 Ines Gallego-Landin1
Ines Gallego-Landin1 Olga Valverde
Olga Valverde
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol., 06 December 2021
Sec. Neuropharmacology
Volume 12 - 2021 | https://doi.org/10.3389/fphar.2021.762738
This article is part of the Research TopicUnraveling Vulnerability Factors in Addiction Drug Use and Potential TreatmentsView all 20 articles
Major depressive disorder is a high-impact, debilitating disease and it is currently considered the most prevalent mental illness. It is associated with disability, as well as increased morbidity and mortality. Despite its significant repercussions in our society, its exact pathophysiology remains unclear and therefore, available antidepressant treatment options are limited and, in some cases, ineffective. In the past years, research has focused on the development of a multifactorial theory of depression. Simultaneously, evidence supporting the role of the endocannabinoid system in the neurobiology of neuropsychiatric diseases has emerged. Studies have shown that the endocannabinoid system strongly impacts neurotransmission, and the neuroendocrine and neuroimmune systems, which are known to be dysfunctional in depressive patients. Accordingly, common antidepressants were shown to have a direct impact on the expression of cannabinoid receptors throughout the brain. Therefore, the relationship between the endocannabinoid system and major depressive disorder is worth consideration. Nevertheless, most studies focus on smaller pieces of what is undoubtedly a larger mosaic of interdependent processes. Therefore, the present review summarizes the existing literature regarding the role of the endocannabinoid system in depression aiming to integrate this information into a holistic picture for a better understanding of the relationship between the two.
Major Depressive Disorder (MDD) is the most common mood disorder characterized by the loss of interest or pleasure in activities, unjustified feelings of worthlessness and presence of depressed mood or aversion to activity (American Psychiatric Association, 2013).
According to the World Health Organization, each year, 25% of the European population suffers from anxiety or depressive disorders (World Health Organization, 2020). Its prevalence has increased by 18% between 2005 and 2015 (Akil et al., 2018) and currently, it constitutes the leading cause of disability. It is calculated that around 80% of MDD patients suffer from some sort of impairment during their daily life (Pratt and Brody, 2008). Importantly, this disorder occurs chronically throughout the lifespan: half of the patients undergoing depressive episodes continue to experience them with increasing frequency and severity over time. Furthermore, untreated MDD is the leading cause of suicide (National Collaborating Centre for Mental Health (UK), 2010) which, simultaneously is the second leading cause of premature death among 15–29 year-old individuals and number three among the 15–44 age group (Bachmann, 2018). On this basis, the impact of MDD in society should not be dismissed.
Currently, the most common treatment for MDD involves administration of antidepressant medications combined with psychiatric or psychological treatment. Although these medications do have positive effects, less than 50% of the patients accomplish full remission after the first pharmacological treatment (Trivedi et al., 2006).
Furthermore, the large-scale societal effects of MDD are highlighted by the responses to global coronavirus disease 2019 (COVID-19). Notably, due to the imposed lockdown, epidemiological studies show an increase in the percentage of people reporting mood disorders (Benke et al., 2020; Morin et al., 2020). Between August 2020 and February 2021, the number of individuals with recent anxiety or depressed symptoms grew by 5% (from 36.4 to 41.5%) (Vahratian et al., 2021). Therefore, the relevance of MDD-related studies is, arguably, more important than ever.
Unfortunately, although many risk factors have been identified to contribute to the development of MDD, the current hypothesized etiologies fail to explain its underlying mechanisms due to the complex interplay of the risk factors and the individual differences in symptomatology. For instance, social, genetic, hormonal or lifestyle factors all play a role and contribute to a higher risk for MDD.
Recently, the field of the endocannabinoids has grown in popularity since the discovery of endogenous cannabinoid receptors in the brain (Matsuda et al., 1990) and their most relevant endocannabinoids ligands. Multiple studies have reported its role on a variety of brain structures and cognitive functions like memory, appetite, metabolism, immune system, mood and sleep (Murillo-Rodriguez et al., 2011; Tanasescu and Constantinescu, 2010; Zanettini et al., 2011).
Although this field of research is relatively novel, there exist millennia-old reports about the use of the Cannabis Sativa plant as a medicinal herb to treat conditions like anxiety and mania (Zuardi, 2006). Currently, in the United States, a cross-sectional survey revealed that depression is the third reported condition (50.3% of users) for usage of therapeutic cannabis (Sexton et al., 2016), and users reported a 86% reduction in symptomatology (Sexton et al., 2016).
In accordance with the increase of anxiety and depressive symptoms during the COVID-19 pandemic, a recent study reported increased cannabis consumption in those who declared usage to cope with depressive symptoms (Bartelet al., 2020). Similarly, during the lockdown, more users increased than decreased cannabis consumption regarding both, frequency and quantity (Laar et al., 2020). This is in line with the observation that cannabis users increase use during times of elevated stress (Kaplan et al., 1986).
On this basis, a potential role for the endocannabinoid system (ECS) as a contributor to the pathophysiology of MDD has been explored (Patel and Hillard, 2009). In fact, ongoing clinical studies are already investigating cannabinoid-based medications as treatment options for MDD (Sarris et al., 2020). Nevertheless, evidence on the use of these substances is scarce and future research is needed.
The present review aims to provide a concise overview of the existing knowledge on the ECS and its influence on the development of MDD. First, a summary of the most relevant and recent findings regarding both phenomena will be presented. Second, and more specific, the article will highlight the potential of the ECS as a unionizing figure between the currently proposed models for the pathophysiology of MDD, focusing on the most recent empirical evidence associating both phenomena as well as the existing knowledge gaps that should be addressed by future research.
MDD is one of the most common mood disorders worldwide. Although the prevalence percentage varies among reports, it is estimated that 3.4–4.4% of the global population suffers from MDD, which translates to 264–322 million people worldwide (World Health Organization, 2017; Dattani et al., 2018). It has been observed to be twice as common in females (5.1%) than in males (3.6%), although the fundamental cause of this gender gap has not been identified. Several impact factors have been proposed, such as: biological differences as well as socioeconomic factors like discrimination and poverty (Belle and Doucet, 2003; Rai et al., 2013). In particular, females also suffer specific forms of MDD like postpartum depression and postmenopausal depression and anxiety. These are associated with alterations in the fluctuation of ovarian hormones, which might directly or indirectly contribute to the elevated prevalence (Albert, 2015). Nonetheless, the underlying mechanisms are unclear and female-specific treatments have not yet been developed.
The most prescribed treatment for MDD is the administration of common antidepressants. Among them, Selective Serotonin Reuptake Inhibitors (SSRIs) are the most widely prescribed (64.2% of US patients in 2015), followed by Serotonin and Norepinephrine Reuptake Inhibitors (SNRIs) (16.4% of patients) (Luo et al., 2020). In fact, approximately 10–35% of patients do not remit from MDD even after several treatment attempts (Nemeroff, 2007; Kubitz et al., 2013).
This lack of availability of reliable medication for such a high-impact, debilitating disease is, in part, due to the inadequacy of current hypothesized etiologies to explain its underlying mechanisms. The above-mentioned available medications are a result of a serendipitous discovery of the antidepressant effects of monoamine oxidase inhibitor (MOAIs) Iproniazid during the 1950’s, which was used at the time against tuberculosis (López-Muñoz and Alamo 2009). Furthermore, also during this decade, imipramine, the first identified tricyclic antidepressant (TCAs), was synthesized for the first time as an antihistaminic medication (López-Muñoz and Alamo 2009). Initially, although academic psychiatrists of the time regarded these drugs as cures for certain severe depressive states, the mechanisms by which these were exerted remained unknown. The first clue arrived with the observation that both classes of antidepressants increased catecholamine levels in the brain via distinct biochemical pathways (Spectoret al., 1960). This leads to the development of the monoamine theory, which inaugurated the modern psychopharmacological era in psychiatry. Shortly, the monoamine theory hypothesized that, since the enhancement of monoamine concentrations in the brain had antidepressant properties, depression itself was a consequence of depletion of centrally available monoamines. This generated a debate over the importance of different monoamines in the etiology of MDD. Two years later, serotonin (5-HT) deficiency was linked to the development of MDD (Coppen, 1967). Thus, after these years, there were two competing monoamine theories which were held for decades, to later evolve into monoamine receptor theories, associating MDD with the alteration of this neurotransmission (Stahl, 1984).
This way, the monoamine hypothesis of MDD dominated our understanding of the pathophysiology of depression and the action of the available antidepressants. Overall, it is reasonable to hypothesize that depressive symptoms are a result of inadequate monoamine neurotransmission. In fact, the best pharmacological treatments for depression, to date, compromise drugs that enhance monoaminergic levels (SSRIs, SNRIs, etc.). Nonetheless, in more recent years, serious limitations were encountered for this theory, which lead to the speculation that factors beyond monoamine imbalance and deficiency must also be involved.
One of the largest challenges in this matter is accounting for the extensive constellation of symptoms exhibited by depressed patients. The Diagnostic Statistical Manual 5 (DSM-5) requires the presence of 5 out of 9 of the described symptoms including anhedonia or depressed mood in order to be diagnosed with MDD (Uher et al., 2014). This implies that a total number of 681 possible combinations of symptoms is contemplated for each patient (Akil et al., 2018). Such variety of symptoms and individual differences among patients are clear indications of the heterogeneity of its pathophysiology (Massart et al., 2012).
Hence, currently, the hypothesis of a unitary construct as the cause of MDD has been discarded. Instead, it is believed that a multifactorial etiology of MDD provides a more complete explanation. In fact, several factors have been long associated with the development of MDD including environmental, but also genetic (Nestler et al., 2002). For example, adverse life events, like early-life stress, are considered one of the greatest risk factors (Torres-Berrío et al., 2019; Nelson and Gabard-Durnam 2020). Alternatively, some genetic influences have also been associated with the heritability of MDD, which is estimated to be approximately 38% (Kendler et al., 2006).
Since then, although several theories have been developed, none of them are able to justify the substantial variability of symptoms and risk factors of MDD patients. Therefore, there is a clear need for more integrative theories that recapitulate potentially altered mechanisms leading to such assorted symptomatology. This would be truly beneficial for the development of novel therapies, which could mitigate the enormous burden that MDD entails for our society.
The ECS is a biological modulatory system present in the central nervous system (CNS) of most vertebrates as well as in peripheric tissues. It consists of two main endocannabinoid receptors (ECRs), their endogenous ligands (endocannabinoids) and a number of specialized enzymes for the synthesis and degradation of said ligands. Furthermore, exogenous cannabinoids have also been detailed, which can be synthetic or natural, namely phytocannabinoids. The following section will shortly describe each component of the ECS as well as the most relevant findings in this area.
The first identified endogenous component of the ECS was the first cannabinoid receptor (CB1) which was discovered and cloned from the cerebral cortex of rats (Matsuda et al., 1990). In the brain, CB1 receptors are the most common G-protein coupled receptor (GPCR), expressed mainly in neurons and varying greatly across brain areas (Mackie, 2005). Nonetheless, although in much lower concentrations, CB1 receptors are also present in astrocytes, oligodendrocytes and microglia (Stella, 2009; Castillo et al., 2012).
Similarly to most GPCRs, CB1 is essentially located in the cell membrane, particularly in presynaptic axon terminals. There, they are activated by ligands released by post-synaptic neurons upon their depolarization resulting in the inhibition of further neurotransmitter release (Szabo et al., 2000). This retrograde function can resolve into short-term depolarization-induced inhibition of excitatory or inhibitory (DSE and DSI, respectively) transmission (Yoshida et al., 2002; Diana and Marty, 2004), or long-lasting forms of neuroplasticity, like long-term depression (LTD) or potentiation (LTP) (Kellogget al., 2009; Silva-Cruz et al., 2017). Furthermore, CB1 can also act pre-synaptically leading to the activation of the mitogen-activated protein kinase (MAPK) pathway (Bouaboula et al., 1995), suggesting its involvement in cell proliferation and death processes in the hippocampus (Derkinderen et al., 2001). Lastly, CB1 has also been found in alternative subcellular localizations with different functionalities from their plasma membrane equivalents, constituting a subpopulation with distinct pharmacological properties (Robledo-Menendez et al., 2021).
Although the molecular mechanisms of CB1 require further investigation, several studies have linked CB1 function to certain behavioral pathways. Furthermore, CB1 knock-out (KO) mice display an anxiogenic phenotype (Ledent et al., 1999; Zimmer et al., 1999; Martin et al., 2002), suggesting a role for the ECS in anxiety. Interestingly, effects of CB1 agonists are broadly described as biphasic, since their activation in different types of cells can lead to opposing effects on behavior (Busquets-Garcia et al., 2018).
The cannabinoid receptor (CB2) was identified and cloned in 1993 (Munro et al., 1993). It was believed that CB2 was only expressed in immune cells, but their presence in the CNS was demonstrated years later (Van Sickle et al., 2005; Ashton et al., 2006). In light of such findings, an immunoregulatory function of CB2 was proposed. Transgenic models of mice lacking CB2 receptors have contributed greatly to the investigation of its immunomodulatory role. Indeed, CB2 KO mice show exacerbated inflammation (Turcotte et al., 2016). Such findings suggest the fundamental role of CB2 receptors in maintaining immune homeostasis across the organism.
In the CNS, CB2 receptors are present in microglia, macrophages, T and B cells, and natural killers (Matias et al., 2002; Klegeris et al., 2003; Graham et al., 2010; Ramirez et al., 2013). They are a key modulator of neurological activities like nociception, neuroinflammation, and neuroprotection (Malan et al., 2001; Cabral et al., 2008), while, at the same time, its activation is devoid of psychotropic effects. Although the cellular mechanisms of CB2 function are mostly unknown. CB2 are involved in neurological functions such as anxiety, impulsive behaviors, and pain (García-Gutiérrez et al., 2012; Navarrete et al., 2012; Han et al., 2013). Nevertheless, more research is required to clarify the function of CB2 in the CNS.
Even though during the past years numerous studies have investigated the ECS, to date, no further cannabinoid receptors have been reported. Although, some results suggest that certain effects of cannabinoids are not regulated by CB1 or CB2 (Brown, 2007), which has generated some debate regarding the potential existence of a third cannabinoid receptor, nonetheless this remains merely a hypothesis.
Following the identification of the endogenous cannabinoid receptors CB1 and CB2, research focused on the study of endogenous ligands. This lead to the discovery of the first cannabinoid-like substance N-arachidonoylethanolamide, also known as anandamide (AEA) (Devane et al., 1992). Additionally, a second endocannabinomimetic compound was isolated: 2-arachidonoylglycerol (2-AG). The discovery of these two endogenous cannabinoids (namely, endocannabinoids) reaffirmed the significance of the cannabinoid receptors and their ligands as mediators of a wide variety of biological mechanisms.
Both molecules are lipophilic structures derived from arachidonic acid (Pacher et al., 2006) and they are mainly generated at postsynaptic neurons. Their synthesis is triggered by an increase in postsynaptic intracellular calcium by itself or combined with the activation of postsynaptic GPCRs (Maejima et al., 2001). Upon release, endocannabinoids bind to CB1 and CB2 receptors in the presynaptic membrane with varying affinity. Specifically, AEA seems to be a high affinity, partial agonist of the CB1 receptor, but almost inactive at CB2, whereas 2-AG behaves as a full agonist at both CB1 and CB2 with moderate to low affinity (Sugiura et al., 2000; Pertwee et al., 2010; Di Marzo and De Petrocellis, 2012).
In the brain, the basal levels of 2-AG are approximately 200-fold higher than those of AEA (Sugiura et al., 2006), which indicates its more prominent effects in the CNS. 2-AG is mostly responsible for retrograde signaling via activation of CB1 receptors (Sugiura et al., 1997). It is considered the major mediator of CB1-induced forms of synaptic plasticity such as DSI and long-term hippocampal GABAergic depression (Wilson and Nicoll, 2001; Kim and Alger, 2004). Furthermore, 2-AG is able to activate CB1 receptors present in astrocytes, which eventually results in glutamate release (Navarrete and Araque, 2008) and therefore, mediating neuron-astrocyte communication.
Regarding AEA, it acts as a retrograde messenger and activates CB1 receptors expressed pre-synaptically in glutamatergic terminals (Grueter et al., 2010). This process results in LTD via suppression of glutamate release. Furthermore, AEA participates in “tonic” suppression of GABAergic transmission in the hippocampus (Kim and Alger, 2010). Besides, AEA can act on intracellular CB1 associated with endosomal and lysosomal compartments.
Overall, it is hypothesized that, regarding CB1, AEA represents the “tonic” signaling molecule regulating basal synaptic transmission, whereas 2-AG represents the “phasic” signal which is activated during sustained neuronal depolarization and is responsible for many forms of synaptic plasticity, at least, in the hippocampus (Castillo et al., 2012; Lee et al., 2015). However, these signaling pathways might differ in other brain regions under different physiological or pathophysiological conditions.
When bound to CB2 receptors, endocannabinoids display a series of effects in immune cell function. Interestingly, there is a pronounced contrast between the effects of AEA and 2-AG on immune regulation. 2-AG was found to regulate mechanisms regarding leukocyte recruitment like chemokine release and cell migration (Turcotte et al., 2016). This implies the existence of positive regulation of the immune system by 2-AG. Alternatively, AEA was shown to downregulate leukocyte functions, such as cytokine release and nitric oxide production (Turcotte et al., 2016). Some studies report an increase in production of anti-inflammatory molecules like interleukin (IL) 10 in cells treated with AEA (Correa et al., 2010; 2011). In fact, CB2 receptor agonists like delta-9-tetrahydrocannabinol (THC) and WIN 55,212-2 have only been shown to cause anti-inflammatory effects on leukocytes (Turcotte et al., 2016).
AEA and 2-AG have also been reported to interact with other receptors in the body. AEA acts as an endogenous ligand for transient receptor potential vanilloid 1 (TRPV1) (Starowicz et al., 2007). At pre-synaptic TRPV1s, AEA directly facilitates glutamate release in the striatum (Musella et al., 2009). Alternatively, post-synaptic activation of TRPV1 by AEA also occurs and results in a reduction of biosynthesis of 2-AG (Maccarrone et al., 2008) which, in turn, might lead to LTD (Chávez et al., 2010). In addition, AEA receptor targets include other GPCRs like GPR55 and GPR119 (Sharir et al., 2012). However, these interactions have not yet been fully studied nor understood.
Other key components of the ECS are the catabolic and anabolic enzymes responsible for the synthesis and degradation of endocannabinoids. Briefly, 2-AG is produced by the enzyme diacylglycerol lipase (DAGL) starting from the compound diacylglycerol (DAG) (Murataevaet al., 2014). Alternatively, the biosynthesis of AEA begins from membrane phospholipid precursor N-acyl-phosphatidylethanolamine (NAPE) and it involves the enzymes calcium-dependent or independent N-acyltransferase (NAT or iNAT, respectively) (Jin et al., 2007; Jin et al., 2009) together with NAPE-specific phospholipase D (NAPE-PLD) (Okamoto et al., 2004). However, the exact mechanisms of its synthesis remain under investigation.
Regarding their catabolism, both molecules follow distinct paths. Upon 2-AG reuptake, it is degraded by enzyme monoacylglycerol lipase (MAGL) (Dinh et al., 2002, 2004) which can be found in presynaptic locations and in axon terminals (Gulyas et al., 2004). On the other hand, AEA is degraded by fatty acid amide hydrolase (FAAH) into arachidonic acid and ethanolamine (McKinney and Cravatt 2005) present, particularly, in postsynaptic terminals (Gulyas et al., 2004).
