Corrigendum: Targeting Myc Interacting Proteins as a Winding Path in Cancer Therapy
 Yihui Zhou
Yihui Zhou Xiaomeng Gao
Xiaomeng Gao Meng Yuan
Meng Yuan Bo Yang
Bo Yang Qiaojun He
Qiaojun He Ji Cao
Ji Cao- 1Zhejiang Province Key Laboratory of Anti-Cancer Drug Research, Institute of Pharmacology and Toxicology, College of Pharmaceutical Sciences, Zhejiang University, Hangzhou, China
- 2The Innovation Institute for Artificial Intelligence in Medicine, Zhejiang University, Hangzhou, China
- 3Cancer Center of Zhejiang University, Hangzhou, China
MYC, as a well-known oncogene, plays essential roles in promoting tumor occurrence, development, invasion and metastasis in many kinds of solid tumors and hematologic neoplasms. In tumors, the low expression and the short half-life of Myc are reversed, cause tumorigenesis. And proteins that directly interact with different Myc domains have exerted a significant impact in the process of Myc-driven carcinogenesis. Apart from affecting the transcription of Myc target genes, Myc interaction proteins also regulate the stability of Myc through acetylation, methylation, phosphorylation and other post-translational modifications, as well as competitive combination with Myc. In this review, we summarize a series of Myc interacting proteins and recent advances in the related inhibitors, hoping that can provide new opportunities for Myc-driven cancer treatment.
1 Introduction
Myc is a multifunctional transcription factor, regulates multiple genes comprised of varieties of cell physiological and pathological processes including proliferation, differentiation, apoptosis and tumorigenesis (Farrell and Sears, 2014). Originally MYC was isolated on chicken cells, and the gene encoding c-Myc was a cellular homolog of v-Myc, which was present in avian myelocytomatosis virus strain 29 causing avian leukemia (Vennstrom et al., 1982). Subsequently other transformed and more specific Myc family members were also identified in mammal tissues, including c-Myc, N-Myc and L-Myc, respectively (Dalla-Favera et al., 1982; Nau et al., 1985; Rickman et al., 2018). Myc family genes have been shown to be differentially expressed in terms of tissue type and developmental stage (Xu et al., 1991). c-Myc only express in tissues with rapid proliferation, while L-Myc and N-Myc often express specifically in tissues that undergoing differentiation (Hirning et al., 1991). Besides, the mice lack of c-Myc or N-Myc all lead to embryonic death (Pirity et al., 2006). In comparison, L-Myc is only unnecessary for gross morphological development, by MYCL knockout mice model. This might be due to the overlapping expression patterns of other Myc has made up for L-Myc deficiency (Hatton et al., 1996).
Although there are three types of Myc and their chromosomal locations are different, they are all homologous proteins, which are highly conserved in gene sequence and have similar structural domains (Chen et al., 2018). Myc has several structure regions that are critical for the biological functions, including the amino-terminal transactivation domain (TAD), central region and the carboxy-terminal basic-helix-loop-helix-leucine zipper (bHLH-LZ) domain (Duffy et al., 2021). bHLH-LZ domain is responsible for dimerization with its essential partner, Myc-associated protein X (Max), and for sequence-particular DNA binding. TAD and central region are main protein-protein interaction (PPI) area, including six highly conserved regions (MB0, MBI, MBII, MBIIIa, MBIIIb, MBIV), termed Myc homology boxes (MBs). MB0 accelerates the transcription by binding to the general transcription factor IIF (TFIIF); MBI controls proteasome-mediated degradation of Myc protein; MBII participates in chromatin remodeling and modification; MBIIIa play a role in gene repression; MBIIIb binds to WD repeat domain 5 (WDR5) as a glue binding on chromatin and MBIV shows potential association with chromatin (including apoptosis, G2 cell arrest) (Baluapuri et al., 2020; Duffy et al., 2021). Through proteomics analysis, more than half of the Myc interactors demand at least one of MBs for binding (Kalkat et al., 2018).
It is now clear that Myc proteins are principal drivers of human tumorigenesis, more than 70% of cancers are related to Myc disorders (Dang et al., 2006; Lancho and Herranz, 2018). Minor alteration of Myc levels can facilitate or prevent oncogenic transformation and tumour progression (Wang T. et al., 2019). Myc binds to the promoters of downstream genes at the RNA polymerase II (RNAPII)-bound and promotes their expression, regulating the increase or decrease of transcription. The carcinogenicity of Myc is that, it can increase the transcription level of high-affinity target genes or even push them to saturation, and can also regulate (up-regulate or down-regulate) low-affinity target genes, transforming normal cells into tumor cells (Baluapuri et al., 2020). MYC gene is activated mainly through amplification and chromosomal translocation rearrangement. It can regulate the expression of a variety of genes related to cell proliferation and metabolic process, and its corresponding genes are also the most common high abundance oncogenes in human cancers (Difilippantonio et al., 2002; Chen et al., 2014). Myc protein is expressed at a low level in proliferating cells and has a very short half-life of only 30 min, after which it is degraded by the ubiquitin proteasome pathway (Thomas and Tansey, 2011). However, this characteristic of Myc is often changed in tumors, prolonged half-life and excessive accumulation are also a major cause of promoting the occurrence of tumors (Wu et al., 2020).
The process of Myc binding to the target chromatin and regulating the transcription level of the target gene is not completed independently. The well-known protein Max, which is first described as Myc-interacting protein (Blackwood and Eisenman, 1991). Max binds to the bHLH-LZ domain of Myc and forms Myc/Max heterodimers to achieve DNA recognition and binding (Cascón and Robledo, 2012). In most chromatin binding and transcriptional regulation, Myc is entirely dependent on heterodimerization with Max (Grandori and Eisenman, 1997; Castell et al., 2018). Deletion of Max destabilizes Myc protein and reduces the expression of Myc-target gene, even eliminates Myc-driven tumorigenesis (Mathsyaraja et al., 2019; Augert et al., 2020). Recent evidence showed, Myc still retained some biological functions without Max, meaning Max was not the only interacting protein that maintains Myc functions (Cascón and Robledo, 2012). Besides of Max, some other critical proteins can interact with Myc as well to regulate physiological processes including transcription activation, transcription repression, chromatin remodeling and ubiquitination degradation, etc.
This review, we concentrate on a number of Myc interacting proteins that contribute to Myc function (Figure 1), and also discussed current inhibitors and strategies targeting the interacting proteins, in the interest of providing new opportunities for Myc-related cancer treatment.
FIGURE 1
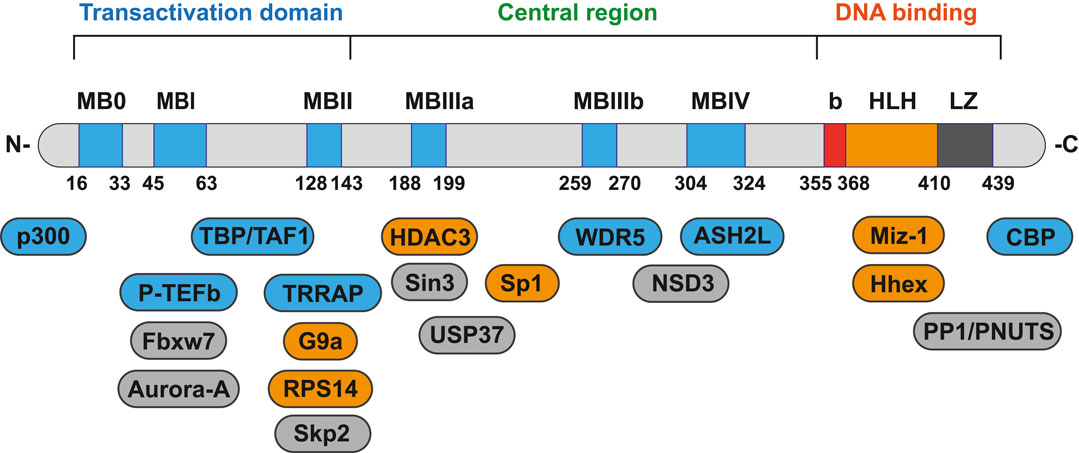
FIGURE 1. Protein-protein interaction on Myc domains.
2 Protein-Protein Interaction Works on Myc Transcriptional Activation
As a transcriptional regulator, Myc affects a wide range of gene transcription levels. Under normal circumstances, the excessive growth and proliferation of Myc-amplified tumors are caused by the transcriptional activation of oncogenes by Myc (Kim et al., 2019). The bHLH-LZ DNA binding domain of Myc binds to chromatin and recruits some cofactor proteins to modify the chromatin or Myc itself, and finally achieve the function of chromatin transcription activation (Tu et al., 2015). In this process, the interacting proteins play a decisive role in coordination with the function of Myc (Figure 2).
FIGURE 2
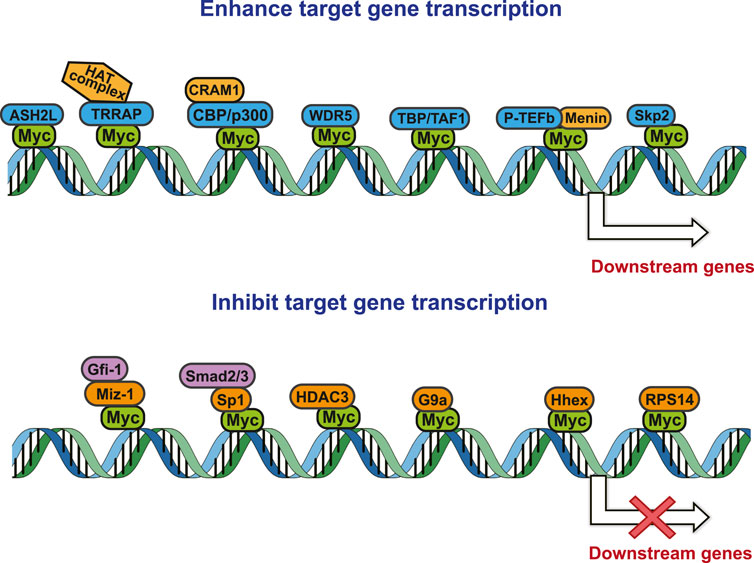
FIGURE 2. Protein-protein interaction works on Myc transcriptional activation and repression.
2.1 Transactivation/Transformation-Domain Associated Protein (TRRAP)
TRRAP is a component of histone-acetylation (HAT) complexes, acts as a scaffold to stabilize (Cogné et al., 2019). Although being part of the phosphoinositide 3-kinase-related kinase (PIKK) family, TRRAP lacks a kinase domain (Elías-Villalobos et al., 2019). It was reported that TRRAP has direct interaction with Myc in the MBⅡ domain, and the recruitment of TRRAP was required for Myc-mediated oncogenic transformation (Nikiforov et al., 2002). In HAT complexes, Tat-interactive protein 60 (Tip60) and General control non-derepressible 5 (Gcn5) work histone acetylase activity, and TRRAP itself doesn’t exert catalytic activity (Liu et al., 2003; Feris et al., 2019). TRRAP links between HAT complexes and Myc, enables the activities of HAT complexes to be recruited and anchored at Myc binding DNA areas in order to stimulate gene expression (McMahon et al., 2000). After recruitment by TRRAP, HAT complexes regulate the modification of histones near promoter and hyperacetylation of lysine residues on terminal of histones, creating an open chromatin environment to promote transcription (Kalkat et al., 2018). Without serum stimulation, for low level of H4 acetylation, Myc alone was inefficient in inducing target genes’ expression (Frank et al., 2001). In addition, reducing the acetylase activity of Tip60 affects the function of the HAT complex, and the level of Myc binding to chromatin will also be weakened (Frank et al., 2003). Therefore, as a cofactor of myc, TRRAP can not only promote the binding of Myc to chromatin, but also open up the nearby chromatin environment to promote transcription.
