
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol., 06 December 2021
Sec. Experimental Pharmacology and Drug Discovery
Volume 12 - 2021 | https://doi.org/10.3389/fphar.2021.773198
 Maria Yu. Zakharova1,2†
Maria Yu. Zakharova1,2† Alexandra A. Kuznetsova3†
Alexandra A. Kuznetsova3† Victoria I. Uvarova4
Victoria I. Uvarova4 Anastasiia D. Fomina4,5
Anastasiia D. Fomina4,5 Liubov I. Kozlovskaya4,7
Liubov I. Kozlovskaya4,7 Elena N. Kaliberda1
Elena N. Kaliberda1 Inna N. Kurbatskaia1
Inna N. Kurbatskaia1 Ivan V. Smirnov1,5
Ivan V. Smirnov1,5 Anatoly A. Bulygin3
Anatoly A. Bulygin3 Vera D. Knorre1
Vera D. Knorre1 Olga S. Fedorova3
Olga S. Fedorova3 Alexandre Varnek6
Alexandre Varnek6 Dmitry I. Osolodkin4,5,7Aydar A. Ishmukhametov4,7
Dmitry I. Osolodkin4,5,7Aydar A. Ishmukhametov4,7 Alexey M. Egorov4,5*
Alexey M. Egorov4,5* Alexander G. Gabibov1,5,8*
Alexander G. Gabibov1,5,8* Nikita A. Kuznetsov3,9*
Nikita A. Kuznetsov3,9*The design of effective target-specific drugs for COVID-19 treatment has become an intriguing challenge for modern science. The SARS-CoV-2 main protease, Mpro, responsible for the processing of SARS-CoV-2 polyproteins and production of individual components of viral replication machinery, is an attractive candidate target for drug discovery. Specific Mpro inhibitors have turned out to be promising anticoronaviral agents. Thus, an effective platform for quantitative screening of Mpro-targeting molecules is urgently needed. Here, we propose a pre–steady-state kinetic analysis of the interaction of Mpro with inhibitors as a basis for such a platform. We examined the kinetic mechanism of peptide substrate binding and cleavage by wild-type Mpro and by its catalytically inactive mutant C145A. The enzyme induces conformational changes of the peptide during the reaction. The inhibition of Mpro by boceprevir, telaprevir, GC-376, PF-00835231, or thimerosal was investigated. Detailed pre–steady-state kinetics of the interaction of the wild-type enzyme with the most potent inhibitor, PF-00835231, revealed a two-step binding mechanism, followed by covalent complex formation. The C145A Mpro mutant interacts with PF-00835231 approximately 100-fold less effectively. Nevertheless, the binding constant of PF-00835231 toward C145A Mpro is still good enough to inhibit the enzyme. Therefore, our results suggest that even noncovalent inhibitor binding due to a fine conformational fit into the active site is sufficient for efficient inhibition. A structure-based virtual screening and a subsequent detailed assessment of inhibition efficacy allowed us to select two compounds as promising noncovalent inhibitor leads of SARS-CoV-2 Mpro.
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) was identified in 2020 as a novel member of the family Coronaviridae (genus Betacoronavirus) (Zhu et al., 2020a). This infectious agent causes coronavirus disease 2019 (COVID-19) and is a big threat to public health worldwide (Dong et al., 2020; Wu et al., 2020). The substantial achievements in the development of COVID-19 vaccines (Calina et al., 2020) as well as the design of neutralizing immunotherapeutics (Shang et al., 2020; Guo et al., 2021; Zhou et al., 2021) are promising milestones in the fight against the pandemic. On the other hand, attainment of stable protection against SARS-CoV-2-induced infection remains the most challenging problem of current life sciences. The development of effective drug discovery strategies and approaches, including the screening of small-molecule antivirals, necessitates in-depth knowledge about molecular and cellular mechanisms of coronavirus infection. This requires thorough research on specific targets of antiviral treatment at the molecular level (V’kovski et al., 2021). Directly acting and SARS-CoV-2–specific antiviral treatments are extremely limited today. A number of small-molecule compounds that may inhibit SARS-CoV-2 replication have been proposed (Zhu et al., 2020b; Khan et al., 2021; Wang et al., 2021). Nevertheless, only one directly acting antiviral nucleoside analog, namely remdesivir, which inhibits the SARS-CoV-2 RNA-dependent RNA polymerase, is currently approved (Pruijssers et al., 2020; Yin et al., 2020). The large 30 kb RNA genome of SARS-CoV-2 contains 13 open reading frames, two of which encode large polyproteins, processed by a 3C-like cysteine protease (main protease, Mpro or 3CLpro) at 11 sites and a papain-like cysteine protease (PLpro) at three sites, resulting in 16 nonstructural proteins, forming the replication complex. Both proteases are essential for the viral life cycle, making them alternate attractive targets for a therapeutic intervention (Cannalire et al., 2020; Rut et al., 2020; Ullrich and Nitsche, 2020; Qiao et al., 2021), and because of the more pronounced role of Mpro in the polyprotein processing, it is considered the primary SARS-CoV-2 enzyme target of directly acting antivirals.
The catalytically active form of Mpro is a homodimer with an extended substrate-binding site and a catalytic Cys145−His41 dyad (Ziebuhr and Siddell, 1999; Gadlage and Denison, 2010; Dai et al., 2020; Zhang et al., 2020; Noske et al., 2021) (Figure 1A). The active site consists of five subpockets: the S0 subpocket is formed mostly by Asn142, Ser144, Cys145, and Leu27; S1 includes Phe140, His163, Glu166, and His172; S2 contains His41, Met49, Arg188, and Asp187; S4 includes Glu166, Leu167, Pro168, Gln189, and Ala191; and S3 is exposed on the outer surface of the active site. Mpro selectively cleaves the −Y−Z−Leu−Gln↓−X sequence, where X is a small amino acid (Ser, Ala, or Gly), Y is a hydrophobic amino acid, and Z is a solvent-exposed amino acid residue (Figure 1B). Such substrate specificity is not shared by any known human protease, implying good potential for high specificity and a limited number of adverse effects of Mpro-targeting antivirals (Zhang et al., 2020).
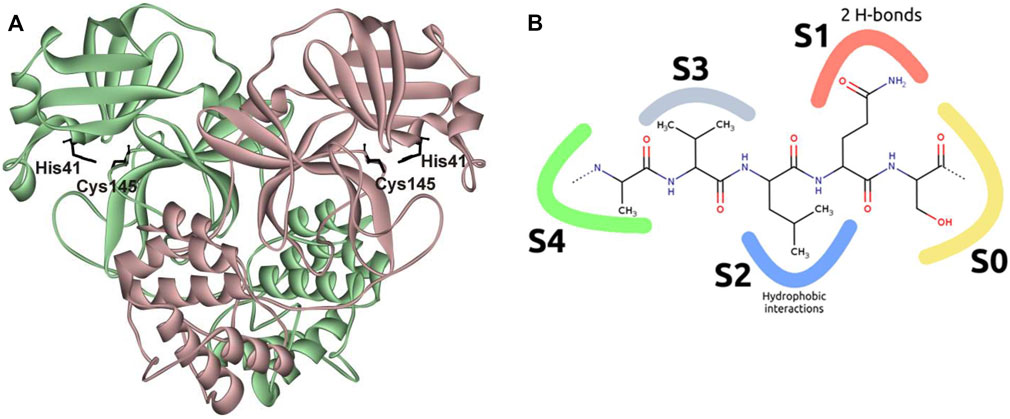
FIGURE 1. Structure of SARS-CoV-2 Mpro. (A) Overview of the structure of an Mpro complex with PF-00835231 (Protein Data Bank [PDB] ID 6XHM (Hoffman et al., 2020); ligand not shown); (B) schematic representation of interactions of the active site with the substrate. The S0 subpocket is formed mostly by Asn142, Ser144, Cys145, and Leu27; S1 includes Phe140, His163, Glu166, and His172; S2 contains His41, Met49, Arg188, and Asp187; S4 includes Glu166, Leu167, Pro168, Gln189, and Ala191; and S3 is exposed on the outer surface of the active site.
Numerous small-molecule screening and drug repurposing programs involve Mpro as the target enzyme (Roe et al., 2021; Vandyck and Deval, 2021; Yang and Yang, 2021). The most prominent inhibitors appear to bear a reactive warhead that can form a covalent bond with Mpro residue Cys145 (several examples are given in Table 1 and Figure 2). Boceprevir and telaprevir, which are approved antiviral drugs targeting the hepatitis C NS3 protease, have emerged as SARS-CoV-2 Mpro inhibitors in numerous drug repurposing campaigns (Lang, 2007; Rotella, 2013). GC-376 has been specifically designed to target feline infectious peritonitis virus (FIPV) Mpro and has a potent antiviral activity against multiple coronaviruses, including MERS-CoV, FIPV, and subsequently SARS-CoV-2 (Kim et al., 2012; Kim et al., 2016; Pedersen et al., 2018; Vuong et al., 2021). One of the most promising compounds, PF-00835231, was initially designed in response to the previous coronavirus epidemic in 2003 as an inhibitor of SARS-CoV Mpro (Hoffman et al., 2020). Recent studies on this compound (Boras et al., 2021; de Vries et al., 2021) confirmed both the antiviral activity against SARS-CoV-2 and Mpro inhibition due to high conservation of the PF-00835231–binding site in Mpro between SARS-CoV and SARS-CoV-2. Thimerosal is an organometallic compound that possesses antibacterial properties due to its capacity to bind thiol groups in proteins, e.g., the catalytic cysteine of Mpro, and came to the fore in an early drug repurposing screen (Geier et al., 2007; Coelho et al., 2020). Although there have been intensive efforts to develop Mpro inhibitors specific for SARS-CoV-2 (Jin et al., 2020a; Jin et al., 2020b; Hoffman et al., 2020; Ma et al., 2020; Yoshino et al., 2020), only PF-07304814 (a prodrug of PF-00835231) and its orally bioavailable analog PF-07321332 have reached clinical trials (Boras et al., 2021; Owen et al., 2021).
