
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 22 December 2021
Sec. Respiratory Pharmacology
Volume 12 - 2021 | https://doi.org/10.3389/fphar.2021.778649
This article is part of the Research Topic Fibrosis in the Respiratory and Digestive Systems View all 50 articles
 Alexandra Nagy1†Tamas Nagy1†Abigel Margit Kolonics-Farkas1Noemi Eszes1Krisztina Vincze1Eniko Barczi1Adam Domonkos Tarnoki2David Laszlo Tarnoki2György Nagy3,4Emese Kiss5,6Pal Maurovich-Horvat2Aniko Bohacs1†
Alexandra Nagy1†Tamas Nagy1†Abigel Margit Kolonics-Farkas1Noemi Eszes1Krisztina Vincze1Eniko Barczi1Adam Domonkos Tarnoki2David Laszlo Tarnoki2György Nagy3,4Emese Kiss5,6Pal Maurovich-Horvat2Aniko Bohacs1† Veronika Müller1*†
Veronika Müller1*†A subset of interstitial lung diseases (ILDs) with autoimmune traits—including connective tissue disease-associated ILD (CTD-ILD) and interstitial pneumonia with autoimmune features (IPAF)—develops progressive fibrosing (PF)-ILD. The aim of our study was to evaluate the clinical characteristics and predictors of longitudinal lung function (LF) changes in autoimmune PF-ILD patients in a real-world setting. All ILD cases with confirmed or suspected autoimmunity discussed by a multidisciplinary team (MDT) between January 2017 and June 2019 (n = 511) were reviewed, including 63 CTD-ILD and 44 IPAF patients. Detailed medical history, LF test, diffusing capacity of the lung for carbon monoxide (DLCO), 6-min walk test (6MWT), blood gas analysis (BGA), and high-resolution computer tomography (HRCT) were performed. Longitudinal follow-up for functional parameters was at least 2 years. Women were overrepresented (70.1%), and the age of the IPAF group was significantly higher as compared to the CTD-ILD group (p < 0.001). Dyspnea, crackles, and weight loss were significantly more common in the IPAF group as compared to the CTD-ILD group (84.1% vs. 58.7%, p = 0.006; 72.7% vs. 49.2%, p = 0.017; 29.6% vs. 4.8%, p = 0.001). Forced vital capacity (FVC) yearly decline was more pronounced in IPAF (53.1 ± 0.3 vs. 16.7 ± 0.2 ml; p = 0.294), while the majority of patients (IPAF: 68% and CTD-ILD 82%) did not deteriorate. Factors influencing progression included malignancy as a comorbidity, anti-SS-A antibodies, and post-exercise pulse increase at 6MWT. Antifibrotic therapy was administered significantly more often in IPAF as compared to CTD-ILD patients (n = 13, 29.5% vs. n = 5, 7.9%; p = 0.007), and importantly, this treatment reduced lung function decline when compared to non-treated patients. Majority of patients improved or were stable regarding lung function, and autoimmune-associated PF-ILD was more common in patients having IPAF. Functional decline predictors were anti-SS-A antibodies and marked post-exercise pulse increase at 6MWT. Antifibrotic treatments reduced progression in progressive fibrosing CTD-ILD and IPAF, emphasizing the need for guidelines including optimal treatment start and combination therapies in this special patient group.
Interstitial lung diseases (ILDs) are a heterogeneous group of lung disorders, with diffuse parenchymal involvement also associated with a relevant morbidity and mortality. The spectrum of ILD is very diverse and the etiology is often idiopathic; however, a significant proportion of patients present with confirmed or possible autoimmune characteristics (Antoniou et al., 2014; Fischer et al., 2015; Martin et al., 2016). Connective tissue diseases (CTDs) are often associated with ILD. Lung involvement may occur in the initial phase of the systemic autoimmune disorder; however, ILD can manifest even before the diagnosis of CTD (Fischer and Du Bois, 2012). The term “interstitial pneumonia with autoimmune features” (IPAF) describes a type of interstitial pneumonias that are clinically and serologically associated with autoimmune characteristics but do not correspond completely to the diagnostic criteria of CTD (Sambataro et al., 2018).
Continuous monitoring of patients is essential to recognize progression (Fisher et al., 2020). The phenotype of progressive fibrosing (PF)-ILD, regardless of the underlying disease, shows common clinical features of lung function decline (Johannson et al., 2021). Worsening of symptoms—mainly dyspnea and cough—is often associated with progression of fibrosis on high-resolution computer tomography (HRCT); however, the definition for PF-ILD is not unitary (Cottin et al., 2018; Cottin et al., 2019; Brown et al., 2020; Kolb and Flaherty, 2021). PF-ILD results in the deterioration of quality of life and leads to early mortality. Forced vital capacity (FVC) and diffusing capacity of the lung for carbon monoxide (DLCO) decline are important and most frequently accepted markers of progression and are predictive factors of mortality (Brown et al., 2020), (Volkmann et al., 2019; Solomon et al., 2016; George et al., 2020).
A multidisciplinary approach is crucial for proper ILD diagnosis and treatment (Grewal et al., 2019). Considering the wide spectrum of disorders among autoimmune ILDs including different CTD-ILDs and even IPAF, it is essential to outline the best therapeutic possibilities for these patients. In addition to immunosuppressive therapy being extensively used, new antifibrotic agents—nintedanib and pirfenidone—also impact on the disease course; however, data on the interaction between these medications are lacking (Johannson et al., 2021; Wollin et al., 2019; Maher et al., 2020; Gao and Moua, 2020). It is challenging to find the best time for the introduction of certain drugs as well as choosing the optimal treatment course and combination for autoimmune-mediated ILDs (Cottin et al., 2019; Flaherty et al., 2019; George et al., 2020).
Our goal was to assess the clinical course of autoimmune ILDs—regarding the PF-ILD phenotype—and to confirm risk factors for progression and potential beneficial therapies in a real-word setting.
Our study is based on retrospective data analysis of ILD patients. Each case was presented and diagnosed by our multidisciplinary team (MDT) including pulmonologists, rheumatologists, radiologists, and pathologists. The ILD-MDT evaluation of the patients was performed at the Department of Pulmonology Semmelweis University between January 2017 and June 2019 (Richeldi et al., 2019).
The diagnosis of CTD was based on the internationally accepted American College of Rheumatology/European League Against Rheumatism Collaborative Initiative (EULAR-ACR) clinical and serologic criteria by rheumatology specialists working at one of the two rheumatology centers in Central Hungary. CTDs included rheumatoid arthritis (RA), systemic sclerosis (SSc), systemic lupus erythematosus (SLE), vasculitis, idiopathic inflammatory myopathies including polymyositis/dermatomyositis (IIM; PM/DM), and other categories [mixed connective tissue disease (MCTD) and undifferentiated connective tissue disease (UCTD)] (Kay and Upchurch, 2012; Van Den Hoogen et al., 2013; Petri et al., 2012; Aringer, 2019; Yates et al., 2016; Lundberg et al., 2017; Ortega-Hernandez and Shoenfeld, 2012; Mosca et al., 2014). The diagnosis of IPAF consisted of clinical, serological, and morphological domains based on the classification criteria proposed by the European Respiratory Society/American Thoracic Society (ERS/ATS) in 2015 (Fischer et al., 2015). All patients were consulted by rheumatologists to exclude manifestations of CTD at the time of diagnosis or in case of clinical suspicion thereafter. None of the IPAF patients developed CTD during follow-up.
