
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 23 November 2021
Sec. Experimental Pharmacology and Drug Discovery
Volume 12 - 2021 | https://doi.org/10.3389/fphar.2021.772296
This article is part of the Research Topic Artificial Intelligence for Drug Design, Development and Therapy View all 20 articles
 Haiping Zhang1†
Haiping Zhang1† Junxin Li2†
Junxin Li2† Konda Mani Saravanan1
Konda Mani Saravanan1 Hao Wu1
Hao Wu1 Zhichao Wang1Du Wu1
Zhichao Wang1Du Wu1 Yanjie Wei1Zhen Lu3Youhai H. Chen3*
Yanjie Wei1Zhen Lu3Youhai H. Chen3* Xiaochun Wan2*
Xiaochun Wan2* Yi Pan1*
Yi Pan1*The TIPE2 (tumor necrosis factor-alpha-induced protein 8-like 2) protein is a major regulator of cancer and inflammatory diseases. The availability of its sequence and structure, as well as the critical amino acids involved in its ligand binding, provides insights into its function and helps greatly identify novel drug candidates against TIPE2 protein. With the current advances in deep learning and molecular dynamics simulation-based drug screening, large-scale exploration of inhibitory candidates for TIPE2 becomes possible. In this work, we apply deep learning-based methods to perform a preliminary screening against TIPE2 over several commercially available compound datasets. Then, we carried a fine screening by molecular dynamics simulations, followed by metadynamics simulations. Finally, four compounds were selected for experimental validation from 64 candidates obtained from the screening. With surprising accuracy, three compounds out of four can bind to TIPE2. Among them, UM-164 exhibited the strongest binding affinity of 4.97 µM and was able to interfere with the binding of TIPE2 and PIP2 according to competitive bio-layer interferometry (BLI), which indicates that UM-164 is a potential inhibitor against TIPE2 function. The work demonstrates the feasibility of incorporating deep learning and MD simulation in virtual drug screening and provides high potential inhibitors against TIPE2 for drug development.
Tumor necrosis factor (TNF)-alpha-induced protein 8 (TNFAIP8 or TIPE) family of proteins is believed to be regulators of innate and adaptive immunity, as well as cell proliferation, inflammation, and cell death (Lou and Liu, 2011; Goldsmith and Chen, 2017; Yuan et al., 2020). As a member of the TNFAIP8 family, TIPE2 protein is mainly expressed in the placenta and lymphoid tissues (Padmavathi et al., 2018). To maintain immune homeostasis, TIPE2 controls immune cell activation, migration, and apoptosis (Luan et al., 2014). TIPE2 is found to promote Fas-mediated T cell apoptosis (Liu MW. et al., 2015). Depending on the cell types involved, TIPE2 may act as a tumor suppressor (Zhang et al., 2016) or enhancer through, for example, MDSCs (myeloid-derived suppressor cells) (Yan et al., 2017, 2020). Therefore, inhibiting TIPE2 in certain cell types has the potential to treat certain cancers, e.g., lung cancer (Bordoloi et al., 2019). Importantly, TIPE2 is a new target of STAT3, and inhibiting its expression in MDSCs helps enhance T cell activation in tumors (Yan et al., 2017). This indicates that inhibitors of TIPE2 may exert an anti-cancer effect through MDSCs, and hence, it would be medically meaningful to discover TIPE2 inhibitors. We previously identified TIPE2 as a new therapeutic target for cancer immunotherapy since it plays a critical role in the functional polarization of MDSCs (Yan et al., 2020). To the best of our knowledge, despite the significance of TIPE2 in preventing some cancers, no compounds have demonstrated inhibition against TIPE2 function to date.
The high-resolution crystal structure of TIPE2 protein (cytoplasmic protein) was solved in 2009 with an uncharacterized fold that is different from the predicted fold of a DED (Zhang et al., 2009). TIPE2 protein is made up of 184 amino acid residues and is mainly made up of helices. It consists of six antiparallel helices in which helix 5 contains a kink (caused by pro153) and is broken into two short helices (helix 5a and 5b), forming the base of the helical bundle. Surprisingly, TIPE2 contains a huge hydrophobic central cavity, which helps in cofactor binding, and this cavity is found to undergo competitive cofactor binding to activate the immune response to maintain immune dynamic balance. From the crystal structures, it is believed that the unique structural features of TIPE2 protein determine its unique biological functions (Zhang et al., 2010). The availability of a high-resolution experimental structure can aid greatly in identifying novel potential drug candidates against TIPE2 protein.
In general, the drug discovery process against a disease takes a long time and is a costly process. The advances in machine learning methods, especially deep learning, will shorten the drug discovery time, and the researchers in this field will develop more and more applications that solve well-defined problems in the near future. Virtual screening is one field that greatly benefits from the development of deep learning; however, Drug–Target (DTA) prediction is the core component of virtual screening, which directly determines its accuracy and efficiency. To improve the performance of DTA prediction, a model based on deep learning has been developed by the Wei research group (Kaushik et al., 2020). In another interesting study, the authors selected KEAP1 protein, a hot protein in the tumor research field, as a virtual screening target for a database containing 1.3 billion ligands (Gorgulla et al., 2020). However, the ability of the VirtualFlow platform developed in this study lies in its tens of thousands of combined CPUs for molecular docking and cloud computing platforms.
In our very recent work, we built a DL model, “DeepBindRG,” for categorizing native-like protein-ligand complex (Zhang et al., 2019b). We used a complex figure-like 2D input representation and ResNet architecture. We also propose a strategy for virtual screening inhibitors against SARS-CoV-2 3C-like protease (Zhang et al., 2020a). Most importantly, we recently demonstrated that a hybrid method can efficiently be used for drug repurposing for SARS-CoV-2 (Zhang et al., 2020b). It first demonstrates that deep learning-based methods combined with MD simulations are promising strategies to aid the drug development process, especially in identifying novel inhibitors or modulators against therapeutic protein targets.
In our present study, we have built a TIPE2-ligand complex model based on the crystal structure of TIPE2. With the binding pocket from the complex model, we have carried out a large-scale virtual screening against three datasets. Compared to our previous drug repurposing virtual screening against RdRp of SARS-CoV-2, we have a very large data size and have carried much more MD simulations. Since TIPE2 is much smaller than RdRp, it allows us to carry out MD simulation of the full protein instead of the simplified pocket MD simulation to achieve higher accuracy in acceptable computational consumption.
The ability of our hybrid virtual screening pipeline developed in this study lies in its highly efficient protein-ligand binding prediction algorithms and several stages of screening strategies, which emphasizes a gradual shift from efficiency on a large scale to accuracy in the later stage. Finally, the authors considered four out of 69 candidates for experimental validation. They found one compound with a low-micromolar binding affinity with TIPE2, which is an excellent candidate for further optimization, and two compounds with a relatively high-micromolar binding affinity. Among them, UM-164 has a binding affinity of 4.97 µM. Our work has shown the promising future of applying deep learning-based methods and MD simulation-related methods in large-scale drug lead discovery.
