 Marie-Anne Morren
Marie-Anne Morren Eric Legius2
Eric Legius2 Smail Hadj-Rabia
Smail Hadj-Rabia Daniel Hohl
Daniel Hohl
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol., 05 January 2022
Sec. Drugs Outcomes Research and Policies
Volume 12 - 2021 | https://doi.org/10.3389/fphar.2021.746664
This article is part of the Research TopicPrevention, Diagnosis and Treatment of Rare DisordersView all 20 articles
Genodermatoses are rare inherited skin diseases that frequently affect other organs. They often have marked effects on wellbeing and may cause early death. Progress in molecular genetics and translational research has unravelled many underlying pathological mechanisms, and in several disorders with high unmet need, has opened the way for the introduction of innovative treatments. One approach is to intervene where cell-signaling pathways are dysregulated, in the case of overactive pathways by the use of selective inhibitors, or when the activity of an essential factor is decreased by augmenting a molecular component to correct disequilibrium in the pathway. Where inflammatory reactions have been induced by a genetically altered protein, another possible approach is to suppress the inflammation directly. Depending on the nature of the genodermatosis, the implicated protein or even on the particular mutation, to correct the consequences or the genetic defect, may require a highly personalised stratagem. Repurposed drugs, can be used to bring about a “read through” strategy especially where the genetic defect induces premature termination codons. Sometimes the defective protein can be replaced by a normal functioning one. Cell therapies with allogeneic normal keratinocytes or fibroblasts may restore the integrity of diseased skin and allogeneic bone marrow or mesenchymal cells may additionally rescue other affected organs. Genetic engineering is expanding rapidly. The insertion of a normal functioning gene into cells of the recipient is since long explored. More recently, genome editing, allows reframing, insertion or deletion of exons or disruption of aberrantly functioning genes. There are now several examples where these stratagems are being explored in the (pre)clinical phase of therapeutic trial programmes. Another stratagem, designed to reduce the severity of a given disease involves the use of RNAi to attenuate expression of a harmful protein by decreasing abundance of the cognate transcript. Most of these strategies are short-lasting and will thus require intermittent life-long administration. In contrast, insertion of healthy copies of the relevant gene or editing the disease locus in the genome to correct harmful mutations in stem cells is more likely to induce a permanent cure. Here we discuss the potential advantages and drawbacks of applying these technologies in patients with these genetic conditions. Given the severity of many genodermatoses, prevention of transmission to future generations remains an important goal including offering reproductive choices, such as preimplantation genetic testing, which can allow selection of an unaffected embryo for transfer to the uterus.
Genodermatoses are rare monogenic diseases that affect less than 1/2000 people, which primarily manifest as skin abnormalities, yet they are commonly associated with systemic symptoms (Aşkın et al., 2020). Some conditions may induce neonatal mortality including Harlequin ichthyosis (HI), severe generalised junctional epidermolysis bullosa (EB), severe hypohidrotic ectodermal dysplasia (HED), Netherton syndrome (NS), as well as ankyloblepharon ectodermal defects cleft lip palate (AEC) syndrome. In many life expectancy is reduced or an enhanced cancer risk is present, as observed in xeroderma pigmentosum (XP), neurofibromatosis type1 (NF1), naevoid basal cell nevus syndrome or junctional (JEB), and dystrophic (DEB) EB. Most of these diseases also markedly affect patient quality of life (Aşkın et al., 2020). While treatment modalities are still limited, some of them evolve rapidly in regard to secondary inflammatory or oncologic consequences. At this moment, treatment options are for most of these diseases restricted to skin and wound care, comprising compounded topical preparations, surgery, symptomatic pain relief, treatment of itching, as well as treatment of complications (Bardhan et al., 2020; Mazereeuw-Hautier et al., 2019; Mazereeuw-Hautier et al., 2019; Bodemer et al., 2021; Lenders and Brand, 2021) like skin cancers (de Andrade et al., 2021). This is complemented with patient education designed to enhance disease understanding and therapy adherence, as well as to teach how to avoid triggering factors, including heat and sun exposure among others (de Andrade et al., 2021; Dufresne et al., 2013). Only very few systemic drugs are available and if so, undesirable effects must be taken into account, given that treatment may be required from birth or an early age on, as in the case of retinoid administration. The socio-economic burden of most diseases is very high. In a survey involving six European countries, the average annual costs for EB varied from country to country. These costs ranged from €9,509 to €49,233 in the reference year 2012, 18% of which were direct medical costs, 74,8% non-healthcare costs (non-healthcare transportation, social care services, and caregiver’s time), and 7.2% related to productivity losses (Angelis et al., 2016).
During the 21st century, a revolution took place that consisted in unravelling the molecular genetic background of genodermatoses, especially with the advent of high throughput sequencing techniques. This enabled the thorough investigation of underlying pathophysiological mechanisms, which in turn opened ways to new treatment modalities. This paper provides an overview of these new treatment strategies. In addition, we review the different ongoing efforts that seek to repair the causative genetic defects, yet without paying too much attention to technical features. Besides, we also present some treatment illustrations, some of which have already entered the clinical trial stage.
Over-activation of the involved pathway may be caused by a gain of function (GOF) of a main protein or, alternatively, by loss of function (LOF) of an inhibiting protein. To illustrate, Costello syndrome is caused by GOF mutations in the Harvey rat sarcoma viral oncogene homolog (HRAS) gene, whereas neurofibromatosis Type 1 is due to LOF mutations in NF1 gene (Walker and Upadhyaya, 2018). Both genetic variants result in an overactive mitogen-activated protein kinase (MAPK) pathway, commonly called RASopathies. Inhibiting this overactive pathway is thus a logical option, with similar drugs possibly used for different RASopathies. However, the overactive RAS proteins resulting from LOF variants in NF1 might activate several downstream pathways. This may be tissue-dependent, as hypothesised in NF-1; as a result, this requires targeting different manifestations, separately. Hence, the optimal treatment for neurofibromas may be different from those involving skeletal abnormalities or cognitive dysfunction (Walker and Upadhyaya, 2018). Fortunately, for many pathways, drugs have already been developed in the framework of other pathologies. Commonly, these are often anti-cancer drugs that may be repurposed for use in genodermatoses, provided that undesirable effects are acceptable.
A recent example is the orphan genodermatosis Olmsted syndrome. It is a rare form of painful mutilating palmoplantar keratoderma (PPK), presenting early in life with periorificial keratotic plaques, which are at times associated with alopecia, along with a risk to develop spinocellular carcinomas later in life. The cause in most patients is a heterozygous GOF mutation in transient receptor potential vanniloid-3 (TRPV3), which encodes Ca2+ permeable channels. The latter form a signalling complex with transforming growth factor-α (TGF-α) and epidermal growth factor receptor (EGFR). Over-activation of the channels stimulates TGF-α release, which in turn is a stimulating ligand for EGFR. This results in an increased channel activity, hence creating a positive feed-back loop. In a proof of concept study, three patients with activating TRPV3 mutations were treated using erlotinib, an EGFR-blocker (Greco et al., 2020). Within 3 months after initiating therapy, PPK had drastically improved and pain disappeared.
Another illustration is Gorlin syndrome, also referred to as nevoid basal cell carcinoma (BCC) syndrome, which is caused by LOF variants in the tumour suppressor patched-1 homolog gene (PTCH1), provoking release of smoothened (SMO) oncogene inhibition. This results in the subsequent re-activation of the Hedgehog-signalling pathway. Vismodegib, which is a SMO inhibitor, was able to reduce not only the tumour burden and induction of new BCC, but also the growth of jaw keratocystic odontogenic tumours in Gorlin patients (Booms et al., 2015).
Intervening within a pathway that is less active or even inactive appears to be more challenging. DNA-repair disorders are caused by a defective DNA-repair pathway that was initially aimed at restoring genomic damage. As a result, naturally occurring DNA lesions are rapidly neutralized. If this pathway is defective, this predisposes to cancer. Only very few therapeutic possibilities are available for these diseases (de Andrade et al., 2021) (Weon and Glass, 2019). Screening of a library comprising Food and Drug Administration (FDA)-approved drugs has shown that the sulfonylureas acetohexamide and glimepiride are able in vitro to enhance viability in XPA cells after UV radiation at rather high dosages (Mazouzi et al., 2017). The exact mechanism is not yet understood. Moreover, preliminary data suggest that nicotinamide or other SIRT-1 inhibitors could be of benefit for XPD/CS patients by decreasing not only UV damage but also preventing neurological degradation. Nevertheless, further studies are needed to confirm these encouraging results (Vélez-Cruz et al., 2013).
Erythropoietic protoporphyria is most often caused by a biallelic loss of function mutation in the gene encoding ferrochelatase, thereby abolishing normal haem production and resulting in the accumulation of protoporphyrin in erythroid cells. The accumulated phototoxic protoporphyrin in superficial vessels is activated by blue light. From early childhood on, severe neuropathic pain, followed by oedema and blistering develops within minutes after sun exposure, which obliges patients to avoid any sun exposure. A recent therapeutic approach is the stimulation of photoprotective eumelanin production by stimulating the melanocortin 1 receptor in melanocytes using its agonist afamelanotide. The latter is a potent analogue of human α-melanocyte-stimulating hormone. Clinically, this approach has been shown to enable symptom-free sun exposure, thereby improving patients quality of life (Langendonk et al., 2015).
The genetic defects underlying genodermatoses likely induce an inflammatory reaction cascade, which contributes to the phenotypic presentation.
The hereditary ichthyoses are a large group of various diseases presenting with hyperkeratosis, scaling, and mostly different degrees of inflammation that involves the entire skin. These conditions are manifest from birth on; in rare cases, they are associated with other organ involvements (Mazereeuw-Hautier et al., 2019). This entity is a diverse group, with more than 50 genes so far identified as the underlying cause. Phenotypically, these conditions present as ichthyosis vulgaris, recessive X-linked, epidermolytic, autosomal recessive congenital ichthyosis (ARCI), and NS, among others. To date, treatment is largely focussed on targeting hyperkeratosis with topical agents and retinoids (Mazereeuw-Hautier et al., 2019). Besides, anti-inflammatory treatments based on corticosteroids and calcineurin inhibitors are commonly employed so as to control inflammation (Mazereeuw-Hautier et al., 2019). More recently, biotherapies targeting the inflammatory process, including TNF-α inhibitors and omalizumab, have been used with some success (Fontao et al., 2011; Yalcin, 2016; Roda et al., 2017).
Surprisingly, a study evaluating the immune dysregulation in patients suffering from ichthyosis identified a large number of ichthyosis subtypes that share IL-23/Th17 skewing in the skin (Paller et al., 2017). This observation offered a more solid pathophysiological basis for new treatment targets. Ustekinumab is a human monoclonal antibody that binds to the p40 subunit common to both Il-12 and Il-23. This agent thus blocks the binding of these cytokines to their receptor, and it has been successfully applied in a patient suffering from erythrodermic ichthyosis caused by a biallelic pathogenic variant in NIPAL4 (Poulton et al., 2019). This agent has also been employed with success in a patient with NS (Volc et al., 2020), and in another patient exhibiting a biallelic mutation in desmoplakin that manifested as ichthyosis (Paller et al., 2018). Different case reports and small series have already been published focussing on Il-17 inhibitors such as sekukinumab (Blanchard and Prose, 2020; Luchsinger et al., 2020; Barbieux et al., 2021) in NS, although sustained improvement could not be obtained in one patient. Given that, after this treatment, a Th2 signature remained (Barbieux et al., 2021), using dupilumab, a blocker of the IL-4 receptor’s alpha-chain, thereby blocking Il-4 and Il-13 cytokines, could be another target for some NS patients, as it has already been tested with encouraging results (Andreasen et al., 2020; Steuer and Cohen, 2020; Süßmuth et al., 2021).
A recent paper identified potential therapeutic targets for Harlequin ichthyosis (HI), which is the most severe form of ichthyosis, and it is often associated with marked neonatal lethality. In an in vitro model, investigators discovered an upregulation of IL-36α and IL-36γ, as well as of STAT1 and its downstream target, consisting of inducible nitric oxide synthase (NOS2). Treatment using either a NOS2-inhibitor or the JAK inhibitor tofacitinib was proven able to restore the lipid barrier in the HI 3D-model (Enjalbert et al., 2020). Drugs blocking either Il-36 or downstream IL-23 and IL-17 would probably be an option that deserves to be considered. Il-36 cytokines are secreted by keratinocytes, representing a danger signal. Hence, we believe this cytokine to be a potential candidate, given that it is one of the earliest cytokines that are being secreted in response to barrier disruption, as seen in ichthyosis.
However, more large-scale studies are now needed, seeking to evaluate the effect of these different biologicals and identify the precise patient profile that would best respond to the different drugs. So far, several trials have already been initiated (see Table 1).
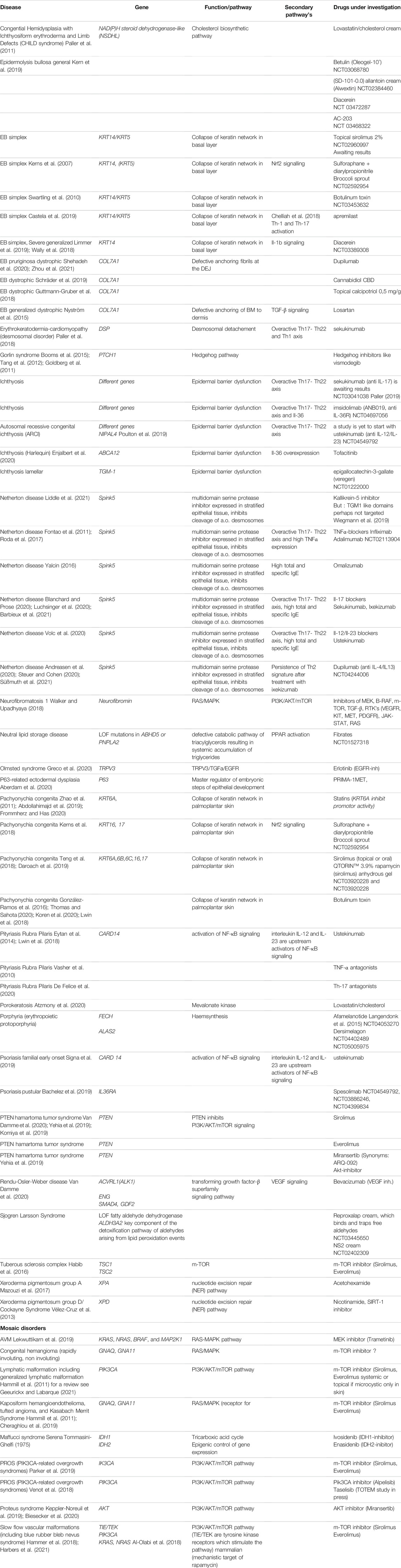
TABLE 1. New and repurposed drugs for different genodermatoses, based on pathophysiology (ongoing trials mentioned in clinicaltrials.gov with NCT).
