 Karolina E. Kolaczynska
Karolina E. Kolaczynska Dino Luethi
Dino Luethi Daniel Trachsel
Daniel Trachsel Marius C. Hoener
Marius C. Hoener Matthias E. Liechti
Matthias E. Liechti
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol., 09 February 2022
Sec. Neuropharmacology
Volume 12 - 2021 | https://doi.org/10.3389/fphar.2021.794254
This article is part of the Research TopicNew Players on the Monoaminergic Field: Relevance to the Mental DisordersView all 7 articles
3,4,5-Trimethoxyphenethylamine (mescaline) is a psychedelic alkaloid found in peyote cactus. Related 4-alkoxy-3,5-dimethoxy-substituted phenethylamines (scalines) and amphetamines (3C-scalines) are reported to induce similarly potent psychedelic effects and are therefore potential novel therapeutics for psychedelic-assisted therapy. Herein, several pharmacologically uninvestigated scalines and 3C-scalines were examined at key monoamine targets in vitro. Binding affinity at human serotonergic 5-HT1A, 5-HT2A, and 5-HT2C, adrenergic α1A and α2A, and dopaminergic D2 receptors, rat and mouse trace amine-associated receptor 1 (TAAR1), and human monoamine transporters were assessed using target specific transfected cells. Furthermore, activation of human 5-HT2A and 5-HT2B receptors, and TAAR1 was examined. Generally, scalines and 3C-scalines bound with weak to moderately high affinity to the 5-HT2A receptor (Ki = 150–12,000 nM). 3C-scalines showed a marginal preference for the 5-HT2A vs the 5-HT2C and 5-HT1A receptors whereas no preference was observed for the scalines. Extending the 4-alkoxy substituent increased 5-HT2A and 5-HT2C receptors binding affinities, and enhanced activation potency and efficacy at the 5-HT2A but not at the 5-HT2B receptor. Introduction of fluorinated 4-alkoxy substituents generally increased 5-HT2A and 5-HT2C receptors binding affinities and increased the activation potency and efficacy at the 5-HT2A and 5-HT2B receptors. Overall, no potent affinity was observed at non-serotonergic targets. As observed for other psychedelics, scalines and 3C-scalines interacted with the 5-HT2A and 5-HT2C receptors and bound with higher affinities (up to 63-fold and 34-fold increase, respectively) when compared to mescaline.
Serotonin [5-hydroxytryptamine, 5-HT (1; Figure 1)] modulates vital central nervous system processes like appetite, sexual activity, memory, attention, or sleep through interactions with various 5-HT receptors (G protein-coupled receptors except for 5-HT3 receptors) (Berger et al., 2009; Pithadia and Jain 2009). Altered 5-HT modulation can lead to several psychiatric conditions like anxiety, depression, or schizophrenia (Rapport et al., 1948). Widely distributed in the central nervous system, the 5-HT2 receptor subtype (5-HT2A, 5-HT2B, and 5-HT2C receptors) is a key pharmacological target for therapeutic drugs including antidepressants, anxiolytics, and antipsychotics (Roth 2011). Due to the lack of selectivity, however, identifying the various roles of each receptor subtype is difficult. In recent years, this issue has been tackled by the synthesis of selective ligands for each receptor isoform. Ligands which show high affinity-binding at the 5-HT2 receptor family but are devoid of subtype selectivity include substituted phenethylamines like 4-bromo-2,5-dimethoxyamphetamine (DOB; 2; Figure 1) (Glennon et al., 1992; Monte et al., 1996; Chambers et al., 2001; Chambers et al., 2002). The 5-HT2A and, albeit to a lesser extent, the 5-HT2C receptor isoforms are both involved in the induction of psychedelic effects associated with classical psychedelics (Figure 1) like lysergic acid diethylamide (LSD; 3) or psilocybin (4) as well as novel derivatives thereof (Glennon et al., 1992; Vollenweider et al., 1998; Chambers et al., 2002; Preller et al., 2016; Rickli et al., 2016; Preller et al., 2017; Luethi and Liechti 2020; Rudin et al., 2021). Both receptors mediate their effects via Gq-protein-mediated activation of phospholipase C (PLC), which catalyzes the hydrolysis of phosphatidylinositol 4,5-biphosphate (PIP2) to diacyl glycerol (DAG) and inositol triphosphate (IP3). This then leads to protein kinase C activation and calcium release to initiate further downstream effects (Roth 2011). Moreover, G protein-independent signaling pathways mediated by β-arrestins are activated and involved in the receptor effects (Allen et al., 2008). In addition to similar signaling mechanisms, selective binding between the two receptors is difficult as the receptor isoforms share a high degree of sequence homology in both the agonistic and antagonistic ligand binding sites (Boess and Martin 1994; Trachsel et al., 2009). For the past five decades, some natural and many synthetic phenethylamines have been examined for their 5-HT2 receptor binding affinities and psychoactive effects (Aldous et al., 1974; Barfknecht and Nichols 1975; Glennon et al., 1982; Trachsel 2003; Trachsel et al., 2013; Rickli et al., 2015; Luethi et al., 2018b; Eshleman et al., 2018; Kolaczynska et al., 2019; Luethi et al., 2019; Luethi and Liechti 2020; Rudin et al., 2021). The large family consisting of more than hundred psychedelics includes 3,4,5-trimethoxyphenethylamine (mescaline; 5) as the prototypical natural lead structure. Psychedelic phenethylamines can be classified into three distinct groups, based on their aryl substitution pattern: the 2,4,5-trisubstituted, the 2,4,6-trisubstituted, and the 3,4,5-trisubstituted compounds. For all three classes, some of the most active compounds contain two methoxy (MeO) groups allocated at the 2- or 3-position and at the 5- or 6-position. Modifications at the crucial 4-position may include small lipophilic substituents such as a Cl, Br, I, MeO, or methyl group, or larger lipophilic substituents such as a propylthio or a methallyloxy group. Currently, the in vitro and in vivo data available are mostly obtained from 2,4,5-trisubstituted derivatives [extensively reviewed in (Trachsel et al., 2013)].
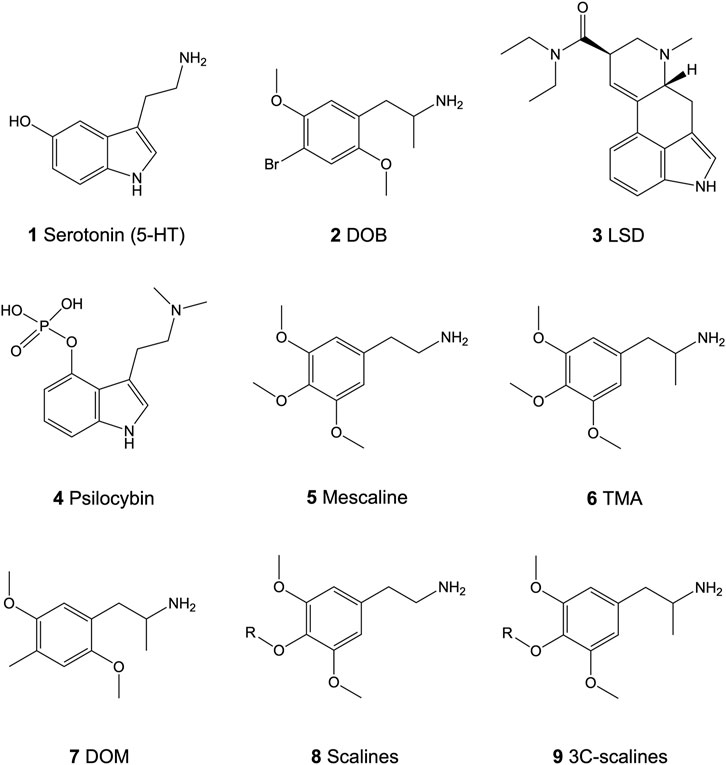
FIGURE 1. Chemical structures of the neurotransmitter serotonin (5-HT; 1) and the psychedelics 4-bromo-2,5-dimethoxyamphetamine (DOB; 2), lysergic acid diethylamide (LSD; 3), psilocybin (4), 3,4,5-trimethoxyphenethylamine (mescaline; 5), 3,4,5-trimethoxyamphetamine (TMA; 6), and 2,5-dimethoxy-4-methyl-amphetamine (DOM; 7), and the core structure of the 4-alkoxy-3,5-dimethoxyphenethylamines (scalines; 8) and 4-alkoxy-3,5-dimethoxyamphetamine (3C-scalines; 9).
Mescaline (5) was discovered as a natural ingredient of the psychoactive cactus peyote and identified as the principle pharmacological agent as early as in 1897 by Arthur Heffter (1898). Psychedelic doses of mescaline lie in the range of 180–360 mg or higher (Shulgin and Shulgin 1991). Mescaline’s α-methyl congener 3,4,5-trimethoxyamphetamine (TMA; 6) was first synthesized in 1947 by P. Hey (1947) with active doses lying in the range of 100–200 mg (Shulgin and Shulgin 1991). Thus far, the thorough investigation of the structure-activity relationship (SAR) of 3,4,5-trisubstituted phenethylamines has been slow largely due to early reports of their relatively weak human potencies. The focus shifted even more to 2,4,5-trisubstituted derivatives when Alexander Shulgin discovered that some of these substances are active at doses well below 10 mg [e.g., DOB (2): 1–3 mg or 2,5-dimethoxy-4-methylamphetamine (7); DOM: 3–10 mg] (Shulgin and Shulgin 1991). None of the more than three dozen of 3,4,5-trisubstituted phenethylamines and related amphetamines predominantly investigated by Shulgin have proven to be fully active at doses below 20 mg (Shulgin and Shulgin 1991). Moreover, the few performed in vitro studies indicated a markedly lower affinity at the 5-HT2A/2C receptors for 3,4,5-trisubstituted derivatives (scalines and 3C-scalines) compared to 2,4,5-trisubstituted phenethylamines. Retrospectively, however, this somewhat unwarranted focus towards the 2,4,5-series may be based on somewhat overhasty and generalized assumptions. Comparing human potencies of 2,5-dimethoxy- and 3,5-dimethoxyphenethylamines including their α-methyl congeners bearing identical 4-substituents revealed a far less distinctive predominance in favor of the 2,4,5-class (Trachsel et al., 2013). Some of the 4-substituents even lead to more potent 3,4,5-trisubstituted derivatives compared to 2,4,5-trisubstituted derivatives. Moreover, there are still numerous 4-substituents remaining to be tested within the 3,4,5-series, which will allow further comparisons and conclusions.