Incorporating deep learning and MD simulation helps achieve the balance between accuracy and efficiency in virtual drug screening. Currently, there are very few works that combined large-scale deep learning and MD in drug screening. Our previous drug repurposed against RdRp was the first attempt (Zhang et al., 2020b), but the compound database is relatively too small to sufficiently demonstrate the power of this hybrid method in large-scale drug screening. This time, we have screened over more than 8 thousand compounds and have carried more than 60 standard MD simulations, which is on a much larger scale compared to screening over ∼1 thousand approved drugs and only 12 pocket MD simulations in the previous work.
A previously developed deep learning and molecular simulation-based hybrid strategy is used for virtual screening against TIPE2 over several compound datasets. The step-by-step virtual screening pipeline results in 69 high potential binding candidates with TIPE2. Among them, four were considered to validate the efficacy.
The TIPE2 sequence was obtained from UniProt Consortium (2021), and the TIPE2-ligand model was constructed by I-TASSER (Zhang, 2008). Here, we intended to model the holo (ligand-bound) conformation, given that the currently available high-resolution crystal structure of TIPE2 is an apo structure. The ligand was taken from the template protein (PDB ID: 4xk8) (Zhou et al., 2008) by the COFACTOR algorithm (Roy et al., 2012) within the I-TASSER using structure comparison and protein-protein networks, later manually shifting the position to avoid atoms clash. We extract the amino acids within 1 nm of the ligand as the binding pocket. The three TargetMol datasets (Targetmol-Approved_Drug_Library, Targetmol-Natural_Compound_Library, and Targetmol-Bioactive_Compound_Library) and a compiled in-house dataset were used as virtual screening libraries.
A deep learning-based method, DFCNN (Dense fully Connected Neural Network), has been developed for predicting protein-drug binding probability (Zhang et al., 2019a) and used in this article for the initial drug screening (Figure 1A). DFCNN has used concise mol2vec representation for the protein pocket and ligand, compared to many other methods. It is not dependent on protein and ligand binding conformation, which has greatly accelerated the prediction efficiency. Moreover, it avoids misleading outcomes caused by wrong predicted binding conformation as other methods often encountered. Here, we have used the training set mean and deviation values for the normalization of testing data. The loaded model weights of DFCNN are from the previously completed training. As described in our previous article (Zhang et al., 2019a), the DFCNN model was trained on a dataset extracted from the PDBbind database (2017 version) (Liu Z. et al., 2015). The input representation is the concatenated molecular vector of protein pocket and ligand, and all the molecular vectors are generated by mol2vec (Jaeger et al., 2018), which is inspired by the word2vec model in natural language processing. The positive data are directly from the experimental known protein-ligand pairs, whereas the negative data are created by cross-combination of proteins and ligands from the PDBbind database. After finishing the training, we obtained a model with well-trained weight, which can be used in future applications. We had written homemade scripts to prepare the model input and model application, respectively. A directly loaded trained DFCNN model is enough to run the prediction; no extra parameter settings are needed. The virtual screening procedure by DFCNN is similar to that in our previous work, which is drug repurposing against RdRp of SARS-CoV-2 (Zhang et al., 2020b). DFCNN achieved an AUC value around 0.91 for the independent testing set, which has a much better performance compared to the other four machine learning methods, that is, SVM (AUC of 0.6729), RandomForest (AUC of 0.8444), xgboost (AUC of 0.8601), and CNN (AUC of 0.8642) (Zhang et al., 2019a). The model is about ∼100,000 times faster than Autodock Vina (Trott and Olson, 2010) in predicting protein-ligand binding probability (range 0–1), mostly due to no need to explore the protein-drug complex conformation. We have previously validated the efficiency and effectiveness of the DFCNN by missing the known active compounds in a dataset of ∼10 million compounds and checking DFCNN’s ability to recall the known active compounds by scoring. The following formula is used:
where
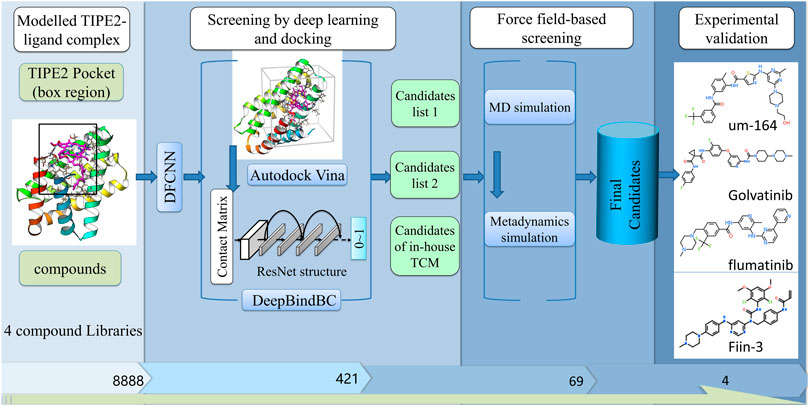
FIGURE 1. The diagram of our screening pipeline. The screening procedures include preliminary screening by deep learning and docking, fine screening by force field-based methods, and fine experimental validation.
DeepBindBC, an in-house deep learning-based software, is used for structure-based drug screening. Unlike the DFCNN, the input of DeepBindBC includes both the physical-chemical information and spatial information between the protein-ligand interfaces (Figure 1A); hence, DeepBindBC can achieve higher accuracy but requires protein-drug complex structure information as input generated by Autodock Vina.
Autodock Vina is used to dock the target with the potential ligands (Steffen et al., 2010). The pocket is determined by the location of the ligand in the template protein. Moreover, we set the cavity volume space at 2.5, 2.5, and 2.5 nm in x, y, and z dimensions from the pocket mass center. AutoDock Tools were used to convert the PDB file format to the PDBQT file format (Morris et al., 2009). The exhaustiveness was set to 8; the num_modes was set to 20, and the energy range was set to 3. The scoring function and optimization algorithm of Autodock Vina has been well discussed in a previous article (Steffen et al., 2010).
The DeepBindBC is a ResNet model trained over the PDBbind database. In DeepBindBC, the protein-ligand interaction interface information will be converted into figure-like metrics, similar to DeepBindRG (Zhang et al., 2019b). By incorporating the cross-docking (docking proteins and ligands from different experimental complexes) conformation as negative training data, DeepBindBC is highly possible to distinguish non-binders.
Further drug screening was carried out by force field-based molecular dynamics (MD) simulations. In this study, we selected 69 compound binding complexes, which were predicted candidates by previous deep learning methods, for MD simulation, respectively. Binding free energy calculation can be estimated by metadynamics simulations to explore whether protein-ligand will bind in solution. Metadynamics relies on the addition of a bias potential to sample the free energy landscape along with a specific collective variable of interest (Laio and Gervasio, 2008; Saleh et al., 2017). The detailed MD simulation and followed metadynamics simulation were illustrated in Supplementary Material Section S1.
The USCF Chimera, VMD, ICM-browserPro, and Discovery Studio Visualizer 2019 were used to generate the structure and visualize the 2D protein-ligand interactions (Humphrey et al., 1996; Pettersen et al., 2004; BIOVIA, 2005; icm_browser_pro, 2020).