Pustular psoriasis, due to a LOF mutation of Il36RN that encodes a receptor antagonist, is another disease candidate for IL-36 antagonists (Gooderham et al., 2019). Interestingly, improvement after one single injection was already observed in a proof-of-concept study involving spesolimab. This compound, which is an Il-36R blocker is investigated in a recent study (NCT02978690), however was, associated with mild to moderate undesirable effects, including infections, fever, or arthralgia, in all the seven patients that were tested. It will certainly be interesting to further assess this compound’s clinical effects and undesirable effects, as well (Bachelez et al., 2019), in larger-scale clinical trials (Choon et al., 2021), (Table1).
Familial pityriasis rubra pilaris (PRP) is commonly caused by heterozygous GOF mutations of CARD14 (Fuchs-Telem et al., 2012), which is a regulator of nuclear factor-κB (NF-κB-) signalling. As this pathway positively regulates Th17 differentiation, and in turn is regulated by TNF-α, indeed treatment with TNF-α inhibitors (Vasher et al., 2010), Th-17 inhibitors (De Felice et al., 2020) and IL12/IL23 inhibitors have been effective treatments (Eytan et al., 2014; Lwin et al., 2018). As the same CARD14 mutations have been associated with familial early-onset psoriasis (Craiglow et al., 2018), ustekinumab, namely an Il12/Il23-antagonist, may prove to be effective in these psoriasis patients (Signa et al., 2019).
However, other genodermatoses could also be candidates for targeting inflammatory mediators, provided that inflammation is actually part of their clinical manifestations. To illustrate, severe generalised EB simplex is a genodermatosis that manifests as skin blistering upon minimal trauma. This condition is caused by heterozygous KRT14 mutations, the pro-inflammatory cytokine Il-1 being strongly expressed. Diacerein is a down-regulator of the Il-1 signalling cascade, which is administered in a cream formulation. This agent has proven to be effective in reducing the number of blisters, without significant undesirable effects (Wally et al., 2018; Limmer et al., 2019). Diacerein is also under investigation for EB which is caused by mutations in other genes. AC-203 is another drug with similar properties, which is currently under investigation (Table 1). Targeting Th-17 cells, as well as TGF-β or Th-2 cytokines represent other therapeutic options (Nyström et al., 2015; Castela et al., 2019; Shehadeh et al., 2020; Zhou et al., 2021).
Table 1 provides a summary of genodermatoses, while also listing the new or repurposed drugs currently under investigation.
Nonsense mutations leading to premature termination codons (PTCs) and truncated inactive or decoyed proteins are estimated to be the cause of about 11% of all monogenic diseases. Repurposed drugs like aminoglycosides favour, during translation, mispairing of near-cognate tRNA at the place of the premature termination, resulting in the incorporation of a different amino-acid into the translated protein. Usually, this transforms a nonsense mutation into a missense mutation, thereby restoring the full-length protein (Figure 1) (Nagel-Wolfrum et al., 2016).
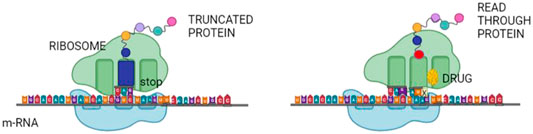
FIGURE 1. Read through therapy. Nonsense mutations in various genes may be repaired by read-through therapy. Small molecules, known as TR-inducing drugs (TRIDs), enable the translation machinery to suppress a nonsense codon, which would stop further translation of the protein. It allows for the synthesis of a full-length protein with no or only minor disruption of the synthesized protein.
In genodermatoses, topical gentamycin administration on skin lesions in Hailey-Hailey disease (HHD) (Kellermayer et al., 2006), hereditary hypotrichosis simplex (Peled et al., 2020), and PPK caused by SERPINB7 mutations (Ohguchi et al., 2018), as well as topical or intradermal (ID) gentamycin administration in dystrophic EB (Woodley et al., 2017) have already been tested, yet with variable results. Moreover, in vitro tests in JEB cells were demonstrated to restore expression of laminin 332, (Lincoln et al., 2018), and of XPC in XP cells (Kuschal et al., 2015).
More studies are presently ongoing including topical application GENTELBULL (NCT04644627), with topical BPM 31510 cream (NCT02793960) in EB; intravenous (IV) administration in both JEB (NCT03526159) and DEB (NCT03392909). Trial NCT03012191 in DEB has been completed, with preliminary results showing some C7 expression in exclusively the skin in IV- or ID-treated patients, yet not in topically-treated patients.
Although these readthrough drugs look promising and could be applied in numerous genetic diseases, currently available compounds exhibit weak (less than 5%) activity in vitro, and in only a fraction of tested patients. Moreover, the oto- and nephrotoxicity of aminoglycosides is a concern in the event of long-term administration, as is the risk of antibiotic resistance if these drugs are topically applied on chronic wounds, such as in EB. There is an ongoing search for new compounds with a good safety profile or for drug combinations [for a review see (Baradaran-Heravi et al., 2016; Nagel-Wolfrum et al., 2016)]. A recent study investigated amlexanox, which has proven to be effective in cultured fibroblasts and keratinocytes from dystrophic EB patients (Atanasova et al., 2017).
Replacement of the protein encoded by the mutated gene is currently under investigation in different genodermatoses (Figure 2A). A proof of concept study using a mice model for dystrophic EB, based on missing Type VII collagen (C7) anchoring fibrils, has demonstrated that injecting normal C7 is indeed able to form anchoring fibrils at the dermo-epidermal junction (DEJ). This ability was demonstrated not only in the skin but also in the oesophagus, lasting for several weeks and preventing the mice from dying (Woodley et al., 2013; Hou et al., 2015).
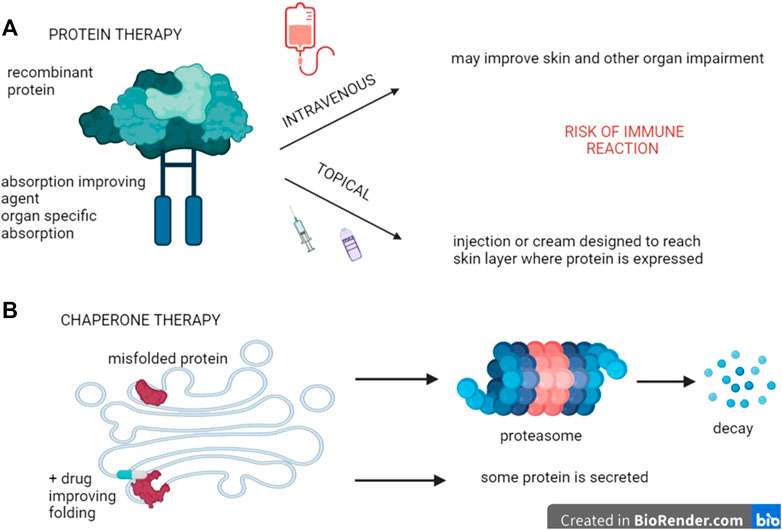
FIGURE 2. Protein therapy. (A) Recombinant proteins can be used to replace dysfunctional ones, provided they are delivered at the site where they are expressed. In genodermatoses, topical application constitutes a possibility that may consist of creams with penetration-improved formulations, intradermal injections, or even intravenous applications if the latter enable the active principle to reach their target environment. (B) In some cases, mutations code for proteins that are misfolded, thereby remaining in the endoplasmatic reticulum. Drugs called chaperones that improve this folding are thus likely to circumvent this problem and restore protein activity to some extent.
Presently, phase 1 and 2 studies in humans are ongoing, administering IV recombinant C7 in recessive DEB (PTR-01) (NCT03752905 and NCT04599881). Immunogenicity does not appear to be a big issue. However, their size and tendency to form aggregates likely limit skin and other tissue homing. The drug has orphan designation.
Similar ongoing studies are focused on transglutaminase-1 (TGM-1)-deficient ARCI. In a skin humanized mouse model, topical application of liposomes containing recombinant human TG1 (rh-TG1) resulted in considerable improvement in ichthyosis phenotype, as well as normalization of the regenerated ARCI skin (Aufenvenne et al., 2013). More recently, such positive outcome was confirmed in a skin equivalent model using thermo-responsive nanogels (tNG) encapsulating TG1 (Witting et al., 2015; Plank et al., 2019).
The timing of protein replacement appears essential, as illustrated in hypohidrotic ectodermal dysplasia (HED), which is a disease that affects several ectodermal structures. Sweat glands, teeth development, and meibomian glands are severely impaired in this condition, with distinctive facial features usually present, as well as sparse blond hair. The condition’s most common cause is a recessive X-linked mutation in ectodysplasin A (EDA) (Schneider et al., 2018). The inability to sweat may be life-threatening due to hyperthermia.
Fc-EDA, which is a fusion protein linking the constant domain of IgG1 and the receptor-binding portion of EDA, was administered prenatally via amniocentesis into the amniotic cavities of three foetuses of two different pregnancies, namely at Week 26 for one and for the twins at Week 26 and Week 31. Sweat glands, meibomian glands, and tooth germs almost normally developed in the new-borns, being more numerous than in their affected older brothers (Schneider et al., 2018). However, when this Fc-EDA was administered to a baby at birth, no effect was observed, which further underlines that administration must be performed when the pathway is active, meaning between Weeks 20–30 during foetal development for sweat glands. Nevertheless, a larger study with EDI200 administered immediately after birth has been completed, while still awaiting results (NCT01775462).
For genetic diseases caused by retention of the defective protein within the endoplasmatic reticulum, due to misfolding and subsequent absence of transport to the Golgi apparatus, chaperone therapy is under investigation, mostly based on low concentrations of competitive inhibitors. By binding to misfolded enzymes and forming stable complexes instead, the transport and further processing into the Golgi apparatus is being restored. As a result, some (variable) enzymatic activity and partial phenotype rescue is rendered possible (Figure 2B). This technique is under investigation for storage diseases like Gaucher and Fabry disease and also in oculo-cutaneous albinism Type A (OCA1A), (Table 2) (Lenders and Brand, 2021; Teramae et al., 2019), while it may also be taken into consideration for keratinopathies (Chamcheu et al., 2011).
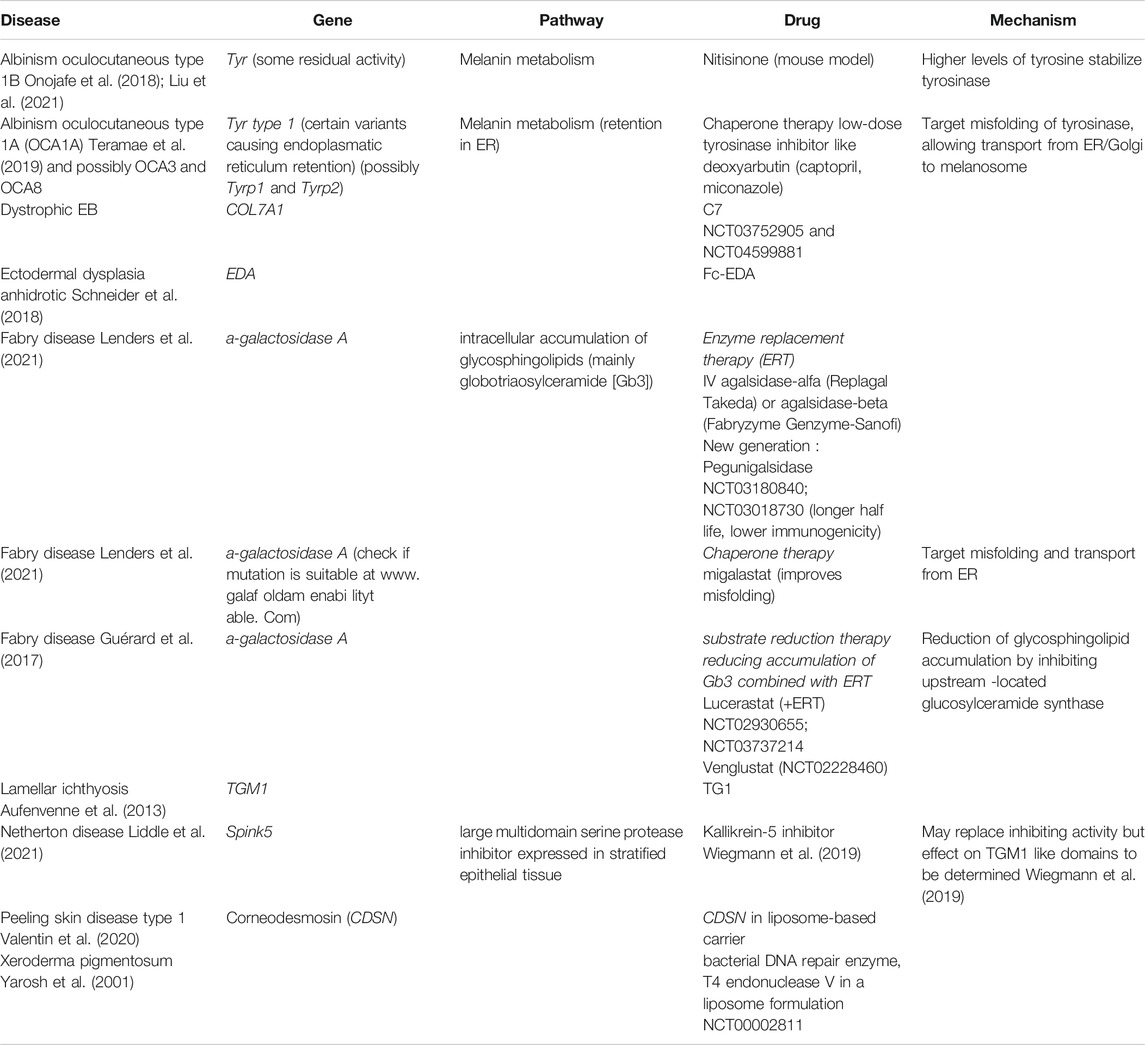
TABLE 2. Treatments under investigation aiming to restore protein expression (ongoing trials mentioned in clinicaltrials.gov with NCT)
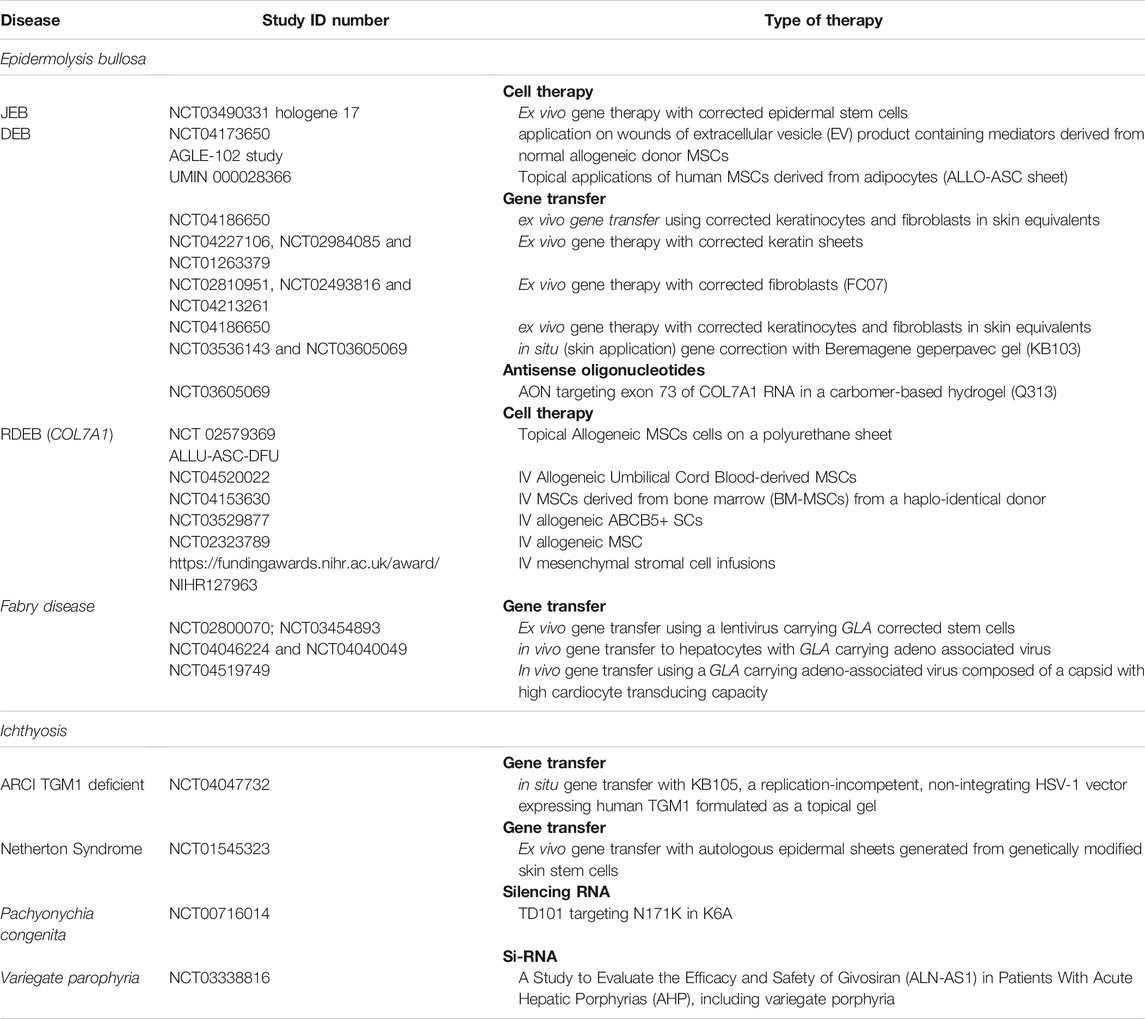
TABLE 3. Cell and gene therapies under investigation for genodermatoses mentioned in clinicaltrials.gov with NCT.
