 Delma Veron
Delma Veron Pardeep K. Aggarwal1†
Pardeep K. Aggarwal1† Gilbert Moeckel
Gilbert Moeckel Alda Tufro
Alda Tufro
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol., 23 February 2022
Sec. Renal Pharmacology
Volume 12 - 2021 | https://doi.org/10.3389/fphar.2021.788886
This article is part of the Research TopicReceptor Biology and Cell Signaling in DiabetesView all 9 articles
Vascular endothelial growth factor-a (VEGF-A) and nitric oxide (NO) are essential for glomerular filtration barrier homeostasis, and are dysregulated in diabetic kidney disease (DKD). While NO availability is consistently low in diabetes, both high and low VEGF-A have been reported in patients with DKD. Here we examined the effect of inducible podocyte VEGF-A knockdown (VEGFKD) in diabetic mice and in endothelial nitric oxide synthase knockout mice (eNOS−/−). Diabetes was induced with streptozotocin using the Animal Models of Diabetic Complications Consortium (AMDCC) protocol. Induction of podocyte VEGFKD led to diffuse glomerulosclerosis, foot process effacement, and GBM thickening in both diabetic mice with intact eNOS and in non-diabetic eNOS−/−:VEGFKD mice. VEGFKD diabetic mice developed mild proteinuria and maintained normal glomerular filtration rate (GFR), associated with extremely high NO and thiol urinary excretion. In eNOS−/−:VEGFKD (+dox) mice severe diffuse glomerulosclerosis was associated with microaneurisms, arteriolar hyalinosis, massive proteinuria, and renal failure. Collectively, data indicate that combined podocyte VEGF-A and eNOS deficiency result in diffuse glomerulosclerosis in mice; compensatory NO and thiol generation prevents severe proteinuria and GFR loss in VEGFKD diabetic mice with intact eNOS, whereas VEGFKD induction in eNOS−/−:VEGFKD mice causes massive proteinuria and renal failure mimicking DKD in the absence of diabetes. Mechanistically, we identify VEGFKD-induced abnormal S-nitrosylation of specific proteins, including β3-integrin, laminin, and S-nitrosoglutathione reductase (GSNOR), as targetable molecular mechanisms involved in the development of advanced diffuse glomerulosclerosis and renal failure.
Diabetic kidney disease (DKD) is a major complication of both type 1 and type 2 diabetes that leads to renal failure, and the single most frequent cause of end-stage renal disease (ESRD) worldwide (Tuttle et al., 2014). An incomplete understanding of the molecular mechanisms that lead to DKD has precluded the development of effective treatments preventing progression to ESRD (Tufro and Veron, 2012; Reidy et al., 2014).
Vascular endothelial growth factor-A (VEGF-A) and nitric oxide (NO) are essential for glomerular filtration barrier homeostasis, and both are disregulated in diabetic nephropathy (Papapetropoulos et al., 1997; Shen et al., 1999; Hohenstein et al., 2006; Tufro and Veron, 2012). Unlike consistently low NO availability in diabetes, both high and low VEGF-A have been observed in patients with DKD (Hohenstein et al., 2006; Baelde et al., 2007; Lindenmeyer et al., 2007). We have shown that podocyte VEGF-A gain-of-function in diabetic mice leads to the development of Kimmelstiel-Wilson-like nodular glomerulosclerosis and massive proteinuria (Veron et al., 2011). Similar glomerular phenotype was reported in eNOS deficient type 1 and type 2 diabetic mouse models (Zhao et al., 2006; Nakagawa et al., 2007). Moreover, we showed that VEGF-A gain-of-function in eNOS KO mice also induces nodular glomerulosclerosis, massive proteinuria and renal failure in the absence of diabetes (Veron et al., 2014). These findings demonstrated that NO deficiency and excess VEGF-A have a synergistic deleterious effect that is necessary and sufficient for the development of nodular glomerulosclerosis, the prototypical glomerular phenotype of human advanced DKD (Veron et al., 2014). Endothelial cell or podocyte VEGF-A knockout causes thrombotic microangiopathy in adult mice (Lee et al., 2007; Eremina et al., 2008). Short term VEGF-A knockdown in podocytes induces acute renal failure and proteinuria associated with endotheliosis, mesangiolysis, and microaneurisms (Veron et al., 2012), and VEGF-A deletion accelerates DKD in a short term diabetes mouse model (Sivaskandarajah et al., 2012).
Here we examined the effect of podocyte VEGF-A knockdown (VEGFKD) in diabetic mice and in eNOS−/−:VEGFKD mice. We determined that in the setting of NO deficiency, caused either by diabetic milieu or eNOS knockout, VEGFKD results in diffuse glomerulosclerosis and proteinuria, mimicking human diabetic diffuse glomerulosclerosis of increasing severity. This phenotype is linked to the generation of NO and thiol mediated by changes in S-nitrosoglutathione reductase (GSNOR) and β3-integrin S-nitrosylation that impairs their activity.
We generated doxycycline-inducible podocyte VEGFKD in eNOS KO mice (eNOS−/−:VEGFKD) by crossbreeding podocin-rtTA:tet-O-siVEGF mice (siVEGF) (Veron et al., 2012) with eNOS−/− (Shesely et al., 1996) (eNOS KO, C57BL/6j-Nos3tm1Unc; The Jackson Laboratory, Bar Harbor, ME), and we backcrossed them >8 generations to a stable FVB background. In this study eNOS−/−:VEGFKD mice were fed standard or doxycycline-containing chow (Harlan-Teklad) for 1 month.
Diabetes was induced in 6- to 8-week-old male siVEGF mice (Veron et al., 2012) (herein called VEGFKD) by intraperitoneal streptozotocin (STZ) using the low dose AMDCC (Animal Models of Diabetic Complications Consortium) protocol, as previously described (Veron et al., 2011; Aggarwal et al., 2015). Random blood glucose concentration >300 mg/dl was confirmed a week after the last STZ injection and every 4 weeks along the experiment. Diabetic VEGFKD (DM-VEGFKD) and non-diabetic (non-DM-VEGFKD) mice were fed standard (−dox) or doxycycline containing chow (+dox) for 12 weeks to induce VEGF-A knockdown. At the end of the study 24 h urine was collected in metabolic cages; blood and kidney samples were obtained under anesthesia, as we previously described (Veron et al., 2011; Veron et al., 2012; Veron et al., 2014). All experimental protocols were approved by the Institutional Animal Care and Use Committee at Yale University School of Medicine.
Random blood glucose was measured by glucose oxidase biosensor (OneTouch Ultra-2; LifeScan), and BP was measured under anesthesia and analyzed using PowerLab/8SP system (Chart; AD Instruments, Colorado Springs, CO, Unites States) as previously described (Veron et al., 2011; Veron et al., 2012). Plasma and urine creatinine were measured by HPLC, and glomerular filtration rate (GFR) was assessed by creatinine clearance. Albuminuria was evaluated by Coomassie blue staining and measured by ELISA (Albuwell-M, Exocell), plasma and urine VEGF-A were quantified by ELISA (R&D), NO was measured by colorimetric assay (Cayman), as previously described (Veron et al., 2014), and urine thiols (Cys and GSH) were measured by fluorometric assay (Cayman), following manufacturers’ protocol.
Kidneys were processed for light microscopy and TEM or frozen in isopentane, mounted in OCT (Sakura). Histology was assessed by hematoxylin/eosin and periodic acid–Schiff’s reagent (PAS) stains. TEM was performed using standard techniques, as previously described (Tsurumi et al., 1997; Veron et al., 2011). A renal pathologist (G.M.) examined all kidney samples by light and TEM, blinded to specimens’ identity (Veron et al., 2011; Veron et al., 2012; Veron et al., 2014; Aggarwal et al., 2015). Morphometric analysis was performed using point counting technique on PAS-stained sections, as previously described (Nakagawa et al., 2007). Glomerulosclerosis, mesangial expansion, mesangiolysis, endothelial injury, interstitial fibrosis, and inflammatory infiltrates were assessed using a semi-quantitative score (Véniant et al., 1994; Gross et al., 2006): 0 = none; 1 = 1–25%; 2 = 26–50%; 3 = 51–75%; 4 = 76–100% of glomerular or section areas, as appropriate (Veron et al., 2011; Veron et al., 2014). Glomerular diameters were measured in 147 ± 8 glomeruli per 5-6 mice/experimental group and glomerular volumes calculated as previously described (Reidy et al., 2009).
Immunohistochemistry (IHC) was performed in frozen kidney sections using primary antibodies against laminin, nephrin, podocin, and S-nitroso-cysteine and appropriate Cy2 and Cy3 fluorescent-tagged secondary antibodies (Jackson ImmunoResearch Laboratories), visualized by confocal microscopy (FluoView 300; Olympus), as previously described (Veron et al., 2011; Veron et al., 2012; Veron et al., 2014). Quantitation of immunofluorescent signals was performed in ≥10 glomeruli/mouse, n ≥ 4/experimental group using ImageJ software (National Institutes of Health, Bethesda, MD), as previously described (Veron et al., 2012; Aggarwal et al., 2015).
Immunoblotting was performed using the following primary antibodies: podocin (P0372, Sigma), nephrin (20R-NP002, Fitzgerald Inc.), laminin (L9393, Sigma), β3-integrin (sc-14009, Santa Cruz), VEGF receptor 2 (2479, Cell Signaling Technologies); actin (A2066. Sigma) or tubulin (Sigma) were used as a loading control. Signals were visualized by chemiluminescence, and quantified using ImageJ software (Veron et al., 2011; Veron et al., 2012; Veron et al., 2014; Aggarwal et al., 2015).