2,4,6-trisubstituted derivatives are even less investigated. However, the available data suggests that there seems to be more shared SAR for these compounds with the 2,4,5 derivatives than with the 3,4,5-trisubstituted series. To be specific, while some of the 2,4,6-trisubstituted derivatives with identical 4-substituent are significantly less potent in human than the 2,4,5-series, the identical 4-substituents lead to the most potent derivatives in both series so far (e.g., 4-Br or 4-Me) (Shulgin and Shulgin 1991; Trachsel et al., 2013). The same could be observed for 5-HT2A/2C receptor interactions (Chambers et al., 2002). Also, conformational restriction of the MeO groups in both the 2,4,5- and 2,4,6-series lead to increased in vitro and in vivo potencies (Monte et al., 1996; Chambers et al., 2002). This is in contrast to the 3,4,5-trisubstituted compounds, where conformational restriction of the MeO groups of the mescaline molecule towards dihydrobenzofurane and tetrahydrobenzodifurane moieties only slightly increased 5-HT2A/2C receptor affinities. However, in contrast to mescaline, they failed to fully substitute in a drug discrimination experiment [training drug: LSD; 3 (Monte et al., 1997)]. The authors of that study concluded that the MeO groups of mescaline might need to be non-constrained in order to conformationally adapt when activating the 5-HT2A receptor. Therefore, the 3,4,5-trisubstituted phenethylamines may show a somewhat different binding mode than 2,4,5- or 2,4,6-trisubstituted compounds, and their functional potency may be of more importance than mere affinity. Another significant structural modifier is the presence of an α-methyl (α-Me) group. This only has a small effect on binding affinity of 2,4,5-trisubstituted derivatives at 5-HT2A/2C receptors for racemic α-Me containing derivatives (amphetamines), since they show similar affinity at the receptor when compared to their equivalent phenethylamine counterparts (Johnson et al., 1990; Glennon et al., 1992; Dowd et al., 2000; Parrish et al., 2005; Kolaczynska et al., 2019). In vivo, introduction of an α-Me group into the 2,4,5-series has noteworthy effects on, e.g., drug discrimination experiments (Glennon et al., 1988; Glennon 1991) or on head-twitch response (Halberstadt et al., 2020), where significantly higher potencies have been observed for racemic α-Me-containing substances. In humans, these α-Me derivatives display up to one order of magnitude increased potency and usually significantly prolonged duration of action compared to their phenethylamine counterparts (Shulgin and Shulgin 1991). This can, to some extent, be explained by an increase in hydrophobic properties and metabolic stability observed for the amphetamines due to monoamine oxidase inhibition by the α-Me group (Glennon et al., 1983; Nichols 1991; Glennon et al., 1992). A stronger intrinsic activity (i.e., maximal response produced when the receptor is bound and activated by the compound) observed for the amphetamines when compared to their phenethylamine counterparts may also explain these SAR since the intrinsic activity plays a key role at the receptor (Nichols et al., 1994; Parrish et al., 2005). It is important to note that the role of configuration of the chiral center in the 2,4,5-series has been extensively investigated in binding studies (Johnson et al., 1987; Sadzot et al., 1989; Parrish et al., 2005; Braden 2007), drug discrimination studies (Glennon 1991), and human experiments (Shulgin and Shulgin 1991). As an overall conclusion, the amphetamines with an R-configuration behaved as the more potent enantiomers (eutomers). They not only showed a higher affinity to the 5-HT2A receptor but also a higher functional potency and functional efficacy (intrinsic activity) than the S-enantiomers. The few human data available revealed a 2-fold increased potency for the R-enantiomers in comparison to the corresponding racemates, and the S-enantiomers contributed only very little to the psychedelic properties. Hitherto, the effect of chirality caused by α-Me introduction on the psychopharmacology of scalines has not been studied.
Both animal and human observations with 2,4,5-trisubstituted derivatives are in strong contrast to what has been observed for scalines and 3C-scalines. The data available for comparison of 3,4,5-trisubstituted phenethylamine derivatives (Figure 1; 8) with their α-Me congeners (Figure 1; 9) showed an only marginal increase in dose potency and a comparable duration of action, with mescaline (5; 180–360 mg; 8–12 h) vs TMA (6; 100–250 mg; 6–8 h) being an exception (Shulgin and Shulgin 1991; Shulgin and Shulgin 1997; Trachsel et al., 2013). Moreover, 3,4,5-tri-O-substituted phenethylamines undergo a different amino oxidase-based metabolism than 2,4,5-tri-O-substituted phenethylamines (e.g., monoamine oxidase) (Clark et al., 1965). This might at least somewhat explain why the introduction of an α-Me group has only little influence on human doses of scalines vs 3C-scalines. With the data available so far, it remains difficult to draw solid conclusions or to apply existing SAR of one of the three different classes to another class. In spite of the many SAR available so far, the effects of 4-position substituents on 5-HT2A/2C receptor interaction properties are not entirely understood and require further investigation. However, the overall experienced psychedelic effects are influenced several factors including agonist-to-antagonist transition, receptor activation potency, or interactions with additional targets (Ray 2010). As mentioned before, significant changes have been achieved with mescaline derivatives (Figure 2) bearing larger carbon chain lengths at the 4-alkoxy position. These derivatives include escaline (15), isoproscaline (IP; 22), proscaline (24), allylescaline (AL; 30), and methallylescaline (MAL; 32). All of these compounds are significantly more potent than mescaline (5) in humans (effective doses ranging from 30 to 80 mg) and have a similar duration of action (8–12 h) (Shulgin and Shulgin 1991; Trachsel et al., 2013). Their psychedelic properties (i.e., the many different aspects of altered perception, mood and cognition, ego dissolution, and transcendence) seem to be changed significantly by modifying the chemical structure, at least based on interpreting the anecdotal data available so far (Trachsel et al., 2013).
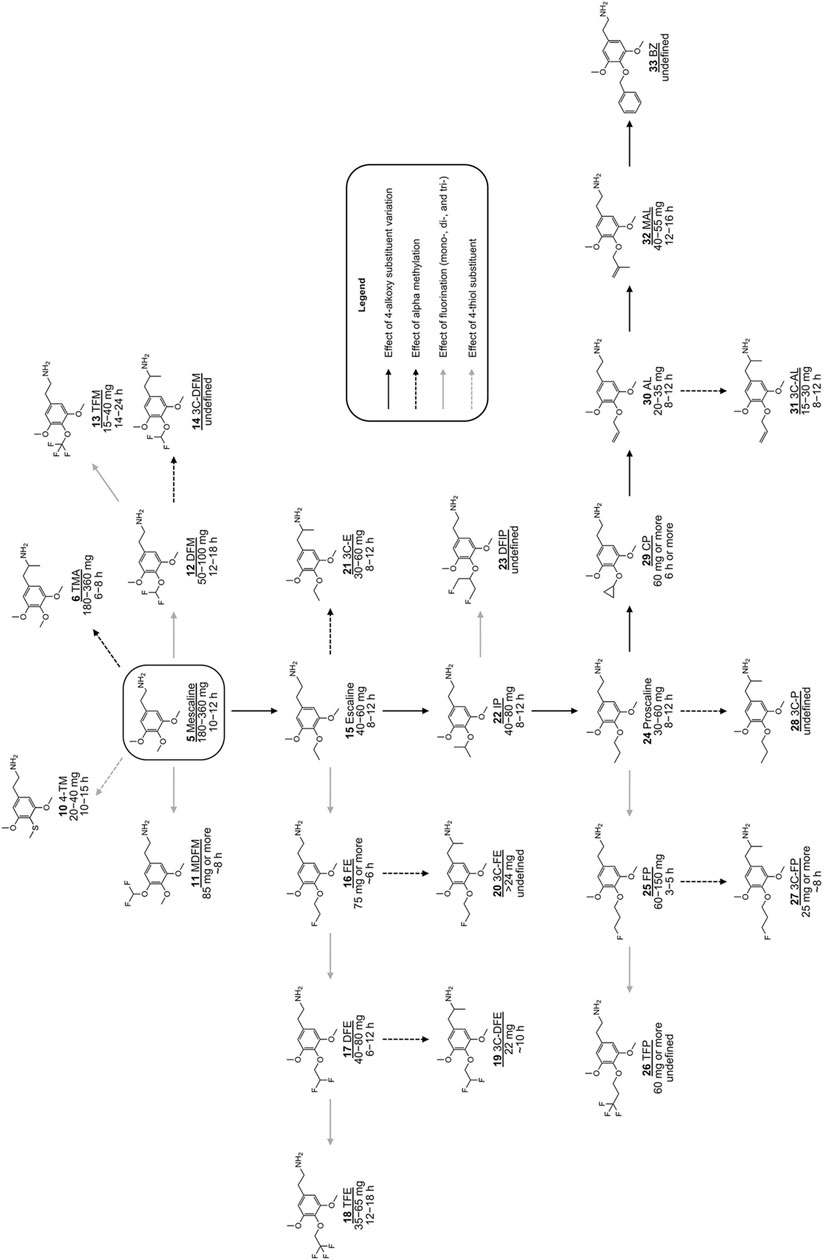
FIGURE 2. Chemical structures of various previously investigated scalines and 3C-scalines (Shulgin and Shulgin 1991; Shulgin and Shulgin 1997; Trachsel et al., 2013). Human oral doses and duration of action were taken from (Shulgin and Shulgin 1991; Shulgin and Shulgin 1997; Trachsel et al., 2013). Compounds tested in vitro in the present investigation are underlined.
The nomenclature for naming derivatives with a structural modification of the 4-substituent of mescaline (5) (i.e., the 4-MeO group) involves a common name in respect to their 4-substituent (Shulgin and Shulgin 1991; Trachsel et al., 2013). For example, the phenethylamine escaline (15) bears a 4-ethoxy group which can be shorted to a single letter, i.e., E. This single letter shortening can also be used to name several derivatives related to escaline, i.e fluoroescaline (FE; 16), which can also be called FE. Furthermore, P stands for proscaline (24; 4-PrO), AL for allylescaline (30; 4-Allyloxy) etc. The α-Me group containing counterparts (amphetamines) are defined as 3C compounds, and this term is simply used as a prefix such as 3C-E (21) or 3C-AL (31) and so on (Shulgin and Shulgin 1991; Shulgin and Shulgin 1997; Trachsel et al., 2013).
