/min by an isocratic pump (Shimadzu LC-10 AD, Kyoto, Japan). The mobile phase consisted of an aqueous solution of 75 mM NaH2PO4, 1.7 mM octane sulfonic acid, 0.3 mM EDTA, acetonitrile 10%, buffered at pH 3.0. Chemicals and reagents were purchased from Sigma Aldrich (Sigma Aldrich Milan Italy). Chromeleon software (version 6.80, Thermo Scientific Dionex, San Donato Milanese, Italy) was used for data analyses. The 5-HT turnover was calculated as 5-HIAA/5-HT ratio. Sample concentrations were expressed as fmol/mg of tissue. A fluorescence detector (emission length, 460 nm; excitation length, 340 nm, Jasco, Tokyo, Japan), after derivatization with o-phthalaldehyde/mercaptopropionic acid, was used to determine GLU, glutamine and GABA hypothalamic concentrations, as previously described (Bove et al., 2018). Chromatographic evaluation was accomplished by using an ODS-3 column (Kinetex 150 mm × 3 mm, ODS 5 μm; Phenomenex, Castel Maggiore-Bologna, Italy) with a gradient mobile phase (50 mM sodium acetate buffer, pH 6.95, with methanol increasing linearly from 2 to 30% (v/v) over 40 min. The flow rate was set at 0.5 ml/min. Data were acquired and integrated by using Borwin software (version 1.50; Jasco). Results are expressed as μM/mg of tissue.
Cys-C levels were quantified in plasmatic samples by using Rat Cys-C (Enzyme-Linked Immunosorbent Assays) ELISA kit (Fine Test, Wuhan Fine Biotech Co., Ltd., Wuhan, Hubei, China), according to manufacturers’ instructions. In order to avoid intra-assay variations, both standards and samples were analyzed in duplicate.
Statistical analyses were performed by using Two-way ANOVA for repeated measures followed by Šídák’s multiple comparisons test or Unpaired Student’s t-test, as required. Welch’s correction was applied to Unpaired Student’s t-test when F test indicated unequal variances. The statistical software used was GraphPad 9.0 for Windows (GraphPad Software, San Diego, CA, United States). For all tests, p-value was set at 0.05. Results are expressed as means ± standard error of the mean (S.E.M.).
In order to evaluate the effects of Aβ administration in response to a noxious mechanical stimulus, we performed the paw pressure test (hyperalgesia-like measurement). Figure 1A showed that the pain threshold, expressed as grams of tolerated weight, was significantly decreased in Aβ-treated rats compared to sham rats (Figure 1A, Two-Way ANOVA for repeated measures, followed by Šídák’s multiple comparisons test, F (3, 42) = 5.039, p < 0.05, time 0: Aβ vs sham). Moreover, we assessed the thermal pain sensitivity in Aβ-treated and sham rats by performing the cold plate test, based on a non-noxious cold stimulation (allodynia-like measurement). Our results showed no differences in cold hypersensitivity, expressed as latency to lick the paws, between the two experimental groups, although a slight trend to reduction could be appreciated (Figure 1B, Two-Way ANOVA for repeated measures, followed by Šídák’s multiple comparisons test, F (3, 36) = 1.599, p > 0.05 n. s., Aβ vs sham).
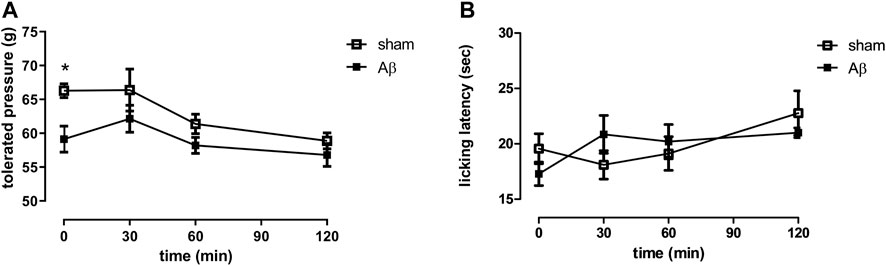
FIGURE 1. Effects of Aβ administration on paw pressure and cold plate tests. (A) Tolerated weight (g) in sham (n = 9) and Aβ-treated (n = 7) animals. Two-way ANOVA for repeated measures followed by Šídák’s multiple comparisons test, *p < 0.05 Aβ vs sham. (B) Licking latency (sec) in sham (n = 10) and Aβ-treated (n = 7) animals. Two-way ANOVA for repeated measures followed by Šídák’s multiple comparisons test, p > 0.05.
Here, we tested the effects of Aβ administration on social memory by carrying out the social recognition test. We found that sham rats reported a significant decrease in time spent sniffing and performing social interaction in the second exposition to a juvenile rats compared to first presentation, while no difference in both sniffing and social interaction between first and second encounter were retrieved in Aβ-treated rats (Figure 2A, Two-Way ANOVA for repeated measures followed by Šídák’s multiple comparisons test, F(1,12) = 41.13, p < 0.0001 sham T2 vs sham T1, Figure 2B, Two-Way ANOVA for repeated measures followed by Šídák’s multiple comparisons test, F(1,12) = 18.85, p < 0.001 sham T2 vs sham T1). In order to evaluate whether the results in time spent performing social exploration might depend from exploratory deficits, we scored total exploratory behaviours and we found no differences between trial 1 and trial 2 in both Aβ-treated and sham groups (Figure 2C, Two-Way ANOVA for repeated measures followed by Šídák’s multiple comparisons test, F(1,12) = 1.109, n. s sham T2 vs sham T1).
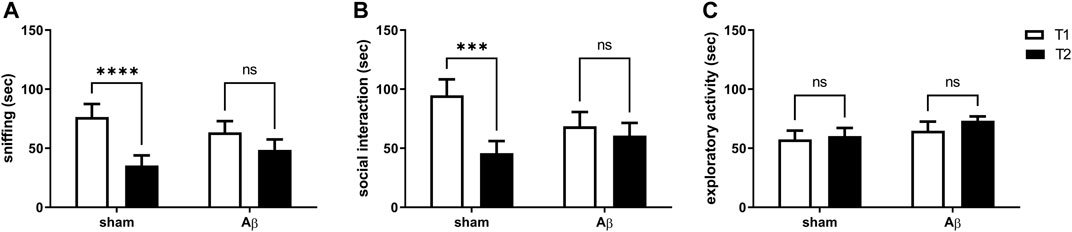
FIGURE 2. Effects of Aβ administration on social recognition paradigm. (A) Sniffing time (sec) in sham (n = 7) and Aβ-treated (n = 7) animals. Two-Way ANOVA for repeated measures followed by Šídák’s multiple comparisons test, ****p < 0.0001 Aβ vs sham. (B) Social interaction time (sec) in sham (n = 7) and Aβ-treated (n = 7) animals. Two-way ANOVA for repeated measures followed by Šídák’s multiple comparisons test, ***p < 0.001 Aβ vs sham. (C) Exploratory activity time (sec) in sham (n = 7) and Aβ-treated (n = 7) animals. Two-way ANOVA for repeated measures followed by Šídák’s multiple comparisons test, p > 0.05.