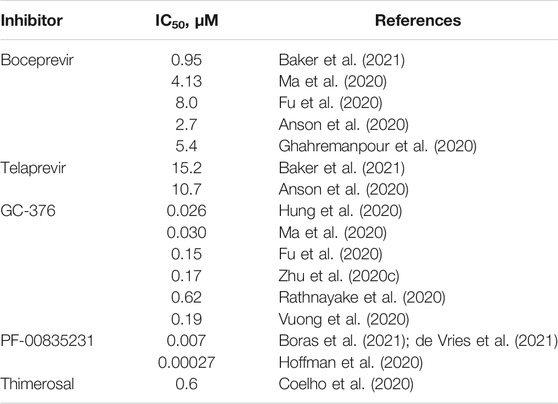
TABLE 1. Mpro 50% inhibitory concentration (IC50) for selected inhibitors.
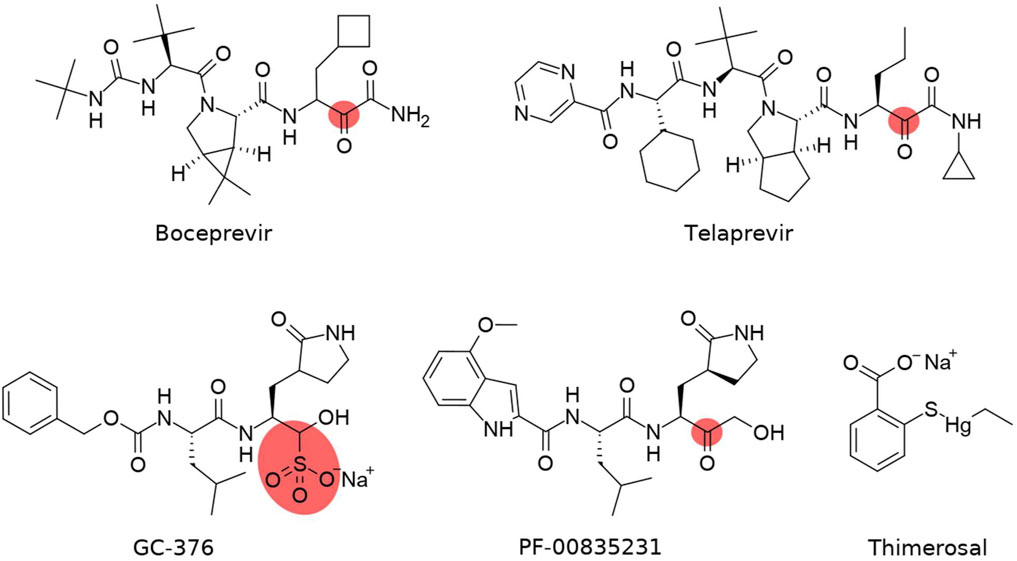
FIGURE 2. Structures of covalent inhibitors of SARS-CoV-2 Mpro. The covalent-binding atom is highlighted. The sulphonate group in GC-376 is cleaved upon Cys145 binding.
According to reported data, the development of specific inhibitors with good binding parameters toward the SARS-CoV-2 main protease looks like a promising strategy against the COVID-19 pandemic. Thus, an effective screening and binding characterization pipeline for such compounds is in high demand (Zaidman et al., 2020). In the present work, we propose a screening platform for this purpose.
Here, we for the first time elucidated the key steps in the mechanism behind the enzyme–substrate and enzyme–inhibitor interactions that ensure specific binding and catalytic transformation. We employed a pre–steady-state kinetic approach. The stopped-flow kinetic analysis of sequential stages of model peptide binding and cleavage by Mpro allowed us to determine the rates of formation of the enzyme–substrate catalytic complex and peptide cleavage. It was found that the interaction of Mpro with PF-00835231 proceeds through two-step binding-complex formation with a subsequent chemical step of covalent bond formation. The strong reversible binding of PF-00835231 in the active site of C145A Mpro clearly indicates that potential inhibitors should have specific structural characteristics to finely fit into the pocket of the enzyme’s active site even without covalent bond formation. Next, structure-based virtual screening of small-molecule noncovalent inhibitors of Mpro was performed. Inhibition efficacy of these compounds was analyzed in the thermal shift assay and steady-state and pre–steady-state kinetic approaches, which enabled us to identify two new noncovalent inhibitors of SARS-CoV-2 Mpro.
A codon-optimized gene coding for full-length SARS-CoV-2 Mpro fused with the 6×His tag at the C terminus and with GST protein at the N terminus in plasmid vector pGEX6p (Zhang et al., 2020) was kindly provided by Prof. Rolf Hilgenfeld. The Mpro gene was flanked by sequences of two protease sites for subsequent excision of native full-length Mpro: a site recognized by Mpro for auto-excision (at the N terminus of the Mpro sequence) and a PreScission™ Pro site immediately before the 6×His tag for its removal, as described in (Zhang et al., 2020). The gene of the C145A Mpro mutant was generated by PCR-mediated site-directed mutagenesis using two overlapping primers and the Mpro pGEX6p vector as a template.
The full-length Mpro protein was overproduced in E. coli BL21 (DE3) and purified as described (Zhang et al., 2020) with minor modifications. Namely, the GST-Mpro fusion protein was subjected to self-processing during E. coli expression to prepare Mpro with the intact N terminus. Mpro fusion with the GST protein was employed to improve the Mpro yield and solubility. Next, Mpro-His was purified by IMAC chromatography on TALON (Clontech) and treated with PreSсission™ Pro (Mpro/PreSсission ratio 100:1) for 48 h at 4 °C for 6×His tag removal and obtaining Mpro with the intact C terminus. Then, a mixture of Mpro with PreSсission™ Pro (which contains a GST tag and His tag) was loaded on a GST-Sepharose (Amersham Biosciences) column and a TALON (Clontech) IMAC column, connected in tandem. Pure Mpro without tags was obtained in a flow-throw fraction. Western blot analysis with anti 6×His antibodies revealed that the 6×His tag was completely removed. Mpro was concentrated up to 10 mg/mL in 50 mM Tris (pH 7.5) and stored at −80°C. C145A Mpro mutant protein was obtained in the same way with an additional step of intact N-terminus generation, because the non-active C145A Mpro form cannot process itself during E.coli expression. For this purpose, IMAC-purified GST-C145A MproHis protein was treated by wild type Mpro (the GST-C145A Mpro-His/Mpro-ratio was 100:1), then the IMAC step was repeated to remove free Mpro. Then C145A MproHis protein was subjected for PreSсission™ Pro cleavage and further procedures as described above.
The kinetic assays were implemented using the FRET substrate (FRET-S), Dabcyl-KTSAVLQ↓SGFRKM-E(Edans)-NH2 (BPS Bioscience, United States), and standard covalent inhibitors GC-376, PF-00835231, boceprevir, telaprevir (Selleckchem, United States), and thimerosal (Serva). FRET-S contains a main-protease cleavage site (indicated by the arrow in the sequence above) and was utilized as the substrate in the FRET-based cleavage assay. Stock solutions of the inhibitors were prepared in DMSO (final concentration 5.0 mM).
Virtual screening was performed in July 2020 via the blind docking approach. The apo-structure of Mpro at room temperature [PDB ID 6WQF (Kneller et al., 2020)] was selected as the one representing the most physiologically relevant conditions. The protein structure was optimized in Chimera (Pettersen et al., 2004) using the Dock Prep tool: solvent molecules were deleted, while hydrogens (taking into account hydrogen bonds) and AMBER ff14SB charges (Maier et al., 2015) were added. Docking was performed in DOCK 6.9 (Allen et al., 2015). The grid box was generated to enclose the orthosteric binding site of chain A.
Docking-based virtual screening was performed on molecules previously selected from ZINC15 (Sterling and Irwin, 2015) as potential novel anticoronavirus compounds (hitlist ZINCVS/novel.-smi_id_frq) using the Generative Topographic Mapping approach (Horvath et al., 2020). Three-dimensional coordinates for the molecules were downloaded in MOL2 format from the ZINC15 website via the “Search Many” feature and used without further processing. Docked compounds were ranked by the values of grid_score for the best scored pose, and 22 of those with grid_score < −50 were designated as primary hits. They were grouped according to the molecule core and assessed for commercial availability, thereby leading to three cores available from a local supplier, Alinda (http://www.alinda.ru). The hitlist was then expanded to include compounds from the same classes with different substituents and grid_score < −45, resulting in an experimental assessment list of 10 compounds (Supplementary Table S1). Purchased compounds were used in experiments without further purification. Stock DMSO solutions were prepared with compound concentration of 5 mM.