At baseline and every follow-up, physical examination was performed, and a detailed medical history was taken with special emphasis on symptoms (dry/productive cough, sputum, and chest pain), respiratory infections, and comorbidities (Barczi et al., 2020). In our clinical routine, studied autoantibodies were anti-nuclear antibodies (ANA), rheumatoid factor (RF), anti-cyclic citrullinated peptide antibodies (ACPA), anti-RNA polymerase, anti-centromere, anti-proliferating cell nuclear antigen (APCNA), anti-Ku, anti-P-ribosomal, anti-cytoplasmatic, anti-cytoskeleton, anti-chromatin, anti-Smith, anti-myeloperoxidase, anti-proteinase-3, anti-Jo-1, anti-SS-A, anti-SS-B, anti-SCL-70, anti-ribonucleoprotein (RNP), and anti-neutrophil cytoplasmic antibodies (ANCA).
Baseline lung HRCT scans, pulmonary function test (PFT), blood gas analysis (BGA), and 6-min walk test (6MWT) were implemented at the time of the ILD diagnosis. Gender-age-physiology (GAP) score was used for clinical severity prediction in CTD-ILD and IPAF (Ryerson et al., 2014).
Confirmed ILDs were classified into four main groups: ILDs with known etiology including mainly confirmed CTD-ILD and hypersensitive pneumonitis (HP) cases; idiopathic interstitial pneumonia (IIP) including idiopathic pulmonary fibrosis (IPF), idiopathic non-specific interstitial pneumonia (iNSIP), and other IIPs; granulomatous diseases; and other rare forms of ILDs according to current guidelines (Travis et al., 2013), (Raghu et al., 2011). IPAF was considered a separate entity; nevertheless, it was included in the first group. The study population selection is summarized in Figure 1. Patients with autoimmune characteristics were divided into two subgroups: CTD-ILD and IPAF patients.
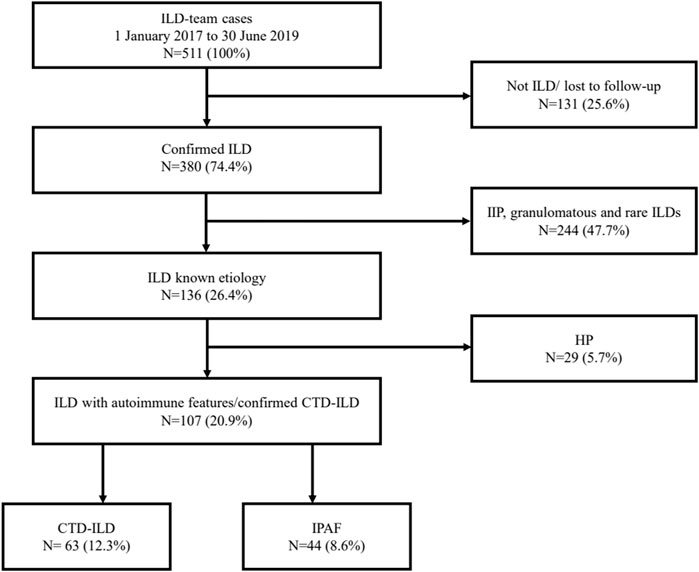
FIGURE 1. Study population. ILD, interstitial lung disease; IIP, idiopathic interstitial pneumonia; HP, hypersensitivity pneumonitis; CTD-ILD, connective tissue disease-associated interstitial lung disease; IPAF, interstitial pneumonia with autoimmune features.
The long-term care included pulmonary and rheumatology controls defined by patients’ disease requirements.
PFT, including the analysis of FVC, forced expiratory volume in 1 s (FEV1), FEV1/FVC, total lung capacity (TLC), was performed according to the standardized protocol at the Department of Pulmonology. Lung diffusion capacity was measured for DLCO using the single-breath CO method, and transfer coefficient of the lung for CO (KLCO) was calculated (PDD-301/s, Piston, Budapest, Hungary). Exercise tolerance was established using the 6MWT. Distance in meters (m), baseline and post-exercise oxygen saturation (SpO2), heart rate, and Borg scale referring to dyspnea were assessed. Arterialized capillary BGA were evaluated at room air temperature (Cobas b 221, Roche, Hungary).
HRCT scan was performed in both inspiration and expiration positions using Philips Ingenuity Core 64 and Philips Brilliance 16 CT scanners. NSIP pattern was divided into cellular and fibrotic subtypes by radiologist experts according to HRCT scans. Radiologic features typically include cellular variant with ground-glass opacities and fine reticular opacities; besides, the fibrotic subtype is characterized predominantly by traction bronchiectasis (Kligerman et al., 2009). In case of usual interstitial pneumonia (UIP), honeycombing with subpleural and basal predominance can be observed. Traction bronchiectasis might be associated with ground-glass opacification. The pattern of probable UIP (pUIP) is characterized by the same abnormalities without honeycombing (Raghu et al., 2011).
Pulmonary follow-up of at least 24 months after ILD diagnosis included measurements of lung function parameters, diffusion capacity, laboratory testing, and BGA controls. At this time point, we recorded the immunosuppressive and/or antifibrotic therapies between the visit intervals. All CTD patients were followed at the respective rheumatology centers.
In our study, PF-ILD was defined as FVC relative yearly decline ≥5% and either deterioration of clinical symptoms or progression of fibrosis on HRCT (Cottin et al., 2018).
Analysis was performed using the GraphPad software (GraphPad Prism 5.0 Software, Inc., La Jolla, CA, United States) and SPSS v25 (IBM Corporation, Armonk, NY, United States). Continuous variables were expressed as mean ± standard deviation. Normality of the data was determined using Kolmogorov–Smirnov test. Differences between groups for continuous data were evaluated in normally distributed data with Student’s t-test; otherwise, Mann–Whitney U-test was used. Chi-squared test and two-tailed Fisher’s exact test were applied for comparing categorical variables. Predictors of progression were analyzed using Cox proportional hazards regression model. All percentage values are expressed for the whole study population or respective subgroups as indicated. A p-value <0.05 was defined as statistically significant.
Patient characteristics are summarized in Table 1. The study population included mainly women. Patients in the IPAF subgroup were significantly older compared to the CTD subgroup. Dyspnea, crackles, and weight loss were significantly more common in the IPAF group as compared to the CTD-ILD group. Subtypes of CTD (n = 63) were, by order of prevalence, SSc (50.8%) RA (20.6%), SLE (9.5%), others (MCTD and UCTD) (9.5%), PM/DM (6.4%), and vasculitis (3.2%). Raynaud’s phenomenon occurred significantly more often in patients with known CTD. LF at baseline is summarized in Table 2. Patients were characterized by mild restrictive functional impairment. There was a slight decrease in TLC and CO diffusion parameters. No differences in LF, 6MWT, or BGA were noted between the two groups.