Recombinant TIPE2 proteins were purchased from the Abcam company and biotinylated by the EZ-Link biotinylation reagent (Thermo Fisher Scientific). Briefly, TIPE2 proteins and biotinylation reagent were mixed with a 1:1 molar ratio and then incubated for 2 h at 4°C. The mixture was purified by 3K MWCO dialysis cassettes (Thermo Fisher Scientific) to remove unreacted biotinylation reagent.
Affinities (KD values) were determined by BLI using an OctetK2 instrument (PALL ForteBio). Biotinylated TIPE2 proteins in PBS were loaded onto Super Streptavidin (SSA) biosensors (ForteBio). Average saturation response levels of 5 nm were observed in 10 min for TIPE2 proteins. PBS with 0.1% BSA, 0.01% Tween-20, and 1% DMSO was prepared as the assay buffer. Sensors with TIPE2 proteins were then washed in assay buffer for 10 min to remove nonspecifically bound proteins and establish stable baselines before starting association-dissociation cycles with small molecules. DMSO only references were included in all assays. Raw kinetic data collected were processed in the Data Analysis software provided by the manufacturer using double reference subtraction in which both DMSO only reference and an inactive reference were subtracted. The resulting data were analyzed based on a 1:1 binding model from which kon and koff values were obtained and then KD values were calculated.
PIP2 (phosphatidylinositol 4,5-bisphosphate) was purchased from the Echelon company. To measure competitive binding to TIPE2 protein between PIP2 and UM-164 by BLI, two concentrations (5 and 25 μM) of UM-164 were added to PBS with 0.01% Tween-20 as two sets of assay buffers, respectively, and the interference patterns from the assay buffer without UM-164 and a biosensor without TIPE2 protein were used as two of controls. TIPE2 proteins (10 μg/ml) in three assay buffers were loaded to Streptavidin (SA) biosensors (ForteBio) for 60 s and flowed 150 μM of PIP2 for the 60 s, respectively. The competitive characteristics were analyzed by the Data Analysis software.
The screening pipeline is illustrated in Figure 1. Here, we first identified the potential ligand binding pocket and then used a deep learning-based method and docking to do a fast preliminary screening. After that, molecular dynamics simulation-based methods were used to further shortlist the results. We have gone over the final candidates and have a preference for those that have a good score and a strong interaction based on a 2D snapshot. Finally, we selected four compounds from the final candidates and carried out experimental validation. However, the other compounds that were not chosen for experiment validation; nevertheless, they have a good chance of binding the TIPE2. The result shows that among the four compounds, one has a low-micromolar binding affinity with a KD value of 4.97 µM, two compounds have a binding affinity with a KD value of 189.1 and 298.3 µM, and one has a weak binding affinity with a KD value of 858,100 µM.
Unlike many other drug screening pipelines, our screening strategy is highly hybrid and takes full usage of different methods’ advantages to achieve a balance between accuracy and efficacy. In the deep learning screening section, we have used the molecular vector-based method DFCNN first since it is highly efficient as it does not rely on docking structure, making it quite suitable for large-scale preliminary drug screening (Zhang et al., 2019a, 2020a, 2020b; Majumdar et al., 2021). The output list from DFCNN was used for docking and structure-based deep learning prediction by the DeepBindBC method (Zhang et al., 2020b), which makes the prediction more robust. In the force field-based screening section, we first carry MD simulations for the predicted protein-ligand complexes from the output of the deep learning screening section. After that, metadynamics simulation was continued from previous results to further examine the binding free energy. The MD simulation results can provide predicted atomic interaction details and binding stability; meanwhile, metadynamics simulation predicted protein-ligand binding free energy landscape. Altogether, this pipeline provides a highly efficient and accurate way to identify inhibitors against TIPE2. The molecular diversity is extremely large; hence, screening over some large databases would be desirable. This pipeline provides important clues that may help to virtual screening over billions of compounds databases while keeping the balance between accuracy and efficacy.
Figure 2 and Supplementary Figure S1 together display the interaction between four selected compounds that interacted with the TIPE2 from the last frame of 100 ns MD simulation. We observed that the UM-164 has two hydrogen bonds, a Pi–Pi stacking, and a halogen interaction with the TIPE2 and has many other non-hydrophobic interactions. The hydrogen bonds are formed with ASP32 and ALA98. The Pi–Pi interaction was formed with PHE150. This strongly supports the experimental result that UM-164 has the strongest binding affinity among the four test cases, with a KD value of 4.97 × 10−06.
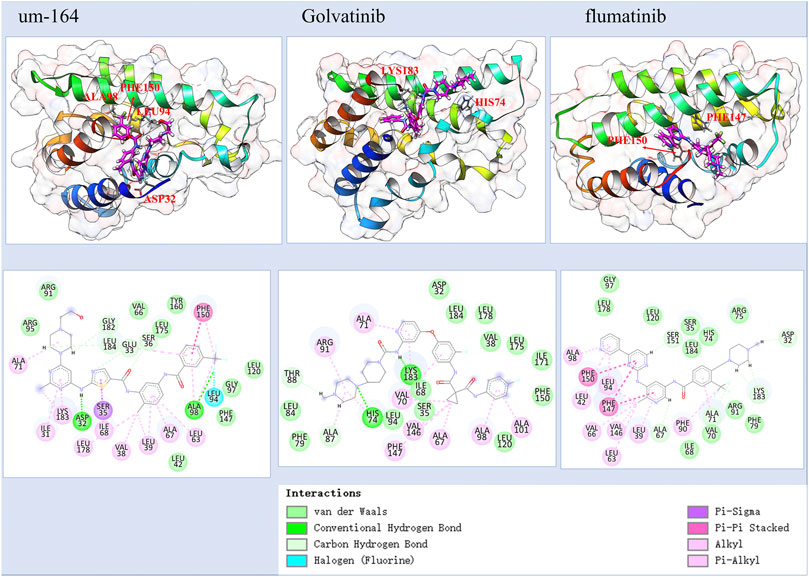
FIGURE 2. The predicted atomic interaction details of the three experimental validated potent compounds, among them, UM-164 has the strongest binding affinity with a KD value of 4.970 µM.
Golvtinib was also predicted to have formed hydrogen bond interaction with HIS74 and LYS183, respectively. The binding of flumatinib seems mostly maintained by Pi–Pi interactions and other types of hydrophobic interactions. It forms Pi–Pi interaction with PHE150 and PHE147, respectively. This supports the findings that golvatinib and flumatinib have binding affinities with KD values of 189.1 and 298.3 µM, respectively. Fiin-3 formed one hydrogen bond with ASP32 and formed Pi–Pi interaction with PHE150, as shown in Supplementary Figure S1; however, experimental results show that it only has a very little reactivity against TIPE2.
In a step-by-step manner, we obtain TIPE2 inhibitor candidates with high predicted values from DFCNN and DeepBindBC and with a low docking score. Sixteen compounds were selected with the criteria of DeepBindBC>0.99, Docking<=−10, and DFCNN>0.99, as shown in Table 1. It is found that Sennoside_A has very high DeepBindBC and DFCNN scores. Meanwhile, it also has a very low docking score, which indicates that the compound has a high potential to bind with the TIPE2. According to the prediction scores, most of the other listed compounds all have a very high chance of binding with TIPE2.
