 Sara Ahmed
Sara Ahmed Alyssa Manning2
Alyssa Manning2 Tanya Parish
Tanya Parish
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 03 January 2022
Sec. Experimental Pharmacology and Drug Discovery
Volume 12 - 2021 | https://doi.org/10.3389/fphar.2021.790583
This article is part of the Research Topic Antimycobacterial Drug Discovery: Molecular Therapeutics and Target Identification View all 7 articles
Mycobacterium tuberculosis is an important global pathogen for which new drugs are urgently required. The ability of the organism to survive and multiply within macrophages may contribute to the lengthy treatment regimen with multiple drugs that are required to cure the infection. We screened the MyriaScreen II diversity library of 10,000 compounds to identify novel inhibitors of M. tuberculosis growth within macrophage-like cells using high content analysis. Hits were selected which inhibited the intramacrophage growth of M. tuberculosis without significant cytotoxicity to infected macrophages. We selected and prioritized compound series based on their biological and physicochemical properties and the novelty of the chemotypes. We identified five chemical classes of interest and conducted limited catalog structure-activity relationship studies to determine their tractability. We tested activity against intracellular and extracellular M. tuberculosis, as well as cytoxicity against murine RAW264.7 and human HepG2 cells. Benzene amide ethers, thiophene carboxamides and thienopyridines were only active against intracellular bacteria, whereas the phenylthiourea series was also active against extracellular bacteria. One member of a phenyl pyrazole series was moderately active against extracellular bacteria. We identified the benzene amide ethers as an interesting series for further work. These new compound classes serve as starting points for the development of novel drugs to target intracellular M. tuberculosis.
Tuberculosis (TB) is the leading cause of death from a bacterial infection worldwide. Despite the availability of chemotherapy, TB is responsible for >10 million infections annually and >1 million deaths (Gordon and Parish, 2018; Global Tuberculosis Report, 2020). The increasing incidence of drug failure for TB due to widespread drug-resistant infections underscores the urgent need for new drugs and new bacterial targets for M. tuberculosis.
M. tuberculosis is an intracellular pathogen which can survive and replicate within macrophages; this property relies on the ability to block normal bactericidal activities and turns the macrophage into a niche for replication (Vandal et al., 2009; Deretic, 2014; VanderVen et al., 2015; Weiss and Schaible, 2015). The physiology and metabolism of M. tuberculosis within macrophages is very different from extracellular bacteria and so the complement of essential genes differs (Sassetti et al., 2001; Sassetti et al., 2003; Sassetti and Rubin, 2003). From a drug discovery perspective, this means that intracellular bacteria have additional essential processes that can be targeted by chemical inhibition (Manjunatha and Smith, 2015; Parish, 2019; Parish, 2020).
Whole cell screening has been broadly used to identify new drug candidates for tuberculosis. In contrast to target-based drug discovery, in phenotypic screens every essential pathway is a potential drug target. This greatly increases the likelihood of finding inhibitors and overcomes the barrier of obtaining cell-penetrant molecules, which is often a limitation of biochemical screens (Payne et al., 2007). However, conditionally essential genes that are required for M. tuberculosis growth within macrophages or in vivo, may be overlooked in phenotypic screens against axenically-cultured M. tuberculosis. High-throughput screening in conditions that closely resemble the intracellular environment have the potential to uncover novel antimicrobials that target these conditionally essential genes, in addition to discovering compounds targeting the host cells to increase bacterial elimination (Manning et al., 2017; Njikan et al., 2018).
High-content imaging screening is a powerful technology to identify active compounds by monitoring both bacterial and macrophage cell numbers simultaneously and in the same wells, thus leading to more reliable data and a quicker assessment of compound attractiveness. We previously developed a fluorescence-based, live-cell, high-content analysis (HCA) assay to examine drug efficacy against intracellular M. tuberculosis (Manning et al., 2017).
In this study, we screened 10,000 diverse small molecules by high throughput HCA and identified active compounds with low cytotoxicity. We selected a number of series for follow up based on their physicochemical properties and the novelty of the chemotypes and identified five chemical classes of interest. We tested available analogs for each series to complete a limited structure-activity relationship study. These new compound classes serve as starting points for the development of new series for antitubercular drug discovery.
Murine RAW 264.7 macrophages (ATCC TIB-71) were grown at 37°C in a humidified atmosphere containing 5% CO2 in RPMI 1640 medium supplemented with 5% fetal bovine serum, 1 mM sodium pyruvate solution, and 2 mM GlutaGro supplement (Corning). HepG2 cells (ATCC HB-8065) were cultured in Dulbecco-modified Eagle medium (DMEM), 10% fetal bovine serum (FBS), and 1× penicillin streptomycin solution (100 U/ml). M. tuberculosis H37Rv-LP (ATCC 25618) constitutively expressing codon-optimized DsRed from plasmid pBlazeC8 (DREAM8) (Carroll et al., 2018) was cultured at 37°C in Middlebrook 7H9 medium containing 10% v/v OADC (oleic acid, dextrose, catalase) supplement (Becton Dickinson) and 0.05% w/v Tween 80 (7H9-Tw-OADC) plus 100 μg/ml hygromycin B.
Compounds were obtained in 384-well plates as 10 mM DMSO stock solutions and tested as described (Manning et al., 2017). Briefly, assay plates were prepared in clear bottom, black 384-well plates with 30 μl cRPMI and 0.6 μl compound in columns 3–22 (final concentration of 10 μM compound, 1% DMSO). 1% DMSO was used as the negative control, 10 mM isoniazid (INH) and 100 μM staurosporine (STA) were included as positive controls (maximum inhibition) for anti-tubercular activity and cytotoxicity respectively. INH and STA were also included in dose response on each plate. RAW264.7 cells were infected with M. tuberculosis DREAM8 as described (Manning et al., 2017) at an MOI of 1 for 24 h and extracellular bacteria removed by washing. Cells were recovered using Accumax, harvested, washed and resuspended in serum-free RPMI; 30 μl of infected cells were dispensed into each well at 3,300 cells/well. Plates were incubated for 72 h, 10 μl of 5X SYBR Green I was added and plates were imaged with an ImageXpress Micro High Content Screening System (Molecular Devices) using a ×4 objective and FITC and Texas Red channels. MetaXpress was used to analyze images. The integrated intensity of M. tuberculosis or macrophages was calculated for each well. Growth inhibition was calculated for each test well by normalizing to the average integrated intensity of the DMSO control wells. IC50 was calculated as the compound concentration required to reduce bacterial growth by 50%. TC50 was calculated as the compound concentration required to reduce macrophage viability by 50%.
Cytotoxicity against HepG2 cells was measured after 72 h. Cells were seeded in 384-well plates at 1,800 cells per well. Compounds were added as a 10-point three-fold serial dilution after 24 h (final assay concentration of 1% DMSO). CellTiter-Glo® reagent (Promega) was added and relative luminescence units (RLU) measured. Data were normalized to the DMSO controls. Curves were fitted using the Levenberg–Marquardt algorithm; TC50 was calculated as the compound concentration required to reduce cell viability by 50%.
Minimum inhibitory concentrations (IC90) against M. tuberculosis were determined in liquid medium as described (Ollinger et al., 2013). Bacterial growth was measured after 5 days by OD590. IC90 was defined as the concentration of compound required to inhibit growth of M. tuberculosis by 90% and was determined using the Levenberg–Marquardt least-squares plot.