A simple substitution of the 4-MeO group on mescaline (5; Figure 2) to a 4-S group, leads to 4-thiomescaline (4-TM; 10), an analogue that has been shown to increase human potency 10-fold compared to 5 (active dose of 10 in humans = 20–40 mg) (Shulgin and Shulgin 1991). Introduction of fluorinated alkyloxy groups onto the 4-position of mescaline (5) has also led to derivatives with increased human potency when compared to 5 (Figure 2). These derivatives include difluoromescaline (DFM; 12) and trifluromescaline (TFM; 13), which have a 4-fold and > 9-fold increase in human potency, respectively. Both substances induce strong psychedelic effects and have significantly longer lasting effects than mescaline, with 13 being among the most potent mescaline-based derivatives synthesized to date (Trachsel 2012). Likewise, several fluorinated derivatives of the aforementioned 4-alkoxy analogues of mescaline have been synthesized (reviewed in (Trachsel 2012)), and initially pharmacologically investigated (Trachsel et al., 2013), e.g., compounds 5–33 (Figure 2).
In the light of the renewed interest in psychedelic substances in research and psychiatric therapy (Nichols 2016; Liechti 2017; Vollenweider and Preller 2020), investigating these derivatives is important to understand how certain structural modifications alter the way a derivative behaves at monoaminergic receptors and to gain further insight into the pharmacological properties of these derivatives. In the present investigation, we determined the receptor binding and activation properties of different mescaline derivatives and their α-Me containing counterparts at human serotonergic, adrenergic, and dopaminergic receptors, and at trace amine-associated receptor 1 (TAAR1). In addition, we explored the binding and inhibition potencies at human monoamine transporters.
Full names and abbreviations of the test compounds are provided in Supplementary Table S1. The 3,5-dimethoxy-4-substituted phenethylamines (mescaline [5], MDFM [11], DFM [12], TFM [13], FE [16], DFE [17], TFE [18], IP [22], DFIP [23], FP [25], TFP [26], CP [29], MAL [32], BZ [33]) and the 3,5-dimethoxy-4-substituted amphetamines (TMA [6], 3C-DFM [14], 3C-DFE [19], 3C-FE [20], 3C-E [21], 3C-FP [27], 3C-P [28], 3C-AL [31]) were synthesized as racemates as previously described (Shulgin and Shulgin 1991; Trachsel 2002; Trachsel 2003; Trachsel et al., 2013), and provided as hydrochloride salts for pharmacological testing by ReseaChem (Burgdorf, Switzerland). Purity of all substances was >98%. [3H]serotonin (80.0 Ci/mmol) was purchased from Anawa (Zurich, Switzerland). [3H]dopamine (30.0 Ci/mmol) and [3H]norepinephrine (13.1 Ci/mmol) were obtained from Perkin-Elmer (Schwerzenbach, Switzerland).
Radioligand binding affinity (Ki) for monoamine receptors and transporters was assessed according to previously described methods (Luethi et al., 2018b). In short, different cell line derived membrane preparations overexpressing respective monoamine receptors (human genes with the exception of rat and mouse TAAR1) or transporter were briefly incubated with corresponding radiolabeled selective ligands at a concentration equal to the dissociation constant Kd. The cell membrane preparations were obtained from Chinese hamster ovary cells (for hα1A adrenergic receptor), Chinese hamster lung cells (for hα2A adrenergic receptor) and HEK 293 cells (for h5-HT1A, h5-HT2A, h5-HT2C, and hD2 receptors, TAAR1, and hNET, hDAT, and hSERT). The specific binding of radioligand to the target site was defined by measuring the difference between total binding and nonspecific binding (calculated in the presence of the respective receptor competitor in excess). This was used to measure the ligand displacement by the substances under investigation.
The following radioligands and their respective competitors were used: 0.90 nM [3H]8-hydroxy-2-(dipropylamine)tetralin (8-OH-DPAT) and 10 μM pindolol (h5-HT1A receptor), 0.40 nM [3H]ketanserin and 10 μM spiperone (h5-HT2A receptor), 1.4 nM [3H]mesulergine and 10 μM mianserin (h5-HT2C receptor), 3.5 nM or 2.4 nM (rat or mouse isoform, respectively) [3H]RO5166017 and 10 μM RO5166017 (TAAR1), 0.11 nM [3H]prazosin and 10 μM chlorpromazine (hα1A adrenergic receptor), 2 nM [3H]rauwolscine and 10 μM phentolamine (hα2 adrenergic receptor), 1.2 nM [3H]spiperone and 10 μM spiperone (dopaminergic hD2 receptor), 2.9 nM N-methyl-[3H]nisoxetine and 10 μM indatraline (hNET), 1.5 nM [3H]citalopram and 10 μM indatraline (hSERT), 3.3 nM [3H]WIN35,428 and 10 μM indatraline (hDAT).
To assess the functional activity at the serotonin 5-HT2A receptor, mouse embryonic fibroblasts (NIH-3T3 cells) expressing human 5-HT2A receptor were seeded at a density of 70,000 cells per 100 μl in poly-D-lysine-coated 96-well plates according to methods previously described by Luethi, Trachsel et al. (2018). In brief, the NIH-3T3 cells were incubated in HEPES-Hank’s Balanced Salt Solution (HBSS) buffer (Gibco) for 1 h at 37°C. Subsequently, the plates were incubated with dye solution (100 μl/well) for 1 h at 37°C (fluorescence imaging plate reader [FLIPR] calcium 5 assay kit; Molecular Devices, Sunnyvale, CA, United States). Twenty-five microliter of test drugs diluted in HEPES-HBSS buffer composed of 250 mM probenecid were added to the plate online. Using nonlinear regression, the rise in fluorescence was measured and EC50 values were calculated from the concentration-response curves. The efficacy was calculated relative to 5-HT activity, which was defined as 100%.
To assess the functional activity at the serotonin 5-HT2B receptors, HEK 293 cells expressing the human 5-HT2B receptor were seeded at a density of 50,000 cells per well in 96-well poly-D-lysine-coated plates overnight at 37°C, according to methods previously described by Luethi, Trachsel et al. (2018). In brief, the HEK 293 cells were incubated overnight at 37°C in high glucose Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen, Zug, Switzerland), 10% fetal calf serum (non-dialyzed, heat-inactivated), 250 mg/L Geneticin and 10 ml/L PenStrep (Gibco). Using snap inversion, the growth medium was removed and 100 μl of calcium indicator Fluo-4 solution (Molecular Probes, Eugene, OR, United States) was added to each well for an incubation time of 45 min at 31°C. Thereafter, the Fluo-4 solution was removed (snap inversion) and subsequently an additional 100 μl of the Fluo-4 solution was added (incubation of 45 min at 31°C). Next, using the EMBLA cell washer, the cells were washed just before testing with HBSS and 20 mM HEPES and exposed to 100 μl of assay buffer. The plate was placed inside the FLIPR and 25 μl of test drugs diluted in assay buffer were added to the plate online. Using nonlinear regression, the rise in fluorescence was measured and EC50 values were calculated from the concentration-response curves. The efficacy was calculated relative to 5-HT activity, which was defined as 100%.
To assess the functional activity at the human TAAR1, HEK 293 cells expressing recombinant human TAAR1 were grown in 250 ml falcon culture flasks containing 30 ml of high glucose DMEM [10% heat inactivated fetal calf serum, 500 μg/ml Geneticin (Gibco, Zug, Switzerland) and 500 μg/ml hygromycin B] at 37°C and 5% CO2/95% air, according to methods previously described by Luethi, Trachsel et al. (2018). Once 80–90% confluency was reached, the cells were collected by removing the medium, washing with PBS and then adding 5 ml of trypsin/EDTA solution for 5 min at 37°C. Next, 45 ml of medium was added and the entire mixture was transferred into a falcon tube. The tube was then centrifuged at room temperature for 3 min at 900 revolutions per minute (rpm). Next, the supernatant was removed in order to resuspend the remaining cell pellet in fresh medium to 5 × 105 cells/ml. Hundred microliter of the cells was transferred into a 96-well plate (80,000 cells/well; BIOCOAT 6640, Becton Dickinson, Allschwil, Switzerland) and incubated for 20 h at 37°C.
For the cAMP assay, the aspirated medium was replaced with 50 μl PBS without Ca2+ and Mg2+ ions. Using snap inversion, the PBS was extracted and the plate was gently tapped against tissue. Next, 90 μl of Krebs-Ringer Bicarbonate buffer (KRB, Sigma-Aldrich) containing 1 mM IBMX was added and incubated for 60 min at 37°C and 5% CO2/95% air. Each test compound was examined in duplicate in a concentration range between 300 pM and 30 μM. A standard curve with a range of cAMP concentrations (0.13 nM–10 μM) was created for each 96-well plate. Each experiment was accompanied with a reference plate that included RO5256390, β-phenylethylamine and p-tyramine. The cells were exposed to either 30 μl of compound solution, 30 μl of β-phenylethylamine (as maximal response), or a basal control in PBS (containing 1 mM IBMX) for 40 min at 37°C. Next, under forceful shaking using black lids, the cells were exposed to 50 μl of 3× detection mix solution (composed of Ru-cAMP Alexa700 anti-cAMP antibody and lysis buffer) for 120 min at room temperature. Using the NanoScan reader (Innovate Optische Messtechnik, Berlin, Germany; 456 nm excitation wavelength; 630 and 700 nm emission wavelengths), the fluorescence was examined and the FRET signal was determined using the following equation; FRET (700 nm)- P × FRET (630 nm), where P = Ru (700 nm)/Ru (630 nm).
To exclude activity of the scalines and 3C-scalines at monoamine transporters at pharmacologically relevant concentrations, a single high drug concentration was examined using HEK 293 cells stably transfected with the human serotonin, norepinephrine or dopamine transporters (hSERT, hNET, or hDAT, respectively) as previously described (Luethi et al., 2018a). In summary, the cells were cultured in DMEM (Gibco, Life Technologies, Zug, Switzerland) containing both 250 μg/ml Geneticin (Gibco) and 10% fetal bovine serum (Gibco). Once the cells were confluent (70–90%) they were detached and resuspended in KRB (Sigma-Aldrich, Buchs, Switzerland) at a density of 3 × 106 cells/ml of buffer. For [3H]dopamine uptake experiments, the buffer additionally contained 0.2 mg/ml ascorbic acid. Hundred microliter of cell suspension per well was added to a round bottom 96-well plate. The cells were then incubated with 25 μl buffer containing the test drug (10 μM), vehicle control (0.1% dimethyl sulfoxide), or transporter-specific inhibitors [10 μM fluoxetine (SERT), 10 μM mazindol (DAT) or 10 μM nisoxetine (NET)] for 10 min by shaking on a rotary shaker (450 rpm) at room temperature. Uptake transport was initiated by adding [3H]serotonin, [3H]dopamine, or [3H]norepinephrine at a final concentration of 5 nM to the mixture. After 10 min, uptake transport was halted by the transfer of 100 μl of the cell mixture to 500 μl microcentrifuge tubes containing 50 μl of 3 M KOH and 200 μl silicon oil (1:1 mixture of silicon oil types AR 20 and 200; Sigma-Aldrich). The tubes were centrifuged for 3 min at 13,200 rpm, to allow the transport of the cells through the silicon oil layer into the KOH layer. The tubes were frozen in liquid nitrogen and the cell pellet was cut into 6 ml scintillation vials (Perkin-Elmer) containing 0.5 ml lysis buffer (1% NP-40, 50 mM NaCl, 0.05 M TRIS-HCl, 5 mM EDTA and deionized water). The samples were shaken for 1 h before 3 ml of scintillation fluid (Ultima Gold, Perkin Elmer, Schwerzenbach, Switzerland) was added. Monoamine uptake was then quantified by liquid scintillation counting on a Packard Tri-Carb Liquid Scintillation Counter 1900 TR. Nonspecific uptake in the presence of selective inhibitors was subtracted from the total counts.