We evaluated GSNOR S-nitrosylation by biotin switch assay in whole kidney lysates using a S-nitrosylated protein detection kit (Cayman Chemical, Co.), as previously described (Veron et al., 2014; Li et al., 2021). Ascorbate was omitted in the labeling step to serve as negative control. We localized kidney S-nitrosylated proteins by IHC, as described (Veron et al., 2014). In situ Proximity Link Assays (PLA) were performed to identify specific S-nitrosylated proteins in kidney frozen sections using laminin rabbit polyclonal antibody (L9393, Sigma-Aldrich) or β3-integrin antibody (sc-14009, Santa Cruz) and S-nitrosocysteine mouse monoclonal antibody (AG Scientific) and Duolink II fluorescence protocol (Olink Bioscience, Uppsala, Sweden) (Söderberg et al., 2006), as previously described (Veron et al., 2014).
Data are expressed as mean ± SEM unless otherwise stated. Statistical significance (p < 0.05) was determined using Prism 8 software by unpaired t test with Welch’s correction and one-way Brown-Forsythe ANOVA to compare two or multiple experimental groups, respectively. Mann-Whitney test was used to analyze non-parametric variables.
Experimental design is shown in Figures 1A,B and general parameters from diabetic and non-diabetic mice are summarized in Table 1. Diabetes caused glomerulomegaly and mild mesangial expansion in uninduced (control) DM-VEGFKD − dox mice (Figures 1D,G, hatched blue bar), consistent with early diabetic kidney disease (Gundersen and Osterby, 1977; Tervaert et al., 2010). Podocyte VEGFKD induction with doxycycline for 12 weeks prevented the development of glomerular hypertrophy in DM-VEGFKD + dox mice (Figures 1F,G, blue bars), and decreased glomerular size in non-diabetic mice (Figures 1C, E, G, white and gray bars). Glomerular size in diabetic VEGFKD (+dox) mice (Figure 1F) was similar to non-diabetic control (− dox) mice (Figure 1C), as quantified in Figure 1G (white bar). However, diabetic mice with podocyte DM-VEGFKD (+dox) revealed diffuse glomerulosclerosis (Figure 1F) with limited inflammatory infiltrate and tubulo-interstitial damage. Table 2 summarizes the semi-quantitative pathology scores comparing diabetic kidneys DM-VEGFKD −dox vs. + dox.
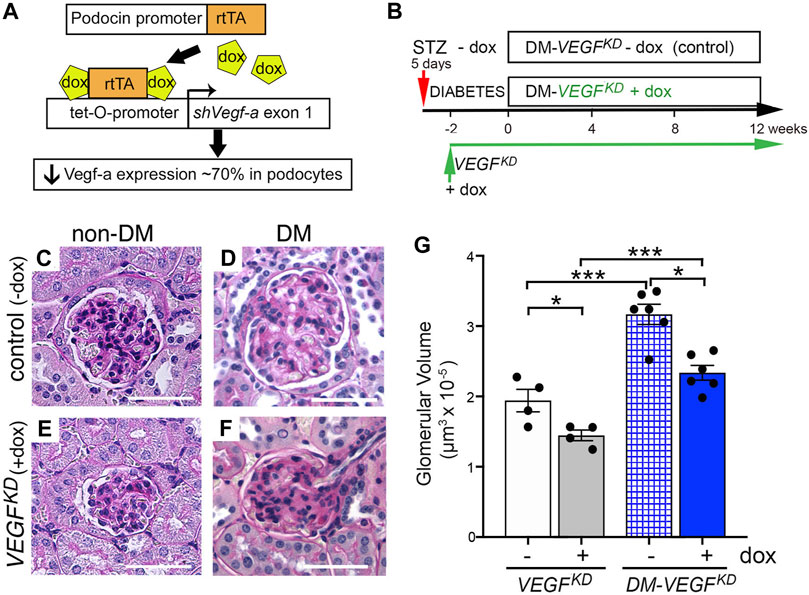
FIGURE 1. Podocyte VEGF knockdown (VEGFKD) prevents glomerular hypertrophy in diabetic mice. (A) VEGFKD Transgenic mouse line carries 4 transgenes: Nphs2-rtTA and tet-0-shVEGF that are activated by doxycycline to synthesize shRNA targeting Vegf-a exon 1, which inhibits expression of all Vegf-a isoforms in podocytes (Veron et al., 2012). (B) VEGFKD mice received STZ (50 mg IP, 5 daily doses) (DM-VEGFKD − dox), STZ + doxycycline (DM-VEGFKD + dox), doxycycline (VEGFKD + dox) or no treatment (VEGFKD − dox). Dox was started a week after STZ, 2 weeks later was considered time 0 (when random blood glucose was steadily elevated) for DM-VEGFKD mice; (C) non-diabetic control glomerulus (VEGFKD − dox) shows normal histology; (D) diabetic control (DM-VEGFKD − dox) glomerulus shows hypertrophy and mesangial expansion; (E) non-diabetic VEGFKD (+ dox) glomerulus is smaller than control (C, − dox); (F) diabetic VEGFKD (+ dox) glomerulus shows diffuse glomerulosclerosis and is smaller than control (D, − dox); Scale bars = 50 μm; (G) quantitation of glomerular size demonstrates significantly smaller glomerular volume in VEGFKD (+ dox) vs. control (−dox) glomeruli from non-diabetic (VEGFKD) and diabetic (DM-VEGFKD) mice; unpaired t-test with Welch’s correction was used; asterisk (*) indicates p < 0.05, (***) indicates p < 0.001; control vs. VEGFKD or non-diabetic vs. diabetic, as indicated; non-DM, non-diabetic mice, DM, diabetic mice; dox, uninduced mice; dox, doxycycline- treated mice.

TABLE 1. General parameters.

TABLE 2. Pathology score.
Glomerular histology was mostly normal in uninduced eNOS−/−:VEGFKD (− dox) mice (Figure 2A), although mild mesangial expansion, endothelial injury, and interstitial fibrosis were observed occasionally. Induction of podocyte VEGFKD (+dox) for 4 weeks in eNOS−/−:VEGFKD mice caused severe diffuse glomerulosclerosis (Figure 2B). Extensive mesangiolysis, microaneurisms, and extracellular matrix expansion were observed (Figures 2B1–3), whereas no glomerular nodules were detected in PAS stained sections from eNOS−/−:VEGFKD (+dox) mice. Significant tubulo-interstitial damage consisting of tubular atrophy and basement membrane thickening, tubular proteinaceous casts, and interstitial lymphocytic infiltrates were also observed in eNOS−/−:VEGFKD (+dox) (Figures 2B4–6) but were not present in eNOS−/−:VEGFKD (−dox) kidneys (Figure 2A) or in diabetic DM-VEGFKD (+dox) kidneys (Figure 1F). A semi-quantitative analysis of the histological abnormalities summarized in Table 2 confirmed these observations. Although the pathology scores revealed mild endothelial injury, mesangial sclerosis, and interstitial fibrosis in eNOS−/−:VEGFKD (−dox) kidneys, the severity and extension of the changes observed in eNOS−/−:VEGFKD (+dox) kidneys were obvious as demonstrated by highly significant score differences in all parameters.
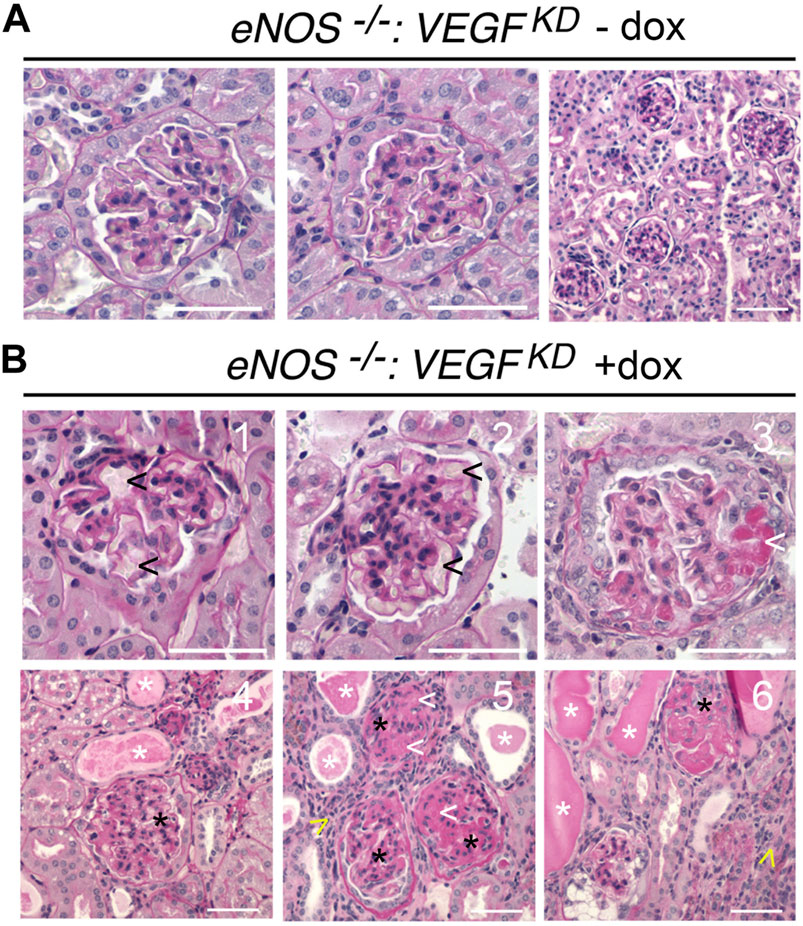
FIGURE 2. Histology of eNOS −/−:VEGFKD kidneys reveals diffuse glomerulosclerosis and mimics advanced DKD: PAS stain representative images: (A) eNOS−/−:VEGFKD − dox glomeruli are normal by light microscopy; (B) eNOS−/−:VEGFKD + dox glomeruli show microaneurisms (1-2, black arrowheads), mesangiolysis (3,5,6, white arrowheads), mesangial expansion (Papapetropoulos et al., 1997; Shen et al., 1999; Reidy et al., 2014; Tuttle et al., 2014), severe mesangial sclerosis (4-6, black asterisks), proteinaceous tubular casts (4-6, white asterisks) and lymphocytic infiltrates (5–6, yellow arrowheads); Scale bars = 50 μm (A, B1-3) and 100 μm (B4-6); PAS: Periodic acid-Schiff stain.