Binding of inhibitors to SARS-CoV-2 Mpro was monitored by the TSA on a QIAGEN Rotor Gene Q Real Time PCR System. An Mpro solution (5 μM) was mixed with 50 μM inhibitor (or 1% DMSO), a dye (ProteOrange Protein Gel Stain; Lumiprobe, Russia; 5X in the final volume) in the buffer solution (20 mM HEPES, pH 6.5, 120 mM NaCl, 0.4 mM EDTA, 4 mM DTT, and 20% of glycerol) to attain the final volume of 25 μL. After 30 min incubation at 30 °C, each sample was heated to 95 °C with 0.05 °C/s increment, and fluorescence was monitored in the green channel (λex/λem = 470/510 nm). Tm was calculated as the maximum of the first derivative of the fluorescence signal in Origin 2017 software (OriginLab).
For these assays, 160 nM Mpro in reaction buffer (20 mM Tris-HCl pH 7.3, 100 mM NaCl, 1.0 mM EDTA, and 1.0 mM DTT) was incubated with or without a tested compound at various concentrations for 30 min at 30 °C. The reaction was initiated by the addition of FRET-S (0–80 μM) in reaction buffer. Substrate is cleaved by Mpro generating a product containing a free Edans group. The dequenching of fluorescence by the cleavage of the substrate catalyzed by Mpro was monitored at 460 nm with excitation at 360 nm on a Thermo Scientific Varioscan plate fluorimeter. The concentration of a fluorescent product was determined according to the calibration curve of free Edans fluorescence covering a concentration range of 0.1–20.0 μM. Initial rates of FRET-S cleavage by Mpro at each inhibitor concentration were computed from reaction kinetic curves of Mpro activity at several substrate concentrations. Kinetic constants (Vmax and KM) were derived by fitting the data to the Michaelis–Menten equation, V = Vmax × [S]/(KM + [S]). After that, kcat was calculated according to the equation kcat = Vmax/[E].
For the preliminary screening of noncovalent Mpro inhibitors, 160 nM Mpro was incubated with 80 μM inhibitor (or 1.6% DMSO) for 30 min at 30 °C in reaction buffer, and then 16 μM FRET-S was added to initiate the reaction (final volume 20 μL). The fluorescence signal of the reaction was monitored for 2 h at λex/λem = 355/460 nm (Thermo Scientific Fluoroskan FLash fluorimeter). The initial rate was calculated by linear regression for the first 10 min of the kinetic progress curves. Residual activities were computed by dividing the initial velocity in the presence of an inhibitor by the initial rate in its absence (DMSO control).
Stopped-flow measurements with fluorescence detection were carried out using a SX.20 stopped-flow spectrometer (Applied Photophysics Ltd., United Kingdom) equipped with a 150-W Xe arc lamp and an optical cell with 2 mm path length. The dead time of the instrument is 1.0 ms. For the analysis of enzyme–substrate interactions, the FRET-S substrate modified with the dye–quencher pair Edans/Dabcyl was utilized. The fluorescence of Edans was excited at λex = 340 nm and monitored at λem > 435 nm as transmitted by filter GG-435 (Schott, Mainz, Germany). The binding of Mpro to PF-00835231 was monitored by means of changes in intrinsic fluorescence intensity of the inhibitor. The excitation wavelength was 300 nm, and the emission was monitored using long-pass wavelength filters at λem > 370 nm (Corion filter LG-370).
The enzyme was placed in one of the instrument’s syringes and rapidly mixed in the reaction chamber with the substrate, inhibitor, or a substrate/inhibitor mixture from another syringe. The concentration of FRET-S in all the experiments was 2.5 μM, while concentrations of Mpro or its C145A mutant were varied from 0.1 to 3.0 μM. The reported concentrations of reactants are those in the reaction chamber after the mixing. All experiments were conducted at 25 °C in the reaction buffer.
Kinetic simulation of the time course of appearance and disappearance of various reaction intermediates was done by solving a system of differential equations in the DynaFit software (BioKin, Pullman, WA) (Kuzmic, 1996) as described before (Zakharova et al., 2009; Zakharova et al., 2017). The fast kinetic analysis combined with fluorimetry detection of conformational changes is a powerful method that may provide detailed information about mechanisms of enzyme–substrate interaction (Kuznetsov and Fedorova, 2016; Kuznetsov et al., 2017; Kladova et al., 2019; Kuznetsova et al., 2020; Kuznetsov and Fedorova, 2020). This approach is based on fluorescence intensity variation in the course of the reaction owing to sequential formation and subsequent transformation of the enzyme–substrate complex. The stopped-flow fluorescence traces were directly fitted to the fluorescence intensity at any reaction time point as the sum of background fluorescence and fluorescence intensity values of each intermediate complex that contribute to the signal.
The software performs numerical integration of a system of ordinary differential equations with subsequent nonlinear least-squares regression analysis. In the evaluated mechanisms, except for the first bimolecular step, all other reactions are first-order. In the fits, we optimized all relevant rate constants for the forward and reverse reactions as well as specific molar response factors for all intermediate complexes.
During the data processing, the kinetic information was obtained from the temporal behavior of the fluorescence intensity, not from the amplitudes of the specific signal contributions. The response factors for different states resulting from the fits were not used for determining equilibrium constants but rather provided additional information on fluorescence intensity variations in different states of the complex.
To thoroughly characterize the kinetic mechanism underlying catalytic cleavage of a peptide, pre–steady-state kinetic assays based on the Förster resonance energy transfer (FRET) effect were performed. The Dabcyl-KTSAVLQSGFRKM-E(Edans)-NH2 peptide armed with a dye–quencher pair was used for FRET measurements (Zhu et al., 2020c; Hoffman et al., 2020). FRET analysis could reveal changes in the distance between the dye and quencher in the processes of peptide penetration into the active site and formation of specific contacts between the side chains of the substrate and the respective binding cavities—that subsequently result in the catalytic state, hydrolysis of the peptide bond, and the release the products.
The C145A substitution led to complete elimination of the catalytic activity of the protease owing to a loss of the catalytic thiol group. The interaction of C145A Mpro with the substrate can lead only to its binding and formation of a preincision complex. Indeed, as shown in Figure 3A, the FRET signal during the interaction of C145A Mpro with the substrate increased up to time point of 5 s. It is possible that the increase in the FRET signal reflects increased distance between the fluorogenic Edans residue and quenching Dabcyl residue owing to peptide stretching in the active site of the protease. An analysis of the kinetic curves suggested that the minimal kinetic mechanism of the interaction between the catalytically inactive mutant and FRET substrate involved one-step equilibrium binding (Scheme 1). The rate constants for the forward and reverse reactions are given in Table 2.
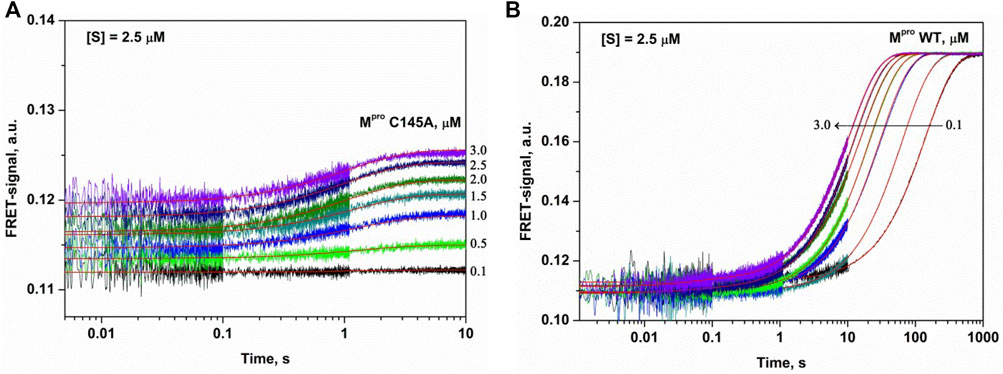
FIGURE 3. Experimental and theoretical (smooth curves) kinetic curves for the FRET signal changes during the interaction of C145A Mpro (A) or WT Mpro (B) with FRET-S. The FRET-S concentration was 2.5 µM, and the enzyme concentration is indicated in the panel.
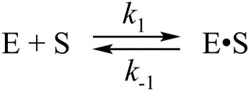
SCHEME 1. The kinetic mechanism of the interaction between C145A Mpro and FRET-S. E: C145A Mpro, S: substrate, E•S: enzyme–substrate complex.

TABLE 2. Rate constants of the interaction of WT Mpro and C145A Mpro with the substrate.
The interaction of WT Mpro with the substrate was slower and proceeded up to 100 s but caused a high amplitude increase in the FRET signal (Figure 3B). Such a growth of the FRET signal most likely reflects a release of the incised peptide products from the complex with the enzyme. Taking into account that the catalytically inactive mutant form revealed one-step binding mechanism, we assumed two-step mechanism of product formation when WT protease interacts with the FRET substrate. Indeed, the kinetic curves were satisfactorily described by Scheme 2, containing one equilibrium stage of substrate binding and one irreversible step of hydrolysis and release of the reaction products (Table 2).
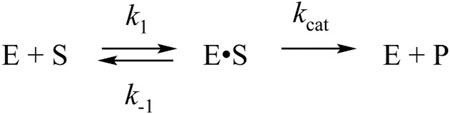
SCHEME 2. The kinetic mechanism of the interaction between WT Mpro and FRET-S. E: WT Mpro, S: substrate, E•S: enzyme-substrate complex, P: product.