Both enzyme replacement and chaperone therapy are similarly being investigated for the treatment of porphyrias. Nevertheless, the major drawback of these approaches consists in their delivery to the targeted organ (Bustad et al., 2021).
Restoring the defective protein by administrating allogeneic cells is another approach in managing genodermatoses (Figure 3). Collagen 7 (C7) is produced by keratinocytes and fibroblasts. Early experiments showed that IV injection of human fibroblasts in athymic mice resulted in the deposition of human C7 that formed anchoring fibrils at the DEJ, yet which occurred in wounded but not in normal mice skin (Woodley et al., 2007). Subsequently, three studies conducted in humans demonstrated ID injection of allogeneic fibroblasts to fasten the healing of wounds in most tested DEB patients (Petrof et al., 2013; Venugopal et al., 2013; Moravvej et al., 2018). Nevertheless, no difference in healing time was observed in comparison with the lesions injected with the vehicle containing albumin and growth factors (Venugopal et al., 2013). Interestingly, the patients who failed in these three studies were unable to express own C7, while the anchoring fibrils seen in the biopsies were rudimentary (Wong et al., 2008). Despite the allogeneic fibroblasts disappearing after 2 weeks, the clinical effect of one single injection lasted 1 month in one study (Petrof et al., 2013), and up to 3 months in another (Moravvej et al., 2018), with C7 expression sustained over 9 months. In contrast with the animal studies, this clearly suggests that the major effect probably consisted of an increase in recipients COL7A1 mRNA and mutant C7 levels, which was induced by paracrine mediators secreted by the allogeneic fibroblasts (Wong et al., 2008), such as heparin-binding epidermal growth factor-like growth factor (HB-EGF) (Nagy et al., 2011).
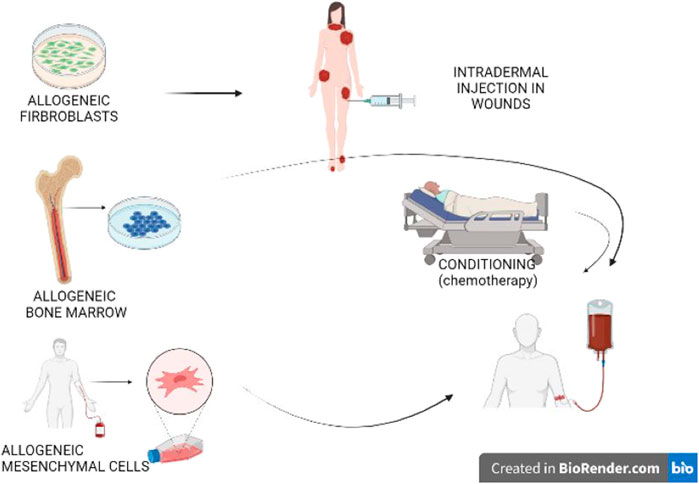
FIGURE 3. Cell therapy. Allogeneic cells secreting the proteins that are dysfunctional within the patient may improve disease severity. Fibroblasts injected in chronic wounds in dystrophic epidermolysis bullosa are currently under investigation. Bone marrow transplantations improve wound healing and systemic symptoms; nevertheless, this technique requires pre-transplantation conditioning, which is associated with a risk of undesirable effects and even mortality. More recent is the use of allogeneic mesenchymal stem cells, which does not require conditioning. Immune reactions are less common when fibroblasts or mesenchymal stem cells are employed, in comparison with hematopoietic stem cells. These reactions are more common in the event of mutations without spontaneous protein expression in the patient.
Allogeneic bone marrow (BM) transplantation is a technique that was primarily designed to treat hematopoietic malignancies. Later on, this technique was found to be effective in reversing the phenotype of some genetic diseases, primarily comprising primary immune-deficiencies. However, this same approach has also been shown to attenuate the muco-cutaneous manifestations and improve the quality of life of severe JEB and DEB (Wagner et al., 2010). By reducing the intensity of the conditioning regimen, essential undesirable effects and mortality could be reduced (Tolar and Wagner, 2013). Further research allowed for unravelling the potential underlying mechanism (Tamai and Uitto, 2016), thereby refining the technique. Release of high mobility group box 1 protein (HMGB1) from necrotic epithelial cells does transfer signals to bone marrow-derived PDGFRa+/CXCR4+ cells. This leads to their migration into circulation, along with subsequent homing to the damaged skin through stromal cell-derived factor 1a (SDF-1a)/CXCR4 axis (Tamai and Uitto, 2016). The most essential action of MSCs is likely the secretion of anti-inflammatory agents, whereas they may also differentiate into fibroblasts and keratinocytes. Transplanted patients are able to receive skin grafts from the same donor, which survive for long periods and outgrow into adjacent wounds, thereby suggesting immune tolerance and outgrowth of stem cells (Ebens et al., 2020).
Three open clinical trials have been conducted to date, involving subjects with RDEB using MSCs from healthy donors. Of these, two were carried out in children (Petrof et al., 2015; El-Darouti et al., 2016) and one in adults (Rashidghamat et al., 2020). In these trials two or three IV injections were administered over a 1-month period, without any HLA matching or preconditioning. Effects on wound healing, pain, and especially pruritus seemed to be rather promising, with only mild undesirable effects observed. The latter were mostly transient, and all were unrelated to MSCs. A major concern, however, has been the development of squamous cell carcinomas (SCC) in two out of 10 adult participants during the study period (Rashidghamat et al., 2020). Nevertheless, the causality of this serious event is still being debated, given that SCCs represent a naturally occurring complication of RDEB. No increase in C7 or anchoring fibrils at the DEJ could be detected in skin biopsies. The improvement in adhesion has been attributed to either an increased expression of other junctional adhesion proteins or a reduction in inflammatory mediators that impair DEJ adhesion. Optimizing the number and frequency of MSC infusions, while addressing specific MSC subpopulations like skin homing ATP-binding or cassette subfamily B member 5 positive (ABCB5) MSC (Riedl et al., 2021) are currently being studied and designed to improve the outcome results. Injection of MSC-derived anti-inflammatory products carried in exosomes (Perdoni et al., 2014; Shabbir et al., 2015; McBride et al., 2018) is similarly under investigation, as are HMGB1 and SDF1 able to attract MSC.
In principle, all genodermatoses are candidates for gene therapy (Ain et al., 2021).
The major obstacle to this process is the efficient, safe, and targeted delivery of the gene (Ain et al., 2021; Bustad et al., 2021). Non-integrating viral vectors like adenoviruses were the first to be applied and have so far been the most widely used. Nevertheless, they carry a restricted cargo potential, which may be a limiting factor for large genes such as NF1 (Walker and Upadhyaya, 2018). Besides, herpesviruses are commonly chosen for in vivo transfection. Retroviruses and the subgenus lentiviruses, as well, have the capacity to integrate into the genome, thereby allowing for stable and long-term gene expression, yet with a risk of being mutagenic at the insertion site.
Alternatives to viruses are lipid based nanoparticles (LNPs) and polymeric nanoparticles. These have the advantage of being able to overcome proteolytic degradation in the skin, while they are devoid of mutagenic risk and permit the transport of larger fragments. Moreover, electroporation, sonoporation, iontophoresis, and microneedles are physical methods that may be applied so as to deliver products into the cells (Ain et al., 2021).
Gene administration may be ex vivo, involving cells harvested from the patient. Gene administration can also occur in vivo while delivering the vector carrying the correct gene to the patient via transfusion. In the last case scenario, the vector must be designed with a specific tropism for the target organ. Another means of administration consist of in situ injection into tissues or topical application, such as on the skin (Papanikolaou and Bosio, 2021).
Gene transfer refers to the process of inserting the correct gene into the cells, in addition to the defective gene, thereby augmenting the alleles for the respective gene. This technique was first used for PID’s (Figure 4). If stem cells are transfected, there is the possibility of life long gene insertion, especially after transfecting with viruses that integrate into the genome.
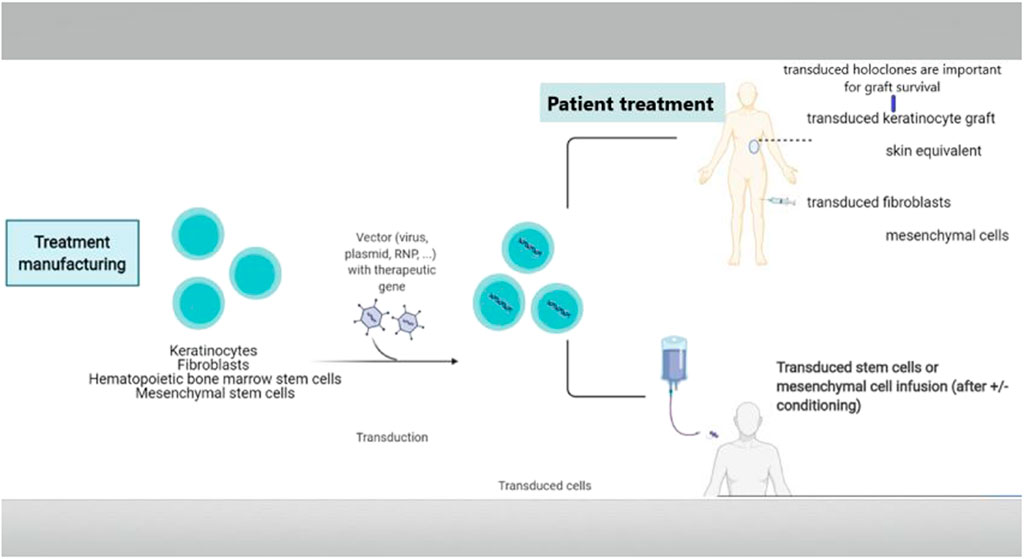
FIGURE 4. Gene replacement therapy. Delivering the correct gene to the patient is called gene augmentation. A vector is used to transduce a recombinant gene into the patient’s cells, including keratinocytes, fibroblasts, hematopoietic stem cells, mesenchymal stem cells, or iPSC. Usually, this is carried out in vitro, which is then followed by transplantation of the transduced cells. Moreover, several vectors even allow for in vivo transduction.
The first genodermatosis studies were carried out ex vivo in TGM1-deficient ARCI keratinocytes (Choate et al., 1996). Later on, an ex vivo investigation was conducted using corrected LAMB3-deficient keratinocytes that originated from a patient suffering from JEB. This was followed by skin grafting over a small area (Mavilio et al., 2006), resulting in long-term graft survival (De Rosa et al., 2014). However, the real breakthrough came up with a JEB patient for whom 80% of the body surface were treated following ex vivo gene transfer to keratinocytes (Hirsch et al., 2017). Cultured autologous keratinocytes were transfected with retroviruses expressing full-length LAMB3. These keratinocytes were subsequently expanded in order to obtain large sheets of keratinocytes. They were then used to graft about 80% of the patient’s body surface (Hirsch et al., 2017). After 21 months, the skin displayed a normal appearance. It was demonstrated that holoclones (colony-forming stem cells with a higher growth potential than a mero- and paraclone, not containing differentiated cells) turned out to be the progenitor cells that were responsible for sustained skin regeneration, as the integration patterns of mero- and paraclones over time resemble more and more those of holoclones.
Important concerns for gene insertion, however, are vector-induced off target effects and malignancy risk, in addition to viral packaging capacity and immune reactions, especially in patients without any protein expression. Therefore, highly branched polyβ‐amino esters (HPAE) appear to be promising alternatives, designed to deliver full length COL7A1 in the form of polyplexes like AP103, which has been granted orphan drug status for DEB treatment.
Similar projects have already been initiated for other conditions, including NS (Di et al., 2011) (Di et al., 2019) (for a review see (Ain et al., 2021) (Jayarajan et al., 2021)), porphyrias (Bustad et al., 2021), and DEB (Eichstadt et al., 2019). For the latter, experiments in engineered epidermal-dermal skin substitutes suggest that expression of the COL7A1 gene in both keratinocytes and fibroblasts is most likely necessary so as to produce structurally normal anchoring fibrils. (Supp et al., 2019). This is presently further evaluated in clinical trials.