Importantly, suppression of FAAH and MAGL leads to an activity prolongation of endocannabinoids (Gaetani et al., 2009) resulting in differential effects. On one hand, blocking anandamide degradation reduces pain, inflammation depression and anxiety (Tanaka et al., 2019; Y.; Wang and Zhang, 2017). On the other hand, blockage of 2-AG degradation leads to hypothermia, hypomotility and analgesia (Long et al., 2009). This observation suggested FAAH as a potential pharmacological target and the development of synthetic inhibitors that could potentiate AEA transmission which might prove beneficial for the treatment of disorders like MDD (Fowler, 2015). A schematic representation summarizing the role of the named elements of the ECS in neurotransmission can be observed in Figure 1.
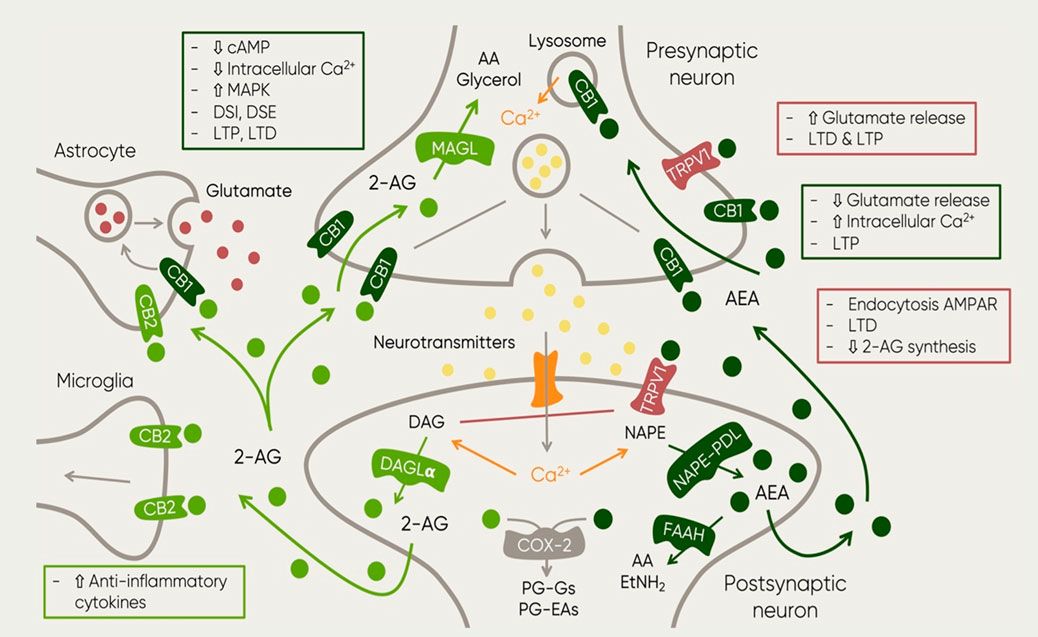
FIGURE 1. Schematic representation of the main elements of the ECS and their function in the context of neurotransmission. The figure also includes the main synthesis and degradation processes of both endocannabinoids: 2-AG and AEA. The first one is mainly synthesized by the enzyme DAG in post-synaptic neurons as a response to increased excitatory activity (Ludanyi et al., 2011). Upon 2-AG reuptake, it is degraded by enzyme monoacylglycero/lipase (MAGL) (Dinh et al., 2002; Dinh et al., 2004) which can be found in presynaptic locations and in axon terminals (Gulyas et al., 2004). This process results in two major byproducts: AA and glycerol (Dinh et al., 2002). AEA on the other hand, it is synthesized by the enzyme NAPE PDL in postsynaptic neurons where it is also degraded by enzyme FAAH (Gulyas et al., 2004). Then, metabolites of AEA and 2-AG undergo further oxidative processes involving cyclooxygenase (COX) and lipoxygenase (LOX) (Vandevoorde and Lambert, 2007). Such process results in the generation of prostag/andins (PGs), in particular, 2-AG and AEA degradation leads to PG-glycerol esters (PG-Gs) and PG-ethanolamides (PG-EAs) (Alhouayek and Muccioli, 2014). The image also shows the receptors at which both endocannabinoids bind. Furthermore, the squares described the resulting mechanisms of the activation of such receptors at different locations. Abbreviations: 2-AG, 2-arachidonoylglycerol; AA, arachidonic acid; AEA, anandamide; cAMP, cyclic adenosine monophosphate; Ca2+, calcium; CB1, cannabinoid receptor 1; CB2, cannabinoid receptor 2; DAG, diacylglycerol; DAGLa; diacylglycerol lipase; DSE, depolarization-induced inhibition of excitatory transmission OSI, depolarization-induced inhibition of inhibitory transmission EtNH21 ethylamide FAAH, fatty acid amide hydro/ase; LTD, long-term depression; LTP, long term potentiation; MAGL, Monoacyclycerol Lipase; MAPK, Mitogen-Activated Protein Kinase; NAPE-POL, NAPE-specific Phospholipase D; NAPE, N-acyl-phosphatidylethanolamine; PG-EAs, prostamides; PG-Gs, prostag/andin-glycero/esters; TRPV1, Transient Receptor Potential Vanilloid.
Phytocannabinoids are cannabinoids produced in the trichomes of the Cannabis plant (Cannabis sativa, Cannabis indica and Cannabis ruderalis). These species contain more than 100 different biologically psychoactive compounds (Aizpurua-Olaizola et al., 2016; Pertwee, 2006). Among them, the most commonly studied due to its strong psychotropic effects is THC. Nonetheless, an increasing number of studies have focused on a different compound named cannabidiol (CBD) since it displays very different pharmacological effects than THC without psychotropic activity. Importantly, later findings unveiled the lack of psychotropic activity of CBD altogether, as well as its absence of reinforcing and addictive properties and a number of characteristics which will advocate for its high potential for therapeutic use (Howlett et al., 2002). Interestingly, the pharmacological effects of CBD go beyond endocannabinoid receptors. For instance, when exerting activity at 5-HTA receptors, CBD induces anxiolytic effects (de Mello Schier et al., 2014; Zanelati et al., 2010). Furthermore, CBD can act as an agonist of vanilloid receptor TRPV1 and therefore influences pain perception, inflammation (Costa et al., 2004). Beyond that, CBD is also known to have affinity for glycine (Ahrens et al., 2009), GABAA (Bakas et al., 2017), adenosine A1 (Gonca and Darıcı, 2015) and nuclear receptors (Scuderi et al., 2014).
In this manner, research then established that THC mainly acts as a partial agonist of CB1 and CB2 (Shahbazi et al., 2020). Alternatively, CBD was found to have reduced binding affinity to both cannabinoid receptors (Pertwee, 2008). However, this compound can also act as a negative allosteric modulator of CB1 and CB2 receptor agonists (Laprairie et al., 2015).
As previously mentioned, MDD is a growing global problem. Unfortunately, only a reduced percentage of people with depression achieve a complete remission. As discussed in previous sections, no hypothesis has been able to explain all the signs and symptoms of MDD, since it is a multifactorial disorder involving multiple interlinked mechanisms. Such interconnectivity of pathophysiological mechanisms is believed to manifest as a constellation of symptoms depicting MDD. These include: dysregulation of the HPA-axis, genetic and environmental factors, neurogenesis, and neuroinflammation (Jesulola et al., 2018). Nevertheless, none of these are able to explain MDD’s intricate pathophysiology, nor its symptomatology by themselves. In fact, it is unclear whether some observed dysregulations are a direct cause or rather a consequence of this disorder. This last challenge obstructs enormously the development of very necessary novel medications or treatments for MDD.
Fortunately, the growing research in the field of the ECS has been opened a promising perspective. As stated in previous sections, the proper interplay between all the elements of the ECS is essential for the homeostatic maintenance of a number of physiological, cognitive, behavioral, and emotional processes (Mechoulam and Parker, 2013). Therefore, when dysregulation of the ECS occurs, cognitive deficits might arise. Particularly, animal research has established a clear symptomatic overlap between ECS alterations and MDD (Hill and Gorzalka, 2005). Importantly, this phenomenon has also been observed in humans. For example, female patients diagnosed with depression presented altered endocannabinoid levels in serum compared to healthy participants (Hill et al., 2008a). Accordingly, the participation of the ECS in the pharmacology of antidepressant drugs has also been reported (Hill et al., 2008b). This evidence indicates a clear role of the ECS in the pathophysiology of MDD in humans. Besides, these ECS alterations seem to occur in a brain/region-dependent manner in depressed individuals, which would explain the variety of symptoms contemplated at the time of diagnosing MDD.
Particularly, the contribution of CB1 receptors to MDD has received broad attention. Animal experiments have established the important role of CB1 as a mood regulator. This can be observed in CB1-KO rodents, which show depressive-like phenotypes (Valverde and Torrens 2012). For example, ablation of CB1 results in anhedonia (Sanchis-Segura et al., 2004), passive stress-coping behavior (Steiner et al., 2008), and higher sensitivity to develop depressive-like symptoms (Haller et al., 2002; Martin et al., 2002).
Furthermore, it is important to consider the evidence regarding the impact of cannabinoid agonist and antagonists administration on MDD. For instance, enhancement of CB1 signaling via administration of HU-210, a potent CB1 agonist, results in antidepressant effects (Hill and Gorzalka, 2005). Additionally, methanandamine, a stable AEA derivative, when applied through stereotaxic injections into the prefrontal cortex (PFC) of rats, was able to induce anxiolytic effects mediated by CB1 (Rubino et al., 2008). Lastly, synthetic agonist WIN55,212-2 was found to induce these effects via modulation of the 5-HT neuronal activity (Bambico et al., 2007). Nevertheless, these results appear to be biphasic, since the application of high doses of CB1 agonists can result in increased anxiety-like behavior. In fact, in humans, functional magnetic resonance imaging (fMRI) revealed that THC can reduce amygdalar reactivity of healthy volunteers exposed to threat signals (Phan et al., 2008), an effect that is commonly observed after administration of anxiolytic drugs like benzodiazepines. In contrast, intravenous administration induced psychotic-like symptoms and anxiety (D’Souza et al., 2004). Therefore, it is hypothesized that oral administration might contribute to lower peak serum concentrations, potentially helping to avoid aversive emotions resulting from high levels of CB1 activation (Moreira et al., 2009).
Accordingly, there is evidence that CB1 antagonism can increase aversive responses in animal models anxiety and depression. The best example is rimonabant, a CB1 antagonist initially marketed as a treatment for depression that resulted in severe adverse psychiatric events in the patients (Mitchell and Morris 2007). Posterior research revealed that, indeed, rimonabant was an anxiogenic substance that not only induced a depressive-like phenotype in laboratory animals, but it also lead to a series of molecular alterations such as decreased serotonin levels in the frontal cortex, reductions in hippocampal cell proliferation and survival, and increased concentrations of pro-inflammatory cytokines (Beyer et al., 2010). Furthermore, AM-251 a different CB1 antagonist, has also been observed to possess anxiogenic properties (Rodgers et al., 2005). In addition, administration of both antagonists; AM-251 and AM-630, significantly reverses the antidepressant effects of WIN55, 212-2 in animals exposed to social-isolation-induced stress (Haj-Mirzaian et al., 2017). Unfortunately, the mechanisms underlying the effects of both CB1 receptor agonists and antagonists remain unknown and must be explored in more detail at the mechanistic level. The supplementary materials include a table summarising the preclinical and clinical studies confirming association between the endocannabinoid system and MDD (see Supplementary Table S1).
Nevertheless, a considerable number of studies has researched the implications of endocannabinoid components in several mechanisms involved in MDD without directly studying their impact on this disorder per se. Hence, this existing literature might be helpful at the time of understanding the role of the ECS in proposed etiologies of MDD, particularly, genetic factors, dysregulation of the HPA-axis, neurogenesis, and neuroinflammation (Jesulola et al., 2018). In the following sections, the present review will focus on the role of the ECS for each theory regarding the pathophysiology of MDD.
Genetic studies are a great source of evidence confirming the role of the ECS in MDD. Together with other genetic factors external to the ECS, epidemiological work of past decades has provided evidence of a certain degree of MDD heritability (Mondimore et al., 2006). Hence, it was hypothesized that variations in certain genes could be substantially responsible for the development of MDD. Nonetheless, up to date, no single genetic mutation is necessary nor sufficient to explain MDD, instead, each gene variation contributes only a reduced fraction of the total risk (Major Depressive Disorder Working Group of the Psychiatric GWAS Consortium et al., 2013).
In humans, genetic studies have investigated single nucleotide polymorphisms in the CB1 gene (CNR1) associated to depressive phenotypes and responses to antidepressants. For example, presence of the G allele of rs806371 CNR1 gene polymorphism is higher in individuals with MDD (Mitjans et al., 2013). On the same line, the G allele of the CNR1rs1049353 polymorphism has been associated to antidepressant resistance (Domschke et al., 2008). Alternatively, the minor C allele of rs2023239 displayed a protective influence against MDD (Icick et al., 2015).
Nevertheless, studies associating gene variants and MDD often encounter inconsistent data. A recent report analyzed several candidate genes previously implicated in high prevalence of MDD, among them, a CNR1 variant (Gonda et al., 2018). It was observed that most polymorphisms, including those in CNR1, showed increasing relevance for MDD in participants with higher exposure to recent negative life events. Possibly, the lack of accountability for environmental factors like stress could bias their contribution to MDD and cause the results to become irreproducible. As a matter of fact, a recent meta-analysis by Kong et al. (2019) revealed no association between CNR1rs1049353 or CNR1 triple repeat with increased risk of MDD when combining all available literature.
In contrast, a significant correlation was found between all four genetic models of the CB2 gene (CNR2) polymorphism CNR2rs2501432 and MDD. This was first observed by Onaivi and collaborators (2008), who reported a significant association between such CNR2 polymorphism and depressed patients. Furthermore, a recent article has linked CNR2 R63Q variation to a greater sensitivity towards childhood trauma and overactivation of the HPA-axis (Lazary et al., 2019). Interestingly, the article also describes the involvement of a FAAH gene polymorphism in the susceptibility to trauma observed in Lazary et al. (2016).
In conclusion, the role of CNR1 in MDD should not be dismissed and further research with wider coverage is needed to evaluate its impact. However, this finding positions CNR2 as a key factor in the development of MDD. Hence, pharmacological modulation of CNR2 ligands might be a potential therapeutic approach for this disorder. Importantly, the association of endocannabinoid receptors to MDD is not sufficient to explain the entirety of its pathophysiology. Therefore, such results should be interpreted with caution, while contemplating the essential role of environmental factors in its etiology.
Stressful life events are considered the main predisposing factor for the development of psychiatry disorders like anxiety and MDD. In mammals, the stress response is mediated by the HPA-axis, resulting in a cascade of events involving a series of hormones; like corticotropin-releasing hormone (CRH) and adrenocorticotropic hormone (ACTH); ultimately resulting in the release of glucocorticoids into the bloodstream (Smith and Vale, 2006). Termination of the stress response is achieved by resetting the HPA-axis via negative feedback mechanisms. On this basis, hyperactivity of the HPA-axis has been suggested as a mechanism leading to stress vulnerability shown by MDD patients. Certainly, these patients show inadequate HPA-axis suppression when exposed to stress or exogenous glucocorticoid administration compared to healthy controls (Gillespie and Nemeroff, 2005; Juruena, 2014). Interestingly, this dysregulation of the HPA-axis seems to be related to endocannabinoid signaling (Hillard et al., 2016).
For example, CB1 receptors are believed to play a major role in HPA-axis regulation. This hypothesis arose from the observation of high basal corticosterone levels and HPA hyperactivity in CB1 KO animal models (Barna et al., 2004). This indicated a clear role for CB1 in the inhibition of the HPA-axis and therefore, in the termination of the stress response. On the other hand, HPA-axis dysregulation can also influence the ECS via alterations in endocannabinoid synthesis and CB1 expression, both involved in stress-related symptoms (Micale and Drago, 2018). This suggests the existence of a bidirectional link between the two systems.
Furthermore, the role of the ECS was investigated in existing stress models associated to anxiety-like behavior in rodents. It was observed that chronic stress induced a reduction of AEA levels in the amygdala and hippocampus (Patel et al., 2005; Wang et al., 2012) via increased activity of FAAH (Navarria et al., 2014; Gray et al., 2015), which is consistent with previous evidence supporting the anxiolytic properties of AEA (Patel and Hillard, 2006). This reduction was attributed to the elevated serum corticosterone content (Hill et al., 2009) through a CRHR1-mediated mechanism (Gray et al., 2016) resulting in the generation of anxiety (Gray et al., 2015). Nevertheless, the obtained results depended largely on the type of stressor. Furthermore, exposure to repeated stressors increased 2-AG content throughout regions in the CNS (Micale and Drago 2018) probably mediated by decreased expression of MAGL (Sumislawski et al., 2011). This is in accordance with the observed decrease in AEA levels following stress exposure, since its interaction with TRPV1 results in downregulation of 2-AG synthesis (Maccarrone et al., 2008). However, this link has not been empirically demonstrated.
Importantly, following chronic unpredictable stress, CB1 undergoes widespread downregulation and desensitization in limbic areas like the hippocampus, hypothalamus, amygdala and nucleus accumbens (Wamsteeker et al., 2010; Wang et al., 2010; Lee and Hill, 2013). In contrast, many studies have found an upregulation of CB1 in PFC (Zoppi et al., 2011; Lee and Hill, 2013). Unfortunately, the exact mechanism by which this CB1 regulation occurs remains under speculation. However, it is consistent with the observation that CB1 activation decreases GABAergic transmission in said limbic areas, contributing to the termination of the stress response (Martin et al., 2002; Hill et al., 2011). Particularly, CB1 blockade leads to increased plasma corticosterone (Wade et al., 2006). Furthermore, the activation of CB1 on noradrenergic signaling represents an important mechanism for stress adaptation (Wyrofsky et al., 2019). Chronic HPA-axis-induced stimulation of noradrenergic areas such as the locus coeruleus increases anxiety and depressive-like behavior (Valentino and Van Bockstaele, 2008). Importantly, hyperactivity of the ECS via CB1 signaling due to chronic stress might lead to increased noradrenergic activity in the PFC and thus, contributing to such behaviors (Wyrofsky et al., 2019).
On this basis, CB1 dysregulation within specific brain areas would lead to a hyperactive HPA-axis, which is precisely what is observed in CB1 KO animals (Daniela Cota et al., 2007; Steiner et al., 2008). CB1 KO animals express higher anxiogenic-like behavior and increased stress-induced ACTH levels, as well as anxiolytic drug resistance (Cota, 2008). Accordingly, hyperactivity of the HPA-axis is one of the most consistent biological evidence in MDD in both clinical and pre-clinical studies (Sánchez et al., 2001; Heim and Nemeroff 2002). This suggests a clear role for CB1 as a direct contributor to the termination of the HPA-mediated stress response and therefore, as a key partaker in the pathophysiology of depressive symptoms.