It should be noted that the rate constants of the substrate binding were not affected by the C145A substitution. Nevertheless, the total binding constant was approximately fivefold higher in the case of the C145A variant owing to a decrease in k−1. These findings suggested that the Cys145 residue influenced the stability of the enzyme–substrate complex but did not affect the rate of peptide binding.
To verify the kinetic scheme and the rate constants calculated by the global fitting procedure, we determined steady-state reaction parameters: Michaelis constant KM and catalytic reaction rate constant kcat (Figure 4). The Michaelis constant KM calculated from the elementary kinetic constants via the formula KM = (kcat + k–1)/k1 (4.6 μM, Table 2) was three- to sixfold lower than the value obtained by the steady-state analysis (28 μM, Figure 4) or reported earlier [14 μM (Hoffman et al., 2020)]. Catalytic constants kcat were similar between the stopped-flow and steady-state analyses, suggesting that the fast kinetic pre–steady-state approach allowed us to determine relevant characteristics of the enzymatic reaction. Fitting of the steady-state data by the Hill equation (n = 2, Supplementary Figure S1) also provides similar constant K50 of 50% enzyme saturation (15.7 μM). The determined Hill coefficient indicates positive cooperativity and is in good agreement with the reported data (Lee et al., 2020; Vuong et al., 2020). However, complication of the pre–steady-state kinetic mechanism (Scheme 1) up to the two-substrate binding model does not provide a better fit of the experimental data, supporting independent action of each active site of the enzyme in the pre–steady-state conditions.
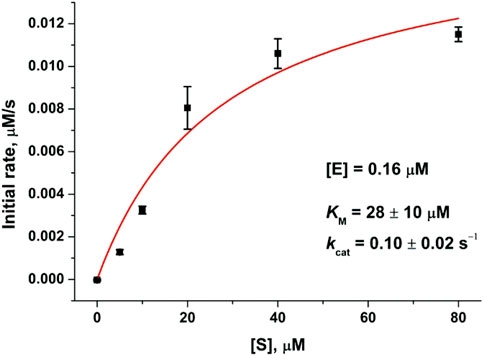
FIGURE 4. Dependence of the initial rate of substrate cleavage by SARS-CoV-2 Mpro. Each data point is an average from at least three independent experiments, the values are presented as the mean ± SE.
The analysis of the PF-00835231 binding kinetics in the course of its interaction with WT or C145A Mpro was performed by the stopped-flow technique with detection of intrinsic florescence intensity of PF-00835231. The association of C145A Mpro with PF-00835231 led to a two-phase increase in the fluorescence intensity up to time point 2 s (Figure 5A). The kinetic curves were satisfactorily described by Scheme 3, which contains two equilibrium stages (Table 3). It is likely that after the formation of the initial complex, there is an additional step of formation of specific interactions between inhibitor moieties and enzyme cavities resulting in full insertion of the inhibitor molecule into the active site of the protease (Figure 6). Of note, the affinity of C145A Mpro for PF-00835231 even without the covalent binding was 2.5 μM, strongly supporting perfect complementarity between the inhibitor and the active site, as first revealed by X-ray crystallography (Hoffman et al., 2020).
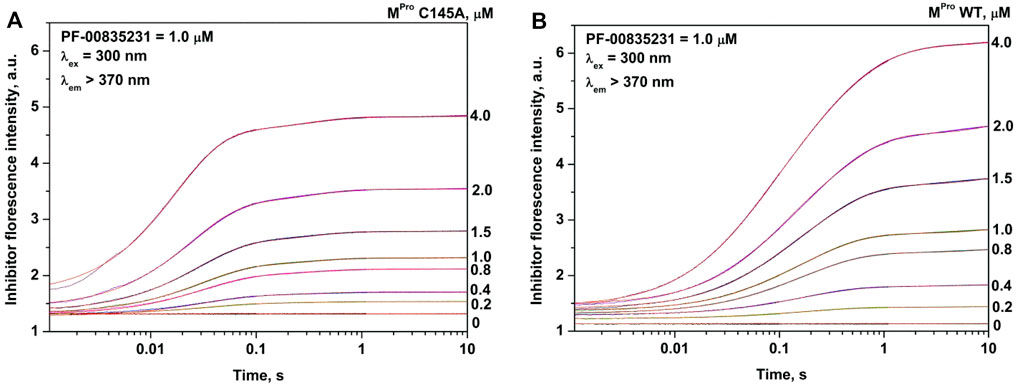
FIGURE 5. Experimental and theoretical (red) kinetic curves of changes in PF-00835231 fluorescence intensity during the interaction with C145A Mpro (A) or WT Mpro (B). The concentration of PF-00835231 was 1.0 µM, and the enzyme concentration is shown in the panel. Substantial overlap of experimental and theoretical kinetic curves indicates good fitting quality and masks visual differences between experimental and theoretical traces.
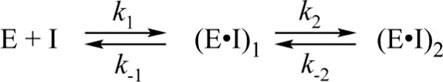
SCHEME 3. The kinetic mechanism of the interaction between C145A Mpro and PF-00835231. E: C145A Mpro, I: PF-00835231, (E•I)i: enzyme–inhibitor noncovalent complexes.
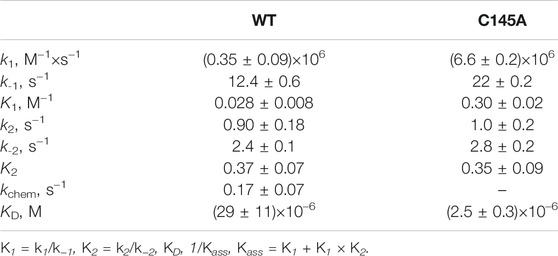
TABLE 3. Rate constants for the interaction of WT Mpro or C145A Mpro with PF-00835231.
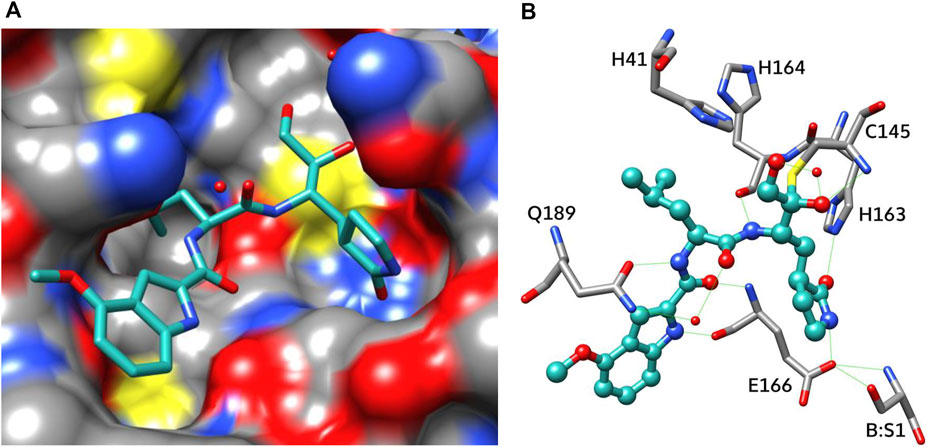
FIGURE 6. Binding mode of PF-00835231 (PDB ID 6XHM): surface representation (A) and an interaction scheme (B). The inhibitor molecule is shown as a ball-and-stick model, water molecules as red balls, and hydrogen bonds as green lines.
The process of interaction of WT Mpro with PF-00835231 is slower (up to time point 10 s) and induces a greater increase in the fluorescence intensity (Figure 5B). It should be noted that in the case of the WT enzyme, the initial phase of the fluorescence intensity increase was significantly slower as compared with C145A Mpro, indicating that the Cys145 residue must play an important role in the formation of the initial complex. As soon as the binding of the inhibitor to C145A Mpro went through two steps, the kinetic curves for the WT enzyme could be described by Scheme 4, which contains two equilibrium steps and one irreversible step (Table 3).
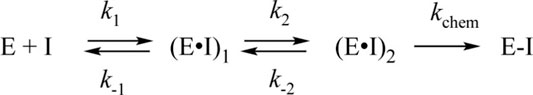
SCHEME 4. The kinetic mechanism of the interaction between WT Mpro and PF-00835231. E: WT Mpro, I: PF-00835231, (E•I)i: enzyme–inhibitor noncovalent complexes, E–I: enzyme–inhibitor covalent complex.
The value of rate constant k1 revealed that the formation of the initial complex is approximately 20-fold faster for C145A Mpro, eventually yielding a 10-fold difference in the equilibrium binding constant between the WT and C145A enzymes. On the other hand, rate constants of the second binding step were very similar between the two enzymes, indicating that the specific interaction of the inhibitor and enzyme is independent of the Cys145 residue. The rate constant for the formation of the covalent bond with the inhibitor was similar to the catalytic constant of peptide cleavage (Tables 2 and 3).
Overall, it can be concluded that noncovalent interaction of WT Mpro and the PF-00835231 inhibitor is not as strong as expected from the nanomolar range of the inhibition constants reported in several studies (Hoffman et al., 2020; Boras et al., 2021; de Vries et al., 2021). Nevertheless, the high efficiency of PF-00835231 is explained by the subsequent covalent modification of the enzyme; this modification significantly stabilizes the enzyme–inhibitor complex.