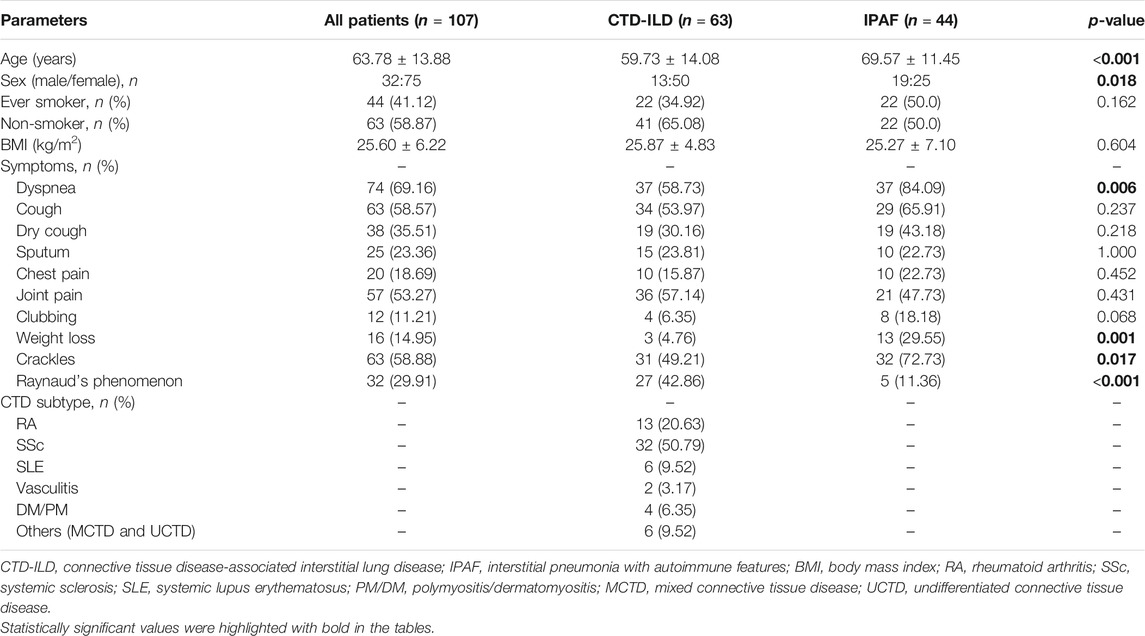
TABLE 1. Patient characteristics.
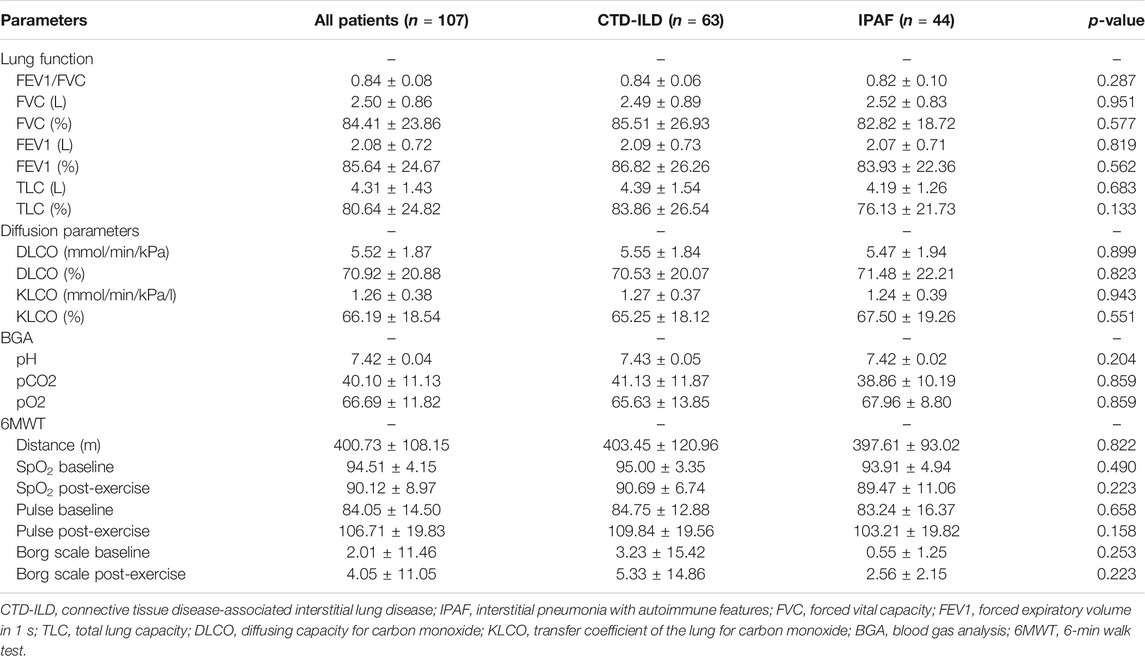
TABLE 2. Functional parameters.
The most common radiological pattern was NSIP; however, significantly more pUIP was noted in IPAF patients. HRCT data are summarized in Table 3. Most frequently confirmed auto-antibodies were ANA, followed by anti-chromatin antibodies and RF, with no differences among the two groups (Table 4).

TABLE 3. HRCT morphological domain.
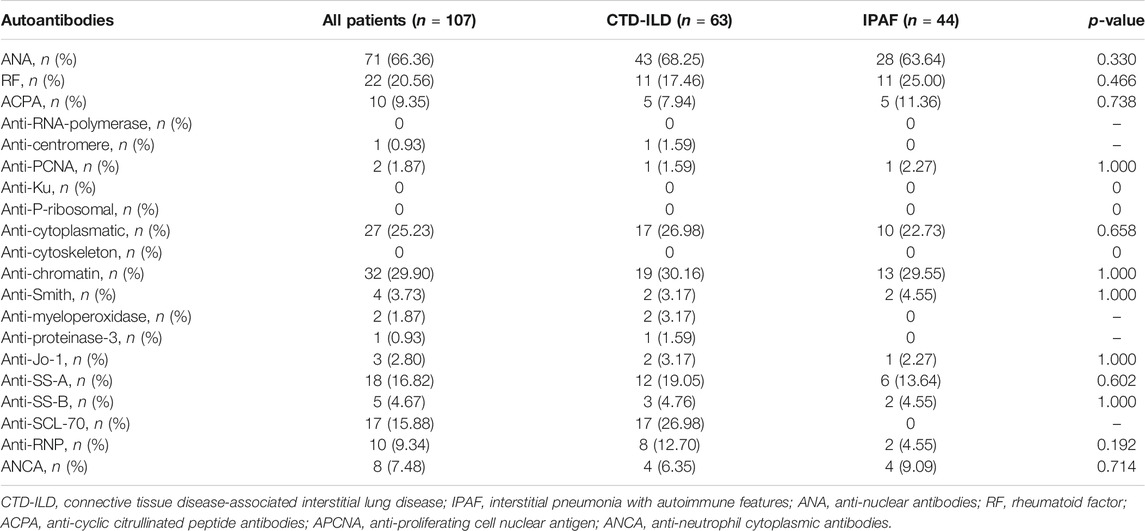
TABLE 4. Autoimmune serology.
Fifty-nine patients had functional data during the 24-month follow-up including 34 CTD-ILD (23.5% males; mean age 58.42 ± 13.01 years) and 25 IPAF (48.0% males; mean age 69.02 ± 12.51 years) patients. Baseline data of CTD-ILD [SSc (55.9%), RA (20.6%), PM/DM (11.8%), SLE (5.9%), and other MCTD and UCTD (5.9%)] and IPAF patients with available functional follow-up did not differ in any parameter from the whole respective group. To estimate mortality, we applied the GAP risk prediction model, which is also validated for non-IPF ILDs (Ryerson et al., 2014). Values were markedly better in the CTD group compared to the IPAF group (1.82 vs. 2.48, p = 0.07).
FVC yearly decline was more dominant in the IPAF group than in the CTD-ILD group (53.1 ± 0.3 ml vs. 16.7 ± 0.2 ml; p = 0.294) (Figure 2A). It is important to note that 68.0% (out of the followed 25 patients) did not deteriorate in the IPAF group as compared to 82.4% (out of followed 34 patients) in the CTD-ILD group (p = 0.200). PF-ILD criteria were met by 14 patients. We also determined the prevalence of PF-ILD in each entity of CTD-ILD: RA (n = 3), SSc (n = 2), other (n = 1), and IPAF (n = 8).