In order to corroborate behavioural results with neurochemical analyses, we quantified NA, DA and 5-HT content in the hypothalamus. Our results show no difference in NA content between the two experimental groups (Figure 3A, Unpaired Student’s t-test with Welch’s correction, n. s., η2= 0.005560 Aβ vs sham), while DA was significantly increased in Aβ injected rats (Figure 3B, Unpaired Student’s t-test with Welch’s correction, p < 0.01 η2= 0.7808, Aβ vs sham). In addition, rats injected with Aβ showed a significant decrease in 5-HT levels (Figure 3C, Unpaired Student’s t-test with Welch’s correction, p < 0.05 η2= 0.5795, Aβ vs sham), while KYN levels were significantly increased (Figure 3D, Unpaired Student’s t-test, p < 0.001 η2 = 0.7456, Aβ vs sham) compared to controls.
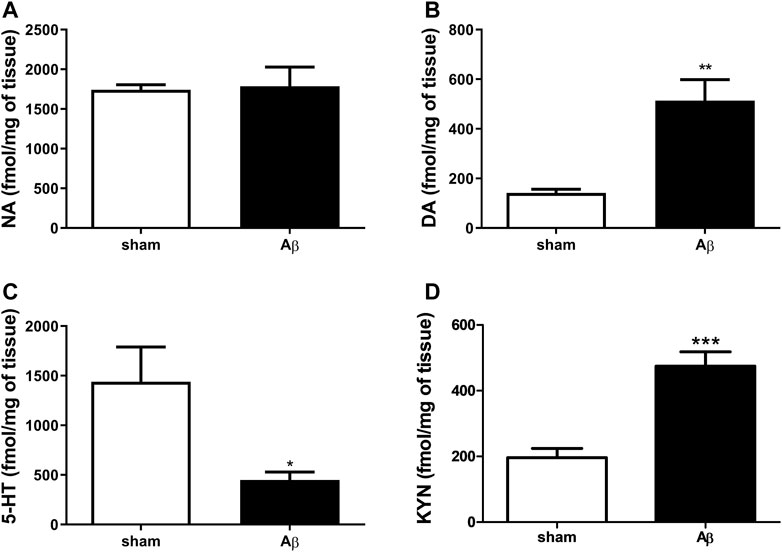
FIGURE 3. Effects of Aβ administration on hypothalamic NA, DA and 5-HT levels. (A) NA (fmol/mg of tissue) in sham (n = 5) and Aβ-treated (n = 6) animals. Unpaired Student’s t-test, p > 0.05. (B) DA (fmol/mg of tissue) in sham (n = 5) and Aβ-treated (n = 6) animals. Unpaired Student’s t-test, **p < 0.01 Aβ vs sham. (C) 5-HT (fmol/mg of tissue) in sham (n = 6) and Aβ-treated (n = 6) animals. Unpaired Student’s t-test, *p < 0.05 Aβ vs sham. (D) KYN (fmol/μl) in sham (n = 5) and Aβ-treated (n = 5) animals. Unpaired Student’s t-test, ***p < 0.001 Aβ vs sham.
We evaluated the effect of Aβ administration on hypothalamic GLU, glutamine and GABA levels. We found an increase in GLU and glutamine contents in Aβ-treated rats (Figures 4A,B, Unpaired Student’s t-test with Welch’s correction, η2= 0.5718 and 0.5628, respectively, p < 0.05 Aβ vs sham). As regarding GABA quantifications, rats administrated with Aβ did not show any difference compared to sham rats (Figure 4C, Unpaired Student’s t-test with Welch’s correction, n. s. η2= 0.1124, Aβ vs sham).
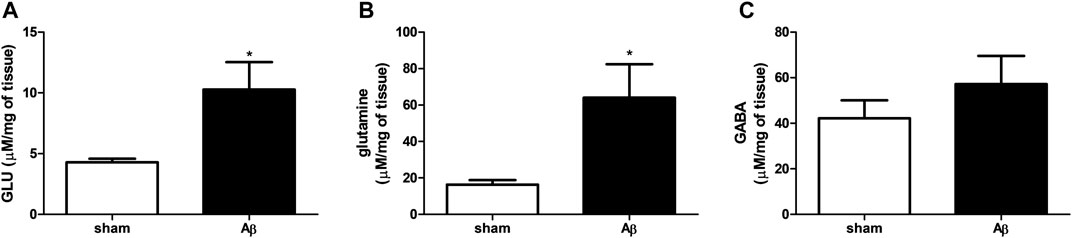
FIGURE 4. Effects of Aβ administration on hypothalamic GLU, glutamine and GABA levels. (A) GLU (μM/mg of tissue) in sham (n = 5) and Aβ-treated (n = 6) animals. Unpaired Student’s t-test, *p < 0.05. (B) Glutamine (μM/mg of tissue) in sham (n = 5) and Aβ-treated (n = 6) animals. Unpaired Student’s t-test, *p < 0.05. (C) GABA (μM/mg of tissue) in sham (n = 5) and Aβ-treated (n = 6) animals. Unpaired Student’s t-test, p > 0.05 Aβ vs sham.
In Figure 5, peripheral alterations after Aβ injection have been determined. In particular, we measured 5-HT and its turnover in plasma samples of Aβ-injected and sham rats, together with KYN, MEL and Cys-C content. Concerning 5-HT, rats treated with Aβ showed a significant decrease in plasmatic levels compared to sham (Figure 5A, Unpaired Student’s t-test with Welch’s correction, η2 = 0.6862, p < 0.05 Aβ vs sham). In addition, an increased 5-HT turnover was found in Aβ-treated rats compared to sham ones (Figure 5B, Unpaired Student’s t-test with Welch’s correction, η2 = 0.9085, p < 0.05 Aβ vs sham). No difference was found between groups in KYN levels (Figure 5C, Unpaired Student’s t-test, η2 = 0.03343, p > 0.05 n. s.), while MEL was significantly reduced in Aβ-treated rats (Figure 5D, Unpaired Student’s t-test, η2 = 0.7742, p < 0.01 Aβ vs sham). As regarding plasma Cys-C content, our results reported a significant increase in animals administrated with Aβ compared to sham rats (Figure 5E, Unpaired Student’s t-test, η2 = 0.5641, p < 0.05 Aβ vs sham).
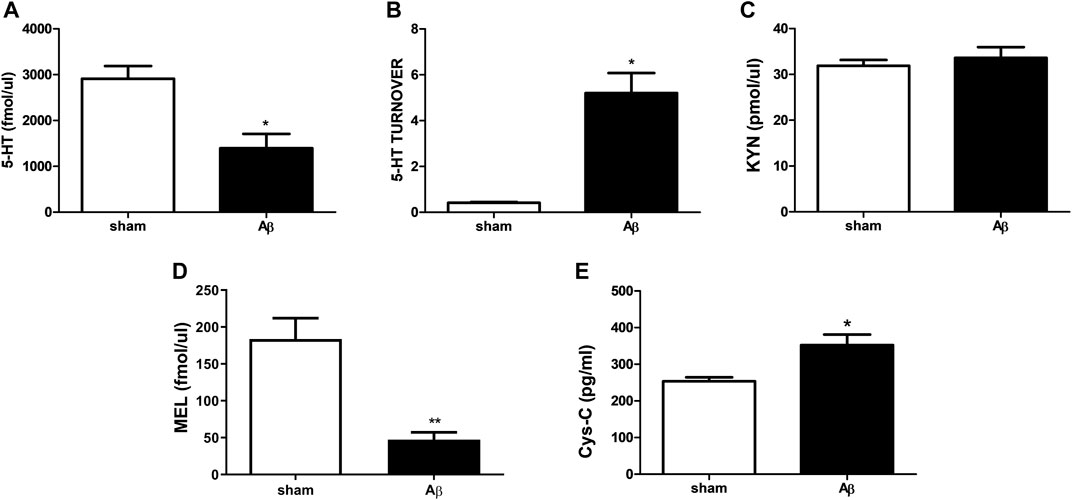
FIGURE 5. Effects of Aβ administration on plasma 5-HT, 5-HT turnover, KYN, MEL and Cys-C levels. (A) 5-HT (fmol/μl) in sham (n = 4) and Aβ-treated (n = 4) animals. Unpaired Student’s t-test, *p < 0.05 Aβ vs sham. (B) 5-HT turnover in sham (n = 4) and Aβ-treated (n = 4) animals. Unpaired Student’s t-test with Welch’s correction, *p < 0.05 Aβ vs sham. (C) KYN (pmol/μl) in sham (n = 5) and Aβ-treated (n = 5) animals. Unpaired Student’s t-test, p > 0.05 Aβ vs sham. (D) MEL (fmol/μl) in sham (n = 5) and Aβ-treated (n = 5) animals. Unpaired Student’s t-test, **p < 0.01 Aβ vs sham. (E) Cys-C (pg/ml) in sham (n = 5) and Aβ-treated (n = 5) animals. Unpaired Student’s t-test, *p < 0.05 Aβ vs sham.