With regards to CB2 receptors, despite their expression in areas involved in the stress response, few studies have explored their implications in HPA-axis signaling. In these, results are contradictory. Some stress-induced depressive-like behavior models have described no changes in CB2 levels, whereas others encountered a decreased CB2 hippocampal expression (Marco et al., 2017). However, unlike CB1, manipulation of CB2 receptors does not alter plasma concentrations of corticosterone after exposure to stress (Zoppi et al., 2014). Such findings speak against a direct relationship between the HPA-axis and CB2 receptors, although it is possible that CB2 might regulate the activity of the HPA-axis through other indirect mechanisms. Indeed, a recent article has linked an existing polymorphism of the gene encoding CB2 to greater sensitivity for childhood trauma and overactivation of the HPA-axis (Lazary et al., 2019). This process seems to be mediated through neuroinflammatory mechanisms influenced by CB2. This observation is in accordance with previous findings showing that CB2 KO animals display exacerbated stress-influenced neuroinflammatory responses (Zoppi et al., 2014).
All together, these findings suggest the involvement of the ECS; particularly, the CB1 receptor and endocannabinoids AEA and 2-AG, as a fundamental regulatory system involved in the termination of the stress response by mediating negative feedbacks controlling HPA-axis’ activity. Therefore, these ECS components have a direct role in anxiety-like behavior and vulnerability to stress, and they must be regarded as important contributors to the pathophysiology of MDD. All the above-mentioned mechanisms are graphically described within the blue area of Figure 2.
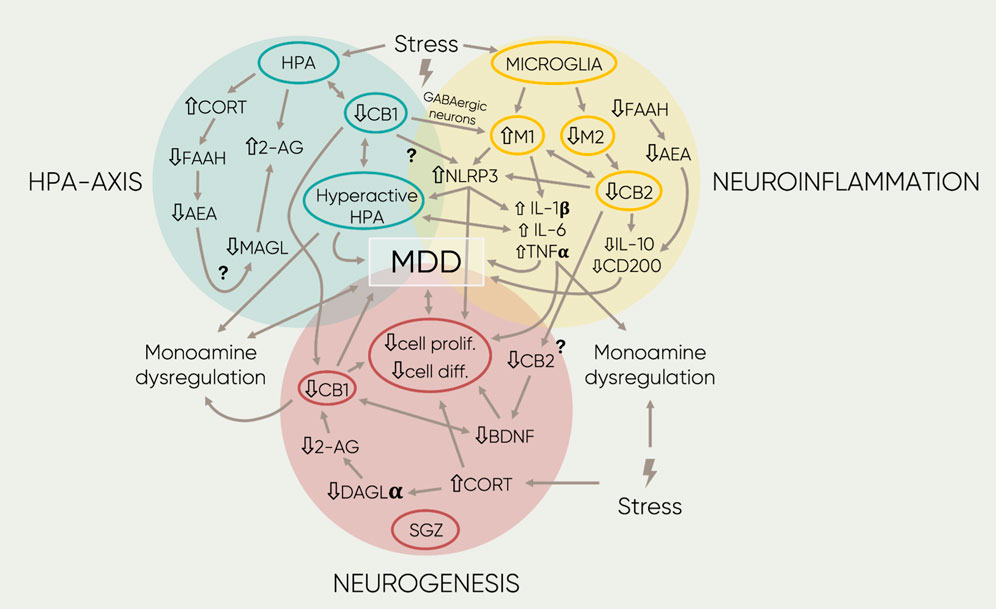
FIGURE 2. Schematic representation of the main findings regarding the association of the ECS and MOD. The four different proposed etiologies of MOD are represented by the three colored circles. Overlaps between circles symbolize interactions between mechanisms. Inside, each circumference highlights the central components in each field. The single-arrowed lines represent direct observed influences between factors whereas the double-arrowed ones, bidirectional links. Next to the text, arrows represent down- or upregulation of the component or signaling mediated by receptors. Furthermore, the lightning symbols stand for different forms of stress (early-life, acute, or chronic stress; see text for more details). Lastly, the question marks beside the arrowed lines indicate potential links that remain to be researched. 2-AG, 2- arachidonoylglycerol; AA, arachidonic acid; AEA, anandamide; BDN F, brain derived neurotropic factor; CB1, cannabinoid receptor 1; CB2, cannabinoid receptor 2; CORT, cortisol/corticosterone, DAGLa, diacylglycerol lipase; diff, differentiation; FAAH, fatty acid amide hydrolase; IL-1,6′, Interleukin 1/l; IL-6, interleukin 6 MAGL, Monoacyclycerol Lipase; Ml, microglia phenotype 1; M2, microglia phenotype 2; N LRP3, family pyrin domain containing 3; pro/if, proliferation; TNFa, tumor necrosis factor a.
Among the other potential altered circuitries in MDD, neuroinflammation has received increasing attention (Troubat et al., 2020). Since the first observations, several studies have described a strong relationship between depressive symptomology and altered presence of pro-inflammatory markers. Hence, research has focused on the involvement of the immune system in the CNS, which consists of glial cells.
As the resident macrophagic cells and main form of innate immune defense in the CNS, microglia were expected to play a crucial role in this relationship. Microglia are responsible for the release of chemokines and cytokines as a response to physical insults or infectious agents. Upon activation, microglia can evolve into two possible phenotypes, known as M1 and M2, which might include specific features under different pathological insults (Ma et al., 2017). The first, M1, is in charge of the initiation of the inflammatory response through the release of pro-inflammatory mediators like IL-1β, IL-6, IL-8 and tumor necrosis factor α (TNFα) (Lively and Schlichter, 2018). Furthermore, they control the recruitment of additional central and peripheral immune cells to the site of infection or brain damage. Alternatively, the second phenotype, M2, is involved in the prevention of M1-induced neuronal damage and toxicity through secretion of anti-inflammatory cytokines like IL-4, IL-10 and transforming growth factor (TGF) (Koscsó et al., 2013). This M1/M2 phenotype theory has been found to be an oversimplification of this cytological process based on experimental evidence (Ransohoff 2016). However, this terminology is useful when characterizing microglia states (pro-inflammatory and neuroprotective) and will continue to be used for the purpose of this review.
Due to their primary role in neuromodulation of the inflammatory response, microglia have become the center of attention as a key component in the inflammatory etiology of MDD (Yirmiya et al., 2015). Evidence supporting these theories is provided by clinical and preclinical research. For instance, elevated levels of proinflammatory cytokines released by microglia are correlated to the severity of depressive symptomatology in MDD patients (Haapakoski et al., 2015). Furthermore, rodents exposed to chronic stress paradigms display microglia hyper-ramification and overactivation in several brain regions (Sugama et al., 2007; Tynan et al., 2010; Hinwood et al., 2013).
Particularly, IL-6 and C-reactive protein (CRP) are the most strongly associated to anhedonia (Felger et al., 2018). In accordance, administration of antidepressants directly impacts microglia signaling and IL-6 production (Hashioka et al., 2007). A more recent study has also linked the imbalance of microglial pro- and anti-inflammatory conditions through a mechanism involving brain-derived growth factor (BDNF), more specifically, via a BDNF-TrkB-dependent pathway in the hippocampus (Liu et al., 2019). Interestingly, the authors found a sex-specific relationship between BDNF and microglial inflammatory biomarkers, a mechanism that could become very important for sex differences in depression. On this basis, some researchers have suggested that MDD could start to be considered a microglial disease (Yirmiya et al., 2015).
The presence of CB2 receptors in microglia switched the attention to the potential role of the ECS in glia-mediated neuroinflammation involved in MDD. CB2 has been described to possess general anti-inflammatory properties. Genetically modified rodents with overexpression of CB2 show reduced stress-induced pro-inflammatory cytokine TNFα and enzyme COX-2 (Zoppi et al., 2014). In parallel, CB2 KO animals display exacerbated neuroinflammatory responses to chronic stress (Zoppi et al., 2014).
CB2 receptors are expressed in microglia in a state-dependent manner (Carlisle et al., 2002). Specifically, they are not present in resting microglia but rather in fully active cells and intermediary stages. In the past decade, its presence has been associated to induction of M2 polarization (Tanaka et al., 2020). For example, administration of synthetic CB2 agonists alleviate neuroinflammation by enhancing the conversion from M1 microglia into the M2 phenotype (Tao et al., 2016; Luo et al., 2018). Importantly, a downregulation of inflammatory cytokines and upregulation of anti-inflammatory mediators were paired to M2 polarization (Tao et al., 2016). These mechanisms were not observed when animals were co-treated with CB2 synthetic antagonists. Such results evidence the seemingly important role of CB2 receptors in microglia-induced neuroprotection in the presence of inflammation.
Furthermore, it appears these anti-inflammatory effects of CB2 might be initiated by AEA. Administration of AEA reduces neuron toxicity by downregulating IL-1B and IL-6 in in vitro activated microglia via CB2 receptors (Malek et al., 2015). At the same time, it exerts neuroprotective properties via production of IL-10 (Correa et al., 2010), and upregulation of CD200 receptor, known to suppress microglial inflammatory response (Manich et al., 2019). Moreover, pharmacological inhibition of FAAH also leads to anti-inflammatory effects in vitro via increased levels of AEA (Tanaka et al., 2019).
Although these observations have been widely described in pharmacologically induced neuroinflammation, the immunological role of the ECS in the context of MDD models is understudied. However, two recent studies have investigated this relationship. First, Chen et al. (2018) reported that FAAH inhibition resulted in alleviation of pro-inflammatory response to acute stress. This is in accordance with previous studies reporting anti-inflammatory properties of AEA. Second, Lisboa et al. (2018) observed that administration of selective cannabinoid agonist WIN55,212-2 was able to reverse social stress-induced neuroinflammation and anxiety-like symptomatology. Although, since named agonist could act on both cannabinoid receptors, whether this action occurred through mediation of CB1 or CB2 is unclear.
Regarding CB1, despite its low expression in glial cells, some studies have shown that this receptor is also able to regulate immune function through distinct mechanisms. For instance, specific CB1 KO of forebrain GABAergic neurons in vivo lead to pro-inflammatory microglia phenotypes without significant cognitive deficits (Ativie et al., 2018). This finding provides evidence of the role of CB1 in neuron-glia communication with a crucial involvement of GABAergic neurons. Furthermore, a recent research work observed that chronic stress increased microglia activation in CB1 KO animals, which correlated with the severity of depressive-like symptoms (Beins, 2020). Hence, it has been hypothesized that CB1 might be an indirect mediator of microglia-induced inflammation. Although, how CB1 signaling can regulate microglial activity remains uncertain and requires further research.
For this, it is important to consider the potential interplay between HPA-axis and microglial neuroinflammation. As mentioned earlier, HPA-axis hyperactivity is observed across animal models of MDD, as well as in human patients. Increased glucocorticoid release has been found to induce an overproduction of pro-inflammatory cytokines through microglia activation (Nair and Bonneau, 2006; Sorrells and Sapolsky, 2007). In the same manner, an immune modulation of the HPA-axis has also been suggested following immunology research of viral infections (Silverman et al., 2005). However, the exact mechanisms underlying chronic stress and microglia activation remain under discussion. A recent article described a potential link between HPA signaling and neuroinflammation. Feng et al. (2019) observed that elevated corticosterone levels after chronic stress increased the release of pro-inflammatory elements like IL-1β and IL-18 in activated hippocampal microglia. Particularly, they observed that this process was mediated by nuclear factor kappa B (NF-κB) and the Nod-like receptor protein (NLRP3) namely, NF-κB-NLRP3 pathway. This study provides a new and valuable insight into the relationship between stress, the HPA-axis and microglia-mediated neuroinflammation.
Although much less investigated, the ECS might also be related to this mechanism. Recent research has found that administration of two phytocannabinoids; CBD and cannabigerol produced anti-inflammatory effects by reducing NF-κB activation among other mechanisms (Mammana et al., 2019). Furthermore, previous work had stablished a neuroprotective role for CB2 via inhibition of the NLRP3 inflammasome in autoimmune encephalomyelitis models (Shao et al., 2014). This process might be mediated via the CB2-induced inhibition of NF-κB activation in microglia (Zoppi et al., 2014). Additionally, CB1-mediated effects of 2-AG have been linked to neuroprotective functions via inhibition of NF-κB (Panikashvili et al., 2005). Likewise, AEA has been described to enhance production of IL-10 in activated microglia and inhibiting NF-κB activation. More recently, in the context of liver inflammatory disease, CB1 receptors have been reported to mediate macrophage NLRP3 expression and inflammation (Yang et al., 2020). This evidence suggests a potential role for the ECS as a bridge in the crosstalk between the HPA-axis and neuroinflammation via the NF-κB-NLRP3 pathway. However, effects of the ECS in modulation of microglia-mediated neuroinflammation as a result of chronic stress, particularly regarding the NLRP3, remains to be studied and requires much further research.
Importantly, a novel field of research has emerged in recent years, focusing on polyunsaturated fatty acids (PUFAs) and their potential as a treatment for MDD. PUFAs are lipid derivatives of omega-3 or omega-6 that act as precursors for endocannabinoids. Particularly, supplementation with two main PUFAs, namely, eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) has been extensively investigated (Mozaffari-Khosravi et al., 2013; Marangell et al., 2003). Indeed a recent meta-analysis confirmed the beneficial effects of omega-3 PUFAs on depression symptoms (Liao et al., 2019). Co-administration of DHA and EPA or EPA alone are able to reduce inflammation through several mechanisms. Firstly, they both can reduce the production of proinflammatory cytokines like TNFα, IL-1β, IL 2 and IL-6 (Caughey et al., 1996). Secondly, they have been described to suppress NF-κB signaling (Horowitz et al., 2015). Lastly, administration of a variety of omega-3 PUFAs reduced inflammation via activation of macrophage autophagy and attenuation of NLRP3 inflammasome (Shen et al., 2017). These findings strongly confirm the endocannabinoid regulation of neuroinflammatory processes, and they provide an opening to a new research field in the context of psychiatric disease like MDD.
Overall, the abovementioned evidence supports the theory that hypofunction of the ECS directly impacts neuro-immune modulatory pathways in the CNS, leading to pro-inflammatory processes mediated by microglia. Specifically, CB2 seems to be directly involved in promotion of anti-inflammatory mechanisms mediated by AEA signaling. Nevertheless, the involvement of CB2 in stress-induced inflammation remains unknown. Alternatively, CB1 has been suggested to indirectly modulate neuro-immune responses by mediating neuron-glia communication in the CNS. Such interactions between the immune system, the ECS and MDD symptomatology are depicted in the yellow area in Figure 2.
Adult neurogenesis is a neurobiological process by which neurons are continually generated within the CNS throughout an organism’s life. Specifically, the subgranular zone (SGZ) of the dentate gyrus of the hippocampus has been established as the primary area involved in this process (Bond et al., 2015). Notably, the exact biological role of neurogenesis in cognition and behavior remains unknown.
Since the discovery of hippocampal atrophy in untreated MDD patients (Sheline et al., 1996), it was hypothesized that loss of neurogenesis in the dentate gyrus could significantly impact the development of this disorder. In fact, successful treatment with antidepressants and most therapies is shown to induce hippocampal neurogenesis (Praag et al., 2000; Malberg and Duman 2003; van). This phenomenon seems to be mediated by BDNF, since it is found to be reduced in animals models of MDD (Autry et al., 2009), while simultaneously recovered by antidepressant administration (Santarelli et al., 2003). However, neurogenesis ablation in experimental animals does not always induce depressive symptoms (Jayatissa et al., 2010). On this line, some antidepressants show neurogenesis-independent mechanisms (David et al., 2009). Due to such conflictive results, it is currently hypothesized that neurogenesis might be a key restorative mechanism for hippocampal structure and function which might indirectly result in alleviation of MDD symptomatology (Hanson et al., 2011). Therefore, when disrupted, it could theoretically participate in the etiology of MDD even though it is unlikely to cause the entire mood disorder. In the present, however, neurogenesis is still considered a significant contributing factor to the pathophysiology of MDD (Jesulola et al., 2018).
During the past decades, research has confirmed the involvement of the ECS in hippocampal proliferation, starting with the extensive expression of endocannabinoid entities in neuronal progenitor cells (Aguado et al., 2006). Specifically, CB1 has been established as a direct mediator of adult neurogenesis. For example, administration of high selective agonists of CB1 promotes neural proliferation in the SGZ (Andres-Mach et al., 2015). Accordingly, this effect was prevented by CB1 synthetic antagonist AM251 and not present in CB1 and CB2 double KO animals (Hutch and Hegg 2016). Furthermore, DAGLα KO rodents exhibited decreased 2-AG levels, as well as impaired neurogenesis in the dentate gyrus (Gao et al., 2010). Therefore, CB1 activation via 2-AG agonism seems to play a major role in this biological process. However, the exact mechanism by which CB1 might induce neurogenesis is under discussion, although some pathways have been described (Zimmermann et al., 2018).
Relevant to depressive-like behavior, CB1 KO mice were observed to be more vulnerable to stress-induced depressive-like responses with a high susceptibility for anhedonia (Martin et al., 2002). This symptomatology was later associated to downregulation of BDNF expression in the hippocampus (Aso et al., 2008). Such findings suggested a clear involvement of both, CB1 and neurogenesis in the development of depressive disorders.
Conversely, administration of CBD prevented detrimental effects of chronic stress and increased hippocampal proliferation via CB1 activation (Wolf et al., 2010; Campos et al., 2013). Another recent study reported that CBD administration could reverse the effects of chronic stress, facilitating neurogenesis and dendritic remodeling (Fogaça et al., 2018). However, whether anxiolytic effects of CBD are directly a consequence of improved neurogenesis or other unrelated endocannabinoid mechanisms cannot be concluded. These observations also occurred when considering endocannabinoids. For example, prevention of 2-AG degradation via blockade of MAGL results in an enhancement of neurogenesis and heightened antidepressant-like effects in mice exposed to chronic stress (Zhang et al., 2015). In addition, DAGLα KO animals displayed reduced endocannabinoid levels in the hippocampus, as well as impaired neurogenesis and anxiety-like behavior (Jenniches et al., 2016). Nonetheless, the cause-consequence relationship between the observed depressive symptoms and loss of neurogenesis cannot be established from this study. In summary, CB1 is hypothesized to contribute to anti-depressive effects paired with neurogenic mechanisms in the hippocampus.
A more complex role of CB2 in adult neurogenesis has been suggested. Unlike CB1 KO, CB2 KO animals display a stable adult neurogenesis (Mensching et al., 2019). Although, these findings are contradictory to previously reported observations indeed showing altered neurogenesis in this strain (Palazuelos et al., 2006). This perhaps could be due to the age difference among rodents used for the studies. Regardless of this contradiction, CB2 agonism has been related to indirect potentiation of neurogenesis through its role as a homeostasis regulator, normalizing processes like apoptosis, oxidative stress, and neuroinflammation (Avraham et al., 2014; Shi et al., 2017). For instance, in an Alzheimer’s disease model, administration of CB2 agonist MDA7 induced neurogenesis together with improved hippocampal synaptic plasticity and regulated microglial activation (Wu et al., 2017). Interestingly, microglia are able to regulate this process via the release of cytokines and chemokines that may act as stimulants or suppressors of neurogenesis (Sato 2015). In this manner, pro-inflammatory mediators like IL-1β can inhibit processes such as cell proliferation and differentiation in the dentate gyrus (Wu et al., 2013). In particular, chronic stress in rodents produces alterations in microglia activation, which contributes to the loss of neurogenesis (Kreisel et al., 2014). Alternatively, microglia can also release BDNF, thereby supporting survival of novel neurons (Ferrini and De Koninck 2013). Whether this mechanism is directly regulated by CB2 remains unclear. Nevertheless, more recently it has been observed that activation of CB2 receptors by selective agonist JWH133 upregulated microglia expression of BDNF in the lateral ventricular tissue (Tang et al., 2017). Though this finding has shed some light on the topic, further research is still required before drawing solid conclusions.