TEM revealed focal foot process effacement, mesangial expansion, and GBM thickening in all diabetic mice (Figures 3A–D), whereas mesangial sclerosis was more extensive in glomeruli from diabetic DM-VEGFKD (+dox) mice (Figure 3C). eNOS−/−:VEGFKD (−dox) kidneys showed normal glomeruliar filtration barrier ultrastructure (Figures 3E,F). In contrast, eNOS−/−:VEGFKD mice with podocyte VEGFKD (+dox) revealed extensive foot process effacement, GBM thickening, severe mesangial sclerosis, and endotheliosis (Figures 3G,H), a more severe phenotype than that observed in diabetic DM-VEGFKD (+dox) kidneys (Figures 3C,D).
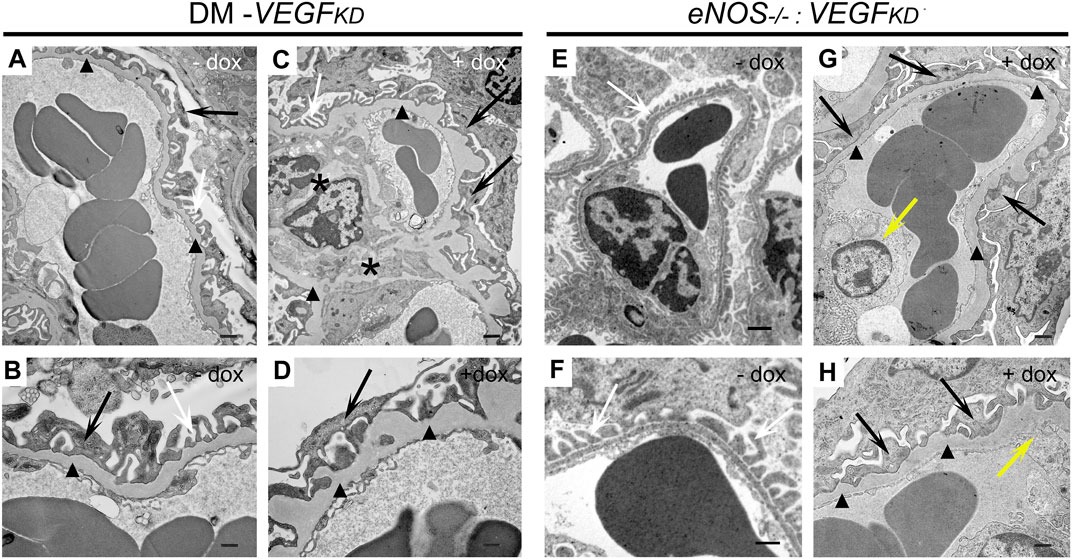
FIGURE 3. Effect of VEGFKD on glomerular ultrastructure of diabetic and eNOS−/−: VEGFKD kidneys. Representative TEM images: (A,B) diabetic control (DM-VEGFKD (− dox) glomerular capillary loop shows GBM thickening (black arrowheads) and partial foot process effacement (FPE: black arrows, normal FP: white arrows); (C,D) diabetic VEGFKD (+ dox) glomerulus shows mesangial sclerosis (black asterisks), extensive FPE (black arrows) and GBM thickening (black arrowheads); (E,F) eNOS−/−:VEGFKD (−dox) glomerulus shows preserved filtration barrier ultrastructure; (G,H) eNOS−/−:VEGFKD (+dox) glomerulus shows massive FPE (black arrows), GBM thickening (black arrowheads) and endotheliosis (yellow arrows). Scale bars: 1 μm in top images (A,C,E,G); 500 nm in bottom images (B,D,F,H); + dox:VEGFKD induction with doxycycline.
Podocyte VEGFKD in eNOS−/−:VEGFKD (+dox) and diabetic mice resulted in nephrin downregulation, assessed by immunoblotting and immunohistochemistry (Figures 4A,B), and we detected similar changes in podocin expression by immunoblot (Figure 4C). Expression of VEGF-A receptor 2 (VEGFR2) was mildly decreased in eNOS−/−:VEGFKD and diabetic mice subjected to VEGFKD (+dox) (Figure 4D). Podocyte VEGFKD resulted in β3-integrin upregulation in eNOS−/−:VEGFKD (+dox) kidneys, whereas β3-integrin protein expression was not altered in diabetic mice with intact eNOS (Figure 4E). Collectively, these changes in protein expression levels suggest dysregulation of the nephrin-VEGFR2-β3-integrin pathway, which is necessary for the structural and functional integrity of the glomerular filtration barrier (Bertuccio et al., 2011; Veron et al., 2012; Aggarwal et al., 2015; Hayek et al., 2017).
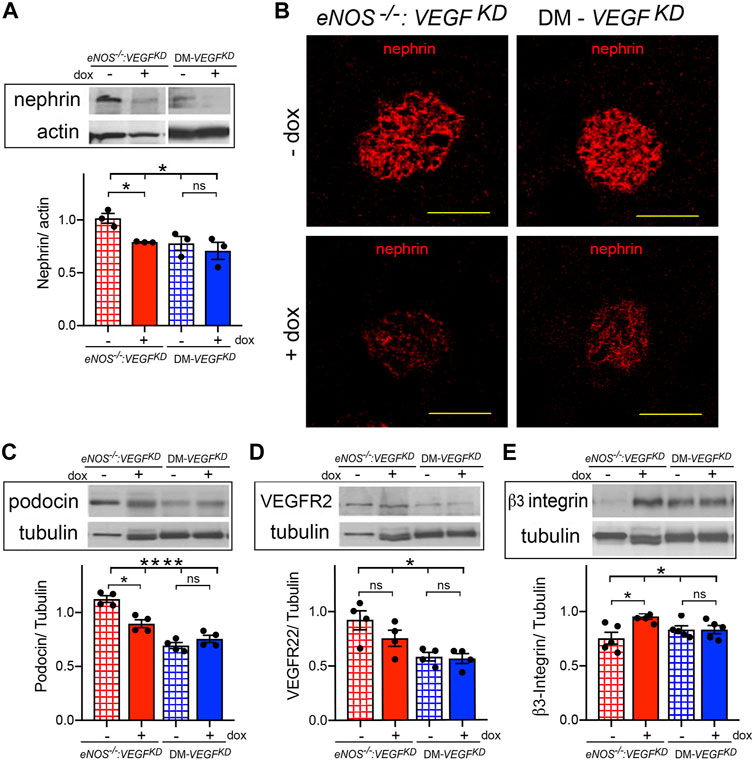
FIGURE 4. Podocyte VEGFKD downregulates nephrin in diabetic and eNOS−/−:VEGFKD mice: (A) WB: show nephrin downregulation in eNOS−/−:VEGFKD + dox and diabetic kidneys (Brown-Forsythe ANOVA, p = 0.047), no significant difference was detected between DM-VEGFKD − dox and + dox (Welch’s t-test) ; (B) IHC: nephin IF signals are clearly decreased in glomeruli from eNOS−/−:VEGFKD (+dox) and DM-VEGFKD (+dox) kidneys; (C) WB: podocin decreased in eNOS−/−:VEGFKD (+dox) and diabetic kidneys (Brown-Forsythe ANOVA, p = 0.0001), no significant difference was detected between DM-VEGFKD − dox and + dox (Welch’s t-test); (D) WB: VEGFR2 decreased in eNOS −/−:VEGFKD (+dox) and diabetic kidneys (Brown-Forsythe ANOVA, p = 0.015) but differences (+dox vs. - dox) were not significant; (E) WB: significant β3-integrin upregulation was detected in eNOS−/−:VEGFKD (+dox) kidneys (Brown-Forsythe ANOVA, p = 0.03). Scale bars = 50 μm, + dox = VEGFKD induction with doxycycline.
Induction of podocyte VEGFKD in eNOS−/−:VEGFKD (+dox) mice caused massive albuminuria >30-fold higher than that measured in uninduced genetically identical mice eNOS−/−:VEGFKD (−dox), (Figures 5A,B, red bars) and ∼15 fold higher than in eNOS−/− mice (data not shown). In contrast, mice with intact eNOS developed mild proteinuria when podocyte VEGFKD (+dox) was induced (Figure 5A, white/gray bars), suggesting that eNOS and VEGF-A deficiency have synergistic effect worsening proteinuria. Surprisingly, podocyte VEGFKD for 12 weeks did not increase albuminuria in diabetic (DM-VEGFKD + dox) mice (Figures 5A,B blue bars). Hypertension was not observed in non-diabetic eNOS−/−: VEGFKD mice (mean BP = 84 ± 2 mmHg vs. 78 ± 2 mmHg, + dox vs. −dox, pNS), as reported in VEGFKD mice with intact eNOS (Veron et al., 2012). Podocyte VEGFKD caused renal failure in eNOS−/−:VEGFKD (+dox) mice (Figure 5C, red bar), whereas it did not significantly alter creatinine clearance in mice with diabetes (DM-VEGFKD) or intact eNOS (VEGFKD) (Figure 5C, blue and gray bars, respectively). Taken together, these findings suggest that eNOS insufficiency and VEGF-A knockdown have additive pathogenic effects leading to renal failure when a compensatory NO source is not available.