A two-step virtual screening procedure was carried out to prioritize commercially available small-molecule compounds as potential Mpro inhibitors. At the first step, an antiviral chemical space (Nikitina et al., 2019) analysis was performed using the Generative Topographic Mapping approach (Horvath et al., 2020). Of 800 million ZINC compounds, 574 were predicted as hits potentially possessing an anticoronaviral activity. Items of this hitlist were then docked into the active-site cavity of room temperature Mpro crystal structure 6WQF (Kneller et al., 2020), and the best compounds were selected according to the scoring function (Figure 7A, Supplementary Table S1). Ten compounds with grid scores less than −50 were designated as primary hits and subjected to the initial screening of Mpro inhibition (Figure 7B). Three compounds—IBS-E0680092, IBS-E0183442, and IBS-E0474913—showed more than 50% enzyme inhibition, whereas IBS-E0530026 manifested a less prominent inhibitory activity.
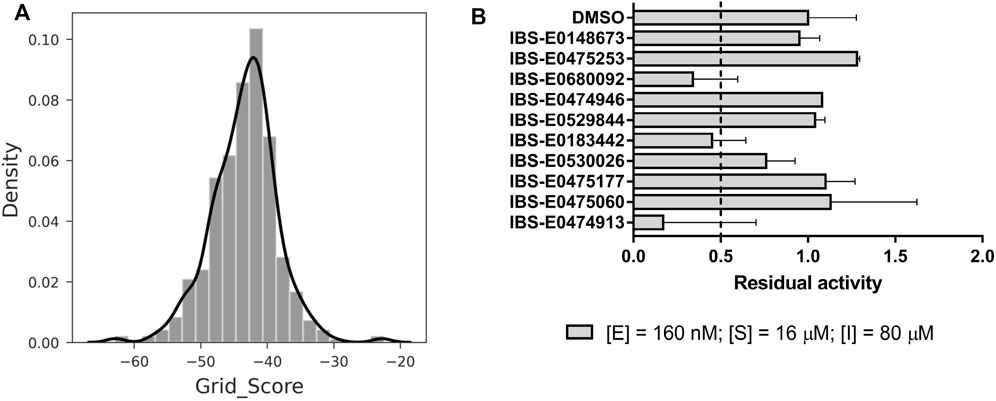
FIGURE 7. Structure-based virtual screening and an assessment of inhibition efficacy. (A) Docking score distribution for Generative Topographic Mapping hits. More negative values correspond to better scores. (B) Preliminary screening of the compounds for Mpro inhibition.
These four compounds were chosen for a more detailed comparison with covalent inhibitors of Mpro. First of all, the protein stabilization by inhibitor binding was compared using thermal shift assay (TSA). TSA allows a direct comparison of protein binding efficiency between small-molecule compounds. A substantial change of protein melting temperature (Tm) may be an indicator of tight binding (Figure 8; Table 4). The binding of covalent inhibitors GC-376, PF-00835231, and boceprevir caused changes of protein Tm, pointing to strong stabilization of the enzyme molecule in the covalent complex with these compounds. On the other hand, the interaction of telaprevir and all noncovalent inhibitors did not result in Tm changes, suggesting that the binding of these compounds with the enzyme is less efficient.
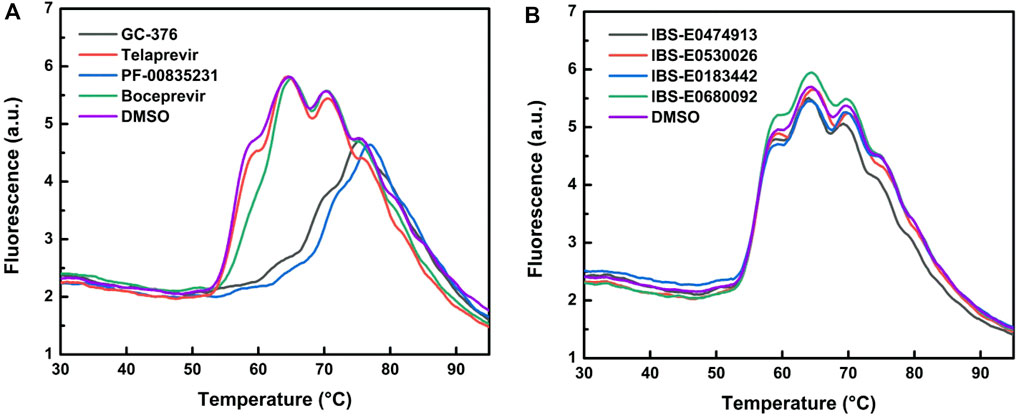
FIGURE 8. Melting curves of WT Mpro in the absence and presence of covalent (A) or noncovalent (B) inhibitors.
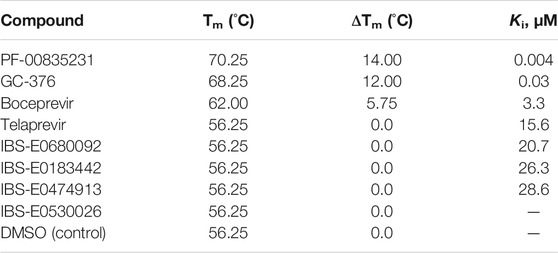
TABLE 4. Changes in the Mpro melting temperature upon the inhibitor binding, and inhibition constants of the tested compounds.
Steady-state analysis of the inhibition efficacy of all the tested compounds revealed (Table 4) that the thermal shift is directly related to inhibition constant Ki. Indeed, the decrease of ΔTm correlated with an increase of the inhibition constant. Moreover, the thermal shift was negligible when Ki approached ∼10–15 μM. Furthermore, the elimination of the Mpro covalent binding with PF-00835231 by means of the C145A mutation also strongly decreased the change in Tm (to ∼2.2°C) as compared to the covalently bound WT adduct (∼14.0°C), consistently with the pre-steady-state data, which yielded an inhibition constant of 2.5 μM (Table 3).
The efficiency of enzymatic hydrolysis of the substrate by Mpro in the presence of a standard inhibitor (boceprevir, telaprevir, GC-376, PF-00835231, or thimerosal; Figure 9A) or a new inhibitor from the virtual screening (Figure 9B) was determined by comparison of the kinetics of the substrate cleavage. The protease was mixed with an inhibitor and kept on ice for 5 min to obtain an enzyme–inhibitor complex. After that, the substrate hydrolysis was initiated by stopped-flow fast mixing of this complex with FRET-S, and the signal was monitored. To estimate the remaining enzymatic activity, the initial slope of the FRET signal increase was calculated (Figure 9C). A comparison of the obtained data allowed us to conclude that both GC-376 and PF-00835231 were the most potent covalent inhibitors of Mpro among the tested ketone-based and organometallic compounds. Boceprevir showed an intermediate level of activity, whereas telaprevir and thimerosal were on the lowest activity tier, in line with the specificity of the design approach and binding-site complementarity for each of these compounds. Indeed, GC-376 and PF-00835231 were specifically designed as coronavirus Mpro inhibitors; boceprevir is less complementary to the binding site but still has a similar molecular size. On the other hand, telaprevir is much larger and engages in fewer specific interactions although it is still able to form a covalent bond with Cys145. Thimerosal, on the contrary, is substantially smaller and does not form specific interactions in the binding site; these features reduce the inhibitory mechanism of thimerosal to pure Cys145 blockage.
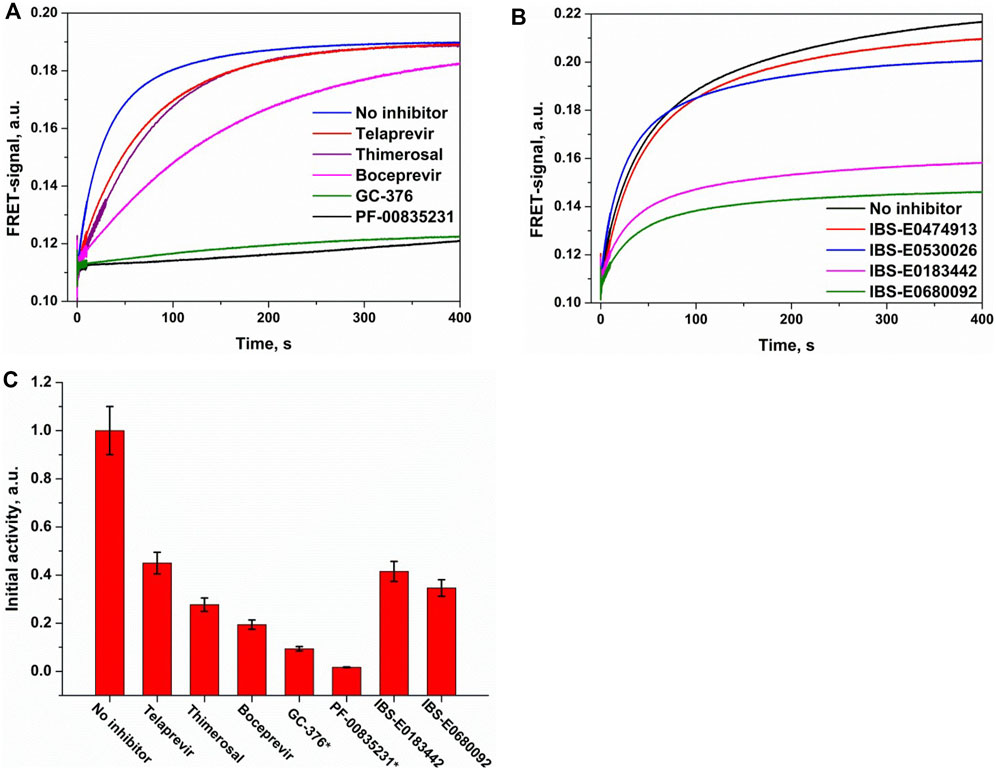
FIGURE 9. Comparative analysis of Mpro inhibition by covalent (A) and noncovalent (B) inhibitors. (C) Residual enzymatic activity calculated from the initial slope of kinetic curves. The activity of free enzyme was normalized to 1.0. FRET-S and enzyme concentrations were 2.5 µM, and the inhibitor concentration was 25 µM except for the compounds marked with an asterisk (2.5 µM).