Neurogenesis is also related to HPA function in pathological conditions. For example, animals with suppressed neurogenesis display a hyperactive HPA-axis associated to depressive-like behaviors (Schloesser et al., 2009). Given the important role of the hippocampus in termination of HPA-mediated stress response, it was hypothesized that disturbances in neurogenesis could negatively alter the negative feedback loop regulating HPA activity. Simultaneously, chronically elevated glucocorticoid concentrations as a result of a hyperactive HPA also leads neurogenesis disruptions (Anacker et al., 2013).
Interestingly, changes induced by stress or inflammation may affect neurogenesis via the NF-κB pathway (Anisman et al., 2008), which is, at the same time, the suggested mechanism bridging HPA activity and neuroinflammatory processes. Furthermore, pro-inflammatory cytokines can additionally stimulate the HPA-axis to release glucocorticoids, which, in turn, suppress neurogenesis (Liu et al., 2003). Hence, it can be concluded that there is a strong interconnection between the ECS and altered mechanisms observed in MDD.
Overall, these results indicate a role for the ECS in modulation of hippocampal cell proliferation and cell differentiation. On one hand, CB1 and 2-AG signaling seem to be essential components in the stimulation of neurogenesis. On the other, CB2 might participate as a key regulator under pathological conditions by exerting neuroprotective mechanisms towards neurons via immune system regulation. These mechanisms are depicted within the red area in Figure 2. In conclusion, the presented data suggest a clear involvement of the ECS in the neuroprotective effects of hippocampal neurogenesis during the development of MDD.
The present work aimed to be an integrative review about the role of the ECS in the pathophysiology of MDD. As previously described, all the currently hypothesized etiologies for MDD, whether they are genetic, neuroendocrine, immunological, or cytogenetic, seem to rely on the correct functioning of endocannabinoid signaling. In fact, a number of interactions between studied factors can already be explained through the involvement of the ECS. Figure 2 aims to summarize all the discussed findings in a cohesive mind map. As it can be observed, the different proposed etiologies of MDD—HPA-axis, genetic factors, neuroinflammation, and neurogenesis—can all be integrated into the context of endocannabinoid signaling. In addition, the figure illustrates the potential links that remain to be explored. Among them, the possible role of the NF-κB-NLRP3 should be highlighted as an important factor in the mediation of the multiple described systems. Moreover, given the intricacy of the mechanisms underlying stress-related conditions, further studies are essential to evaluate the role of different players, such as TRPV1 receptors and other GPCRs; diverse neuronal subpopulations, i.e., GABAergic vs glutamatergic; and even considering brain regions, which could interact with each other to regulate mood and cognitive aspects involved in MDD.
This image is a clear example of the complex nature of MDD as well as indication of the current need for interdisciplinary work. Furthermore, it has been described that a sole etiology or a unitary construct as the cause of MDD cannot explain the constellation of symptoms exhibited by depressed patients. Hence, the unification of diverse research fields must occur in order to advance in our comprehension of MDD and other complex etiologies like schizophrenia, post-traumatic stress disorder, or autism spectrum disorder.
As a matter of fact, the complexity of the ECS might be useful when searching for connections between pathological pathways. Similarly to MDD, its function cannot be presented as a list of individual items, but it should be regarded as a wide interconnected network. One that is sensitive to environmental factors, such as stress, which might threaten the integrity of brain homeostasis. It is evident that the correct functioning of the ECS is imperative for maintaining mental health. However, despite being an ever-growing acclaimed research field, further preclinical and clinical studies are crucial for a better understanding of its mechanisms. For instance, the role of GPR55 as an expanded endocannabinoid receptor remains controversial. Moreover, GPR18 has not been investigated, although its presence in microglia suggests a potential role as a neuro-immune regulator. A recent review discusses in depth the so called “expanded” ECS or endocannabinoidome, including a number of elements overlapping with pathways attributed to the ECS (Cristino et al., 2020). The work of Cristino et al. (2020) certainly illustrates the complexity of cannabinoid mechanisms and further highlights the importance of interdisciplinary work at the time of researching this phenomenon.
Such complexity deeply challenges the development of cannabinoid-based therapies. Their characteristic chemical promiscuity can give rise to unexpected side effects. For instance, based on in vitro and in vivo studies, it was believed that synthetic inhibitors of FAAH could be a potential treatment for depressive and anxiety disorders (Gunduz-Cinar et al., 2013). Therefore, a variety of them were synthesized and tested clinically. Nonetheless, a vast number of them were quickly suspended due to their devastating side effects. Particularly, a famous case reported severe adverse effects in 5 patients and at least one death during a drug trial in France (Kaur et al., 2016). Furthermore, rimonabant, a selective CB1 agonist developed as a treatment against obesity succeeded in clinical trials but had to be withdrawn from the market 3 years later due to its high risk of severe psychiatric disorders including anxiety and suicidal ideations (Christensen et al., 2007; Moreira and Crippa, 2009).
In this line of research, it is imperative to mention the large attention received by a particular cannabinoid substance, CBD. This phytocannabinoid present in Cannabis sativa is a natural negative allosteric modular of the CB1 and CB2 receptors. Although its precise mechanism is unclear, CBD has been described to exert neuroprotective and anti-inflammatory effects (Lastres-Becker et al., 2005). Its best described mechanisms of action are: inhibition of FAAH, therefore enhancing AEA signaling (De Petrocellis et al., 2011); and regulation of microglia migration and activation (Martín-Moreno et al., 2011). However, it is important to consider that CBD exhibits more than 65 identified molecular targets across the body (Elsaid and Le Foll, 2020). Such pharmacological complexity makes CBD an interesting candidate for therapeutic research. In fact, CBD has been shown to decrease anxiety-like behavior in animal models of MDD (Hen-Shoval et al., 2018; Xu et al., 2019). In particular, CBD can act via CB1 receptors and induce hippocampal neurogenesis associated with anxiolytic effects (Campos et al., 2013; Luján and Valverde, 2020). Similarly, in humans, 62% of self-users of CBD commercial formulations report using this drug in order to treat a medical condition (Corroon and Phillips, 2018). Specifically, consumers are utilizing CBD as a therapy for multiple psychiatric conditions like anxiety and depression, but also pain and sleep disorders. Furthermore, it is well tolerated, non-addictive, and its use is safe (Iffland and Grotenhermen, 2017; World Health Organization and others, 2017). Nevertheless, the efficacy of CBD as a treatment for depression has not been clinically confirmed and remains under investigation. A recent meta-analysis yielding 924 records from clinical trials found that there were no studies investigating the efficacy of CBD by assessing depressive symptoms as the primary outcome (Pinto et al., 2020). Therefore, they concluded that the potential therapeutic properties of CBD as a treatment for MDD needs much further research. In fact, a number of clinical trials are now starting to test CBD as an adjunctive treatment for depressive disorders and, hopefully, the results will be reported in the coming years. Curiously, this year a one case-report described a successful CBD treatment in an adolescent suffering from multiple substance use disorder, social phobia, narcissistic personality disorder, and severe depression (Laczkovics et al., 2020). Researchers reported that the patient was administered CBD upon treatment with antidepressants, after which they showed improvement regarding depressive and anxiety symptoms. Although, this study compromises only one patient, this observation is a quite promising outcome shedding a light on the therapeutic potential of CBD as a treatment for MDD.
Furthermore, in the last 20 years a novel pharmacological treatment has received increasing attention. Ketamine is a chiral compound that displays rapid antidepressant properties after a single intravenous (IV) dose (Murrough et al., 2013). Unfortunately, the exact mechanism by which ketamine exerts its antidepressant effects remains under discussion, although some possible pathways have been proposed. For instance, ketamine seems to inhibit the activation of the HPA-axis signaling via blocking enzyme expression and activity of the adrenal gland (Besnier et al., 2017). Furthermore, ketamine administration has also been linked to increased hippocampal neurogenesis associated to its antidepressant effects (Yamada and Jinno, 2019). Recently, the involvement of the endocannabinoid system in its mechanisms of action has been described (Ferreira et al., 2018; Khakpai et al., 2019). In particular, Ferreira et al. (2018) describes that nociceptive actions of ketamine are connected to AEA release and CB1 activation. These findings reinforce the importance of the ECS in the pathophysiology of MDD and highlight the need for integrative research when studying intricate, heterogenic illnesses like neuropsychiatric disorders.
It is also worth mentioning a novel field of research that has emerged in recent years involving another potential disrupted mechanism contributing to MDD’s pathophysiology. The gut microbiota has been shown to impact a series of physiological mechanisms including cognition and behavior. In fact, a number of microbiome alterations have been correlated to quality of life of MDD patients (Valles-Colomer et al., 2019). However, currently no consensus has emerged about which bacterial strains are more relevant for MDD (Cheung et al., 2019). Importantly, expression of CB1 has also been observed in the gastrointestinal tract (GI), both in the enteric nervous system but also in non-neuronal cells forming the intestinal mucosa (Izzo and Sharkey 2010). Relevant to MDD animal models, Zoppi et al. (2012) reported that CB1 exerted a protective role in the colon of rodents exposed to stress paradigms. Unfortunately, this line of research has not been extensively explored probably due to the complexity of all three mechanisms involved: the ECS, MDD, and the microbiome.
Overall, the synthesis of the above mentioned “puzzle pieces” further supports the claim made throughout this entire paper: the ECS is a very complex system with a variety of functions, which we are only beginning to understand. This, once again, serves as a reminder that narrow and focused research will not be able to uncover its intricate workings, and that of course, interdisciplinary work needs to be prioritized.
The present review provides a concise overview of the involvement of the ECS in a number of theorized etiologies of MDD such as: genetic factors, hyperactivation of the HPA-axis, dysregulation of immune response mediated by microglia, and loss of neurogenesis. In this manner, epidemiological studies have associated both CB1 and CB2 gene polymorphisms to the development of MDD. Importantly, these associations by themselves are not sufficient to explain the entirety of its pathophysiology due to the essential role played by environmental factors. Furthermore, the ECS is responsible for the termination of HPA-axis signaling after exposure to stress. Particularly, downregulation of CB1 receptors, as a result of prolonged increase of glucocorticoid concentrations induced by chronic stress, is associated to a hyperactive HPA-axis. Hence, endocannabinoid regulation plays an important role in vulnerability to stress, a major risk factor for MDD. Alternatively, CB2 seems to be a primary contributor to the neuroinflammatory etiology of MDD. Evidence supports the role of CB2 in microglia-mediated neuroprotection and anti-inflammatory function. Specifically, AEA-dependent CB2 receptor activation promotes the shift towards immunosuppressive phenotype of microglia. Hence, alterations in CB2 expression, AEA concentrations and FAAH activity could be a trigger of depressive symptomatology. Interestingly, CB1 seems responsible for the communication between HPA-related mechanisms and CB2-mediated microglial activation. Therefore, its contribution to the immune function should not be dismissed. Lastly, neurogenesis is also described as an important mechanism underlying the pathophysiology of MDD. Potential involvement of CB1 as a direct contributor to neurogenetic pathways has been suggested. On the other hand, CB2 seems to be responsible for microglia-induced stimulation and depression of hippocampal cell proliferation. Importantly, HPA-axis activity can also regulate neurogenesis and vice versa. This demonstrates the great level of inseparability between the components of the endocannabinoid system and the underlying circuits of major depressive disorder, as well as the need of integrative work that can integrate interdisciplinary findings to achieve a better understanding of heterogenic, multifactorial disorders.
Overall, this review proposes the ECS as a unitary entity of the most important recognized pathways leading to major depressive disorder. It also emphasizes the need for interdisciplinary studies that provide new scopes about its pathophysiology in order to develop more efficient therapies for this devastating disease.
IG-L, AG-B, AC-Z, and OV were responsible for the study concept and design. All authors drafted the manuscript and approved the final version for publication.
This work was supported by Ministerio de Economía y Competitividad: PID 2019-104077-RB-100/AEI/10.3389/fphar.2021.762738, Ministerio de Sanidad, Asuntos Sociales e Igualdad (Retic-ISCIII-RD/16/0017/0010-FEDER and Plan Nacional Sobre Drogas (#2018/007). AG-B received a FI-AGAUR grant from the Generalitat de Catalunya (2019FI_B0081). The Department of Experimental and Health Sciences (UPF) is a “Unidad de Excelencia María de Maeztu” funded by the AEI (CEX 2018-000792-M).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2021.762738/full#supplementary-material
Aguado, T., Palazuelos, J., Monory, K., Stella, N., Cravatt, B., Lutz, B., et al. (2006). The Endocannabinoid System Promotes Astroglial Differentiation by Acting on Neural Progenitor Cells. J. Neurosci. 26 (5), 1551–1561. doi:10.1523/JNEUROSCI.3101-05.2006
Ahrens, J., Demir, R., Leuwer, M., de la Roche, J., Krampfl, K., Foadi, N., et al. (2009). The Nonpsychotropic Cannabinoid Cannabidiol Modulates and Directly Activates Alpha-1 and Alpha-1-Beta Glycine Receptor Function. Pharmacology 83 (4), 217–222. doi:10.1159/000201556
Aizpurua-Olaizola, O., Soydaner, U., Öztürk, E., Schibano, D., Simsir, Y., Navarro, P., et al. (2016). Evolution of the Cannabinoid and Terpene Content during the Growth of Cannabis Sativa Plants from Different Chemotypes. J. Nat. Prod. 79 (2), 324–331. doi:10.1021/acs.jnatprod.5b00949
Akil, H., Gordon, J., Hen, R., Javitch, J., Mayberg, H., McEwen, B., et al. (2018). Treatment Resistant Depression: A Multi-Scale, Systems Biology Approach. Neurosci. Biobehav Rev. 84 (January), 272–288. doi:10.1016/j.neubiorev.2017.08.019
Albert, P. (2015). Why Is Depression More Prevalent in Women? J. Psychiatry Neurosci. 40 (4), 219–221. doi:10.1503/jpn.150205
American Psychiatric Association (2013). Diagnostic and Statistical Manual of Mental Disorders. Fifth Edition. Virginia, United States: American Psychiatric Association. doi:10.1176/appi.books.9780890425596
Anacker, C., Cattaneo, A., Luoni, A., Musaelyan, K., Zunszain, P. A., Milanesi, E., et al. (2013). Glucocorticoid-Related Molecular Signaling Pathways Regulating Hippocampal Neurogenesis. Neuropsychopharmacology 38 (5), 872–883. doi:10.1038/npp.2012.253
Andres-Mach, M., Haratym-Maj, A., Zagaja, M., Rola, R., Maj, M., Chrościńska-Krawczyk, M., et al. (2015). ACEA (A Highly Selective Cannabinoid CB1 Receptor Agonist) Stimulates Hippocampal Neurogenesis in Mice Treated with Antiepileptic Drugs. Brain Res. 1624 (October), 86–94. doi:10.1016/j.brainres.2015.07.028
Anisman, H., Merali, Z., and Hayley, S. (2008). Neurotransmitter, Peptide and Cytokine Processes in Relation to Depressive Disorder: Comorbidity between Depression and Neurodegenerative Disorders. Prog. Neurobiol. 85 (1), 1–74. doi:10.1016/j.pneurobio.2008.01.004
Ashton, J. C., Friberg, D., Darlington, C. L., and Smith, P. F. (2006). Expression of the Cannabinoid CB2 Receptor in the Rat Cerebellum: An Immunohistochemical Study. Neurosci. Lett. 396 (2), 113–116. doi:10.1016/j.neulet.2005.11.038
Aso, E., Ozaita, A., Valdizán, E. M., Ledent, C., Pazos, A., Maldonado, R., et al. (2008). BDNF Impairment in the Hippocampus Is Related to Enhanced Despair Behavior in CB1 Knockout Mice. J. Neurochem. 105 (2), 565–572. doi:10.1111/j.1471-4159.2007.05149.x
Ativie, F., Komorowska, J. A., Beins, E., Albayram, Ö., Zimmer, T., Zimmer, A., et al. (2018). Cannabinoid 1 Receptor Signaling on Hippocampal GABAergic Neurons Influences Microglial Activity. Front. Mol. Neurosci. 11, 295. doi:10.3389/fnmol.2018.00295
Autry, A. E., Adachi, M., Cheng, P., and Monteggia, L. M. (2009). Gender-Specific Impact of Brain-Derived Neurotrophic Factor Signaling on Stress-Induced Depression-like Behavior. Biol. Psychiatry 66 (1), 84–90. doi:10.1016/j.biopsych.2009.02.007
Avraham, H. K., Jiang, S., Fu, Y., Rockenstein, E., Makriyannis, A., Zvonok, A., et al. (2014). The Cannabinoid CB₂ Receptor Agonist AM1241 Enhances Neurogenesis in GFAP/Gp120 Transgenic Mice Displaying Deficits in Neurogenesis. Br. J. Pharmacol. 171 (2), 468–479. doi:10.1111/bph.12478
Bachmann, S. (2018). Epidemiology of Suicide and the Psychiatric Perspective. Int. J. Environ. Res. Public Health 15 (7). doi:10.3390/ijerph15071425
Bakas, T., van Nieuwenhuijzen, P. S., Devenish, S. O., McGregor, I. S., Arnold, J. C., and Chebib, M. (2017). The Direct Actions of Cannabidiol and 2-Arachidonoyl Glycerol at GABAA Receptors. Pharmacol. Res. 119 (May), 358–370. doi:10.1016/j.phrs.2017.02.022
Bambico, F. R., Katz, N., Debonnel, G., Gobbi, G., and Gobbi, Gabriella. (2007). Cannabinoids Elicit Antidepressant-like Behavior and Activate Serotonergic Neurons through the Medial Prefrontal Cortex. J. Neurosci. 27 (43), 11700–11711. doi:10.1523/JNEUROSCI.1636-07.2007
Barna, I., Zelena, D., Arszovszki, A. C., and Ledent, C. (2004). The Role of Endogenous Cannabinoids in the Hypothalamo-Pituitary-Adrenal Axis Regulation: In Vivo and In Vitro Studies in CB1 Receptor Knockout Mice. Life Sci. 75 (24), 2959–2970. doi:10.1016/j.lfs.2004.06.006
Bartel, S. J., Sherry, S. B., and Stewart, S. H. (2020). Self-Isolation: A Significant Contributor to Cannabis Use during the COVID-19 Pandemic. Subst. Abus 41 (0), 409–412. doi:10.1080/08897077.2020.1823550
Beins, Eva. Carolina. (2020). The Role of the Endocannabinoid System in Stress-Related Disorders and Neuroimmune Communication. Available at: https://bonndoc.ulb.uni-bonn.de/xmlui/handle/20.500.11811/8344.