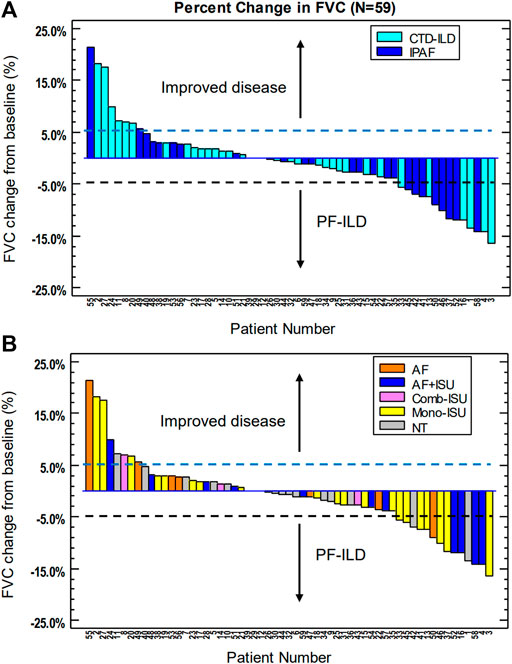
FIGURE 2. Longitudinal follow-up of CTD-ILD and IPAF patients: percent change in FVC. (A) Changes according to treatment; (B) respective patients according to underlying disease. CTD-ILD, connective tissue disease-associated interstitial lung disease; IPAF, interstitial pneumonia with autoimmune features; FVC, forced vital capacity; PF-ILD, [progressive fibrosing ILD; AF, antifibrotic treatment; AF + ISU, antifibrotic treatment with immunosuppressive agent; Comb-ISU, combined immunosuppressive treatment; Mono-ISU, one immunosuppressive agent; NT, no treatment].
Factors influencing rapid progression qualifying as PF-ILD included malignancy as a comorbidity, ANA, anti-SS-A antibodies, and post-exercise pulse increase at the 6MWT (Table 5). Malignancy was diagnosed in seven patients (two males and five females) including CML (1), lung (2), ovarian (1), breast (1), esophageal (1), and laryngeal cancer (1). There was no correlation between HRCT pattern (UIP, pUIP, fibrotic, or cellular NSIP) and progression. Detailed data were not included, as no relationship was present.
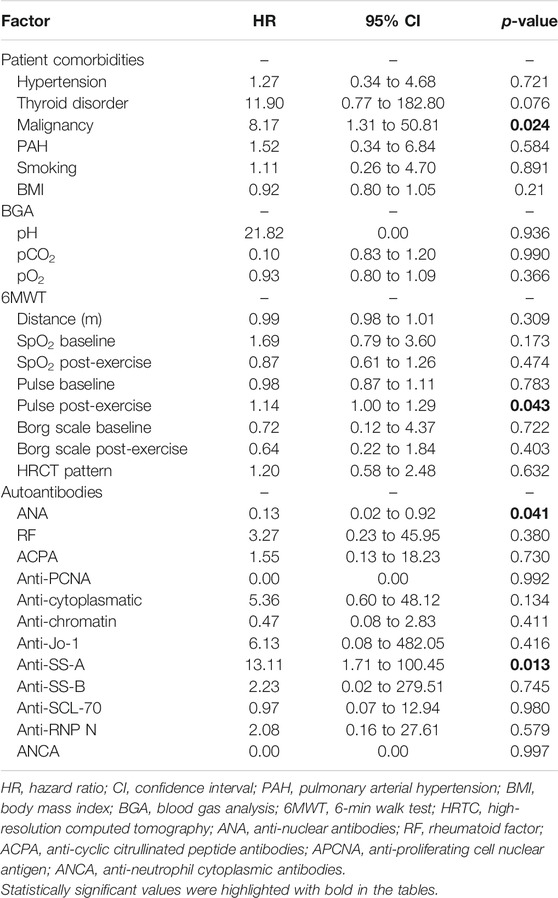
TABLE 5. Factors influencing progression of autoimmune ILDs.
During the follow-up period, 16 patients (CTD-ILD n = 11; IPAF n = 5) did not receive any treatment. Conventional immunosuppressive (ISU) therapies including corticosteroids, rituximab, mycophenolate mofetil, azathioprine, cyclophosphamide, and methotrexate were the initial medical treatment in 36 cases (CTD-ILD n = 22; IPAF n = 14). Mono or combined ISU therapies were appropriate during follow-up period for 25 patients (CTD-ILD n = 18; IPAF n = 7). In some cases, when antifibrotic therapy was given, progressive phenotype was observed. Patients showing progressive phenotype are those whose ISU therapy was supplemented with antifibrotic therapies such as nintedanib and pirfenidone. Antifibrotic drugs were administered significantly more often in IPAF as compared to CTD-ILD (n = 13 vs. n = 5; p = 0.007). The majority of these patients (72.2% on antifibrotic treatment) represented stable lung function or improvement following treatment introduction. Individual functional change according to therapy is summarized in Figure 2B. Antifibrotic treatment (pirfenidone 801 mg tid n = 2; nintedanib 150 mg bid n = 17, including one patient who switched to pirfenidone due to elevated liver enzymes)-related adverse events—all grade 1 and transient—included gastrointestinal symptoms, mainly nausea and vomiting, diarrhea, and heartburn. Most of them were solved by dosage reduction and supportive medications. Elevated liver enzymes were only observed in one patient and was resolved after changing to another antifibrotic drug. Unfortunately, during follow-up, nine patients with mono or combined ISU therapy developed PF-ILD according to our criteria, four (CTD-ILD n = 2; IPAF n = 2) of them had anti-SS-A antibody positivity and five patients (CTD-ILD n = 3; IPAF n = 2) had post-exercise pulse increase.
We presented the first single-center real-life data analyzing the functional progression of autoimmune ILDs. A small proportion of CTD-ILD and IPAF patients deteriorated (13.1% of the whole population) over the observed period, which is similar to other international data (Simpson et al., 2021). Most of the patients were stable, and remarkably, eight patients had even ≥5% FVC improvement due to therapy out of 59 followed.
Our data are the first to show ILD distribution of cases from an Eastern European country. Out of the 511 cases presented to the ILD team, 20.9% were CTD-ILD or IPAF, which is very similar to international data (Oldham et al., 2016; Sambataro et al., 2019). CTD-ILD did mainly include SSc (50.8%) and RA (20.6%) patients, also in line with previously published numbers (Oliveira et al., 2020; Sambataro et al., 2018).
IPAF is mainly considered as a research entity with an autoimmune profile and affected 25 patients in our study. Assessment by rheumatology specialists and serological testing were always performed to confirm or exclude CTD in these cases (Fischer et al., 2015; Sambataro et al., 2018; Raghu et al., 2018). However, there is no international agreement on which serological tests are required at the first encounter with the patient (Jee et al., 2017). The serological pattern in IPAF patients was consistent with the current classification criteria (Fischer et al., 2015; Sambataro et al., 2018).
The most common radiological pattern among IPAF patients was pUIP, which correlates with the data of Oldham et al. (2016); however, it contradicts prospective international data where NSIP was the most frequent pattern (Ahmad et al., 2017; Sambataro et al., 2018). In a retrospective study, UIP and non-UIP IPAF had a similar chance to transform into specific autoimmune diseases; thus, the role of the morphological domain of IPAF is questionable (Sambataro et al., 2020). HRCT evaluation is not homogenous among ILD expert radiologists and might have contributed partially to these differences (Walsh et al., 2016), (Widell and Lidén, 2020). Additionally, IPAF is not a homogenous entity, as it may be very similar to CTD-ILD or in contrast to IPF (Oldham et al., 2016; Ferri et al., 2016).