All calculations and analyses were performed using Prism 7.0a (GraphPad, San Diego, CA, United States). IC50 values of the radioligand binding were determined by calculating nonlinear regression curves for a one-site model using at least three independent 10-point concentration-response curves for each substance. The Ki values correspond to the dissociative constant for the inhibitor and were calculated using the Cheng-Prusoff equation. Nonlinear regression concentration-response curves were used to determine EC50 values for 5-HT2A and 5-HT2B receptor activation. Maximal activation activity (efficacy) is expressed relative to the activity of 5-HT, which was set to 100%. Monoamine uptake of four independent experiments was compared to control using 1-way ANOVA analysis of variance followed by a Dunett’s multiple-comparison test. Monoamine uptake of MDMA was included as comparison. Receptor affinity binding (Ki) < 50 nM was defined as high affinity binding, Ki < 500 nM as moderate affinity binding, while Ki > 1,000 nM was defined as low affinity binding. Activation efficacy (max %) < 85% was defined as partial agonism while max % > 85% was defined as full agonism.
The 5-HT receptor binding affinities and activation potencies of the examined derivatives are listed in Table 1. The classical psychedelics LSD and 2C-B were previously tested using the same assays and included for comparison (Rickli et al., 2016; Luethi et al., 2018b). Among the phenethylamines, mescaline, MDFM, DFM TFM, CP, MAL, and BZ (Figure 2, structures 5, 11, 12, 13, 29, 32 and 33, respectively) were the only compounds that bound to the 5-HT1A receptor, albeit only in the lower micromolar range (Ki = 1.6–6.7 μM). In contrast, none of the 3C-scalines (Figure 2, structures 6, 14, 19, 20, 21, 27, 28, and 31, respectively) bound to the 5-HT1A receptor at the concentrations tested (Ki > 5,600 nM).
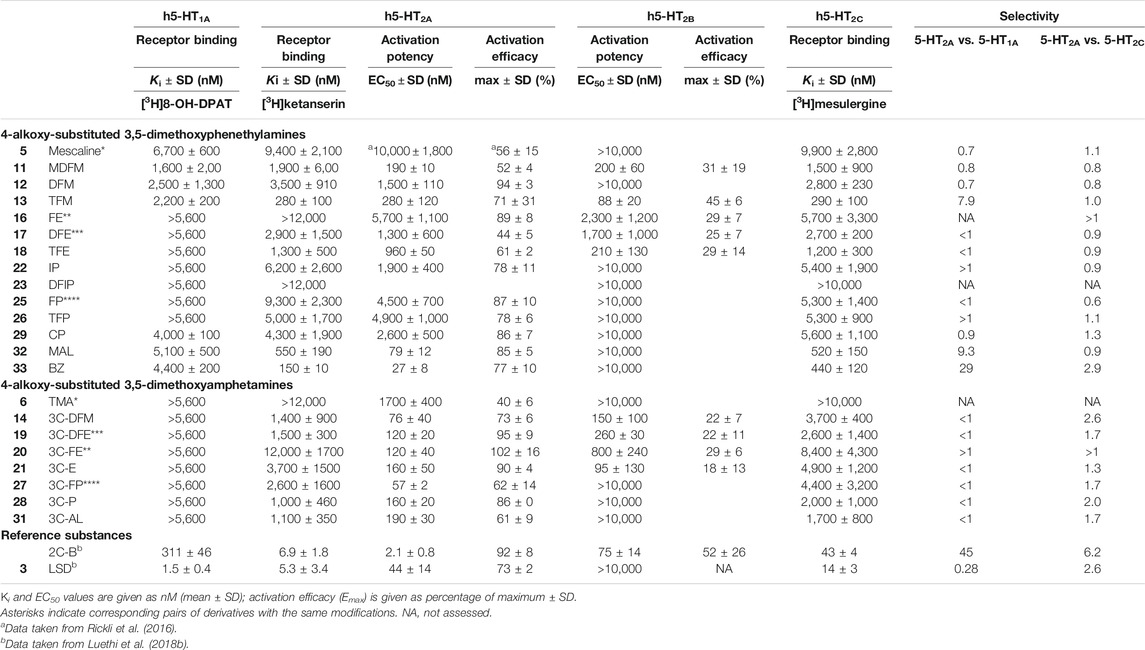
TABLE 1. Serotonin receptor binding affinities and activation potencies of 4-alkoxy-substituted 3,5-dimethoxyphenethylamines and amphetamines.
The fluorinated and bulky substituted phenethylamines TFM (13), MAL (32), and BZ (33) bound relatively potently to the 5-HT2A receptor in the submicromolar range (Ki = 150–550 nM). The remaining compounds (structures 11, 12, 17, 18, 22, 25, 26, and 29) bound in the micromolar range (Ki = 1,300–9,400 nM) with the exception of FE and DFIP (16 and 23; Ki > 12,000 nM). Compounds 5, 11, 13, 17, 18, 22, 26, and 33 were 5-HT2A receptor partial agonists with EC50 values in the range of 27–10,000 nM and activation efficacies of 44–78%. DFM (12), FE (16), FP (25), CP (29), and MAL (32) activated the 5-HT2A receptor as a full agonists with EC50 values in the range of 79–5,700 nM and activation efficacy of 85–94%.
The amphetamines bound to the 5-HT2A receptor in the micromolar range (structures 14, 19, 21, 27, 28, and 31; Ki = 1,000–3,700 nM) with the exception of TMA and 3C-FE (6 and 20; Ki > 12,000 nM). The amphetamines activated the receptor as with EC50 values in the range of 57–1,700 nM. Some amphetamines (6, 14, 27, and 31) activated the receptor as partial agonists with activation efficacies of 40–73%. 3C-DFE (12), 3C-FE (13), 3C-E (14), and 3C-P (31) were full agonists at the 5-HT2A receptor (activation efficacy = 86–102%).
The fluorinated phenethylamines MDFM (11), TFM (13), and TFE (18) activated the 5-HT2B receptor in the submicromolar range (EC50 = 88–210 nM), while FE (16) and DFE (17) activated the receptor in the micromolar range (EC50 = 1,700–2,300 nM). All of these compounds were relatively low efficacy partial agonists at the 5-HT2B receptor (activation efficacy = 25–45%). The remaining phenethylamines did not activate the 5-HT2B receptor (EC50 > 10,000 nM). The amphetamine derivatives 3C-DFM (14), 3C-DFE (19), 3C-FE (20), and 3C-E (21) activated the 5-HT2B receptor in the submicromolar range (EC50 = 95–800 nM) as low efficacy partial agonists with activation efficacy in the range of 18–29%. The remaining amphetamine derivatives did not activate the 5-HT2B receptor (EC50 > 10,000 nM).
Most compounds bound to the 5-HT2C receptor with micromolar affinity (Ki = 1,200–9,900 nM). Exception to this were the phenethylamines TFM (13), MAL (32), and BZ (33), which bound to the 5-HT2C receptor with submicromolar affinity (Ki = 290–520 nM) and DFIP (23), which did not bind to the 5-HT2C receptor (Ki > 10,000 nM).
Monoamine receptor and transporter binding affinities are listed in Table 2. None of the examined compounds activated the human TAAR1 (EC50 > 10,000 nM). At the rat TAAR1, most phenethylamine derivatives bound within a micromolar range (Ki = 1,000–3,000 nM) with the exception of DFM (12), TFM (13), TFP (26), and BZ (33), which bound at submicromolar concentrations (Ki = 110–910 nM). FE (16), IP (22), and DFIP (23) did not bind to the rat TAAR1 at the concentrations tested (Ki > 4,000 nM). The amphetamine derivative 3C-DFM (14) bound with a Ki of 380 nM to the rat TAAR1. TMA (6), 3C-DFE (19), 3C-P (28), and 3C-AL (31) bound in the micromolar range (Ki = 3,200–3,900 nM); for 3C-FE (20), 3C-E (21), and 3C-FP (27) no binding was observed at the rat TAAR1 at examined concentrations (Ki > 4,700 nM).
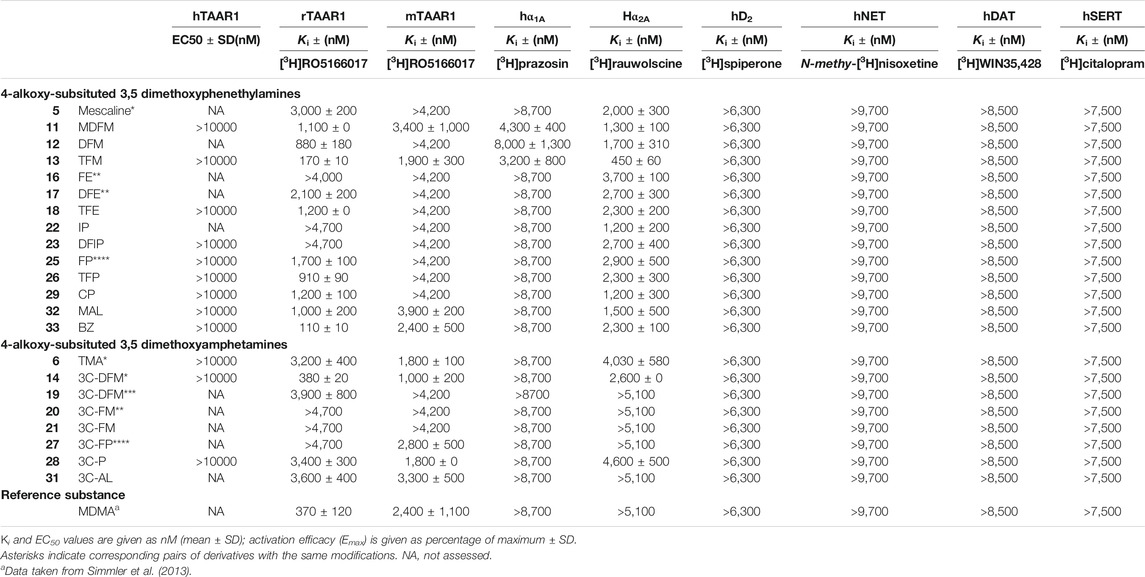
TABLE 2. Non-serotonergic receptor and transporter binding affinities of 4-alkoxy-subsituted 3,5-dimethoxyphenethylamines and amphetamines.