Belle, D., and Doucet, J. (2003). Poverty, Inequality, and Discrimination as Sources of Depression Among U.S. Women. Psychol. Women Q. 27 (2), 101–113. doi:10.1111/1471-6402.00090
Benke, C., Autenrieth, L. K., Asselmann, E., and Pané-Farré, C. A. (2020). Lockdown, Quarantine Measures, and Social Distancing: Associations with Depression, Anxiety and Distress at the Beginning of the COVID-19 Pandemic Among Adults from Germany. Psychiatry Res. 293 (November), 113462. doi:10.1016/j.psychres.2020.113462
Besnier, E., Clavier, T., Tonon, M. C., Selim, J., Lefevre-Scelles, A., Morin, F., et al. (2017). Ketamine and Etomidate Down-Regulate the Hypothalamic-Pituitary-Adrenal Axis in an Endotoxemic Mouse Model. Anesthesiology 127 (2), 347–354. doi:10.1097/ALN.0000000000001704
Beyer, C. E., Dwyer Piesla, J. M, Piesla, M. J., Platt, B. J., Shen, R., Rahman, Z., et al. (2010). Ru Shen, Zia Rahman, Karen Chan, et alDepression-like Phenotype Following Chronic CB1 Receptor Antagonism. Neurobiol. Dis. 39 (2), 148–155. doi:10.1016/j.nbd.2010.03.020
Bond, A. M., Ming, G. L., and Song, H. (2015). Adult Mammalian Neural Stem Cells and Neurogenesis: Five Decades Later. Cell Stem Cell 17 (4), 385–395. doi:10.1016/j.stem.2015.09.003
Bouaboula, M., Poinot-Chazel, C., Bourrié, B., Canat, X., Calandra, B., Rinaldi-Carmona, M., et al. (1995). Activation of Mitogen-Activated Protein Kinases by Stimulation of the Central Cannabinoid Receptor CB1. Biochem. J. 312 ( Pt 2) (Pt 2), 637–641. doi:10.1042/bj3120637
Brown, A. J. (2007). Novel Cannabinoid Receptors. Br. J. Pharmacol. 152 (5), 567–575. doi:10.1038/sj.bjp.0707481
Busquets-Garcia, A., Bains, J., and Marsicano, G. (2018). CB1 Receptor Signaling in the Brain: Extracting Specificity from Ubiquity. Neuropsychopharmacology 43 (1), 4–20. doi:10.1038/npp.2017.206
Cabral, G. A., Raborn, E. S., Griffin, L., Dennis, J., and Marciano-Cabral, F. (2008). CB2 Receptors in the Brain: Role in Central Immune Function. Br. J. Pharmacol. 153 (2), 240–251. doi:10.1038/sj.bjp.0707584
Campos, A. C., Ortega, Z., Palazuelos, J., Fogaça, M. V., Aguiar, D. C., Díaz-Alonso, J., et al. (2013). The Anxiolytic Effect of Cannabidiol on Chronically Stressed Mice Depends on Hippocampal Neurogenesis: Involvement of the Endocannabinoid System. Int. J. Neuropsychopharmacol. 16 (6), 1407–1419. doi:10.1017/S1461145712001502
Carlisle, S. J., Marciano-Cabral, F., Staab, A., Ludwick, C., and Cabral, G. A. (2002). Differential Expression of the CB2 Cannabinoid Receptor by Rodent Macrophages and Macrophage-like Cells in Relation to Cell Activation. Int. Immunopharmacol 2 (1), 69–82. doi:10.1016/s1567-5769(01)00147-3
Castillo, P. E., Younts, T. J., Chávez, A. E., and Hashimotodani, Y. (2012). Endocannabinoid Signaling and Synaptic Function. Neuron 76 (1), 70–81. doi:10.1016/j.neuron.2012.09.020
Caughey, G. E., Mantzioris, E., Gibson, R. A., Cleland, L. G., and James, M. J. (1996). The Effect on Human Tumor Necrosis Factor Alpha and Interleukin 1 Beta Production of Diets Enriched in N-3 Fatty Acids from Vegetable Oil or Fish Oil. Am. J. Clin. Nutr. 63 (1), 116–122. doi:10.1093/ajcn/63.1.116
Chávez, A. E., Chiu, C. Q., and Castillo, P. E. (2010). TRPV1 Activation by Endogenous Anandamide Triggers Postsynaptic Long-Term Depression in Dentate Gyrus. Nat. Neurosci. 13 (12), 1511–1518. doi:10.1038/nn.2684
ChenCortina, H. C., Spiers, J. G., Sernia, C., and Lavidis, N. A. (2018). Inhibition of Fatty Acid Amide Hydrolase by PF-3845 Alleviates the Nitrergic and Proinflammatory Response in Rat Hippocampus Following Acute Stress. Int. J. Neuropsychopharmacol. 21 (8), 786–795. doi:10.1093/ijnp/pyy033
Cheung, S. G., GoldenthalGoldenthal, A. R., Uhlemann, A. C., Mann, J. J., Miller, J. M., and Sublette, M. E. (2019). Systematic Review of Gut Microbiota and Major Depression. Front. Psychiatry 10, 34. doi:10.3389/fpsyt.2019.00034
Christensen, R., Kristensen, P. K., Bartels, E. M., Bliddal, H., and Astrup, A. (2007). Efficacy and Safety of the Weight-Loss Drug Rimonabant: A Meta-Analysis of Randomised Trials. The Lancet 370 (9600), 1706–1713. doi:10.1016/S0140-6736(07)61721-8
Coppen, A. (1967). The Biochemistry of Affective Disorders. Br. J. Psychiatry 113 (504), 1237–1264. doi:10.1192/bjp.113.504.1237
Correa, F., Hernangómez, M., Mestre, L., Loría, F., Spagnolo, A., Docagne, F., et al. (2010). Anandamide Enhances IL-10 Production in Activated Microglia by Targeting CB(2) Receptors: Roles of ERK1/2, JNK, and NF-KappaB. Glia 58 (2), 135–147. doi:10.1002/glia.20907
Correa, F., Hernangómez-Herrero, M., Mestre, L., Loría, F., Docagne, F., and Guaza, C. (2011). The Endocannabinoid Anandamide Downregulates IL-23 and IL-12 Subunits in a Viral Model of Multiple Sclerosis: Evidence for a Cross-Talk between IL-12p70/IL-23 Axis and IL-10 in Microglial Cells. Brain Behav. Immun. 25 (4), 736–749. doi:10.1016/j.bbi.2011.01.020
Corroon, J., and Phillips, J. A. (2018). A Cross-Sectional Study of Cannabidiol Users. Cannabis Cannabinoid Res. 3 (1), 152–161. doi:10.1089/can.2018.0006
Costa, B., Giagnoni, G., Franke, C., Trovato, A. E., and Colleoni, M. (2004). Vanilloid TRPV1 Receptor Mediates the Antihyperalgesic Effect of the Nonpsychoactive Cannabinoid, Cannabidiol, in a Rat Model of Acute Inflammation. Br. J. Pharmacol. 143 (2), 247–250. doi:10.1038/sj.bjp.0705920
Cota, D., Steiner, M. A., Marsicano, G., Cervino, C., Herman, J. P., Grübler, Y., et al. (2007). Requirement of Cannabinoid Receptor Type 1 for the Basal Modulation of Hypothalamic-Pituitary-Adrenal Axis Function. Endocrinology 148 (4), 1574–1581. doi:10.1210/en.2005-1649
Cota, D. (2008). The Role of the Endocannabinoid System in the Regulation of Hypothalamic-Pituitary-Adrenal Axis Activity. J. Neuroendocrinol 20 Suppl 1 (Suppl. 1May), 35–38. doi:10.1111/j.1365-2826.2008.01673.x
Cristino, L., Bisogno, T., and Di Marzo, V. (2020). Cannabinoids and the Expanded Endocannabinoid System in Neurological Disorders. Nat. Rev. Neurol. 16 (1), 9–29. doi:10.1038/s41582-019-0284-z
D'Souza, D. C., Perry, E., MacDougall, L., Ammerman, Y., Cooper, T., Wu, Y.-t., et al. (2004). The Psychotomimetic Effects of Intravenous Delta-9-Tetrahydrocannabinol in Healthy Individuals: Implications for Psychosis. Neuropsychopharmacol 29 (8), 1558–1572. doi:10.1038/sj.npp.1300496
Dattani, S., Ritchie, H., and Roser, M. (2018). Mental Health. Our World in Data. Available at: https://ourworldindata.org/mental-health.
David, D. J., Samuels, B. A., Rainer, Q., Wang, J.-W., Marsteller, D., Mendez, I., et al. (2009). Neurogenesis-Dependent and -Independent Effects of Fluoxetine in an Animal Model of Anxiety/DepressionBehavioral Effects of Fluoxetine in an Animal Model of Anxiety/Depression Are Mediated by Both Neurogenesis-dependent and Independent Mechanisms. Neuron 62 (4), 479–493. doi:10.1016/j.neuron.2009.04.017
De Petrocellis, L., Ligresti, A., Moriello, A. S., Allarà, M., Bisogno, T., Petrosino, S., et al. (2011). Effects of Cannabinoids and Cannabinoid-Enriched Cannabis Extracts on TRP Channels and Endocannabinoid Metabolic Enzymes. Br. J. Pharmacol. 163 (7), 1479–1494. doi:10.1111/j.1476-5381.2010.01166.x
Derkinderen, P., Ledent, C., Parmentier, M., and Girault, J. A. (2001). Cannabinoids Activate P38 Mitogen-Activated Protein Kinases through CB1 Receptors in Hippocampus. J. Neurochem. 77 (3), 957–960. doi:10.1046/j.1471-4159.2001.00333.x
Devane, W. A., Hanus, L., Breuer, A., Pertwee, R. G., Stevenson, L. A., Griffin, G., et al. (1992). Isolation and Structure of a Brain Constituent that Binds to the Cannabinoid Receptor. Science 258 (5090), 1946–1949. doi:10.1126/science.1470919
Di Marzo, V., and De Petrocellis, L. (2012). Why Do Cannabinoid Receptors Have More Than One Endogenous Ligand? Philos. Trans. R. Soc. Lond. B Biol. Sci. 367 (1607), 3216–3228. doi:10.1098/rstb.2011.0382
Diana, M. A., and Marty, A. (2004). Endocannabinoid-Mediated Short-Term Synaptic Plasticity: Depolarization-Induced Suppression of Inhibition (DSI) and Depolarization-Induced Suppression of Excitation (DSE). Br. J. Pharmacol. 142 (1), 9–19. doi:10.1038/sj.bjp.0705726
Dinh, T. P., Carpenter, D., Leslie, F. M., Freund, T. F., Katona, I., Sensi, S. L., et al. (2002). Brain Monoglyceride Lipase Participating in Endocannabinoid Inactivation. Proc. Natl. Acad. Sci. U S A. 99 (16), 10819–10824. doi:10.1073/pnas.152334899
Dinh, T. P., Kathuria, S., and Piomelli, D. (2004). RNA Interference Suggests a Primary Role for Monoacylglycerol Lipase in the Degradation of the Endocannabinoid 2-Arachidonoylglycerol. Mol. Pharmacol. 66 (5), 1260–1264. doi:10.1124/mol.104.002071
Domschke, K., Dannlowski, U., Ohrmann, P., Lawford, B., Bauer, J., Kugel, H., et al. (2008). Cannabinoid Receptor 1 (CNR1) Gene: Impact on Antidepressant Treatment Response and Emotion Processing in Major Depression. Eur. Neuropsychopharmacol. 18 (10), 751–759. doi:10.1016/j.euroneuro.2008.05.003
Elsaid, S., and Le Foll, B. (2020). The Complexity of Pharmacology of Cannabidiol (CBD) and its Implications in the Treatment of Brain Disorders. Neuropsychopharmacology 45 (1), 229–230. doi:10.1038/s41386-019-0518-1
Felger, J. C., Haroon, E., Patel, T. A., Goldsmith, D. R., Wommack, E. C., Woolwine, B. J., et al. (2018). What Does Plasma CRP Tell Us about Peripheral and Central Inflammation in Depression? Mol. Psychiatry 25 (June), 1301–1311. doi:10.1038/s41380-018-0096-3
Ferreira, R. C. M., Castor, M. G. M., Piscitelli, F., Di Marzo, V., Duarte, I. D. G., and Romero, T. R. L. (2018). The Involvement of the Endocannabinoid System in the Peripheral Antinociceptive Action of Ketamine. J. Pain 19 (5), 487–495. doi:10.1016/j.jpain.2017.12.002
Ferrini, F., and De Koninck, Y. (2013). Microglia Control Neuronal Network Excitability via BDNF Signalling. Neural Plast. 2013 (September), 429815. doi:10.1155/2013/429815
Fogaça, M. V., Campos, A. C., Coelho, L. D., Duman, R. S., and Guimarães, F. S. (2018). The Anxiolytic Effects of Cannabidiol in Chronically Stressed Mice Are Mediated by the Endocannabinoid System: Role of Neurogenesis and Dendritic Remodeling. Neuropharmacology 135, 22–33. doi:10.1016/j.neuropharm.2018.03.001
Fowler, C. J. (2015). The Potential of Inhibitors of Endocannabinoid Metabolism as Anxiolytic and Antidepressive Drugs-A Practical View. Eur. Neuropsychopharmacol. 25 (6), 749–762. doi:10.1016/j.euroneuro.2015.02.005
Gaetani, S., Dipasquale, P., Romano, A., Righetti, L., Cassano, T., Piomelli, D., et al. (2009). The Endocannabinoid System as a Target for Novel Anxiolytic and Antidepressant Drugs. Int. Rev. Neurobiol. 85, 57–72. doi:10.1016/S0074-7742(09)85005-8
Gao, Y., Vasilyev, D. V., Goncalves, M. B., Howell, F. V., Hobbs, C., Reisenberg, M., et al. (2010). Loss of Retrograde Endocannabinoid Signaling and Reduced Adult Neurogenesis in Diacylglycerol Lipase Knock-Out Mice. J. Neurosci. 30 (6), 2017–2024. doi:10.1523/JNEUROSCI.5693-09.2010
García-Gutiérrez, M. S., García-Bueno, B., Zoppi, S., Leza, J. C., Manzanares, J., and Manzanares, Jorge. (2012). Chronic Blockade of Cannabinoid CB2 Receptors Induces Anxiolytic-like Actions Associated with Alterations in GABAA Receptors. Br. J. Pharmacol. 165 (4), 951–964. doi:10.1111/j.1476-5381.2011.01625.x
Gillespie, C. F., and Nemeroff, C. B. (2005). Hypercortisolemia and Depression. Psychosom Med. 67 Suppl 1 (Suppl. 1June), S26–S28. doi:10.1097/01.psy.0000163456.22154.d2
Gonca, E., and Darıcı, F. (2015). The Effect of Cannabidiol on Ischemia/Reperfusion-Induced Ventricular Arrhythmias: The Role of Adenosine A1 Receptors. J. Cardiovasc. Pharmacol. Ther. 20 (1), 76–83. doi:10.1177/1074248414532013
Gonda, X., Hullam, G., Antal, P., Eszlari, N., Petschner, P., Hökfelt, T. G., et al. (2018). Significance of Risk Polymorphisms for Depression Depends on Stress Exposure. Sci. Rep. 8 (1), 3946. doi:10.1038/s41598-018-22221-z
Graham, E. S., Angel, C. E., Schwarcz, L. E., Dunbar, P. R., and Glass, M. (2010). Detailed Characterisation of CB2 Receptor Protein Expression in Peripheral Blood Immune Cells from Healthy Human Volunteers Using Flow Cytometry. Int. J. Immunopathol Pharmacol. 23 (1), 25–34. doi:10.1177/039463201002300103
Gray, J. M., Vecchiarelli, H. A., Morena, M., Lee, T. T., Hermanson, D. J., Kim, A. B., et al. (2015). Corticotropin-Releasing Hormone Drives Anandamide Hydrolysis in the Amygdala to Promote Anxiety. J. Neurosci. 35 (9), 3879–3892. doi:10.1523/JNEUROSCI.2737-14.2015
Gray, J. M., Wilson, C. D., Lee, T. T., Pittman, Q. J., Deussing, J. M., Hillard, C. J., et al. (2016). Sustained Glucocorticoid Exposure Recruits Cortico-Limbic CRH Signaling to Modulate Endocannabinoid Function. Psychoneuroendocrinology 66 (April), 151–158. doi:10.1016/j.psyneuen.2016.01.004
Grueter, B. A., Brasnjo, G., and Malenka, R. C. (2010). Postsynaptic TRPV1 Triggers Cell Type-specific Long-Term Depression in the Nucleus Accumbens. Nat. Neurosci. 13 (12), 1519–1525. doi:10.1038/nn.2685
Gulyas, A. I., Cravatt, B. F., Bracey, M. H., Dinh, T. P., Piomelli, D., Boscia, F., et al. (2004). Segregation of Two Endocannabinoid-Hydrolyzing Enzymes into Pre- and Postsynaptic Compartments in the Rat Hippocampus, Cerebellum and Amygdala. Eur. J. Neurosci. 20 (2), 441–458. doi:10.1111/j.1460-9568.2004.03428.x
Gunduz-CinarHill, O., Hill, M. N., McEwen, B. S., and Holmes, A. (2013). Amygdala FAAH and Anandamide: Mediating Protection and Recovery from Stress. Trends Pharmacol. Sci. 34 (11), 637–644. doi:10.1016/j.tips.2013.08.008
Haapakoski, R., Mathieu, J., Ebmeier, K. P., Alenius, H., and Kivimäki, M. (2015). Cumulative Meta-Analysis of Interleukins 6 and 1β, Tumour Necrosis Factor α and C-Reactive Protein in Patients with Major Depressive Disorder. Brain Behav. Immun. 49 (October), 206–215. doi:10.1016/j.bbi.2015.06.001
Haj-Mirzaian, A., Amini-Khoei, H., Khoei, Amini., Haj-Mirzaian, A., Amiri, S., Ghesmati, M., et al. (2017). Activation of Cannabinoid Receptors Elicits Antidepressant-like Effects in a Mouse Model of Social Isolation Stress. Brain Res. Bull. 130 (April), 200–210. doi:10.1016/j.brainresbull.2017.01.018
Haller, J., Bakos, N., Szirmay, M., Ledent, C., and Freund, T. F. (2002). The Effects of Genetic and Pharmacological Blockade of the CB1 Cannabinoid Receptor on Anxiety. Eur. J. Neurosci. 16 (7), 1395–1398. doi:10.1046/j.1460-9568.2002.02192.x
Han, S., Thatte, J., Buzard, D. J., and Jones, R. M. (2013). Therapeutic Utility of Cannabinoid Receptor Type 2 (CB(2)) Selective Agonists. J. Med. Chem. 56 (21), 8224–8256. doi:10.1021/jm4005626
Hanson, N. D., Owens, M. J., and Nemeroff, C. B. (2011). Depression, Antidepressants, and Neurogenesis: A Critical Reappraisal. Neuropsychopharmacology 36 (13), 2589–2602. doi:10.1038/npp.2011.220
Hashioka, S., Klegeris, A., Monji, A., Kato, T., Sawada, M., McGeer, P. L., et al. (2007). Antidepressants Inhibit Interferon-Gamma-Induced Microglial Production of IL-6 and Nitric Oxide. Exp. Neurol. 206 (1), 33–42. doi:10.1016/j.expneurol.2007.03.022
Heim, C., and Nemeroff, C. B. (2002). Neurobiology of Early Life Stress: Clinical Studies. Semin. Clin. Neuropsychiatry 7 (2), 147–159. doi:10.1053/scnp.2002.33127