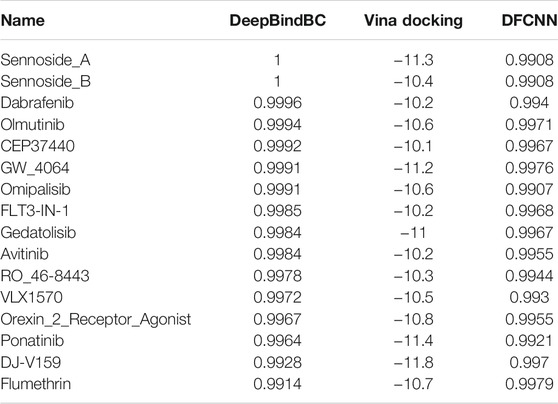
TABLE 1. The potential inhibitors of TIPE2 from the three TargetMol datasets (DeepBindBC>0.99, Vina Docking<=−10, and DFCNN>0.99). The table was ranked by DeepBindBC score in descending order.
Considering that in some cases the high docking score may be due to dominant non-specific hydrophobic interactions, we also selected candidates with criteria of DeepBindBC>0.99, Docking<=−8.5, and DFCNN>0.998, which resulted in extra 24 compounds, as shown in Table 2. We found that many compounds are high potential binders of TIPE2 according to DeepBindBC and DFCNN. For instance, the UM-164 has a DeepBindBC score of 0.9968 and a DFCNN of 0.9994.
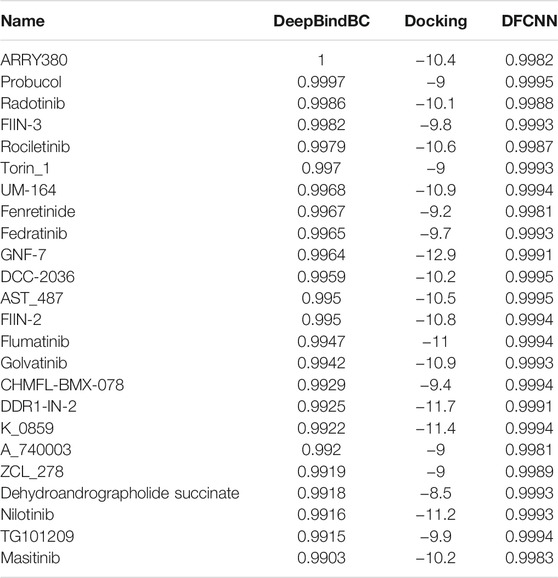
TABLE 2. The potential inhibitors of TIPE2 from the TargetMol dataset (DeepBindBC>0.99, Docking<=−8.5, and DFCNN>0.998, excluding compounds in Table 1). The table was ranked by docking score in descending order.
At the same time, we also obtained 22 candidates from the in-house TCM dataset, with the criteria of DeepBindBC>0.9, Docking<=−6, and DFCNN>0.9. Since the TCM ingredients are an important source of many drug leads, it would be attractive if there are some active compounds against TIPE2 by binding. Among the 22 TCM ingredients in Supplementary Table S1, kurarinone and astaxanthin have a high chance of binding with TIPE2 according to the prediction scores of DeepBindBC and DFCNN; both scores are above 0.99. Their docking scores are −9.4 and −10.8, respectively, which support their potential high-affinity binding.
To further validate the ligand binding stability and confirm the atomistic interaction details of the above-selected candidates with the TIPE2, a 100 ns molecular dynamics simulation was carried out for each selected complex. The binding stability was mainly estimated by RMSD of the calculated ligand and protein C alpha carbons from the simulation trajectory. The top eight stable ligand RMSD from list 1 are shown in Supplementary Figure S2. The hydrogen bond numbers over the simulation time were also shown. The detailed atomic interaction of these eight complexes from the last frame of MD simulation is displayed in Figure 3. The RMSD of the top eight most stable ligands from list 2 are shown in Supplementary Figure S3A. The corresponding hydrogen bonding number with the protein along the simulation time is shown in Supplementary Figure S3B.
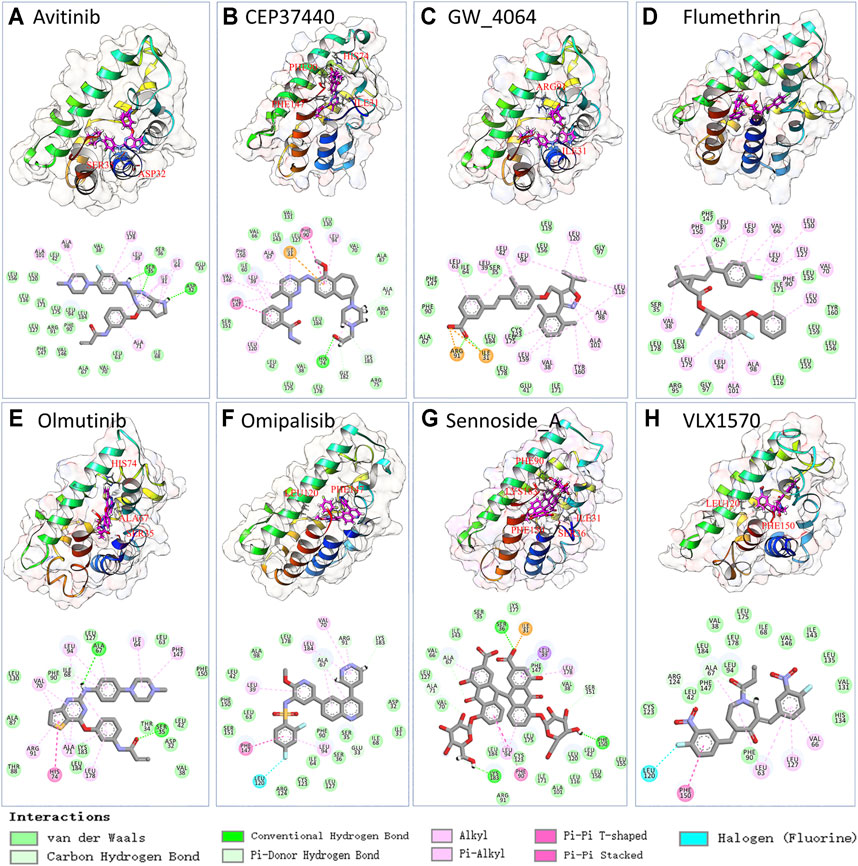
FIGURE 3. The snapshot and 2D interaction of last frame conformation from the 100 ns MD simulations trajectories of candidate list 1.
As too many potentially inhibiting compounds were found in our research, we clustered them into six groups and focused on the representative compounds of each group to better present the results, as shown in Figure 4. The compounds in the same cluster show very similar structural conformation and physical-chemical properties. It was found that cluster 1 and cluster 2 contain a large number of predicted candidates, while the representative structure of these two clusters shows that hydrophobic interaction has a dominant contribution. The representative structures of clusters 3, 5, and 6 have formed hydrogen bonds with the TIPE2 according to the 2D interaction plot for the last frame of MD simulation. Furthermore, we displayed snapshots of TIPE2 and six representative ligands with detailed atomic interactions from the last frame of MD simulation in Figure 5, respectively. It can be seen that the six representatives have accommodated well with the TIPE2 pockets, which indicates that all the six representatives have a high chance of binding with the TIPE2.