An assessment of relative efficacy of Mpro inhibition by the noncovalent binders revealed that two of them, IBS-E0183442 and IBS-E0680092, significantly inhibit the enzymatic activity, albeit less potently than the covalent inhibitors. This observation supports possible further inhibitor optimization via introduction of additional chemical groups to improve specific interactions as well as indicates that such a pre–steady-state analysis may be a good platform for rapid low-cost screening of small molecule compounds to reveal their inhibitory potential.
Even though the SARS-CoV-2 main protease is an attractive target for a therapeutic intervention into COVID-19, only an extremely small number of compounds are currently known with inhibitory properties toward this enzyme. Therefore, the design of specific inhibitors as well as the development of effective screening systems for such compounds are urgently needed. In the present work, for the first time, we report a pre–steady-state kinetic analysis of sequential stages of a model peptide’s binding and cleavage by WT Mpro. The fast-kinetics approach enables determining the rates of formation of an enzyme–substrate catalytic complex and rates of peptide cleavage by Mpro on the basis of the FRET effect. An interaction of the catalytically inactive mutant enzyme (C145A Mpro) with the same substrate revealed that the enzyme normally induces conformational changes in the peptide during the complex formation. Our findings suggest that the binding of a peptide substrate in the active site of this protease proceeds through a single reversible stage in the kinetic scheme. A collision of the enzyme and peptide substrate rapidly gives rise to the catalytic complex in which site-specific cleavage of the peptide takes place.
The efficiency of enzymatic hydrolysis of the peptide substrate by Mpro in the presence of one suitable inhibitor was determined by a pre–steady-state kinetic analysis. In this study, the most promising covalent inhibitors of SARS-CoV-2 Mpro such as PF-00835231, GC-376, boceprevir, and telaprevir were tested. Thimerosal was also used because it is an organometallic binder of the catalytic cysteine of Mpro. As expected, among all the tested covalent inhibitors, PF-00835231 was the most effective. It turned out that the interaction of Mpro with PF-00835231 involves two steps of reversible binding-complex formation with subsequent covalent binding step (Scheme 4). In this scheme it is likely that after the formation of the initial complex, there is an additional step of formation of specific interactions between the inhibitor and the enzyme pocket, resulting in the proper placement of the inhibitor in the active site of the protease. The irreversible step of Scheme 4 corresponds to the covalent bond formation between Cys145 of the enzyme and the inhibitor molecule. Indeed, the interaction of PF-00835231 in the active site of C145A Mpro, which is incapable of a covalent binding with this compound, revealed that there are only two reversible steps of binding-complex formation. This result meant that potential inhibitory compounds should have certain pharmacophoric features for a fine conformational fit and must engage in specific interactions in the active-site pocket of the enzyme rather than contain specific warheads forming a covalent bond with the protein. Then, we performed a structure–guided selection, which yielded four small molecule-weight promising noncovalent inhibitors of Mpro. The inhibition efficacy of these compounds was analyzed by a TSA and steady-state and pre–steady-state kinetic approaches, which helped us to identify two new noncovalent inhibitors of SARS-CoV-2 Mpro.
Overall, we developed a platform for low-cost rapid quantitative estimation of the type and magnitude of inhibition for prospective inhibitors of the SARS-CoV-2 main protease. The analysis of kinetics using the WT enzyme together with its catalytically inactive mutant, C145A, enabled us to identify the mechanism of action of the inhibitors and gave an opportunity to hypothesize the therapeutic potential of a model drug. This approach allows us to work with various mutants of the enzyme as well as different types of inhibitors (specific to allosteric or active sites) and thus may be regarded as a technique supported by proof of concept in a target-based drug assessment prior to preclinical studies. The high sensitivity of the method and its ability to provide precise quantitative data will help us to discriminate relevant compounds by their kinetic properties. If the dynamics of the interaction of an enzyme with an inhibitor are crucial for the therapeutic potential of the drug, then the proposed technique will give a unique opportunity to accept/reject the compounds selected by means of molecular docking simulations.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
MZ, AK, VU, AF, LK, EK, IK, AB, and AV conducted the experiments; IS, DO, AE, AG, and NK conceived and designed the experiments; VK, OF, DO, AE, AG, and NK analyzed the data; OF, AI, AE, AG, and NK contributed the reagents, materials, and/or analytical tools; and AE, AG, and NK wrote the paper.
This research was supported by the RFBR Grant #20-04-60468 as well as by State Research Funding for FSASI “Chumakov FSC R&D IBP RAS” (Institute of Poliomyelitis): project “Development of inhibitors for SARS-CoV-2 main protease”. The part of the work with stopped-flow kinetics was specifically funded by Russian-State-funded project #121112900214-2 for Institute of Chemical Biology and Fundamental Medicine “Inhibitors of main protease of coronavirus SARS-CoV-2”.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Drs. Igor Rodin, Dmitry Vasilenko, and Ivan Plyushchenko for the assessment of thimerosal purity.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2021.773198/full#supplementary-material
Allen, W. J., Balius, T. E., Mukherjee, S., Brozell, S. R., Moustakas, D. T., Lang, P. T., et al. (2015). DOCK 6: Impact of New Features and Current Docking Performance. J. Comput. Chem. 36, 1132–1156. doi:10.1002/JCC.23905
Anson, B. J., Chapman, M. E., Lendy, E. K., Pshenychnyi, S., D’Aquila, R. T., Satchell, K. J. F., et al. (2020). Broad-spectrum Inhibition of Coronavirus Main and Papain-like Proteases by HCV Drugs. Research Square. doi:10.21203/RS.3.RS-26344/V1
Baker, J. D., Uhrich, R. L., Kraemer, G. C., Love, J. E., and Kraemer, B. C. (2021). A Drug Repurposing Screen Identifies Hepatitis C Antivirals as Inhibitors of the SARS-CoV2 Main Protease. PLoS One 16, e0245962. doi:10.1371/JOURNAL.PONE.0245962
Boras, B., Jones, R. M., Anson, B. J., Arenson, D., Aschenbrenner, L., Bakowski, M. A., et al. (2021). Discovery of a Novel Inhibitor of Coronavirus 3CL Protease for the Potential Treatment of COVID-19. bioRxiv, 293498. doi:10.1101/2020.09.12.293498
Calina, D., Docea, A. O., Petrakis, D., Egorov, A. M., Ishmukhametov, A. A., Gabibov, A. G., et al. (2020). Towards Effective COVID-19 V-accines: Updates, P-erspectives and C-hallenges (Review). Int. J. Mol. Med. 46, 3–16. doi:10.3892/IJMM.2020.4596
Cannalire, R., Cerchia, C., Beccari, A. R., Di Leva, F. S., and Summa, V. (2020). Targeting SARS-CoV-2 Proteases and Polymerase for COVID-19 Treatment: State of the Art and Future Opportunities. J. Med. Chem. 1, acs.jmedchem.0c01140. doi:10.1021/ACS.JMEDCHEM.0C01140