Genome editing is a recent type of genetic engineering, which is still largely in the preclinical phase. With this new technology, DNA is inserted, deleted, modified, or replaced in the genome of a living organism (Figure 5), thereby enabling more precise interventions. Except for base editors that are treated later in the paper, gene editing consists of carrying out double-strand breaks using nucleases including zinc finger nucleases (ZFNs), mega-nucleases, transcription-activator like effector nucleases (TALEN), as well as the most recently introduced clustered regularly interspaced short palindromic repeats (CRISPR/Cas9 and his mutant CRISPR/Cas9 variant). These double-strand brakes (DSB) will likely be repaired by taking advantage of the cell’s intrinsic repair pathways, using either the fast, yet error prone non-homologous end joining (NHEJ), or the homology directed repair (HDR). While the latter appears to be less efficient, it is certainly more precise, enabling the precise incorporation of desired sequence alterations. These nucleases must be designed in a way that enables them to precisely target the DNA sequence (or RNA) designed to be altered, which is carried out with tandem arrays of 33–35 amino acid repeats that either bind a specific nucleotide in TALEN or a guide RNA complementary to the targeted DNA in CRISPR [for a review see (March et al., 2018)]. A vector is required to enable the tools to enter into the cells. The choice of therapeutic strategy largely depends on the mutation type to be corrected [for a review see (March et al., 2020)].
FIGURE 5. Gene editing. Gene editing offers diverse possibilities adapted to the genetic defect to be corrected. A vector is then required to enter the tools into the cells. The first step is to carry out a double strand break using nucleases. These breaks will be repaired by the error prone non-homologous end joining (NHEJ) with loss of a small number of nucleotides, which may result in non-expression of the aberrant protein. This option represents a therapeutic opportunity for diseases where haploinsufficiency carries an advantage in the case of over-expression of structurally aberrant proteins, which occurs in most keratinopathies. It may also have the potential of reframing in the case of indels or of deleting a codon or exon with the mutation. Repair by the homology directed repair (HDR) pathway enables incorporating the desired sequence alterations based on a donor DNA template with a normal sequence.
of pathogenic alleles can be obtained by generating a single DSB, which is then followed by error-prone NHEJ-based repair, thereby inducing indels into the target gene. This frequently provokes frameshifts within the target allele, likely to induce premature termination codons (PTCs) followed by nonsense mediated m-RNA decay (NMD), without any pathogenic protein formation.
This technique is specially suitable for managing dominant-negative genetic variants causing skin diseases (March et al., 2020), which is the case for most keratinopathies, in which the phenotype expression results from the abnormal protein produced via a modified allele, yet not from a protein reduction, should the allele be haplo-insufficient.
Two proof-of-concept studies based on a similar, non-allele-specific protocol, were developed. The first was focused in EB simplex with mutant KRT5, targeting exon 1 of wild type and mutant KRT5 alleles (Aushev et al., 2017); the second concerned epidermolytic ichthyosis (EI), using a TALEN nuclease, designed to target a region of KRT10, upstream of a PTC known to induce a genetic knockout (March et al., 2019). Nevertheless, this technique required that, after editing, a selection of those keratinocyte clones where only the mutated allele was disrupted was performed, these may then be further expanded in culture. Both studies obtained high gene disruption percentages both in immortalized (Aushev et al., 2017; March et al., 2019) and in mutant keratinocytes from patients (March et al., 2019). Due to the selection required, this technique is only suitable for ex vivo procedures.
In addition, two mutation-specific targeting studies were conducted. Of these, one was an in vivo study conducted in KRT9 mutant transgenic mice using CRISPR-Cas9, which turned out to be successful (Luan et al., 2018). Indeed, KRT9 mutations are involved in epidermolytic PPK. A second study was conducted ex vivo in dominant DEB. Induced pluripotent stem cells (iPSCs) were successfully transduced, with loss of expression of the aberrant C7 obtained (Shinkuma et al., 2016). To be effective, a careful design is needed, which should seek complete destruction of the affected allele, thereby resulting in a complete absence of the aberrant protein.
Restoration of the reading frame may constitute a solution for frameshift inducing variants, provided that the amino-acid divergence induced by the correction results in a semi-functional protein. In this case, the same technique as for disruption may be applied, given that approximately one-third of all nuclease-induced indels lead to reading frame restoration. This approach appears useful for recessive mutations where expression of slightly modified protein variants replacing null variants is likely to provide some therapeutic alleviation. Preclinical studies have been conducted in the setting of recessive DEB. Their results demonstrated that the c.6527insC mutation of COL7A1, which is rather common in Spanish patients, appears suited for exon reframing (Chamorro et al., 2016; Mencía et al., 2018) as well as the c.5819delC, (Takashima et al., 2019).
Dual CRISPR/Cas9 targeting (with CRISPR/Cas9 sgRNA) can be used for highly efficient excision of the intervening sequence. Nevertheless, this technology carries a high risk for off target damage. Hence, an extremely careful design is required, thereby enabling this technology to exclusively target the desired DNA sequence comprising the pathogenic variant. Nevertheless, this technology appears particularly promising, especially if hotspot mutations are present in a particular exon. Indeed, this provokes protein decay and, if skipping of the exon gives rise to a functional shortened protein, this results in a less severe clinical phenotype. Preclinical studies deleting exon 80 have been carried out in vivo in COL7A1 c.6485G > A mut/mut mice (Wu et al., 2017) and in keratinocytes of Spanish patients with the recurrent c.6527insC as well (Bonafont et al., 2019). Exon skipping with antisense oligonucleotides (see next session) however is already in a more advanced developmental stage, whereas this technology is not associated with the possibility to result in permanent recovery.
Exon/gene insertion approaches utilize HDR machinery, requiring a repair template that harbours left and right homology arms, which bind with the DNA fragment surrounding the insertion place, so as to enable precise insertion of large correct DNA fragments. In comparison with gene augmentation, in which incorporation of receptor DNA occurs at random, these techniques carry the advantage of abrogating the risk of insertional mutagenesis and transgene expression. Because HDR is only active during late S/G2 replication phase, this technique appears to be poorly efficient, thus requiring selection and cultivation of correctly repaired cells afterwards. However, in theory, there is no restriction on either disease or mutation.
Preclinical studies have been performed for DEB (Sebastiano et al., 2014; Osborn et al., 2018), but in JEB as well (Benati et al., 2018).
Directly reverting a disease-causing variant in a gene via a single-nucleotide change may be reached using homologous recombination. This is a personalized, mutation specific repair, based on a repair template during HDR, but without integrating the whole exon. The template can serve for different mutations that cover the complementary template.
This technique has been evaluated in XP fibroblast cell lines carrying a homozygous deletion in exon 9 of the XPC gene (Dupuy et al., 2013). Although a correction in only 2.5% of the cells was attained, the investigators suggested this may be sufficient for clinical efficacy. Other preclinical studies demonstrating the feasibility of this technique were conducted in recessive DEB (Izmiryan et al., 2016; Izmiryan et al., 2018; Kocher et al., 2019), along with another study demonstrating its feasibility in EBS (Kocher et al., 2017).
Base editing is a new technology that enables the direct, irreversible conversion of a specific DNA base into another at a targeted genomic locus. Current base editors contain a catalytically impaired CRISPR–Cas nuclease (that cannot make DSBs), serving to guide the binding, which is fused to a single-stranded DNA deaminase enzyme and, in some cases, to proteins that manipulate the DNA repair machinery (Anzalone et al., 2020). Hence, this technique does not rely on double-stranded DNA breaks. Instead, this approach uses enzymes designed to precisely rearrange some of the atoms in one of the four bases that structure either DNA or RNA. Two main classes of base editors have been developed to date including cytosine base editors (CBEs), which catalyze the conversion of C•G base pairs into T•A base pairs, in addition to adenine base editors (ABEs), which catalyze A•T–G•C conversions. Upon Cas binding, hybridization of the guide RNA spacer to the target DNA strand causes displacement of the PAM-containing genomic DNA strand to form a ssDNA R-loop (Anzalone et al., 2020), while within this loop, bases can be edited. A first proof of concept study was conducted in RDEB, providing promising results (Osborn et al., 2020). The technique’s advantage is that it is template independent, giving rise to fewer indels, while enabling high correction efficacy. However, there is a risk for off target damage. As a result, this technique is only suitable for point mutations, provided they can be targeted using CRISPR-Casp. Base editing is suitable not only for correcting single base mutations but also for gene disruption (March et al., 2020).
Nature sometimes corrects a disease phenotype, and this phenomenon is called revertant mosaicism. It is not rare and well known in plant biology. In humans, this phenomenon appears to be common in self-regenerating organs like skin, blood, and liver, being particularly frequent in certain diseases. Concerning the skin, this condition was first described in JEB, presenting as patches of normal texture and tan, which never blister (Jonkman et al., 1997), with at least 35% of JEB carrying revertant patches (Jonkman and Pasmooij, 2012). Subsequently, this entity has been detected in dystrophic and simplex EB, Kindler syndrome, ichthyosis with confetti (IWC), keratitis-ichthyosis-deafness syndrome, dyskeratosis congenita, Bloom syndrome [reviewed in Jonkman and Pasmooij (2012), Pasmooij et al. (2012a)], and loricrin keratoderma, as well (Suzuki et al., 2019). In addition, the condition is commonly found in primary immunodeficiency disorders like Wiskott-Aldrich syndrome (Davis et al., 2010). The mechanisms underlying revertant mosaicism appear to be rather diverse, including gene conversion, intragenic crossover, mitotic recombination, back mutation, and second-site mutations (such as indels) (Pasmooij et al., 2012a). Ichthyosis with confetti (IWC) is caused by frameshift mutations, affecting the tail domain of the affected protein, either K10 or K1. The characteristic confetti lesions are caused by loss of heterozygosity caused by mitotic recombination (Guerra et al., 2015). Hence, there are mutations that promote reversion. In DEB and JEB patients with different small normal patches, genetic analysis revealed distinct gene conversion events (Pasmooij et al., 2005; Pasmooij et al., 2012b). Given this scenario, corrected cells would exhibit a proliferation advantage, leading locally to complete or partial phenotype reversion (Lai-Cheong et al., 2011). The triggers inducing these second hits are not yet well understood, with Sun exposure suggested for IWC. This spontaneous occurring phenomenon offers excellent therapeutic opportunities if these revertant cells expand in vivo, which is referred to as natural gene therapy. The technique’s advantages are its lack of immunogenicity and rejection risk; in addition, it does not require expensive genetic engineering techniques that are associated with a genotoxicity risk. Punch grafts (Gostyński et al., 2014) or cultured epidermal autografts (Matsumura et al., 2019) from a donor site with reverted skin onto wounds would, indeed, be the simplest method. Their survival on long-term likely depends on the presence of a sufficient number of reverted cells within the culture and, as learned from the skin graft trial using transgenic autologous stem cells in JEB (Hirsch et al., 2017), as well, on the presence of a sufficient number of holoclone stem cells, which were not seen in most biopsies taken. Reprogramming different cell types into iPSCs (Tolar et al., 2014) offers further opportunities. Tolar et al. (Tolar et al., 2014) succeeded in reprogramming revertant keratinocytes into iPSC and letting them proliferate. Subsequently, they succeeded in deriving these iPSC into hematopoietic cells, as well as epidermis-like keratinocyte layers, with C7 deposition. This outcome does open novel possibilities as these revertant hematopoietic cells could be employed for autologous HCT, without any need for pre-transplant conditioning, and with less toxicity. Moreover, as the iPSC are an inexhaustible source of keratinocytes, this technology offers tremendous possibilities in the context of local wound healing in patients carrying spontaneous revertant patches (Figure 6).
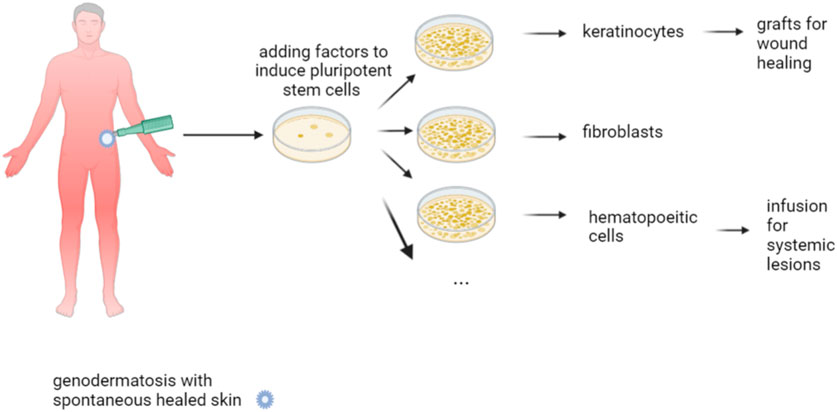
FIGURE 6. Natural gene therapy. Natural gene therapy is based on the appearance of normal skin within genetic aberrant one. This is caused by newly occurring mutations within genetically aberrant skin, which correct the defect, at least to some extent. This phenomenon of revertant mosaicism can be exploited for therapy by grafting this revertant skin into wounds. Reprogramming the revertant keratinocytes into iPSC and proliferation in culture do offer an inexhaustible source of cells, which can then be converted into keratinocytes or hematopoietic cells that are either transplanted or transfused into the patient.
Different techniques are being developed using exogenously-administered RNA-molecules or manipulating endogenous mRNA through editing or readthrough [for a review see (Bornert et al., 2017)]. It is an alternative technique for genome editing aimed at the same type of restoration on protein level and applicable in vivo. Given that the changes are performed at the RNA level, they are, however, not long-lasting.
Small interfering RNAs (si-RNA) are small double-stranded RNAs, which target mRNA for degradation and abolishing protein production (knock-down), by using the endogenous RNA silencing complex (RISC) (Uitto et al., 2012; Bornert et al., 2017). It constitutes an alternative to gene disruption using editing techniques. To date, most research has been conducted in the field of keratinopathies. In pachyonychia congenita gene disruption was proven feasible using a mutation specific design, while destroying the mutated allele (Leachman et al., 2010). The injected lesions displayed less hyperkeratosis, however the pain the injections caused definitely restricted its use. To overcome limitations of mutation specificity, another approach consisting of eliminating both mutated and wild type mRNA has also been considered. This approach can be used if keratins are interchangeable, such as the three paralogs KRT6A, KRT6B, and KRT6C encoding the three K6 isomorphs, including K6a, K6b, and K6c, all of which are expressed in palmoplantar skin (Leachman et al., 2010). Recently, a study has been completed, involving three patients with a mutation in KRT6A targeted by the si-RNA TD101 (NCT00716014). This knock-down strategy is also under investigation for dominant dystrophic EB (Pendaries et al., 2012). A si-RNA, targeting the enzyme aminolevulinic acid synthase 1 (Givosiran), which catalyses the formation of delta-aminolevulinic acid, is also currently available. This is meant to prevent repetitive acute abdominal pain attacks occurring in certain porphyria subtypes, including some cutaneous forms like variegate porphyria, where acute abdominal pain is a presenting sign. The common mechanism is the accumulation of haem synthesis intermediates, mainly comprising delta-aminolaevulinic acid (Bustad et al., 2021).