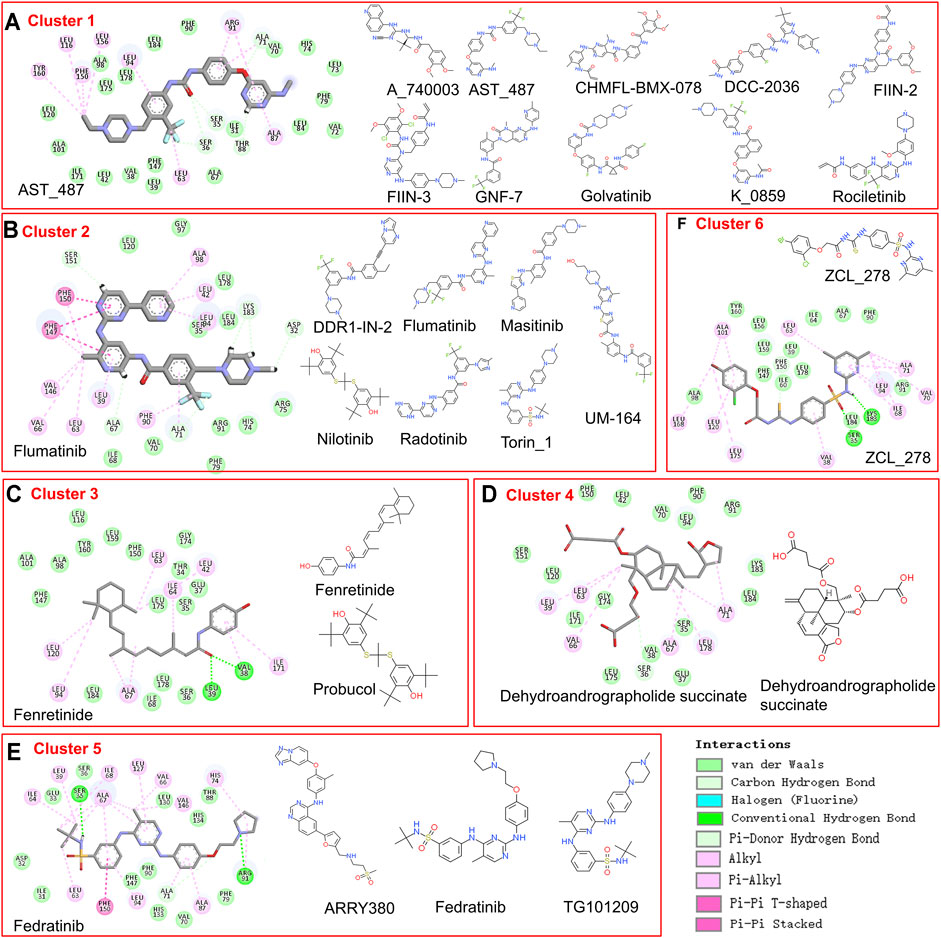
FIGURE 4. The compounds in six clusters of list 2 and the 2D interaction of each representative compound with the binding pocket.
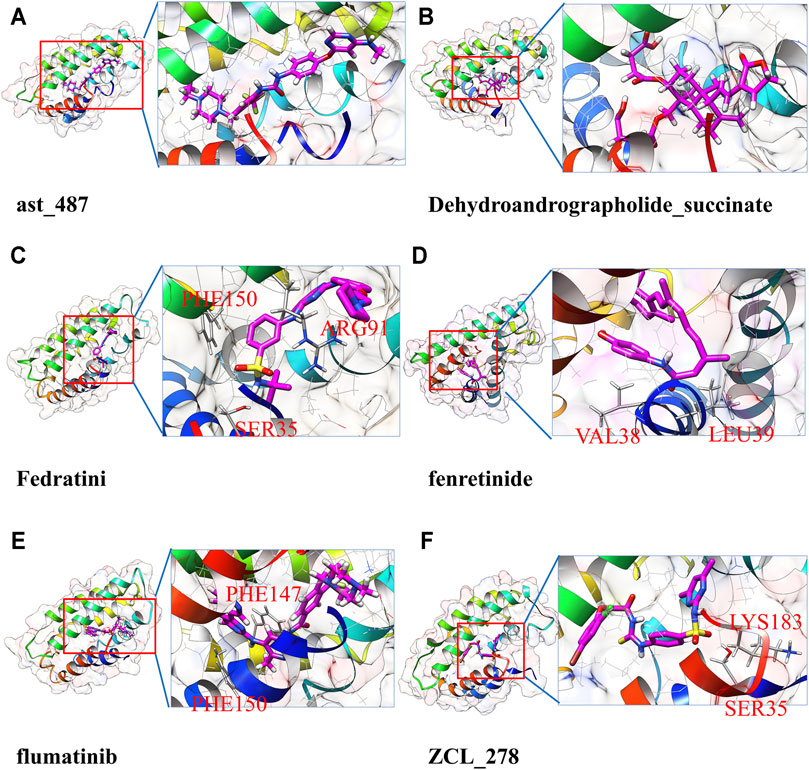
FIGURE 5. Detailed atomic interaction between TIPE2 and six representative ligands of candidate’s list 2 in the last frame of MD simulation.
To more accurately estimate the binding preference of the selected ligand candidates to TIPE2, we have examined the calculated free binding free energy landscape from the metadynamics simulation. Most candidates in list 1 and list 2 were high potential binders according to the calculated binding free energy, and their binding free energy landscape was shown in Figure 6. Among the seven selected candidates from the in-house TCM dataset, four ingredients show favorable binding with the TIPE2 according to the free energy landscape from the metadynamics simulation, as shown in Supplementary Figure S4.
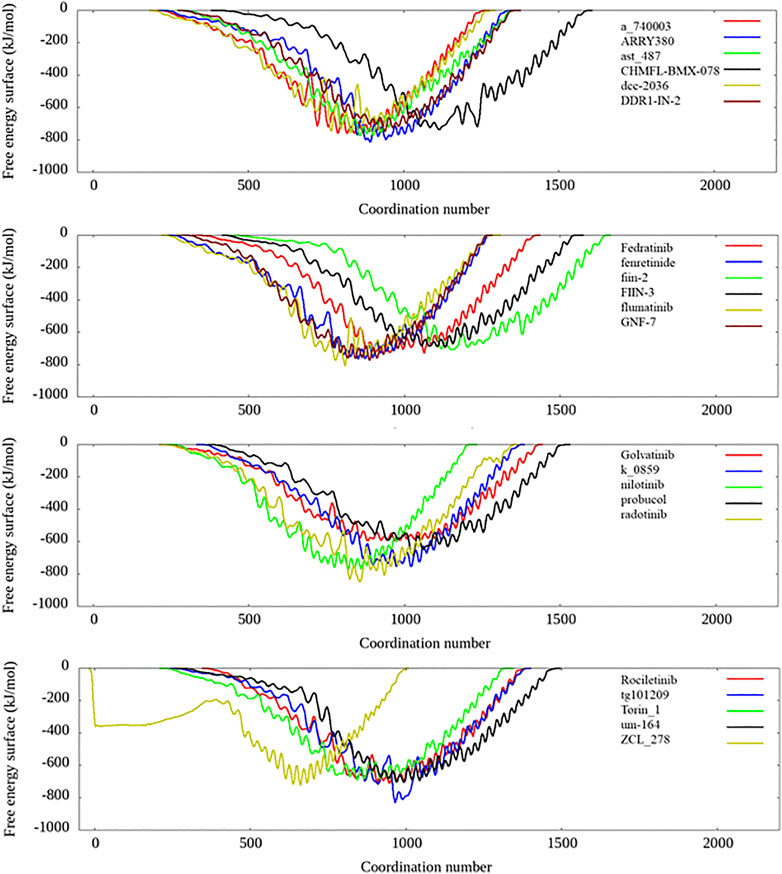
FIGURE 6. The free energy landscape of several candidates from candidate lists 1 and 2.