As shown in Figure 6, urinary concentrations of 5-HT and its turnover, KYN and MEL after Aβ or its vehicle injection were determined. Concerning 5-HT, rats treated with Aβ showed a significant decrease in its plasmatic levels compared to sham (Figure 6A, Unpaired Student’s t-test with Welch’s correction, η2 = 0.7247, p < 0.05 Aβ vs sham). In addition, an increased 5-HT turnover was found in Aβ-treated rats compared to control (Figure 6B, Unpaired Student’s t-test with Welch’s correction, η2 = 0.8237, p < 0.05 Aβ vs sham). No difference was found between groups in KYN levels (Figure 6C, Unpaired Student’s t-test, η2 = 0.5078, p > 0.05 n. s.), while MEL was significantly reduced in Aβ-treated rats (Figure 6D, Unpaired Student’s t-test, η2 = 0.4647, p < 0.05 Aβ vs sham).
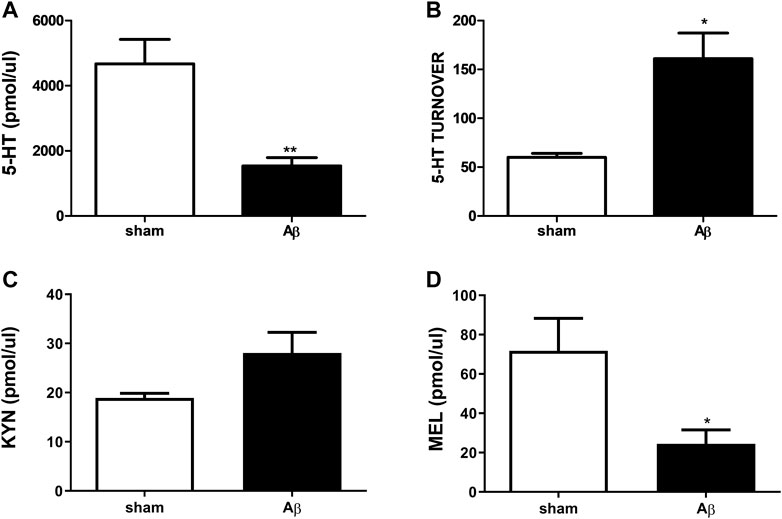
FIGURE 6. Effects of Aβ administration on urinary 5-HT, 5-HT turnover, KYN and MEL. (A) 5-HT (pmol/μl) in sham (n = 4) and Aβ-treated (n = 4) animals. Unpaired Student’s t-test, **p < 0.01 Aβ vs sham. (B) 5-HT turnover in sham (n = 4) and Aβ-treated (n = 4) animals. Unpaired Student’s t-test with Welch’s correction, *p < 0.05 Aβ vs sham. (C) KYN (pmol/μl) in sham (n = 5) and Aβ-treated (n = 5) animals. Unpaired Student’s t-test, p > 0.05 Aβ vs sham. (D) MEL (pmol/μl) in sham (n = 5) and Aβ-treated (n = 5) animals. Unpaired Student’s t-test, *p < 0.05 Aβ vs sham.
In the present work, we aimed at evaluating if the rat model of Aβ-induced toxicity might represent a nonclinical model able to provide reliably probabilistic estimates of translation into human biology, according to the need of precision medicine.
Here, we demonstrated showed that in rats exposed to central administration of Aβ peptide, a rodent model we have previously shown validated useful for mimicking the symptomatology of early phase AD (Colaianna et al., 2010; Morgese et al., 2014), also showed impairment in social memory, along with reduced 5-HT content and enhanced GLU, DA and KYN at hypothalamic level. In addition, increased mechanonociception, but not thermal nociception, was evident after central Aβ administration.
This event might likely be due to increased inflammation caused by the peptide. Indeed, mechanonociception seems to be specifically dependent on phospholipase A2 (PLA2) activation, arachidonic acid release and production of pro-inflammatory cyclooxygenase (COX) derivatives (Meller, 1994). In keeping with this hypothesis, we have previously shown, in the same animal model, that the icv injection of Aβ peptide increased the expression of the inducible COX-2 enzyme (Mhillaj et al., 2018). Furthermore, it has been shown that expression of COX-2 as well as prostaglandin E2, can be enhanced after phosphorylation of MAPKs and NF-κB induced by oxidative stress (Onodera et al., 2015). In our experience, we have found that Aβ-induced toxicity is also occurring through the increased oxidative biomarker production and NF-κB expression (Morgese et al., 2021a; Morgese et al., 2021b), therefore we can speculate that alterations in oxidative and inflammatory status migh represent putative factors that predispose Aβ-treated rats to increased mechanical pain susceptibility.
Over the past few decades, a pivotal role for GLU in pain sensation and transmission has been increasingly proposed, and, in this regard, novel pain medications targeting this excitatory system are in the spotlight. Thus, the role played by supraspinal mGluRs to pain modulation, including the descending pain pathway that involves also the hypothalamus, is an emerging field of study (Pereira and Goudet, 2018). In this regard, it is well known that neural populations of the descending inhibitory pain pathway originate in the amygdala and in the hypothalamus and project to the spinal cord and other relevant areas (Rea et al., 2007). Glutamate and glutamatergic receptor activation in this area exerts a fine tune on the release of hypothalamic factors, including Substance P, crucial for pain chronicity (Zieglgansberger, 2019), and GABA. Thus, this model may be also suitable for the evaluation of novel drugs acting on these targets, future investigations are surely warranted.