Antisense oligonucleotides (AONs) are presently considered an attractive class of compounds. This is a personalized treatment, given that the oligonucleotides are designed to target specific disease-causing mutations, if localized on in frame exons. AONs hybridize during pre-RNA splicing, while hiding it, which results in skipping of the exon with the mutation (Bornert et al., 2017). An internally deleted protein is thereby produced, which is aimed to be still functional, hence applicable for diseases where the presence of a shorter protein provokes a less severe phenotype, as compared to its (partially) absence (Figure 7). Exon skipping is a technique that reached clinical application in Duchennes muscular dystrophy. Pre-clinial studies have so far been conducted in DEB, given that the COL7A1 gene is considered to be a good candidate, owing to its large size, counting 117 exons. Indeed, most of its exons are rather short and primarily in frame, enabling them to be removed without disturbing the open reading frame and with conservation of most of the protein. Moreover, a study focusing on clinical and molecular DEB patient data revealed that recessive variants in exons that are affected by exon skipping, as part of natural low level alternative splicing events, generally represent relatively mild phenotypes within the clinical RDEB spectrum, whereas for dominant pathogenic variants in exons undergoing natural exon skipping, it seems to make less difference (Bremer et al., 2019). In DEB models, it has been proven for exons 13, 70, 73, 80, and 105 that skipping of one exon allows for the formation of a slightly shorter, still functional protein that should improve the phenotype (Goto et al., 2006; Bornert et al., 2016; Bremer et al., 2016; Bornert et al., 2021). A first study using an AON targeting exon 73 in a carbomer-based hydrogel (Q313) in both dominant and recessive DEB is actually recruiting patients (NCT03605069). Systemic administration, as practiced in Duchennes muscular dystrophy, would offer the possibility to additionally treat extra-cutaneous symptoms, provided such application is tolerated on the long-term (Bremer et al., 2016). Exon skipping is also under investigation in the settings of COL17 A1 mutations causing junctional EB (Condrat et al., 2018). Proof of its feasibility is the spontaneous occurrence of areas of normal skin in a JEB patient being a carrier of compound heterozygous (a frameshift and a nonsense) pathogenic variants in COL17 A1. This “normal” skin showed an additional splice mutation, resulting in skipping of the exon and formation of a truncated, yet functioning, protein (Kowalewski et al., 2016). As AONs are rather short-lived, repeat treatments are likely necessary, whereas there are still some concerns with respect to long-term safety (Hilhorst et al., 2019; Nguyen et al., 2019).
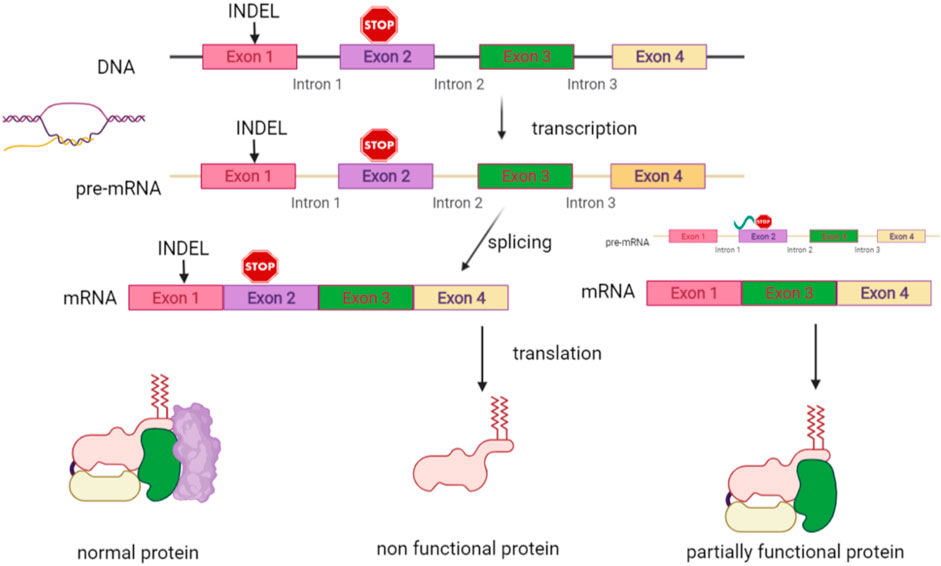
FIGURE 7. RNA-based therapies. Interference is additionally possible at RNA level. Antisense oligonucleotides (AONs) are designed to hybridize during pre-RNA splicing to the region of the exon with a mutation. This results in skipping of this exon and production of an internally deleted protein, which is designed to be still functional. It is applicable to diseases where the presence of a shorter protein provokes a less severe phenotype than in the case of its totally or partially absence.
Mutations creating a new splice site may likewise be a good target for AONs, especially if located deeply intronic, enabling specific splicing without off target damage. These mutations prevent the splicing machinery to recognize the cryptic splice site and, as demonstrated in a preclinical study in NF1, enable restoring normal splicing (Pros et al., 2009). An AON targeting the hypomorphic FECH polymorphism IVS3-48C > T, which redirects the cryptic to the physiologic splice site, is currently under investigation for EPP (Erwin and Balwani, 2021).
Spliceosome-mediated RNA trans-splicing (SMaRT) is a technique replacing the pre-mRNA fragment, containing the disease-causing variant, thereby resulting in a hybrid full-length wild-type mRNA (Bornert et al., 2017). Using the cell’s splicing machinery, an exogenous piece of mRNA replaces the fragment containing the pathogenic variant, starting from the splice site preceding the mutation. Preclinical studies have been conducted in an in vitro disease model, using fibroblasts from EB simplex with muscular dystrophy patients, carrying variants in the plectin gene. In this model, wild type allele expression could be increased by 58.7% (Wally et al., 2008). Proof of concept was similarly obtained in a DEB model (Fritsch et al., 2009; Murauer et al., 2011; Morgan et al., 2013).
Micro RNAs (miRNAs), which remain untranslated, are endogenous post-transcriptional modulators of gene expression with critical functions in health and disease. Most probably, together with DNA/RNA methylation, they represent an important epigenetic factor regulating gene function (López-Jiménez and Andrés-León, 2021) (Wagner et al., 2021). There is some evidence that they might explain at least to some extent, several phenotypic variations of genodermatoses within individuals harboring the same genetic background. A recent study revealed miR-125b to be upregulated in HHD patients suffering from clinical symptoms (Manca et al., 2011). The authors suggest that oxidative stress-mediated induction of miR-125b (Manca et al., 2011) plays a specific role in the HHD pathogenesis, specifically by regulating the expression of proteins involved in Notch signaling. Indeed, this pathway plays a key role in keratinocyte proliferation and differentiation and possibly in clinical HHD manifestations, as well. In EB, miRNAs play a major role in fibrosis (miR-29b and miR-145) and SCC development (miR-10b) (Wagner et al., 2021). In recessive DEB, the miR-29 levels are decreased, resulting in increased miR-29 target mRNAs, including pro-fibrotic extracellular matrix collagens. It is likely to be an essential regulatory mechanism for C7 production and TGF-β-mediated fibrosis inhibition (Vanden Oever et al., 2016). MiRNAs are a recent research topic, which could bring upon novel therapeutic options in the future.
Other research topics concerning RNA-based techniques are transcript transfer, which introduces in vitro transcribed mRNA into the cell in order to translate it into wild type proteins, as well as the use of mRNA transcripts encoding nucleases for genome editing (Bornert et al., 2017; Bustad et al., 2021).
It is crucial that each family with an affected member receives appropriate genetic counselling regarding the variability of the disease phenotype, its genetic transmission, and the possibilities of prenatal or preimplantation genetic testing. Special educational programmes are currently available for several genodermatosis groups. The support provided by a psychologist and by patient organisations are extremely valuable and are designed to accompany the families that are confronted with such rare diseases.
Despite all these upcoming treatment possibilities, prevention of recurrence in future pregnancies remains crucial, based on techniques of prenatal or preimplantation genetic testing, with the implantation of a non-affected embryo as an option. However, this domain is beyond the scope of this article, but we refer to comprehensive reviews on these topics (Vermeesch et al., 2016; Jónsson et al., 2018; Antonarakis, 2019; Che et al., 2020).
Genodermatoses have long been considered as diseases for which effective treatments were almost non-existent, with very little hope for inventive progression. However, given that the underlying mutations are progressively unravelled and along with translational research generated, more possibilities are currently at the horizon.
These mutations may cause dysfunction of biochemical pathways thereby offering opportunities to interact with dysfunctional paths. One approach consists of repurposing drugs, which are already well-known and have been investigated for other pathologies. If inflammatory pathways are triggered, targeted biological therapies can be applied.
However, modern technologies are additionally offering new inventive tools, enabling us to intervene with the real cause of genodermatoses and thus either restore or replace the affected gene or gene-product. Some of these new techniques likely enable very precise corrections. Besides, if DNA in stem cells can be corrected, then the dream of definitive correction comes close to reality. The first, in vivo trials, using genetically-engineered cells and tissues are currently under way. These sophisticated technologies are only possible in the event that a molecular genetic diagnosis is available. Moreover, these techniques require dedicated and specialised competences, subsequent care and follow-up. For some genodermatoses, such competences are only available in certain centres. Therefore, collaboration across Europe appears crucial, allowing us to offer our patients the best possible care, with the support of the European Union. More information is to be found here: https://ec.europa.eu/health/cross_border_care/overview_en.
In this article, we have shed light over different new avenues that are currently being investigated. Each of the above-mentioned genodermatoses poses its own challenges, in regard to its way of transmission, mutation type and localisation, and altered protein function. With respect to genome editing and RNA interference, these approaches constitute personalised therapies for which feasibility, lack of undesirable effects, and off target damage, in addition to cost/effectiveness, are likely to play a role in determining which of interventions is likely to be made available for clinical use.
Therefore, information on these conditions and prevention of these, at times, severe and life threatening diseases that dramatically impair the quality of life of the patients and their families, are of interest. An increasing number of centres offer the possibility of patient follow-up at specialised consultations for genodermatoses. This is instrumental in enabling a better follow-up, superior information, earlier diagnosis of potential complications, as well as the possibility to be directed to new treatment options that are at the horizon. While the future is not yet bright, there is at least some sunshine.