2.2 cAMP-Response-Element-Binding Protein (CBP/p300)
Acetyltransferases p300 and CBP are multifunctional transcriptional co-activators, belonging to lysine acetyltransferases (KATs) family. Due to their extensive sequence homology and functional similarity, they are defined as a whole: CBP/p300 (Weinert et al., 2018). CBP/p300 contains a catalytic domain KAT to acetylate target proteins, and a recognition domain bromodomain (BRD) to bind with the acetylated proteins. For this reason, CBP/p300 can not only be recruited by MYC to modify chromatin acetylation, but also regulate the acetylation level of Myc itself. Six lysine residues in Myc are direct substrates of p300, and acetylated Myc could interact with promoter binding factors as Miz-1 effectively (Zhang et al., 2005). A recent study reported that p300 binds to c-Myc N-terminus and recruit co-activator-associated arginine methyltransferase 1 (CARM1), in which CARM1-p300-c-Myc-Max (CPCM) transcriptional complex controls the transcription of CUL4A/4B (Lu et al., 2020). Interestingly, CBP binds to the carboxy-terminal region of c-Myc without transactivating activity. This modification is no need MBII, indicating that this function is independent of TRRAP (Vervoorts et al., 2003).
2.3 WD Repeat Domain 5 (WDR5)
With highly conserved WD40 repeat-containing protein, WDR5 is indispensable for appropriate regulation of multi-cellular processes (Guarnaccia and Tansey, 2018). WDR5 protein comprises seven WD40 repeat domains, folding into a seven-bladed propeller structure with several exposed surfaces (Lu et al., 2018). WDR5 mainly exists in the histone lysine methyltransferase subclass 2 (KMT2) enzymes and the non-specific lethal (NSL) complex (Guarnaccia et al., 2021). On account of unusual structure and exposed surfaces, WDR5 forms multiprotein complexes, including with Myc. Acting as a cofactor, WDR5 contributes to the recruitment of Myc to chromatin, and directly combines with Myc on its shallow hydrophobic cleft (Thomas et al., 2019). Myc interacts with WDR5 via an evolutionarily conserved MBIIIb domain, and the core amino acid sequence is “-EEIDVV-” (Thomas et al., 2015b). Otherwise, WDR5 controls Myc target gene transcription by inducing demethylation and subsequently acetylation of H3K27 (Ullius et al., 2014). Myc-WDR5 interaction stabilizes Myc/Max dimer on the promotor of pivotal protumorigenic target genes, accelerating the process of gene transcription (Thomas et al., 2015a). WDR5 could interact with the MBIIIb motif of c-Myc and facilitate Myc-induced HIF1-α transcription, therefore promoting the EMT, invasion and metastasis of cholangiocarcinoma (CCA) (Chen et al., 2021). Myc also maintains the DNA replication in pancreatic ductal adenocarcinoma (PDAC) cells appropriately through interacting with WDR5 (Carugo et al., 2016).
2.4 TATA-Binding Protein (TBP)
TBP is an essential component of the transcription initiation complex TFIID, participating in most gene expression processes in eukaryotes (Bhuiyan and Timmers, 2019). TBP and Myc have been reported to interact at two sites, both of which are located in the TAD domain of Myc. TBP combines at 115–124 amino acids of Myc, and TBP-associated factor 1 (TAF1) at 98–111 (Wei et al., 2019). Studies have shown that the Myc-TBP interaction enhanced gene transcription by regulating the energy distribution upon the transcription initiation complex assembly (Wei et al., 2019). TBP stimulates the transcriptional activation of Myc and enhances the functional characteristics of Myc target genes (Barrett et al., 2005).
2.5 Positive Transcription Elongation Factor b (P-TEFb)
P-TEFb is a transcription factor that stimulates transcription elongation by RNAPII, and functions through directly interacting with various cellular transcription factors, leading to a variety of inflammatory diseases and tumors (Fujinaga, 2020). P-TEFb is composed of the cyclin-dependent kinase 9 (Cdk9) and its regulatory subunit cyclin T. Cdk9 in P-TEFb can phosphorylate the C-terminal domain (CTD) of RNAPII (Kanazawa et al., 2003). While Cyclin T1 binds to Myc at the highly conserved region MBI, promoting the function of Myc to activate the cad promoter (Eberhardy and Farnham, 2002). Menin interacts with TAD domain of Myc and cyclin T1, and subsequently enhances Myc-mediated transcription via P-TEFb (Wu et al., 2017). The cooperation between P-TEFb and Myc also requires the Ski-interacting protein (SKIP), an mRNA elongation and splicing factor (Brès et al., 2009).
2.6 Set1/Ash2 Histone Methyltransferase Complex Subunit ASH2 (ASH2L)
ASH2L is a transcriptional regulator, as part of the KMT2 complex it is involved in methylation and dimethylation at “Lys-4” of histone H3. Research showed, ASH2L and Myc directly interacted in vitro and existed chromatin co-location. Two distinct domains in Myc play to ASH2L binding, 263–350 amino acids directly and bHLH-LZ domain indirectly (Ullius et al., 2014). Since both ASH2L and WDR5 are subunits of KMT2 complex, Myc does not recruit ASH2L to participate in chromatin binding, so the interaction between Myc and ASH2L may be guided by WDR5. Knockdown of ASH2L affects transcription of Myc target genes (Ullius et al., 2014).
3 Protein-Protein Interaction Works on Myc Transcriptional Repression
Tumor occurrence is often accompanied by mutations and abnormal expressions of proto-oncogenes as well as tumor suppressor genes. Upon regulating target genes and promoting cancer progression, Myc not only promotes the transcription of oncogenes, but also suppresses the transcription of tumor suppressor genes. During the tumor-promoting process, the MBII domain and bHLH-LZ domain are necessary for Myc to inhibit transcription, and there are numerous interacting proteins helpful to exert this function. Besides, there are interacting proteins binding to other Myc domains, which can also affect this process (Figure 2).
3.1 Myc Interacting Zinc Finger Protein 1 (Miz-1)
Miz-1, a transcription factor containing BTB/POZ domain, can come into play as an activator or repressor depending on its binding partners (Möröy et al., 2011). Recent research suggested that the transcriptional activities of c-Myc can be reversed once associated with Miz-1. Miz-1 competes with Max to form a complex with c-Myc through the b-HLH-LZ domain (between 12th and 13th zinc finger) (Bédard et al., 2017). Miz-1 can interact with zinc-finger (ZF) transcriptional repressor growth factor independence 1 (Gfi-1) and Myc, form a ternary complex at the cyclin dependent kinase inhibitor (CDKN) promoter (including CDKN1A and CDKN2B), and repress CDKN synergistically (Basu et al., 2009; Liu et al., 2010; Aesoy et al., 2014). Myc is directly recruited by Miz-1 to the cell cycle inhibitors p15INK4B and p21CIP1 promoter, inhibits tumour suppressor p53 and favours the initiation of apoptosis (Seoane et al., 2002; Qi et al., 2017). The Mad family is known as an endogenous transcription suppressor of Myc due to its interaction with Max, Mad4 also is suppressed by Miz1-Myc complex (Quéva et al., 1998). In addition, c-Myc contributes to Wnt inhibitory factor-1 (WIF-1) transcriptional repression in a Miz-1-dependent manner (Licchesi et al., 2010). In leukemia stem cells (LSCs), Myc-Miz-1 interaction represses the expression of CCAAT/enhancer-binding protein α (Cebpα) and Cebpδ, accelerating the self-renewal of LSCs (Zhang et al., 2020). Ablation of the Miz-1 POZ domain conduces to treatment of leukemias and lymphomas, chemotherapy more effective with targeting Miz-1 (Ross et al., 2019).
3.2 Specificity Protein 1 (Sp1)
Sp1 is a significant transcription factor, through specific binding to GC-rich DNA sequences, regulates the expression of polytype genes (Vizcaíno et al., 2015). For promoting the transcription of tumor-related growth factors, Sp1 expressed high level in kinds of tumors and associated with poor prognosis (Beishline and Azizkhan-Clifford, 2015). By interacting with Myc on central region (143–352), Sp1-Myc can repress p21 transcription, thus covering the p21-mediated cell cycle checkpoint (Gartel et al., 2001). Myc can also bind to the Sp1/Myc overlapping site, inhibits the promoter activity and endogenous mRNA expression of BRD7 (Liu et al., 2008). Through the Sp1-Smad complex at the promoter of CDKN2B, Smad2 and Smad3 can directly interact with Myc. Thus affect the transcriptional activity of Sp1 and Sp1-Smad-dependent transcription of the CDKN2B (Feng et al., 2002; 2016).
3.3 Histone Deacetylase 3 (HDAC3)
As a member of the Class I HDAC family, HDAC3 assists the acetyl groups removed on histone and non-histone, repressing gene transcription by promoting chromatin contraction (Dávalos-Salas et al., 2019). HDAC3 interacts with Myc through the MBIIIa domain (Kurland and Tansey, 2008), and subsequently reduces miR-15a/16-1 level in mantle cell lymphoma (MCL) by anchoring at the two promoters of the miR-15a/16-1 cluster gene, DLEU2, and exerting repressive function (Zhang et al., 2012). Tumor necrosis factor receptor-associated factor 6 (TRAF6) can ubiquitinate HDAC3 and lead to the dissociation of HDAC3 from the c-Myc, and then promote human hepatocarcinogenesis (Wu et al., 2020). HDAC3-Myc induces FOXA2 transcriptional repression through its regulation on FOXA2-mediated FTO/m6A/MYC axis, leading to the development of gastric cancer (Yang et al., 2021).
3.4 G9a
G9a is a primary enzyme that catalyzes the methylation of histone 3 lysine 9 (H3K9) and histone 3 lysine 27 (H3K27), playing a crucial role in diverse biological processes and human diseases (Chen et al., 2017; Cao et al., 2019). The MBII region has been identified essential for Myc-G9a interaction, which could promote breast tumor growth by inhibiting gene transcription. Without G9a, H3K9me2 level decreased at Myc-repressed gene promoters, and reduced Myc binding loci (Tu et al., 2018). Meanwhile, depletion of G9a in vivo suppresses Myc-dependent tumor growth. Deficiency of G9a reduces c-Myc binding activity to promoters and inhibits glioblastoma cell proliferation and tumorigenesis ability (Ke et al., 2020). Dual EZH2 and G9a inhibition suppresses multiple myeloma (MM) cell proliferation through the IRF4-Myc axis (Ishiguro et al., 2021). It is worth mentioning that Myc-G9a repress gene transcription in Miz-1-independent manner, this reminds that G9a is necessary for Myc chromatin-binding and gene repression (Tu et al., 2018).
3.5 Haematopoietically Expressed Homeobox (Hhex)
Hhex is a transcriptional repression regulator mainly in charge of organismal development and hematopoiesis (Goh et al., 2020). Hhex can regulate the proliferation level of NK cells and cooperate with the corepressor transducin-like enhancer of Split3 (Tle3) to promote memory B cells (MBCs) development (Laidlaw et al., 2020). In addition to the positive regulation of normal cells, Hhex also negatively regulate the differentiation and function of Treg cells via inhibition of Foxp3 (Jang et al., 2019). Recent research has shown that Hhex was able to interact with the bHLH-LZ region of c-Myc. Hhex overexpression limits the transcription activation, hyperproliferation, metabolism activity and transformation characteristic of Myc oncogenic activities by disrupting Myc/Max formation (Marfil et al., 2015). It is foreseeable that Hhex could be used as a new negative regulator of Myc to inhibit its carcinogenic ability.