We were interested in identifying new anti-tubercular molecules with activity against intracellular bacteria. We used our previously validated high throughput high content screen to test the MyriaScreen II library of 10,000 diverse molecules which is comprised of ∼60% singletons with diverse structural groups (TimTec/Sigma Aldrich). Molecules were tested at a single concentration of 10 µM (Figure 2A). We monitored cytotoxicity to the macrophages in the same wells to exclude false positives (since the bacteria are unable to replicate if the macrophages are dead). We identified 308 compounds with >70% bacterial growth inhibition; of these we discarded 134 with significant macrophage toxicity, defined as <70% macrophage survival (Figure 1, Figure 2B). This left 174 hit compounds that were non-cytotoxic but restricted the growth of M. tuberculosis in macrophages, a hit rate of 1.7% (Figure 1).
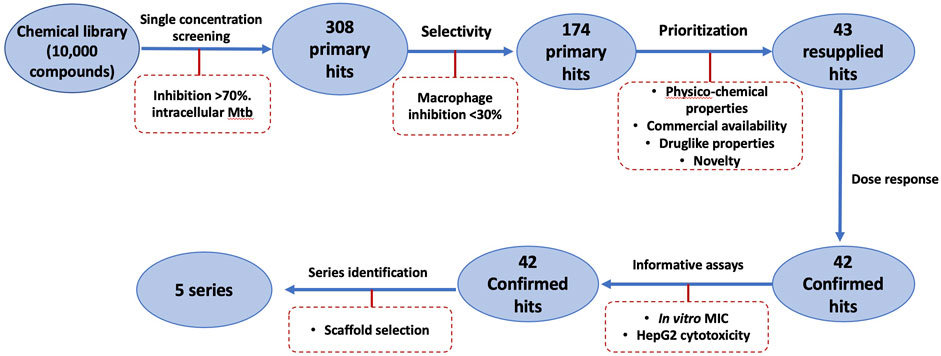
FIGURE 1. Schematic of High Throughput Screening (HTS) of a library of small molecules.
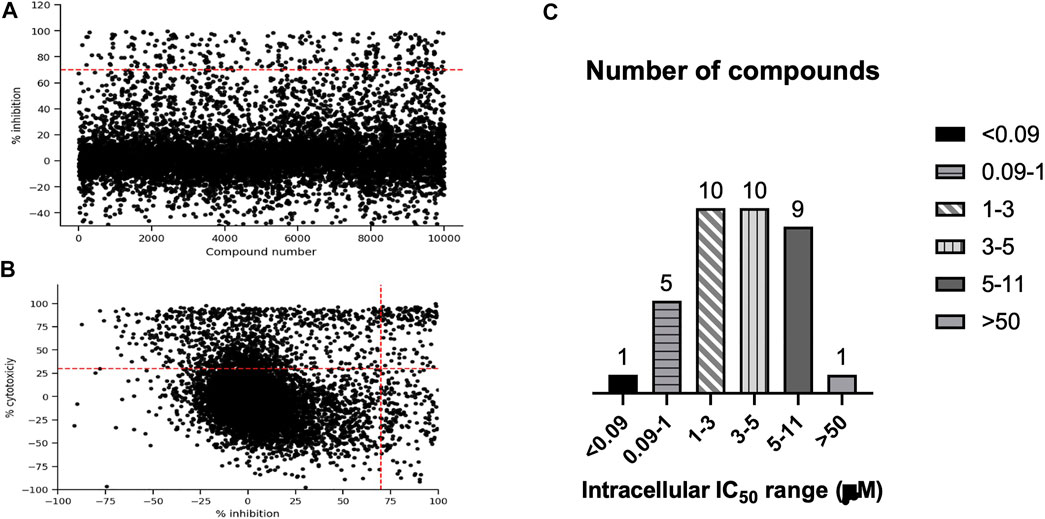
FIGURE 2. High content, high throughput screen against intracellular M. tuberculosis. (A). Compounds were tested for activity against RAW264.7 cells infected with M. tuberculosis at an MOI of 1. The primary screen was conducted using a single concentration of each molecule at 10 1.1 M. Inhibition of bacterial growth and macrophage viability were determined for each compound after 72 h (A). Anti-tubercular activity measured as % inhibition of bacterial growth. (B) Cytotoxicity (% viability) was plotted compared to inhibition of bacterial growth (% Inhibition). Compounds in the top right quadrant were eliminated from the hit list due to cytotoxicity. (C) Selected hit compounds were purchased and tested in dose response to determine IC50 (concentration required to inhibit bacterial growth by 50%). Number of compounds in each group are noted.
We saw some negative inhibition values in the screen, which is not unusual. Compound fluorescence will lead to negative inhibition and is seen more often in the green end of the spectrum than the red. Cytotoxicity can lead to increased signal from the bacterial populaton, presumably because the bacteria are released into the medium and no longer intracellular; we see this effect with staurosporine. Changes in growth of RAW264.7 cells could also be a factor, since uninfected cells will grow, whereas the infected cells do not tend to grow; if compounds are very active, this could lead to increased macrophage growth.
We analyzed the 174 resulting hits and prioritized hit compounds based on their physicochemical properties including molecular weight (MW), calculated partition coefficient (cLogP), novelty, structural diversity, chemical tractability, and commercial availability. We selected 43 compounds for further analysis (Figure 1, Figure 3); we purchased new batches of compounds and determined the activity using a dose response format (Figure 2C). Seven compounds exhibited cytotoxicity against eukaryotic cells that hampered our ability to determine their anti-bacterial activity and were excluded from further study (Table 1). The IC50 of the remaining 36 hits was determined (Figure 3C); of these all but one (35/36) had confirmed activity with IC50 < 50 µM against intracellular M. tuberculosis (Table 1). The majority of the hits (Mori et al., 2015) had an IC50 <5 μM, and 6 compounds were potent with an IC50 < 1 µM (Figure 3C). The majority of the compounds had no cytotoxicity (TC50 > 50 µM); a few compounds were cytotoxic at higher concentrations, but retained good selectivity (Table 1). The IC50 for INH against M. tuberculosis was 0.23 ± 0.08 µM (n = 192) and for STA against the RAW264.7 cells was 0.0034 ± 0.0013 µM (n = 112).