M-AM has written the original manuscript, EL wrote the chapter on prevention strategies, EL, FG, SH-R, CB, and DH reviewed the manuscript. All figures were Created with BioRender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abdollahimajd, F., Rajabi, F., Shahidi-Dadras, M., Saket, S., Youssefian, L., Vahidnezhad, H., et al. (2019). Pachyonychia Congenita: a Case Report of a Successful Treatment With Rosuvastatin in a Patient With a KRT6A Mutation. Br. J. Dermatol. 181 (3), 584–586. doi:10.1111/bjd.17276
Aberdam, E., Roux, L. N., Secrétan, P. H., Boralevi, F., Schlatter, J., Morice-Picard, F., et al. (2020). Improvement of Epidermal Covering on AEC Patients With Severe Skin Erosions by PRIMA-1MET/APR-246. Cell Death Dis. 11 (1), 30. doi:10.1038/s41419-020-2223-8
Ain, Q. U., Campos, E. V. R., Huynh, A., Witzigmann, D., and Hedtrich, S. (2021). Gene Delivery to the Skin - How Far Have We Come. Trends Biotechnol. 39 (5), 474–487. doi:10.1016/j.tibtech.2020.07.012
Al-Olabi, L., Polubothu, S., Dowsett, K., Andrews, K. A., Stadnik, P., Joseph, A. P., et al. (2018). Mosaic RAS/MAPK Variants Cause Sporadic Vascular Malformations Which Respond to Targeted Therapy. J. Clin. Invest. 128 (4), 5185–5508. doi:10.1172/JCI124649
Andreasen, T. H., Karstensen, H. G., Duno, M., Lei, U., Zachariae, C., and Thyssen, J. P. (2020). Successful Treatment with Dupilumab of an Adult with Netherton Syndrome. Clin. Exp. Dermatol. 45 (7), 915–917. doi:10.1111/ced.14317
Angelis, A., Kanavos, P., López-Bastida, J., Linertová, R., Oliva-Moreno, J., Serrano-Aguilar, P., et al. (2016). Social/economic Costs and Health-Related Quality of Life in Patients with Epidermolysis Bullosa in Europe. Eur. J. Health Econ. 17 (Suppl. 1), 31–42. doi:10.1007/s10198-016-0783-4
Antonarakis, S. E. (2019). Carrier Screening for Recessive Disorders. Nat. Rev. Genet. 20 (9), 549–561. doi:10.1038/s41576-019-0134-2
Anzalone, A. V., Koblan, L. W., and Liu, D. R. (2020). Genome Editing with CRISPR-Cas Nucleases, Base Editors, Transposases and Prime Editors. Nat. Biotechnol. 38 (7), 824–844. doi:10.1038/s41587-020-0561-9
Aşkın, Ö., Engin, B., Gencebay, G., and Tüzün, Y. (2020). A Multistep Approach to the Diagnosis of Rare Genodermatoses. Clin. Dermatol. 38 (4), 399–407. doi:10.1016/j.clindermatol.2020.03.009
Atanasova, V. S., Jiang, Q., Prisco, M., Gruber, C., Piñón Hofbauer, J., Chen, M., et al. (2017). Amlexanox Enhances Premature Termination Codon Read-Through in COL7A1 and Expression of Full Length Type VII Collagen: Potential Therapy for Recessive Dystrophic Epidermolysis Bullosa. J. Invest. Dermatol. 137 (9), 1842–1849. doi:10.1016/j.jid.2017.05.011
Atzmony, L., Lim, Y. H., Hamilton, C., Leventhal, J. S., Wagner, A., Paller, A. S., et al. (2020). Topical Cholesterol/Lovastatin for the Treatment of Porokeratosis: A Pathogenesis-Directed Therapy. J. Am. Acad. Dermatol. 82 (1), 123–131. doi:10.1016/j.jaad.2019.08.043
Aufenvenne, K., Larcher, F., Hausser, I., Duarte, B., Oji, V., Nikolenko, H., et al. (2013). Topical Enzyme-Replacement Therapy Restores Transglutaminase 1 Activity and Corrects Architecture of Transglutaminase-1-Deficient Skin Grafts. Am. J. Hum. Genet. 93 (4), 620–630. doi:10.1016/j.ajhg.2013.08.003
Aushev, M., Koller, U., Mussolino, C., Cathomen, T., and Reichelt, J. (2017). Traceless Targeting and Isolation of Gene-Edited Immortalized Keratinocytes from Epidermolysis Bullosa Simplex Patients. Mol. Ther. Methods Clin. Dev. 6, 112–123. doi:10.1016/j.omtm.2017.06.008
Bachelez, H., Choon, S. E., Marrakchi, S., Burden, A. D., Tsai, T. F., Morita, A., et al. (2019). Inhibition of the Interleukin-36 Pathway for the Treatment of Generalized Pustular Psoriasis. N. Engl. J. Med. 380 (10), 981–983. doi:10.1056/NEJMc1811317
Baradaran-Heravi, A., Balgi, A. D., Zimmerman, C., Choi, K., Shidmoossavee, F. S., Tan, J. S., et al. (2016). Novel Small Molecules Potentiate Premature Termination Codon Readthrough by Aminoglycosides. Nucleic Acids Res. 44 (14), 6583–6598. doi:10.1093/nar/gkw638
Barbieux, C., Bonnet des Claustres, M., de la Brassinne, M., Bricteux, G., Bagot, M., Bourrat, E., et al. (2021). Duality of Netherton Syndrome Manifestations and Response to Ixekizumab. J. Am. Acad. Dermatol. 84 (5), 1476–1480. doi:10.1016/j.jaad.2020.07.054
Bardhan, A., Bruckner-Tuderman, L., Chapple, I. L. C., Fine, J. D., Harper, N., Has, C., et al. (2020). Epidermolysis Bullosa. Nat. Rev. Dis. Primers 6 (1), 78. doi:10.1038/s41572-020-0210-0
Benati, D., Miselli, F., Cocchiarella, F., Patrizi, C., Carretero, M., Baldassarri, S., et al. (2018). CRISPR/Cas9-Mediated In Situ Correction of LAMB3 Gene in Keratinocytes Derived from a Junctional Epidermolysis Bullosa Patient. Mol. Ther. 26 (11), 2592–2603. doi:10.1016/j.ymthe.2018.07.024
Biesecker, L. G., Edwards, M., O'Donnell, S., Doherty, P., MacDougall, T., Tith, K., et al. (2020). Clinical Report: One Year of Treatment of Proteus Syndrome With Miransertib (ARQ 092). Cold Spring Harb Mol. Case Stud. 6 (1), a004549. doi:10.1101/mcs.a004549
Blanchard, S. K., and Prose, N. S. (2020). Successful Use of Secukinumab in Netherton Syndrome. JAAD Case Rep. 6 (6), 577–578. doi:10.1016/j.jdcr.2020.04.025
Bodemer, C., Steijlen, P., Mazereeuw-Hautier, J., and O'Toole, E. A. (2021). Treatment of Hereditary Palmoplantar Keratoderma: a Review by Analysis of the Literature. Br. J. Dermatol. 184 (3), 393–400. doi:10.1111/bjd.19144
Bonafont, J., Mencía, Á., García, M., Torres, R., Rodríguez, S., Carretero, M., et al. (2019). Clinically Relevant Correction of Recessive Dystrophic Epidermolysis Bullosa by Dual sgRNA CRISPR/Cas9-Mediated Gene Editing. Mol. Ther. 27 (5), 986–998. doi:10.1016/j.ymthe.2019.03.007
Booms, P., Harth, M., Sader, R., and Ghanaati, S. (2015). Vismodegib Hedgehog-Signaling Inhibition and Treatment of Basal Cell Carcinomas as Well as Keratocystic Odontogenic Tumors in Gorlin Syndrome. Ann. Maxillofac. Surg. 5 (1), 14–19. doi:10.4103/2231-0746.161049
Bornert, O., Hogervorst, M., Nauroy, P., Bischof, J., Swildens, J., Athanasiou, I., et al. (2021). QR-313, an Antisense Oligonucleotide, Shows Therapeutic Efficacy for Treatment of Dominant and Recessive Dystrophic Epidermolysis Bullosa: A Preclinical Study. J. Invest. Dermatol. 141 (4), 883–e6. doi:10.1016/j.jid.2020.08.018
Bornert, O., Kühl, T., Bremer, J., van den Akker, P. C., Pasmooij, A. M., and Nyström, A. (2016). Analysis of the Functional Consequences of Targeted Exon Deletion in COL7A1 Reveals Prospects for Dystrophic Epidermolysis Bullosa Therapy. Mol. Ther. 24 (7), 1302–1311. doi:10.1038/mt.2016.92
Bornert, O., Peking, P., Bremer, J., Koller, U., van den Akker, P. C., Aartsma-Rus, A., et al. (2017). RNA-based Therapies for Genodermatoses. Exp. Dermatol. 26 (1), 3–10. doi:10.1111/exd.13141
Bremer, J., Bornert, O., Nyström, A., Gostynski, A., Jonkman, M. F., Aartsma-Rus, A., et al. (2016). Antisense Oligonucleotide-Mediated Exon Skipping as a Systemic Therapeutic Approach for Recessive Dystrophic Epidermolysis Bullosa. Mol. Ther. Nucleic Acids 5, e379. doi:10.1038/mtna.2016.87
Bremer, J., van der Heijden, E. H., Eichhorn, D. S., Meijer, R., Lemmink, H. H., Scheffer, H., et al. (2019). Natural Exon Skipping Sets the Stage for Exon Skipping as Therapy for Dystrophic Epidermolysis Bullosa. Mol. Ther. Nucleic Acids 18, 465–475. doi:10.1016/j.omtn.2019.09.009
Bustad, H. J., Kallio, J. P., Vorland, M., Fiorentino, V., Sandberg, S., Schmitt, C., et al. (2021). Acute Intermittent Porphyria: An Overview of Therapy Developments and Future Perspectives Focusing on Stabilisation of HMBS and Proteostasis Regulators. Ijms 22 (2), 675. doi:10.3390/ijms22020675
Castela, E., Tulic, M. K., Rozières, A., Bourrat, E., Nicolas, J. F., Kanitakis, J., et al. (2019). Epidermolysis Bullosa Simplex Generalized Severe Induces a T Helper 17 Response and Is Improved by Apremilast Treatment. Br. J. Dermatol. 180 (2), 357–364. doi:10.1111/bjd.16897
Chamcheu, J. C., Navsaria, H., Pihl-Lundin, I., Liovic, M., Vahlquist, A., and Törmä, H. (2011). Chemical Chaperones Protect Epidermolysis Bullosa Simplex Keratinocytes from Heat Stress-Induced Keratin Aggregation: Involvement of Heat Shock Proteins and MAP Kinases. J. Invest. Dermatol. 131 (8), 1684–1691. doi:10.1038/jid.2011.93
Chamorro, C., Mencía, A., Almarza, D., Duarte, B., Büning, H., Sallach, J., et al. (2016). Gene Editing for the Efficient Correction of a Recurrent COL7A1 Mutation in Recessive Dystrophic Epidermolysis Bullosa Keratinocytes. Mol. Ther. Nucleic Acids 5, e307. doi:10.1038/mtna.2016.19
Che, H., Villela, D., Dimitriadou, E., Melotte, C., Brison, N., Neofytou, M., et al. (2020). Noninvasive Prenatal Diagnosis by Genome-wide Haplotyping of Cell-free Plasma DNA. Genet. Med. 22 (5), 962–973. doi:10.1038/s41436-019-0748-y
Chelliah, M. P., Zinn, Z., Khuu, P., and Teng, J. M. C. (2018). Self-Initiated Use of Topical Cannabidiol Oil for Epidermolysis Bullosa. Pediatr. Dermatol. 35, e224–e227.
Cheraghlou, S., Lim, Y., and Choate, K. (2019). Genetic Investigation of Childhood Vascular Tumor Biology Reveals Pathways for Therapeutic Intervention. F1000Res. 8, 590. doi:10.12688/f1000research.16160.1
Choate, K. A., Kinsella, T. M., Williams, M. L., Nolan, G. P., and Khavari, P. A. (1996). Transglutaminase 1 Delivery to Lamellar Ichthyosis Keratinocytes. Hum. Gene Ther. 7 (18), 2247–2253. doi:10.1089/hum.1996.7.18-2247
Choon, S. E., Lebwohl, M. G., Marrakchi, S., Burden, A. D., Tsai, T. F., Morita, A., et al. (2021). Study Protocol of the Global Effisayil 1 Phase II, Multicentre, Randomised, Double-Blind, Placebo-Controlled Trial of Spesolimab in Patients with Generalized Pustular Psoriasis Presenting with an Acute Flare. BMJ Open 11 (3), e043666. doi:10.1136/bmjopen-2020-043666
Condrat, I., He, Y., Cosgarea, R., and Has, C. (2018). Junctional Epidermolysis Bullosa: Allelic Heterogeneity and Mutation Stratification for Precision Medicine. Front. Med. (Lausanne) 5, 363. doi:10.3389/fmed.2018.00363
Craiglow, B. G., Boyden, L. M., Hu, R., Virtanen, M., Su, J., Rodriguez, G., et al. (2018). CARD14-associated Papulosquamous Eruption: A Spectrum Including Features of Psoriasis and Pityriasis Rubra Pilaris. J. Am. Acad. Dermatol. 79 (3), 487–494. doi:10.1016/j.jaad.2018.02.034
Davis, B. R., Yan, Q., Bui, J. H., Felix, K., Moratto, D., Muul, L. M., et al. (2010). Somatic Mosaicism in the Wiskott-Aldrich Syndrome: Molecular and Functional Characterization of Genotypic Revertants. Clin. Immunol. 135 (1), 72–83. doi:10.1016/j.clim.2009.12.011
Daroach, M., Dogra, S., Bhattacharjee, R., Tp, A., and Mahajan, R. (2019). Pachyonychia Congenita Responding Favorably to a Combination of Surgical and Medical Therapies. Dermatol. Ther. 32 (5), e13045. doi:10.1111/dth.13045
de Andrade, F. A. G., Cavalcanti, C. E. O., Isoldi, F. C., and Ferreira, L. M. (2021). Therapeutics of Xeroderma Pigmentosum: A PRISMA-Compliant Systematic Review. Indian J. Dermatol. Venereol. Leprol. 87, 176–189. doi:10.25259/IJDVL_431_19
De Felice, C., Graceffa, D., Morrone, A., and Bonifati, C. (2020). Familial Pityriasis Rubra Pilaris Successfully Treated with Brodalumab. Int. J. Dermatol. 59 (7), 885–887. doi:10.1111/ijd.14857
De Rosa, L., Carulli, S., Cocchiarella, F., Quaglino, D., Enzo, E., Franchini, E., et al. (2014). Long-Term Stability and Safety of Transgenic Cultured Epidermal Stem Cells in Gene Therapy of Junctional Epidermolysis Bullosa. Stem Cel Rep. 2 (1), 1–8. doi:10.1016/j.stemcr.2013.11.001
Di, W. L., Larcher, F., Semenova, E., Talbot, G. E., Harper, J. I., Del Rio, M., et al. (2011). Ex-vivo Gene Therapy Restores LEKTI Activity and Corrects the Architecture of Netherton Syndrome-Derived Skin Grafts. Mol. Ther. 19 (2), 408–416. doi:10.1038/mt.2010.201
Di, W. L., Lwin, S. M., Petrova, A., Bernadis, C., Syed, F., Farzaneh, F., et al. (2019). Generation and Clinical Application of Gene-Modified Autologous Epidermal Sheets in Netherton Syndrome: Lessons Learned from a Phase 1 Trial. Hum. Gene Ther. 30 (9), 1067–1078. doi:10.1089/hum.2019.049
Dufresne, H., Hadj-Rabia, S., Taïeb, C., and Bodemer, C. (2013). Importance of Therapeutic Patient Education in Ichthyosis: Results of a Prospective Single Reference center Study. Orphanet J. Rare Dis. 8 (1), 113. doi:10.1186/1750-1172-8-113
Dupuy, A., Valton, J., Leduc, S., Armier, J., Galetto, R., Gouble, A., et al. (2013). Targeted Gene Therapy of Xeroderma Pigmentosum Cells Using Meganuclease and TALEN™. PLoS ONE 8 (11), e78678. doi:10.1371/journal.pone.0078678
Ebens, C. L., McGrath, J. A., Riedl, J. A., Keith, A. R., Lilja, G., Rusch, S., et al. (2020). Immune Tolerance of Allogeneic Haematopoietic Cell Transplantation Supports Donor Epidermal Grafting of Recessive Dystrophic Epidermolysis Bullosa Chronic Wounds. Br. J. Dermatol. 184 (6), 1161–1169. doi:10.1111/bjd.19503
Eichstadt, S., Barriga, M., Ponakala, A., Teng, C., Nguyen, N. T., Siprashvili, Z., et al. (2019). Phase 1/2a Clinical Trial of Gene-Corrected Autologous Cell Therapy for Recessive Dystrophic Epidermolysis Bullosa. JCI Insight 4 (19), e130554. doi:10.1172/jci.insight.130554
El-Darouti, M., Fawzy, M., Amin, I., Abdel Hay, R., Hegazy, R., Gabr, H., et al. (2016). Treatment of Dystrophic Epidermolysis Bullosa with Bone Marrow Non-hematopoeitic Stem Cells: a Randomized Controlled Trial. Dermatol. Ther. 29 (2), 96–100. doi:10.1111/dth.12305
Enjalbert, F., Dewan, P., Caley, M. P., Jones, E. M., Morse, M. A., Kelsell, D. P., et al. (2020). 3D Model of Harlequin Ichthyosis Reveals Inflammatory Therapeutic Targets. J. Clin. Invest. 130 (9), 4798–4810. doi:10.1172/JCI132987
Erwin, A. L., and Balwani, M. (2021). Porphyrias in the Age of Targeted Therapies. Diagnostics (Basel) 11 (10), 1795. doi:10.3390/diagnostics11101795
Eytan, O., Sarig, O., Sprecher, E., and van Steensel, M. A. (2014). Clinical Response to Ustekinumab in Familial Pityriasis Rubra Pilaris Caused by a Novel Mutation in CARD14. Br. J. Dermatol. 171 (2), 420–422. doi:10.1111/bjd.12952
Fontao, L., Laffitte, E., Briot, A., Kaya, G., Roux-Lombard, P., Fraitag, S., et al. (2011). Infliximab Infusions for Netherton Syndrome: Sustained Clinical Improvement Correlates with a Reduction of Thymic Stromal Lymphopoietin Levels in the Skin. J. Invest. Dermatol. 131 (9), 1947–1950. doi:10.1038/jid.2011.124
Fritsch, A., Spassov, S., Elfert, S., Schlosser, A., Gache, Y., Meneguzzi, G., et al. (2009). Dominant-negative Effects of COL7A1 Mutations Can Be Rescued by Controlled Overexpression of Normal Collagen VII. J. Biol. Chem. 284 (44), 30248–30256. doi:10.1074/jbc.M109.045294
Frommherz, L., and Has, C. (2020). Successful Treatment of Pachyonychia Congenita With Rosuvastatin. J. Eur. Acad. Dermatol. Venereol. 34 (9), e480–e482. doi:10.1111/jdv.16393
Fuchs-Telem, D., Sarig, O., van Steensel, M. A., Isakov, O., Israeli, S., Nousbeck, J., et al. (2012). Familial Pityriasis Rubra Pilaris Is Caused by Mutations in CARD14. Am. J. Hum. Genet. 91 (1), 163–170. doi:10.1016/j.ajhg.2012.05.010
Geeurickx, M., and Labarque, V. (2021). A Narrative Review of the Role of Sirolimus in the Treatment of Congenital Vascular Malformations. J. Vasc. Surg. Venous Lymphat Disord. 9, 1321. doi:10.1016/j.jvsv.2021.03.001
Goldberg, L. H., Landau, J. M., Moody, M. N., Kazakevich, N., Holzer, A. M., and Myers, A. (2011). Resolution of Odontogenic Keratocysts of the Jaw in Basal Cell Nevus Syndrome With GDC-0449. Arch. Dermatol. 147 (7), 839–841. doi:10.1001/archdermatol.2011.50
González-Ramos, J., Sendagorta-Cudós, E., González-López, G., Mayor-Ibarguren, A., Feltes-Ochoa, R., and Herranz-Pinto, P. (2016). Efficacy of Botulinum Toxin in Pachyonychia Congenita Type 1: Report of Two New Cases. Dermatol. Ther. 29 (1), 32–36.