Hen-Shoval, D., Amar, S., Shbiro, L., Smoum, R., Haj, C. G., Mechoulam, R., et al. (2018). Acute Oral Cannabidiolic Acid Methyl Ester Reduces Depression-like Behavior in Two Genetic Animal Models of Depression. Behav. Brain Res. 351 (October), 1–3. doi:10.1016/j.bbr.2018.05.027
Hill, M. N., and Gorzalka, B. B. (2005). Is There a Role for the Endocannabinoid System in the Etiology and Treatment of Melancholic Depression? Behav. Pharmacol. 16 (5–6), 333–352. doi:10.1097/00008877-200509000-00006
Hill, M. N., and Gorzalka, B. B. (2005). Pharmacological Enhancement of Cannabinoid CB1 Receptor Activity Elicits an Antidepressant-like Response in the Rat Forced Swim Test. Eur. Neuropsychopharmacol. 15 (6), 593–599. doi:10.1016/j.euroneuro.2005.03.003
Hill, M. N., HoHillard, W. S., Hillard, C. J., and Gorzalka, B. B. (2008).Differential Effects of the Antidepressants Tranylcypromine and Fluoxetine on Limbic Cannabinoid Receptor Binding and Endocannabinoid Contents. J. Neural Transm. (Vienna) 115, 1673–1679. doi:10.1007/s00702-008-0131-7
Hill, M. N., McLaughlin, R. J. Bin. Pan., Karatsoreos, B. Christopher, Fitzgerald, M. L., Roberts, C. J., Lee, T. T., et al. (2011). Recruitment of Prefrontal Cortical Endocannabinoid Signaling by Glucocorticoids Contributes to Termination of the Stress Response. J. Neurosci. 31 (29), 10506–10515. doi:10.1523/JNEUROSCI.0496-11.2011
Hill, M. N., Miller, G. E., Ho, W. S., Gorzalka, B. B., and Hillard, C. J. (2008). Serum Endocannabinoid Content Is Altered in Females with Depressive Disorders: A Preliminary Report. Pharmacopsychiatry 41 (2), 48–53. doi:10.1055/s-2007-993211
Hillard, C. J., Beatka, M., and Sarvaideo., J. (2016). Endocannabinoid Signaling and the Hypothalamic-Pituitary-Adrenal Axis. Compr. Physiol. 7 (1), 1–15. doi:10.1002/cphy.c160005
Hinwood, M., Tynan, Ross. J., Tynan, J. L., Beynon, S. B., Day, T. A., and Walker, F. R. (20131991). Chronic Stress Induced Remodeling of the Prefrontal Cortex: Structural Re-organization of Microglia and the Inhibitory Effect of Minocycline. Cereb. Cortex 23 (8), 1784–1797. doi:10.1093/cercor/bhs151
Horowitz, M. A., Wertz, J., Zhu, D., Cattaneo, A., Musaelyan, K., Nikkheslat, N., et al. (2015). Antidepressant Compounds Can Be Both Pro- and Anti-inflammatory in Human Hippocampal Cells. Int. J. Neuropsychopharmacol. 18 (3). doi:10.1093/ijnp/pyu076
Howlett, A. C., Barth, F., Bonner, T. I., Cabral, G., Casellas, P., Devane, W. A., et al. (2002). International Union of Pharmacology. XXVII. Classification of Cannabinoid Receptors. Pharmacol. Rev. 54 (2), 161–202. doi:10.1124/pr.54.2.161
Hutch, C. R., and Hegg, C. C. (2016). Cannabinoid Receptor Signaling Induces Proliferation but Not Neurogenesis in the Mouse Olfactory Epithelium. Neurogenesis (Austin) 3 (1), e1118177. doi:10.1080/23262133.2015.1118177
Icick, R., Peoc'h, K., Karsinti, E., Ksouda, K., Hajj, A., Bloch, V., et al. (2015). A Cannabinoid Receptor 1 Polymorphism Is Protective against Major Depressive Disorder in Methadone-Maintained Outpatients. Am. J. Addict. 24 (7), 613–620. doi:10.1111/ajad.12273
Iffland, K., and Grotenhermen, F. (2017). An Update on Safety and Side Effects of Cannabidiol: A Review of Clinical Data and Relevant Animal Studies. Cannabis Cannabinoid Res. 2 (1), 139–154. doi:10.1089/can.2016.0034
Izzo, A. A., and Sharkey, K. A. (2010). Cannabinoids and the Gut: New Developments and Emerging Concepts. Pharmacol. Ther. 126 (1), 21–38. doi:10.1016/j.pharmthera.2009.12.005
Jayatissa, M. N., Henningsen, K., Nikolajsen, G., West, M. J., and Wiborg, O. (2010). A Reduced Number of Hippocampal Granule Cells Does Not Associate with an Anhedonia-like Phenotype in a Rat Chronic Mild Stress Model of Depression. Stress 13 (2), 95–105. doi:10.3109/10253890902951786
Jenniches, I., Ternes, S., Albayram, O., Otte, D. M., Bach, K., Bindila, L., et al. (2016). Anxiety, Stress, and Fear Response in Mice with Reduced Endocannabinoid Levels. Biol. Psychiatry 79 (10), 858–868. doi:10.1016/j.biopsych.2015.03.033
Jesulola, E., Micalos, P., and Baguley, I. J. (2018). Understanding the Pathophysiology of Depression: From Monoamines to the Neurogenesis Hypothesis Model - Are We There yet? Behav. Brain Res. 341 (April), 79–90. doi:10.1016/j.bbr.2017.12.025
Jin, X. H., Okamoto, Y., Morishita, J., Tsuboi, K., Tonai, T., and Ueda, N. (2007). Discovery and Characterization of a Ca2+-independent Phosphatidylethanolamine N-Acyltransferase Generating the Anandamide Precursor and its Congeners. J. Biol. Chem. 282 (6), 3614–3623. doi:10.1074/jbc.M606369200
Jin, X. H., Uyama, T., Wang, J., Okamoto, Y., Tonai, T., and Ueda, N. (2009). cDNA Cloning and Characterization of Human and Mouse Ca(2+)-independent Phosphatidylethanolamine N-Acyltransferases. Biochim. Biophys. Acta 1791 (1), 32–38. doi:10.1016/j.bbalip.2008.09.006
Juruena, M. F. (2014). Early-Life Stress and HPA Axis Trigger Recurrent Adulthood Depression. Epilepsy Behav. 38 (September), 148–159. doi:10.1016/j.yebeh.2013.10.020
Kaplan, H. B., Martin, S. S., Johnson, R. J., and Robbins, C. A. (1986). Escalation of Marijuana Use: Application of a General Theory of Deviant Behavior. J. Health Soc. Behav. 27 (1), 44–61. doi:10.2307/2136502
Kaur, R., Sidhu, P., and Singh, S. (2016). What Failed BIA 10-2474 Phase I Clinical Trial? Global Speculations and Recommendations for Future Phase I Trials. J. Pharmacol. Pharmacother. 7 (3), 120–126. doi:10.4103/0976-500X.189661
Kellogg, R., Mackie, K., and Straiker, A. (2009). Cannabinoid CB1 Receptor-dependent Long-Term Depression in Autaptic Excitatory Neurons. J. Neurophysiol. 102 (2), 1160–1171. doi:10.1152/jn.00266.2009
Kendler, K. S., Gatz, M., Gardner, C. O., and Pedersen, N. L. (2006). A Swedish National Twin Study of Lifetime Major Depression. Am. J. Psychiatry 163 (1), 109–114. doi:10.1176/appi.ajp.163.1.109
Khakpai, F., Ebrahimi-Ghiri, M., Alijanpour, S., and Zarrindast, M. R. (2019). Ketamine-Induced Antidepressant like Effects in Mice: A Possible Involvement of Cannabinoid System. Biomed. Pharmacother. 112 (April), 108717. doi:10.1016/j.biopha.2019.108717
Kim, J., and Alger, B. E. (2004). Inhibition of Cyclooxygenase-2 Potentiates Retrograde Endocannabinoid Effects in Hippocampus. Nat. Neurosci. 7 (7), 697–698. doi:10.1038/nn1262
Kim, J., and Alger, B. E. (2010). Reduction in Endocannabinoid Tone Is a Homeostatic Mechanism for Specific Inhibitory Synapses. Nat. Neurosci. 13 (5), 592–600. doi:10.1038/nn.2517
Klegeris, A., Bissonnette, C. J., and McGeer, P. L. (2003). Reduction of Human Monocytic Cell Neurotoxicity and Cytokine Secretion by Ligands of the Cannabinoid-type CB2 Receptor. Br. J. Pharmacol. 139 (4), 775–786. doi:10.1038/sj.bjp.0705304
Koscsó, B., Csóka, B., Kókai, E., Németh, Z. H., Pacher, P., Virág, L., et al. (2013). Adenosine Augments IL‐10‐induced STAT3 Signaling in M2c Macrophages. J. Leukoc. Biol. 94 (6), 1309–1315. doi:10.1189/jlb.0113043
Kreisel, T., Frank, M. G., Licht, T., Reshef, R., Ben-Menachem-Zidon, O., Baratta, M. V., et al. (2014). Dynamic Microglial Alterations Underlie Stress-Induced Depressive-like Behavior and Suppressed Neurogenesis. Mol. Psychiatry 19 (6), 699–709. doi:10.1038/mp.2013.155
Kubitz, N., Mehra, M., Potluri, R. C., Garg, N., and Cossrow, N. (2013). Characterization of Treatment Resistant Depression Episodes in a Cohort of Patients from a US Commercial Claims Database. PLOS ONE 8 (10), e76882. doi:10.1371/journal.pone.0076882
Laczkovics, C., Kothgassner, O. D., Felnhofer, A., and Klier, C. M. (2020). Cannabidiol Treatment in an Adolescent with Multiple Substance Abuse, Social Anxiety and Depression. Neuropsychiatr, 35, 31–34. doi:10.1007/s40211-020-00334-0
Laprairie, R. B., Bagher, A. M., Kelly, M. E., and Denovan-Wright, E. M. (2015). Cannabidiol Is a Negative Allosteric Modulator of the Cannabinoid CB1 Receptor. Br. J. Pharmacol. 172 (20), 4790–4805. doi:10.1111/bph.13250
Lastres-Becker, I., Molina-Holgado, Francisco., Molina-Holgado, F., Ramos, J. A., Mechoulam, R., and Fernández-Ruiz, J. (2005). Cannabinoids Provide Neuroprotection against 6-hydroxydopamine Toxicity In Vivo and In Vitro: Relevance to Parkinson's Disease. Neurobiol. Dis. 19 (1), 96–107. doi:10.1016/j.nbd.2004.11.009
Lazary, J., Eszlari, N., Juhasz, G., and Bagdy, G. (2019). A Functional Variant of CB2 Receptor Gene Interacts with Childhood Trauma and FAAH Gene on Anxious and Depressive Phenotypes. J. Affect Disord. 257 (October), 716–722. doi:10.1016/j.jad.2019.07.083
Lazary, J., Eszlari, N., Juhasz, G., and Bagdy, G. (2016). Genetically Reduced FAAH Activity May Be a Risk for the Development of Anxiety and Depression in Persons with Repetitive Childhood Trauma. Eur. Neuropsychopharmacol. 26 (6), 1020–1028. doi:10.1016/j.euroneuro.2016.03.003
Ledent, C., Valverde, O., Cossu, G., Petitet, F., Aubert, J. F., Beslot, F., et al. (1999). Unresponsiveness to Cannabinoids and Reduced Addictive Effects of Opiates in CB1 Receptor Knockout Mice. Science 283 (5400), 401–404. doi:10.1126/science.283.5400.401
Lee, S. H., Ledri, M., Tóth, B., Marchionni, I., Henstridge, C. M., Dudok, B., et al. (2015). Multiple Forms of Endocannabinoid and Endovanilloid Signaling Regulate the Tonic Control of GABA Release. J. Neurosci. 35 (27), 10039–10057. doi:10.1523/JNEUROSCI.4112-14.2015
Lee, T. T. Y., and Hill, M. N. (2013). Age of Stress Exposure Modulates the Immediate and Sustained Effects of Repeated Stress on Corticolimbic Cannabinoid CB1 Receptor Binding in Male Rats. Neuroscience 249 (September), 106–114. doi:10.1016/j.neuroscience.2012.11.017
Liao, Y., Xie, B., Zhang, H., He, Q., Guo, L., Subramaniapillai, M., et al. (2019). Efficacy of Omega-3 PUFAs in Depression: A Meta-Analysis. Transl Psychiatry 9 (1), 190–199. doi:10.1038/s41398-019-0515-5
Lisboa, S. F., Niraula, A., Resstel, L. B., Guimaraes, F. S., Godbout, J. P., and Sheridan, J. F. (2018). Repeated Social Defeat-Induced Neuroinflammation, Anxiety-like Behavior and Resistance to Fear Extinction Were Attenuated by the Cannabinoid Receptor Agonist WIN55,212-2. Neuropsychopharmacology 43 (9), 1924–1933. doi:10.1038/s41386-018-0064-2
Liu, L.-L., Li, J.-M., Su, W.-J., Wang, B., and Jiang, C.-L. (2019). Sex Differences in Depressive-like Behaviour May Relate to Imbalance of Microglia Activation in the Hippocampus. Brain Behav. Immun. 81 (October), 188–197. doi:10.1016/j.bbi.2019.06.012
Lively, S., and Schlichter, L. C. (2018). Microglia Responses to Pro-inflammatory Stimuli (LPS, IFNγ+TNFα) and Reprogramming by Resolving Cytokines (IL-4, IL-10). Front Cel Neurosci 12, 215. doi:10.3389/fncel.2018.00215
Long, J. Z., NomuraNomura, Robert E. VannWalentiny, D. K. D. Matthew., Vann, R. E., Walentiny, D. M., Booker, L., Jin, X., et al. (2009). Dual Blockade of FAAH and MAGL Identifies Behavioral Processes Regulated by Endocannabinoid Crosstalk In Vivo. Proc. Natl. Acad. Sci. U S A. 106 (48), 20270–20275. doi:10.1073/pnas.0909411106
Lopez-Munoz, F., and Alamo, C. (2009). Monoaminergic Neurotransmission: The History of the Discovery of Antidepressants from 1950s until Today. Cpd 15 (14), 1563–1586. doi:10.2174/138161209788168001
Luján, M. Á., and Valverde., O. (2020). The Pro-neurogenic Effects of Cannabidiol and its Potential Therapeutic Implications in Psychiatric Disorders. Front. Behav. Neurosci. 14 (June), 109. doi:10.3389/fnbeh.2020.00109
Luo, X. Q., Li, A., Yang, X., Xiao, X., Hu, R., Wang, T. W., et al. (2018). Paeoniflorin Exerts Neuroprotective Effects by Modulating the M1/M2 Subset Polarization of Microglia/Macrophages in the Hippocampal CA1 Region of Vascular Dementia Rats via Cannabinoid Receptor 2. Chin. Med. 13 (1), 14. doi:10.1186/s13020-018-0173-1
Luo, Y., Kataoka, Y., Ostinelli, E. G., Cipriani, A., and Furukawa, T. A. (2020). National Prescription Patterns of Antidepressants in the Treatment of Adults with Major Depression in the US between 1996 and 2015: A Population Representative Survey Based Analysis. Front. Psychiatry 11. doi:10.3389/fpsyt.2020.00035
Ma, Y., Wang, J., Wang, Y., and Yang, G. Y. (2017). The Biphasic Function of Microglia in Ischemic Stroke. Prog. Neurobiol. 157 (October), 247–272. doi:10.1016/j.pneurobio.2016.01.005
Maccarrone, M., Rossi, S., Bari, M., De Chiara, V., Fezza, F., Musella, A., et al. (2008). Anandamide Inhibits Metabolism and Physiological Actions of 2-Arachidonoylglycerol in the Striatum. Nat. Neurosci. 11 (2), 152–159. doi:10.1038/nn2042
Mackie, K. (2005). Distribution of Cannabinoid Receptors in the Central and Peripheral Nervous System. Handb Exp. Pharmacol. 168, 299–325. doi:10.1007/3-540-26573-2_10
Maejima, T., Hashimoto, K., Yoshida, T., Aiba, A., and Kano, M. (2001). Presynaptic Inhibition Caused by Retrograde Signal from Metabotropic Glutamate to Cannabinoid Receptors. Neuron 31 (3), 463–475. doi:10.1016/s0896-6273(01)00375-0
Malan, T. P., Ibrahim, M. M., Deng, H., Liu, Q., Mata, H. P., Vanderah, T., et al. (2001). CB2 Cannabinoid Receptor-Mediated Peripheral Antinociception. Pain 93 (3), 239–245. doi:10.1016/s0304-3959(01)00321-9
Malberg, J. E., and Duman, R. S. (2003). Cell Proliferation in Adult Hippocampus Is Decreased by Inescapable Stress: Reversal by Fluoxetine Treatment. Neuropsychopharmacology 28 (9), 1562–1571. doi:10.1038/sj.npp.1300234
Malek, N., Popiolek-Barczyk, K., Mika, J., Przewlocka, B., and Starowicz, K. (20152015). Anandamide, Acting viaCB2Receptors, Alleviates LPS-Induced Neuroinflammation in Rat Primary Microglial Cultures. Neural Plasticity 2015, 1–10. doi:10.1155/2015/130639
Mammana, S., Cavalli, E., Gugliandolo, A., Silvestro, S., Pollastro, F., Bramanti, P., et al. (2019). Could the Combination of Two Non-psychotropic Cannabinoids Counteract Neuroinflammation? Effectiveness of Cannabidiol Associated with Cannabigerol. Medicina (Kaunas) 55 (11), 747. doi:10.3390/medicina55110747
Manich, G., Recasens, M., Valente, T., Almolda, B., González, B., and Castellano, B. (2019). Role of the CD200-Cd200r Axis during Homeostasis and Neuroinflammation. Neuroscience 405 (May), 118–136. doi:10.1016/j.neuroscience.2018.10.030
Marangell, L. B., Martinez, J. M., Zboyan, H. A., Kertz, B., Kim, H. F., and Puryear, L. J. (2003). A Double-Blind, Placebo-Controlled Study of the Omega-3 Fatty Acid Docosahexaenoic Acid in the Treatment of Major Depression. Am. J. Psychiatry 160 (5), 996–998. doi:10.1176/appi.ajp.160.5.996
Marco, E. M., Ballesta, J. A., Irala, C., Hernández, M.-D., Serrano, M. E., Mela, V., et al. (2017). Javier Antonio Ballesta, Carlos Irala, María-Donina Hernández, María Elisa Serrano, Virginia Mela, Meritxell López-Gallardo, and María-Paz ViverosSex-dependent Influence of Chronic Mild Stress (CMS) on Voluntary Alcohol Consumption; Study of Neurobiological Consequences. Pharmacol. Biochem. Behav. 152 (January), 68–80. doi:10.1016/j.pbb.2016.11.005
Martin, M., Ledent, C., Parmentier, M., Maldonado, R., and Valverde, O. (2002). Involvement of CB1 Cannabinoid Receptors in Emotional Behaviour. Psychopharmacology (Berl) 159 (4), 379–387. doi:10.1007/s00213-001-0946-5
Martín-Moreno, A. M., Reigada, D., Ramírez, B. G., Mechoulam, R., Innamorato, N., Cuadrado, A., et al. (2011). Cannabidiol and Other Cannabinoids Reduce Microglial Activation In Vitro and In Vivo: Relevance to Alzheimer's Disease. Mol. Pharmacol. 79 (6), 964–973. doi:10.1124/mol.111.071290