At the mouse TAAR1, the phenethylamine derivatives MDFM (11), TFM (13), MAL (32), and BZ (33) bound in the micromolar range (Ki = 1,900–3,900 nM) while none of the remaining derivatives bound to the receptor (Ki > 4,200 nM). The amphetamine derivatives TMA (6), 3C-DFM (14), 3C-DFE (19), 3C-P (28), and 3C-AL (31) bound in the micromolar concentration range to the mouse TAAR1 (Ki = 1,000–3,300 nM); 3C-DFE (19), 3C-FE (20), and 3C-E (21) did not bind at examined concentrations (Ki > 4,200 nM).
At the adrenergic α1A receptor, the phenethylamine derivatives MDFM (11), DFM (12), and TFM (13) were the only derivatives showing any affinities at tested concentrations (Ki = 3,200–8,000 nM). At the α2A receptor, all phenethylamine derivatives bound in the micromolar range (Ki = 1,200–3,700 nM) except for TFM (13), which bound with moderate affinity (Ki = 450 nM). The only amphetamine derivatives that bound to the α2A receptors were TMA (6), 3C-DFM (14), and 3C-P (28) (Ki = 2,600–4,600 nM).
None of the compounds examined bound to the dopaminergic D2 receptor (Ki > 6,300 nM) or any of the monoamine transporters (Ki > 7,500 nM). Furthermore, none of the investigated compounds significantly inhibited any of the monoamine uptake transporters (IC50 > 10,000 nM).
Taken from the extensive SAR of 2,4,5-trisubstituted derivatives, small lipophilic substituents at the 4-position of 2,5-dimethoxy substituted phenethylamines and amphetamines lead to derivatives that have agonistic properties, while derivatives with large lipophilic substituents at the 4-position lead to antagonistic effects at the 5-HT2A/2C receptors (Dowd et al., 2000). Furthermore, hydrophilic substituents at the 4-position attenuate 5-HT2A receptor affinity and in vivo potency (Nelson et al., 1999; Braden 2007). In line with functional properties of ligands with lipophilic 4-substituents, a similar trend could be observed when reviewing the active doses of these compounds as psychedelics in man; when surpassing a certain steric bulkiness, compounds tend to lose their psychedelic properties (Shulgin and Shulgin 1991; Trachsel et al., 2013). Thus, smaller lipophilic 4-substituents not only yield agonists/partial agonists but also lead to the most potent psychedelics.
Thus far, the few in vitro investigated 3,4,5-trisubstituted phenethylamines and amphetamines have been shown to have the lowest 5-HT2A receptor affinities among psychedelic phenethylamines (Monte et al., 1997; Parker et al., 2008; Trachsel et al., 2013) when compared to the 2,4,5-trisubsituted and 2,4,6-trisubsituted phenethylamines. However, initial SAR investigations of a series of 4-alkoxysubstituted 3,5-dimethoxyphenethylamines and their α-methyl congeners revealed a similar trend in that more lipophilic 4-substituents lead to higher affinities (Parker et al., 2008; Trachsel et al., 2013). Similarly, 3,5-dimethoxy derivatives with more lipophilic 4-substituents also lead to more potent compounds in man, when not surpassing a certain steric bulkiness (Shulgin and Shulgin 1991; Trachsel et al., 2013).
In the present investigation, only a few phenethylamine derivatives (MDFM; 11, DFM; 12, TFM; 13, CP; 29, MAL; 32, and BZ; 33) slightly augmented the binding affinity at the 5-HT1A receptor when compared to mescaline (5). None of the α-Me-containing compounds showed affinities at this receptor subtype (Ki > 5,600 nM), indicating that the 5-HT1A receptor does not tolerate this steric expansion in 3,4,5-trisubstituted amphetamines. This is in line with other α-Me-containing compounds like 2,4,5-trisubstituted amphetamines (Kolaczynska et al., 2019).
All tested phenethylamine derivatives, except for DFIP (23), displayed an increased affinity at the 5-HT2A receptor compared to mescaline (5). However, these 5-HT2A receptor interactions were less potent when compared to other psychedelic phenethylamines (for instance NBOMe or 2,4,5-trisubstituted derivatives), which bind in the low nanomolar range (Rickli et al., 2015; Luethi et al., 2018b; Kolaczynska et al., 2019; Luethi et al., 2019). This is in line with what has been observed so far for 3,4,5-trisubstituted phenethylamines (Monte et al., 1997; Trachsel et al., 2013). Nowadays, it is well established that phenethylamine psychedelics induce their psychoactive effects mainly by agonistic action at the 5-HT2A receptor (Glennon et al., 1992; Monte et al., 1996; Chambers et al., 2001; Chambers et al., 2002). However, different downstream signaling cascades, biased agonism, and other pharmacological targets may contribute to the subjective effects. Mescaline (5) binds and activates the 5-HT2A receptor as partial agonist with low potency in vitro (Monte et al., 1997; Rickli et al., 2016). Nevertheless, in vivo, it induces intense and long lasting psychedelic effects if applied at high doses (Shulgin and Shulgin 1991). This suggests that low affinity binding to the receptor does not exclude marked psychoactivity in vivo when the corresponding compound is ingested at an adequate dose. In fact, it has been shown that binding affinity serves as a marker of the clinical doses needed to induce such effects (Luethi and Liechti 2018). However, for 3,4,5-substituted derivatives additional pharmacological interactions or targets may significantly contribute to the overall psychedelic effects observed in humans. Furthermore, it is important to note that 5-HT2A agonists have a higher apparent affinity for receptors labeled with an agonist as displacement ligand compared to an antagonist displacement ligand (Sleight et al., 1996). Therefore, the apparent affinity of 3,4,5-substituted phenethylamines and amphetamines for the 5-HT2A receptor depends on the intrinsic efficacy of the radioligand used. This complicates the correlation of Ki values, which were assessed using an antagonistic labelling setup, with psychoactive doses.
The most promising modifications resulting in increased affinity at the 5-HT2A receptor were 4-trifluoromethoxy (TFM, 13), 4-methallyloxy (MAL, 32), and 4-benzyloxy (BZ, 33) substituents, resulting in 17- to 63-fold higher affinities. The aforementioned derivatives except for 33 are known to be active in humans and show up to 9-fold higher potency when compared to 5 (Shulgin and Shulgin 1991; Trachsel 2002; Trachsel 2003; Trachsel et al., 2013). Since the amphetamine homolog 3C-BZ induces psychedelic effects similar to LSD (3) or TMA (6) (Shulgin and Shulgin 1991), BZ (33) may induce psychedelic effects as well, based on its similar structure and high binding affinity at the 5-HT2A receptor.
Similar to the investigated phenethylamines, all examined structural modifications on the amphetamine derivatives increased affinity at the 5-HT2A receptor when compared to TMA (6). Most derivatives bound in the micromolar range and showed at least a 3-fold increase in 5-HT2A receptor affinity compared with 6. 3C-P (4-propyloxy substituent; 28) and 3C-AL (4-allyloxy substituent; 31) were the most potent amphetamine derivatives, showing at least a 10-fold increase in 5-HT2A receptor affinity, equivalent to the binding observed for highly potent phenethylamine derivatives such as TFE (18) (Trachsel et al., 2013). The phenethylamine analogs of 28 and 31, namely proscaline (24) and AL (30), respectively, are among the most potent phenethylamines in the 3,4,5-series (Shulgin and Shulgin 1991). Previous research and the present study suggest that α-methyl containing congers bind with slightly higher affinity to the 5-HT2A receptor and show slightly greater activation potency (Shulgin and Shulgin 1991; Trachsel et al., 2013). This would suggest 28 and 3C-AL (31) to be relatively potent psychedelics. In fact, it has been reported that 31 is active in humans with doses lying in the range of 15–30 mg (Trachsel et al., 2013).
Similar to the 5-HT2A receptor binding, all phenethylamine derivatives, except for DFIP (23), had substituents that improved the affinity at the 5-HT2C receptor (Ki = 290–5,700 nM) when compared to 5 (Ki = 9,900 nM). The increase in 5-HT2C receptor affinity compared to 5 was 2- to 8-fold for most derivatives. Exceptions were TFM (13), MAL (32), and BZ (33), which displayed submicromolar affinity at the 5-HT2C receptor (Ki = 290–520 nM), with 19- to 34-fold higher affinity than 5. Similarly, the amphetamine derivatives showed increased binding to the 5-HT2C receptor (Ki = 1,700–8,400 nM) compared to TMA (6) (Ki > 10,000 nM).
Most of the 3,4,5-substituted phenethylamine and amphetamine derivatives had moderate to high preference for the 5-HT2A over the 5-HT1A receptor (up to 29-fold 5-HT2A vs 5-HT1A binding ratio), similar to psychedelic 2C derivatives investigated earlier (Rickli et al., 2015; Luethi et al., 2018b; Kolaczynska et al., 2019; Luethi et al., 2019). A minority of the substances were either slightly more selective for the 5-HT1A receptor or non-selective.
Overall, the tested derivatives showed similar affinities at the 5-HT2A and 5-HT2C receptors, with some compounds being slightly more selective for one or the other receptor subtype. This is not uncommon and has been observed for most of the many investigated ligands with a substituted phenethylamine or amphetamine pharmacophore in past (Glennon et al., 1992; Monte et al., 1996; Chambers et al., 2001; Chambers et al., 2002). Notably though, based on extensive SAR investigations, a few agonists with a remarkable 5-HT2A vs 5-HT2C receptor selectivity have been designed. However, these compounds were not simple phenethylamines but a conformationally restricted phenethylamine derivative (2A vs 2C selectivity of 124) (Juncosa et al., 2013) and a N-(2-hydroxybenzyl) substituted phenethylamine (2A vs 2C selectivity in the range of 52–81, depending on the assay type) (Jensen et al., 2017). Both 5-HT2A and 5-HT2C receptor affinities have been shown to correlate with clinical potency of psychedelics (Luethi and Liechti 2018). However, assessed affinity values and observed trends might potentially differ if instead of using antagonists, agonists would be used as displacement ligands (Sleight et al., 1996; Colom et al., 2019). Moreover, 5-HT2A/2C interactions are not the only factors that influence potency in humans and pharmacokinetics may potentially have a significant impact on in vivo effects. Namely, interactions with other monoamine receptors, lipophilicity, receptor activation, functional selectivity, and metabolism via cytochrome P450 enzymes or amine oxidases could also play a role.