Gooderham, M. J., Van Voorhees, A. S., and Lebwohl, M. G. (2019). An Update on Generalized Pustular Psoriasis. Expert Rev. Clin. Immunol. 15 (9), 907–919. doi:10.1080/1744666X.2019.1648209
Gostyński, A., Pasmooij, A. M., and Jonkman, M. F. (2014). Successful Therapeutic Transplantation of Revertant Skin in Epidermolysis Bullosa. J. Am. Acad. Dermatol. 70 (1), 98–101. doi:10.1016/j.jaad.2013.08.052
Goto, M., Sawamura, D., Nishie, W., Sakai, K., McMillan, J. R., Akiyama, M., et al. (2006). Targeted Skipping of a Single Exon Harboring a Premature Termination Codon Mutation: Implications and Potential for Gene Correction Therapy for Selective Dystrophic Epidermolysis Bullosa Patients. J. Invest. Dermatol. 126 (12), 2614–2620. doi:10.1038/sj.jid.5700435
Greco, C., Leclerc-Mercier, S., Chaumon, S., Doz, F., Hadj-Rabia, S., Molina, T., et al. (2020). Use of Epidermal Growth Factor Receptor Inhibitor Erlotinib to Treat Palmoplantar Keratoderma in Patients with Olmsted Syndrome Caused by TRPV3 Mutations. JAMA Dermatol. 156 (2), 191–195. doi:10.1001/jamadermatol.2019.4126
Guérard, N., Morand, O., and Dingemanse, J. (2017). Lucerastat, an Iminosugar With Potential as Substrate Reduction Therapy for Glycolipid Storage Disorders: Safety, Tolerability, and Pharmacokinetics in Healthy Subjects. Orphanet J. Rare Dis. 12 (1), 9.
Guerra, L., Diociaiuti, A., El Hachem, M., Castiglia, D., and Zambruno, G. (2015). Ichthyosis with Confetti: Clinics, Molecular Genetics and Management. Orphanet J. Rare Dis. 10 (1), 115. doi:10.1186/s13023-015-0336-4
Guttmann-Gruber, C., Tockner, B., Scharler, C., Hüttner, C., Common, J. E., Tay, A. S. L., et al. (2018). Low-Dose Calcipotriol Can Elicit Wound Closure, Anti-Microbial, and Anti-Neoplastic Effects in Epidermolysis Bullosa Keratinocytes. Sci. Rep. 8 (1), 13430. doi:10.1038/s41598-018-31823-6
Habib, S. L., Al-Obaidi, N. Y., Nowacki, M., Pietkun, K., Zegarska, B., Kloskowski, T., et al. (2016). Is mTOR Inhibitor Good Enough for Treatment All Tumors in TSC Patients? J. Cancer. 7 (12), 1621–1631. doi:10.7150/jca.14747
Hammer, J., Seront, E., Duez, S., Dupont, S., Van Damme, A., Schmitz, S., et al. (2018). Sirolimus Is Efficacious in Treatment for Extensive And/or Complex Slow-Flow Vascular Malformations: a Monocentric Prospective Phase II Study. Orphanet J. Rare Dis. 13 (1), 191. doi:10.1186/s13023-018-0934-z
Hammill, A. M., Wentzel, M., Gupta, A., Nelson, S., Lucky, A., Elluru, R., et al. (2011). Sirolimus for the Treatment of Complicated Vascular Anomalies in Children. Pediatr. Blood Cancer. 57 (6), 1018–1024. doi:10.1002/pbc.23124
Harbers, V. E. M., Rongen, G. A. P. J. M., van der Vleuten, C. J. M., Verhoeven, B. H., de Laat, P. C. J., van der Horst, C. M. A. M., et al. (2021). Patients with Congenital Low-Flow Vascular Malformation Treated with Low Dose Sirolimus. Adv Ther [Internet]. [cited 2021 May 24]. Available at: https://link.springer.com/10.1007/s12325-021-01758-y.
Hilhorst, N., Spanoudi-Kitrimi, I., Goemans, N., and Morren, M. A. (2019). Injection Site Reactions after Long-Term Subcutaneous Delivery of Drisapersen: a Retrospective Study. Eur. J. Pediatr. 178 (2), 253–258. doi:10.1007/s00431-018-3272-1
Hirsch, T., Rothoeft, T., Teig, N., Bauer, J. W., Pellegrini, G., De Rosa, L., et al. (2017). Regeneration of the Entire Human Epidermis Using Transgenic Stem Cells. Nature 551 (7680), 327–332. doi:10.1038/nature24487
Hou, Y., Guey, L. T., Wu, T., Gao, R., Cogan, J., Wang, X., et al. (2015). Intravenously Administered Recombinant Human Type VII Collagen Derived from Chinese Hamster Ovary Cells Reverses the Disease Phenotype in Recessive Dystrophic Epidermolysis Bullosa Mice. J. Invest. Dermatol. 135 (12), 3060–3067. doi:10.1038/jid.2015.291
Izmiryan, A., Danos, O., and Hovnanian, A. (2016). Meganuclease-Mediated COL7A1 Gene Correction for Recessive Dystrophic Epidermolysis Bullosa. J. Invest. Dermatol. 136 (4), 872–875. doi:10.1016/j.jid.2015.11.028
Izmiryan, A., Ganier, C., Bovolenta, M., Schmitt, A., Mavilio, F., and Hovnanian, A. (2018). Ex Vivo COL7A1 Correction for Recessive Dystrophic Epidermolysis Bullosa Using CRISPR/Cas9 and Homology-Directed Repair. Mol. Ther. Nucleic Acids 12, 554–567. doi:10.1016/j.omtn.2018.06.008
Jayarajan, V., Kounatidou, E., Qasim, W., and Di, W. L. (2021). Ex Vivo gene Modification Therapy for Genetic Skin Diseases-Recent Advances in Gene Modification Technologies and Delivery. Exp. Dermatol. 30 (7), 887–896. doi:10.1111/exd.14314
Jonkman, M. F., and Pasmooij, A. M. (2012). Realm of Revertant Mosaicism Expanding. J. Invest. Dermatol. 132 (3), 514–516. doi:10.1038/jid.2011.445
Jonkman, M. F., Scheffer, H., Stulp, R., Pas, H. H., Nijenhuis, M., Heeres, K., et al. (1997). Revertant Mosaicism in Epidermolysis Bullosa Caused by Mitotic Gene Conversion. Cell 88 (4), 543–551. doi:10.1016/s0092-8674(00)81894-2
Jónsson, H., Sulem, P., Arnadottir, G. A., Pálsson, G., Eggertsson, H. P., Kristmundsdottir, S., et al. (2018). Multiple Transmissions of De Novo Mutations in Families. Nat. Genet. 50 (12), 1674–1680.
Kellermayer, R., Szigeti, R., Keeling, K. M., Bedekovics, T., and Bedwell, D. M. (2006). Aminoglycosides as Potential Pharmacogenetic Agents in the Treatment of Hailey-Hailey Disease. J. Invest. Dermatol. 126 (1), 229–231. doi:10.1038/sj.jid.5700031
Keppler-Noreuil, K. M., Sapp, J. C., Lindhurst, M. J., Darling, T. N., Burton-Akright, J., Bagheri, M., et al. (2019). Pharmacodynamic Study of Miransertib in Individuals With Proteus Syndrome. Am. J. Hum. Genet. 104 (3), 484–491. doi:10.1016/j.ajhg.2019.01.015
Kern, J. S., Schwieger-Briel, A., Löwe, S., Sumeray, M., Davis, C., and Martinez, A. E. (2019). Oleogel-S10 Phase 3 Study "EASE" for Epidermolysis Bullosa: Study Design and Rationale. Trials. 20 (1), 350. doi:10.1186/s13063-019-3362-z
Kerns, M. L., DePianto, D., Dinkova-Kostova, A. T., Talalay, P., and Coulombe, P. A. (2007). Reprogramming of Keratin Biosynthesis by Sulforaphane Restores Skin Integrity in Epidermolysis Bullosa Simplex. Proc. Natl. Acad. Sci. U S A. 104 (36), 14460–14465. doi:10.1073/pnas.0706486104
Kerns, M. L., Hakim, J. M. C., Zieman, A., Lu, R. G., and Coulombe, P. A. (2018). Sexual Dimorphism in Response to an NRF2 Inducer in a Model for Pachyonychia Congenita. J. Invest. Dermatol. 138 (5), 1094–1100. doi:10.1016/j.jid.2017.09.054
Kocher, T., Peking, P., Klausegger, A., Murauer, E. M., Hofbauer, J. P., Wally, V., et al. (2017). Cut and Paste: Efficient Homology-Directed Repair of a Dominant Negative KRT14 Mutation via CRISPR/Cas9 Nickases. Mol. Ther. 25 (11), 2585–2598. doi:10.1016/j.ymthe.2017.08.015
Kocher, T., Wagner, R. N., Klausegger, A., Guttmann-Gruber, C., Hainzl, S., Bauer, J. W., et al. (2019). Improved Double-Nicking Strategies for COL7A1-Editing by Homologous Recombination. Mol. Ther. Nucleic Acids 18, 496–507. doi:10.1016/j.omtn.2019.09.011
Komiya, T., Blumenthal, G. M., DeChowdhury, R., Fioravanti, S., Ballas, M. S., Morris, J., et al. (2019). A Pilot Study of Sirolimus in Subjects With Cowden Syndrome or Other Syndromes Characterized by Germline Mutations in PTEN. Oncologist. 24 (12), 1510–e1265. doi:10.1634/theoncologist.2019-0514
Koren, A., Sprecher, E., Reider, E., and Artzi, O. (2020). A Treatment Protocol for Botulinum Toxin Injections in the Treatment of Pachyonychia Congenita-Associated Keratoderma. Br. J. Dermatol. 182 (3), 671–677. doi:10.1111/bjd.18169
Kowalewski, C., Bremer, J., Gostynski, A., Wertheim-Tysarowska, K., Wozniak, K., Bal, J., et al. (2016). Amelioration of Junctional Epidermolysis Bullosa Due to Exon Skipping. Br. J. Dermatol. 174 (6), 1375–1379. doi:10.1111/bjd.14374
Kuschal, C., Khan, S. G., Enk, B., DiGiovanna, J. J., and Kraemer, K. H. (2015). Readthrough of Stop Codons by Use of Aminoglycosides in Cells from Xeroderma Pigmentosum Group C Patients. Exp. Dermatol. 24 (4), 296–297. doi:10.1111/exd.12655
Lai-Cheong, J. E., McGrath, J. A., and Uitto, J. (2011). Revertant Mosaicism in Skin: Natural Gene Therapy. Trends Mol. Med. 17 (3), 140–148. doi:10.1016/j.molmed.2010.11.003
Langendonk, J. G., Balwani, M., Anderson, K. E., Bonkovsky, H. L., Anstey, A. V., Bissell, D. M., et al. (2015). Afamelanotide for Erythropoietic Protoporphyria. N. Engl. J. Med. 373 (1), 48–59. doi:10.1056/NEJMoa1411481
Leachman, S. A., Hickerson, R. P., Schwartz, M. E., Bullough, E. E., Hutcherson, S. L., Boucher, K. M., et al. (2010). First-in-human Mutation-Targeted siRNA Phase Ib Trial of an Inherited Skin Disorder. Mol. Ther. 18 (2), 442–446. doi:10.1038/mt.2009.273
Lekwuttikarn, R., Lim, Y. H., Admani, S., Choate, K. A., and Teng, J. M. C. (2019). Genotype-Guided Medical Treatment of an Arteriovenous Malformation in a Child. JAMA Dermatol. 155 (2), 256–257. doi:10.1001/jamadermatol.2018.4653
Lenders, M., and Brand, E. (2021). Fabry Disease: The Current Treatment Landscape. Drugs 81 (6), 635–645. doi:10.1007/s40265-021-01486-1
Liddle, J., Beneton, V., Benson, M., Bingham, R., Bouillot, A., Boullay, A-B., et al. (2021). A Potent and Selective Kallikrein-5 Inhibitor Delivers High Pharmacological Activity in Netherton Syndrome Patient Skin. J. Invest. Dermatol. 141 (9), 2272–2279. doi:10.1016/j.jid.2021.01.029
Limmer, A. L., Nwannunu, C. E., Shah, R., Coleman, K., Patel, R. R., Mui, U. N., et al. (2019). Topical Diacerein Ointment for Epidermolysis Bullosa Simplex: A Review. Skin Ther. Lett 24 (3), 7–9.