Interestingly, hypothalamic dopaminergic tone was increased in Aβ-treated rats. Previous reports have evaluted hypothalamic-spinal dopaminergic system on pain modulation, however the role played by this neurotrasmitter in such a context has not been completely unravelled. Indeed, activation of DA receptors seems to differently interact with nociception and in particular, D2-like agonism has been linked to analgesia specific to mechanonociception, but not thermonociception (Almanza et al., 2015). It is worth to note that, in patients early diagnosed with AD, reduction of D2 receptors has been reported in brain areas associated with emotional control, such as the hippocampus, but also in brain areas deputed to control motor coordination such as the nigrostriatal pathways (Pan et al., 2019). Therefore, we can speculate that such an increase in DA can be related to reduced D2 receptors, leading to enhanced DA tone and higher D1 activation. On the other hand, Aβ has been shown to modulate the release of factors implicated in the response to stress, such as corticotropin releasing factor (CRF) (Lv et al., 2020). Elevated glucocorticoids can trigger dopamine release (Czyrak et al., 2003; Belujon and Grace, 2017), while GLU can induce CRF release (Herman et al., 2004). In addition, stress response is correlated with Aβ release and the peptide, in turn, controls HPA functioning (Morgese et al., 2017). In addition, along with the well defined decline of cognitive and emotional domains, also functions under hypothalamic control are dysfunctional in these patients, such as the disruption of circadian rhythms (Baloyannis et al., 2018). Indeed, reduced levels of MEL were identified at both plasma and urine in our model. This evaluation, in the light of individuating novel peripheral biomarkers useful for monitoring AD progression or efficacy of used AD medications might represents a valid advantage also for the purpose of precision medicine. In particular, MEL is a multifunctional hormone involved not only in the regulation of circadian rhythm, but also with recognized anti-oxidant and anti-amyloidogenic (He et al., 2010) properties, proposed as a possible factor for contrasting AD progression (Hossain et al., 2019). Indeed, MEL has been suggested as an early biomarker for detecting the first stages of AD (Wu et al., 2003; Hossain et al., 2019). Furthermore, MEL was shown to suppressed NF-κB, an ubiquitary located transcriptional effector of inflammatory mediators, whose activation lead to higher prostaglandin and nitric oxide levels contributing to the development of hyperalgesia (Petho and Reeh, 2012). In this regard, we have very recently shown, in mice, that the icv injection of Aβ led to increased expression of this factor (Morgese et al., 2021b). In addition, MEL can prevent IL-6 release NF-κB-induced in Aβ-treated brain slices (Lau et al., 2012) and memory loss secondary to NF-κB activation (Shen et al., 2007). Pro-inflammatory biomarkers, such as cytokines, are able of modulating neuronal activity by promoting the release of neuroactive molecules from glia or the endothelium, including GLU (Vezzani and Viviani, 2015). Furthermore, chronic inflammation is able to decrease central 5-HT tone (Couch et al., 2013) also in condition of pseudoinflammation, an inflammatory state in absence of pathogens, associated with enhanced Aβ production and depressive-like behavior in rats (Morgese et al., 2020). Lower cerebral 5-HT levels leads to alteration in functioning of descending pain pathways, indicating that impairment in mood and emotions mediated by brainstem areas are important in determining the levels of pain. In particular, the emotional and autonomic aspects of nociceptive processing crucially implicated the parabrachial area. This important supraspinal target relays to the hypothalamus and amygdala, thus driving the descending modulatory pathways through the rostroventral medulla, resulting in a spino–bulbo–spinal loop. Indeed, the effectiveness of pain relievers depends on 5-HT levels and represents a mechanism whereby emotions can alter pain perception (Suzuki et al., 2004). As confirmation, pharmacological modulation that correspond to higher 5-HT tone has been shown to be useful for reverting pain-like behaviors in pertinent animal models (Bobinski et al., 2015). Indeed, in our model, we found that 5-HT was reduced in the hypothalamus of treated rats. Therefore, such different outcomes, retrieved in the behavioral paradigms used, might depend on the selective activation of glutamatergic and serotonergic pathways associated with inflammatory state. In this regard, it has been reported that inflammation (Tu et al., 2005), as well as increased corticosterone levels (Luo et al., 2017), can lead to enhaced metabolism of tryptophan toward KYN production at hypothalamic level. Moreover, it has been reported that KYN metabolites can directly act on GLU receptors, thus influencing excitatory neurotransmission and stimulating an increase in GLU release (Schwarcz, 2016). In addition, plasma and urinary content of 5-HT and 5-HT turnover were reduced in our model. This kind of alterations have been reported also in depressed patients (Mitani et al., 2006) and might represent a possible non invasive biomarkers for monitoring drug efficacy.
In keeping with this hypothesis, Aβ administration was able to alter other relevant biological peripheral parameters, such as Cys-C (Zhai et al., 2016), involved in both depression and AD (Sundelof et al., 2008; Minev et al., 2010). This molecule is a cysteine protease inhibitor, expressed either centrally (Lofberg and Grubb, 1979) or peripherally. This enzyme has been proposed as a biomarker of renal insufficiency and it has been shown to correlate with depressive symptoms in the elderly with normal renal function (Minev et al., 2010). Accordingly, a greater Cys-C levels were associated with an increased risk of new diagnosis of depression in subjects with normal renal function (Li H. et al., 2019). Furthermore, cognitive decline and AD are associated with renal impairment in human (Zhang et al., 2020) and in animal model of AD (Nakagawa et al., 2017). In further support of this evidence, it has been shown that Cys-C and Aβ peptide co-localize in brains of AD patients (Sastre et al., 2004) and polymorphisms of the Cys-C gene are associated with an increased risk for AD (Finckh et al., 2000; Chuo et al., 2007). Furthermore Cys-C levels directly correlate with cognitive impairment in the elderly (Yaffe et al., 2008). Our data are in line with these clinical evidences, considering that higher systemic Cys-C levels were found here and impairment of memory were previuosly shown in our model (Mhillaj et al., 2018). Cys-C was reported to be higher also in chronic pain patients and persistent pain can induce the synthesis and the release of Cys-C in dorsal spinal cord able to outflow to the brain (Mannes et al., 2003).
In conclusion, we found that in our model of depressive-like phenotype, induced by central administration of Aβ peptide, a state of enhanced pain perception was present. This increased susceptibility to mechanical pain was accompanied by impaired central and peripheral neurotransmission. In turn, also alterations in peripheral biomarkers, both linked to depression and AD, were evidenced. These outcomes also support the hypothesis that this animal model can be a suitable tool for investigating comorbid conditions associated with the early phase AD.
After a long search for new drugs for this condition, AD is still a fatal global epidemic. However, research in recent decades has focused its attention on the development of drugs capable of counteracting this complex pathology without seeking specific treatments for specific symptoms. This animal model could therefore result in a useful tool displaying some AD endophenotypes on which a targeted pharmacological strategy could be pursued in a tailored manner.
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by The Italian Ministry of Health, experimental protocol B2EF8.18-aut.954-2017-PR.
MGM, LDCM, MB, SS, PT, CG and LT designed the research. MB, LDCM, SD, EM, VS, ALC, SS, PT and MGM performed the research. MB, PT, SS, LDCM, EM, VS, ALC and MGM analyzed the data. MGM, LCDM, MB, PT, CG, SS and LT wrote the manuscript. All authors revised and contributed to the article and approved the submitted version.