3.6 Ribosomal Protein S14 (RPS14)
The demonstration of haploinsufficiency of RPS14 is recognized one of the reasons for p53 activation, and RSP14 is also associated with cellular senescence (Rhoads and Roufa, 1991; Boultwood, 2011). Recent research found that RPS14 affected the transcription function of Myc. RPS14 interacts with MBII and the bHLH-LZ domains of the oncoprotein c-Myc, and prevents the recruitment of Myc-cofactor TRRAP (Zhou et al., 2013). RPS14 not only directly inhibits c-Myc transcriptional activity, but also reduces c-Myc mRNA level (Zhou et al., 2013).
4 Protein-Protein Interaction Works on Myc Protein Stability
Myc is unstable in cells, with short half-life of ∼30 min (Cattoretti, 2013; De Melo et al., 2017). The degradation of Myc is mainly dependent on the phosphorylation of serine-62 and threonine-58 in MBI region by cyclin B/Cdk1 and Gsk3 sequentially, and both of these two residues are often mutated in cancer (Yada et al., 2004). The phosphorylation of Ser62 and Thr58 touches off protein phosphatase 2A (PP2A)-mediated Ser62 dephosphorylation (Mudgapalli et al., 2019). In normal cells, the most important way to control Myc levels is through the targeted degradation of the ubiquitin-proteasome system (UPS). UPS consists of ubiquitin (Ub), ubiquitin activase (E1), ubiquitin-conjugating enzyme (E2), ubiquitin ligase (E3), proteasome and its substrate (Asmamaw et al., 2020). The substrate K48 site was ubiquitinated by E1, E2 and E3, and the ubiquitinated protein was degraded by proteasomes. In MYC-driven cancers, due to the mutation or overexpression of Myc, the proteasome is not enough to degrade Myc any more, leading to excessive accumulation of Myc and eventual tumorigenesis (Bahram et al., 2000). According to existing research, some interacting proteins have been reported to affect the phosphorylation modification of Myc protein, and subsequently affect the degradation of Myc through Fbxw7-mediated ubiquitination modification (Figure 3).
FIGURE 3
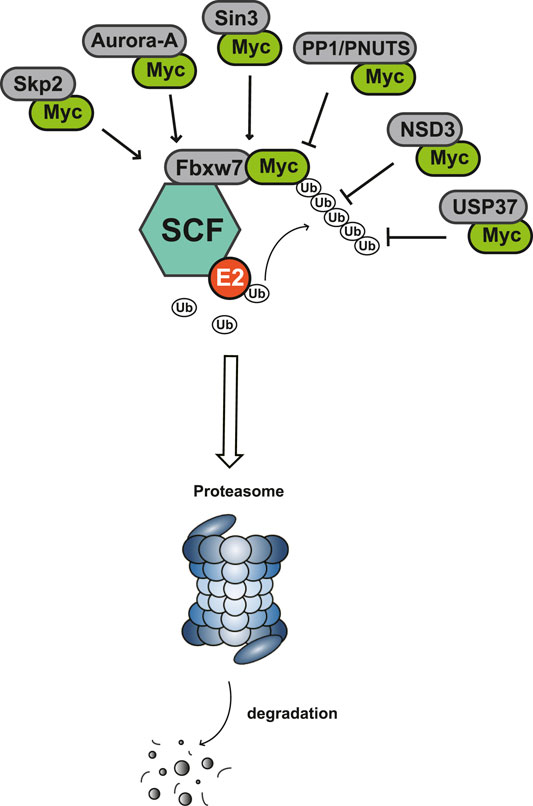
FIGURE 3. Protein-protein interaction works on Myc degradation.
4.1 F-Box With 7 Tandem WD40 (Fbxw7)
The Fbxw7 encoded by FBXW7 is one of the crucial components of Skp1-Cullin1-F-box (SCF) complex, which targets proteins for UPS degradation (Sailo et al., 2019). Fbxw7 interacting and subsequently destabilizing with Myc relies on the phosphorylation of MBI: modifying Myc with K48-linked ubiquitin chains, leading to poly-ubiquitylation and the degradation of Myc through UPS (Welcker et al., 2004; Yada et al., 2004). In embryonic stem cell, Fbxw7 controls its differentiation by degrading c-Myc (Buckley et al., 2012). Loss of Fbxw7 cooperating with activated Akt to induce c-Myc-dependent cholangiocarcinogenesis in mice (Wang J. et al., 2019). In T cell acute lymphoblastic leukemia, Fbxw7 mutations affect the half-life of c-Myc and strengthen leukemia initiating cell activity (King et al., 2013). In addition, deubiquitinating enzyme (DUB) USP9X antagonizes Fbxw7 ubiquitylation to regulate Fbw7 protein stability, reduces c-Myc and alleviates tumor progression (Khan et al., 2018). And DUB USP28 stabilizes c-Myc may also via Fbxw7 complex (Gersch et al., 2019).
4.2 S-phase Kinase-Associated Protein 2 (Skp2)
Skp2 was discovered as a partner of the CDK2 complex at first, but identified as the F-box-binding component of the SCF complex later. Skp2 triggers c-Myc ubiquitylation through directly interacting with the MBII (Hydbring et al., 2017). The interaction of Skp2-Myc occurs at stages from G1 to S phase in normal lymphocytes (von der Lehr et al., 2003). Interestingly, Skp2 is a transcriptional co-activator for Myc as well, considered to be an essential component for recognizing Myc activation domain and activating Myc target genes (Kim et al., 2003). Therefore, Skp2 has positive effect in the interaction with Myc from two aspects, which is achieved by combining with different Myc domains.
4.3 Aurora-A
Aurora-A is a serine/threonine kinase of the Aurora kinase family, including Aurora-A, Aurora-B, and Aurora-C (Yan et al., 2016). Aurora-A is a powerful oncogene that has been reported to promote tumor proliferation, invasion and metastasis through mitosis and other ways (Li et al., 2018; Lin et al., 2020). Aurora-A interacts with N-Myc on both sides of MBI, upon which the ubiquitin ligase Fbxw7 complexes also bind with N-Myc (Richards et al., 2016). Aurora-A-N-Myc protects N-Myc from proteasomal degradation mediated by the Fbxw7, thus inhibits N-Myc degradation and stabilizes the protein level of N-Myc (Otto et al., 2009). On the other hand, high level of Aurora-A enhances the expression and transcriptional activity of c-Myc, and c-Myc can regulate the transcription level of Aurora-A in turn (den Hollander et al., 2010; Yang et al., 2010). Therefore, Aurora-A is likely to be an important Myc stability regulator, which can also affect the transcriptional activation ability of Myc.
4.4 Protein Phosphatase 1 (PP1)/Protein Phosphatase-1 Nuclear-Targeting Subunit (PNUTS)
PP1 is a Ser/Thr phosphatase, and PNUTS is a regulatory subunit of PP1 (Wang F. et al., 2019). PP1 catalyzes the dephosphorylation of more than half of phosphorylated serine and threonine in cells (Bertolotti, 2018). The binding area of PP1/PNUTS with Myc is still uncertain, but it can be observed that the enrichment of Myc-Max and Myc-PP1/PNUTS on Myc target gene promoters (Dingar et al., 2018). By proximity ligation assay (PLA), endogenic Myc-PNUTS interaction was defineded (Dingar et al., 2018). Inhibition of PP1/PNUTS induced the hyperphosphorylation of Myc, causing degradation by the classical SCF-Fbxw7 pathway (Dingar et al., 2018). In addition, PNUTS knockdown resulted in decreased N-Myc protein, and repressed the progression of MYCN-amplified neuroblastoma (Tee et al., 2020). So PP1/PNUTS is also asignificant assistant of Myc’s carcinogenic process.
4.5 Sin3
Sin3 is a transcriptional repressor with a similar structure of the helix-loop-helix dimerization domain from Myc (Kadamb et al., 2013). Sin3 forms a complex with HDAC, thus regulates histone deacetylation and gene transcription (Banks et al., 2020). Sin3 includes Sin3a and Sin3b, both of which can interact with Myc (Yang et al., 2012; Garcia-Sanz et al., 2014). Sin3b interacts with Myc at amino acids 186–203, belonging to the MBIIIa domain, and recruits HDAC1 to exert the deacetylase activity (Garcia-Sanz et al., 2014). However, Sin3 itself is not associated with Myc target gene down-regulation, only inducing the degradation of Myc, while the transcriptional repression of Myc needs to combine with Mad-Max or Mxi1-Max complexes (Harper et al., 1996).
4.6 Nuclear Receptor Binding SET Domain Protein 3 (NSD3)
NSD3 is a histone lysine methyltransferase, identified as a Myc cofactor (Li et al., 2017). A noncatalytic isoform of NSD3, named NSD3S, shows specially stabilization of Myc half-life. NSD3S binds directly with Myc domain between MBIII and MBIV, and NSD3S residues 389–404 plays a functional role in it. NSD3S suppresses the FBXW7 activity by interacting with Myc, increases Myc half-life and transcriptional function (Gonzalez-Pecchi et al., 2020).
4.7 Ubiquitin-Specific Protease 37 (USP37)
USPs that may regulate c-Myc stability, like USP9X and USP28, stabilizes c-Myc via Fbxw7 (Popov et al., 2007; Khan et al., 2018). USP37 as a novel deubiquitinating enzyme (DUB) that binds c-Myc directly to stabilize it. USP37 binds with Myc MBIII domain, stabilizes c-Myc from polyubiquitination-mediated degradation independent of Fbxw7 (Pan et al., 2015). In lung cancers, USP37 expression is upregulated and positively correlated with Myc, suggests that USP37-Myc inhibitors may be a therapeutic strategy for lung cancer.
5 Inhibitor Progression of Protein-Protein Interaction With Myc
Myc inhibitors designed based on protein-protein interactions have been studied. In addition, in the process of research on other proteins inhibitors that existed directly Myc-interaction, it has also been found to have an impact on the function of Myc and the stability of the protein. These inhibitors may be a new weapon against the oncogene MYC (Table 1).
TABLE 1

TABLE 1. Inhibitors and functions of Myc interaction proteins.
5.1 Targeting TRRAP-Myc Interaction
As TRRAP is an essential gene, mutation or deletion of TRRAP leads to early embryonic lethality or poor embryonic development (Iwanami et al., 2009; Shukla et al., 2011). Due to the importance of TRRAP in the organism, knocking out or degrading TRRAP is not a good way to treat Myc-amplified tumors (Leduc et al., 2014). Blocking or interrupting the PPIs between TRRAP and Myc can inhibit the transcriptional activation of Myc. What’s more, MBII is interaction interface of TRRAP and Myc, both form of a structurally-stable conformation, thus the development of Myc-PPIs inhibitors targeting the MBII domain is an effective strategy (Feris et al., 2019). Besides, ribosomal proteins L11 shows inhibition on c-Myc -induced transcription and cell proliferation by competing with TRRAP upon binding to MBII (Dai et al., 2007a; Dai et al., 2007b). Silencing of L11 increased the expression level of Myc (Jung et al., 2016). Therefore, TRRAP-Myc inhibitors can be designed based on the L11 protein structure.
5.2 Targeting CBP/p300-Myc Interaction
Targeting lysine acetyltransferases CBP/p300 is an effective strategy, small molecule inhibitors that target some of these PPIs domains have been developed. Aiming at the bromodomain of CBP/p300, inhibition probe CPI-637 strongly inhibits MYC expression (Taylor et al., 2016). Inhibitors like NEO2734 and NEO1132 targeting both BET and CBP/p300 proteins could induce the depletion of Myc and inhibition of multiple myeloma growth (Spriano et al., 2020). Sensitivity to the dual inhibitors was only in connection with Myc protein expression levels (Ryan et al., 2021).