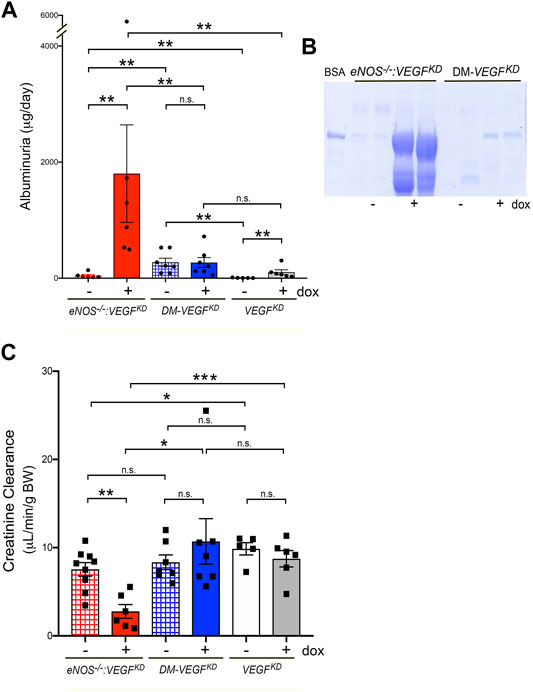
FIGURE 5. Podocyte VEGFKD causes massive proteinuria and renal failure in eNOS−/−:VEGFKD mice. (A) Induction of VEGFKD in eNOS−/−:VEGFKD (+dox) mice (red bar) increases albuminuria ∼30 fold higher than in control eNOS−/−:VEGFKD (− dox) (**, p = 0.0022) but does not change albuminuria in diabetic mice (DM-VEGFKD + dox, blue bar) (n.s., p = 0.9015); VEGFKD causes mild albuminuria in non-diabetic mice (VEGFKD + dox, gray bar) compared to controls (VEGFKD − dox, white bar) (**, p = 0.0043), Mann-Whitney test. (B) SDS PAGE/Coomassie stain shows severe albuminuria in eNOS−/−:VEGFKD + dox and milder albuminuria in diabetic VEGFKD + dox mice; BSA = bovine serum albumin marker, urine volume loading was normalized to creatinine. (C) Creatinine clearance decreases upon VEGFKD induction in eNOS−/−:VEGFKD + dox mice (red bar) to ∼1/3 of control eNOS−/−:VEGFKD − dox (***, p = 0.0009), but is not significantly altered in diabetic mice (DM-VEGFKD − dox and + dox, hatched/blue bars) (n.s., p = 0.4114) or VEGFKD in non-diabetic mice (VEGFKD − dox and + dox, white/gray bars) (n.s., p = 0.359) with intact eNOS; induced eNOS−/−:VEGFKD (+dox) mice had significantly lower Creat Cl than diabetic VEGFKD + dox and non-diabetic VEGFKD + dox mice (*, p = 0.02 and ***, p = 0.0007, respectively).
To gain insight into the availability of NO and VEGF-A systemically and at the glomerular filtration barrier we measured VEGF-A and NO in plasma and urine. We determined that plasma VEGF-A and urinary excretion are similarly elevated in eNOS−/−:VEGFKD mice, irrespective of podocyte VEGFKD (Figures 6A,B, red bars). In diabetic mice podocyte VEGFKD decreased plasma VEGF-A (Figure 6A, blue bar), which remained significantly higher (∼2-fold) than in non-diabetic mice (Figure 6A, white/gray bars). Urine VEGF-A excretion was not altered in diabetic mice, irrespective of podocyte VEGFKD (Figure 6A, blue bars). Conversely, podocyte VEGFKD in non-diabetic mice with intact eNOS significantly decreased VEGF-A excretion (Figure 6B, white/gray bars).
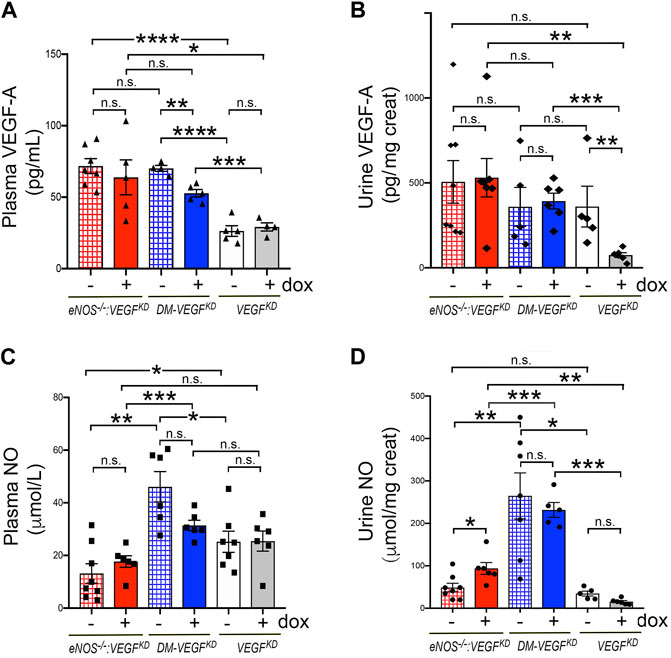
FIGURE 6. Effect of VEGFKD on circulating and urine VEGF-A and NO in diabetic and eNOS−/−:VEGFKD mice. (A) plasma VEGF-A is similarly elevated in eNOS−/−:VEGFKD mice (red bars) irrespectively of VEGFKD, as compared to non-diabetic eNOS intact mice (VEGFKD − dox, white bar) (****, p =<0.0001) or VEGFKD + dox mice (gray bar) (*, p < 0.005); in diabetic mice VEGFKD (DM-VEGFKD + dox, blue bar) significantly decreases circulating VEGF-A (**, p = 0.0013), but all diabetic mice have plasma VEGF-A >2-fold higher than non-diabetic mice with intact eNOS (VEGFKD, white/gray bars) (****, p < 0.0001 and ***, p = 0.0006). (B) Urine VEGF-A: podocyte VEGFKD does not alter VEGF-A excretion in eNOS−/−:VEGFKD (red bars) or diabetic mice (blue bars); VEGFKD significantly inhibits VEGF-A excretion in non-diabetic mice (VEGFKD + dox, gray bar) (**, p = 0.0043). (C) Plasma NO: podocyte VEGFKD (+ dox) does not significantly alter plasma NO in any experimental group; plasma NO is lower in eNOS−/−:VEGFKD (red bars) than diabetic (blue bars) (**, p = 0.001 and ***, p = 0.0009) and non-diabetic mice with intact eNOS (white bar) (*, p = 0.047); plasma NO is higher in diabetic (DM-VEGFKD − dox, hatched blue bar) than in non-diabetic mice (VEGFKD − dox, white bar) (*, p = 0.015) and VEGFKD abrogates this change (DM-VEGFKD + dox, blue bar). (D) Urine NO: VEGFKD increases NO excretion in eNOS−/−:VEGFKD + dox mice (*, p = 0.0272, red bar); all diabetic mice (blue bars) have several fold higher NO excretion than non-diabetic mice (white/gray bars), irrespectively of VEGFKD.
As expected, NO plasma level was low in eNOS−/−:VEGFKD mice (Figure 6C, red bars). In mice with intact eNOS, NO plasma level was higher in diabetic (blue bars) than in non-diabetic mice (white bar), but VEGFKD did not significantly decrease plasma NO in any experimental group (Figure 6C). Surprisingly, NO urinary excretion was similar in non-diabetic mice with deficient or intact eNOS, VEGFKD increased NO excretion two-fold in eNOS−/−:VEGFKD mice, whereas NO excretion increased dramatically (>6 fold) in diabetic mice, irrespective of podocyte VEGFKD (Figure 6D). No correlation was detected between VEGF-A and NO plasma levels in any experimental group, nor between VEGF-A or NO and albuminuria or creatinine clearance. These findings suggest that urinary NO excretion is not determined only by eNOS or VEGF-A and that the diabetic milieu elicits higher systemic NO and increases NO excretion in the urine, involving additional factors.
S-nitrosoglutathione (GSNO) is the major source of cellular NO not generated by NOS (Liu et al., 2001). GSNO reductase (GSNOR) deletion or decreased activity results in GSNO accumulation and promotes protein S-nitrosylation (Liu et al., 2001; Guerra et al., 2016; Stomberski et al., 2019). In turn, GSNOR activity is controlled by its S-nitrosylation (Brown-Steinke et al., 2010; Guerra et al., 2016). We determined that kidney GSNOR protein expression is not significantly altered by eNOS−/−, VEGFKD or diabetes (Figure 7A). In contrast, VEGFKD significantly decreased GSNOR S-nitrosylation in eNOS−/−:VEGFKD (+dox) and in DM-VEGFKD (+dox) kidneys, as detected by biotin-shift assay (BST) (Figure 7B), This SNO-GSNOR reduction could decrease GSNOR activity and lead to GSNO accumulation, providing an alternate NO source (and might mitigate glomerular damage). To assess the effect of decreased SNO-GSNOR and reductase activity, we measured thiol excretion in the urine. Cys thiol excretion was similar in non-diabetic mice with deficient (eNOS−/−:VEGFKD − dox) or intact eNOS (VEGFKD − dox), but increased ∼2.5 fold when VEGFKD was induced in eNOS−/−:VEGFKD (+ dox) mice (Figure 7C). In contrast, Cys thiol excretion was >6-fold higher in diabetic mice irrespective of VEGFKD (DM-VEGFKD −dox or + dox), than in non-diabetic mice (eNOS−/−:VEGFKD − dox or VEGFKD − dox) (Figure 7C). Urine GSH thiol excretion represented ∼40% of Cys-thiols in every experimental group, it was increased two-fold in eNOS−/−:VEGFKD (+ dox) mice and ∼5-fold in diabetic DM-VEGFKD mice than in non-diabetic mice (Supplementary Figure S1). The remarkable increase in urine thiol excretion reflects GSNO accumulation mediated by decreased SNO-GSNOR, NO generation, and protein S-nitrosylation.