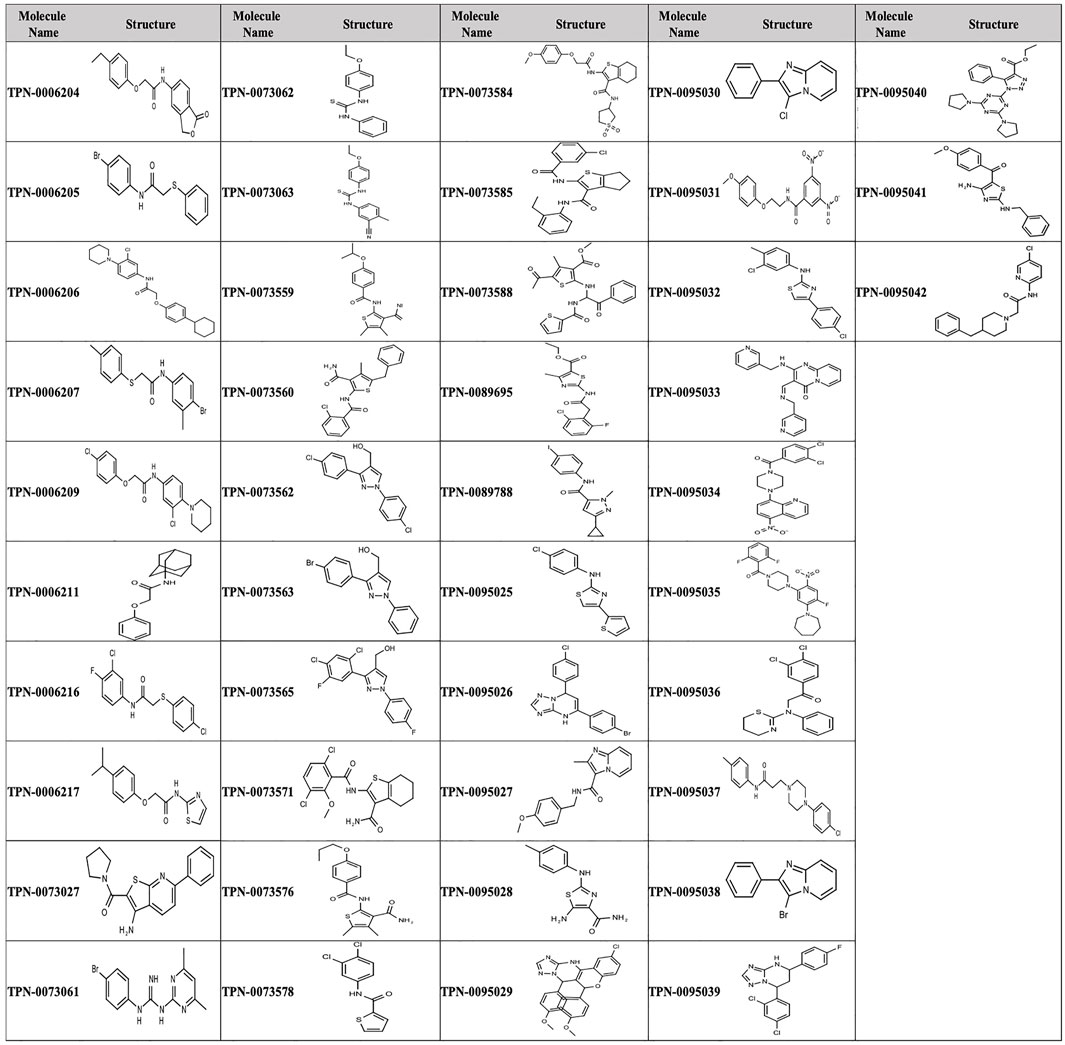
FIGURE 3. Structure of primary hits selected for follow up.
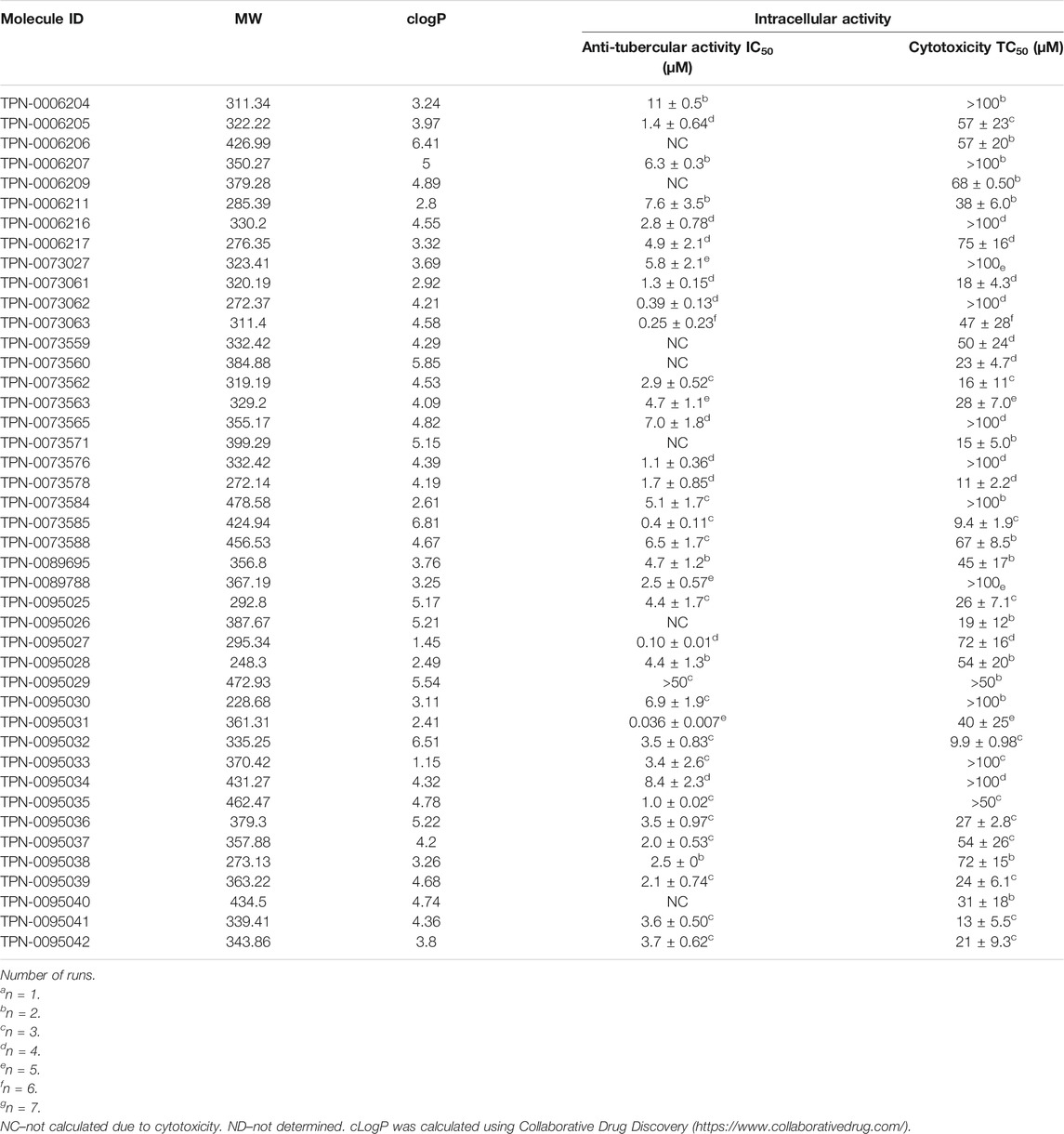
TABLE 1. Activity of primary hits.
We further evaluated cytotoxicity using the HepG2 cell line cultured with either glucose or galactose in the medium (Table 2). HepG2 cells cultured with galactose rely on mitochondrial oxidative phosphorylation rather than glycolysis which increases their susceptibility to mitochondrial toxicants allowing us to identify compounds with mitochondrial toxicity (Kamalian et al., 2015). The majority of the compounds showed no difference in cytotoxicity between the two conditions with the exception of compound TPN-0073571 which had increased cytotoxicity against HepG2 cells in galactose (ratio of TC50glu/TC50gal of ∼4) suggesting that this compound may impair mitochondrial function. Interestingly the compound showed similar cytotoxicity against the murine macrophages (TC50 = 15 ± 5 µM). Overall, cytotoxicity against the two cell lines (murine RAW 264.7 and human HepG2 cells) was comparable (Table 2).