Treatment resulted in lung function improvement, especially in CTD-ILD. Variation of disease course is well known in SSc, where patients can have a rapid progression, stability of disease, and even improvement. Our data confirmed that most patients’ lung function remained stable over the 2-year period; some of them even improved similarly to the Scleroderma Lung Study (SLS) I and II trials and SENSCIS (Volkmann et al., 2017), (Vonk et al., 2021).
An important new finding and interesting consideration of our study is the identification of new possible prognostic factors for PF-ILD in autoimmune-mediated ILDs including ANA and anti-SS-A antibodies, post-exercise pulse increase at 6MWT, and malignancy. Anti-SS-A antibodies such as Ro52 and Ro60 are often used in autoimmune disease diagnosis. Based on literature data, isolated anti-SS-A/Ro60+ is independently associated with SLE. Detection of anti-SS-A/Ro52+ has a prognostic importance in SSc-associated ILD and diagnostic value in PM/DM (Robbins et al., 2019; Hudson et al., 2012; Dugar et al., 2010; Menéndez et al., 2013). Previous small cohort studies have proven that in anti-synthetase syndrome or inflammatory myopathy, anti-SS-A antibody-positive individuals develop more severe ILD including more extensive pulmonary fibrosis and decreased LF. Additionally, these patients are less responsive to immunosuppressive therapies (La Corte et al., 2006; Váncsa et al., 2009). Literature about the diagnostic utility of separated anti-SS-A antibodies is heterogeneous (Hervier et al., 2009; Langguth et al., 2007; Robbins et al., 2019). According to the official recommendation for IPAF by ATS/ERS, in serological domain, Ro60 and Ro52 antibodies are not separated (Fischer et al., 2015). Therefore, we analyzed mixed anti-SS-A level.
Another predictor of progression was post-exercise pulse increase at the 6MWT. The connection between heart rate and 6MWT has not been studied profoundly before in CTD-ILD and IPAF patients; however an association has been found to be a prognostic marker in IPF (Holland et al., 2013). Although, chronotropic response abnormality cannot be certainly established due to various comorbidities and medication history regarding beta blockers being inaccessible (Sanges et al., 2017). The third variable for confirmed faster progression of PF-ILD in our patients was malignancy. Malignancy as a comorbidity is a serious complication associated with ILDs, especially in those showing progression as published previously in our previous study (Barczi et al., 2020).
Defining progression is a difficult task, as for CTD patients several treatment possibilities are open for their underlying disease. According to recent studies in IPF and CTD-ILD patients, a decrease in DLCO is proposed in the definition of PF-ILD (Khanna et al., 2015; Volkmann et al., 2019; Wong et al., 2020; Cottin et al., 2018). Inclusion criteria for PF-ILD subjects in the INBUILD (Efficacy and Safety of Nintedanib in Patients With PF-ILD) trial included DLCO of at least 30% and less than 80% predicted (Brown et al., 2020; Flaherty et al., 2019). Low baseline DLCO is also a clinically meaningful risk factor for acute exacerbations (Wong et al., 2020). In our study, patients had decreased DLCO; however, we did not find any correlation between progression and DLCO change.
We provided real-world data on the treatment and functional outcome for these special patient groups. Therapy in CTD-ILD changes according to underlying disease, while no therapy guidance for IPAF is available (Sambataro et al., 2018; Gao and Moua, 2020). PF-ILD is much more of a disease phenotype than a diagnosis. Timely initiation of antifibrotic therapy slows the progression of the disease (Johannson et al., 2021). In our study, the ILD team recommended antifibrotic treatment to patients with a rapid progression and to those with IPF characteristics. More patients with IPAF and progression were offered this therapy than CTD-ILD patients showing PF-ILD phenotype, mainly due to the fact that the antifibrotic nintedanib was only approved for PF-ILD based on the data of the INBUILD trial in 2020 (Flaherty et al., 2019; Wells et al., 2020; European Medicines Agency, 2019).
Antifibrotic treatment did stabilize lung function in the majority of our patients. PF-ILD was detected in nine patients (CTD-ILD n = 4; IPAF n = 5) who did not receive antifibrotics including 44.4% with anti-SS-A positivity and 55.5% with post-exercise pulse increase, emphasizing the need for possible extension of antifibrotic treatment. Data on the effectivity of combination therapy using different immunosuppressive treatments with antifibrotics is lacking. In real life, patients under immunosuppressive or immunomodulatory therapy are not excluded from additional antifibrotic therapy. However, in the INBUILD study, restricted therapies were only applied after 6 months of deterioration (Cottin et al., 2021). Similarly, SSc-ILD treatment outcome of SENSCIS secondary analysis showed that mycophenolate mofetil and nintedanib co-treated patients did benefit the most from treatment; however, the study was not powered for combination treatment effectivity (Distler et al., 2019; Highland et al., 2021). After applying the combination of different immunosuppressive treatments with antifibrotics, two-thirds of patients experienced mild adverse events in our cohort. Safety and tolerability profile was consistent with the product label and similar to our previously published data (Barczi et al., 2019). In our patients, 67% experienced an adverse event, similar to the INBUILD trial, where diarrhea was observed in 67%, followed by nausea (29%) (Flaherty et al., 2019). The single grade 3 adverse event of liver enzyme increase needing drug discontinuation was resolved by changing to another antifibrotic agent. Acute exacerbations are serious complications of ILDs (Suzuki et al., 2020; Kolb et al., 2018). Unfortunately, our data were not available to analyze these effects on progression.
In conclusion, the majority of autoimmune-associated ILDs including CTD-ILD and IPAF might be stable or even improve due to proper combination therapy. Patients receiving antifibrotic treatment were less likely to deteriorate and fulfill criteria for PF-ILD. Progression was associated with anti-SS-A antibodies, post-exercise pulse increase at 6MWT, and concomitant malignancies—patients presenting with these parameters should be followed more closely. Antifibrotic treatment was effective in stabilizing functional decline, and the drugs confirmed a safety and tolerability profile consistent with the product label. More data is needed in a real-world setting to identify optimal combination therapies and timing for initiation of antifibrotics in CTD-ILD and IPAF patients. Stable lung function might be a result of the relatively short observation period, and more longitudinal data are awaited.
The main limitation of our study includes the retrospective single-center design and limited number of patients. Further prospective studies need to evaluate this special subgroup of ILD patients to develop guidelines for optimal treatment start and combination therapies.
On the other hand, our data are the first to represent ILD distribution of cases from an Eastern European country. Our study is based on long-term longitudinal follow-up of ILD patients with autoimmune characteristics. Disease population covered the two main rheumatology centers in the region of Central Hungary.
The original contributions presented in the study are included in the article; further inquiries can be directed to the corresponding author.
Ethical review and approval were required for the study on human participants in accordance with the local legislation and institutional requirements. The study protocol of retrospective data analysis was approved by the Ethical Committee of Semmelweis University in accordance with the Declaration of Helsinki.
AN, TN, and VM contributed to the conception and design of the study, organized the database, performed the statistical analysis, and wrote the manuscript. All the included authors were treating physicians or radiologists, also members of the multidisciplinary ILD team at Semmelweis University and contributed with data, drafting and review of the manuscript. All authors contributed to the article and approved the submitted version.