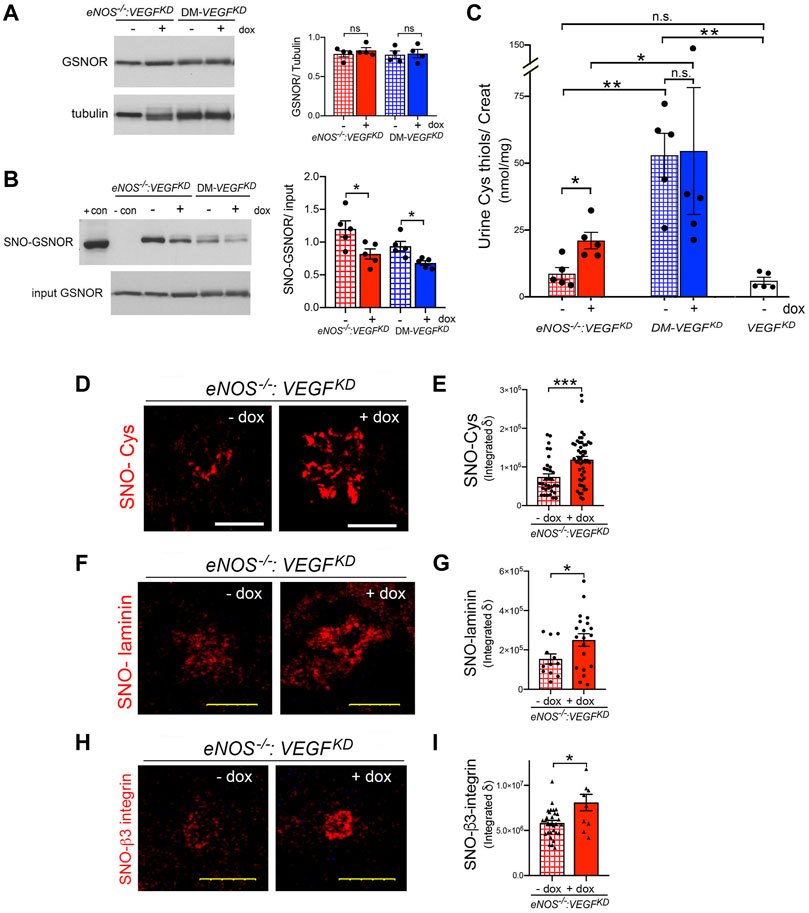
FIGURE 7. Podocyte VEGFKD induces thiol-mediated mechanisms in diabetic and eNOS−/−:VEGFKD mice. (A) WB: Kidney GSNOR expression is not altered by diabetes or VEGFKD, tubulin is shown as loading control. (B) GSNOR S-nitrosylation (SNO-GSNOR) detected by BST: VEGFKD (+dox) decreases SNO-GSNOR in eNOS−/−:VEGFKD and diabetic kidneys; podocyte and kidney lysates are used as SNO positive and negative BST controls, respectively, input shows equal loading for BST. (C) Urine Cys thiol excretion (normalized to creatinine): podocyte VEGFKD increases ∼2.5 fold Cys thiol excretion in eNOS−/−:VEGFKD + dox mice (red bar) (*, p = 0.013); diabetic mice (blue bars), irrespective of VEGFKD, have ∼6-fold higher Cys thiol excretion than uninduced non-diabetic mice with intact eNOS (VEGFKD − dox, white bar) (**, p = 0.004) or eNOS−/−:VEGFKD − dox (hatched red bar) (**, p = 0.004). (D) IHC: podocyte VEGFKD (+ dox) increases S-nitrosylation of glomerular proteins in eNOS−/−:VEGFKD kidneys, SNO-Cys quantification is shown in (E), (****, p = 0.0003). (F) PLA: shows that podocyte VEGFKD (+ dox) increases laminin S-nitrosylation (SNO-laminin) in eNOS−/−:VEGFKD kidneys, SNO-laminin PLA quantification is shown in (G) (*, p = 0.025). (H) PLA: shows β3-integrin S-nitrosylation (SNO-β3-integrin) in eNOS−/−:VEGFKD glomeruli, which is increased by podocyte VEGFKD (+dox); SNO-β3-integrin quantification is shown in (I) (*, p = 0.036).
We examined whether VEGFKD (+dox) alters S-nitrosylation of glomerular proteins in eNOS−/−:VEGFKD mice. Using immunohistochemistry we determined that S-nitrosylated proteins localized to glomeruli are significantly increased in eNOS−/−:VEGFKD kidneys with VEGFKD (+dox) as compared to uninduced eNOS−/−:VEGFKD (− dox) kidneys (Figures 7D,E), indicating that podocyte VEGFKD promotes S-nitrosylation.
Specific S-nitrosylated proteins were detected in situ using proximity link assays (PLA). Glomerular laminin S-nitrosylation (SNO-laminin) was increased significantly in eNOS−/−:VEGFKD mice with VEGFKD (+dox) as compared to uninduced eNOS−/−:VEGFKD (− dox) mice (Figures 7F,G). Consistent with the PLA findings, immunohistochemical S-nitrosylation signals (Cys-SNO) partially co-localized with glomerular laminin were also increased in eNOS−/−:VEGFKD (+ dox) mice (Figure S2).
Next, we examined β3-integrin, a transmembrane protein critically involved in maintaining glomerular filtration barrier integrity (Veron et al., 2012; Hayek et al., 2017), whose activity and signaling are known to be downregulated by S-nitrosylation (Walsh et al., 2007). Using in situ PLA we detected β3-integrin S-nitrosylation in glomeruli (Figure 7H). Quantitation of β3-integrin PLA signals revealed that S-nitrosylated β3-integrin (SNO-β3-integrin) is increased in eNOS−/−:VEGFKD (+dox) kidneys as compared to uninduced eNOS−/−:VEGFKD (−dox) kidneys (Figure 7I). Collectively, our findings suggest that enhanced S-nitrosylation of β3-integrin and laminin may contribute to the development of diffuse glomerulosclerosis in eNOS−/−:VEGFKD (+dox) mice, a phenotype that mimics human advanced DKD with low VEGF.
This study demonstrates that in the setting of bioavailable NO deficiency, caused by diabetic milieu or by eNOS knockout, podocyte VEGF-A knockdown results in diffuse glomerulosclerosis and proteinuria of increasing severity, leading to renal failure in eNOS−/−:VEGFKD mice. We show that podocyte VEGFKD and eNOS−/− induce severe diffuse glomerulosclerosis in the absence of diabetic milieu. Podocyte VEGFKD in diabetic mice prevents diabetes-induced glomerulomegaly but causes diabetic diffuse glomerulosclerosis. Mechanistically, we show that compensatory local NO and thiols generation prevent severe proteinuria and GFR loss in diabetic mice with intact eNOS, and we identify abnormal S-nitrosylation of specific proteins, including GSNOR, laminin, and β3-integrin, as novel molecular pathways potentially involved in advanced diffuse glomerulosclerosis.
High circulating VEGF-A in diabetic mice stimulates NOS leading to NO production, protecting the integrity of the glomerular endothelium and attenuating functional abnormalities of the glomerular filtration barrier (Du et al., 2001; Nakagawa, 2008; Tufro and Veron, 2012). VEGF-A is a survival factor for all glomerular cell types and stimulates endothelial and mesangial cell proliferation (Tsurumi et al., 1997; Feliers et al., 2005; Guan et al., 2006; Lee et al., 2007), and thereby mediates glomerular hypertrophy and angiogenesis in DKD (Farquhar et al., 1959; Stout et al., 1993; Flyvbjerg et al., 2002; Veron et al., 2010; Veron et al., 2011; Tufro and Veron, 2012). Here we show that in diabetic mice podocyte VEGFKD abrogates VEGF-A-mediated glomerular hypertrophy, leading to diffuse glomerulosclerosis with modest albuminuria and normal creatinine clearance. In contrast, eNOS−/−:VEGFKD mice have a decreased ability to increase NO when podocyte VEGFKD is induced, despite similarly elevated circulating VEGF-A, thereby becoming more susceptible than diabetic mice to deleterious effects of local VEGFKD, resulting in mesangiolysis, extensive podocyte foot process effacement, GBM thickening, and a notably severe diffuse glomerulosclerosis phenotype reminiscent of advanced diabetic diffuse glomerulosclerosis (Farquhar et al., 1959; Tsurumi et al., 1997; Nakagawa, 2008). Moreover, VEGFKD and eNOS deficiency have a synergistic effect exacerbating proteinuria (>15 fold either individual genotype) and leading to renal failure, consistent with the more severe morphologic phenotype.
Previous studies demonstrated that glomerular hypertrophy and hyperfiltration occurring in diabetic mice are VEGF-A dependent (Flyvbjerg et al., 2002; Veron et al., 2010; Veron et al., 2011; Tufro and Veron, 2012), and showed that short term podocyte VEGF knockdown results in decreased glomerular size in non-diabetic mice (Veron et al., 2012). Here we extend this observation documenting that long term podocyte VEGF-A knockdown leads to significant decrease in glomerular size in non-diabetic mice and abrogates the glomerulomegaly typically observed in diabetic mice. Diabetic mice with podocyte VEGFKD developed diffuse glomerulosclerosis associated with inflammatory infiltrates and no evidence of endothelial injury or thrombotic microangiopathy (TMA). This phenotype is partially similar to that described in diabetic VEGF-A knockout mice (Sivaskandarajah et al., 2012), suggesting a dose effect of VEGF-A loss-of-function. Most mouse models of DKD show glomerular hypertrophy, mesangial, and extracellular matrix expansion (reviewed in (Brosius et al., 2009; Alpers and Hudkins, 2011). In contrast, few mouse models show advanced diabetic nodular glomerulosclerosis (Zhao et al., 2006; Nakagawa et al., 2007; Hudkins et al., 2010; Alpers and Hudkins, 2011; Veron et al., 2011; Takahashi and Harris, 2014; Aggarwal et al., 2015) or diabetic diffuse glomerulosclerosis (Alpers and Hudkins, 2011; Sivaskandarajah et al., 2012). To our knowledge, the mechanisms leading to such distinct glomerular lesions remain undefined.
eNOS KO mice are susceptible to developing renal failure in the setting of diabetes (Zhao et al., 2006; Nakagawa et al., 2007; Hudkins et al., 2010; Kakoki et al., 2010; Alpers and Hudkins, 2011; Yuen et al., 2012; Takahashi and Harris, 2014), reduced renal mass (Nakayama et al., 2009), and VEGF-A gain-of-function (Veron et al., 2014). We have previously shown that podocyte VEGF-A gain-of-function in eNOS KO mice causes massive proteinuria and renal failure (Veron et al., 2014), not unlike those described here in eNOS−/−:VEGFKD + dox mice, illustrating that a relatively narrow range “normal” VEGF-A expression and signaling at the glomerular filtration barrier are required to maintain GFR and selective permeability, as has been previously observed in other genetic and experimental models (Eremina et al., 2008; Sivaskandarajah et al., 2012; Yuen et al., 2012). Despite the similar functional consequences of podocyte VEGF-A gain-of-function and knockdown in eNOS−/− mice, their morphologic phenotypes are strikingly different and parallel two histologic variants of DKD described in humans: nodular or diffuse glomerulosclerosis, respectively (Farquhar et al., 1959; Stout et al., 1993). These mouse models provide the opportunity to examine the molecular pathogenic mechanisms leading to nodular or diffuse glomerulosclerosis, which are poorly understood in humans.