This research was funded by PRIN 2017 code 2017YZF7MA to LT from Italian Ministry of Education, University and Research (MIUR), PRIN 2017 code 2017AY8BP4 to SS from MIUR and Research for Innovation (REFIN) from Apulia Region code OE9C2692 to MB.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Almanza, A., Simón-Arceo, K., Coffeen, U., Fuentes-García, R., Contreras, B., Pellicer, F., et al. (2015). A D2-like Receptor Family Agonist Produces Analgesia in Mechanonociception but Not in Thermonociception at the Spinal Cord Level in Rats. Pharmacol. Biochem. Behav. 137, 119–125. doi:10.1016/j.pbb.2015.08.013
Baloyannis, S. J., Mavroudis, I., Mitilineos, D., Baloyannis, I. S., and Costa, V. G. (2018). The Hypothalamus in Alzheimer’s Disease. London, UK: IntechOpen, 76–97. doi:10.5772/intechopen.81475
Bao, A. M., and Swaab, D. F. (2019). The Human Hypothalamus in Mood Disorders: The HPA axis in the center. IBRO Rep. 6, 45–53. doi:10.1016/j.ibror.2018.11.008
Belujon, P., and Grace, A. A. (2017). Dopamine System Dysregulation in Major Depressive Disorders. Int. J. Neuropsychopharmacol. 20, 1036–1046. doi:10.1093/ijnp/pyx056
Blardi, P., De Lalla, A., Leo, A., Auteri, A., Iapichino, S., Di Muro, A., et al. (2002). Serotonin and Fluoxetine Levels in Plasma and Platelets after Fluoxetine Treatment in Depressive Patients. J. Clin. Psychopharmacol. 22, 131–136. doi:10.1097/00004714-200204000-00005
Bobinski, F., Ferreira, T. A. A., Córdova, M. M., Dombrowski, P. A., Da Cunha, C., Santo, C. C. D. E., et al. (2015). Role of Brainstem Serotonin in Analgesia Produced by Low-Intensity Exercise on Neuropathic Pain after Sciatic Nerve Injury in Mice. Pain 156, 2595–2606. doi:10.1097/j.pain.0000000000000372
Bove, M., Mhillaj, E., Tucci, P., Giardino, I., Schiavone, S., Morgese, M. G., et al. (2018). Effects of N-3 PUFA Enriched and N-3 PUFA Deficient Diets in Naïve and Aβ-Treated Female Rats. Biochem. Pharmacol. 155, 326–335. doi:10.1016/j.bcp.2018.07.017
Bushnell, M. C., Ceko, M., and Low, L. A. (2013). Cognitive and Emotional Control of Pain and its Disruption in Chronic Pain. Nat. Rev. Neurosci. 14, 502–511. doi:10.1038/nrn3516
Chuo, L. J., Sheu, W. H., Pai, M. C., and Kuo, Y. M. (2007). Genotype and Plasma Concentration of Cystatin C in Patients with Late-Onset Alzheimer Disease. Dement Geriatr. Cogn. Disord. 23, 251–257. doi:10.1159/000100021
Colaianna, M., Tucci, P., Zotti, M., Morgese, M. G., Schiavone, S., Govoni, S., et al. (2010). Soluble Beta Amyloid(1-42): a Critical Player in Producing Behavioural and Biochemical Changes Evoking Depressive-Related State? Br. J. Pharmacol. 159, 1704–1715. doi:10.1111/j.1476-5381.2010.00669.x
Couch, Y., Martin, C. J., Howarth, C., Raley, J., Khrapitchev, A. A., Stratford, M., et al. (2013). Systemic Inflammation Alters central 5-HT Function as Determined by Pharmacological MRI. Neuroimage 75, 177–186. doi:10.1016/j.neuroimage.2013.02.046
Cox, V. C., and Valenstein, E. S. (1965). Attenuation of Aversive Properties of Peripheral Shock by Hypothalamic Stimulation. Science 149, 323–325. doi:10.1126/science.149.3681.323
Czyrak, A., Maćkowiak, M., Chocyk, A., Fijał, K., and Wedzony, K. (2003). Role of Glucocorticoids in the Regulation of Dopaminergic Neurotransmission. Pol. J. Pharmacol. 55, 667–674.
Defrin, R., Amanzio, M., De Tommaso, M., Dimova, V., Filipovic, S., Finn, D. P., et al. (2015). Experimental Pain Processing in Individuals with Cognitive Impairment: Current State of the Science. Pain 156, 1396–1408. doi:10.1097/j.pain.0000000000000195
Deuis, J. R., Dvorakova, L. S., and Vetter, I. (2017). Methods Used to Evaluate Pain Behaviors in Rodents. Front. Mol. Neurosci. 10, 284. doi:10.3389/fnmol.2017.00284
Di Cesare Mannelli, L., Marcoli, M., Micheli, L., Zanardelli, M., Maura, G., Ghelardini, C., et al. (2015). Oxaliplatin Evokes P2X7-dependent Glutamate Release in the Cerebral Cortex: A Pain Mechanism Mediated by Pannexin 1. Neuropharmacology 97, 133–141. doi:10.1016/j.neuropharm.2015.05.037
Dowben, J. S., Grant, J. S., and Keltner, N. L. (2013). Ketamine as an Alternative Treatment for Treatment-Resistant Depression. Perspect. Psychiatr. Care 49, 2–4. doi:10.1111/ppc.12006
Ernberg, M., Voog, U., Alstergren, P., Lundeberg, T., and Kopp, S. (2000). Plasma and Serum Serotonin Levels and Their Relationship to Orofacial Pain and Anxiety in Fibromyalgia. J. Orofac Pain 14, 37–46.
Farhanchi, A., Karkhanei, B., Amani, N., Aghajanloo, M., Khanlarzadeh, E., and Emami, Z. (2018). Association of Serum Serotonin and Pain in Patients with Chronic Low Back Pain before and after Spinal Surgery. Pain Res. Treat. 2018, 4901242. doi:10.1155/2018/4901242
Ferrari, C., and Sorbi, S. (2021). The Complexity of Alzheimer's Disease: an Evolving Puzzle. Physiol. Rev. 101, 1047–1081. doi:10.1152/physrev.00015.2020
Finckh, U., Von Der Kammer, H., Velden, J., Michel, T., Andresen, B., Deng, A., et al. (2000). Genetic Association of a Cystatin C Gene Polymorphism with Late-Onset Alzheimer Disease. Arch. Neurol. 57, 1579–1583. doi:10.1001/archneur.57.11.1579
Findley, C. A., Bartke, A., Hascup, K. N., and Hascup, E. R. (2019). Amyloid Beta-Related Alterations to Glutamate Signaling Dynamics during Alzheimer's Disease Progression. ASN Neuro 11, 1759091419855541. doi:10.1177/1759091419855541
Hayley, S., and Litteljohn, D. (2013). Neuroplasticity and the Next Wave of Antidepressant Strategies. Front Cel Neurosci 7, 218. doi:10.3389/fncel.2013.00218
He, H., Dong, W., and Huang, F. (2010). Anti-amyloidogenic and Anti-apoptotic Role of Melatonin in Alzheimer Disease. Curr. Neuropharmacol 8, 211–217. doi:10.2174/157015910792246137
Herman, J. P., Mueller, N. K., and Figueiredo, H. (2004). Role of GABA and Glutamate Circuitry in Hypothalamo-Pituitary-Adrenocortical Stress Integration. Ann. N. Y Acad. Sci. 1018, 35–45. doi:10.1196/annals.1296.004