Coelho, C., Gallo, G., Campos, C. B., Hardy, L., and Würtele, M. (2020). Biochemical Screening for SARS-CoV-2 Main Protease Inhibitors. PLoS One 15, e0240079. doi:10.1371/JOURNAL.PONE.0240079
Dai, W., Zhang, B., Jiang, X. M., Su, H., Li, J., Zhao, Y., et al. (2020). Structure-based Design of Antiviral Drug Candidates Targeting the SARS-CoV-2 Main Protease. Science 368, 1331–1335. doi:10.1126/SCIENCE.ABB4489
de Vries, M., Mohamed, A. S., Prescott, R. A., Valero-Jimenez, A. M., Desvignes, L., O’Connor, R., et al. (2021). A Comparative Analysis of SARS-CoV-2 Antivirals Characterizes 3CLpro Inhibitor PF-00835231 as a Potential New Treatment for COVID-19. J. Virol. 95, 1. doi:10.1128/JVI.01819-20
Dong, E., Du, H., and Gardner, L. (2020). An Interactive Web-Based Dashboard to Track COVID-19 in Real Time. Lancet Infect. Dis. 20, 533–534. doi:10.1016/S1473-3099(20)30120-1
Fu, L., Ye, F., Feng, Y., Yu, F., Wang, Q., Wu, Y., et al. (2020). Both Boceprevir and GC376 Efficaciously Inhibit SARS-CoV-2 by Targeting its Main Protease. Nat. Commun. 11, 4417–4418. doi:10.1038/s41467-020-18233-x
Gadlage, M. J., and Denison, M. R. (2010). Exchange of the Coronavirus Replicase Polyprotein Cleavage Sites Alters Protease Specificity and Processing. J. Virol. 84, 6894–6898. doi:10.1128/JVI.00752-10
Geier, D. A., Sykes, L. K., and Geier, M. R. (2007). A Review of Thimerosal (Merthiolate) and its Ethylmercury Breakdown Product: Specific Historical Considerations Regarding Safety and Effectiveness. J. Toxicol. Environ. Health B Crit. Rev. 10, 575–596. doi:10.1080/10937400701389875
Ghahremanpour, M. M., Tirado-Rives, J., Deshmukh, M., Ippolito, J. A., Zhang, C. H., de Vaca, I. C., et al. (2020). Identification of 14 Known Drugs as Inhibitors of the Main Protease of SARS-CoV-2. bioRxiv 11, 2526–2533. doi:10.1021/ACSMEDCHEMLETT.0C0052110.1101/2020.08.28.271957
Guo, Y., Huang, L., Zhang, G., Yao, Y., Zhou, H., Shen, S., et al. (2021). A SARS-CoV-2 Neutralizing Antibody with Extensive Spike Binding Coverage and Modified for Optimal Therapeutic Outcomes. Nat. Commun. 12, 1–11. doi:10.1038/s41467-021-22926-2
Hoffman, R. L., Kania, R. S., Brothers, M. A., Davies, J. F., Ferre, R. A., Gajiwala, K. S., et al. (2020). Discovery of Ketone-Based Covalent Inhibitors of Coronavirus 3CL Proteases for the Potential Therapeutic Treatment of COVID-19. J. Med. Chem. 63, 12725–12747. doi:10.1021/ACS.JMEDCHEM.0C01063
Horvath, D., Orlov, A., Osolodkin, D. I., Ishmukhametov, A. A., Marcou, G., and Varnek, A. (2020). A Chemographic Audit of Anti-coronavirus Structure-Activity Information from Public Databases (ChEMBL). Mol. Inform. 39, 2000080. doi:10.1002/MINF.202000080
Hung, H. C., Ke, Y. Y., Huang, S. Y., Huang, P. N., Kung, Y. A., Chang, T. Y., et al. (2020). Discovery of M Protease Inhibitors Encoded by SARS-CoV-2. Antimicrob. Agents Chemother. 64, 1. doi:10.1128/AAC.00872-20
Jin, Z., Du, X., Xu, Y., Deng, Y., Liu, M., Zhao, Y., et al. (2020a). Structure of Mpro from SARS-CoV-2 and Discovery of its Inhibitors. Nature 582, 289–293. doi:10.1038/s41586-020-2223-y
Jin, Z., Zhao, Y., Sun, Y., Zhang, B., Wang, H., Wu, Y., et al. (2020b). Structural Basis for the Inhibition of SARS-CoV-2 Main Protease by Antineoplastic Drug Carmofur. Nat. Struct. Mol. Biol. 27, 529–532. doi:10.1038/s41594-020-0440-6
Khan, S., Attar, F., Bloukh, S. H., Sharifi, M., Nabi, F., Bai, Q., et al. (2021). A Review on the Interaction of Nucleoside Analogues with SARS-CoV-2 RNA Dependent RNA Polymerase. Int. J. Biol. Macromol. 181, 605–611. doi:10.1016/J.IJBIOMAC.2021.03.112
Kim, Y., Liu, H., Galasiti Kankanamalage, A. C., Weerasekara, S., Hua, D. H., Groutas, W. C., et al. (2016). Reversal of the Progression of Fatal Coronavirus Infection in Cats by a Broad-Spectrum Coronavirus Protease Inhibitor. Plos Pathog. 12, e1005531. doi:10.1371/JOURNAL.PPAT.1005531
Kim, Y., Lovell, S., Tiew, K. C., Mandadapu, S. R., Alliston, K. R., Battaile, K. P., et al. (2012). Broad-spectrum Antivirals against 3C or 3C-like Proteases of Picornaviruses, Noroviruses, and Coronaviruses. J. Virol. 86, 11754–11762. doi:10.1128/JVI.01348-12
Kladova, O. A., Grin, I. R., Fedorova, O. S., Kuznetsov, N. A., and Zharkov, D. O. (2019). Conformational Dynamics of Damage Processing by Human DNA Glycosylase NEIL1. J. Mol. Biol. 431, 1098–1112. doi:10.1016/j.jmb.2019.01.030
Kneller, D. W., Phillips, G., O'Neill, H. M., Jedrzejczak, R., Stols, L., Langan, P., et al. (2020). Structural Plasticity of SARS-CoV-2 3CL Mpro Active Site Cavity Revealed by Room Temperature X-ray Crystallography. Nat. Commun. 11, 3202–3206. doi:10.1038/s41467-020-16954-7
Kuzmic, P. (1996). Program DYNAFIT for the Analysis of Enzyme Kinetic Data: Application to HIV Proteinase. Anal. Biochem. 237, 260–273. doi:10.1006/abio.1996.0238
Kuznetsov, N. A., and Fedorova, O. S. (2020). Kinetic Milestones of Damage Recognition by DNA Glycosylases of the Helix-hairpin-Helix Structural Superfamily. Adv. Exp. Med. Biol. 1241, 1–18. doi:10.1007/978-3-030-41283-8_1
Kuznetsov, N. A., and Fedorova, O. S. (2016). Thermodynamic Analysis of Fast Stages of Specific Lesion Recognition by DNA Repair Enzymes. Biochemistry (Mosc) 81, 1136–1152. doi:10.1134/S0006297916100114
Kuznetsov, N. A., Kiryutin, A. S., Kuznetsova, A. A., Panov, M. S., Barsukova, M. O., Yurkovskaya, A. V., et al. (2017). The Formation of Catalytically Competent Enzyme-Substrate Complex Is Not a Bottleneck in Lesion Excision by Human Alkyladenine DNA Glycosylase. J. Biomol. Struct. Dyn. 35, 950–967. doi:10.1080/07391102.2016.1171800
Kuznetsova, A. A., Fedorova, O. S., and Kuznetsov, N. A. (2020). Lesion Recognition and Cleavage of Damage-Containing Quadruplexes and Bulged Structures by DNA Glycosylases. Front. Cell Dev. Biol. 8, 1462. doi:10.3389/FCELL.2020.595687
Lang, L. (2007). Combination Therapy with Telaprevir and Pegylated Interferon Suppresses Both Wild-type and Resistant Hepatitis C Virus. Gastroenterology 132, 5–6. doi:10.1053/J.GASTRO.2006.12.011
Lee, J., Worrall, L. J., Vuckovic, M., Rosell, F. I., Gentile, F., Ton, A. T., et al. (2020). Crystallographic Structure of Wild-type SARS-CoV-2 Main Protease Acyl-Enzyme Intermediate with Physiological C-Terminal Autoprocessing Site. Nat. Commun. 11, 5877. doi:10.1038/s41467-020-19662-4
Ma, C., Sacco, M. D., Hurst, B., Townsend, J. A., Hu, Y., Szeto, T., et al. (2020). Boceprevir, GC-376, and Calpain Inhibitors II, XII Inhibit SARS-CoV-2 Viral Replication by Targeting the Viral Main Protease. Cell Res 30, 678–692. doi:10.1038/s41422-020-0356-z
Maier, J. A., Martinez, C., Kasavajhala, K., Wickstrom, L., Hauser, K. E., and Simmerling, C. (2015). ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theor. Comput. 11, 3696–3713. doi:10.1021/ACS.JCTC.5B00255
Nikitina, A. A., Orlov, A. A., Kozlovskaya, L. I., Palyulin, V. A., and Osolodkin, D. I. (2019). Enhanced Taxonomy Annotation of Antiviral Activity Data from ChEMBL. Database (Oxford) 2019, bay 139. doi:10.1093/DATABASE/BAY139
Noske, G. D., Nakamura, A. M., Gawriljuk, V. O., Fernandes, R. S., Lima, G. M. A., Rosa, H. V. D., et al. (2021). A Crystallographic Snapshot of SARS-CoV-2 Main Protease Maturation Process. J. Mol. Biol. 433, 167118. doi:10.1016/J.JMB.2021.167118
Owen, D. R., Allerton, C. M. N., Anderson, A. S., Aschenbrenner, L., Avery, M., Berritt, S., et al. (2021). An Oral SARS-CoV-2 Mpro Inhibitor Clinical Candidate for the Treatment of COVID-19. medRxiv 2021, 21261232. doi:10.1126/science.abl4784