To measure the binding affinities of these small molecules, bio-layer interferometry using recombinant TIPE2 proteins was performed. The bio-layer interferometry (BLI) technique is extremely valuable and one of the most authoritative methods to estimate protein-ligand binding affinity (Zhou et al., 2018; Abdul Azeez et al., 2019; Maji et al., 2019). As shown in Figure 7, UM-164 was able to bind to the TIPE2 protein in appreciable potency, with a KD value of 4.97 µM, while Fedratinib and Golvatinib were bound to TIPE2 proteins with KD values of 298 and 189 μM, respectively. Nevertheless, FIIN-3 exhibited a lack of reactivity against TIPE2 protein.
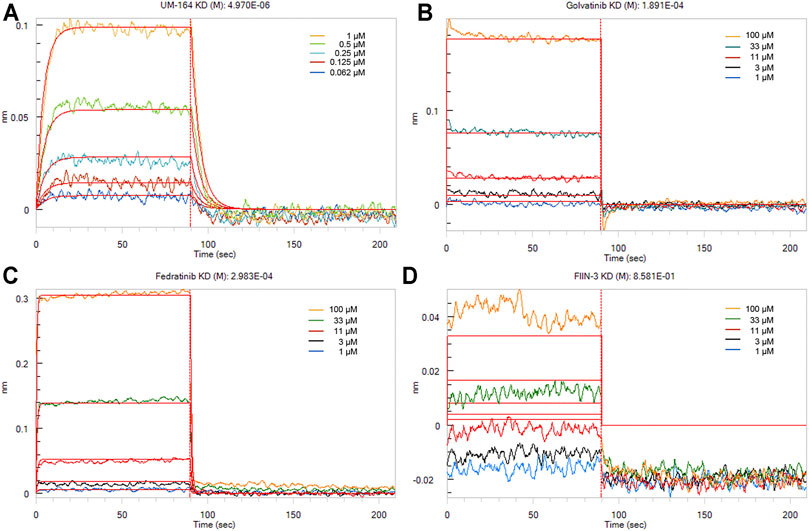
FIGURE 7. The binding affinities of small molecules with TIPE2. TIPE2 proteins were loaded onto SSA biosensors for BLI analysis and incubated with gradient concentrations of UM-164 (A), Golvatinib (B), Fedratinib (C), and FIIN-3 (D), respectively. The raw binding curves at different concentrations are shown in five kinds of color, and red curves are the best global fits to the data used to calculate the KD values.
To assess the ability of UM-164 to inhibit the interaction between TIPE2 and PIP2, competitive binding assays were performed by BLI. Immobilized biotinylated TIPE2 proteins were saturated with PIP2 in the presence of 5 and 25 μM UM-164 or in the absence of UM-164, respectively. Compared with 5 μM UM-164 and without UM-164, when 25 μM UM-164 was added to the assay buffer, a dramatic reduction of PIP2 bound to TIPE2 protein was observed (Figure 8) possibly because UM-164 and PIP2 may recognize the same or overlapped site on TIPE2 protein, or allosteric effects, and therefore UM-164 was able to interfere with the binding of TIPE2 and PIP2. These results indicate that UM-164 is a potential inhibitor against TIPE2 function.
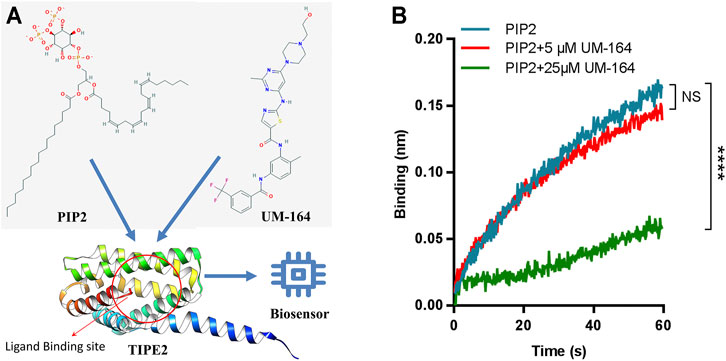
FIGURE 8. Competitive binding to TIPE2 protein between PIP2 and UM-164. (A) Diagram of PIP2 and UM-164 competitive binding toTIPE2. (B) The competitive BLI results. For BLI assays, immobilized biotinylated TIPE2 proteins were bound to PIP2 in the presence of 5 μM UM-164 (red) and 25 μM UM-164 (green) and without UM-164 (blue), respectively, and then respective binding signals of PIP2 were shown. ****: compared with the group without UM-164, p < 0.0001 (unpaired t-test). NS, no significance.
Constructing a computational-aided drug screening pipeline can greatly facilitate the process of finding drug leads. Finding effective inhibitors with limited experiments would be a challenging task. We have already performed virtual screening with a hybrid method of deep learning and molecular dynamics simulation in our previous work. Together with a previous SARS-Cov2 drug repurposing work, we further show the workable and high efficiency of the hybrid screening strategy. With the high efficiency of deep learning and relative accuracy of MD simulation, our drug screening pipeline demonstrated good performance. Deep learning and docking-based methods can greatly narrow down the candidate pool with affordable computational cost. Since the TIPE2 has a relatively small size and a large ligand binding cavity, it is possible for us to run relatively large-scale MD simulations and metadynamics simulations that can lead to a more accurate prediction.
MD simulations provide more accurate atomic details of the protein-ligand interaction, making the prediction much more explainable, and help gain insights into the mechanisms of ligand binding. Taking the MD simulation part alone, it is the first time that we carried more than 60 MD simulations for screening inhibitors against a given protein target. Thanks to the increasingly available computational resources nowadays, the MD simulation would play a much important role in future drug screening.
It is the first time that a low-nanomolar affinity binder of TIPE2 (UM-164) has been discovered, and its binding is strong enough to block the binding of native ligand PIP2, indicating that the UM-164 can inhibit TIPE2’s function. To explore which kinds of ligands prefer binding with the TIPE2, we have carefully checked the atomic interaction of candidates binding with TIPE2. We also provide a large number of TIPE2 inhibitor candidates, which can greatly promote the future discovery of new inhibitors by future experimental validation. Together, it may promote the future development of novel drugs that may cure lung cancer.