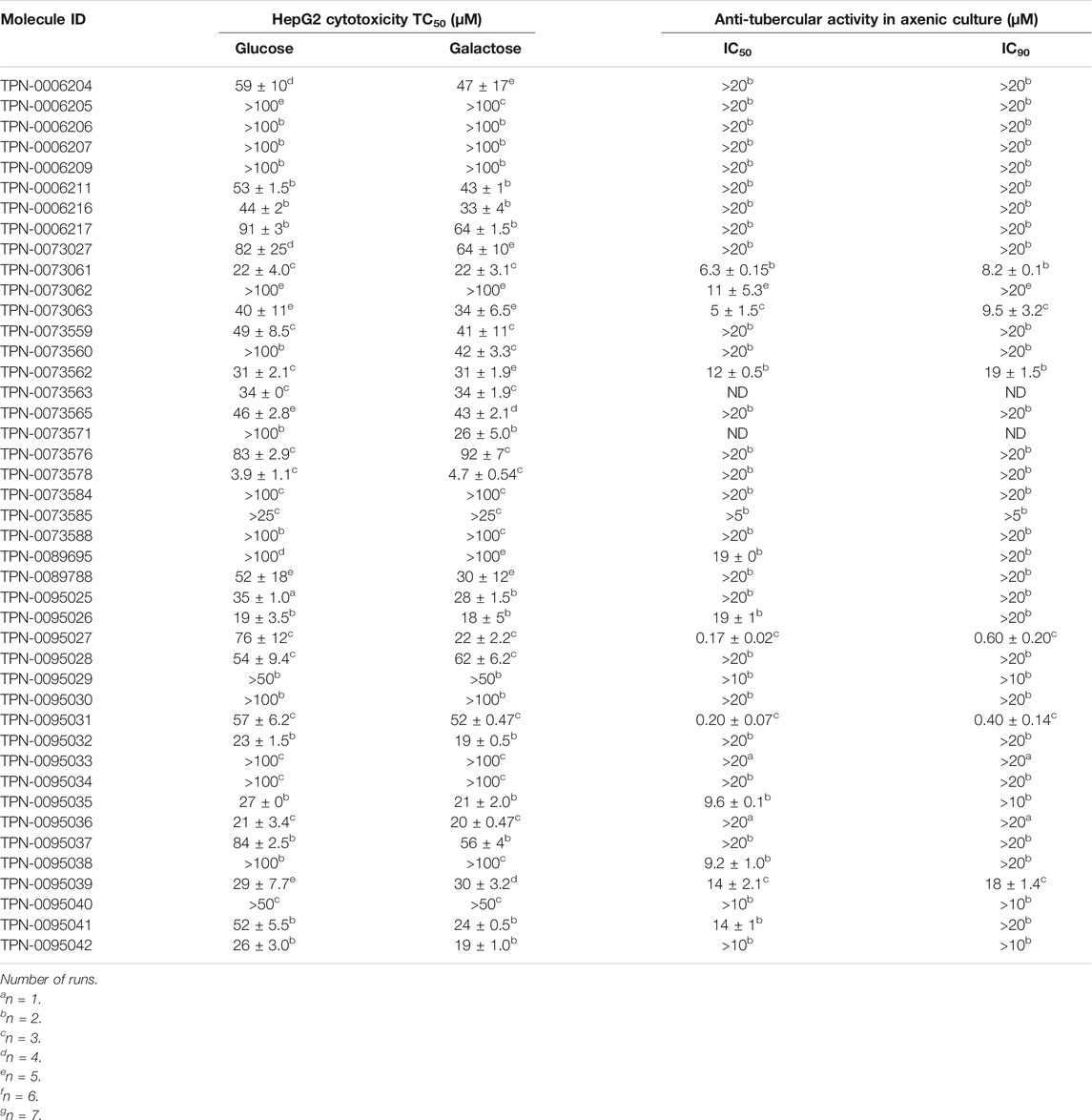
TABLE 2. Cytotoxicity and activity against extracellular M. tuberculosis.
We wanted to determine if our compounds were selective for intracellular bacteria, so we determined the IC90 against M. tuberculosis in axenic culture. Only 12 compounds had IC50 > 20 µM against extracellular M. tuberculosis, with six of these having an IC90 of >20 µM (Table 2). None of the compounds were more active against M. tuberculosis growing in axenic culture, with the extracellular/intracellular IC50 ratios ranging from 4 to 28.
We performed structural clustering on the set of 174 hits in order to identify common and potentially privileged scaffolds. Compounds were grouped into series sharing the same scaffold/chemotype, each featuring 2 to 8 compounds with 8 singletons. Scaffolds used in the substructure searching, along with their frequency in the hit list and the entire library are depicted in Table 3.

TABLE 3. Series selected for follow-up.
The prioritization of compounds was not based solely on biological potency, but focus was placed on the quality/structural novelty of each chemotype, along with their physicochemical properties. We highlighted five chemotypes that were of interest for further development. A representative compound from each series is shown in Figure 4.
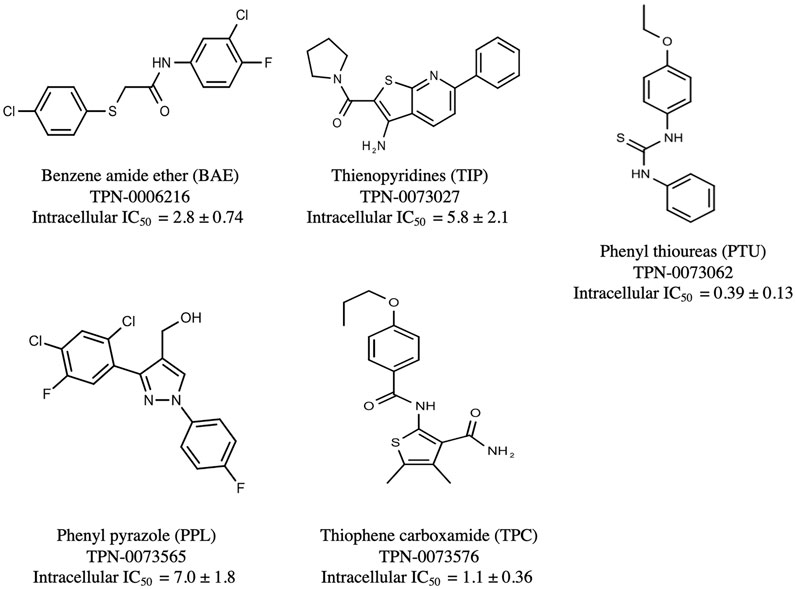
FIGURE 4. Representative molecules from selected series.
We identified 15 compounds with a benzene amide ether scaffold in the primary screen, of which 10 were active (defined as >70% inhibition of bacterial replication) (Table 3). The BAE series has not previously been identified in phenotypic screens against replicating M. tuberculosis, although one compound from this series (6) had activity against non-replicating M. tuberculosis (Christophe et al., 2009). In addition, compound 12 was identified as an inhibitor of M. tuberculosis with an IC90 of 6.9 μg/ml (20). We purchased 13 commercially-available compounds and determined intracellular activity against M. tuberculosis, although we did not see activity up to 20 μM. Eleven compounds had activity against intracellular M. tuberculosis (Table 4). Two compounds (4 and 7) were cytotoxic to macrophages and we could not calculate activity.

TABLE 4. Catalog structure-activity relationship of the BAE series.
NC–not calculated due to cytotoxicity. cLogP was calculated using Collaborative Drug Discovery (https://www.collaborativedrug.com/).
Eight compounds showed good intracellular activity (IC50 <10 µM). There was a ten-fold difference between the most potent compound (3, IC50 = 1.4 μM) and the least potent compound (13, IC50 = 13 μM). Compounds generally showed low cytotoxicity against both RAW264.7 cells and HepG2 cells with either TC50 >100 μM or a selectivity index of >50 (TC50/IC50). Compound 3 and compound 12 were the most potent (IC50 = 1.4 µM and IC50 = 2.4 µM respectively). Although, compound 3 had some cytotoxicity, the selectivity index was 40. Compound 12 was not cytotoxic, with TC50 >100 µM and a selectivity index of >42. None of the compounds were active against replicating M. tuberculosis in axenic medium (IC90 >20 μM) demonstrating clear selectivity for intracellular bacteria.
We were able to determine some elements of the structure activity relationship (SAR) for this series using the limited compound set available. Compounds with a sulfur atom as a linker were generally more potent than those with an oxygen as a linker, with a range of 1.4–6.3 µM compared to 4.9–13 µM. The closest comparison was between 13 with the oxygen linker (IC50 = 4.9 µM) and compounds 3 and 12 (IC50 = 1.4 and 2.4 µM respectively). In addition, the two molecules with significant toxicity to macrophage cells contained the oxygen linker (4 band 7). Four compounds (1, 3, 5 and 12) with one or two substitution of halogen atoms on the amide phenyl ring (R2) exhibited good intracellular activity.