TN is supported by the “Development of scientific workshops of medical, health sciences and pharmaceutical educations” (EFOP‐3.6.3‐VEKOP‐16‐2017‐00009). AK-F and EB were supported by the Hungarian Respiratory Society.
VM, AB, KV, NE received consultation fees from Boehringer Ingelheim during the last 5 years.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Ahmad, K., Barba, T., Gamondes, D., Ginoux, M., Khouatra, C., Spagnolo, P., et al. (2017). Interstitial Pneumonia with Autoimmune Features: Clinical, Radiologic, and Histological Characteristics and Outcome in a Series of 57 Patients. Respir. Med. 123, 56–62. doi:10.1016/j.rmed.2016.10.017
Antoniou, K. M., Margaritopoulos, G. A., Tomassetti, S., Bonella, F., Costabel, U., and Poletti, V. (2014). Interstitial Lung Disease. Eur. Respir. Rev. 23, 40–54. doi:10.1183/09059180.00009113
Aringer, M. (2019). EULAR/ACR Classification Criteria for SLE. Semin. Arthritis Rheum. 49, S14–S17. doi:10.1016/j.semarthrit.2019.09.009
Barczi, E., Nagy, T., Starobinski, L., Kolonics-Farkas, A., Eszes, N., Bohacs, A., et al. (2020). Impact of Interstitial Lung Disease and Simultaneous Lung Cancer on Therapeutic Possibilities and Survival. Thorac. Cancer 11, 1911–1917. doi:10.1111/1759-7714.13481
Barczi, E., Starobinski, L., Kolonics-Farkas, A., Eszes, N., Bohacs, A., Vasakova, M., et al. (2019). Long-Term Effects and Adverse Events of Nintedanib Therapy in Idiopathic Pulmonary Fibrosis Patients with Functionally Advanced Disease. Adv. Ther. 36, 1221–1232. doi:10.1007/s12325-019-00906-9
Brown, K. K., Martinez, F. J., Walsh, S. L. F., Thannickal, V. J., Prasse, A., Schlenker-Herceg, R., et al. (2020). The Natural History of Progressive Fibrosing Interstitial Lung Diseases. Eur. Respir. J. 55. doi:10.1183/13993003.00085-2020
Cottin, V., Hirani, N. A., Hotchkin, D. L., Nambiar, A. M., Ogura, T., Otaola, M., et al. (2018). Presentation, Diagnosis and Clinical Course of the Spectrum of Progressive-Fibrosing Interstitial Lung Diseases. Eur. Respir. Rev. 27. doi:10.1183/16000617.0076-2018
Cottin, V., Wollin, L., Fischer, A., Quaresma, M., Stowasser, S., and Harari, S. (2019). Fibrosing Interstitial Lung Diseases: Knowns and Unknowns. Eur. Respir. Rev. 28, 1–9. doi:10.1183/16000617.0100-2018
Cottin, V., Richeldi, L., Rosas, I., Otaola, M., Song, J. W., Tomassetti, S., et al. (2021). Nintedanib and Immunomodulatory Therapies in Progressive Fibrosing Interstitial Lung Diseases. Respir. Res. 22, 1–9. doi:10.1186/s12931-021-01668-1
Distler, O., Highland, K. B., Gahlemann, M., Azuma, A., Fischer, A., Mayes, M. D., et al. (2019). Nintedanib for Systemic Sclerosis-Associated Interstitial Lung Disease. N. Engl. J. Med. 380, 2518–2528. doi:10.1056/nejmoa1903076
Dugar, M., Cox, S., Limaye, V., Gordon, T. P., and Roberts-Thomson, P. J. (2010). Diagnostic Utility of Anti-ro52 Detection in Systemic Autoimmunity. Postgrad. Med. J. 86, 79–82. doi:10.1136/pgmj.2009.089656
European Medicines Agency (2019). Assessment Report - OFEV. Proced. No. EMEA/H/C/003821/II/0027 31, 16–33. doi:10.4324/9781351201117-4
Ferri, C., Manfredi, A., Sebastiani, M., Colaci, M., Giuggioli, D., Vacchi, C., et al. (2016). Interstitial Pneumonia with Autoimmune Features and Undifferentiated Connective Tissue Disease: Our Interdisciplinary Rheumatology-Pneumology Experience, and Review of the Literature. Autoimmun. Rev. 15, 61–70. doi:10.1016/j.autrev.2015.09.003
Fischer, A., Antoniou, K. M., Brown, K. K., Cadranel, J., Corte, T. J., Du Bois, R. M., et al. (2015). An Official European Respiratory Society/American Thoracic Society Research Statement: Interstitial Pneumonia with Autoimmune Features. Eur. Respir. J. 46, 976–987. doi:10.1183/13993003.00150-2015
Fischer, A., and Du Bois, R. (2012). Interstitial Lung Disease in Connective Tissue Disorders. Lancet 380, 689–698. doi:10.1016/S0140-6736(12)61079-4
Fisher, J. H., Johannson, K. A., Assayag, D., Morisset, J., Boer, K. d., Manganas, H., et al. (2020). Long-term Monitoring of Patients with Fibrotic Interstitial Lung Disease: A Canadian Thoracic Society Position Statement. Can. J. Respir. Crit. Care Sleep Med. 4, 147–155. doi:10.1080/24745332.2020.1796206
Flaherty, K. R., Wells, A. U., Cottin, V., Devaraj, A., Walsh, S. L. F., Inoue, Y., et al. (2019). Nintedanib in Progressive Fibrosing Interstitial Lung Diseases. N. Engl. J. Med. 381, 1718–1727. doi:10.1056/nejmoa1908681
Gao, Y., and Moua, T. (2020). Treatment of the Connective Tissue Disease-Related Interstitial Lung Diseases: A Narrative Review. Mayo Clin. Proc. 95, 554–573. doi:10.1016/j.mayocp.2019.07.007
George, P. M., Spagnolo, P., Kreuter, M., Altinisik, G., Bonifazi, M., Martinez, F. J., et al. (2020). Progressive Fibrosing Interstitial Lung Disease: Clinical Uncertainties, Consensus Recommendations, and Research Priorities. Lancet Respir. Med. 8, 925–934. doi:10.1016/S2213-2600(20)30355-6
Grewal, J. S., Morisset, J., Fisher, J. H., Churg, A. M., Bilawich, A. M., Ellis, J., et al. (2019). Role of a Regional Multidisciplinary Conference in the Diagnosis of Interstitial Lung Disease. Ann. Am. Thorac. Soc. 16, 455–462. doi:10.1513/AnnalsATS.201811-794OC
Hervier, B., Rimbert, M., Colonna, F., Hamidou, M. A., and Audrain, M. (2009). Clinical Significance of Anti-Ro/SSA-52 kDa Antibodies: a Retrospective Monocentric Study. Rheumatology (Oxford) 48, 964–967. doi:10.1093/rheumatology/kep145
Highland, K. B., Distler, O., Kuwana, M., Allanore, Y., Assassi, S., Azuma, A., et al. (2021). Efficacy and Safety of Nintedanib in Patients with Systemic Sclerosis-Associated Interstitial Lung Disease Treated with Mycophenolate: a Subgroup Analysis of the SENSCIS Trial. Lancet Respir. Med. 9, 96–106. doi:10.1016/S2213-2600(20)30330-1
Holland, A. E., Hill, C. J., Glaspole, I., Goh, N., Dowman, L., and McDonald, C. F. (2013). Impaired Chronotropic Response to 6-min Walk Test and Reduced Survival in Interstitial Lung Disease. Respir. Med. 107, 1066–1072. doi:10.1016/j.rmed.2013.04.002