Holck, A., Wolkowitz, O. M., Mellon, S. H., Reus, V. I., Nelson, J. C., Westrin, Å., et al. (2019). Plasma Serotonin Levels Are Associated with Antidepressant Response to SSRIs. J. Affect Disord. 250, 65–70. doi:10.1016/j.jad.2019.02.063
Hossain, M. F., Uddin, M. S., Uddin, G. M. S., Sumsuzzman, D. M., Islam, M. S., Barreto, G. E., et al. (2019). Melatonin in Alzheimer's Disease: A Latent Endogenous Regulator of Neurogenesis to Mitigate Alzheimer's Neuropathology. Mol. Neurobiol. 56, 8255–8276. doi:10.1007/s12035-019-01660-3
Huang, Y., Huang, W., Wei, J., Yin, Z., and Liu, H. (2021). Increased Serum Cystatin C Levels Were Associated with Depressive Symptoms in Patients with Type 2 Diabetes. Diabetes Metab. Syndr. Obes. 14, 857–863. doi:10.2147/DMSO.S295088
Ishii, M., and Iadecola, C. (2015). Metabolic and Non-cognitive Manifestations of Alzheimer's Disease: The Hypothalamus as Both Culprit and Target of Pathology. Cell Metab 22, 761–776. doi:10.1016/j.cmet.2015.08.016
Kim, J. Y., Tillu, D. V., Quinn, T. L., Mejia, G. L., Shy, A., Asiedu, M. N., et al. (2015). Spinal Dopaminergic Projections Control the Transition to Pathological Pain Plasticity via a D1/D5-Mediated Mechanism. J. Neurosci. 35, 6307–6317. doi:10.1523/JNEUROSCI.3481-14.2015
Kumar, A. M., Sevush, S., Kumar, M., Ruiz, J., and Eisdorfer, C. (1995). Peripheral Serotonin in Alzheimer's Disease. Neuropsychobiology 32, 9–12. doi:10.1159/000119205
Lau, W. W., Ng, J. K., Lee, M. M., Chan, A. S., and Wong, Y. H. (2012). Interleukin-6 Autocrine Signaling Mediates Melatonin MT(1/2) Receptor-Induced STAT3 Tyr(705) Phosphorylation. J. Pineal Res. 52, 477–489. doi:10.1111/j.1600-079X.2011.00965.x
Lautenbacher, S., and Krieg, J. C. (1994). Pain Perception in Psychiatric Disorders: a Review of the Literature. J. Psychiatr. Res. 28, 109–122. doi:10.1016/0022-3956(94)90023-x
Li, C., Liu, S., Lu, X., and Tao, F. (2019a). Role of Descending Dopaminergic Pathways in Pain Modulation. Curr. Neuropharmacol 17, 1176–1182. doi:10.2174/1570159X17666190430102531
Li, H., Wang, A., Qi, G., Guo, J., Li, X., Wang, W., et al. (2019b). Cystatin C and Risk of New-Onset Depressive Symptoms Among Individuals with a normal Creatinine-Based Estimated Glomerular Filtration Rate: A Prospective Cohort Study. Psychiatry Res. 273, 75–81. doi:10.1016/j.psychres.2019.01.009
Li, J. X. (2015). Pain and Depression Comorbidity: a Preclinical Perspective. Behav. Brain Res. 276, 92–98. doi:10.1016/j.bbr.2014.04.042
Lin, L., Huang, Q. X., Yang, S. S., Chu, J., Wang, J. Z., and Tian, Q. (2013). Melatonin in Alzheimer's Disease. Int. J. Mol. Sci. 14, 14575–14593. doi:10.3390/ijms140714575
Löfberg, H., and Grubb, A. O. (1979). Quantitation of Gamma-Trace in Human Biological Fluids: Indications for Production in the central Nervous System. Scand. J. Clin. Lab. Invest. 39, 619–626. doi:10.3109/00365517909108866
Luo, G. Q., Liu, L., Gao, Q. W., Wu, X. N., Xiang, W., and Deng, W. T. (2017). Mangiferin Prevents Corticosterone-Induced Behavioural Deficits via Alleviation of Oxido-Nitrosative Stress and Down-Regulation of Indoleamine 2,3-dioxygenase (Ido) Activity. Neurol. Res. 39, 709–718. doi:10.1080/01616412.2017.1310705
Lv, J., Chen, L., Zhu, N., Sun, Y., Pan, J., Gao, J., et al. (2020). Beta Amyloid-Induced Time-dependent Learning and Memory Impairment: Involvement of HPA axis Dysfunction. Metab. Brain Dis. 35, 1385–1394. doi:10.1007/s11011-020-00613-3
Ma, J., Gao, Y., Jiang, L., Chao, F. L., Huang, W., Zhou, C. N., et al. (2017). Fluoxetine Attenuates the Impairment of Spatial Learning Ability and Prevents Neuron Loss in Middle-Aged APPswe/PSEN1dE9 Double Transgenic Alzheimer's Disease Mice. Oncotarget 8, 27676–27692. doi:10.18632/oncotarget.15398
Malejko, K., Huss, A., Schönfeldt-Lecuona, C., Braun, M., and Graf, H. (2020). Emotional Components of Pain Perception in Borderline Personality Disorder and Major Depression-A Repetitive Peripheral Magnetic Stimulation (rPMS) Study. Brain Sci. 10, 905. doi:10.3390/brainsci10120905
Mannes, A. J., Martin, B. M., Yang, H. Y., Keller, J. M., Lewin, S., Gaiser, R. R., et al. (2003). Cystatin C as a Cerebrospinal Fluid Biomarker for Pain in Humans. Pain 102, 251–256. doi:10.1016/s0304-3959(02)00403-7
Mathiasen, J. R., and Dicamillo, A. (2010). Social Recognition Assay in the Rat. Curr. Protoc. Neurosci. 8, Unit 8 5I. doi:10.1002/0471142301.ns0805is53
Meller, S. T. (1994). Thermal and Mechanical Hyperalgesia. APS J. 3, 215–231. doi:10.1016/s1058-9139(05)80269-4
Mhillaj, E., Morgese, M. G., Tucci, P., Furiano, A., Luongo, L., Bove, M., et al. (2018). Celecoxib Prevents Cognitive Impairment and Neuroinflammation in Soluble Amyloid β-treated Rats. Neuroscience 372, 58–73. doi:10.1016/j.neuroscience.2017.12.046
Minev, E., Unruh, M., Shlipak, M. G., Simsonick, E., Yaffe, K., Leak, T. S., et al. (2010). Association of Cystatin C and Depression in Healthy Elders: the Health, Aging and Body Composition Study. Nephron Clin. Pract. 116, c241–6. doi:10.1159/000317205
Mitani, H., Shirayama, Y., Yamada, T., and Kawahara, R. (2006). Plasma Levels of Homovanillic Acid, 5-hydroxyindoleacetic Acid and Cortisol, and Serotonin Turnover in Depressed Patients. Prog. Neuropsychopharmacol. Biol. Psychiatry 30, 531–534. doi:10.1016/j.pnpbp.2005.11.021
Morgese, M. G., Bove, M., Francavilla, M., Schiavone, S., Dimonte, S., Colia, A. L., et al. (2021a). Sublingual AKBA Exerts Antidepressant Effects in the Aβ-Treated Mouse Model. Biomolecules 11, 686. doi:10.3390/biom11050686
Morgese, M. G., Colaianna, M., Mhillaj, E., Zotti, M., Schiavone, S., D'antonio, P., et al. (2015). Soluble Beta Amyloid Evokes Alteration in Brain Norepinephrine Levels: Role of Nitric Oxide and Interleukin-1. Front. Neurosci. 9, 428. doi:10.3389/fnins.2015.00428