The Kimmelstiel-Wilson-like nodular glomerulosclerosis reported in eNOS−/− mice with excess glomerular VEGF-A is associated with decreased laminin S-nitrosylation (Veron et al., 2014). Here we demonstrate that the severe diffuse glomerulosclerosis observed in eNOS−/−:VEGFKD (+dox) mice is associated with increased S-nitrosylation of glomerular proteins. As opposed to loss of laminin S-nitrosylation in the setting of excess VEGF-A (Veron et al., 2014), podocyte VEGF-A knockdown increased laminin S-nitrosylation in eNOS−/−:VEGFKD (+dox) mice associates with severe diffuse glomerulosclerosis, suggesting that laminin nitrosylation might prevent the development of glomerular nodules, probably by regulating the secretion or polymerization of 521-laminin heterotrimers (Cheng et al., 1997).
Reversible S-nitrosylation of specific Cys residues, like Tyr phosphorylation, regulates protein-protein interactions and modulates protein function (Stamler et al., 2001; Hess and Stamler, 2012). We have recently shown that diabetic milieu dysregulates S-nitrosylation of other relevant podocyte proteins: myosin9A, RhoA and actin, activating RhoA and disrupting podocyte function in a partially reversible manner (Li et al., 2021). Thus, we examined additional S-nitrosylated proteins expressed in the kidney. GSNOR is a ubiquitous denitrosylase whose function is regulated by S-nitrosylation (Liu et al., 2001; Guerra et al., 2016; Stomberski et al., 2019). De-nitrosylation reduces GSNOR enzymatic activity in mouse cells and tissues (Brown-Steinke et al., 2010) and leads to GSNO accumulation, representing a major source of NO independent of NOS (Liu et al., 2001; Stomberski et al., 2019), although in vitro purified GSNOR or plant extracts decrease reductase activity upon exposure to NO donors (Guerra et al., 2016). GSNOR decreased activity was recently reported in type 2 diabetes patients and was shown to contribute to hepatic insulin resistance in an obesity mouse model (Qian et al., 2018). We determined that SNO-GSNOR was significantly decreased in eNOS−/−:VEGFKD (+dox) and diabetic VEGFKD (+ dox) mice. Consistent with GSNOR de-nitrosylation, we detected several fold increase in urine NO, GSH-, and Cys-thiols excretion in eNOS−/−:VEGFKD (+dox) and DM-VEGFKD (+dox) diabetic mice. The precise cellular origin of urine NO and thiols (ultrafiltrate, glomerular, or tubular cells) remains to be determined. We posit that GSNOR de-nitrosylation underlies the compensatory mechanism providing an alternative NO source in diabetic and eNOS−/−:VEGFKD (+dox) mice (Figure 8). This compensatory mechanism may support normal renal function and relatively low albuminuria in DM-VEGFKD (+dox) mice, but does not prevent the development of diffuse glomerulosclerosis. The SNO-GSNOR mediated alternate source of NO supports renal function in eNOS−/−:VEGFKD (− dox) mice, but it fails to do so when podocyte VEGFKD is induced (+ dox), leading to massive proteinuria and renal failure, as well as severe diffuse glomerulosclerosis, suggesting incomplete compensation or an additional VEGFKD related pathway, including iNOS activation, which we have not evaluated. GSNOR function is influenced by subcellular localization and modulated by VEGF and NOS signaling (Stomberski et al., 2019).
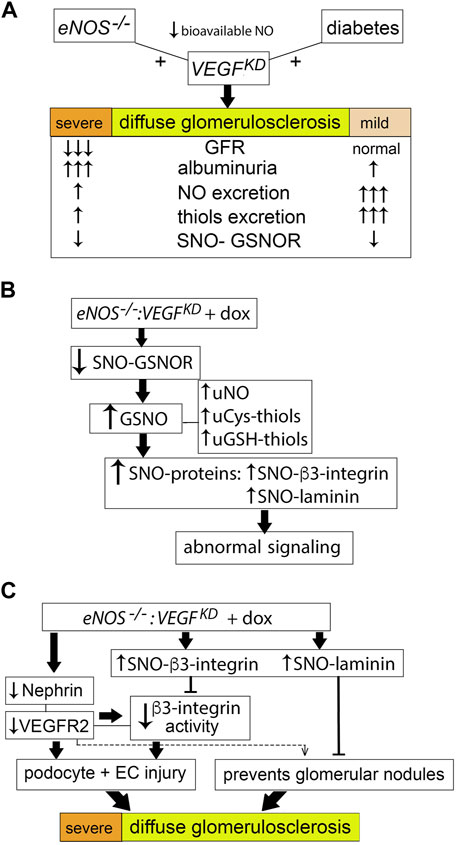
FIGURE 8. Proposed model of podocyte VEGFKD driven diffuse glomerulosclerosis in DM-VEGFKD and eNOS:VEGFKD mice. (A) Strong compensatory NO and thiol generation prevents GFR loss, attenuates proteinuria and diffuse glomerulosclerosis in diabetic VEGFKD mice, while limitation of this compensatory mechanism in eNOS:VEGFKD mice worsens the renal phenotype, leading to renal failure. (B) Reduced GSNOR S-nitrosylation increases GSNO and promotes increased S-nitrosylation of proteins, altering their signaling pathways: (C) decreased nephrin and VEGFR2 signaling and high SNO-β3-integrin inhibit β3-integrin activity leading to podocyte and endothelial cell injury; high SNO-laminin and low VEGFR2 signaling may contribute to the severe diffuse glomerulosclerosis described herein in eNOS:VEGFKD + dox mice.
The novel finding that VEGFKD increases β3-integrin S-nitrosylation in eNOS−/− glomeruli might be linked to diffuse glomerulosclerosis. Laminin-521, the mature GBM laminin, binds αvβ3-integrin through interaction between α5-laminin and β3-integrin, transducing FGF and VEGF signals (Genersch et al., 2003). S-nitrosylation of β3-integrin causes conformational changes that lead to decreased integrin signaling (Walsh et al., 2007). β3-integrin S-nitrosylation in endothelial cells induces loss of integrin activity (Walsh et al., 2007; Robinson et al., 2009). We previously showed that VEGFKD decreases αvβ3-integrin activity in non-diabetic kidneys and cultured podocytes (Veron et al., 2012). Here we find that VEGFKD increases glomerular β3-integrin S-nitrosylation in eNOS−/−:VEGFKD (+dox) mice, likely decreasing β3-integrin signaling. Decreased β3-integrin inside-out activation disrupts nephrin-VEGFR2-β3 integrin signaling in podocytes (Bertuccio et al., 2011; Veron et al., 2012), as well as VEGFR2-β3 integrin signaling in endothelial cells (Robinson et al., 2009), leading to podocyte and endothelial injury, and eventually to diffuse glomerulosclerosis, as observed in eNOS−/−:VEGFKD (+ dox) mice. (Figure 8C). Whether increased S-nitrosylation impairs binding of β3-integrin and laminin-521 remains to be determined. Both decreased (Yoo et al., 2015) and increased (Maile et al., 2014) β3-integrin activity have been implicated as a mechanism of diabetic kidney disease, suggesting a context dependent role. Blockade of αvβ3-integrin activity by a monoclonal antibody improved early markers of diabetic nephropathy in pigs (Maile et al., 2014) probably by interfering with excessive VEGF-A signaling (Robinson et al., 2009; Bertuccio et al., 2011). Thus, we propose that in the setting of VEGFKD and NO deficiency, low β3-integrin activity associated with increased S-nitrosylation of β3-integrin and laminin impair growth and survival signals, resulting in severe glomerular filtration barrier disruption, leading to massive proteinuria and renal failure (Figures 8B,C).
Collectively, these findings suggest that S-nitrosylation contributes to the tight regulation of glomerular homeostasis by modulating several important signaling pathways in DKD models. Our findings support a model whereby laminin S-nitrosylation is instrumental to prevent glomerular nodule development, while GSNOR denitrosylation and increased β3-integrin S-nitrosylation lead to diffuse glomerulosclerosis in the setting of low podocyte VEGF-A.
Further studies are needed to address several limitations of this study: evaluate diabetic eNOS−/−:VEGFKD mice, perform a broad molecular phenotyping, confirm in cultured glomerular cell types the S-nitrosylation abnormalities identified in eNOS−/−:VEGFKD + dox kidneys and assess SNO-protein dysregulation in diabetic mice. Such additional studies will provide insight into how S-nitrosylation modulates several signaling pathways that are critical for glomerular homeostasis in DKD.