It seems that lipophilicity did not correlate with anti-tubercular activity. Although the most active compounds (3 and 12) had cLogP of 3.97 and 4.55 respectively, compound 13 with cLogP of 3.32 was only two-fold less active and compounds with cLogP <3 still retained good activity (IC50 <10 µM). An electron-withdrawing moiety on the para position of the amide phenyl group (R2) appears to be an essential requirement for good anti-mycobacterial activity. The amide phenyl ring (R2) is not critical to maintain antimycobacterial activity, since compounds 8 and 12 with saturated rings. Both compounds 8 and 10 with adamantane and piperidine groups respectively, had good activity (IC50 = 7.6 µM and IC50 = 6.6 µM). Further exploration of the length of the carbon chain or inclusion of other heteroatoms would expand the SAR, but such molecules are not available commerically and would require chemical synthesis.
We identified 6 compounds with a phenyl thiourea scaffold in the primary screen, of which 4 were active (Table 3). We purchased 5 commercially-available compounds and determined intracellular activity against M. tuberculosis. Four compounds had potent activity against intracellular M. tuberculosis (Table 5). One compound (18) was cytotoxic and we could not calculate activity. One compound (17) had significant cytotoxicity against macrophages (TC50 of 1.8 µM) and was also toxic for HepG2 cells (TC50 of 5.5 µM). The remaining three compounds had excellent potency (0.30–1.3 µM) and good selectivity. Compound 14 with a guanidine functionality as the core had higher cytotoxicity (SI = 14) than the two potent molecules (15 and 16) with the thiourea core (SI = >250 and >150 respectively).
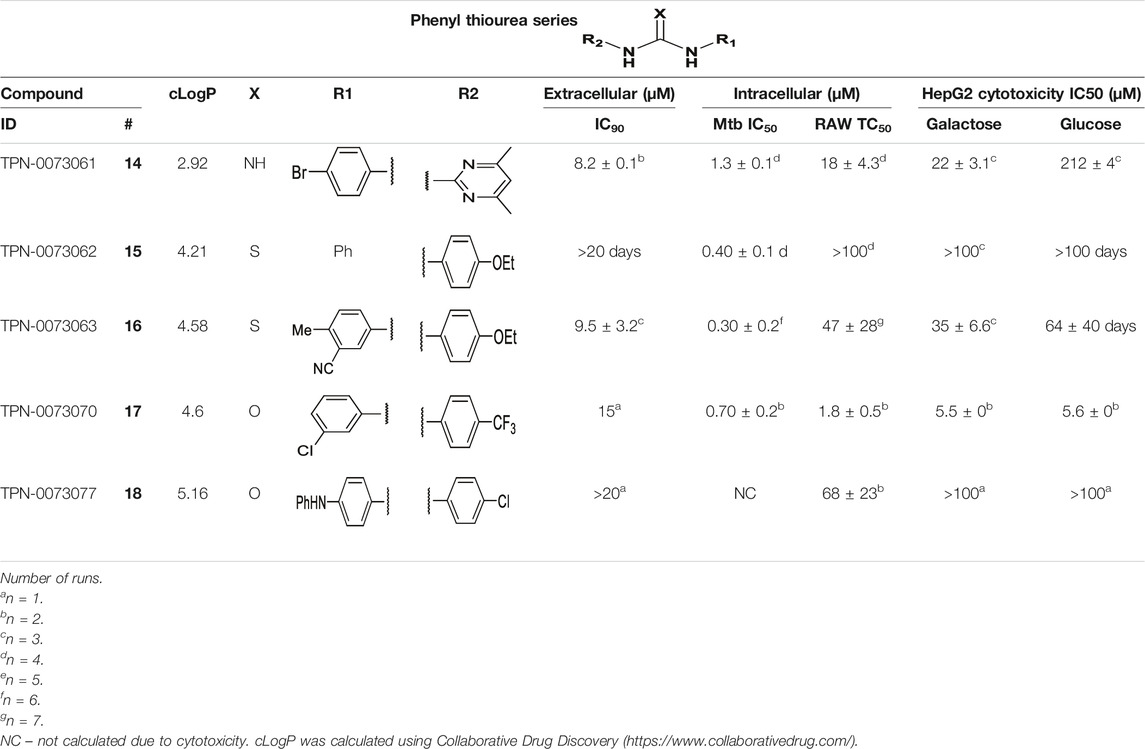
TABLE 5. Catalog structure-activity relationship of the PTU series.
NC–not calculated due to cytotoxicity. cLogP was calculated using Collaborative Drug Discovery (https://www.collaborativedrug.com/).
In contrast to the BAE series, the PTU molecules were also active against extracellular bacteria; 3 of the 5 molecules had IC90 < 20 µM. In general, PTUs were 5–10-fold more active against intracellular bacteria. To the best of our knowledge, this is the first report of these compounds as antitubercular agents although other thiourea derivatives have antitubercular activity (Liav et al., 2008; Plutín et al., 2016; Ghorab et al., 2017; Doğan et al., 2020).
We identified 6 compounds with a phenyl pyrazole scaffold in the primary screen, of which 5 were active (Table 3). We purchased 4 commercially-available compounds and determined intracellular activity against M. tuberculosis. All four compounds (19–22) had activity against intracellular M. tuberculosis (Table 6). Although the compounds demonstrated some cytotoxicity against HepG2 cells and/or macrophages, the selectivity index was good (>5). These sets of compounds have 1,3,4-trisubstituted pyrazoles where C-4 bears a hydroxymethyl group. Compound 19 was the most potent with good intracellular activity (IC50 = 2.9 µM) and >30-fold selectivity against HepG2 cells. Compound 21 also showed weak activity (IC50 = 18.5 µM) against axenically-grown M. tuberculosis.
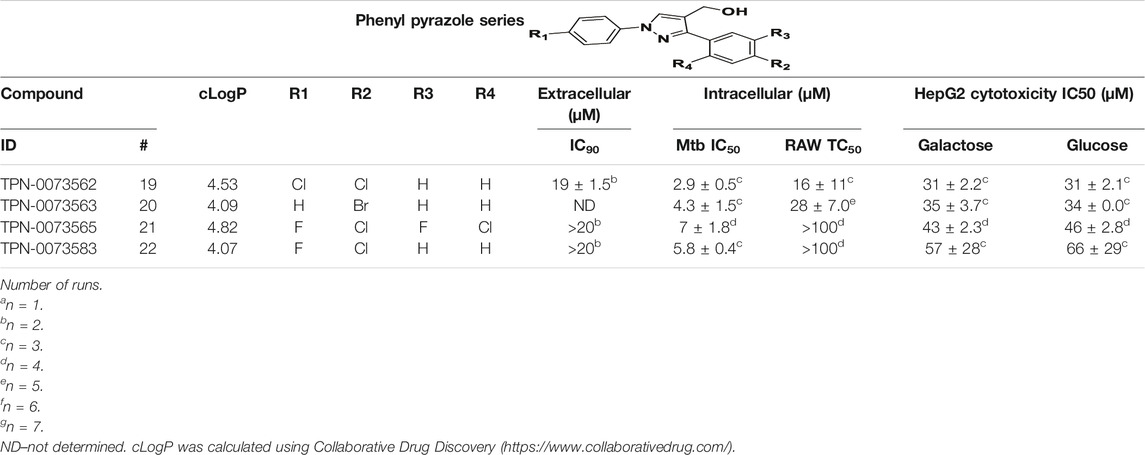
TABLE 6. Catalog structure-activity relationship of the PPL series.