Compared to ligand-based drug screening, such as LigandScout (Wolber and Langer, 2005), our pipeline can select potential drugs with a much wider chemical space, which greatly promotes real novel drug development. As we can see from Figure 3, the potential TIPE2 binding ligands found by our method are diverse and have been divided into several categories. Compounds with quite different structures can bind into the same target, while traditional screening-based on ligand similarity cannot achieve this. Candidates with different structures provided the chance to develop diversified drugs.
With the help of the hybrid drug screening pipeline, we found 69 drug candidates against TIPE2 function. We selected four candidates for final experimental tests, in which UM-164 could bind to TIPE2 with low-micromolar affinity (KD = 4.97 µM). In addition, Golvalinib and Fedratinib were bound to TIPE2 with KD values of 189.1 and 298.3 µM, respectively. Only FIIN-3 has a weak binding affinity with a KD value of 8.581 × 10−1 M. This greatly expanded the available inhibitors for TIPE2, which is a potential novel drug target related to cancer and inflammation. Moreover, the high success rate of the hybrid drug screening pipeline indicates a huge potential for the implementation of similar methods in the drug discovery of other targets. The active compounds we found and their possible derivatives have the potential to facilitate drug development for TIPE2. The detailed interaction between those inhibitors and TIPE2 also provides insight into understanding the binding mechanism and drug design and refinement.
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding authors.
HZ and YP designed the study. HZ, KS, HW, ZC, and DW performed computations and data analyses. JL and ZL performed the bio-layer interferometry (BLI) and competitive BLI experiments. All authors contributed to writing the manuscript. YP, YC, XW, and YW supervised the study. All authors read and approved the final manuscript.
This work was supported in part by the Shenzhen KQTD Project (No. KQTD20200820113106007) (YP), National Key R&D Program of China (2019YFA0906100) (XW), Key-Area Research and Development Program of Guangdong Province (2019B020201014) (XW), Guangdong Basic and Applied Basic Research Foundation (2020A1515110840) (JL), and the Shenzhen Basic Research Fund under Grant No. JCYJ20190807170801656 (JL). This study was supported in part by the National Key Research and Development Program of China under Grant No. 2018YFB0204403 (YW) and Research Funding of Shenzhen (JCYJ201803053000708) (XW). This work was partly supported by Strategic Priority CAS Project XDB38000000 (YW), National Science Foundation of China under Grant No. U1813203 (YW), the Shenzhen Basic Research Fund under Grants Nos. KQTD20200820113106007 (YW) and RCYX2020071411473419 (YW), and CAS Key Lab under Grant No. 2011DP173015 (YW).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2021.772296/full#supplementary-material
Abdul Azeez, K. R., Chatterjee, S., Yu, C., Golub, T. R., Sobott, F., and Elkins, J. M. (2019). Structural Mechanism of Synergistic Activation of Aurora Kinase B/C by Phosphorylated INCENP. Nat. Commun. 10, 3166. doi:10.1038/s41467-019-11085-0
Bordoloi, D., Banik, K., Padmavathi, G., Vikkurthi, R., Harsha, C., Roy, N. K., et al. (2019). TIPE2 Induced the Proliferation, Survival, and Migration of Lung Cancer Cells through Modulation of Akt/mTOR/NF-Κb Signaling Cascade. Biomolecules 9, 836. doi:10.3390/biom9120836
Goldsmith, J. R., and Chen, Y. H. (2017). Regulation of Inflammation and Tumorigenesis by the TIPE Family of Phospholipid Transfer Proteins. Cell. Mol. Immunol. 14, 482–487. doi:10.1038/cmi.2017.4
Gorgulla, C., Boeszoermenyi, A., Wang, Z. F., Fischer, P. D., Coote, P. W., Padmanabha Das, K. M., et al. (2020). An Open-Source Drug Discovery Platform Enables Ultra-large Virtual Screens. Nature 580, 663–668. doi:10.1038/s41586-020-2117-z
Humphrey, W., Dalke, A., and Schulten, K. (1996). VMD: Visual Molecular Dynamics. J. Mol. Graph. 14 (33–8), 33–38. doi:10.1016/0263-7855(96)00018-5
icm_browser_pro (2020). icm_browser_pro. Available at: http://www.molsoft.com/icm_browser_pro.html.
Jaeger, S., Fulle, S., and Turk, S. (2018). Mol2vec: Unsupervised Machine Learning Approach with Chemical Intuition. J. Chem. Inf. Model. 58, 27–35. doi:10.1021/acs.jcim.7b00616
Kaushik, A. C., Mehmood, A., Dai, X., and Wei, D. Q. (2020). A Comparative Chemogenic Analysis for Predicting Drug-Target Pair via Machine Learning Approaches. Sci. Rep. 10, 6870. doi:10.1038/s41598-020-63842-7
Laio, A., and Gervasio, F. L. (2008). Metadynamics: a Method to Simulate Rare Events and Reconstruct the Free Energy in Biophysics, Chemistry and Material Science. Rep. Prog. Phys. 71, 126601. doi:10.1088/0034-4885/71/12/126601
Liu, M. W., Su, M. X., Zhang, W., Zhang, L. M., Wang, Y. H., and Qian, C. Y. (2015a). Rhodiola Rosea Suppresses Thymus T-Lymphocyte Apoptosis by Downregulating Tumor Necrosis Factor-α-Induced Protein 8-like-2 in Septic Rats. Int. J. Mol. Med. 36, 386–398. doi:10.3892/ijmm.2015.2241
Liu, Z., Li, Y., Han, L., Li, J., Liu, J., Zhao, Z., et al. (2015b). PDB-wide Collection of Binding Data: Current Status of the PDBbind Database. Bioinformatics 31, 405–412. doi:10.1093/bioinformatics/btu626
Lou, Y., and Liu, S. (2011). The TIPE (TNFAIP8) Family in Inflammation, Immunity, and Cancer. Mol. Immunol. 49, 4–7. doi:10.1016/j.molimm.2011.08.006
Luan, Y. Y., Dong, N., Xie, M., Xiao, X. Z., and Yao, Y. M. (2014). The Significance and Regulatory Mechanisms of Innate Immune Cells in the Development of Sepsis. J. Interferon Cytokine Res. 34, 2–15. doi:10.1089/jir.2013.0042
Maji, B., Gangopadhyay, S. A., Lee, M., Shi, M., Wu, P., Heler, R., et al. (2019). A High-Throughput Platform to Identify Small-Molecule Inhibitors of CRISPR-Cas9. Cell 177, 1067–1079. doi:10.1016/j.cell.2019.04.009