In summary, VEGFKD in eNOS−/−:VEGFKD mice causes renal failure, massive proteinuria, and severe diffuse glomerulosclerosis in the absence of diabetes. VEGFKD in diabetic mice with intact eNOS prevents diabetes-induced glomerulomegaly, causes diabetic diffuse glomerulosclerosis, and compensatory NO generation attenuates proteinuria and prevents GFR loss. Together, these models are reminiscent of human DKD phenotypes associated with low VEGF-A expression (Baelde et al., 2007; Lindenmeyer et al., 2007). Mechanistically, VEGFKD in eNOS−/−:VEGFKD mice induces increased glomerular β3-integrin S-nitrosylation, likely disrupting nephrin-VEGFR2-β3-integrin signaling (Genersch et al., 2003; Robinson et al., 2009; Bertuccio et al., 2011; Veron et al., 2012). Our observations highlight a potentially targetable novel regulatory pathway that protects the glomerular filtration barrier up to a point in mouse models that mimic human DKD.
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by Institutional Animal Care and Use Committee at Yale University School of Medicine.
DV, PA, and QL performed experiments, DV, GM, MK, and AT analyzed data, AT designed the experiments and wrote the article. All authors revised and approved the article.
This work was supported by National Institutes of Health grants RO1-DK59333, RO1-DK098824, and RO1-DK109434 (A.T.) and National Institutes of Health grant P30-DK079310 George M. O’Brien Kidney Center at Yale.
Author PA is currently employed by the company Janssen Biopharma.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Heino Velazquez, Ph.D. (George M. O’Brien Kidney Center at Yale) for blood pressure measurements.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2021.788886/full#supplementary-material
Aggarwal, P. K., Veron, D., Thomas, D. B., Siegel, D., Moeckel, G., Kashgarian, M., et al. (2015). Semaphorin3a Promotes Advanced Diabetic Nephropathy. Diabetes 64, 1743–1759. doi:10.2337/db14-0719
Alpers, C. E., and Hudkins, K. L. (2011). Mouse Models of Diabetic Nephropathy. Curr. Opin. Nephrol. Hypertens. 20, 278–284. doi:10.1097/MNH.0b013e3283451901
Baelde, H. J., Eikmans, M., Lappin, D. W., Doran, P. P., Hohenadel, D., Brinkkoetter, P. T., et al. (2007). Reduction of VEGF-A and CTGF Expression in Diabetic Nephropathy Is Associated with Podocyte Loss. Kidney Int. 71, 637–645. doi:10.1038/sj.ki.5002101
Bertuccio, C., Veron, D., Aggarwal, P. K., Holzman, L., and Tufro, A. (2011). Vascular Endothelial Growth Factor Receptor 2 Direct Interaction with Nephrin Links VEGF-A Signals to Actin in Kidney Podocytes. J. Biol. Chem. 286, 39933–39944. doi:10.1074/jbc.M111.241620
Brosius, F. C., Alpers, C. E., Bottinger, E. P., Breyer, M. D., Coffman, T. M., Gurley, S. B., et al. (2009). Mouse Models of Diabetic Nephropathy. J. Am. Soc. Nephrol. 20, 2503–2512. doi:10.1681/ASN.2009070721
Brown-Steinke, K., deRonde, K., Yemen, S., and Palmer, L. A. (2010). Gender Differences in S-Nitrosoglutathione Reductase Activity in the Lung. PLoS ONE 5, e14007. doi:10.1371/journal.pone.0014007
Cheng, Y. S., Champliaud, M. F., Burgeson, R. E., Marinkovich, M. P., and Yurchenco, P. D. (1997). Self-assembly of Laminin Isoforms. J. Biol. Chem. 272, 31525–31532. doi:10.1074/jbc.272.50.31525
Du, X. L., Edelstein, D., Dimmeler, S., Ju, Q., Sui, C., and Brownlee, M. (2001). Hyperglycemia Inhibits Endothelial Nitric Oxide Synthase Activity by Posttranslational Modification at the Akt Site. J. Clin. Invest. 108, 1341–1348. doi:10.1172/JCI11235
Eremina, V., Jefferson, J. A., Kowalewska, J., Hochster, H., Haas, M., Weisstuch, J., et al. (2008). VEGF Inhibition and Renal Thrombotic Microangiopathy. N. Engl. J. Med. 358, 1129–1136. doi:10.1056/NEJMoa0707330
Farquhar, M. G., Hopper, J., and Moon, H. D. (1959). Diabetic Glomerulosclerosis: Electron and Light Microscopic Studies. Am. J. Pathol. 35, 721–753. PMC1934823.
Feliers, D., Chen, X., Akis, N., Choudhury, G. G., Madaio, M., and Kasinath, B. S. (2005). VEGF Regulation of Endothelial Nitric Oxide Synthase in Glomerular Endothelial Cells. Kidney Int. 68, 1648–1659. doi:10.1111/j.1523-1755.2005.00575.x
Flyvbjerg, A., Dagnaes-Hansen, F., De Vriese, A. S., Schrijvers, B. F., Tilton, R. G., and Rasch, R. (2002). Amelioration of Long-Term Renal Changes in Obese Type 2 Diabetic Mice by a Neutralizing Vascular Endothelial Growth Factor Antibody. Diabetes 51, 3090–3094. doi:10.2337/diabetes.51.10.3090
Genersch, E., Ferletta, M., Virtanen, I., Haller, H., and Ekblom, P. (2003). Integrin Alphavbeta3 Binding to Human Alpha5-Laminins Facilitates FGF-2- and VEGF-Induced Proliferation of Human ECV304 Carcinoma Cells. Eur. J. Cel Biol 82, 105–117. doi:10.1078/0171-9335-00297
Gross, M. L., Koch, A., Mühlbauer, B., Adamczak, M., Ziebart, H., Drescher, K., et al. (2006). Renoprotective Effect of a Dopamine D3 Receptor Antagonist in Experimental Type II Diabetes. Lab. Invest. 86, 262–274. doi:10.1038/labinvest.3700383
Guan, F., Villegas, G., Teichman, J., Mundel, P., and Tufro, A. (2006). Autocrine VEGF-A System in Podocytes Regulates Podocin and its Interaction with CD2AP. Am. J. Physiol. Ren. Physiol 291, F422–F428. doi:10.1152/ajprenal.00448.2005
Guerra, D., Ballard, K., Truebridge, I., and Vierling, E. (2016). S-nitrosation of Conserved Cysteines Modulates Activity and Stability of S-Nitrosoglutathione Reductase (GSNOR). Biochemistry 55, 2452–2464. doi:10.1021/acs.biochem.5b01373
Gundersen, H. J., and Osterby, R. (1977). Glomerular Size and Structure in Diabetes Mellitus. II. Late Abnormalities. Diabetologia 13, 43–48. doi:10.1007/BF00996326
Hayek, S. S., Koh, K. H., Grams, M. E., Wei, C., Ko, Y. A., Li, J., et al. (2017). A Tripartite Complex of suPAR, APOL1 Risk Variants and αvβ3 Integrin on Podocytes Mediates Chronic Kidney Disease. Nat. Med. 23, 945–953. doi:10.1038/nm.4362
Hess, D. T., and Stamler, J. S. (2012). Regulation by S-Nitrosylation of Protein post-translational Modification. J. Biol. Chem. 287, 4411–4418. doi:10.1074/jbc.R111.285742
Hohenstein, B., Hausknecht, B., Boehmer, K., Riess, R., Brekken, R. A., and Hugo, C. P. (2006). Local VEGF Activity but Not VEGF Expression Is Tightly Regulated during Diabetic Nephropathy in Man. Kidney Int. 69, 1654–1661. doi:10.1038/sj.ki.5000294
Hudkins, K. L., Pichaiwong, W., Wietecha, T., Kowalewska, J., Banas, M. C., Spencer, M. W., et al. (2010). BTBR Ob/Ob Mutant Mice Model Progressive Diabetic Nephropathy. J. Am. Soc. Nephrol. 21, 1533–1542. doi:10.1681/ASN.2009121290
Kakoki, M., Sullivan, K. A., Backus, C., Hayes, J. M., Oh, S. S., Hua, K., et al. (2010). Lack of Both Bradykinin B1 and B2 Receptors Enhances Nephropathy, Neuropathy, and Bone mineral Loss in Akita Diabetic Mice. Proc. Natl. Acad. Sci. U S A. 107, 10190–10195. doi:10.1073/pnas.1005144107
Lee, S., Chen, T. T., Barber, C. L., Jordan, M. C., Murdock, J., Desai, S., et al. (2007). Autocrine VEGF Signaling Is Required for Vascular Homeostasis. Cell 130, 691–703. doi:10.1016/j.cell.2007.06.054
Li, Q., Veron, D., and Tufro, A. (2021). S-nitrosylation of RhoGAP myosin9A Is Altered in Advanced Diabetic Kidney Disease. Front. Med. (Lausanne) 8, 679518. doi:10.3389/fmed.2021.679518
Lindenmeyer, M. T., Kretzler, M., Boucherot, A., Berra, S., Yasuda, Y., Henger, A., et al. (2007). Interstitial Vascular Rarefaction and Reduced VEGF-A Expression in Human Diabetic Nephropathy. J. Am. Soc. Nephrol. 18, 1765–1776. doi:10.1681/ASN.2006121304
Liu, L., Hausladen, A., Zeng, M., Que, L., Heitman, J., and Stamler, J. S. (2001). A Metabolic Enzyme for S-Nitrosothiol Conserved from Bacteria to Humans. Nature 410, 490–494. doi:10.1038/35068596