We identified 8 compounds with a thiophene carboxamide scaffold in the primary screen, of which all 8 were active (Table 3). We purchased 8 commercially-available compounds (23–30) and determined intracellular activity against M. tuberculosis. Five compounds had activity against intracellular M. tuberculosis with good acitivity of IC50 < 10 µM (Table 7). Three compounds (23–25) were cyotoxic and we could not calculate activity.
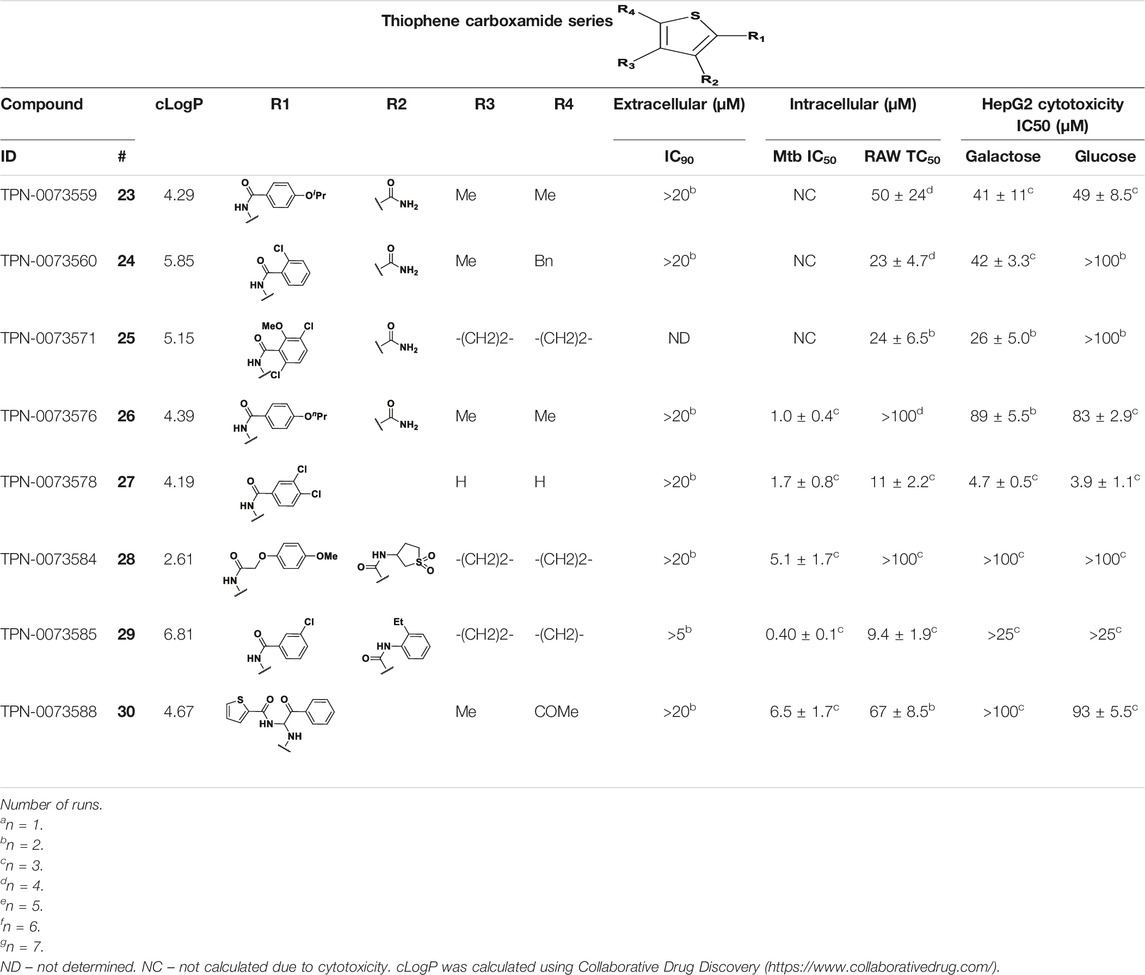
TABLE 7. Catalog structure-activity relationship of the TPC series.
Three compounds (26, 27 and 29) showed good intracellular potency with IC50 < 5 μM. All except 26 and 28 were cytotoxic against macropahges with IC50 ranging from 9 to 67 μM; compound 27 was the most toxic and also showed cytotoxicity against HepG2 cells (IC50 = 4.7 μM). However, despite this, compounds 26, 28, 29, and 30 al l had good selectivity (SI > 10)—values for HepG2 were >80, 20, >60 and 15 respectively. Two compounds (24 and 25) were more cytotoxic against HepG2 cells in galactose media (ratio ∼4) suggesting that they may impair mitochondrial function. However, these were also toxic against macrophages and were not considered active. None of the compounds had any activity against extracellular M. tuberculosis.
We identified 8 compounds with a thiophene carboxamide scaffold in the primary screen, of which 2 were active (Table 3).
We purchased 7 commercially-available compounds (31–37) and determined intracellular activity against M. tuberculosis. Analogs varied in terms of substitution on the pyridine ring but all possessed the amino group at the C-3 position and a carboxamide substitution on the thiophene ring. Four compounds had activity against intracellular M. tuberculosis (Table 8), but only two had activity <10 μM (compounds 34 and 36). Compound 34 bearing a ketone moiety had weak activity. Two compounds (31 and 35) were not active. Compounds were selective, but had no activity against axenically cultured bacteria.
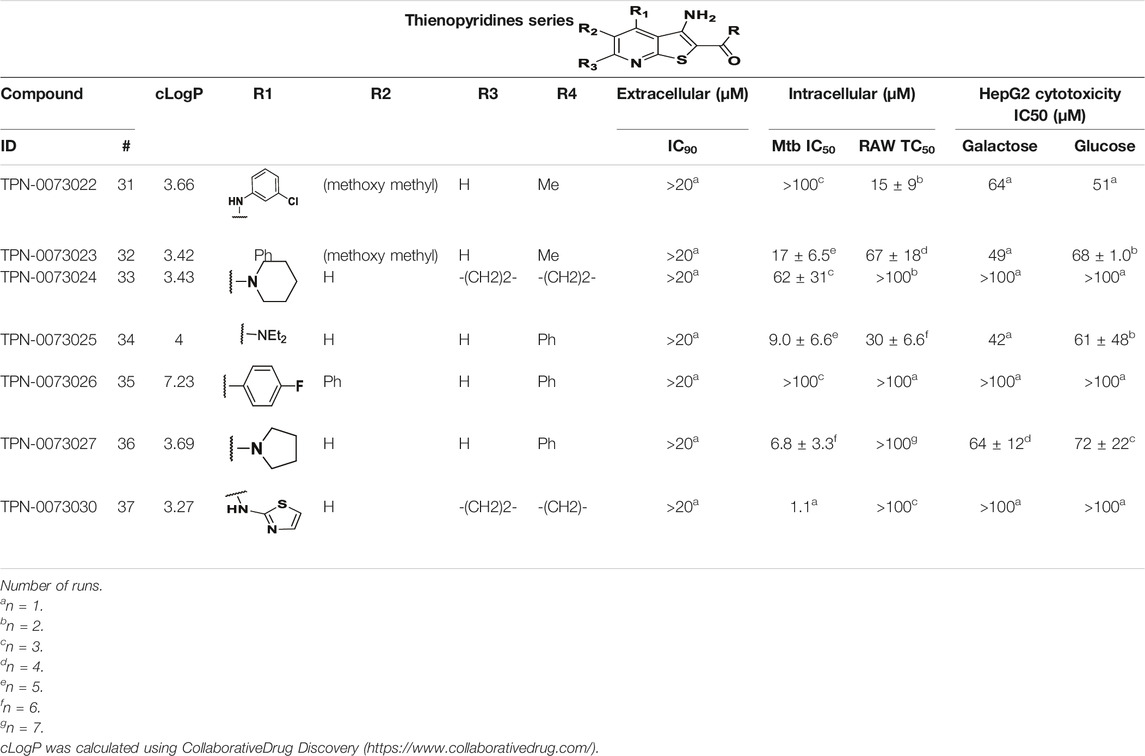
TABLE 8. Catalog structure-ctivity relationship of the TIP series.