5.3 Targeting WDR5-Myc Interaction
Not only c-Myc, all Myc family members could interact with WDR5. In MYCN-amplified neuroblastomas, WDR5 functions as a core cofactor participating in transcriptional activation and tumorigenesis under the guidance of N-Myc. Clinically, high expression of WDR5 in neuroblastoma were a valid indicator of unfavorable prognosis (Sun et al., 2015). It is suggested that the strategy of inhibiting Myc through WDR5 can be adopted to treat a variety of malignant tumors (Thomas et al., 2015b). WDR5 has two main active pockets, a hydrophobic cleft: WDR5 binding motif (WBM) and an arginine-binding pocket: WDR5 interaction (WIN) site (Macdonald et al., 2019; Bryan et al., 2020). A preponderant small molecule inhibitor of the WDR5-Myc interaction based on WDR5 WBM-site structure is compound 12 (Chacón Simon et al., 2020). This compound disrupted the WDR5-Myc interaction in cell lysates, and co-IP in HEK293 cells showed a ∼4-fold reduction of the WDR5-Myc with treating compound 12. Besides, a novel WDR5 WIN site antagonist containing a dihydroisoquinolinone bicyclic core is designed, named compound 16 (Tian et al., 2020). Compound 16 reduces Myc recruitment to chromatin and inhibits Myc–driven cancer proliferation.
5.4 Targeting P-TEFb-Myc Interaction
The development of Cdk9 inhibitors is an advantageous strategy for the P-TEFb-Myc interaction. Up to now, multiple Cdk9 inhibitors have been developed, some of which can affect the transcription function of Myc, weaken the stability of Myc and promote Myc degradation. Peptidomimetic lead compounds, KL-1 and KL-2, downregulates Myc and transcriptional regulated by Myc by destroying the P-TEFb complex (Liang et al., 2018). A clinical inhibitor of Cdk9 and Cdk2, CYC065, can hinder the transcriptional activation of N-Myc by inhibiting the Cdk9 in P-TEFb complex, realizing the therapeutic effect on MYCN-amplified neuroblastoma (Poon et al., 2020). Atuveciclib (BAY 1143572) is a highly selective P-TEFb/Cdk9 inhibitor, which inhibits the phosphorylation of RNAPII and reduces Myc level (Lücking et al., 2017; Narita et al., 2017). UNC10112785 is a potent Cdk9 inhibitor, that destabilizes Myc and induces the substantial loss of Myc protein in KRAS-mutant pancreatic cancer (Blake et al., 2019). A Cdk7/9 inhibitor SNS-032 represses the c-Myc-dependent transcription of RhoA gene, inhibiting liver metastasis in uveal melanoma (Zhang et al., 2019). However, long-term inhibition of Cdk9 may also lead to a compensatory increase in Myc expression and recruit more P-TEFb to Myc target genes in the end (Lu et al., 2015). This suggests that we need to use combination therapy for long-term treatment of tumors when targeting Cdk9.
5.5 Targeting HDAC-Myc Interaction
Histone deacetylase inhibitors (HDACi) is a kind of anti-tumor drug with great development potential. HDAC is that can target Myc mainly selectively inhibit HDAC1 and HDAC3. The HDACi Vorinostat (SAHA) and Entinostat (MS27-275) are effective against leukemic cells, which could induce c-Myc acetylation at lysine 323 and disrupt Myc’s transcriptional repression, finally inducing TRAIL expression and apoptosis (Nebbioso et al., 2017). Panobinostat (LBH589) is a pan-HDACi, which could reduce Myc protein level in human AML cell lines (Beyer et al., 2019). The HDAC3 inhibitor RGFP966 and HDAC1/2 inhibitor depsipeptide (FK228) remit Myc-mediated transcriptional repression of the miR-15 and let-7 families in malignant cells, inducing apoptosis as a result (Konstantinopoulos et al., 2006; Adams et al., 2016).
5.6 Targeting Aurora-A-Myc Interaction
For the reason that the presence of Aurora-A increases the stability of Myc, inhibitors targeting Aurora-A can promote the degradation of Myc and achieve the effect of tumor inhibition. An Aurora-A inhibitor CD532 breaks the native conformation of Aurora-A and drives the degradation of N-Myc in N-Myc-driven cancers (Gustafson et al., 2014). The stronger evidence is that CD532 can cause cells blocking entry into S-phase and lead a subsequent G0/G1 arrest, which is a phenomenon of damaged Myc function (Gustafson et al., 2014). Alisertib is an oral Aurora kinase inhibitor, that has entered clinical trials for a variety of diseases (DuBois et al., 2018; O'Connor et al., 2019; Gay et al., 2020). Alisertib consistently disrupted the N-Myc-Aurora-A complex in vitro, thus inhibited N-Myc signaling and suppressed tumor growth (Beltran et al., 2019). CCT137690 is a potent inhibitor of Aurora kinases, which could dose-dependent reduce N-Myc protein level in Rhabdomyosarcoma (RMS) cells (Ommer et al., 2020).
6 Discussion
There are ample evidences that manifests targeting Myc could form the element of extensively effective anti-cancer therapies. However, due to the flat structure of Myc, there is no binding pocket for moleculars, making the idea of directly inhibiting Myc difficult to become a reality. Some researchers have tried to exploit small molecule drugs to break the Myc/Max interaction, but the feasibility is limited. One difficulty is that there is extensive contact of the bHLH-LZ domain. And a large number of transcription factors share this motif. Therefore, it is arduous to separately inhibit Myc/Max heterodimer without causing off-target side effects on other transcription factors bound to bHLH-LZ. Eventually produce great toxic side effects on normal cells.
What’s more, it turns out that Myc’s recognition of target genes not only depend on the interaction with Max. Model shows that in terms of the affinity of Myc/Max dimers to DNA, about 90% of Myc binding cases in cells cannot be interpreted, and it has been shown that many nucleoproteins can promote Myc recruitment to its target genes (Lorenzin et al., 2016). Some of these recruited proteins interact directly with Myc, and the other proteins form a protein complex which participate in the regulation of Myc function and protein stability. More and more studies have shown that there are many transcription cofactors, which either affect or even determine the transcription of target genes by Myc through protein post-translational modification, or change protein conformation, or compete for protein binding sites. If inhibitors can be designed based on such protein-protein interactions, targeting Myc interacting proteins can achieve the goal of curing Myc-amplified tumors. From the information we summarized in this review, we can see that a variety of small molecule and peptide inhibitors have shown more or less effect on the protein expression level, degradation level, and target gene transcription level of Myc. Some inhibitors were designed from the beginning to destroy the protein interaction of Myc protein. They have indeed achieved certain results in preclinical or clinical trials, which can effectively inhibit tumor growth and promote tumor apoptosis.
Of course, these inhibitors also face some problems. First, for the reason that the targeted proteins are in charge of multiple physiological functions in cells, the inhibition of these proteins may also have an impact on other cell functions. Second, whether the inhibitor can accurately target the Myc-interacting protein complex and how selective it is, remain to be verified. Third, due to the powerful ability of Myc itself, although the capacity of a single inhibitor was strong, will there be any compensation or replacement, making the final therapeutic effect insignificant? These problems have yet to be resolved.
On the whole, the most important point is that targeting the direct PPIs between Myc and other cofactor proteins is an effective and feasible strategy for the treatment of diseases caused by Myc in spite of existing thorny problems mentioned above. We believe that targeting Myc interacting proteins could become a winding path in Myc-associated cancer therapy in the future.
Author Contributions
Concept design: JC, QH, and XG; YZ, XG, MY, and JC wrote the manuscript; JC, QH, and BY directed the study.
Funding
This work was supported by grants from National Natural Science Foundation of China (No. 81872885 to QH), Zhejiang Provincial Natural Science Foundation (No. Y18H310001 to JC) and Fundamental Research Funds for the Central Universities.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling Editor declared a past co-authorship with one of the authors QH.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adams, C. M., Hiebert, S. W., and Eischen, C. M. (2016). Myc Induces miRNA-Mediated Apoptosis in Response to HDAC Inhibition in Hematologic Malignancies. Cancer Res. 76 (3), 736–748. doi:10.1158/0008-5472.CAN-15-1751
Aesoy, R., Gradin, K., Aasrud, K. S., Hoivik, E. A., Ruas, J. L., Poellinger, L., et al. (2014). Regulation of CDKN2B Expression by Interaction of Arnt with Miz-1-Aa Basis for Functional Integration between the HIF and Myc Gene Regulatory Pathways. Mol. Cancer 13, 54. doi:10.1186/1476-4598-13-54
Asmamaw, M. D., Liu, Y., Zheng, Y. C., Shi, X. J., and Liu, H. M. (2020). Skp2 in the Ubiquitin-Proteasome System: A Comprehensive Review. Med. Res. Rev. 40 (5), 1920–1949. doi:10.1002/med.21675
Augert, A., Mathsyaraja, H., Ibrahim, A. H., Freie, B., Geuenich, M. J., Cheng, P. F., et al. (2020). MAX Functions as a Tumor Suppressor and Rewires Metabolism in Small Cell Lung Cancer. Cancer cell 38 (1), 97–e7. doi:10.1016/j.ccell.2020.04.016