Massart, R., Mongeau, R., and Lanfumey, L. (2012). Beyond the Monoaminergic Hypothesis: Neuroplasticity and Epigenetic Changes in a Transgenic Mouse Model of Depression. Philos. Trans. R. Soc. Lond. B Biol. Sci. 367 (1601), 2485–2494. doi:10.1098/rstb.2012.0212
Matias, I., Pochard, P., Orlando, P., Salzet, M., Pestel, J., and Di Marzo, V. (2002). Presence and Regulation of the Endocannabinoid System in Human Dendritic Cells. Eur. J. Biochem. 269 (15), 3771–3778. doi:10.1046/j.1432-1033.2002.03078.x
Matsuda, L. A., Lolait, S. J., Brownstein, M. J., Young, A. C., and Bonner, T. I. (1990). Structure of a Cannabinoid Receptor and Functional Expression of the Cloned CDNA. Nature 346 (6284), 561–564. doi:10.1038/346561a0
McKinney, M. K., and Cravatt, B. F. (2005). Structure and Function of Fatty Acid Amide Hydrolase. Annu. Rev. Biochem. 74, 411–432. doi:10.1146/annurev.biochem.74.082803.133450
Mechoulam, R., and Parker, L. A. (2013). The Endocannabinoid System and the Brain. Annu. Rev. Psychol. 64 (1), 21–47. doi:10.1146/annurev-psych-113011-143739
Mensching, L., Djogo, N., Keller, C., Rading, S., and Karsak, M. (2019). Stable Adult Hippocampal Neurogenesis in Cannabinoid Receptor CB2 Deficient Mice. Int. J. Mol. Sci. 20 (15), 3759. doi:10.3390/ijms20153759
Micale, V., and Drago, F. (2018). Endocannabinoid System, Stress and HPA Axis. Eur. J. Pharmacol. 834 (September), 230–239. doi:10.1016/j.ejphar.2018.07.039
Mitchell, P. B., and Morris., M. J. (2007). Depression and Anxiety with Rimonabant. Lancet 370 (9600), 1671–1672. doi:10.1016/S0140-6736(07)61705-X
Mitjans, M., Serretti, A., Fabbri, C., Gastó, C., Catalán, R., Fañanás, L., et al. (2013). Screening Genetic Variability at the CNR1 Gene in Both Major Depression Etiology and Clinical Response to Citalopram Treatment. Psychopharmacology (Berl) 227 (3), 509–519. doi:10.1007/s00213-013-2995-y
Mondimore, F. M., Zandi, P. P., Mackinnon, D. F., McInnis, Melvin. G., Erin, B., Miller, M. G., et al. (2006). Familial Aggregation of Illness Chronicity in Recurrent, Early-Onset Major Depression Pedigrees. Am. J. Psychiatry 163 (9), 1554–1560. doi:10.1176/ajp.2006.163.9.1554
Moreira, F. A., Grieb, M., and Lutz., B. (2009). Central Side-Effects of Therapies Based on CB1 Cannabinoid Receptor Agonists and Antagonists: Focus on Anxiety and Depression. Best Pract. Res. Clin. Endocrinol. Metab. 23 (1), 133–144. doi:10.1016/j.beem.2008.09.003
Moreira, F. A., and CrippaCrippa, J. A. S. (2009). The Psychiatric Side-Effects of Rimonabant. Rev. Bras. Psiquiatr. 31 (2), 145–153. doi:10.1590/s1516-44462009000200012
Morin, C. M., Carrier, J., Bastien, C., and Godbout, R. (2020). Sleep and Circadian Rhythm in Response to the COVID-19 Pandemic. Can. J. Public Health 111, 654–657. doi:10.17269/s41997-020-00382-7
Mozaffari-Khosravi, H., Yassini-Ardakani, M., Karamati, M., and Shariati-Bafghi, S.-E. (2013). Eicosapentaenoic Acid versus Docosahexaenoic Acid in Mild-To-Moderate Depression: A Randomized, Double-Blind, Placebo-Controlled Trial. Eur. Neuropsychopharmacol. 23 (7), 636–644. doi:10.1016/j.euroneuro.2012.08.003
Munro, S., Thomas, K. L., and Abu-Shaar, M. (1993). Molecular Characterization of a Peripheral Receptor for Cannabinoids. Nature 365 (6441), 61–65. doi:10.1038/365061a0
Murataeva, N., Straiker, A., and Mackie, K. (2014). Parsing the Players: 2-Arachidonoylglycerol Synthesis and Degradation in the CNS. Br. J. Pharmacol. 171 (6), 1379–1391. doi:10.1111/bph.12411
Murillo-Rodriguez, E., Poot-Ake, A., Arias-Carrion, O., Pacheco-Pantoja, E., de la Fuente-Ortegon, A., and Arankowsky-Sandoval, G. (2011). The Emerging Role of the Endocannabinoid System in the Sleep-Wake Cycle Modulation. Cnsamc 11 (3), 189–196. doi:10.2174/187152411798047780
Murrough, J. W., Iosifescu, D. V., Chang, L. C. Rayan, Al Jurdi, R. K. Andrew, Green, C. E., Perez, A. M., et al. (2013). Antidepressant Efficacy of Ketamine in Treatment-Resistant Major Depression: A Two-Site Randomized Controlled Trial. Am. J. Psychiatry 170 (10), 1134–1142. doi:10.1176/appi.ajp.2013.13030392
Musella, A., De Chiara, V., Rossi, S., Prosperetti, C., Bernardi, G., Maccarrone, M., et al. (2009). TRPV1 Channels Facilitate Glutamate Transmission in the Striatum. Mol. Cel Neurosci 40 (1), 89–97. doi:10.1016/j.mcn.2008.09.001
Nair, A., and Bonneau, R. H. (2006). Stress-Induced Elevation of Glucocorticoids Increases Microglia Proliferation through NMDA Receptor Activation. J. Neuroimmunol 171 (1), 72–85. doi:10.1016/j.jneuroim.2005.09.012
National Collaborating Centre for Mental Health (Uk) (2010) Depression: The Treatment and Management of Depression in Adults. in National Institute for Health and Clinical Excellence: Guidance. Updated Edition (Leicester (UK): British Psychological Society).
Navarrete, F., Pérez-OrtizPérez-Ortiz, J. M., and Manzanares, J. (2012). Cannabinoid CB₂ Receptor-Mediated Regulation of Impulsive-like Behaviour in DBA/2 Mice. Br. J. Pharmacol. 165 (1), 260–273. doi:10.1111/j.1476-5381.2011.01542.x
Navarrete, M., and Araque, A. (2008). Endocannabinoids Mediate Neuron-Astrocyte Communication. Neuron 57 (6), 883–893. doi:10.1016/j.neuron.2008.01.029
Navarria, A., Tamburella, A., Iannotti, F. A., Micale, V., Camillieri, G., Gozzo, L., et al. (2014). The Dual Blocker of FAAH/TRPV1 N-Arachidonoylserotonin Reverses the Behavioral Despair Induced by Stress in Rats and Modulates the HPA-Axis. Pharmacol. Res. 87 (September), 151–159. doi:10.1016/j.phrs.2014.04.014
Nelson, C. A., and Gabard-Durnam, L. J. (2020). Early Adversity and Critical Periods: Neurodevelopmental Consequences of Violating the Expectable Environment. Trends Neurosci. 43 (3), 133–143. doi:10.1016/j.tins.2020.01.002
Nemeroff, Charles. B. (2007). Prevalence and Management of Treatment-Resistant Depression. J. Clin. Psychiatry 68 (Suppl. 8), 17–25.
Nestler, E. J., Barrot, M., DiLeone, R. J., Eisch, A. J., Gold, S. J., and Monteggia, L. M. (2002). Neurobiology of Depression. Neuron 34 (1), 13–25. doi:10.1016/s0896-6273(02)00653-0
Okamoto, Y., Morishita, J., Tsuboi, K., Tonai, T., and Ueda, N. (2004). Molecular Characterization of a Phospholipase D Generating Anandamide and its Congeners. J. Biol. Chem. 279 (7), 5298–5305. doi:10.1074/jbc.M306642200
Onaivi, E. S., Ishiguro, H., Gong, J. P., Patel, S., Meozzi, P. A., Myers, L., et al. (2008). Brain Neuronal CB2 Cannabinoid Receptors in Drug Abuse and Depression: From Mice to Human Subjects. PLoS One 3 (2), e1640. doi:10.1371/journal.pone.0001640
Pacher, P., Bátkai, S., and Kunos, G. (2006). The Endocannabinoid System as an Emerging Target of Pharmacotherapy. Pharmacol. Rev. 58 (3), 389–462. doi:10.1124/pr.58.3.2
Palazuelos, J., Aguado, T., Egia, A., Mechoulam, R., Guzmán, M., and Galve-Roperh, I. (2006). Non-Psychoactive CB2 Cannabinoid Agonists Stimulate Neural Progenitor Proliferation. FASEB J. 20 (13), 2405–2407. doi:10.1096/fj.06-6164fje
Panikashvili, D., Mechoulam, R., Beni, S. M., Alexandrovich, A., and Shohami, E. (2005). CB1 Cannabinoid Receptors Are Involved in Neuroprotection via NF-Kappa B Inhibition. J. Cereb. Blood Flow Metab. 25 (4), 477–484. doi:10.1038/sj.jcbfm.9600047
Patel, S., and Hillard, C. J. (2006). Pharmacological Evaluation of Cannabinoid Receptor Ligands in a Mouse Model of Anxiety: Further Evidence for an Anxiolytic Role for Endogenous Cannabinoid Signaling. J. Pharmacol. Exp. Ther. 318 (1), 304–311. doi:10.1124/jpet.106.101287
Patel, S., and Hillard, C. J. (2009). Role of Endocannabinoid Signaling in Anxiety and Depression. Curr. Top. Behav. Neurosci. 1, 347–371. doi:10.1007/978-3-540-88955-7_14
Patel, S., Roelke, C. T., Rademacher, D. J., and Hillard, C. J. (2005). Inhibition of Restraint Stress-Induced Neural and Behavioural Activation by Endogenous Cannabinoid Signalling. Eur. J. Neurosci. 21 (4), 1057–1069. doi:10.1111/j.1460-9568.2005.03916.x
Pertwee, R. G., Howlett, A. C., Abood, M. E., Alexander, S. P., Di Marzo, V., Elphick, M. R., et al. (2010). International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid Receptors and Their Ligands: beyond CB₁ and CB₂. Pharmacol. Rev. 62 (4), 588–631. doi:10.1124/pr.110.003004
Pertwee, R. G., and Roger, G. (2006). Cannabinoid Pharmacology: The First 66 Years. Br. J. Pharmacol. 147 Suppl 1 (Suppl. 1), S163–S171. doi:10.1038/sj.bjp.0706406
Pertwee, R. G. (2008). The Diverse CB1 and CB2 Receptor Pharmacology of Three Plant Cannabinoids: delta9-tetrahydrocannabinol, Cannabidiol and delta9-tetrahydrocannabivarin. Br. J. Pharmacol. 153 (2), 199–215. doi:10.1038/sj.bjp.0707442
Phan, K. L., Angstadt, M., Golden, J., Onyewuenyi, I., Popovska, A., and de Wit, H. (2008). Mike Angstadt, Jamie Golden, Ikechukwu Onyewuenyi, Ana Popovska, and Harriet de WitCannabinoid Modulation of Amygdala Reactivity to Social Signals of Threat in Humans. J. Neurosci. 28 (10), 2313–2319. doi:10.1523/JNEUROSCI.5603-07.2008
Pinto, J. V., Saraf, G., Frysch, C., Vigo, D., Keramatian, K., Chakrabarty, T., et al. (2020). Cannabidiol as a Treatment for Mood Disorders: A Systematic Review: Le cannabidiol comme traitement des troubles de l'humeur: une revue systématique. Can. J. Psychiatry 65 (4), 213–227. doi:10.1177/0706743719895195
Pratt, L. A., and Brody, D. J. (2008). Depression in the United States Household Population, 2005-2006. NCHS Data Brief 18 (September)), 1–8.
Rai, D., Zitko, P., Jones, K., Lynch, J., and Araya, R. (2013). Country- and Individual-Level Socioeconomic Determinants of Depression: Multilevel Cross-National Comparison. Br. J. Psychiatry 202 (3), 195–203. doi:10.1192/bjp.bp.112.112482
Ramirez, S. H., Reichenbach, N. L., Fan, S., Rom, S., Merkel, S. F., Wang, X., et al. (2013). Attenuation of HIV-1 Replication in Macrophages by Cannabinoid Receptor 2 Agonists. J. Leukoc. Biol. 93 (5), 801–810. doi:10.1189/jlb.1012523
Ransohoff, R. M. (2016). A Polarizing Question: Do M1 and M2 Microglia Exist? Nat. Neurosci. 19 (8), 987–991. doi:10.1038/nn.4338
Major Depressive Disorder Working Group of the Psychiatric GWAS Consortium Ripke, Stephan., Ripke, S., WrayLewis, N. R. Steven. P. Hamilton., Lewis, C. M., Hamilton, S. P., Weissman, M. M., et al. (2013). A Mega-Analysis of Genome-wide Association Studies for Major Depressive Disorder. Mol. Psychiatry 18 (4), 497–511. doi:10.1038/mp.2012.21
Robledo-Menendez, A., Vella, M., Grandes, P., and Soria-Gomez, E. (2021). Cannabinoid Control of Hippocampal Functions: The where Matters. FEBS J. doi:10.1111/febs.15907
Rodgers, R. J., Evans, P. M., and Murphy, A. (2005). Anxiogenic Profile of AM-251, a Selective Cannabinoid CB1 Receptor Antagonist, in Plus-Maze-Naïve and Plus-Maze-Experienced Mice. Behav. Pharmacol. 16 (5–6), 405–413. doi:10.1097/00008877-200509000-00013
Rubino, T., Realini, N., Castiglioni, C., Guidali, C., Viganó, D., Marras, E., et al. (2008). Role in Anxiety Behavior of the Endocannabinoid System in the Prefrontal Cortex. Cereb. Cortex 18 (6), 1292–1301. doi:10.1093/cercor/bhm161
Sánchez, M. M., Ladd, C. O., and Plotsky, P. M. (2001). Early Adverse Experience as a Developmental Risk Factor for Later Psychopathology: Evidence from Rodent and Primate Models. Dev. Psychopathol 13 (3), 419–449. doi:10.1017/s0954579401003029
Sanchis-Segura, C., Cline, B. H., Marsicano, G., Lutz, B., and Spanagel, R. (2004). Reduced Sensitivity to Reward in CB1 Knockout Mice. Psychopharmacology (Berl) 176 (2), 223–232. doi:10.1007/s00213-004-1877-8
Santarelli, L., Saxe, M., Gross, C., Surget, A., Battaglia, F., Dulawa, S., et al. (2003). Requirement of Hippocampal Neurogenesis for the Behavioral Effects of Antidepressants. Science 301 (5634), 805–809. doi:10.1126/science.1083328
Sarris, J., Sinclair, J., Karamacoska, D., Davidson, M., and Firth, J. (2020). Medicinal Cannabis for Psychiatric Disorders: A Clinically-Focused Systematic Review. BMC Psychiatry 20 (January), 24. doi:10.1186/s12888-019-2409-8
Sato, K. (2015). Effects of Microglia on Neurogenesis. Glia 63 (8), 1394–1405. doi:10.1002/glia.22858
Schier, A., Ribeiro, N., Coutinho, D., Machado, S., Arias-Carrion, O., Crippa, J., et al. (2014). Antidepressant-like and Anxiolytic-like Effects of Cannabidiol: A Chemical Compound of Cannabis Sativa. Cnsnddt 13 (6), 953–960. doi:10.2174/1871527313666140612114838
Schloesser, R. J., Manji, H. K., and Martinowich., K. (2009). Suppression of Adult Neurogenesis Leads to an Increased Hypothalamo-Pituitary-Adrenal axis Response. Neuroreport 20 (6), 553–557. doi:10.1097/WNR.0b013e3283293e59
Scuderi, C., Steardo, L., and Esposito, G. (2014). Cannabidiol Promotes Amyloid Precursor Protein Ubiquitination and Reduction of Beta Amyloid Expression in SHSY5YAPP+ Cells through PPARγ Involvement. Phytother Res. 28 (7), 1007–1013. doi:10.1002/ptr.5095
Sexton, M., Cuttler, C., Finnell, J. S., and Mischley, L. K. (2016). A Cross-Sectional Survey of Medical Cannabis Users: Patterns of Use and Perceived Efficacy. Cannabis Cannabinoid Res. 1 (1), 131–138. doi:10.1089/can.2016.0007
Shahbazi, F., Grandi, V., Banerjee, A., and Trant, J. F. (2020). Cannabinoids and Cannabinoid Receptors: The Story So Far. IScience 23 (7), 101301. doi:10.1016/j.isci.2020.101301
Shao, B.-Z., Wei, W., Ke, P., Xu, Z.-Q., Zhou, J.-X., and Liu, C. (2014). Activating Cannabinoid Receptor 2 Alleviates Pathogenesis of Experimental Autoimmune Encephalomyelitis via Activation of Autophagy and Inhibiting NLRP3 Inflammasome. CNS Neurosci. Ther. 20 (12), 1021–1028. doi:10.1111/cns.12349
Sharir, H., Console-Bram, L., Mundy, C., Popoff, S. N., Kapur, A., Abood, M. E., et al. (2012). The Endocannabinoids Anandamide and Virodhamine Modulate the Activity of the Candidate Cannabinoid Receptor GPR55. J. Neuroimmune Pharmacol. 7 (4), 856–865. doi:10.1007/s11481-012-9351-6
Sheline, Y. I., Wang, P. W., Gado, M. H., Csernansky, J. G., and Vannier, M. W. (1996). Hippocampal Atrophy in Recurrent Major Depression. Proc. Natl. Acad. Sci. U S A. 93 (9), 3908–3913. doi:10.1073/pnas.93.9.3908
Shen, L., Yang, Y., Ou, T., Key, C. C., Tong, S. H., Sequeira, R. C., et al. (2017). Dietary PUFAs Attenuate NLRP3 Inflammasome Activation via Enhancing Macrophage Autophagy. J. Lipid Res. 58 (9), 1808–1821. doi:10.1194/jlr.M075879
Shi, J., Cai, Q., Zhang, J., He, X., Liu, Y., Zhu, R., et al. (2017). AM1241 Alleviates MPTP-Induced Parkinson's Disease and Promotes the Regeneration of DA Neurons in PD Mice. Oncotarget 8 (40), 67837–67850. doi:10.18632/oncotarget.18871
Silva-Cruz, A., Carlström, M., Ribeiro, J. A., and Sebastião, A. M. (2017). Dual Influence of Endocannabinoids on Long-Term Potentiation of Synaptic Transmission. Front. Pharmacol. 8, 921. doi:10.3389/fphar.2017.00921
Silverman, M. N., PearcePearce, B. D. Christine. A. Biron., Biron, C. A., and Miller, A. H. (2005). Immune Modulation of the Hypothalamic-Pituitary-Adrenal (HPA) Axis during Viral Infection. Viral Immunol. 18 (1), 41–78. doi:10.1089/vim.2005.18.41
Smith, S. M., and Vale., W. W. (2006). The Role of the Hypothalamic-Pituitary-Adrenal Axis in Neuroendocrine Responses to Stress. Dialogues Clin. Neurosci. 8 (4), 383–395.