In general, we observed the following SAR in regards to affinity at the investigated 5-HT receptor subtypes: an extension of the carbon chain or fluorination of the 4-alkyloxy moiety in the 3,4,5-substituted series moderately increased the binding affinity at the 5-HT1A receptor for some phenethylamine derivatives. This effect was previously observed with 4-alkoxy substituted 2,5-dimethoxyphenethylamines and 2,5-dimethoxyamphetamines (Kolaczynska et al., 2019), where similar structural modifications had little effect on the 5-HT1A receptor affinity. In contrast and in line with previous studies, extension of the carbon chain at the 4-alkyloxy moiety enhanced binding affinity for the derivatives tested within the scope of this study (Dowd et al., 2000; Luethi et al., 2018b; Kolaczynska et al., 2019). The number of fluorine atoms at the 4-alkyloxy moiety proportionally increased the binding affinity at the 5-HT2A and 5-HT2C receptor (e.g., affinities at the 5-HT2A receptor; mescaline Ki = 9,400 nM > DFM Ki = 3,500 nM > TFM Ki = 280 nM). The presence of an α-Me group had only little and mixed effects on the compounds with the same substituents (mescaline [5] vs TMA [6], FP [25] vs 3C-FP [27], FE [16] vs 3C-FE [20], or DFE [17] vs 3C-DFE [19]). A previous investigation of some of the herein investigated derivatives revealed that introduction of an α-Me group causes slight increases in binding affinity at the 5-HT2A but not 5-HT2C receptors (Trachsel et al., 2013). Affinities assessed in the present study differ slightly from previously reported data, likely explained by differences in used assays and cell lines (Trachsel et al., 2013).
The derivatives with high 5-HT2A receptor affinities (Ki < 1,000 nM), such as TFM (13), MAL (32), and BZ (33), also displayed high activation potency (EC50 in the range of 27–280 nM). Structures 13, and 33 were found to be partial agonists (efficacy < 85%) and 32 had an activation efficacy of 85%, suggesting full agonist properties. In accordance to these in vitro findings, potent psychedelic effects have been described for TFM (13) and MAL (32) (Shulgin and Shulgin 1991; Trachsel et al., 2013), suggesting 33 to be potentially psychedelic in humans. The remaining substances were less potent partial to full 5-HT2A agonists. However, for various substances a discrepancy between binding and activation was observed (i.e., activation potency was distinctively higher than affinity). It has previously been described that unlike receptor binding values, activation potency assessed with a Ca2+ mobilization assay does not necessarily correlate with the potency of the drug (Luethi and Liechti 2018). Functional assays based on other signaling events, for instance IP formation or β-arrestin recruitment, might better predict the clinical potency of scalines and 3C-scalines. In addition to 5-HT2A/2C receptor activity, the head twitch response is an established method to predict the activity and potency of psychedelics (Halberstadt et al., 2011; Halberstadt and Geyer 2014; Halberstadt et al., 2019; Halberstadt et al., 2020). Halberstadt et al. (2019) recently showed that 3C-E (21) and 3C-P (28) induced a head twitch response with almost identical potency. Thus, 28 may induce psychedelic effects in humans at similar doses as 21 (Halberstadt et al., 2019).
Among all tested substances, the only potent partial 5-HT2B agonists were the phenethylamine derivatives MDFM (11), TFM (13), and TFE (18) (EC50 of 88–210 nM), and the amphetamine derivatives 3C-DFM (14), 3C-DFE (19), 3C-FE (20), and 3C-E (21) (95–800 nM). However, these substances were low efficacy partial agonists (EC50 = 18–45%). Endocardial fibrosis has been associated with 5-HT2B activation and is therefore a potential adverse effect to consider for chronic use of substances interacting with this receptor (Rothman et al., 2000; Droogmans et al., 2007; Roth 2007; Doly et al., 2008; Elangbam et al., 2008; Bhattacharyya et al., 2009; Huang et al., 2009; Elangbam 2010; Dawson and Moffatt 2012). As psychedelics are typically not used chronically, endocardial fibrosis is an unlikely adverse effect for users of such substances despite a potential interaction with the 5-HT2B receptor subtype (Luethi et al., 2021).
None of the investigated phenethylamine and amphetamine derivatives interacted with the human TAAR1, the D2 receptor, or monoamine uptake transporters. It is unclear, however, whether co-expression of different receptors would alter a substance’s response at these targets. Still, some derivatives bound to the rat TAAR1 with moderate to high affinity and some substances additionally showed low affinity at the mouse TAAR1. These results confirm the previously observed TAAR1 affinity rank order (rat > mouse > human TAAR1) (Wainscott et al., 2007; Lewin et al., 2008; Rickli et al., 2015; Simmler et al., 2016; Luethi et al., 2018b; Kolaczynska et al., 2019; Luethi et al., 2019). TAAR1 has been shown to negatively modulate monoaminergic neurotransmission (Lindemann et al., 2008; Revel et al., 2011) but the lack of human TAAR1 activation calls into question the relevance of TAAR1 in the mechanism of action of scalines and 3C-scalines. All phenethylamines moderately to weakly interacted with the α2A receptor (Ki = 450–3,700 nM) but only MDFM (11) and TFM (13) bound to the α1A receptor (Ki = 3,200–4,300 nM). Among the amphetamines, only TMA (6), 3C-DFM (14), and 3C-P (28) bound to the α2A receptor (Ki = 2,600–4,600 nM) whereas no binding to the α1A receptor was observed. This is in line with a previously reported higher α2A vs α1A receptor selectivity observed for psychedelic 2,4,5-substituted phenethylamines (2C derivatives) (Rickli et al., 2015; Luethi et al., 2018b; Kolaczynska et al., 2019; Luethi et al., 2019). As observed for the 5-HT2A and 5-HT2C receptors, binding affinity at non-serotonergic receptors increased proportionally to the number of fluorine atoms (e.g., affinities at rTAAR1: mescaline Ki = 3,000 nM > DFM Ki = 880 nM > TFM Ki = 170 nM; DFE Ki = 2,100 nM > TFE Ki = 1,200 nM; FP Ki = 1,700 nM > TFP Ki = 910 nM).
In the present investigation, we pharmacologically examined a series of 4-alkoxy-substituted 3,5-dimethoxyphenethylamines (scalines) and 4-alkoxy-substituted 3,5-dimethoxy-amphetamines (3C-scalines) in vitro. Psychedelic activity in humans has been reported for several of the tested compounds but detailed information on their monoaminergic interactions was hitherto lacking. Overall, the tested compounds interacted from moderate to high potency with the 5-HT2A receptors and to a slightly lesser extent, with the 5-HT2C receptors. Additionally, various compounds bound to adrenergic α1A and α2A receptors, which may therefore modulate the pharmacodynamics together with serotonergic receptor activation. Compared to mescaline (5), various structural modifications of the 4-alkoxy substituent, including introduction of fluorine substituents, increased the 5-HT2A and 5-HT2C receptor affinities. Mescaline (5) has recently regained interest as therapeutic agent in psychiatry. The results of the present study suggest therapeutic potential for several novel mescaline derivatives as well.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
KEK, DT, DL, and MEL designed the research. KEK and MCH performed the research. KEK and MEL analyzed the data. KEK, DT, DL, and MEL wrote the manuscript.
This work was supported by the Federal Office of Public Health (Grant No. 16.921318). DL was supported by a postdoctoral fellowship from the Swiss National Science Foundation (Grant No. P400PM_191032).
DT is an employee of ReseaChem GmbH and MH is an employee of F. Hoffmann-La Roche. ML is a consultant for Mind Medicine, Inc. Knowhow associated with these substances investigated in this work is owned by Mind Medicine, Inc. Mind Medicine, Inc. had no role in financing, planning, or conducting the present research or the present publication.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2021.794254/full#supplementary-material
Aldous, F. A., Barrass, B. C., Brewster, K., Buxton, D. A., Green, D. M., Pinder, R. M., et al. (1974). Structure-activity Relationships in Psychotomimetic Phenylalkylamines. J. Med. Chem. 17 (10), 1100–1111. doi:10.1021/jm00256a016
Allen, J. A., Yadav, P. N., and Roth, B. L. (2008). Insights into the Regulation of 5-HT2A Serotonin Receptors by Scaffolding Proteins and Kinases. Neuropharmacology 55 (6), 961–968. doi:10.1016/j.neuropharm.2008.06.048
Barfknecht, C. F., and Nichols, D. E. (1975). Correlation of Psychotomimetic Activity of Phenethylamines and Amphetamines with 1-Octanol-Water Partition Coefficients. J. Med. Chem. 18 (2), 208–210. doi:10.1021/jm00236a023
Berger, M., Gray, J. A., and Roth, B. L. (2009). The Expanded Biology of Serotonin. Annu. Rev. Med. 60, 355–366. doi:10.1146/annurev.med.60.042307.110802
Bhattacharyya, S., Schapira, A. H., Mikhailidis, D. P., and Davar, J. (2009). Drug-induced Fibrotic Valvular Heart Disease. Lancet 374 (9689), 577–585. doi:10.1016/S0140-6736(09)60252-X
Boess, F. G., and Martin, I. L. (1994). Molecular Biology of 5-HT Receptors. Neuropharmacology 33 (3-4), 275–317. doi:10.1016/0028-3908(94)90059-0
Braden, M. R. (2007). Towards a Biophysical Understanding of Hallucinogen Action. PhD. Indiana: Purdue University.