Bahram, F., von der Lehr, N., Cetinkaya, C., and Larsson, L. G. (2000). c-Myc Hot Spot Mutations in Lymphomas Result in Inefficient Ubiquitination and Decreased Proteasome-Mediated Turnover. Blood 95 (6), 2104–2110. doi:10.1182/blood.v95.6.2104
Baluapuri, A., Wolf, E., and Eilers, M. (2020). Target Gene-independent Functions of MYC Oncoproteins. Nat. Rev. Mol. Cell Biol 21 (5), 255–267. doi:10.1038/s41580-020-0215-2
Banks, C. A. S., Zhang, Y., Miah, S., Hao, Y., Adams, M. K., Wen, Z., et al. (2020). Integrative Modeling of a Sin3/HDAC Complex Sub-structure. Cell Rep 31 (2), 107516. doi:10.1016/j.celrep.2020.03.080
Barrett, J. F., Lee, L. A., and Dang, C. V. (2005). Stimulation of Myc Transactivation by the TATA Binding Protein in Promoter-Reporter Assays. BMC Biochem. 6, 7. doi:10.1186/1471-2091-6-7
Basu, S., Liu, Q., Qiu, Y., and Dong, F. (2009). Gfi-1 Represses CDKN2B Encoding p15INK4B through Interaction with Miz-1. Proc. Natl. Acad. Sci. U S A. 106 (5), 1433–1438. doi:10.1073/pnas.0804863106
Bédard, M., Maltais, L., Montagne, M., and Lavigne, P. (2017). Miz-1 and Max Compete to Engage C-Myc: Implication for the Mechanism of Inhibition of C-Myc Transcriptional Activity by Miz-1. Proteins 85 (2), 199–206. doi:10.1002/prot.25214
Beishline, K., and Azizkhan-Clifford, J. (2015). Sp1 and the 'hallmarks of Cancer'. FEBS J. 282 (2), 224–258. doi:10.1111/febs.13148
Beltran, H., Oromendia, C., Danila, D. C., Montgomery, B., Hoimes, C., Szmulewitz, R. Z., et al. (2019). A Phase II Trial of the Aurora Kinase A Inhibitor Alisertib for Patients with Castration-Resistant and Neuroendocrine Prostate Cancer: Efficacy and Biomarkers. Clin. Cancer Res. 25 (1), 43–51. doi:10.1158/1078-0432.CCR-18-1912
Bertolotti, A. (2018). The Split Protein Phosphatase System. Biochem. J. 475 (23), 3707–3723. doi:10.1042/BCJ20170726
Beyer, M., Romanski, A., Mustafa, A. M., Pons, M., Büchler, I., Vogel, A., et al. (2019). HDAC3 Activity Is Essential for Human Leukemic Cell Growth and the Expression of β-catenin, MYC, and WT1. Cancers (Basel) 11 (10). doi:10.3390/cancers11101436
Bhuiyan, T., and Timmers, H. T. M. (2019). Promoter Recognition: Putting TFIID on the Spot. Trends Cell Biol 29 (9), 752–763. doi:10.1016/j.tcb.2019.06.004
Blackwood, E. M., and Eisenman, R. N. (1991). Max: a helix-loop-helix Zipper Protein that Forms a Sequence-specific DNA-Binding Complex with Myc. Science 251 (4998), 1211–1217. doi:10.1126/science.2006410
Blake, D. R., Vaseva, A. V., Hodge, R. G., Kline, M. P., Gilbert, T. S. K., Tyagi, V., et al. (2019). Application of a MYC Degradation Screen Identifies Sensitivity to CDK9 Inhibitors in KRAS-Mutant Pancreatic Cancer. Sci. Signal. 12 (590). doi:10.1126/scisignal.aav7259
Boultwood, J. (2011). The Role of Haploinsufficiency of RPS14 and P53 Activation in the Molecular Pathogenesis of the 5q- Syndrome. Pediatr. Rep. 3, e10. doi:10.4081/pr.2011.s2.e10
Brès, V., Yoshida, T., Pickle, L., and Jones, K. A. (2009). SKIP Interacts with C-Myc and Menin to Promote HIV-1 Tat Transactivation. Mol. Cell 36 (1), 75–87. doi:10.1016/j.molcel.2009.08.015
Bryan, A. F., Wang, J., Howard, G. C., Guarnaccia, A. D., Woodley, C. M., Aho, E. R., et al. (2020). WDR5 Is a Conserved Regulator of Protein Synthesis Gene Expression. Nucleic Acids Res. 48 (6), 2924–2941. doi:10.1093/nar/gkaa051
Buckley, S. M., Aranda-Orgilles, B., Strikoudis, A., Apostolou, E., Loizou, E., Moran-Crusio, K., et al. (2012). Regulation of Pluripotency and Cellular Reprogramming by the Ubiquitin-Proteasome System. Cell stem cell 11 (6), 783–798. doi:10.1016/j.stem.2012.09.011
Cao, H., Li, L., Yang, D., Zeng, L., Yewei, X., Yu, B., et al. (2019). Recent Progress in Histone Methyltransferase (G9a) Inhibitors as Anticancer Agents. Eur. J. Med. Chem. 179, 537–546. doi:10.1016/j.ejmech.2019.06.072
Carugo, A., Genovese, G., Seth, S., Nezi, L., Rose, J. L., Bossi, D., et al. (2016). In Vivo Functional Platform Targeting Patient-Derived Xenografts Identifies WDR5-Myc Association as a Critical Determinant of Pancreatic Cancer. Cell Rep 16 (1), 133–147. doi:10.1016/j.celrep.2016.05.063
Cascón, A., and Robledo, M. (2012). MAX and MYC: a Heritable Breakup. Cancer Res. 72 (13), 3119–3124. doi:10.1158/0008-5472.CAN-11-3891
Castell, A., Yan, Q., Fawkner, K., Hydbring, P., Zhang, F., Verschut, V., et al. (2018). A Selective High Affinity MYC-Binding Compound Inhibits MYC:MAX Interaction and MYC-dependent Tumor Cell Proliferation. Sci. Rep. 8 (1), 10064. doi:10.1038/s41598-018-28107-4
Cattoretti, G. (2013). MYC Expression and Distribution in normal Mature Lymphoid Cells. J. Pathol. 229 (3), 430–440. doi:10.1002/path.4141
Chacón Simon, S., Wang, F., Thomas, L. R., Phan, J., Zhao, B., Olejniczak, E. T., et al. (2020). Discovery of WD Repeat-Containing Protein 5 (WDR5)-MYC Inhibitors Using Fragment-Based Methods and Structure-Based Design. J. Med. Chem. 63 (8), 4315–4333. doi:10.1021/acs.jmedchem.0c00224
Chen, B. J., Wu, Y. L., Tanaka, Y., and Zhang, W. (2014). Small Molecules Targeting C-Myc Oncogene: Promising Anti-cancer Therapeutics. Int. J. Biol. Sci. 10 (10), 1084–1096. doi:10.7150/ijbs.10190
Chen, H., Liu, H., and Qing, G. (2018). Targeting Oncogenic Myc as a Strategy for Cancer Treatment. Signal. Transduct Target. Ther. 3, 5. doi:10.1038/s41392-018-0008-7
Chen, T., Li, K., Liu, Z., Liu, J., Wang, Y., Sun, R., et al. (2021). WDR5 Facilitates EMT and Metastasis of CCA by Increasing HIF-1α Accumulation in Myc-dependent and Independent Pathways. Mol. Ther. 29, 2134–2150. doi:10.1016/j.ymthe.2021.02.017
Chen, W. L., Sun, H. P., Li, D. D., Wang, Z. H., You, Q. D., and Guo, X. K. (2017). G9a - an Appealing Antineoplastic Target. Curr. Cancer Drug Targets 17 (6), 555–568. doi:10.2174/1568009616666160512145303
Cogné, B., Ehresmann, S., Beauregard-Lacroix, E., Rousseau, J., Besnard, T., Garcia, T., et al. (2019). Missense Variants in the Histone Acetyltransferase Complex Component Gene TRRAP Cause Autism and Syndromic Intellectual Disability. Am. J. Hum. Genet. 104 (3), 530–541. doi:10.1016/j.ajhg.2019.01.010
Dai, M. S., Arnold, H., Sun, X. X., Sears, R., and Lu, H. (2007a). Inhibition of C-Myc Activity by Ribosomal Protein L11. EMBO J. 26 (14), 3332–3345. doi:10.1038/sj.emboj.7601776
Dai, M. S., Sears, R., and Lu, H. (2007b). Feedback Regulation of C-Myc by Ribosomal Protein L11. Cell Cycle 6 (22), 2735–2741. doi:10.4161/cc.6.22.4895
Dalla-Favera, R., Bregni, M., Erikson, J., Patterson, D., Gallo, R. C., and Croce, C. M. (1982). Human C-Myc Onc Gene Is Located on the Region of Chromosome 8 that Is Translocated in Burkitt Lymphoma Cells. Proc. Natl. Acad. Sci. U S A. 79 (24), 7824–7827. doi:10.1073/pnas.79.24.7824
Dang, C. V., O'Donnell, K. A., Zeller, K. I., Nguyen, T., Osthus, R. C., and Li, F. (2006). The C-Myc Target Gene Network. Semin. Cancer Biol. 16 (4), 253–264. doi:10.1016/j.semcancer.2006.07.014
Dávalos-Salas, M., Montgomery, M. K., Reehorst, C. M., Nightingale, R., Ng, I., Anderton, H., et al. (2019). Deletion of Intestinal Hdac3 Remodels the Lipidome of Enterocytes and Protects Mice from Diet-Induced Obesity. Nat. Commun. 10 (1), 5291. doi:10.1038/s41467-019-13180-8
De Melo, J., Kim, S. S., Lourenco, C., and Penn, L. Z. (2017). Lysine-52 Stabilizes the MYC Oncoprotein through an SCFFbxw7-independent Mechanism. Oncogene 36 (49), 6815–6822. doi:10.1038/onc.2017.268
den Hollander, J., Rimpi, S., Doherty, J. R., Rudelius, M., Buck, A., Hoellein, A., et al. (2010). Aurora Kinases A and B Are Up-Regulated by Myc and Are Essential for Maintenance of the Malignant State. Blood 116 (9), 1498–1505. doi:10.1182/blood-2009-11-251074
Difilippantonio, M. J., Petersen, S., Chen, H. T., Johnson, R., Jasin, M., Kanaar, R., et al. (2002). Evidence for Replicative Repair of DNA Double-Strand Breaks Leading to Oncogenic Translocation and Gene Amplification. J. Exp. Med. 196 (4), 469–480. doi:10.1084/jem.20020851
Dingar, D., Tu, W. B., Resetca, D., Lourenco, C., Tamachi, A., De Melo, J., et al. (2018). MYC Dephosphorylation by the PP1/PNUTS Phosphatase Complex Regulates Chromatin Binding and Protein Stability. Nat. Commun. 9 (1), 3502. doi:10.1038/s41467-018-05660-0
DuBois, S. G., Mosse, Y. P., Fox, E., Kudgus, R. A., Reid, J. M., McGovern, R., et al. (2018). Phase II Trial of Alisertib in Combination with Irinotecan and Temozolomide for Patients with Relapsed or Refractory Neuroblastoma. Clin. Cancer Res. 24 (24), 6142–6149. doi:10.1158/1078-0432.CCR-18-1381
Duffy, M. J., O'Grady, S., Tang, M., and Crown, J. (2021). MYC as a Target for Cancer Treatment. Cancer Treat. Rev. 94, 102154. doi:10.1016/j.ctrv.2021.102154
Eberhardy, S. R., and Farnham, P. J. (2002). Myc Recruits P-TEFb to Mediate the Final Step in the Transcriptional Activation of the Cad Promoter. J. Biol. Chem. 277 (42), 40156–40162. doi:10.1074/jbc.M207441200
Elías-Villalobos, A., Fort, P., and Helmlinger, D. (2019). New Insights into the Evolutionary Conservation of the Sole PIKK Pseudokinase Tra1/TRRAP. Biochem. Soc. Trans. 47 (6), 1597–1608. doi:10.1042/BST20180496
Farrell, A. S., and Sears, R. C. (2014). MYC Degradation. Cold Spring Harb Perspect. Med. 4 (3). doi:10.1101/cshperspect.a014365