Identification of new anti-tubercular agents that are active against emerging MDR and XDR-TB is critical. The identification of drugs that inhibit new targets will contribute to developing effective drug regimens for drug-resistant strains. M. tuberculosis is an intracellular pathogen which can replicate within macrophages where the set of essential genes will differ from extracellular bacteria. Thus screening compounds against intracellular bacteria has the potential to identify compounds that inhibit novel targets.
We screened a diversity library of small molecules in order to find novel chemical matter that targets intracellular M. tuberculosis. We identified a large number of hits with a hit rate of 1.7%. This is comparable to the hit rate we see with other whole cell screens and in other groups. We were able to follow up on several series to conduct limited catalog SAR as a first evaluation of tractability. The majority of the hit series we followed up from in this screen were more active against intracellular bacteria and in some cases lacked any extracellular activity. Since we focused on identifying new scaffolds, this was largely expected, since scaffolds with known anti-tubercular activity from previous phenotypic screens would have been deprioritized in our selection process.
Previous studies had identified compounds that restrict M. tuberculosis growth within the context of macrophages and it was demonstrated that these compounds inhibited the novel target decaprenyl-phosphoribose epimerase (DprE1) which catalyses the synthesis of decaprenyl-phospho-arabinose (Christophe et al., 2009). In addition, compounds which selectively inhibit M. tuberculsois growth within the macrophages with unknown mechanism of action were found. Other studies identified host-targeted small molecules that restrict the intracellular growth of M. tuberculosis (Stanley et al., 2014). Our series do not overalp with those previously identified.
For the five series we prioritized, this is the first report of antimycobacterial activity of the majority of these compounds. Other works have reported compounds with similar substructures, but our series are structurally distinct overall. We identified a novel series (PTU) containing a thiourea group. To the best of our knowledge, this is the first report of this structural class of compounds as antitubercular agents although other thiourea derivatives have antitubercular activity (Liav et al., 2008; Plutín et al., 2016; Ghorab et al., 2017; Doğan et al., 2020). We identified a novel phenyl pyrazole series. Previous work has identified similar pyrazole-containing compounds with anti-tubercular activity that target MmpL3. In these studies, a 1,3,5-trisubstituted pyrazole as a central core had good activity against axenically-cultured M. tuberculosis (Poce et al., 2019). Our molecules differ in that they only have weak activity against extracellular bacteria. The molecules we identify here are phenyl pyrazoles which have not been reported previously to have activity against either intracellular or extracellular bacteria. We identified a novel thiophene carboxamide series. To the best of our knowledge, this is the first report of these compounds as antitubercular agents. Our molecules are not structurally similar to the previously reported thiophene carboxamide prodrug (Mori et al., 2015), although both contain a thiophene group and an amide group, the linkages and other features of the molecules are different.
This highlights the power of the screen assay in identifying novel compounds that are conditionally active against intracellular M. tuberculosis. These conditionally-active compounds would be overlooked in other screens against extracellular bacteria. There is accumulating evidence that intracellular bacteria are both heterogenous and antibiotic tolerant (Vijay et al., 2017; Adams et al., 2019; Mandal et al., 2019; Mishra et al., 2019; Nair et al., 2019), and this may be one reason why drug treatment to achieve sterilization takes >4 months. The inclusion of compounds which specifically target intracellular bacteria in a drug regiment could be one approach to shorten therapy. In support of this, GSK286, a molecule identified from intracellular screens targeting cholesterol metabolism, is in phase II clinical trials.
Compounds that are only active against intracellular bacteria could be targeting pathways that are only essential for growth within the macrophages. Alternatively, they could target host pathways that enhance bacterial clearance or could be accumulated and/or metabolized within the macrophage. Further work to identify targets and mode of action is warranted on series with attractive properties.
Early identification of potentially toxic compounds during library screening has a significant impact on both the cost and the success rate of the drug discovery process. We were able to identify cytotoxic compounds rapidly and to exclude compounds that may look active due to killing the macrophages rather than anti-bacterial activity. Out of the 308 hits that we identified as mycobacterial growth inhibitors, we excluded 134 molecules with significant toxicity to the macrophages representing almost a third of the hits. We also saw that compounds had comparable cytotoxicity against both macrophage and HepG2 cell lines, demonstrating the power of this assay in predicting potential cytotoxicity of the compounds early in the screening process.
We identified five novel and tractable chemical scaffolds with promising activity against intracellular M. tuberculosis using a high throughput, high content screen. The mechanism of action and targets remains to be characterized for each series. However, these series represent attractive starting points for lead generation and full structure-activity relationship studies.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
All authors contributed to conception and design of the study. SA, AM, LF, and YO conducted the experimental work. All authors analyzed the data. SA and TP wrote the first draft of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Research reported in this publication was supported by NIAID of the National Institutes of Health under award number R01AI132634. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Sultan Chowdhury and Sammy Njikan for technical assistance and useful discussion.
Adams, K. N., Verma, A. K., Gopalaswamy, R., Adikesavalu, H., Singhal, D. K., Tripathy, S., et al. (2019). Diverse Clinical Isolates of Mycobacterium tuberculosis Develop Macrophage-Induced Rifampin Tolerance. J. Infect. Dis. 219 (10), 1554–1558. doi:10.1093/infdis/jiy710
Carroll, P., Muwanguzi-Karugaba, J., and Parish, T. (2018). Codon-optimized DsRed Fluorescent Protein for Use in Mycobacterium tuberculosis. BMC Res. Notes 11 (1), 685. doi:10.1186/s13104-018-3798-3
Christophe, T., Jackson, M., Jeon, H. K., Fenistein, D., Contreras-Dominguez, M., Kim, J., et al. (2009). High Content Screening Identifies Decaprenyl-Phosphoribose 2' Epimerase as a Target for Intracellular Antimycobacterial Inhibitors. Plos Pathog. 5 (10), e1000645. doi:10.1371/journal.ppat.1000645
Deretic, V. (2014). Autophagy in Tuberculosis. Cold Spring Harb Perspect. Med. 4 (11), a018481. doi:10.1101/cshperspect.a018481
Doğan, Ş. D., Gündüz, M. G., Doğan, H., Krishna, V. S., Lherbet, C., and Sriram, D. (2020). Design and Synthesis of Thiourea-Based Derivatives as Mycobacterium tuberculosis Growth and Enoyl Acyl Carrier Protein Reductase (InhA) Inhibitors. Eur. J. Med. Chem. 199, 112402. doi:10.1016/j.ejmech.2020.112402
Ghorab, M. M., El-Gaby, M. S. A., Soliman, A. M., Alsaid, M. S., Abdel-Aziz, M. M., and Elaasser, M. M. (2017). Synthesis, Docking Study and Biological Evaluation of Some New Thiourea Derivatives Bearing Benzenesulfonamide Moiety. Chem. Cent. J. 11 (1), 42. doi:10.1186/s13065-017-0271-7
Global Tuberculosis Report (2020). Global Tuberculosis Report 2020 [Internet]. Available at: https://www.who.int/publications-detail-redirect/9789240013131 (Accessed 6 Oct, 2021).