Pedersen, N. C., Kim, Y., Liu, H., Galasiti Kankanamalage, A. C., Eckstrand, C., Groutas, W. C., et al. (2018). Efficacy of a 3C-like Protease Inhibitor in Treating Various Forms of Acquired Feline Infectious Peritonitis. J. Feline Med. Surg. 20, 378–392. doi:10.1177/1098612X17729626
Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., et al. (2004). UCSF Chimera-Aa Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 25, 1605–1612. doi:10.1002/jcc.20084
Pruijssers, A. J., George, A. S., Schäfer, A., Leist, S. R., Gralinksi, L. E., Dinnon, K. H., et al. (2020). Remdesivir Inhibits SARS-CoV-2 in Human Lung Cells and Chimeric SARS-CoV Expressing the SARS-CoV-2 RNA Polymerase in Mice. Cell Rep 32, 107940. doi:10.1016/J.CELREP.2020.107940
Qiao, J., Li, Y.-S., Zeng, R., Liu, F.-L., Luo, R.-H., Huang, C., et al. (2021). SARS-CoV-2 M Pro Inhibitors with Antiviral Activity in a Transgenic Mouse Model. Science 371, 1374–1378. doi:10.1126/SCIENCE.ABF1611
Rathnayake, A. D., Zheng, J., Kim, Y., Perera, K. D., Mackin, S., Meyerholz, D. K., et al. (2020). 3C-like Protease Inhibitors Block Coronavirus Replication In Vitro and Improve Survival in MERS-CoV-Infected Mice. Sci. Transl. Med. 12, 5332. doi:10.1126/SCITRANSLMED.ABC5332
Roe, M. K., Junod, N. A., Young, A. R., Beachboard, D. C., and Stobart, C. C. (2021). Targeting Novel Structural and Functional Features of Coronavirus Protease Nsp5 (3CLpro, Mpro) in the Age of COVID-19. J. Gen. Virol. 102, 1. doi:10.1099/JGV.0.001558
Rotella, D. P. (2013). The Discovery and Development of Boceprevir. Expert Opin. Drug Discov. 8, 1439–1447. doi:10.1517/17460441.2013.843525
Rut, W., Lv, Z., Zmudzinski, M., Patchett, S., Nayak, D., Snipas, S. J., et al. (2020). Activity Profiling and crystal Structures of Inhibitor-Bound SARS-CoV-2 Papain-like Protease: A Framework for Anti-COVID-19 Drug Design. Sci. Adv. 6, eabd4596. doi:10.1126/SCIADV.ABD4596
Shang, J., Ye, G., Shi, K., Wan, Y., Luo, C., Aihara, H., et al. (2020). Structural Basis of Receptor Recognition by SARS-CoV-2. Nature 581, 221–224. doi:10.1038/S41586-020-2179-Y
Sterling, T., and Irwin, J. J. (2015). ZINC 15--Ligand Discovery for Everyone. J. Chem. Inf. Model. 55, 2324–2337. doi:10.1021/ACS.JCIM.5B00559
Ullrich, S., and Nitsche, C. (2020). The SARS-CoV-2 Main Protease as Drug Target. Bioorg. Med. Chem. Lett. 30, 127377. doi:10.1016/J.BMCL.2020.127377
Vandyck, K., and Deval, J. (2021). Considerations for the Discovery and Development of 3-chymotrypsin-like Cysteine Protease Inhibitors Targeting SARS-CoV-2 Infection. Curr. Opin. Virol. 49, 36–40. doi:10.1016/J.COVIRO.2021.04.006
V’kovski, P., Kratzel, A., Steiner, S., Stalder, H., Thiel, V., V’kovski, P., et al. (2021). Coronavirus Biology and Replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 19, 155–170. doi:10.1038/s41579-020-00468-6
Vuong, W., Fischer, C., Khan, M. B., van Belkum, M. J., Lamer, T., Willoughby, K. D., et al. (2021). Improved SARS-CoV-2 Mpro Inhibitors Based on Feline Antiviral Drug GC376: Structural Enhancements, Increased Solubility, and Micellar Studies. Eur. J. Med. Chem. 222, 113584. doi:10.1016/J.EJMECH.2021.113584
Vuong, W., Khan, M. B., Fischer, C., Arutyunova, E., Lamer, T., Shields, J., et al. (2020). Feline Coronavirus Drug Inhibits the Main Protease of SARS-CoV-2 and Blocks Virus Replication. Nat. Commun. 11, 4282–4288. doi:10.1038/s41467-020-18096-2
Wang, Y., Anirudhan, V., Du, R., Cui, Q., Rong, L., Y, W., et al. (2021). RNA-dependent RNA Polymerase of SARS-CoV-2 as a Therapeutic Target. J. Med. Virol. 93, 300–310. doi:10.1002/JMV.26264
Wu, F., Zhao, S., Yu, B., Chen, Y. M., Wang, W., Song, Z. G., et al. (2020). A New Coronavirus Associated with Human Respiratory Disease in China. Nature 579, 265–269. doi:10.1038/s41586-020-2008-3
Yang, H., and Yang, J. (2021). A Review of the Latest Research on Mpro Targeting SARS-COV Inhibitors. RSC Med. Chem. 12, 1026–1036. doi:10.1039/D1MD00066G
Yin, W., Mao, C., Luan, X., Shen, D. D., Shen, Q., Su, H., et al. (2020). Structural Basis for Inhibition of the RNA-dependent RNA Polymerase from SARS-CoV-2 by Remdesivir. Science 368, 1499–1504. doi:10.1126/SCIENCE.ABC1560
Yoshino, R., Yasuo, N., and Sekijima, M. (2020). Identification of Key Interactions between SARS-CoV-2 Main Protease and Inhibitor Drug Candidates. Sci. Rep. 10, 12493–12498. doi:10.1038/s41598-020-69337-9
Zaidman, D., Gehrtz, P., Filep, M., Fearon, D., Prilusky, J., Duberstein, S., et al. (2020). An Automatic Pipeline for the Design of Irreversible Derivatives Identifies a Potent SARS-CoV-2 Mpro Inhibitor. bioRxiv 21, 299776. doi:10.1101/2020.09.21.299776
Zakharova, M. Y., Kuznetsov, N. A., Dubiley, S. A., Kozyr, A. V., Fedorova, O. S., Chudakov, D. M., et al. (2009). Substrate Recognition of Anthrax Lethal Factor Examined by Combinatorial and Pre-steady-state Kinetic Approaches. J. Biol. Chem. 284, 17902–17913. doi:10.1074/jbc.M807510200
Zakharova, M. Y., Kuznetsova, A. A., Kaliberda, E. N., Dronina, M. A., Kolesnikov, A. V., Kozyr, A. V., et al. (2017). Evolution of Inhibitor-Resistant Natural Mutant Forms of HIV-1 Protease Probed by Pre-steady State Kinetic Analysis. Biochimie 142, 125–134. doi:10.1016/j.biochi.2017.08.014
Zhang, L., Lin, D., Sun, X., Curth, U., Drosten, C., Sauerhering, L., et al. (2020). Crystal Structure of SARS-CoV-2 Main Protease Provides a Basis for Design of Improved α-ketoamide Inhibitors. Science 368, 409–412. doi:10.1126/SCIENCE.ABB3405
Zhou, X., Wang, H., Ji, Q., Du, M., Liang, Y., Li, H., et al. (2021). Molecular Deconvolution of the Neutralizing Antibodies Induced by an Inactivated SARS-CoV-2 Virus Vaccine. Protein Cell 12 (10), 818–823. doi:10.1007/S13238-021-00840-Z
Zhu, N., Zhang, D., Wang, W., Li, X., Yang, B., Song, J., et al. (2020a). A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 382, 727–733. doi:10.1056/NEJMOA2001017
Zhu, W., Chen, C. Z., Gorshkov, K., Xu, M., Lo, D. C., and Zheng, W. (2020b). RNA-dependent RNA Polymerase as a Target for COVID-19 Drug Discovery. SLAS Discov. 25, 1141–1151. doi:10.1177/2472555220942123
Zhu, W., Xu, M., Chen, C. Z., Guo, H., Shen, M., Hu, X., et al. (2020c). Identification of SARS-CoV-2 3CL Protease Inhibitors by a Quantitative High-Throughput Screening. ACS Pharmacol. Transl. Sci. 3, 1008–1016. doi:10.1021/ACSPTSCI.0C00108
Keywords: SARS-CoV-2, main protease, pre-steady-state kinetics, substrate cleavage, inhibitor binding, molecular docking, stopped-flow
Citation: Zakharova MY, Kuznetsova AA, Uvarova VI, Fomina AD, Kozlovskaya LI, Kaliberda EN, Kurbatskaia IN, Smirnov IV, Bulygin AA, Knorre VD, Fedorova OS, Varnek A, Osolodkin DI, Ishmukhametov AA, Egorov AM, Gabibov AG and Kuznetsov NA (2021) Pre-Steady-State Kinetics of the SARS-CoV-2 Main Protease as a Powerful Tool for Antiviral Drug Discovery. Front. Pharmacol. 12:773198. doi: 10.3389/fphar.2021.773198
Received: 09 September 2021; Accepted: 29 October 2021;
Published: 06 December 2021.
Edited by:
Salvatore Salomone, University of Catania, ItalyReviewed by:
Joanne Lemieux, University of Alberta, CanadaCopyright © 2021 Zakharova, Kuznetsova, Uvarova, Fomina, Kozlovskaya, Kaliberda, Kurbatskaia, Smirnov, Bulygin, Knorre, Fedorova, Varnek, Osolodkin, Ishmukhametov, Egorov, Gabibov and Kuznetsov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexey M. Egorov, YWxleC5tLmVnb3JvdkBnbWFpbC5jb20=; Alexander G. Gabibov, Z2FiaWJvdkBteC5pYmNoLnJ1; Nikita A. Kuznetsov, bmlraXRhLmt1em5ldHNvdkBuaWJvY2gubnNjLnJ1
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.