Majumdar, S., Nandi, S. K., Ghosal, S., Ghosh, B., Mallik, W., Roy, N. D., et al. (2021). Deep Learning-Based Potential Ligand Prediction Framework for COVID-19 with Drug-Target Interaction Model. Cogn. Comput., 1–13. doi:10.1007/s12559-021-09840-x
Morris, G. M., Huey, R., Lindstrom, W., Sanner, M. F., Belew, R. K., Goodsell, D. S., et al. (2009). AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 30, 2785–2791. doi:10.1002/jcc.21256
Padmavathi, G., Banik, K., Monisha, J., Bordoloi, D., Shabnam, B., Arfuso, F., et al. (2018). Novel Tumor Necrosis Factor-α Induced Protein Eight (TNFAIP8/TIPE) Family: Functions and Downstream Targets Involved in Cancer Progression. Cancer Lett. 432, 260–271. doi:10.1016/j.canlet.2018.06.017
Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., et al. (2004). UCSF Chimera-Aa Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 25, 1605–1612. doi:10.1002/jcc.20084
Roy, A., Yang, J., and Zhang, Y. (2012). COFACTOR: an Accurate Comparative Algorithm for Structure-Based Protein Function Annotation. Nucleic Acids Res. 40, W471–W477. doi:10.1093/nar/gks372
Saleh, N., Ibrahim, P., Saladino, G., Gervasio, F. L., and Clark, T. (2017). An Efficient Metadynamics-Based Protocol to Model the Binding Affinity and the Transition State Ensemble of G-Protein-Coupled Receptor Ligands. J. Chem. Inf. Model. 57, 1210–1217. doi:10.1021/acs.jcim.6b00772
Steffen, C., Thomas, K., Huniar, U., Hellweg, A., Rubner, O., and Schroer, A. (2010). TmoleX--a Graphical User Interface for TURBOMOLE. J. Comput. Chem. 31, 2967–2970. doi:10.1002/jcc.21576
Trott, O., and Olson, A. J. (2010). AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 31, 455–461. doi:10.1002/jcc.21334
UniProt Consortium (2021). UniProt: the Universal Protein Knowledgebase in 2021. Nucleic Acids Res. 49, D480–D489. doi:10.1093/nar/gkaa1100
Wolber, G., and Langer, T. (2005). LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters. J. Chem. Inf. Model. 45, 160–169. doi:10.1021/ci049885e
Yan, D., Wang, J., Sun, H., Zamani, A., Zhang, H., Chen, W., et al. (2020). TIPE2 Specifies the Functional Polarization of Myeloid-Derived Suppressor Cells during Tumorigenesis. J. Exp. Med. 217. doi:10.1084/jem.20182005
Yan, D., Hong, Wang., hui, J., and Wanchun, X. (2017). TIPE2 Is a Novel Direct Target of STAT3 in MDSC and Inhibition of its Expression on MDSC Enhanced T Cell Activation in Tumor. J. Immunol. 198, 205. Available at: http://www.jimmunol.org/content/198/1_Supplement/205.3.abstract.
Yuan, Z., Zhang, B., Zhuang, J., Yang, X., Yu, J., Xu, Q., et al. (2020). Chlorotoxin Fusion Protein Regulates miR-374a and TNFAIP8 Expression and Inhibits Glioma Cell Proliferation and Promotes Apoptosis. Cytotechnology 72, 685–694. doi:10.1007/s10616-020-00411-w
Zhang, H., Liao, L., Cai, Y., Hu, Y., and Wang, H. (2019a). IVS2vec: A Tool of Inverse Virtual Screening Based on Word2vec and Deep Learning Techniques. Methods 166, 57–65. doi:10.1016/j.ymeth.2019.03.012
Zhang, H., Liao, L., Saravanan, K. M., Yin, P., and Wei, Y. (2019b). DeepBindRG: a Deep Learning Based Method for Estimating Effective Protein-Ligand Affinity. PeerJ 7, e7362. doi:10.7717/peerj.7362
Zhang, H., Saravanan, K. M., Yang, Y., Hossain, M. T., Li, J., Ren, X., et al. (2020a). Deep Learning Based Drug Screening for Novel Coronavirus 2019-nCov. Interdiscip. Sci. 12, 368–376. doi:10.1007/s12539-020-00376-6
Zhang, H., Yang, Y., Li, J., Wang, M., Saravanan, K. M., Wei, J., et al. (2020b). A Novel Virtual Screening Procedure Identifies Pralatrexate as Inhibitor of SARS-CoV-2 RdRp and it Reduces Viral Replication In Vitro. Plos Comput. Biol. 16, e1008489. doi:10.1371/journal.pcbi.1008489
Zhang, S., Zhang, Y., Wei, X., Zhen, J., Wang, Z., Li, M., et al. (2010). Expression and Regulation of a Novel Identified TNFAIP8 Family Is Associated with Diabetic Nephropathy. Biochim. Biophys. Acta 1802, 1078–1086. doi:10.1016/j.bbadis.2010.08.003
Zhang, X., Wang, J., Fan, C., Li, H., Sun, H., Gong, S., et al. (2009). Crystal Structure of TIPE2 Provides Insights into Immune Homeostasis. Nat. Struct. Mol. Biol. 16, 89–90. doi:10.1038/nsmb.1522
Zhang, Y. (2008). I-TASSER Server for Protein 3D Structure Prediction. BMC Bioinformatics 9, 40. doi:10.1186/1471-2105-9-40
Zhang, Z., Liu, L., Liu, C., Cao, S., Zhu, Y., and Mei, Q. (2016). TIPE2 Suppresses the Tumorigenesis, Growth and Metastasis of Breast Cancer via Inhibition of the AKT and P38 Signaling Pathways. Oncol. Rep. 36, 3311–3316. doi:10.3892/or.2016.5192
Zhou, P., She, Y., Dong, N., Li, P., He, H., Borio, A., et al. (2018). Alpha-kinase 1 Is a Cytosolic Innate Immune Receptor for Bacterial ADP-Heptose. Nature 561, 122–126. doi:10.1038/s41586-018-0433-3
Zhou, Y., Webber, S. E., Murphy, D. E., Li, L.-S., Dragovich, P. S., Tran, C. V., et al. (2008). Novel HCV NS5B Polymerase Inhibitors Derived from 4-(1′,1′-Dioxo-1′,4′-Dihydro-1′λ6-Benzo[1′,2′,4′]thiadiazin-3′-Yl)-5-Hydroxy-2h-Pyridazin-3-Ones. Part 1: Exploration of 7′-substitution of Benzothiadiazine. Bioorg. Med. Chem. Lett. 18, 1413–1418. doi:10.1016/j.bmcl.2008.01.007
Keywords: TIPE2, UM-164, virtual screening, deep learning, molecular dynamics simulation
Citation: Zhang H, Li J, Saravanan KM, Wu H, Wang Z, Wu D, Wei Y, Lu Z, Chen YH, Wan X and Pan Y (2021) An Integrated Deep Learning and Molecular Dynamics Simulation-Based Screening Pipeline Identifies Inhibitors of a New Cancer Drug Target TIPE2. Front. Pharmacol. 12:772296. doi: 10.3389/fphar.2021.772296
Received: 07 September 2021; Accepted: 02 November 2021;
Published: 23 November 2021.
Edited by:
Santiago Gómez-Ruiz, Rey Juan Carlos University, SpainReviewed by:
Jacob Durrant, University of Pittsburgh, United StatesCopyright © 2021 Zhang, Li, Saravanan, Wu, Wang, Wu, Wei, Lu, Chen, Wan and Pan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yi Pan, eWkucGFuQHNpYXQuYWMuY24=; Xiaochun Wan, eGMud2FuQHNpYXQuYWMuY24=; Youhai H. Chen, eWguY2hlbkBzaWF0LmFjLmNu
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.