Chambers, J. J., Kurrasch-Orbaugh, D. M., and Nichols, D. E. (2002). Translocation of the 5-alkoxy Substituent of 2,5-dialkoxyarylalkylamines to the 6-position: Effects on 5-HT2A/2C Receptor Affinity. Bioorg. Med. Chem. Lett. 12 (15), 1997–1999. doi:10.1016/s0960-894x(02)00306-2
Chambers, J. J., Kurrasch-Orbaugh, D. M., Parker, M. A., and Nichols, D. E. (2001). Enantiospecific Synthesis and Pharmacological Evaluation of a Series of Super-potent, Conformationally Restricted 5-HT2A/2C Receptor Agonists. J. Med. Chem. 44 (6), 1003–1010. doi:10.1021/jm000491y
Clark, L. C., Benington, F., and Morin, R. D. (1965). The Effects of Ring-Methoyoxyl Groups on Biological Deamination of Phenethylamines. J. Med. Chem. 8, 353–355. doi:10.1021/jm00327a016
Colom, M., Vidal, B., and Zimmer, L. (2019). Is There a Role for GPCR Agonist Radiotracers in PET Neuroimaging? Front. Mol. Neurosci. 12, 255. doi:10.3389/fnmol.2019.00255
Dawson, P., and Moffatt, J. D. (2012). Cardiovascular Toxicity of Novel Psychoactive Drugs: Lessons from the Past. Prog. Neuropsychopharmacol. Biol. Psychiatry 39 (2), 244–252. doi:10.1016/j.pnpbp.2012.05.003
Doly, S., Valjent, E., Setola, V., Callebert, J., Hervé, D., Launay, J. M., et al. (2008). Serotonin 5-HT2B Receptors Are Required for 3,4-Methylenedioxymethamphetamine-Induced Hyperlocomotion and 5-HT Release In Vivo and In Vitro. J. Neurosci. 28 (11), 2933–2940. doi:10.1523/JNEUROSCI.5723-07.2008
Dowd, C. S., Herrick-Davis, K., Egan, C., DuPre, A., Smith, C., Teitler, M., et al. (2000). 1-[4-(3-Phenylalkyl)phenyl]-2-aminopropanes as 5-HT2A Partial Agonists. J. Med. Chem. 43 (16), 3074–3084. doi:10.1021/jm9906062
Droogmans, S., Cosyns, B., D'Haenen, H., Creeten, E., Weytjens, C., Franken, P. R., et al. (2007). Possible Association between 3,4-methylenedioxymethamphetamine Abuse and Valvular Heart Disease. Am. J. Cardiol. 100 (9), 1442–1445. doi:10.1016/j.amjcard.2007.06.045
Elangbam, C. S. (2010). Drug-induced Valvulopathy: an Update. Toxicol. Pathol. 38 (6), 837–848. doi:10.1177/0192623310378027
Elangbam, C. S., Job, L. E., Zadrozny, L. M., Barton, J. C., Yoon, L. W., Gates, L. D., et al. (2008). 5-hydroxytryptamine (5HT)-Induced Valvulopathy: Compositional Valvular Alterations Are Associated with 5HT2B Receptor and 5HT Transporter Transcript Changes in Sprague-Dawley Rats. Exp. Toxicol. Pathol. 60 (4-5), 253–262. doi:10.1016/j.etp.2008.03.005
Eshleman, A. J., Wolfrum, K. M., Reed, J. F., Kim, S. O., Johnson, R. A., and Janowsky, A. (2018). Neurochemical Pharmacology of Psychoactive Substituted N-Benzylphenethylamines: High Potency Agonists at 5-HT2A Receptors. Biochem. Pharmacol. 158, 27–34. doi:10.1016/j.bcp.2018.09.024
Glennon, R. A. (1991). Discriminative Stimulus Properties of Hallucinogens and Related Designer Drugs. NIDA Res. Monogr. 116, 25–44. doi:10.1037/e496182006-003
Glennon, R. A., Raghupathi, R., Bartyzel, P., Teitler, M., and Leonhardt, S. (1992). Binding of Phenylalkylamine Derivatives at 5-HT1C and 5-HT2 Serotonin Receptors: Evidence for a Lack of Selectivity. J. Med. Chem. 35 (4), 734–740. doi:10.1021/jm00082a014
Glennon, R. A., Titeler, M., and Lyon, R. A. (1988). A Preliminary Investigation of the Psychoactive Agent 4-Bromo-2,5-Dimethoxyphenethylamine: a Potential Drug of Abuse. Pharmacol. Biochem. Behav. 30 (3), 597–601. doi:10.1016/0091-3057(88)90071-8
Glennon, R. A., Young, R., Benington, F., and Morin, R. D. (1982). Behavioral and Serotonin Receptor Properties of 4-substituted Derivatives of the Hallucinogen 1-(2,5-Dimethoxyphenyl)-2-Aminopropane. J. Med. Chem. 25 (10), 1163–1168. doi:10.1021/jm00352a013
Glennon, R. A., Young, R., and Jacyno, J. M. (1983). Indolealkylamine and Phenalkylamine Hallucinogens. Effect of Alpha-Methyl and N-Methyl Substituents on Behavioral Activity. Biochem. Pharmacol. 32 (7), 1267–1273. doi:10.1016/0006-2952(83)90281-2
Halberstadt, A. L., Chatha, M., Chapman, S. J., and Brandt, S. D. (2019). Comparison of the Behavioral Effects of Mescaline Analogs Using the Head Twitch Response in Mice. J. Psychopharmacol. 33 (3), 406–414. doi:10.1177/0269881119826610
Halberstadt, A. L., Chatha, M., Klein, A. K., Wallach, J., and Brandt, S. D. (2020). Correlation between the Potency of Hallucinogens in the Mouse Head-Twitch Response Assay and Their Behavioral and Subjective Effects in Other Species. Neuropharmacology 167, 107933. doi:10.1016/j.neuropharm.2019.107933
Halberstadt, A. L., and Geyer, M. A. (2014). Effects of the Hallucinogen 2,5-Dimethoxy-4-Iodophenethylamine (2C-I) and Superpotent N-Benzyl Derivatives on the Head Twitch Response. Neuropharmacology 77, 200–207. doi:10.1016/j.neuropharm.2013.08.025
Halberstadt, A. L., Koedood, L., Powell, S. B., and Geyer, M. A. (2011). Differential Contributions of Serotonin Receptors to the Behavioral Effects of Indoleamine Hallucinogens in Mice. J. Psychopharmacol. 25 (11), 1548–1561. doi:10.1177/0269881110388326
Heffter, A. (1898). Ueber Pellote. Archiv für experimentelle Pathologie und Pharmakologie 40 (5-6), 385–429. doi:10.1007/bf01825267
Hey, P. (1947). The Synthesis of a New Homologue of Mescaline. Q. J. Pharm. Pharmacol. 20 (2), 129–134.
Huang, X. P., Setola, V., Yadav, P. N., Allen, J. A., Rogan, S. C., Hanson, B. J., et al. (2009). Parallel Functional Activity Profiling Reveals Valvulopathogens Are Potent 5-hydroxytryptamine(2B) Receptor Agonists: Implications for Drug Safety Assessment. Mol. Pharmacol. 76 (4), 710–722. doi:10.1124/mol.109.058057
Jensen, A. A., McCorvy, J. D., Leth-Petersen, S., Bundgaard, C., Liebscher, G., Kenakin, T. P., et al. (2017). Detailed Characterization of the In Vitro Pharmacological and Pharmacokinetic Properties of N-(2-hydroxybenzyl)-2,5-dimethoxy-4-cyanophenylethylamine (25CN-NBOH), a Highly Selective and Brain-Penetrant 5-HT2A Receptor Agonist. J. Pharmacol. Exp. Ther. 361 (3), 441–453. doi:10.1124/jpet.117.239905
Johnson, M. P., Hoffman, A. J., Nichols, D. E., and Mathis, C. A. (1987). Binding to the Serotonin 5-HT2 Receptor by the Enantiomers of 125I-DOI. Neuropharmacology 26 (12), 1803–1806. doi:10.1016/0028-3908(87)90138-9
Johnson, M. P., Mathis, C. A., Shulgin, A. T., Hoffman, A. J., and Nichols, D. E. (1990). [125I]-2-(2,5-dimethoxy-4-iodophenyl)aminoethane ([125I]-2c-I) as a Label for the 5-HT2 Receptor in Rat Frontal Cortex. Pharmacol. Biochem. Behav. 35 (1), 211–217. doi:10.1016/0091-3057(90)90228-a
Juncosa, J. I., Hansen, M., Bonner, L. A., Cueva, J. P., Maglathlin, R., McCorvy, J. D., et al. (2013). Extensive Rigid Analogue Design Maps the Binding Conformation of Potent N-Benzylphenethylamine 5-HT2A Serotonin Receptor Agonist Ligands. ACS Chem. Neurosci. 4 (1), 96–109. doi:10.1021/cn3000668
Kolaczynska, K. E., Luethi, D., Trachsel, D., Hoener, M. C., and Liechti, M. E. (2019). Receptor Interaction Profiles of 4-Alkoxy-Substituted 2,5-dimethoxyphenethylamines and Related Amphetamines. Front. Pharmacol. 10, 1423. doi:10.3389/fphar.2019.01423
Lewin, A. H., Navarro, H. A., and Mascarella, S. W. (2008). Structure-activity Correlations for Beta-Phenethylamines at Human Trace Amine Receptor 1. Bioorg. Med. Chem. 16 (15), 7415–7423. doi:10.1016/j.bmc.2008.06.009
Liechti, M. E. (2017). Modern Clinical Research on LSD. Neuropsychopharmacology 42 (11), 2114–2127. doi:10.1038/npp.2017.86
Lindemann, L., Meyer, C. A., Jeanneau, K., Bradaia, A., Ozmen, L., Bluethmann, H., et al. (2008). Trace Amine-Associated Receptor 1 Modulates Dopaminergic Activity. J. Pharmacol. Exp. Ther. 324 (3), 948–956. doi:10.1124/jpet.107.132647
Luethi, D., Kolaczynska, K. E., Docci, L., Krähenbühl, S., Hoener, M. C., and Liechti, M. E. (2018a). Pharmacological Profile of Mephedrone Analogs and Related New Psychoactive Substances. Neuropharmacology 134 (Pt A), 4–12. doi:10.1016/j.neuropharm.2017.07.026
Luethi, D., and Liechti, M. E. (2020). Designer Drugs: Mechanism of Action and Adverse Effects. Arch. Toxicol. 94 (4), 1085–1133. doi:10.1007/s00204-020-02693-7
Luethi, D., and Liechti, M. E. (2018). Monoamine Transporter and Receptor Interaction Profiles In Vitro Predict Reported Human Doses of Novel Psychoactive Stimulants and Psychedelics. Int. J. Neuropsychopharmacol. 21 (10), 926–931. doi:10.1093/ijnp/pyy047
Luethi, D., Trachsel, D., Hoener, M. C., and Liechti, M. E. (2018b). Monoamine Receptor Interaction Profiles of 4-Thio-Substituted Phenethylamines (2C-T Drugs). Neuropharmacology 134 (Pt A), 141–148. doi:10.1016/j.neuropharm.2017.07.012