Feng, X. H., Liang, Y. Y., Liang, M., Zhai, W., and Lin, X. (2002). Direct Interaction of C-Myc with Smad2 and Smad3 to Inhibit TGF-Beta-Mediated Induction of the CDK Inhibitor p15(Ink4B). Mol. Cell 9 (1), 133–143. doi:10.1016/s1097-2765(01)00430-0
Feng, X. H., Liang, Y. Y., Liang, M., Zhai, W., and Lin, X. (2016). Direct Interaction of C-Myc with Smad2 and Smad3 to Inhibit TGF-β-Mediated Induction of the CDK Inhibitor p15(Ink4B). Mol. Cell 62 (6), 152. doi:10.1016/j.molcel.2016.08.02710.1016/j.molcel.2016.03.026
Feris, E. J., Hinds, J. W., and Cole, M. D. (2019). Formation of a Structurally-Stable Conformation by the Intrinsically Disordered MYC:TRRAP Complex. PloS one 14 (12), e0225784. doi:10.1371/journal.pone.0225784
Frank, S. R., Parisi, T., Taubert, S., Fernandez, P., Fuchs, M., Chan, H. M., et al. (2003). MYC Recruits the TIP60 Histone Acetyltransferase Complex to Chromatin. EMBO Rep. 4 (6), 575–580. doi:10.1038/sj.embor.embor861
Frank, S. R., Schroeder, M., Fernandez, P., Taubert, S., and Amati, B. (2001). Binding of C-Myc to Chromatin Mediates Mitogen-Induced Acetylation of Histone H4 and Gene Activation. Genes Dev. 15 (16), 2069–2082. doi:10.1101/gad.906601
Fujinaga, K. (2020). P-TEFb as A Promising Therapeutic Target. Molecules 25 (4). doi:10.3390/molecules25040838
Garcia-Sanz, P., Quintanilla, A., Lafita, M. C., Moreno-Bueno, G., García-Gutierrez, L., Tabor, V., et al. (2014). Sin3b Interacts with Myc and Decreases Myc Levels. J. Biol. Chem. 289 (32), 22221–22236. doi:10.1074/jbc.M113.538744
Gartel, A. L., Ye, X., Goufman, E., Shianov, P., Hay, N., Najmabadi, F., et al. (2001). Myc Represses the p21(WAF1/CIP1) Promoter and Interacts with Sp1/Sp3. Proc. Natl. Acad. Sci. U S A. 98 (8), 4510–4515. doi:10.1073/pnas.081074898
Gay, C. M., Zhou, Y., Lee, J. J., Tang, X. M., Lu, W., Wistuba, I. I., et al. (2020). A Phase II Trial of Alisertib (MLN8237) in Salvage Malignant Mesothelioma. Oncologist 25 (10), e1457–e1463. doi:10.1634/theoncologist.2020-0610
Gersch, M., Wagstaff, J. L., Toms, A. V., Graves, B., Freund, S. M. V., and Komander, D. (2019). Distinct USP25 and USP28 Oligomerization States Regulate Deubiquitinating Activity. Mol. Cell 74 (3), 436–e7. doi:10.1016/j.molcel.2019.02.030
Goh, W., Scheer, S., Jackson, J. T., Hediyeh-Zadeh, S., Delconte, R. B., Schuster, I. S., et al. (2020). Hhex Directly Represses BIM-dependent Apoptosis to Promote NK Cell Development and Maintenance. Cell Rep 33 (3), 108285. doi:10.1016/j.celrep.2020.108285
Gonzalez-Pecchi, V., Kwan, A. K., Doyle, S., Ivanov, A. A., Du, Y., and Fu, H. (2020). NSD3S Stabilizes MYC through Hindering its Interaction with FBXW7. J. Mol. Cell Biol 12 (6), 438–447. doi:10.1093/jmcb/mjz098
Grandori, C., and Eisenman, R. N. (1997). Myc Target Genes. Trends Biochem. Sci. 22 (5), 177–181. doi:10.1016/s0968-0004(97)01025-6
Guarnaccia, A. D., Rose, K. L., Wang, J., Zhao, B., Popay, T. M., Wang, C. E., et al. (2021). Impact of WIN Site Inhibitor on the WDR5 Interactome. Cell Rep 34 (3), 108636. doi:10.1016/j.celrep.2020.108636
Guarnaccia, A. D., and Tansey, W. P. (2018). Moonlighting with WDR5: A Cellular Multitasker. J. Clin. Med. 7 (2). doi:10.3390/jcm7020021
Gustafson, W. C., Meyerowitz, J. G., Nekritz, E. A., Chen, J., Benes, C., Charron, E., et al. (2014). Drugging MYCN through an Allosteric Transition in Aurora Kinase A. Cancer cell 26 (3), 414–427. doi:10.1016/j.ccr.2014.07.015
Harper, S. E., Qiu, Y., and Sharp, P. A. (1996). Sin3 Corepressor Function in Myc-Induced Transcription and Transformation. Proc. Natl. Acad. Sci. U S A. 93 (16), 8536–8540. doi:10.1073/pnas.93.16.8536
Hatton, K. S., Mahon, K., Chin, L., Chiu, F. C., Lee, H. W., Peng, D., et al. (1996). Expression and Activity of L-Myc in normal Mouse Development. Mol. Cell Biol 16 (4), 1794–1804. doi:10.1128/mcb.16.4.1794
Hirning, U., Schmid, P., Schulz, W. A., Rettenberger, G., and Hameister, H. (1991). A Comparative Analysis of N-Myc and C-Myc Expression and Cellular Proliferation in Mouse Organogenesis. Mech. Dev. 33 (2), 119–125. doi:10.1016/0925-4773(91)90078-k
Hydbring, P., Castell, A., and Larsson, L. G. (2017). MYC Modulation Around the CDK2/p27/SKP2 Axis. Genes (Basel) 8 (7). doi:10.3390/genes8070174
Ishiguro, K., Kitajima, H., Niinuma, T., Maruyama, R., Nishiyama, N., Ohtani, H., et al. (2021). Dual EZH2 and G9a Inhibition Suppresses Multiple Myeloma Cell Proliferation by Regulating the Interferon Signal and IRF4-MYC axis. Cell Death Discov 7 (1), 7. doi:10.1038/s41420-020-00400-0
Iwanami, N., Okada, M., Hoa, V. Q., Seo, Y., Mitani, H., Sasaki, T., et al. (2009). Ethylnitrosourea-induced Thymus-Defective Mutants Identify Roles of KIAA1440, TRRAP, and SKIV2L2 in Teleost Organ Development. Eur. J. Immunol. 39 (9), 2606–2616. doi:10.1002/eji.200939362
Jang, S. W., Hwang, S. S., Kim, H. S., Kim, M. K., Lee, W. H., Hwang, S. U., et al. (2019). Homeobox Protein Hhex Negatively Regulates Treg Cells by Inhibiting Foxp3 Expression and Function. Proc. Natl. Acad. Sci. U S A. 116 (51), 25790–25799. doi:10.1073/pnas.1907224116
Jung, J. H., Kim, M. J., Lee, H., Lee, J., Kim, J., Lee, H. J., et al. (2016). Farnesiferol C Induces Apoptosis via Regulation of L11 and C-Myc with Combinational Potential with Anticancer Drugs in Non-small-cell Lung Cancers. Sci. Rep. 6, 26844. doi:10.1038/srep26844
Kadamb, R., Mittal, S., Bansal, N., Batra, H., and Saluja, D. (2013). Sin3: Insight into its Transcription Regulatory Functions. Eur. J. Cell Biol 92 (8-9), 237–246. doi:10.1016/j.ejcb.2013.09.001
Kalkat, M., Resetca, D., Lourenco, C., Chan, P. K., Wei, Y., Shiah, Y. J., et al. (2018). MYC Protein Interactome Profiling Reveals Functionally Distinct Regions that Cooperate to Drive Tumorigenesis. Mol. Cell 72 (5), 836–e7. doi:10.1016/j.molcel.2018.09.031
Kanazawa, S., Soucek, L., Evan, G., Okamoto, T., and Peterlin, B. M. (2003). c-Myc Recruits P-TEFb for Transcription, Cellular Proliferation and Apoptosis. Oncogene 22 (36), 5707–5711. doi:10.1038/sj.onc.1206800
Ke, X. X., Zhang, R., Zhong, X., Zhang, L., and Cui, H. (2020). Deficiency of G9a Inhibits Cell Proliferation and Activates Autophagy via Transcriptionally Regulating C-Myc Expression in Glioblastoma. Front Cell Dev Biol 8, 593964. doi:10.3389/fcell.2020.593964
Khan, O. M., Carvalho, J., Spencer-Dene, B., Mitter, R., Frith, D., Snijders, A. P., et al. (2018). The Deubiquitinase USP9X Regulates FBW7 Stability and Suppresses Colorectal Cancer. J. Clin. Invest. 128 (4), 1326–1337. doi:10.1172/JCI97325
Kim, D., Brocker, C. N., Takahashi, S., Yagai, T., Kim, T., Xie, G., et al. (2019). Keratin 23 Is a Peroxisome Proliferator-Activated Receptor Alpha-dependent, MYC-Amplified Oncogene that Promotes Hepatocyte Proliferation. Hepatology 70 (1), 154–167. doi:10.1002/hep.30530
Kim, S. Y., Herbst, A., Tworkowski, K. A., Salghetti, S. E., and Tansey, W. P. (2003). Skp2 Regulates Myc Protein Stability and Activity. Mol. Cell 11 (5), 1177–1188. doi:10.1016/s1097-2765(03)00173-4
King, B., Trimarchi, T., Reavie, L., Xu, L., Mullenders, J., Ntziachristos, P., et al. (2013). The Ubiquitin Ligase FBXW7 Modulates Leukemia-Initiating Cell Activity by Regulating MYC Stability. Cell 153 (7), 1552–1566. doi:10.1016/j.cell.2013.05.041
Konstantinopoulos, P. A., Vandoros, G. P., and Papavassiliou, A. G. (2006). FK228 (Depsipeptide): a HDAC Inhibitor with Pleiotropic Antitumor Activities. Cancer Chemother. Pharmacol. 58 (5), 711–715. doi:10.1007/s00280-005-0182-5
Kurland, J. F., and Tansey, W. P. (2008). Myc-mediated Transcriptional Repression by Recruitment of Histone Deacetylase. Cancer Res. 68 (10), 3624–3629. doi:10.1158/0008-5472.CAN-07-6552
Laidlaw, B. J., Duan, L., Xu, Y., Vazquez, S. E., and Cyster, J. G. (2020). The Transcription Factor Hhex Cooperates with the Corepressor Tle3 to Promote Memory B Cell Development. Nat. Immunol. 21 (9), 1082–1093. doi:10.1038/s41590-020-0713-6
Lancho, O., and Herranz, D. (2018). The MYC Enhancer-Ome: Long-Range Transcriptional Regulation of MYC in Cancer. Trends Cancer 4 (12), 810–822. doi:10.1016/j.trecan.2018.10.003
Leduc, C., Chemin, G., Puget, N., Sawan, C., Moutahir, M., Herceg, Z., et al. (2014). Tissue-specific Inactivation of HAT Cofactor TRRAP Reveals its Essential Role in B Cells. Cell Cycle 13 (10), 1583–1589. doi:10.4161/cc.28560
Li, M., Gao, K., Chu, L., Zheng, J., and Yang, J. (2018). The Role of Aurora-A in Cancer Stem Cells. Int. J. Biochem. Cell Biol 98, 89–92. doi:10.1016/j.biocel.2018.03.007
Li, Z., Ivanov, A. A., Su, R., Gonzalez-Pecchi, V., Qi, Q., Liu, S., et al. (2017). The OncoPPi Network of Cancer-Focused Protein-Protein Interactions to Inform Biological Insights and Therapeutic Strategies. Nat. Commun. 8, 14356. doi:10.1038/ncomms14356
Liang, K., Smith, E. R., Aoi, Y., Stoltz, K. L., Katagi, H., Woodfin, A. R., et al. (2018). Targeting Processive Transcription Elongation via SEC Disruption for MYC-Induced Cancer Therapy. Cell 175 (3), 766–e17. doi:10.1016/j.cell.2018.09.027
Licchesi, J. D., Van Neste, L., Tiwari, V. K., Cope, L., Lin, X., Baylin, S. B., et al. (2010). Transcriptional Regulation of Wnt Inhibitory Factor-1 by Miz-1/c-Myc. Oncogene 29 (44), 5923–5934. doi:10.1038/onc.2010.322
Lin, X., Xiang, X., Hao, L., Wang, T., Lai, Y., Abudoureyimu, M., et al. (2020). The Role of Aurora-A in Human Cancers and Future Therapeutics. Am. J. Cancer Res. 10 (9), 2705–2729.