Hudson, M., Pope, J., Mahler, M., Tatibouet, S., Steele, R., Baron, M., et al. (2012). Clinical Significance of Antibodies to Ro52/TRIM21 in Systemic Sclerosis. Arthritis Res. Ther. 14, R50. doi:10.1186/ar3763
Jee, A. S., Adelstein, S., Bleasel, J., Keir, G. J., Nguyen, M., Sahhar, J., et al. (2017). Role of Autoantibodies in the Diagnosis of Connective-Tissue Disease ILD (CTD-ILD) and Interstitial Pneumonia with Autoimmune Features (IPAF). J. Clin. Med. 6, 51. doi:10.3390/jcm6050051
Johannson, K. A., Chaudhuri, N., Adegunsoye, A., and Wolters, P. J. (2021). Series Interstitial Lung Disease 2021 3 Treatment of Fibrotic Interstitial Lung Disease : Current Approaches and Future Directions. Lancet 6736, 1–11. doi:10.1016/S0140-6736(21)01826-2
Kay, J., and Upchurch, K. S. (2012). ACR/EULAR 2010 Rheumatoid Arthritis Classification Criteria. Rheumatology (Oxford) 51 Suppl 6, vi5–9. doi:10.1093/rheumatology/kes279
Khanna, D., Mittoo, S., Aggarwal, R., Proudman, S. M., Dalbeth, N., Matteson, E. L., et al. (2015). Connective Tissue Disease-Associated Interstitial Lung Diseases (CTD-ILD) - Report from OMERACT CTD-ILD Working Group. J. Rheumatol. 42, 2168–2171. doi:10.3899/jrheum.141182
Kligerman, S. J., Groshong, S., Brown, K. K., and Lynch, D. A. (2009). Nonspecific Interstitial Pneumonia: Radiologic, Clinical, and Pathologic Considerations. Radiographics 29, 73–87. doi:10.1148/rg.291085096
Kolb, M., Bondue, B., Pesci, A., Miyazaki, Y., Song, J. W., Bhatt, N. Y., et al. (2018). Acute Exacerbations of Progressive-Fibrosing Interstitial Lung Diseases. Eur. Respir. Rev. 27, 1–8. doi:10.1183/16000617.0071-2018
Kolb, M. R., and Flaherty, K. R. (2021, The Justification for the Progressive Fibrotic Phenotype, 27). 363–367. doi:doi:10.1097/MCP.0000000000000803
La Corte, R., Lo Mo Naco, A., Locaputo, A., Dolzani, F., and Trotta, F. (2006). In Patients with Antisynthetase Syndrome the Occurrence of Anti-ro/SSA Antibodies Causes a More Severe Interstitial Lung Disease. Autoimmunity 39, 249–253. doi:10.1080/08916930600623791
Langguth, D. M., Morris, S., Clifford, L., Wilson, R. J., Neil, J., Hogan, P. G., et al. (2007). Specific Testing for "isolated" Anti-52 kDa SSA/Ro Antibodies during Standard Anti-extractable Nuclear Antigen Testing Is of Limited Clinical Value. J. Clin. Pathol. 60, 670–673. doi:10.1136/jcp.2006.040360
Lundberg, I. E., Tjärnlund, A., Bottai, M., Werth, V. P., Pilkington, C., Visser, M., et al. (2017). 2017 European League against Rheumatism/American College of Rheumatology Classification Criteria for Adult and Juvenile Idiopathic Inflammatory Myopathies and Their Major Subgroups. Ann. Rheum. Dis. 76, 1955–1964. doi:10.1136/annrheumdis-2017-211468
Maher, T. M., Corte, T. J., Fischer, A., Kreuter, M., Lederer, D. J., Molina-Molina, M., et al. (2020). Pirfenidone in Patients with Unclassifiable Progressive Fibrosing Interstitial Lung Disease: a Double-Blind, Randomised, Placebo-Controlled, Phase 2 Trial. Lancet Respir. Med. 8, 147–157. doi:10.1016/S2213-2600(19)30341-8
Martin, M. D., Chung, J. H., and Kanne, J. P. (2016). Idiopathic Pulmonary Fibrosis. J. Thorac. Imaging 31, 127–139. doi:10.1097/RTI.0000000000000204
Menéndez, A., Gómez, J., Escanlar, E., Caminal-Montero, L., and Mozo, L. (2013). Clinical Associations of Anti-SSA/Ro60 and Anti-Ro52/TRIM21 Antibodies: Diagnostic Utility of Their Separate Detection. Autoimmunity 46, 32–39. doi:10.3109/08916934.2012.732131
Mosca, M., Tani, C., Vagnani, S., Carli, L., and Bombardieri, S. (2014). The Diagnosis and Classification of Undifferentiated Connective Tissue Diseases. J. Autoimmun. 48-49, 50–52. doi:10.1016/j.jaut.2014.01.019
Oldham, J. M., Adegunsoye, A., Valenzi, E., Lee, C., Witt, L., Chen, L., et al. (2016). Characterisation of Patients with Interstitial Pneumonia with Autoimmune Features. Eur. Respir. J. 47, 1767–1775. doi:10.1183/13993003.01565-2015
Oliveira, R. P., Ribeiro, R., Melo, L., Grima, B., Oliveira, S., and Alves, J. D. (2020). Connective Tissue Disease-Associated Interstitial Lung Disease. Pulmonology, 6–11. doi:10.1016/j.pulmoe.2020.01.004
Ortega-Hernandez, O. D., and Shoenfeld, Y. (2012). Mixed Connective Tissue Disease: An Overview of Clinical Manifestations, Diagnosis and Treatment. Best Pract. Res. Clin. Rheumatol. 26, 61–72. doi:10.1016/j.berh.2012.01.009
Petri, M., Orbai, A. M., Alarcón, G. S., Gordon, C., Merrill, J. T., Fortin, P. R., et al. (2012). Derivation and Validation of the Systemic Lupus International Collaborating Clinics Classification Criteria for Systemic Lupus Erythematosus. Arthritis Rheum. 64, 2677–2686. doi:10.1002/art.34473
Raghu, G., Collard, H. R., Egan, J. J., Martinez, F. J., Behr, J., Brown, K. K., et al. (2011). An Official ATS/ERS/JRS/ALAT Statement: Idiopathic Pulmonary Fibrosis: Evidence-Based Guidelines for Diagnosis and Management. Am. J. Respir. Crit. Care Med. 183, 788–824. doi:10.1164/rccm.2009-040GL
Raghu, G., Remy-Jardin, M., Myers, J. L., Richeldi, L., Ryerson, C. J., Lederer, D. J., et al. (2018). Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 198, e44–68. doi:10.1164/rccm.201807-1255ST
Richeldi, L., Launders, N., Martinez, F., Walsh, S. L. F., Myers, J., Wang, B., et al. (2019). The Characterisation of Interstitial Lung Disease Multidisciplinary Team Meetings: A Global Study. ERJ Open Res. 5. doi:10.1183/23120541.00209-2018
Robbins, A., Hentzien, M., Toquet, S., Didier, K., Servettaz, A., Pham, B. N., et al. (2019). Diagnostic Utility of Separate Anti-ro60 and Anti-Ro52/TRIM21 Antibody Detection in Autoimmune Diseases. Front. Immunol. 10, 444. doi:10.3389/fimmu.2019.00444
Ryerson, C. J., Vittinghoff, E., Ley, B., Lee, J. S., Mooney, J. J., Jones, K. D., et al. (2014). Predicting Survival across Chronic Interstitial Lung Disease: The ILD-GAP Model. Chest 145, 723–728. doi:10.1378/chest.13-1474