Morgese, M. G., Schiavone, S., Bove, M., Colia, A. L., Dimonte, S., Tucci, P., et al. (2021b). N-3 PUFA Prevent Oxidative Stress in a Rat Model of Beta-Amyloid-Induced Toxicity. Pharmaceuticals (Basel) 14, 339. doi:10.3390/ph14040339
Morgese, M. G., Schiavone, S., Bove, M., Mhillaj, E., Tucci, P., and Trabace, L. (2018). Sub-chronic Celecoxib Prevents Soluble Beta Amyloid-Induced Depressive-like Behaviour in Rats. J. Affect Disord. 238, 118–121. doi:10.1016/j.jad.2018.05.030
Morgese, M. G., Schiavone, S., Maffione, A. B., Tucci, P., and Trabace, L. (2020). Depressive-like Phenotype Evoked by Lifelong Nutritional omega-3 Deficiency in Female Rats: Crosstalk Among Kynurenine, Toll-like Receptors and Amyloid Beta Oligomers. Brain Behav. Immun. 87, 444–454. doi:10.1016/j.bbi.2020.01.015
Morgese, M. G., Schiavone, S., and Trabace, L. (2017). Emerging Role of Amyloid Beta in Stress Response: Implication for Depression and Diabetes. Eur. J. Pharmacol. 817, 22–29. doi:10.1016/j.ejphar.2017.08.031
Morgese, M. G., and Trabace, L. (2019). Monoaminergic System Modulation in Depression and Alzheimer's Disease: A New Standpoint? Front. Pharmacol. 10, 483. doi:10.3389/fphar.2019.00483
Morgese, M. G., Tucci, P., Colaianna, M., Zotti, M., Cuomo, V., Schiavone, S., et al. (2014). Modulatory Activity of Soluble Beta Amyloid on HPA axis Function in Rats. Curr. Pharm. Des. 20, 2539–2546. doi:10.2174/13816128113199990500
Moriarty, O., Mcguire, B. E., and Finn, D. P. (2011). The Effect of Pain on Cognitive Function: a Review of Clinical and Preclinical Research. Prog. Neurobiol. 93, 385–404. doi:10.1016/j.pneurobio.2011.01.002
Mura, E., Lanni, C., Preda, S., Pistoia, F., Sarà, M., Racchi, M., et al. (2010). Beta-amyloid: a Disease Target or a Synaptic Regulator Affecting Age-Related Neurotransmitter Changes? Curr. Pharm. Des. 16, 672–683. doi:10.2174/138161210790883723
Nakagawa, T., Hasegawa, Y., Uekawa, K., and Kim-Mitsuyama, S. (2017). Chronic Kidney Disease Accelerates Cognitive Impairment in a Mouse Model of Alzheimer's Disease, through Angiotensin II. Exp. Gerontol. 87, 108–112. doi:10.1016/j.exger.2016.11.012
Nous, A., Engelborghs, S., and Smolders, I. (2021). Melatonin Levels in the Alzheimer's Disease Continuum: a Systematic Review. Alzheimers Res. Ther. 13, 52. doi:10.1186/s13195-021-00788-6
Onodera, Y., Teramura, T., Takehara, T., Shigi, K., and Fukuda, K. (2015). Reactive Oxygen Species Induce Cox-2 Expression via TAK1 Activation in Synovial Fibroblast Cells. FEBS Open Bio 5, 492–501. doi:10.1016/j.fob.2015.06.001
Pacini, A., Micheli, L., Maresca, M., Branca, J. J. V., Mcintosh, J. M., Ghelardini, C., et al. (2016). The α9α10 Nicotinic Receptor Antagonist α-conotoxin RgIA Prevents Neuropathic Pain Induced by Oxaliplatin Treatment. Exp. Neurol. 282, 37–48. doi:10.1016/j.expneurol.2016.04.022
Pan, X., Kaminga, A. C., Wen, S. W., Wu, X., Acheampong, K., and Liu, A. (2019). Dopamine and Dopamine Receptors in Alzheimer's Disease: A Systematic Review and Network Meta-Analysis. Front. Aging Neurosci. 11, 175. doi:10.3389/fnagi.2019.00175
Paredes, S., Cantillo, S., Candido, K. D., and Knezevic, N. N. (2019). An Association of Serotonin with Pain Disorders and its Modulation by Estrogens. Int. J. Mol. Sci. 20, 5729. doi:10.3390/ijms20225729
Paul-Savoie, E., Potvin, S., Daigle, K., Normand, E., Corbin, J. F., Gagnon, R., et al. (2011). A Deficit in Peripheral Serotonin Levels in Major Depressive Disorder but Not in Chronic Widespread Pain. Clin. J. Pain 27, 529–534. doi:10.1097/AJP.0b013e31820dfede
Pereira, V., and Goudet, C. (2018). Emerging Trends in Pain Modulation by Metabotropic Glutamate Receptors. Front. Mol. Neurosci. 11, 464. doi:10.3389/fnmol.2018.00464
Petho, G., and Reeh, P. W. (2012). Sensory and Signaling Mechanisms of Bradykinin, Eicosanoids, Platelet-Activating Factor, and Nitric Oxide in Peripheral Nociceptors. Physiol. Rev. 92, 1699–1775. doi:10.1152/physrev.00048.2010
Pomara, N., and Sidtis, J. J. (2010). Brain Neurotoxic Amyloid-Beta Peptides: Their Potential Role in the Pathophysiology of Depression and as Molecular Therapeutic Targets. Br. J. Pharmacol. 161, 768–770. doi:10.1111/j.1476-5381.2010.00948.x
Preda, S., Govoni, S., Lanni, C., Racchi, M., Mura, E., Grilli, M., et al. (2008). Acute Beta-Amyloid Administration Disrupts the Cholinergic Control of Dopamine Release in the Nucleus Accumbens. Neuropsychopharmacology 33, 1062–1070. doi:10.1038/sj.npp.1301485
Puopolo, M. (2019). The Hypothalamic-Spinal Dopaminergic System: a Target for Pain Modulation. Neural Regen. Res. 14, 925–930. doi:10.4103/1673-5374.250567
Rea, K., Roche, M., and Finn, D. P. (2007). Supraspinal Modulation of Pain by Cannabinoids: the Role of GABA and Glutamate. Br. J. Pharmacol. 152, 633–648. doi:10.1038/sj.bjp.0707440
Rosales-Corral, S. A., Acuña-Castroviejo, D., Coto-Montes, A., Boga, J. A., Manchester, L. C., Fuentes-Broto, L., et al. (2012). Alzheimer's Disease: Pathological Mechanisms and the Beneficial Role of Melatonin. J. Pineal Res. 52, 167–202. doi:10.1111/j.1600-079X.2011.00937.x
Sastre, M., Calero, M., Pawlik, M., Mathews, P. M., Kumar, A., Danilov, V., et al. (2004). Binding of Cystatin C to Alzheimer's Amyloid Beta Inhibits In Vitro Amyloid Fibril Formation. Neurobiol. Aging 25, 1033–1043. doi:10.1016/j.neurobiolaging.2003.11.006
Savaskan, E., Ayoub, M. A., Ravid, R., Angeloni, D., Fraschini, F., Meier, F., et al. (2005). Reduced Hippocampal MT2 Melatonin Receptor Expression in Alzheimer's Disease. J. Pineal Res. 38, 10–16. doi:10.1111/j.1600-079X.2004.00169.x
Schiavone, S., Tucci, P., Mhillaj, E., Bove, M., Trabace, L., and Morgese, M. G. (2017). Antidepressant Drugs for Beta Amyloid-Induced Depression: A New Standpoint? Prog. Neuropsychopharmacol. Biol. Psychiatry 78, 114–122. doi:10.1016/j.pnpbp.2017.05.004
Schwarcz, R. (2016). Kynurenines and Glutamate: Multiple Links and Therapeutic Implications. Adv. Pharmacol. 76, 13–37. doi:10.1016/bs.apha.2016.01.005