Liu, H., Zhou, M., Luo, X., Zhang, L., Niu, Z., Peng, C., et al. (2008). Transcriptional Regulation of BRD7 Expression by Sp1 and C-Myc. BMC Mol. Biol. 9, 111. doi:10.1186/1471-2199-9-111
Liu, Q., Basu, S., Qiu, Y., Tang, F., and Dong, F. (2010). A Role of Miz-1 in Gfi-1-Mediated Transcriptional Repression of CDKN1A. Oncogene 29 (19), 2843–2852. doi:10.1038/onc.2010.48
Liu, X., Tesfai, J., Evrard, Y. A., Dent, S. Y., and Martinez, E. (2003). c-Myc Transformation Domain Recruits the Human STAGA Complex and Requires TRRAP and GCN5 Acetylase Activity for Transcription Activation. J. Biol. Chem. 278 (22), 20405–20412. doi:10.1074/jbc.M211795200
Lorenzin, F., Benary, U., Baluapuri, A., Walz, S., Jung, L. A., von Eyss, B., et al. (2016). Different Promoter Affinities Account for Specificity in MYC-dependent Gene Regulation. eLife 5. doi:10.7554/eLife.15161
Lu, H., Xue, Y., Xue, Y., Yu, G. K., Arias, C., Lin, J., et al. (2015). Compensatory Induction of MYC Expression by Sustained CDK9 Inhibition via a BRD4-dependent Mechanism. eLife 4, e06535. doi:10.7554/eLife.06535
Lu, K., Tao, H., Si, X., and Chen, Q. (2018). The Histone H3 Lysine 4 Presenter WDR5 as an Oncogenic Protein and Novel Epigenetic Target in Cancer. Front. Oncol. 8, 502. doi:10.3389/fonc.2018.00502
Lu, W., Yang, C., He, H., and Liu, H. (2020). The CARM1-P300-C-Myc-Max (CPCM) Transcriptional Complex Regulates the Expression of CUL4A/4B and Affects the Stability of CRL4 E3 Ligases in Colorectal Cancer. Int. J. Biol. Sci. 16 (6), 1071–1085. doi:10.7150/ijbs.41230
Lücking, U., Scholz, A., Lienau, P., Siemeister, G., Kosemund, D., Bohlmann, R., et al. (2017). Identification of Atuveciclib (BAY 1143572), the First Highly Selective, Clinical PTEFb/CDK9 Inhibitor for the Treatment of Cancer. ChemMedChem 12 (21), 1776–1793. doi:10.1002/cmdc.201700447
Macdonald, J. D., Chacón Simon, S., Han, C., Wang, F., Shaw, J. G., Howes, J. E., et al. (2019). Discovery and Optimization of Salicylic Acid-Derived Sulfonamide Inhibitors of the WD Repeat-Containing Protein 5-MYC Protein-Protein Interaction. J. Med. Chem. 62 (24), 11232–11259. doi:10.1021/acs.jmedchem.9b01411
Marfil, V., Blazquez, M., Serrano, F., Castell, J. V., and Bort, R. (2015). Growth-promoting and Tumourigenic Activity of C-Myc Is Suppressed by Hhex. Oncogene 34 (23), 3011–3022. doi:10.1038/onc.2014.240
Mathsyaraja, H., Freie, B., Cheng, P. F., Babaeva, E., Catchpole, J. T., Janssens, D., et al. (2019). Max Deletion Destabilizes MYC Protein and Abrogates Eµ-Myc Lymphomagenesis. Genes Dev. 33 (17-18), 1252–1264. doi:10.1101/gad.325878.119
McMahon, S. B., Wood, M. A., and Cole, M. D. (2000). The Essential Cofactor TRRAP Recruits the Histone Acetyltransferase hGCN5 to C-Myc. Mol. Cell Biol 20 (2), 556–562. doi:10.1128/mcb.20.2.556-562.2000
Möröy, T., Saba, I., and Kosan, C. (2011). The Role of the Transcription Factor Miz-1 in Lymphocyte Development and Lymphomagenesis-Binding Myc Makes the Difference. Semin. Immunol. 23 (5), 379–387. doi:10.1016/j.smim.2011.09.001
Mudgapalli, N., Nallasamy, P., Chava, H., Chava, S., Pathania, A. S., Gunda, V., et al. (2019). The Role of Exosomes and MYC in Therapy Resistance of Acute Myeloid Leukemia: Challenges and Opportunities. Mol. Aspects Med. 70, 21–32. doi:10.1016/j.mam.2019.10.001
Narita, T., Ishida, T., Ito, A., Masaki, A., Kinoshita, S., Suzuki, S., et al. (2017). Cyclin-dependent Kinase 9 Is a Novel Specific Molecular Target in Adult T-Cell Leukemia/lymphoma. Blood 130 (9), 1114–1124. doi:10.1182/blood-2016-09-741983
Nau, M. M., Brooks, B. J., Battey, J., Sausville, E., Gazdar, A. F., Kirsch, I. R., et al. (1985). L-myc, a New Myc-Related Gene Amplified and Expressed in Human Small Cell Lung Cancer. Nature 318 (6041), 69–73. doi:10.1038/318069a0
Nebbioso, A., Carafa, V., Conte, M., Tambaro, F. P., Abbondanza, C., Martens, J., et al. (2017). c-Myc Modulation and Acetylation Is a Key HDAC Inhibitor Target in Cancer. Clin. Cancer Res. 23 (10), 2542–2555. doi:10.1158/1078-0432.CCR-15-2388
Nikiforov, M. A., Chandriani, S., Park, J., Kotenko, I., Matheos, D., Johnsson, A., et al. (2002). TRRAP-dependent and TRRAP-independent Transcriptional Activation by Myc Family Oncoproteins. Mol. Cell Biol 22 (14), 5054–5063. doi:10.1128/mcb.22.14.5054-5063.2002
O'Connor, O. A., Özcan, M., Jacobsen, E. D., Roncero, J. M., Trotman, J., Demeter, J., et al. (2019). Randomized Phase III Study of Alisertib or Investigator's Choice (Selected Single Agent) in Patients with Relapsed or Refractory Peripheral T-Cell Lymphoma. J. Clin. Oncol. 37 (8), 613–623. doi:10.1200/JCO.18.00899
Ommer, J., Selfe, J. L., Wachtel, M., O'Brien, E. M., Laubscher, D., Roemmele, M., et al. (2020). Aurora A Kinase Inhibition Destabilizes PAX3-FOXO1 and MYCN and Synergizes with Navitoclax to Induce Rhabdomyosarcoma Cell Death. Cancer Res. 80 (4), 832–842. doi:10.1158/0008-5472.CAN-19-1479
Otto, T., Horn, S., Brockmann, M., Eilers, U., Schüttrumpf, L., Popov, N., et al. (2009). Stabilization of N-Myc Is a Critical Function of Aurora A in Human Neuroblastoma. Cancer cell 15 (1), 67–78. doi:10.1016/j.ccr.2008.12.005
Pan, J., Deng, Q., Jiang, C., Wang, X., Niu, T., Li, H., et al. (2015). USP37 Directly Deubiquitinates and Stabilizes C-Myc in Lung Cancer. Oncogene 34 (30), 3957–3967. doi:10.1038/onc.2014.327
Pirity, M., Blanck, J. K., and Schreiber-Agus, N. (2006). Lessons Learned from Myc/Max/Mad Knockout Mice. Curr. Top. Microbiol. Immunol. 302, 205–234. doi:10.1007/3-540-32952-8_8
Poon, E., Liang, T., Jamin, Y., Walz, S., Kwok, C., Hakkert, A., et al. (2020). Orally Bioavailable CDK9/2 Inhibitor Shows Mechanism-Based Therapeutic Potential in MYCN-Driven Neuroblastoma. J. Clin. Invest. 130 (11), 5875–5892. doi:10.1172/JCI134132
Popov, N., Wanzel, M., Madiredjo, M., Zhang, D., Beijersbergen, R., Bernards, R., et al. (2007). The Ubiquitin-specific Protease USP28 Is Required for MYC Stability. Nat. Cell Biol 9 (7), 765–774. doi:10.1038/ncb1601
Qi, Y., Li, X., Chang, C., Xu, F., He, Q., Zhao, Y., et al. (2017). Ribosomal Protein L23 Negatively Regulates Cellular Apoptosis via the RPL23/Miz-1/c-Myc Circuit in Higher-Risk Myelodysplastic Syndrome. Sci. Rep. 7 (1), 2323. doi:10.1038/s41598-017-02403-x
Quéva, C., Hurlin, P. J., Foley, K. P., and Eisenman, R. N. (1998). Sequential Expression of the MAD Family of Transcriptional Repressors during Differentiation and Development. Oncogene 16 (8), 967–977. doi:10.1038/sj.onc.1201611
Rhoads, D. D., and Roufa, D. J. (1991). Molecular Evolution of the Mammalian Ribosomal Protein Gene, RPS14. Mol. Biol. Evol. 8 (4), 503–514. doi:10.1093/oxfordjournals.molbev.a040665
Richards, M. W., Burgess, S. G., Poon, E., Carstensen, A., Eilers, M., Chesler, L., et al. (2016). Structural Basis of N-Myc Binding by Aurora-A and its Destabilization by Kinase Inhibitors. Proc. Natl. Acad. Sci. U S A. 113 (48), 13726–13731. doi:10.1073/pnas.1610626113
Rickman, D. S., Schulte, J. H., and Eilers, M. (2018). The Expanding World of N-MYC-Driven Tumors. Cancer Discov. 8 (2), 150–163. doi:10.1158/2159-8290.CD-17-0273
Ross, J., Rashkovan, M., Fraszczak, J., Joly-Beauparlant, C., Vadnais, C., Winkler, R., et al. (2019). Deletion of the Miz-1 POZ Domain Increases Efficacy of Cytarabine Treatment in T- and B-ALL/Lymphoma Mouse Models. Cancer Res. 79 (16), 4184–4195. doi:10.1158/0008-5472.CAN-18-3038
Ryan, K. R., Giles, F., and Morgan, G. J. (2021). Targeting Both BET and CBP/EP300 Proteins with the Novel Dual Inhibitors NEO2734 and NEO1132 Leads to Anti-tumor Activity in Multiple Myeloma. Eur. J. Haematol. 106 (1), 90–99. doi:10.1111/ejh.13525
Sailo, B. L., Banik, K., Girisa, S., Bordoloi, D., Fan, L., Halim, C. E., et al. (2019). FBXW7 in Cancer: What Has Been Unraveled Thus Far?. Cancers (Basel) 11 (2). doi:10.3390/cancers11020246
Seoane, J., Le, H. V., and Massagué, J. (2002). Myc Suppression of the p21(Cip1) Cdk Inhibitor Influences the Outcome of the P53 Response to DNA Damage. Nature 419 (6908), 729–734. doi:10.1038/nature01119
Shukla, V., Cuenin, C., Dubey, N., and Herceg, Z. (2011). Loss of Histone Acetyltransferase Cofactor Transformation/transcription Domain-Associated Protein Impairs Liver Regeneration after Toxic Injury. Hepatology 53 (3), 954–963. doi:10.1002/hep.24120
Spriano, F., Gaudio, E., Cascione, L., Tarantelli, C., Melle, F., Motta, G., et al. (2020). Antitumor Activity of the Dual BET and CBP/EP300 Inhibitor NEO2734. Blood Adv. 4 (17), 4124–4135. doi:10.1182/bloodadvances.2020001879
Sun, Y., Bell, J. L., Carter, D., Gherardi, S., Poulos, R. C., Milazzo, G., et al. (2015). WDR5 Supports an N-Myc Transcriptional Complex that Drives a Protumorigenic Gene Expression Signature in Neuroblastoma. Cancer Res. 75 (23), 5143–5154. doi:10.1158/0008-5472.CAN-15-0423