Sambataro, G., Sambataro, D., Torrisi, S. E., Vancheri, A., Colaci, M., Pavone, M., et al. (2019). Clinical, Serological and Radiological Features of a Prospective Cohort of Interstitial Pneumonia with Autoimmune Features (IPAF) Patients. Respir. Med. 150, 154–160. doi:10.1016/j.rmed.2019.03.011
Sambataro, G., Sambataro, D., Torrisi, S. E., Vancheri, A., Pavone, M., Rosso, R., et al. (2018). State of the Art in Interstitial Pneumonia with Autoimmune Features: A Systematic Review on Retrospective Studies and Suggestions for Further Advances. Eur. Respir. Rev. 27. doi:10.1183/16000617.0139-2017
Sambataro, G., Vancheri, A., Torrisi, S. E., Colaci, M., Pavone, M., Libra, A., et al. (2020). The Morphological Domain Does Not Affect the Rate of Progression to Defined Autoimmune Diseases in Patients with Interstitial Pneumonia with Autoimmune Features. Chest 157, 238–242. doi:10.1016/j.chest.2019.08.2175
Sanges, S., Giovannelli, J., Sobanski, V., Morell-Dubois, S., Maillard, H., Lambert, M., et al. (2017). Factors Associated with the 6-minute Walk Distance in Patients with Systemic Sclerosis. Arthritis Res. Ther. 19, 279–289. doi:10.1186/s13075-017-1489-4
Simpson, T., Barratt, S. L., Beirne, P., Chaudhuri, N., Crawshaw, A., Crowley, L. E., et al. (2021). The burden of Progressive Fibrotic Interstitial Lung Disease across the UK. Eur. Respir. J. 58. doi:10.1183/13993003.00221-2021
Solomon, J. J., Chung, J. H., Cosgrove, G. P., Demoruelle, M. K., Fernandez-Perez, E. R., Fischer, A., et al. (2016). Predictors of Mortality in Rheumatoid Arthritis-Associated Interstitial Lung Disease. Eur. Respir. J. 47, 588–596. doi:10.1183/13993003.00357-2015
Suzuki, A., Kondoh, Y., Brown, K. K., Johkoh, T., Kataoka, K., Fukuoka, J., et al. (2020). Acute Exacerbations of Fibrotic Interstitial Lung Diseases. Respirology 25, 525–534. doi:10.1111/resp.13682
Travis, W. D., Costabel, U., Hansell, D. M., King, T. E., Lynch, D. A., Nicholson, A. G., et al. (2013). An Official American Thoracic Society/European Respiratory Society Statement: Update of the International Multidisciplinary Classification of the Idiopathic Interstitial Pneumonias. Am. J. Respir. Crit. Care Med. 188, 733–748. doi:10.1164/rccm.201308-1483ST
Van Den Hoogen, F., Khanna, D., Fransen, J., Johnson, S. R., Baron, M., Tyndall, A., et al. (2013). 2013 Classification Criteria for Systemic Sclerosis: An American College of Rheumatology/European League against Rheumatism Collaborative Initiative. Ann. Rheum. Dis. 72, 1747–1755. doi:10.1002/art.3809810.1136/annrheumdis-2013-204424
Váncsa, A., Csípo, I., Németh, J., Dévényi, K., Gergely, L., and Dankó, K. (2009). Characteristics of Interstitial Lung Disease in SS-A positive/Jo-1 Positive Inflammatory Myopathy Patients. Rheumatol. Int. 29, 989–994. doi:10.1007/s00296-009-0884-9
Volkmann, E. R., Tashkin, D. P., Li, N., Roth, M. D., Khanna, D., Hoffmann-Vold, A. M., et al. (2017). Mycophenolate Mofetil versus Placebo for Systemic Sclerosis-Related Interstitial Lung Disease: An Analysis of Scleroderma Lung Studies I and II. ARTHRITIS Rheumatol. 69, 1451–1460. doi:10.1002/art.40114
Volkmann, E. R., Tashkin, D. P., Sim, M., Li, N., Goldmuntz, E., Keyes-Elstein, L., et al. (2019). Short-term Progression of Interstitial Lung Disease in Systemic Sclerosis Predicts Long-Term Survival in Two Independent Clinical Trial Cohorts. Ann. Rheum. Dis. 78, 122–130. doi:10.1136/annrheumdis-2018-213708
Vonk, M. C., Walker, U. A., Volkmann, E. R., Kreuter, M., Johnson, S. R., and Allanore, Y. (2021). Natural Variability in the Disease Course of SSC-ILD: Implications for Treatment. Eur. Respir. Rev. 30. doi:10.1183/16000617.0340-2020
Walsh, S. L., Calandriello, L., Sverzellati, N., Wells, A. U., and Hansell, D. M. (2016). Interobserver Agreement for the ATS/ERS/JRS/ALAT Criteria for a UIP Pattern on CT. Thorax 71, 45–51. doi:10.1136/thoraxjnl-2015-207252
Wells, A. U., Flaherty, K. R., Brown, K. K., Inoue, Y., Devaraj, A., Richeldi, L., et al. (2020). Nintedanib in Patients with Progressive Fibrosing Interstitial Lung Diseases-Subgroup Analyses by Interstitial Lung Disease Diagnosis in the INBUILD Trial: a Randomised, Double-Blind, Placebo-Controlled, Parallel-Group Trial. Lancet Respir. Med. 8, 453–460. doi:10.1016/S2213-2600(20)30036-9
Widell, J., and Lidén, M. (2020). Interobserver Variability in High-Resolution CT of the Lungs. Eur. J. Radiol. Open 7, 100228. doi:10.1016/j.ejro.2020.100228
Wollin, L., Distler, J. H. W., Redente, E. F., Riches, D. W. H., Stowasser, S., Schlenker-Herceg, R., et al. (2019). Potential of Nintedanib in Treatment of Progressive Fibrosing Interstitial Lung Diseases. Eur. Respir. J. 54. doi:10.1183/13993003.00161-2019
Wong, A. W., Ryerson, C. J., and Guler, S. A. (2020). Progression of Fibrosing Interstitial Lung Disease. Respir. Res. 21, 32–10. doi:10.1186/s12931-020-1296-3
Keywords: autoimmune disease, progressive fibrosing interstitial lung disease (PF-ILD), connective tissue disease (CTD), interstitial pneumonia with autoimmune features (IPAF), treatment, antifibrotics
Citation: Nagy A, Nagy T, Kolonics-Farkas AM, Eszes N, Vincze K, Barczi E, Tarnoki AD, Tarnoki DL, Nagy G, Kiss E, Maurovich-Horvat P, Bohacs A and Müller V (2021) Autoimmune Progressive Fibrosing Interstitial Lung Disease: Predictors of Fast Decline. Front. Pharmacol. 12:778649. doi: 10.3389/fphar.2021.778649
Received: 17 September 2021; Accepted: 28 October 2021;
Published: 22 December 2021.
Edited by:
Jian Gao, Shanghai Children’s Medical Center, ChinaReviewed by:
Qiuhongl Li, Tongji University, ChinaCopyright © 2021 Nagy, Nagy, Kolonics-Farkas, Eszes, Vincze, Barczi, Tarnoki, Tarnoki, Nagy, Kiss, Maurovich-Horvat, Bohacs and Müller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Veronika Müller, bXVsbGVyLnZlcm9uaWthQG1lZC5zZW1tZWx3ZWlzLXVuaXYuaHU=
†These authors have contributed equally to this work and share first and last authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.