Sorrells, S. F., and Sapolsky, R. M. (2007). An Inflammatory Review of Glucocorticoid Actions in the CNS. Brain Behav. Immun. 21 (3), 259–272. doi:10.1016/j.bbi.2006.11.006
Spector, S., Shore, P. A., and Brodie, B. B. (1960). Biochemical and Pharmacological Effects of the Monoamine Oxidase Inhibitors, Iproniazid, 1-Phenyl-2-Hydrazinopropane (JB 516) and 1-Phenyl-3-Hydrazinobutane (JB 835). J. Pharmacol. Exp. Ther. 128 (January), 15–21.
Stahl, S. M. (1984). Regulation of Neurotransmitter Receptors by Desipramine and Other Antidepressant Drugs: The Neurotransmitter Receptor Hypothesis of Antidepressant Action. J. Clin. Psychiatry 45 (10 Pt 2), 37–45.
Starowicz, K., Nigam, S., and Di Marzo, V. (2007). Biochemistry and Pharmacology of Endovanilloids. Pharmacol. Ther. 114 (1), 13–33. doi:10.1016/j.pharmthera.2007.01.005
Steiner, M. A., Wanisch, K., Monory, K., Marsicano, G., Borroni, E., Bächli, H., et al. (2008). Impaired Cannabinoid Receptor Type 1 Signaling Interferes with Stress-Coping Behavior in Mice. Pharmacogenomics J. 8 (3), 196–208. doi:10.1038/sj.tpj.6500466
Stella, N. (2009). Endocannabinoid Signaling in Microglial Cells. Neuropharmacology 56 Suppl 1 (Suppl. 1), 244–253. doi:10.1016/j.neuropharm.2008.07.037
Sugama, S., Fujita, M., Hashimoto, M., and Conti, B. (2007). Stress Induced Morphological Microglial Activation in the Rodent Brain: Involvement of Interleukin-18. Neuroscience 146 (3), 1388–1399. doi:10.1016/j.neuroscience.2007.02.043
Sugiura, T., Kishimoto, S., Oka, S., and Gokoh, M. (2006). Biochemistry, Pharmacology and Physiology of 2-Arachidonoylglycerol, an Endogenous Cannabinoid Receptor Ligand. Prog. Lipid Res. 45 (5), 405–446. doi:10.1016/j.plipres.2006.03.003
Sugiura, T., Kodaka, T., Kondo, S., Tonegawa, T., Nakane, S., Kishimoto, S., et al. (1997). Inhibition by 2-arachidonoylglycerol, a Novel Type of Possible Neuromodulator, of the Depolarization-Induced Increase in Intracellular Free Calcium in Neuroblastoma X Glioma Hybrid NG108-15 Cells. Biochem. Biophys. Res. Commun. 233 (1), 207–210. doi:10.1006/bbrc.1997.6425
Sugiura, T., Kondo, S., Kishimoto, S., Miyashita, T., Nakane, S., Kodaka, T., et al. (2000). Evidence that 2-Arachidonoylglycerol but Not N-Palmitoylethanolamine or Anandamide Is the Physiological Ligand for the Cannabinoid CB2 Receptor. Comparison of the Agonistic Activities of Various Cannabinoid Receptor Ligands in HL-60 Cells. J. Biol. Chem. 275 (1), 605–612. doi:10.1074/jbc.275.1.605
Sumislawski, J. J., Ramikie, T. S., and Patel., S. (2011). Reversible Gating of Endocannabinoid Plasticity in the Amygdala by Chronic Stress: A Potential Role for Monoacylglycerol Lipase Inhibition in the Prevention of Stress-Induced Behavioral Adaptation. Neuropsychopharmacology 36 (13), 2750–2761. doi:10.1038/npp.2011.166
Szabo, B., Wallmichrath, I., Mathonia, P., and Pfreundtner, C. (2000). Cannabinoids Inhibit Excitatory Neurotransmission in the Substantia Nigra Pars Reticulata. Neuroscience 97 (1), 89–97. doi:10.1016/S0306-4522(00)00036-1
Tanaka, M., Sackett, S., and Zhang, Y. (2020). Endocannabinoid Modulation of Microglial Phenotypes in Neuropathology. Front. Neurol. 11, 87. doi:10.3389/fneur.2020.00087
Tanaka, M., Yagyu, K., Sackett, S., and Zhang, Y. (2019). Anti-Inflammatory Effects by Pharmacological Inhibition or Knockdown of Fatty Acid Amide Hydrolase in BV2 Microglial Cells. Cells 8 (5), 491. doi:10.3390/cells8050491
Tanasescu, R., and Constantinescu, C. S. (2010). Cannabinoids and the Immune System: An Overview. Immunobiology 215 (8), 588–597. doi:10.1016/j.imbio.2009.12.005
Tang, J., Miao, H., Jiang, B., Chen, Q., Tan, L., Tao, Y., et al. (2017). A Selective CB2R Agonist (JWH133) Restores Neuronal Circuit After Germinal Matrix Hemorrhage In The Preterm Via CX3CR1+ Microglia. Neuropharmacol. 119, 157–169. doi:10.1016/j.neuropharm.2017.01.027
Tao, Y., Li, L., Jiang, B., Feng, Z., Yang, L., Tang, J., et al. (2016). Cannabinoid Receptor-2 Stimulation Suppresses Neuroinflammation by Regulating Microglial M1/M2 Polarization through the CAMP/PKA Pathway in an Experimental GMH Rat Model. Brain Behav. Immun. 58 (November), 118–129. doi:10.1016/j.bbi.2016.05.020
Torres-Berrío, Angélica., Issler, Orna., Parise, Eric. M., and Nestler, Eric. J. (2019). Unraveling the Epigenetic Landscape of Depression: Focus on Early Life Stress. Dialogues Clin. Neurosci. 21 (4), 341–357. doi:10.31887/DCNS.2019.21.4/enestler
Trivedi, M. H., Rush, A. J., Wisniewski, S. R., Nierenberg, A. A., Warden, D., Ritz, L., et al. (2006). Evaluation of Outcomes with Citalopram for Depression Using Measurement-Based Care in STAR*D: Implications for Clinical Practice. Am. J. Psychiatry 163 (1), 28–40. doi:10.1176/appi.ajp.163.1.28
Troubat, R., Barone, P., Leman, S., Desmidt, T., Cressant, A., Atanasova, B., et al. (2020). Neuroinflammation and Depression: A Review. Eur. J. Neurosci. 53, 151–171. doi:10.1111/ejn.14720
Turcotte, C., Blanchet, M. R., Laviolette, M., and Flamand, N. (2016). The CB2 Receptor and its Role as a Regulator of Inflammation. Cell Mol Life Sci 73 (23), 4449–4470. doi:10.1007/s00018-016-2300-4
Tynan, R. J., Naicker, S., Hinwood, M., Nalivaiko, E., Buller, K. M., Pow, D. V., et al. (2010). Chronic Stress Alters the Density and Morphology of Microglia in a Subset of Stress-Responsive Brain Regions. Brain Behav. Immun. 24 (7), 1058–1068. doi:10.1016/j.bbi.2010.02.001
Uher, R., Payne, J. L., Pavlova, B., and Perlis, R. H. (2014). Major Depressive Disorder in Dsm-5: Implications for Clinical Practice and Research of Changes from Dsm-Iv. Depress. Anxiety 31 (6), 459–471. doi:10.1002/da.22217
Vahratian, A., Blumberg, S. J., Terlizzi, E. P., and Schiller, J. S. (2021). Symptoms of Anxiety or Depressive Disorder and Use of Mental Health Care Among Adults during the COVID-19 Pandemic - United States, August 2020-February 2021. MMWR Morb Mortal Wkly Rep. 70 (13), 490–494. doi:10.15585/mmwr.mm7013e2
Valentino, R. J., and Van Bockstaele., E. (2008). Convergent Regulation of Locus Coeruleus Activity as an Adaptive Response to Stress. Eur. J. Pharmacol. 583 (2), 194–203. doi:10.1016/j.ejphar.2007.11.062
Valles-Colomer, M., Falony, G., Darzi, Y., Tigchelaar, E. F., Wang, J., Tito, R. Y., et al. (2019). The Neuroactive Potential of the Human Gut Microbiota in Quality of Life and Depression. Nat. Microbiol. 4 (4), 623–632. doi:10.1038/s41564-018-0337-x
Valverde, O., and Torrens, M. (2012). CB1 Receptor-Deficient Mice as a Model for Depression. Neuroscience 204 (March), 193–206. doi:10.1016/j.neuroscience.2011.09.031
van Laar, M. W., Oomen, C. J. A., Vercoulen, E., Freeman, T. P., and Hall, W. D. (2020). Cannabis and COVID-19: Reasons for Concern. Front. Psychiatry 11. doi:10.3389/fpsyt.2020.601653
van Praag, H., Kempermann, G., and Gage, F. H. (2000). Neural Consequences of Environmental Enrichment. Nat. Rev. Neurosci. 1 (3), 191–198. doi:10.1038/35044558
Van Sickle, M. D., Duncan, M., Kingsley, P. J., Mouihate, A., Urbani, P., Mackie, K., et al. (2005). Identification and Functional Characterization of Brainstem Cannabinoid CB 2 ReceptorsIdentification and Functional Characterization of Brainstem Cannabinoid CB2 Receptors. Science 310 (5746), 329–332. doi:10.1126/science.1115740
Wade, M. R., Degroot, A., and Nomikos, G. G. (2006). Cannabinoid CB1 Receptor Antagonism Modulates Plasma Corticosterone in Rodents. Eur. J. Pharmacol. 551 (1–3), 162–167. doi:10.1016/j.ejphar.2006.08.083
Wamsteeker, J. I., Kuzmiski, J. B., and Bains, J. S. (2010). Repeated Stress Impairs Endocannabinoid Signaling in the Paraventricular Nucleus of the Hypothalamus. J. Neurosci. 30 (33), 11188–11196. doi:10.1523/JNEUROSCI.1046-10.2010
Wang, M., Hill, M. N., Zhang, L., Gorzalka, B. B., Hillard, C. J., and Alger, B. E. (2012). Acute Restraint Stress Enhances Hippocampal Endocannabinoid Function via Glucocorticoid Receptor Activation. J. Psychopharmacol. 26 (1), 56–70. doi:10.1177/0269881111409606
Wang, W., Sun, D., Pan, B., Roberts, C. J., Sun, X., Hillard, C. J., et al. (2010). Deficiency in Endocannabinoid Signaling in the Nucleus Accumbens Induced by Chronic Unpredictable Stress. Neuropsychopharmacology 35 (11), 2249–2261. doi:10.1038/npp.2010.99
Wang, Y., and Zhang, X. (2017). FAAH Inhibition Produces Antidepressant-like Efforts of Mice to Acute Stress via Synaptic Long-Term Depression. Behav. Brain Res. 324 (May), 138–145. doi:10.1016/j.bbr.2017.01.054
Wilson, R. I., and Nicoll., Roger. A. (2001). “Endogenous Cannabinoids Mediate Retrograde Signalling at Hippocampal Synapses.” Nature 410 (6828), 588–592. doi:10.1038/35069076
Wolf, Susanne. A. (2010). “Cannabinoid Receptor CB1 Mediates Baseline and Activity-Induced Survival of New Neurons in Adult Hippocampal Neurogenesis.” Cell Commun. Signaling: CCS 8 (June), 12. doi:10.1186/1478-811X-8-12
World Health Organization and others (2017). Cannabidiol (CBD) Pre-review Report Agenda Item 5.2. Geneva: Expert Committee on Drug Dependence Thirty-Ninth Meeting.
World Health Organization (2017). Depression and Other Common Mental Disorders: Global Health Estimates. Geneva. Available at: https://apps.who.int/iris/bitstream/handle/10665/254610/WHO-MSD-MER-2017.2-eng.pdf.
World Health Organization (2020). Depression in Europe: Facts and Figures.” Depression in Europe: Facts and Figures. Available at: https://www.euro.who.int/en/health-topics/noncommunicable-diseases/mental-health/news/news/2012/10/depression-in-europe/depression-in-europe-facts-and-figures.
Wu, Jiang., Hocevar, Mark., Foss, Joseph. F., Bie, Bihua., and Naguib, Mohamed. (2017). Activation of CB2 Receptor System Restores Cognitive Capacity and Hippocampal Sox2 Expression in a Transgenic Mouse Model of Alzheimer’s Disease. Eur. J. Pharmacol. 811 (September), 12–20. doi:10.1016/j.ejphar.2017.05.044
Wu, Michael. D., Montgomery, Sara. L., Escalera, Fatima. Rivera., Olschowka, John. A., and Kerry O’Banion, M. (2013). Sustained IL-1β Expression Impairs Adult Hippocampal Neurogenesis Independent of IL-1 Signaling in Nestin+ Neural Precursor Cells. Brain Behav. Immun. 32 (August), 9–18. doi:10.1016/j.bbi.2013.03.003
Wyrofsky, Ryan. R., Reyes, Beverly. A. S., Zhang, Xiao-Yan., Bhatnagar, Seema., Kirby, Lynn. G., and Van Bockstaele, Elisabeth. J. (2019). Endocannabinoids, Stress Signaling, and the Locus Coeruleus-Norepinephrine System. Neurobiol. Stress 11 (November), 100176. doi:10.1016/j.ynstr.2019.100176
Xu, Chen., Chang, Tanran., Du, Yaqi., Yu, Chaohui., Tan, Xin., and Li, Xiangdong. (2019). Pharmacokinetics of Oral and Intravenous Cannabidiol and its Antidepressant-like Effects in Chronic Mild Stress Mouse Model. Environ. Toxicol. Pharmacol. 70 (August), 103202. doi:10.1016/j.etap.2019.103202
Yamada, Jun., and Jinno, Shozo. (2019). Potential Link between Antidepressant-like Effects of Ketamine and Promotion of Adult Neurogenesis in the Ventral Hippocampus of Mice. Neuropharmacology 158 (November), 107710. doi:10.1016/j.neuropharm.2019.107710
Yang, Le., Tian, Lei., Zhang, Zhi., Zhou, Xuan., Ji, Xiaofang., Liu, Fuquan., et al. (2020). Cannabinoid Receptor 1/MiR-30b-5p Axis Governs Macrophage NLRP3 Expression and Inflammasome Activation in Liver Inflammatory Disease. Mol. Ther. - Nucleic Acids 20 (June), 725–738. doi:10.1016/j.omtn.2020.04.010
Yirmiya, Raz., Rimmerman, Neta., and Ronen, Reshef. (2015). Depression as a Microglial Disease. Trends Neurosciences, Neuroimmunology 38 (10), 637–658. doi:10.1016/j.tins.2015.08.001
Yoshida, Takayuki., Hashimoto, Kouichi., Zimmer, Andreas., Maejima, Takashi., Araishi, Kenji., and Kano, Masanobu. (2002). The Cannabinoid CB1 Receptor Mediates Retrograde Signals for Depolarization-Induced Suppression of Inhibition in Cerebellar Purkinje Cells. J. Neurosci. 22 (5), 1690–1697. doi:10.1523/JNEUROSCI.22-05-01690.2002
Zanelati, T. V., Biojone, C., Moreira, F. A., Guimarães, F. S., and SâmiaJoca, R. L. (2010). Antidepressant-like Effects of Cannabidiol in Mice: Possible Involvement of 5-HT1A Receptors. Br. J. Pharmacol. 159 (1), 122–128. doi:10.1111/j.1476-5381.2009.00521.x
Zanettini, Claudio., Panlilio, Leigh. V., Aliczki, Manó., Goldberg, Steven. R., Haller, Jozsef., and Yasar, Sevil. (2011). Effects of Endocannabinoid System Modulation on Cognitive and Emotional Behavior. Front. Behav. Neurosci. 5. doi:10.3389/fnbeh.2011.00057
Zhang, Zhen., Wang, Wei., Zhong, Peng., Liu, Sarah. J., Long, Jonathan. Z., Zhao, Li., et al. (2015). Blockade of 2-Arachidonoylglycerol Hydrolysis Produces Antidepressant-like Effects and Enhances Adult Hippocampal Neurogenesis and Synaptic Plasticity. Hippocampus 25 (1), 16–26. doi:10.1002/hipo.22344
Zimmer, A., Zimmer, A. M., Hohmann, A. G., Herkenham, M., Tom, I., and Bonner, T. I. (1999). Increased Mortality, Hypoactivity, and Hypoalgesia in Cannabinoid CB1 Receptor Knockout Mice. Proc. Natl. Acad. Sci. 96 (10), 5780. doi:10.1073/pnas.96.10.5780
Zimmermann, Tina., Maroso, Mattia., Beer, Annika., Baddenhausen, Sarah., Ludewig, Susann., Fan, Wenqiang., et al. (2018). Neural Stem Cell Lineage-specific Cannabinoid Type-1 Receptor Regulates Neurogenesis and Plasticity in the Adult Mouse Hippocampus. Cereb. Cortex 28 (12), 4454–4471. doi:10.1093/cercor/bhy258
Zoppi, Silvia., BeatrizNievas, G. Pérez., José, L., Madrigal, M., Manzanares, Jorge., Leza, Juan. C., et al. (2011). Regulatory Role of Cannabinoid Receptor 1 in Stress-Induced Excitotoxicity and Neuroinflammation. Neuropsychopharmacol. Official Publ. Am. Coll. Neuropsychopharmacol. 36 (4), 805–818. doi:10.1038/npp.2010.214
Zoppi, Silvia., Madrigal, José. L. M., BeatrizPérez-Nievas, G., Marín-Jiménez, Ignacio., Caso, Javier. R., Alou, Luis., et al. (2012). Endogenous Cannabinoid System Regulates Intestinal Barrier Function In Vivo through Cannabinoid Type 1 Receptor Activation. Am. J. PhysiologyGastrointestinal Liver Physiol. 302 (5), G565–G571. doi:10.1152/ajpgi.00158.2011
Zoppi, S., Madrigal, J. L., Caso, J. R., García‐Gutiérrez, M. S., Manzanares, J., Leza, J. C., et al. (2014). Regulatory Role of the Cannabinoid CB2 Receptor in Stress-Induced Neuroinflammation in Mice. Br. J. Pharmacol. 171 (11), 2814–2826. doi:10.1111/bph.12607
Keywords: endocannabinoid system, HPA-axis, major depressive disorder, neurogenesis, neuroinflammation
Citation: Gallego-Landin I, García-Baos A, Castro-Zavala A and Valverde O (2021) Reviewing the Role of the Endocannabinoid System in the Pathophysiology of Depression. Front. Pharmacol. 12:762738. doi: 10.3389/fphar.2021.762738
Received: 22 August 2021; Accepted: 11 November 2021;
Published: 06 December 2021.
Edited by:
Sandra Montagud Romero, University of Zaragoza, SpainReviewed by:
Anna Serefko, Medical University of Lublin, PolandCopyright © 2021 Gallego-Landin, García-Baos, Castro-Zavala and Valverde. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Olga Valverde, b2xnYS52YWx2ZXJkZUB1cGYuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.