Maile, L. A., Busby, W. H., Gollahon, K. A., Flowers, W., Garbacik, N., Garbacik, S., et al. (2014). Blocking Ligand Occupancy of the αVβ3 Integrin Inhibits the Development of Nephropathy in Diabetic Pigs. Endocrinology 155, 4665–4675. doi:10.1210/en.2014-1318
Nakagawa, T., Sato, W., Glushakova, O., Heinig, M., Clarke, T., Campbell-Thompson, M., et al. (2007). Diabetic Endothelial Nitric Oxide Synthase Knockout Mice Develop Advanced Diabetic Nephropathy. J. Am. Soc. Nephrol. 18, 539–550. doi:10.1681/ASN.2006050459
Nakagawa, T. (2008). Uncoupling of VEGF with NO as a Mechanism for Diabetic Nephropathy. Diabetes Res. Clin. Pract. 82 Suppl 1, S67–S69. doi:10.1016/j.diabres.2008.09.030
Nakayama, T., Sato, W., Kosugi, T., Zhang, L., Campbell-Thompson, M., Yoshimura, A., et al. (2009). Endothelial Injury Due to eNOS Deficiency Accelerates the Progression of Chronic Renal Disease in the Mouse. Am. J. Physiol. Ren. Physiol 296, F317–F327. doi:10.1152/ajprenal.90450.2008
Papapetropoulos, A., García-Cardeña, G., Madri, J. A., and Sessa, W. C. (1997). Nitric Oxide Production Contributes to the Angiogenic Properties of Vascular Endothelial Growth Factor in Human Endothelial Cells. J. Clin. Invest. 100, 3131–3139. doi:10.1172/JCI119868
Qian, Q., Zhang, Z., Orwig, A., Chen, S., Ding, W. X., Xu, Y., et al. (2018). S-nitrosoglutathione Reductase Dysfunction Contributes to Obesity-Associated Hepatic Insulin Resistance via Regulating Autophagy. Diabetes 67 (2), 193–207. doi:10.2337/db17-0223
Reidy, K., Kang, H. M., Hostetter, T., and Susztak, K. (2014). Molecular Mechanisms of Diabetic Kidney Disease. J. Clin. Invest. 124, 2333–2340. doi:10.1172/JCI72271
Reidy, K. J., Villegas, G., Teichman, J., Veron, D., Shen, W., Jimenez, J., et al. (2009). Semaphorin3a Regulates Endothelial Cell Number and Podocyte Differentiation during Glomerular Development. Development 136, 3979–3989. doi:10.1242/dev.037267
Robinson, S. D., Reynolds, L. E., Kostourou, V., Reynolds, A. R., da Silva, R. G., Tavora, B., et al. (2009). Alphav Beta3 Integrin Limits the Contribution of Neuropilin-1 to Vascular Endothelial Growth Factor-Induced Angiogenesis. J. Biol. Chem. 284, 33966–33981. doi:10.1074/jbc.M109.030700
Shen, B. Q., Lee, D. Y., and Zioncheck, T. F. (1999). Vascular Endothelial Growth Factor Governs Endothelial Nitric-Oxide Synthase Expression via a KDR/Flk-1 Receptor and a Protein Kinase C Signaling Pathway. J. Biol. Chem. 274, 33057–33063. doi:10.1074/jbc.274.46.33057
Shesely, E. G., Maeda, N., Kim, H. S., Desai, K. M., Krege, J. H., Laubach, V. E., et al. (1996). Elevated Blood Pressures in Mice Lacking Endothelial Nitric Oxide Synthase. Proc. Natl. Acad. Sci. U S A. 93, 13176–13181. doi:10.1073/pnas.93.23.13176
Sivaskandarajah, G. A., Jeansson, M., Maezawa, Y., Eremina, V., Baelde, H. J., and Quaggin, S. E. (2012). Vegfa Protects the Glomerular Microvasculature in Diabetes. Diabetes 61, 2958–2966. doi:10.2337/db11-1655
Söderberg, O., Gullberg, M., Jarvius, M., Ridderstråle, K., Leuchowius, K. J., Jarvius, J., et al. (2006). Direct Observation of Individual Endogenous Protein Complexes In Situ by Proximity Ligation. Nat. Methods 3, 995–1000. doi:10.1038/nmeth947
Stamler, J. S., Lamas, S., and Fang, F. C. (2001). Nitrosylation. The Prototypic Redox-Based Signaling Mechanism. Cell 106, 675–683. doi:10.1016/s0092-8674(01)00495-0
Stomberski, C. T., Hess, D. T., and Stamler, J. S. (2019). Protein S-Nitrosylation: Determinants of Specificity and Enzymatic Regulation of S-Nitrosothiol-Based Signaling. Antioxid. Redox Signal. 30 (10), 1331–1351. doi:10.1089/ars.2017.7403
Stout, L. C., Kumar, S., and Whorton, E. B. (1993). Focal Mesangiolysis and the Pathogenesis of the Kimmelstiel-Wilson Nodule. Hum. Pathol. 24, 77–89. doi:10.1016/0046-8177(93)90066-p
Takahashi, T., and Harris, R. C. (2014). Role of Endothelial Nitric Oxide Synthase in Diabetic Nephropathy: Lessons from Diabetic eNOS Knockout Mice. J. Diabetes Res. 2014, 590541–541. doi:10.1155/2014/590541
Tervaert, T. W., Mooyaart, A. L., Amann, K., Cohen, A. H., Cook, H. T., Drachenberg, C. B., et al. (2010). Pathologic Classification of Diabetic Nephropathy. J. Am. Soc. Nephrol. 21, 556–563. doi:10.1681/ASN.2010010010
Tsurumi, Y., Murohara, T., Krasinski, K., Chen, D., Witzenbichler, B., Kearney, M., et al. (1997). Reciprocal Relation between VEGF and NO in the Regulation of Endothelial Integrity. Nat. Med. 3, 879–886. doi:10.1038/nm0897-879
Tufro, A., and Veron, D. (2012). VEGF and Podocytes in Diabetic Nephropathy. Semin. Nephrol. 32, 385–393. doi:10.1016/j.semnephrol.2012.06.010
Tuttle, K. R., Bakris, G. L., Bilous, R. W., Chiang, J. L., de Boer, I. H., Goldstein-Fuchs, J., et al. (2014). Diabetic Kidney Disease: a Report from an ADA Consensus Conference. Diabetes Care 37, 2864–2883. doi:10.2337/dc14-1296
Véniant, M., Heudes, D., Clozel, J. P., Bruneval, P., and Ménard, J. (1994). Calcium Blockade versus ACE Inhibition in Clipped and Unclipped Kidneys of 2K-1C Rats. Kidney Int. 46, 421–429. doi:10.1038/ki.1994.290
Veron, D., Aggarwal, P. K., Velazquez, H., Kashgarian, M., Moeckel, G., and Tufro, A. (2014). Podocyte-specific VEGF-A Gain of Function Induces Nodular Glomerulosclerosis in eNOS Null Mice. J. Am. Soc. Nephrol. 25, 1814–1824. doi:10.1681/ASN.2013070752
Veron, D., Bertuccio, C. A., Marlier, A., Reidy, K., Garcia, A. M., Jimenez, J., et al. (2011). Podocyte Vascular Endothelial Growth Factor (Vegf₁₆₄) Overexpression Causes Severe Nodular Glomerulosclerosis in a Mouse Model of Type 1 Diabetes. Diabetologia 54, 1227–1241. doi:10.1007/s00125-010-2034-z
Veron, D., Reidy, K. J., Bertuccio, C., Teichman, J., Villegas, G., Jimenez, J., et al. (2010). Overexpression of VEGF-A in Podocytes of Adult Mice Causes Glomerular Disease. Kidney Int. 77, 989–999. doi:10.1038/ki.2010.64
Veron, D., Villegas, G., Aggarwal, P. K., Bertuccio, C., Jimenez, J., Velazquez, H., et al. (2012). Acute Podocyte Vascular Endothelial Growth Factor (VEGF-A) Knockdown Disrupts alphaVbeta3 Integrin Signaling in the Glomerulus. PLoS One 7, e40589. doi:10.1371/journal.pone.0040589
Walsh, G. M., Leane, D., Moran, N., Keyes, T. E., Forster, R. J., Kenny, D., et al. (2007). S-Nitrosylation of Platelet alphaIIbbeta3 as Revealed by Raman Spectroscopy. Biochemistry 46, 6429–6436. doi:10.1021/bi0620712
Yoo, T. H., Pedigo, C. E., Guzman, J., Correa-Medina, M., Wei, C., Villarreal, R., et al. (2015). Sphingomyelinase-like Phosphodiesterase 3b Expression Levels Determine Podocyte Injury Phenotypes in Glomerular Disease. J. Am. Soc. Nephrol. 26, 133–147. doi:10.1681/ASN.2013111213
Yuen, D. A., Stead, B. E., Zhang, Y., White, K. E., Kabir, M. G., Thai, K., et al. (2012). eNOS Deficiency Predisposes Podocytes to Injury in Diabetes. J. Am. Soc. Nephrol. 23, 1810–1823. doi:10.1681/ASN.2011121170
Keywords: diabetic kidney disease, VEGF knockdown, diffuse glomerulosclerosis, S-nitrosylation, β3-integrin, laminin, GSNOR
Citation: Veron D, Aggarwal PK, Li Q, Moeckel G, Kashgarian M and Tufro A (2022) Podocyte VEGF-A Knockdown Induces Diffuse Glomerulosclerosis in Diabetic and in eNOS Knockout Mice. Front. Pharmacol. 12:788886. doi: 10.3389/fphar.2021.788886
Received: 03 October 2021; Accepted: 13 December 2021;
Published: 23 February 2022.
Edited by:
Keizo Kanasaki, Faculty of Medicine Shimane University, JapanReviewed by:
Carlamaria Zoja, Mario Negri Pharmacological Research Institute (IRCCS), ItalyCopyright © 2022 Veron, Aggarwal, Li, Moeckel, Kashgarian and Tufro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alda Tufro, YWxkYS50dWZyb0B5YWxlLmVkdQ==
†Present address: Delma Veron, Facultad de Ciencias de la Salud, Universidad Estatal de Milagro, Milagro, EcuadorPardeep K. Aggarwal, Janssen Biopharma
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.