Luethi, D., Widmer, R., Trachsel, D., Hoener, M. C., and Liechti, M. E. (2019). Monoamine Receptor Interaction Profiles of 4-Aryl-Substituted 2,5-dimethoxyphenethylamines (2C-BI Derivatives). Eur. J. Pharmacol. 855, 103–111. doi:10.1016/j.ejphar.2019.05.014
Luethi, D., Liechti, M. E., Maroteaux, L., and Monassier, L. (2021). 5-HT2B Receptors: From Molecular Biology to Clinical Applications. Cham: Springer International Publishing, 277–289. doi:10.1007/978-3-030-55920-5_16Drugs of Abuse Affecting 5-HT2B Receptors
Monte, A. P., Marona-Lewicka, D., Parker, M. A., Wainscott, D. B., Nelson, D. L., and Nichols, D. E. (1996). Dihydrobenzofuran Analogues of Hallucinogens. 3. Models of 4-substituted (2,5-dimethoxyphenyl)alkylamine Derivatives with Rigidified Methoxy Groups. J. Med. Chem. 39 (15), 2953–2961. doi:10.1021/jm960199j
Monte, A. P., Waldman, S. R., Marona-Lewicka, D., Wainscott, D. B., Nelson, D. L., Sanders-Bush, E., et al. (1997). Dihydrobenzofuran Analogues of Hallucinogens. 4. Mescaline Derivatives. J. Med. Chem. 40 (19), 2997–3008. doi:10.1021/jm970219x
Nelson, D. L., Lucaites, V. L., Wainscott, D. B., and Glennon, R. A. (1999). Comparisons of Hallucinogenic Phenylisopropylamine Binding Affinities at Cloned Human 5-HT2A, 5-HT2B and 5-HT2C Receptors. Naunyn-schmiedeberg's Arch. Pharmacol. 359 (1), 1–6. doi:10.1007/pl00005315
Nichols, D. E., Frescas, S., Marona-Lewicka, D., Huang, X., Roth, B. L., Gudelsky, G. A., et al. (1994). 1-(2,5-Dimethoxy-4-(trifluoromethyl)phenyl)-2-aminopropane: a Potent Serotonin 5-HT2A/2C Agonist. J. Med. Chem. 37 (25), 4346–4351. doi:10.1021/jm00051a011
Parker, M. A., Kurrasch, D. M., and Nichols, D. E. (2008). The Role of Lipophilicity in Determining Binding Affinity and Functional Activity for 5-HT2A Receptor Ligands. Bioorg. Med. Chem. 16 (8), 4661–4669. doi:10.1016/j.bmc.2008.02.033
Parrish, J. C., Braden, M. R., Gundy, E., and Nichols, D. E. (2005). Differential Phospholipase C Activation by Phenylalkylamine Serotonin 5-HT2A Receptor Agonists. J. Neurochem. 95 (6), 1575–1584. doi:10.1111/j.1471-4159.2005.03477.x
Pithadia, A. B., and Jain, S. M. (2009). 5-Hydroxytryptamine Receptor Subtypes and Their Modulators with Therapeutic Potentials. J. Clin. Med. Res. 1 (2), 72–80. doi:10.4021/jocmr2009.05.1237
Preller, K. H., Herdener, M., Pokorny, T., Planzer, A., Kraehenmann, R., Stämpfli, P., et al. (2017). The Fabric of Meaning and Subjective Effects in LSD-Induced States Depend on Serotonin 2A Receptor Activation. Curr. Biol. 27 (3), 451–457. doi:10.1016/j.cub.2016.12.030
Preller, K. H., Pokorny, T., Hock, A., Kraehenmann, R., Stämpfli, P., Seifritz, E., et al. (2016). Effects of Serotonin 2A/1A Receptor Stimulation on Social Exclusion Processing. Proc. Natl. Acad. Sci. U S A. 113 (18), 5119–5124. doi:10.1073/pnas.1524187113
Rapport, M. M., Green, A. A., and Page, I. H. (1948). Serum Vasoconstrictor, Serotonin; Isolation and Characterization. J. Biol. Chem. 176 (3), 1243–1251. doi:10.1016/s0021-9258(18)57137-4
Ray, T. S. (2010). Psychedelics and the Human Receptorome. PLOS ONE 5 (2), e9019. doi:10.1371/journal.pone.0009019
Revel, F. G., Moreau, J. L., Gainetdinov, R. R., Bradaia, A., Sotnikova, T. D., Mory, R., et al. (2011). TAAR1 Activation Modulates Monoaminergic Neurotransmission, Preventing Hyperdopaminergic and Hypoglutamatergic Activity. Proc. Natl. Acad. Sci. U S A. 108 (20), 8485–8490. doi:10.1073/pnas.1103029108
Rickli, A., Luethi, D., Reinisch, J., Buchy, D., Hoener, M. C., and Liechti, M. E. (2015). Receptor Interaction Profiles of Novel N-2-Methoxybenzyl (NBOMe) Derivatives of 2,5-Dimethoxy-Substituted Phenethylamines (2C Drugs). Neuropharmacology 99, 546–553. doi:10.1016/j.neuropharm.2015.08.034
Rickli, A., Moning, O. D., Hoener, M. C., and Liechti, M. E. (2016). Receptor Interaction Profiles of Novel Psychoactive Tryptamines Compared with Classic Hallucinogens. Eur. Neuropsychopharmacol. 26 (8), 1327–1337. doi:10.1016/j.euroneuro.2016.05.001
Roth, B. L. (2007). Drugs and Valvular Heart Disease. N. Engl. J. Med. 356 (1), 6–9. doi:10.1056/NEJMp068265
Roth, B. L. (2011). Irving Page Lecture: 5-HT2A Serotonin Receptor Biology: Interacting Proteins, Kinases and Paradoxical Regulation. Neuropharmacology 61 (348), 348–354. doi:10.1016/j.neuropharm.2011.01.012
Rothman, R. B., Baumann, M. H., Savage, J. E., Rauser, L., McBride, A., Hufeisen, S. J., et al. (2000). Evidence for Possible Involvement of 5-HT(2B) Receptors in the Cardiac Valvulopathy Associated with Fenfluramine and Other Serotonergic Medications. Circulation 102 (23), 2836–2841. doi:10.1161/01.cir.102.23.2836
Rudin, D., Liechti, M. E., and Luethi, D. (2021). Molecular and Clinical Aspects of Potential Neurotoxicity Induced by New Psychoactive Stimulants and Psychedelics. Exp. Neurol. 343, 113778. doi:10.1016/j.expneurol.2021.113778
Sadzot, B., Baraban, J. M., Glennon, R. A., Lyon, R. A., Leonhardt, S., Jan, C. R., et al. (1989). Hallucinogenic Drug Interactions at Human Brain 5-HT2 Receptors: Implications for Treating LSD-Induced Hallucinogenesis. Psychopharmacology (Berl) 98 (4), 495–499. doi:10.1007/BF00441948
Shulgin, A., and Shulgin, A. (1991). PiHKAL: A Chemical Love Story. Berkeley, California: Transform Press.
Shulgin, A. T., and Shulgin, A. (1997). TiHKAL: The Continuation. Berkeley, California: Transform Press.
Simmler, L. D., Buchy, D., Chaboz, S., Hoener, M. C., and Liechti, M. E. (2016). In Vitro characterization of Psychoactive Substances at Rat, Mouse, and Human Trace Amine-Associated Receptor 1. J. Pharmacol. Exp. Ther. 357 (1), 134–144. doi:10.1124/jpet.115.229765
Simmler, L. D., Buser, T. A., Donzelli, M., Schramm, Y., Dieu, L. H., Huwyler, J., et al. (2013). Pharmacological Characterization of Designer Cathinones In Vitro. Br J Pharmacol. 168 (2), 458–470. doi:10.1111/j.1476-5381.2012.02145
Sleight, A. J., Stam, N. J., Mutel, V., and Vanderheyden, P. M. (1996). Radiolabelling of the Human 5-HT2A Receptor with an Agonist, a Partial Agonist and an Antagonist: Effects on Apparent Agonist Affinities. Biochem. Pharmacol. 51 (1), 71–76. doi:10.1016/0006-2952(95)02122-1
Trachsel, D. (2012). Fluorine in Psychedelic Phenethylamines. Drug Test. Anal. 4 (7-8), 577–590. doi:10.1002/dta.413
Trachsel, D., Nichols, D. E., Kidd, S., Hadorn, M., and Baumberger, F. (2009). 4-aryl-substituted 2,5-dimethoxyphenethylamines: Synthesis and Serotonin 5-HT2A Receptor Affinities. Chem. Biodivers 6 (5), 692–704. doi:10.1002/cbdv.200800235
Trachsel, D., Lehmann, D., and Enzensperger, C. (2013). Phenethylamine: Von der Struktur zur Funktion. Solothurn: Nachtschatten Verlag Ag.
Trachsel, D. (2002). Synthese von neuen (Phenylalkyl)aminen zur Untersuchung von Struktur-Aktivitatsbeziehungen, Mitteilung 1: Mescalin Derivative. Hca 85 (9), 3019–3026. doi:10.1002/1522-2675(200209)85:9<3019:aid-hlca3019>3.0.co;2-4
Trachsel, D. (2003). Synthese von neuen (Phenylalkyl)aminen zur Untersuchung von Struktur-Aktivitätsbeziehungen. Mitteilung 2. Hca 86 (7), 2610–2619. doi:10.1002/hlca.200390210
Vollenweider, F. X., and Preller, K. H. (2020). Psychedelic Drugs: Neurobiology and Potential for Treatment of Psychiatric Disorders. Nat. Rev. Neurosci. 21 (11), 611–624. doi:10.1038/s41583-020-0367-2
Vollenweider, F. X., Vollenweider-Scherpenhuyzen, M. F., Bäbler, A., Vogel, H., and Hell, D. (1998). Psilocybin Induces Schizophrenia-like Psychosis in Humans via a Serotonin-2 Agonist Action. Neuroreport 9 (17), 3897–3902. doi:10.1097/00001756-199812010-00024
Wainscott, D. B., Little, S. P., Yin, T., Tu, Y., Rocco, V. P., He, J. X., et al. (2007). Pharmacologic Characterization of the Cloned Human Trace Amine-Associated Receptor1 (TAAR1) and Evidence for Species Differences with the Rat TAAR1. J. Pharmacol. Exp. Ther. 320 (1), 475–485. doi:10.1124/jpet.106.112532
Keywords: phenethylamine, psychedelic, mescaline, scalines, 3C-scalines, fluorination
Citation: Kolaczynska KE, Luethi D, Trachsel D, Hoener MC and Liechti ME (2022) Receptor Interaction Profiles of 4-Alkoxy-3,5-Dimethoxy-Phenethylamines (Mescaline Derivatives) and Related Amphetamines. Front. Pharmacol. 12:794254. doi: 10.3389/fphar.2021.794254
Received: 13 October 2021; Accepted: 01 December 2021;
Published: 09 February 2022.
Edited by:
Damiana Leo, University of Mons, BelgiumReviewed by:
Stefano Espinoza, Italian Institute of Technology (IIT), ItalyCopyright © 2022 Kolaczynska, Luethi, Trachsel, Hoener and Liechti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthias E. Liechti, bWF0dGhpYXMubGllY2h0aUB1c2IuY2g=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.