Gordon, S. V., and Parish, T. (2018). Microbe Profile: Mycobacterium tuberculosis: Humanity's Deadly Microbial Foe. Microbiology (Reading) 164 (4), 437–439. doi:10.1099/mic.0.000601
Kamalian, L., Chadwick, A. E., Bayliss, M., French, N. S., Monshouwer, M., Snoeys, J., et al. (2015). The Utility of HepG2 Cells to Identify Direct Mitochondrial Dysfunction in the Absence of Cell Death. Toxicol. Vitro 29 (4), 732–740. doi:10.1016/j.tiv.2015.02.011
Liav, A., Angala, S. K., Brennan, P. J., and Jackson, M. (2008). N-D-aldopentofuranosyl-N'-[p-(isoamyloxy)phenyl]-thiourea Derivatives: Potential Anti-TB Therapeutic Agents. Bioorg. Med. Chem. Lett. 18 (8), 2649–2651. doi:10.1016/j.bmcl.2008.03.033
Mandal, S., Njikan, S., Kumar, A., Early, J. V., and Parish, T. (2019). The Relevance of Persisters in Tuberculosis Drug Discovery. Microbiology (Reading) 165 (5), 492–499. doi:10.1099/mic.0.000760
Manjunatha, U. H., and Smith, P. W. (2015). Perspective: Challenges and Opportunities in TB Drug Discovery from Phenotypic Screening. Bioorg. Med. Chem. 23 (16), 5087–5097. doi:10.1016/j.bmc.2014.12.031
Manning, A. J., Ovechkina, Y., McGillivray, A., Flint, L., Roberts, D. M., and Parish, T. (2017). A High Content Microscopy Assay to Determine Drug Activity against Intracellular Mycobacterium tuberculosis. Methods 127, 3–11. doi:10.1016/j.ymeth.2017.03.022
Mishra, R., Kohli, S., Malhotra, N., Bandyopadhyay, P., Mehta, M., Munshi, M., et al. (2019). Targeting Redox Heterogeneity to Counteract Drug Tolerance in Replicating Mycobacterium tuberculosis. Sci. Transl Med. 11 (518), eaaw6635. doi:10.1126/scitranslmed.aaw6635
Mori, G., Chiarelli, L. R., Esposito, M., Makarov, V., Bellinzoni, M., Hartkoorn, R. C., et al. (2015). Thiophenecarboxamide Derivatives Activated by EthA Kill Mycobacterium tuberculosis by Inhibiting the CTP Synthetase PyrG. Chem. Biol. 22 (7), 917–927. doi:10.1016/j.chembiol.2015.05.016
Nair, R. R., Sharan, D., Sebastian, J., Swaminath, S., and Ajitkumar, P. (2019). Heterogeneity of ROS Levels in Antibiotic-Exposed Mycobacterial Subpopulations Confers Differential Susceptibility. Microbiology (Reading) 165 (6), 668–682. doi:10.1099/mic.0.000797
Njikan, S., Manning, A. J., Ovechkina, Y., Awasthi, D., and Parish, T. (2018). High Content, High-Throughput Screening for Small Molecule Inducers of NF-Κb Translocation. PLoS One 13 (6), e0199966. doi:10.1371/journal.pone.0199966
Ollinger, J., Bailey, M. A., Moraski, G. C., Casey, A., Florio, S., Alling, T., et al. (2013). A Dual Read-Out Assay to Evaluate the Potency of Compounds Active against Mycobacterium tuberculosis. PLoS One 8 (4), e60531. doi:10.1371/journal.pone.0060531
Parish, T. (2020). In Vitro drug Discovery Models for Mycobacterium tuberculosis Relevant for Host Infection. Expert Opin. Drug Discov. 15 (3), 349–358. doi:10.1080/17460441.2020.1707801
Parish, T. (2019). Steps to Address Anti-microbial Drug Resistance in Today's Drug Discovery. Expert Opin. Drug Discov. 14 (2), 91–94. doi:10.1080/17460441.2019.1550481
Payne, D. J., Gwynn, M. N., Holmes, D. J., and Pompliano, D. L. (2007). Drugs for Bad Bugs: Confronting the Challenges of Antibacterial Discovery. Nat. Rev. Drug Discov. 6 (1), 29–40. doi:10.1038/nrd2201
Plutín, A. M., Alvarez, A., Mocelo, R., Ramos, R., Castellano, E. E., da Silva, M. M., et al. (2016). Anti-Mycobacterium tuberculosis Activity of Platinum(II)/N,N-disubstituted-N′-acyl Thiourea Complexes. Inorg. Chem. Commun. 63, 74–80.
Poce, G., Consalvi, S., Venditti, G., Alfonso, S., Desideri, N., Fernandez-Menendez, R., et al. (2019). Novel Pyrazole-Containing Compounds Active against Mycobacterium tuberculosis. ACS Med. Chem. Lett. 10 (10), 1423–1429. doi:10.1021/acsmedchemlett.9b00204
Sassetti, C. M., Boyd, D. H., and Rubin, E. J. (2001). Comprehensive Identification of Conditionally Essential Genes in Mycobacteria. Proc. Natl. Acad. Sci. U S A. 98 (22), 12712–12717. doi:10.1073/pnas.231275498
Sassetti, C. M., Boyd, D. H., and Rubin, E. J. (2003). Genes Required for Mycobacterial Growth Defined by High Density Mutagenesis. Mol. Microbiol. 48 (1), 77–84. doi:10.1046/j.1365-2958.2003.03425.x
Sassetti, C. M., and Rubin, E. J. (2003). Genetic Requirements for Mycobacterial Survival during Infection. Proc. Natl. Acad. Sci. U S A. 100 (22), 12989–12994. doi:10.1073/pnas.2134250100
Stanley, S. A., Barczak, A. K., Silvis, M. R., Luo, S. S., Sogi, K., Vokes, M., et al. (2014). Identification of Host-Targeted Small Molecules that Restrict Intracellular Mycobacterium tuberculosis Growth. Plos Pathog. 10 (2), e1003946. doi:10.1371/journal.ppat.1003946
Vandal, O. H., Nathan, C. F., and Ehrt, S. (2009). Acid Resistance in Mycobacterium tuberculosis. J. Bacteriol. 191 (15), 4714–4721. doi:10.1128/JB.00305-09
VanderVen, B. C., Fahey, R. J., Lee, W., Liu, Y., Abramovitch, R. B., Memmott, C., et al. (2015). Novel Inhibitors of Cholesterol Degradation in Mycobacterium tuberculosis Reveal How the Bacterium's Metabolism Is Constrained by the Intracellular Environment. Plos Pathog. 11 (2), e1004679. doi:10.1371/journal.ppat.1004679
Vijay, S., Vinh, D. N., Hai, H. T., Ha, V. T. N., Dung, V. T. M., Dinh, T. D., et al. (2017). Influence of Stress and Antibiotic Resistance on Cell-Length Distribution in Mycobacterium tuberculosis Clinical Isolates. Front. Microbiol. 8, 2296. doi:10.3389/fmicb.2017.02296
Keywords: high throughput screening, mycobacteria, anti-tubercular, drug discovery, high content microscopy, macrophage infection, intracellular activity
Citation: Ahmed S, Manning A, Flint L, Awasthi D, Ovechkina Y and Parish T (2022) Identification of Novel Chemical Scaffolds that Inhibit the Growth of Mycobacterium tuberculosis in Macrophages. Front. Pharmacol. 12:790583. doi: 10.3389/fphar.2021.790583
Received: 07 October 2021; Accepted: 22 November 2021;
Published: 03 January 2022.
Edited by:
Gert Kruger, University of KwaZulu-Natal, South AfricaReviewed by:
Verlaine Joy Timms, The University of Newcastle, AustraliaCopyright © 2022 Ahmed, Manning, Flint, Awasthi, Ovechkina and Parish. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tanya Parish, dGFueWEucGFyaXNoQHNlYXR0bGVjaGlsZHJlbnMub3Jn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.