Taylor, A. M., Côté, A., Hewitt, M. C., Pastor, R., Leblanc, Y., Nasveschuk, C. G., et al. (2016). Fragment-Based Discovery of a Selective and Cell-Active Benzodiazepinone CBP/EP300 Bromodomain Inhibitor (CPI-637). ACS Med. Chem. Lett. 7 (5), 531–536. doi:10.1021/acsmedchemlett.6b00075
Tee, A. E., Ciampa, O. C., Wong, M., Fletcher, J. I., Kamili, A., Chen, J., et al. (2020). Combination Therapy with the CDK7 Inhibitor and the Tyrosine Kinase Inhibitor Exerts Synergistic Anticancer Effects against MYCN-Amplified Neuroblastoma. Int. J. Cancer 147 (7), 1928–1938. doi:10.1002/ijc.32936
Thomas, L. R., Adams, C. M., Wang, J., Weissmiller, A. M., Creighton, J., Lorey, S. L., et al. (2019). Interaction of the Oncoprotein Transcription Factor MYC with its Chromatin Cofactor WDR5 Is Essential for Tumor Maintenance. Proc. Natl. Acad. Sci. U S A. 116 (50), 25260–25268. doi:10.1073/pnas.1910391116
Thomas, L. R., Foshage, A. M., Weissmiller, A. M., and Tansey, W. P. (2015a). The MYC-WDR5 Nexus and Cancer. Cancer Res. 75 (19), 4012–4015. doi:10.1158/0008-5472.CAN-15-1216
Thomas, L. R., and Tansey, W. P. (2011). Proteolytic Control of the Oncoprotein Transcription Factor Myc. Adv. Cancer Res. 110, 77–106. doi:10.1016/B978-0-12-386469-7.00004-9
Thomas, L. R., Wang, Q., Grieb, B. C., Phan, J., Foshage, A. M., Sun, Q., et al. (2015b). Interaction with WDR5 Promotes Target Gene Recognition and Tumorigenesis by MYC. Mol. Cell 58 (3), 440–452. doi:10.1016/j.molcel.2015.02.028
Tian, J., Teuscher, K. B., Aho, E. R., Alvarado, J. R., Mills, J. J., Meyers, K. M., et al. (2020). Discovery and Structure-Based Optimization of Potent and Selective WD Repeat Domain 5 (WDR5) Inhibitors Containing a Dihydroisoquinolinone Bicyclic Core. J. Med. Chem. 63 (2), 656–675. doi:10.1021/acs.jmedchem.9b01608
Tu, W. B., Helander, S., Pilstål, R., Hickman, K. A., Lourenco, C., Jurisica, I., et al. (2015). Myc and its Interactors Take Shape. Biochim. Biophys. Acta 1849 (5), 469–483. doi:10.1016/j.bbagrm.2014.06.002
Tu, W. B., Shiah, Y. J., Lourenco, C., Mullen, P. J., Dingar, D., Redel, C., et al. (2018). MYC Interacts with the G9a Histone Methyltransferase to Drive Transcriptional Repression and Tumorigenesis. Cancer cell 34 (4), 579–e8. doi:10.1016/j.ccell.2018.09.001
Ullius, A., Lüscher-Firzlaff, J., Costa, I. G., Walsemann, G., Forst, A. H., Gusmao, E. G., et al. (2014). The Interaction of MYC with the Trithorax Protein ASH2L Promotes Gene Transcription by Regulating H3K27 Modification. Nucleic Acids Res. 42 (11), 6901–6920. doi:10.1093/nar/gku312
Vennstrom, B., Sheiness, D., Zabielski, J., and Bishop, J. M. (1982). Isolation and Characterization of C-Myc, a Cellular Homolog of the Oncogene (V-myc) of Avian Myelocytomatosis Virus Strain 29. J. Virol. 42 (3), 773–779. doi:10.1128/JVI.42.3.773-779.1982
Vervoorts, J., Lüscher-Firzlaff, J. M., Rottmann, S., Lilischkis, R., Walsemann, G., Dohmann, K., et al. (2003). Stimulation of C-MYC Transcriptional Activity and Acetylation by Recruitment of the Cofactor CBP. EMBO Rep. 4 (5), 484–490. doi:10.1038/sj.embor.embor821
Vizcaíno, C., Mansilla, S., and Portugal, J. (2015). Sp1 Transcription Factor: A Long-Standing Target in Cancer Chemotherapy. Pharmacol. Ther. 152, 111–124. doi:10.1016/j.pharmthera.2015.05.008
von der Lehr, N., Johansson, S., Wu, S., Bahram, F., Castell, A., Cetinkaya, C., et al. (2003). The F-Box Protein Skp2 Participates in C-Myc Proteosomal Degradation and Acts as a Cofactor for C-Myc-Regulated Transcription. Mol. Cell 11 (5), 1189–1200. doi:10.1016/s1097-2765(03)00193-x
Wang, F., Wang, L., Fisher, L. A., Li, C., Wang, W., and Peng, A. (2019a). Phosphatase 1 Nuclear Targeting Subunit (PNUTS) Regulates Aurora Kinases and Mitotic Progression. Mol. Cancer Res. 17 (1), 10–19. doi:10.1158/1541-7786.MCR-17-0670
Wang, J., Wang, H., Peters, M., Ding, N., Ribback, S., Utpatel, K., et al. (2019b). Loss of Fbxw7 Synergizes with Activated Akt Signaling to Promote C-Myc Dependent Cholangiocarcinogenesis. J. Hepatol. 71 (4), 742–752. doi:10.1016/j.jhep.2019.05.027
Wang, T., Cai, B., Ding, M., Su, Z., Liu, Y., and Shen, L. (2019c). c-Myc Overexpression Promotes Oral Cancer Cell Proliferation and Migration by Enhancing Glutaminase and Glutamine Synthetase Activity. Am. J. Med. Sci. 358 (3), 235–242. doi:10.1016/j.amjms.2019.05.014
Wei, Y., Resetca, D., Li, Z., Johansson-Åkhe, I., Ahlner, A., Helander, S., et al. (2019). Multiple Direct Interactions of TBP with the MYC Oncoprotein. Nat. Struct. Mol. Biol. 26 (11), 1035–1043. doi:10.1038/s41594-019-0321-z
Weinert, B. T., Narita, T., Satpathy, S., Srinivasan, B., Hansen, B. K., Schölz, C., et al. (2018). Time-Resolved Analysis Reveals Rapid Dynamics and Broad Scope of the CBP/p300 Acetylome. Cell 174 (1), 231–e12. doi:10.1016/j.cell.2018.04.033
Welcker, M., Orian, A., Jin, J., Grim, J. E., Grim, J. A., Harper, J. W., et al. (2004). The Fbw7 Tumor Suppressor Regulates Glycogen Synthase Kinase 3 Phosphorylation-dependent C-Myc Protein Degradation. Proc. Natl. Acad. Sci. U S A. 101 (24), 9085–9090. doi:10.1073/pnas.0402770101
Wu, G., Yuan, M., Shen, S., Ma, X., Fang, J., Zhu, L., et al. (2017). Menin Enhances C-Myc-Mediated Transcription to Promote Cancer Progression. Nat. Commun. 8, 15278. doi:10.1038/ncomms15278
Wu, H., Yang, T. Y., Li, Y., Ye, W. L., Liu, F., He, X. S., et al. (2020). Tumor Necrosis Factor Receptor-Associated Factor 6 Promotes Hepatocarcinogenesis by Interacting with Histone Deacetylase 3 to Enhance C-Myc Gene Expression and Protein Stability. Hepatology 71 (1), 148–163. doi:10.1002/hep.30801
Xu, L., Morgenbesser, S. D., and DePinho, R. A. (1991). Complex Transcriptional Regulation of Myc Family Gene Expression in the Developing Mouse Brain and Liver. Mol. Cell Biol 11 (12), 6007–6015. doi:10.1128/mcb.11.12.6007
Yada, M., Hatakeyama, S., Kamura, T., Nishiyama, M., Tsunematsu, R., Imaki, H., et al. (2004). Phosphorylation-dependent Degradation of C-Myc Is Mediated by the F-Box Protein Fbw7. EMBO J. 23 (10), 2116–2125. doi:10.1038/sj.emboj.7600217
Yan, M., Wang, C., He, B., Yang, M., Tong, M., Long, Z., et al. (2016). Aurora-A Kinase: A Potent Oncogene and Target for Cancer Therapy. Med. Res. Rev. 36 (6), 1036–1079. doi:10.1002/med.21399
Yang, S., He, S., Zhou, X., Liu, M., Zhu, H., Wang, Y., et al. (2010). Suppression of Aurora-A Oncogenic Potential by C-Myc Downregulation. Exp. Mol. Med. 42 (11), 759–767. doi:10.3858/emm.2010.42.11.077
Yang, W., Yang, X., David, G., and Dorsey, J. F. (2012). Dissecting the Complex Regulation of Mad4 in Glioblastoma Multiforme Cells. Cancer Biol. Ther. 13 (13), 1339–1348. doi:10.4161/cbt.21814
Yang, Z., Jiang, X., Zhang, Z., Zhao, Z., Xing, W., Liu, Y., et al. (2021). HDAC3-dependent Transcriptional Repression of FOXA2 Regulates FTO/m6A/MYC Signaling to Contribute to the Development of Gastric Cancer. Cancer Gene Ther. 28 (1-2), 141–155. doi:10.1038/s41417-020-0193-8
Zhang, J., Liu, S., Ye, Q., and Pan, J. (2019). Transcriptional Inhibition by CDK7/9 Inhibitor SNS-032 Abrogates Oncogene Addiction and Reduces Liver Metastasis in Uveal Melanoma. Mol. Cancer 18 (1), 140. doi:10.1186/s12943-019-1070-7
Zhang, K., Faiola, F., and Martinez, E. (2005). Six Lysine Residues on C-Myc Are Direct Substrates for Acetylation by P300. Biochem. Biophys. Res. Commun. 336 (1), 274–280. doi:10.1016/j.bbrc.2005.08.075
Zhang, L., Li, J., Xu, H., Shao, X., Fu, L., Hou, Y., et al. (2020). Myc-Miz1 Signaling Promotes Self-Renewal of Leukemia Stem Cells by Repressing Cebpα and Cebpδ. Blood 135 (14), 1133–1145. doi:10.1182/blood.2019001863
Zhang, X., Chen, X., Lin, J., Lwin, T., Wright, G., Moscinski, L. C., et al. (2012). Myc Represses miR-15a/miR-16-1 Expression through Recruitment of HDAC3 in Mantle Cell and Other Non-hodgkin B-Cell Lymphomas. Oncogene 31 (24), 3002–3008. doi:10.1038/onc.2011.470
Keywords: MYC, cancer therapy, interaction protein, transcriptional regulation, post-translational modification, inhibitors
Citation: Zhou Y, Gao X, Yuan M, Yang B, He Q and Cao J (2021) Targeting Myc Interacting Proteins as a Winding Path in Cancer Therapy. Front. Pharmacol. 12:748852. doi: 10.3389/fphar.2021.748852
Received: 28 July 2021; Accepted: 10 September 2021;
Published: 29 September 2021.
Edited by:
Fanfan Zhou, The University of Sydney, AustraliaReviewed by:
Yanfeng Wang, Beijing Institute of Technology, ChinaIvana Samarzija, Rudjer Boskovic Institute, Croatia
Feng Wang, Beijing Institute of Technology, China
Copyright © 2021 Zhou, Gao, Yuan, Yang, He and Cao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qiaojun He, cWlhb2p1bmhlQHpqdS5lZHUuY24=; Ji Cao, Y2Fvamk4OEB6anUuZWR1LmNu
†These authors have contributed equally to this work