Shen, Y., Zhang, G., Liu, L., and Xu, S. (2007). Suppressive Effects of Melatonin on Amyloid-Beta-Induced Glial Activation in Rat hippocampus. Arch. Med. Res. 38, 284–290. doi:10.1016/j.arcmed.2006.10.007
Sun, Y., Drevets, W., Turecki, G., and Li, Q. S. (2020). The Relationship between Plasma Serotonin and Kynurenine Pathway Metabolite Levels and the Treatment Response to Escitalopram and Desvenlafaxine. Brain Behav. Immun. 87, 404–412. doi:10.1016/j.bbi.2020.01.011
Sundelöf, J., Arnlöv, J., Ingelsson, E., Sundström, J., Basu, S., Zethelius, B., et al. (2008). Serum Cystatin C and the Risk of Alzheimer Disease in Elderly Men. Neurology 71, 1072–1079. doi:10.1212/01.wnl.0000326894.40353.93
Suzuki, R., Rygh, L. J., and Dickenson, A. H. (2004). Bad News from the Brain: Descending 5-HT Pathways that Control Spinal Pain Processing. Trends Pharmacol. Sci. 25, 613–617. doi:10.1016/j.tips.2004.10.002
Thompson, T., Correll, C. U., Gallop, K., Vancampfort, D., and Stubbs, B. (2016). Is Pain Perception Altered in People with Depression? A Systematic Review and Meta-Analysis of Experimental Pain Research. J. Pain 17, 1257–1272. doi:10.1016/j.jpain.2016.08.007
Torta, R. G., and Munari, J. (2010). Symptom Cluster: Depression and Pain. Surg. Oncol. 19, 155–159. doi:10.1016/j.suronc.2009.11.007
Trabace, L., Kendrick, K. M., Castrignanò, S., Colaianna, M., De Giorgi, A., Schiavone, S., et al. (2007). Soluble Amyloid Beta1-42 Reduces Dopamine Levels in Rat Prefrontal Cortex: Relationship to Nitric Oxide. Neuroscience 147, 652–663. doi:10.1016/j.neuroscience.2007.04.056
Tu, H., Rady, P. L., Juelich, T., Smith, E. M., Tyring, S. K., and Hughes, T. K. (2005). Cytokine Regulation of Tryptophan Metabolism in the Hypothalamic-Pituitary-Adrenal (HPA) axis: Implications for Protective and Toxic Consequences in Neuroendocrine Regulation. Cell Mol Neurobiol 25, 673–680. doi:10.1007/s10571-005-4007-1
Tucci, P., Mhillaj, E., Morgese, M. G., Colaianna, M., Zotti, M., Schiavone, S., et al. (2014). Memantine Prevents Memory Consolidation Failure Induced by Soluble Beta Amyloid in Rats. Front. Behav. Neurosci. 8, 332. doi:10.3389/fnbeh.2014.00332
Vercruysse, P., Vieau, D., Blum, D., Petersén, Å., and Dupuis, L. (2018). Hypothalamic Alterations in Neurodegenerative Diseases and Their Relation to Abnormal Energy Metabolism. Front. Mol. Neurosci. 11, 2. doi:10.3389/fnmol.2018.00002
Vezzani, A., and Viviani, B. (2015). Neuromodulatory Properties of Inflammatory Cytokines and Their Impact on Neuronal Excitability. Neuropharmacology 96, 70–82. doi:10.1016/j.neuropharm.2014.10.027
Wood, P. B. (2008). Role of central Dopamine in Pain and Analgesia. Expert Rev. Neurother 8, 781–797. doi:10.1586/14737175.8.5.781
Wu, J., Khan, G. M., and Nichols, R. A. (2007). Dopamine Release in Prefrontal Cortex in Response to Beta-Amyloid Activation of Alpha7 * Nicotinic Receptors. Brain Res. 1182, 82–89. doi:10.1016/j.brainres.2007.08.079
Wu, Y. H., Feenstra, M. G., Zhou, J. N., Liu, R. Y., Toranõ, J. S., Van Kan, H. J., et al. (2003). Molecular Changes Underlying Reduced Pineal Melatonin Levels in Alzheimer Disease: Alterations in Preclinical and Clinical Stages. J. Clin. Endocrinol. Metab. 88, 5898–5906. doi:10.1210/jc.2003-030833
Xie, Y., Liu, P. P., Lian, Y. J., Liu, H. B., and Kang, J. S. (2019). The Effect of Selective Serotonin Reuptake Inhibitors on Cognitive Function in Patients with Alzheimer's Disease and Vascular Dementia: Focusing on Fluoxetine with Long Follow-Up Periods. Signal. Transduct Target. Ther. 4, 30. doi:10.1038/s41392-019-0064-7
Yaffe, K., Lindquist, K., Shlipak, M. G., Simonsick, E., Fried, L., Rosano, C., et al. (2008). Cystatin C as a Marker of Cognitive Function in Elders: Findings from the Health ABC Study. Ann. Neurol. 63, 798–802. doi:10.1002/ana.21383
Yang, S., and Chang, M. C. (2019). Chronic Pain: Structural and Functional Changes in Brain Structures and Associated Negative Affective States. Int. J. Mol. Sci. 20, 3130. doi:10.3390/ijms20133130
Zeng, Q., Huang, Z., Wei, L., Fang, J., and Lin, K. (2019). Correlations of Serum Cystatin C Level and Gene Polymorphism with Vascular Cognitive Impairment after Acute Cerebral Infarction. Neurol. Sci. 40, 1049–1054. doi:10.1007/s10072-019-03777-8
Zhai, J. L., Ge, N., Zhen, Y., Zhao, Q., and Liu, C. (2016). Corticosteroids Significantly Increase Serum Cystatin C Concentration without Affecting Renal Function in Symptomatic Heart Failure. Clin. Lab. 62, 203–207. doi:10.7754/clin.lab.2015.150701
Zhang, C. Y., He, F. F., Su, H., Zhang, C., and Meng, X. F. (2020). Association between Chronic Kidney Disease and Alzheimer's Disease: an Update. Metab. Brain Dis. 35, 883–894. doi:10.1007/s11011-020-00561-y
Zieglgänsberger, W. (2019). Substance P and Pain Chronicity. Cell Tissue Res 375, 227–241. doi:10.1007/s00441-018-2922-y
Keywords: pain, amyloid beta, depression, serotonin, kynurenine, melatonin, glutamate, precision medicine
Citation: Morgese MG, Bove M, Di Cesare Mannelli L, Schiavone S, Colia AL, Dimonte S, Mhillaj E, Sikora V, Tucci P, Ghelardini C and Trabace L (2022) Precision Medicine in Alzheimer’s Disease: Investigating Comorbid Common Biological Substrates in the Rat Model of Amyloid Beta-Induced Toxicity. Front. Pharmacol. 12:799561. doi: 10.3389/fphar.2021.799561
Received: 21 October 2021; Accepted: 01 December 2021;
Published: 03 January 2022.
Edited by:
Salvatore Salomone, University of Catania, ItalyReviewed by:
Shaoqiu He, Johns Hopkins Medicine, United StatesCopyright © 2022 Morgese, Bove, Di Cesare Mannelli, Schiavone, Colia, Dimonte, Mhillaj, Sikora, Tucci, Ghelardini and Trabace. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Grazia Morgese, bWFyaWFncmF6aWEubW9yZ2VzZUB1bmlmZy5pdA==
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.