Lincoln, V., Cogan, J., Hou, Y., Hirsch, M., Hao, M., Alexeev, V., et al. (2018). Gentamicin Induces LAMB3 Nonsense Mutation Readthrough and Restores Functional Laminin 332 in Junctional Epidermolysis Bullosa. Proc. Natl. Acad. Sci. U S A. 115 (28), E6536–E6545. doi:10.1073/pnas.1803154115
Liu, S., Kuht, H. J., Moon, E. H., Maconachie, G. D. E., and Thomas, M. G. (2021). Current and Emerging Treatments for Albinism. Surv. Ophthalmol. 66 (2), 362–377. doi:10.1016/j.survophthal.2020.10.007
López-Jiménez, E., and Andrés-León, E. (2021). The Implications of ncRNAs in the Development of Human Diseases. Non-Coding RNA. 7 (1), 17. doi:10.3390/ncrna7010017
Luan, X. R., Chen, X. L., Tang, Y. X., Zhang, J. Y., Gao, X., Ke, H. P., et al. (2018). CRISPR/Cas9-Mediated Treatment Ameliorates the Phenotype of the Epidermolytic Palmoplantar Keratoderma-like Mouse. Mol. Ther. Nucleic Acids 12, 220–228. doi:10.1016/j.omtn.2018.05.005
Luchsinger, I., Knöpfel, N., Theiler, M., Bonnet des Claustres, M., Barbieux, C., Schwieger-Briel, A., et al. (2020). Secukinumab Therapy for Netherton Syndrome. JAMA Dermatol. 156 (8), 907–911. doi:10.1001/jamadermatol.2020.1019
Lwin, S. M., Hsu, C. K., Liu, L., Huang, H. Y., Levell, N. J., and McGrath, J. A. (2018). Beneficial Effect of Ustekinumab in Familial Pityriasis Rubra Pilaris with a New Missense Mutation in CARD14. Br. J. Dermatol. 178 (4), 969–972. doi:10.1111/bjd.15462
Manca, S., Magrelli, A., Cialfi, S., Lefort, K., Ambra, R., Alimandi, M., et al. (2011). Oxidative Stress Activation of miR-125b Is Part of the Molecular Switch for Hailey-Hailey Disease Manifestation. Exp. Dermatol. 20 (11), 932–937. doi:10.1111/j.1600-0625.2011.01359.x
March, O. P., Kocher, T., and Koller, U. (2020). Context-Dependent Strategies for Enhanced Genome Editing of Genodermatoses. Cells 9 (1), 112. doi:10.3390/cells9010112
March, O. P., Lettner, T., Klausegger, A., Ablinger, M., Kocher, T., Hainzl, S., et al. (2019). Gene Editing-Mediated Disruption of Epidermolytic Ichthyosis-Associated KRT10 Alleles Restores Filament Stability in Keratinocytes. J. Invest. Dermatol. 139 (8), 1699–e6. doi:10.1016/j.jid.2019.03.1146
March, O. P., Reichelt, J., and Koller, U. (2018). Gene Editing for Skin Diseases: Designer Nucleases as Tools for Gene Therapy of Skin Fragility Disorders. Exp. Physiol. 103 (4), 449–455. doi:10.1113/EP086044
Matsumura, W., Fujita, Y., Shinkuma, S., Suzuki, S., Yokoshiki, S., Goto, H., et al. (2019). Cultured Epidermal Autografts from Clinically Revertant Skin as a Potential Wound Treatment for Recessive Dystrophic Epidermolysis Bullosa. J. Invest. Dermatol. 139 (10), 2115–e11. doi:10.1016/j.jid.2019.03.1155
Mavilio, F., Pellegrini, G., Ferrari, S., Di Nunzio, F., Di Iorio, E., Recchia, A., et al. (2006). Correction of Junctional Epidermolysis Bullosa by Transplantation of Genetically Modified Epidermal Stem Cells. Nat. Med. 12 (12), 1397–1402. doi:10.1038/nm1504
Mazereeuw‐Hautier, J., Hernández‐Martín, A., O’Toole, E. A., Bygum, A., Amaro, C., Aldwin, M., et al. (2019). Management of Congenital Ichthyoses: European Guidelines of Care, Part Two. Br. J. Dermatol. 180 (3), 484–495.
Mazereeuw-Hautier, J., Vahlquist, A., Traupe, H., Bygum, A., Amaro, C., Aldwin, M., et al. (2019). Management of Congenital Ichthyoses: European Guidelines of Care, Part One. Br. J. Dermatol. 180 (2), 272–281. doi:10.1111/bjd.17203
Mazouzi, A., Battistini, F., Moser, S. C., Ferreira da Silva, J., Wiedner, M., Owusu, M., et al. (2017). Repair of UV-Induced DNA Damage Independent of Nucleotide Excision Repair Is Masked by MUTYH. Mol. Cel 68 (4), 797–e7. doi:10.1016/j.molcel.2017.10.021
McBride, J. D., Rodriguez-Menocal, L., Candanedo, A., Guzman, W., Garcia-Contreras, M., and Badiavas, E. V. (2018). Dual Mechanism of Type VII Collagen Transfer by Bone Marrow Mesenchymal Stem Cell Extracellular Vesicles to Recessive Dystrophic Epidermolysis Bullosa Fibroblasts. Biochimie 155, 50–58. doi:10.1016/j.biochi.2018.04.007
Mencía, Á., Chamorro, C., Bonafont, J., Duarte, B., Holguin, A., Illera, N., et al. (2018). Deletion of a Pathogenic Mutation-Containing Exon of COL7A1 Allows Clonal Gene Editing Correction of RDEB Patient Epidermal Stem Cells. Mol. Ther. Nucleic Acids 11, 68–78. doi:10.1016/j.omtn.2018.01.009
Moravvej, H., Abdollahimajd, F., Naseh, M. H., Piravar, Z., Abolhasani, E., Mozafari, N., et al. (2018). Cultured Allogeneic Fibroblast Injection vs. Fibroblasts Cultured on Amniotic Membrane Scaffold for Dystrophic Epidermolysis Bullosa Treatment. Br. J. Dermatol. 179 (1), 72–79. doi:10.1111/bjd.16338
Morgan, C. P., Allen, D. S. I., Millington-Ward, S., O'Dwyer, G. E., Palfi, A., and Jane Farrar, G. (2013). A Mutation-independent Therapeutic Strategy for Dominant Dystrophic Epidermolysis Bullosa. J. Invest. Dermatol. 133 (12), 2793–2796. doi:10.1038/jid.2013.241
Murauer, E. M., Gache, Y., Gratz, I. K., Klausegger, A., Muss, W., Gruber, C., et al. (2011). Functional Correction of Type VII Collagen Expression in Dystrophic Epidermolysis Bullosa. J. Invest. Dermatol. 131 (1), 74–83. doi:10.1038/jid.2010.249
Nagel-Wolfrum, K., Möller, F., Penner, I., Baasov, T., and Wolfrum, U. (2016). Targeting Nonsense Mutations in Diseases with Translational Read-Through-Inducing Drugs (TRIDs). BioDrugs 30 (2), 49–74. doi:10.1007/s40259-016-0157-6
Nagy, N., Almaani, N., Tanaka, A., Lai-Cheong, J. E., Techanukul, T., Mellerio, J. E., et al. (2011). HB-EGF Induces COL7A1 Expression in Keratinocytes and Fibroblasts: Possible Mechanism Underlying Allogeneic Fibroblast Therapy in Recessive Dystrophic Epidermolysis Bullosa. J. Invest. Dermatol. 131 (8), 1771–1774. doi:10.1038/jid.2011.85
Nguyen, A. L., Morren, M. A., and van Doorn, R. (2019). Severe Lipodystrophy Following Prolonged Subcutaneous Administration of Antisense Oligonucleotides. JAMA Dermatol. 155 (9), 1084–1086. doi:10.1001/jamadermatol.2019.1404
Nyström, A., Thriene, K., Mittapalli, V., Kern, J. S., Kiritsi, D., Dengjel, J., et al. (2015). Losartan Ameliorates Dystrophic Epidermolysis Bullosa and Uncovers New Disease Mechanisms. EMBO Mol. Med. 7 (9), 1211–1228. doi:10.15252/emmm.201505061
Ohguchi, Y., Nomura, T., Suzuki, S., Takeda, M., Miyauchi, T., Mizuno, O., et al. (2018). Gentamicin-Induced Readthrough and Nonsense-Mediated mRNA Decay of SERPINB7 Nonsense Mutant Transcripts. J. Invest. Dermatol. 138 (4), 836–843. doi:10.1016/j.jid.2017.10.014
Onojafe, I. F., Megan, L. H., Melch, M. G., Aderemi, J. O., Alur, R. P., Abu-Asab, M. S., et al. (2018). Minimal Efficacy of Nitisinone Treatment in a Novel Mouse Model of Oculocutaneous Albinism, Type 3. Invest. Ophthalmol. Vis. Sci. 59 (12), 4945–4952. doi:10.1167/iovs.16-20293
Osborn, M. J., Lees, C. J., McElroy, A. N., Merkel, S. C., Eide, C. R., Mathews, W., et al. (2018). CRISPR/Cas9-Based Cellular Engineering for Targeted Gene Overexpression. Int. J. Mol. Sci. 19 (4), 946. doi:10.3390/ijms19040946
Osborn, M. J., Newby, G. A., McElroy, A. N., Knipping, F., Nielsen, S. C., Riddle, M. J., et al. (2020). Base Editor Correction of COL7A1 in Recessive Dystrophic Epidermolysis Bullosa Patient-Derived Fibroblasts and iPSCs. J. Invest. Dermatol. 140 (2), 338–e5. doi:10.1016/j.jid.2019.07.701
Paller, A. S. (2019). Profiling Immune Expression to Consider Repurposing Therapeutics for the Ichthyoses. J. Invest. Dermatol. 139 (3), 535–540. doi:10.1016/j.jid.2018.08.027
Paller, A. S., van Steensel, M. A., Rodriguez-Martín, M., Sorrell, J., Heath, C., Crumrine, D., et al. (2011). Pathogenesis-Based Therapy Reverses Cutaneous Abnormalities in an Inherited Disorder of Distal Cholesterol Metabolism. J. Invest. Dermatol. 131 (11), 2242–2248. doi:10.1038/jid.2011.189
Paller, A. S., Czarnowicki, T., Renert-Yuval, Y., Holland, K., Huynh, T., Sadlier, M., et al. (2018). The Spectrum of Manifestations in Desmoplakin Gene (DSP) Spectrin Repeat 6 Domain Mutations: Immunophenotyping and Response to Ustekinumab. J. Am. Acad. Dermatol. 78 (3), 498–e2. doi:10.1016/j.jaad.2017.10.026
Paller, A. S., Renert-Yuval, Y., Suprun, M., Esaki, H., Oliva, M., Huynh, T. N., et al. (2017). An IL-17-dominant Immune Profile Is Shared across the Major Orphan Forms of Ichthyosis. J. Allergy Clin. Immunol. 139 (1), 152–165. doi:10.1016/j.jaci.2016.07.019
Papanikolaou, E., and Bosio, A. (2021). The Promise and the Hope of Gene Therapy. Front. Genome Ed. 3, 618346. doi:10.3389/fgeed.2021.618346
Pasmooij, A. M., Jonkman, M. F., and Uitto, J. (2012). Revertant Mosaicism in Heritable Skin Diseases: Mechanisms of Natural Gene Therapy. Discov. Med. 14 (76), 167–179.
Pasmooij, A. M., Nijenhuis, M., Brander, R., and Jonkman, M. F. (2012). Natural Gene Therapy May Occur in All Patients with Generalized Non-herlitz Junctional Epidermolysis Bullosa with COL17A1 Mutations. J. Invest. Dermatol. 132 (5), 1374–1383. doi:10.1038/jid.2011.477
Pasmooij, A. M., Pas, H. H., Deviaene, F. C., Nijenhuis, M., and Jonkman, M. F. (2005). Multiple Correcting COL17A1 Mutations in Patients with Revertant Mosaicism of Epidermolysis Bullosa. Am. J. Hum. Genet. 77 (5), 727–740. doi:10.1086/497344
Peled, A., Samuelov, L., Sarig, O., Bochner, R., Malki, L., Pavlovsky, M., et al. (2020). Treatment of Hereditary Hypotrichosis Simplex of the Scalp with Topical Gentamicin. Br. J. Dermatol. 183 (1), 114–120. doi:10.1111/bjd.18718
Pendaries, V., Gasc, G., Titeux, M., Tonasso, L., Mejía, J. E., and Hovnanian, A. (2012). siRNA-Mediated Allele-specific Inhibition of Mutant Type VII Collagen in Dominant Dystrophic Epidermolysis Bullosa. J. Invest. Dermatol. 132 (6), 1741–1743. doi:10.1038/jid.2012.11
Perdoni, C., McGrath, J. A., and Tolar, J. (2014). Preconditioning of Mesenchymal Stem Cells for Improved Transplantation Efficacy in Recessive Dystrophic Epidermolysis Bullosa. Stem Cel Res Ther 5 (6), 121. doi:10.1186/scrt511
Petrof, G., Lwin, S. M., Martinez-Queipo, M., Abdul-Wahab, A., Tso, S., Mellerio, J. E., et al. (2015). Potential of Systemic Allogeneic Mesenchymal Stromal Cell Therapy for Children with Recessive Dystrophic Epidermolysis Bullosa. J. Invest. Dermatol. 135 (9), 2319–2321. doi:10.1038/jid.2015.158
Petrof, G., Martinez-Queipo, M., Mellerio, J. E., Kemp, P., and McGrath, J. A. (2013). Fibroblast Cell Therapy Enhances Initial Healing in Recessive Dystrophic Epidermolysis Bullosa Wounds: Results of a Randomized, Vehicle-Controlled Trial. Br. J. Dermatol. 169 (5), 1025–1033. doi:10.1111/bjd.12599
Plank, R., Yealland, G., Miceli, E., Lima Cunha, D., Graff, P., Thomforde, S., et al. (2019). Transglutaminase 1 Replacement Therapy Successfully Mitigates the Autosomal Recessive Congenital Ichthyosis Phenotype in Full-Thickness Skin Disease Equivalents. J. Invest. Dermatol. 139 (5), 1191–1195. doi:10.1016/j.jid.2018.11.002
Poulton, C., Gration, D., Murray, K., Baynam, G., and Halbert, A. (2019). Autosomal Recessive Congenital Ichthyosis Due to Homozygous Variants in NIPAL4 with a Dramatic Response to Ustekinumab. Pediatr. Dermatol. 36 (6), 1002–1003. doi:10.1111/pde.13995
PROMISE Working Group Parker, V. E. R., Keppler-Noreuil, K. M., Faivre, L., Luu, M., Oden, N. L., De Silva, L., et al. (2019). Safety and Efficacy of Low-Dose Sirolimus in the PIK3CA-Related Overgrowth Spectrum. Genet. Med. 21 (5), 1189–1198. doi:10.1038/s41436-018-0297-9
Pros, E., Fernández-Rodríguez, J., Canet, B., Benito, L., Sánchez, A., Benavides, A., et al. (2009). Antisense Therapeutics for Neurofibromatosis Type 1 Caused by Deep Intronic Mutations. Hum. Mutat. 30 (3), 454–462. doi:10.1002/humu.20933
Rashidghamat, E., Kadiyirire, T., Ayis, S., Petrof, G., Liu, L., Pullabhatla, V., et al. (2020). Phase I/II Open-Label Trial of Intravenous Allogeneic Mesenchymal Stromal Cell Therapy in Adults with Recessive Dystrophic Epidermolysis Bullosa. J. Am. Acad. Dermatol. 83 (2), 447–454. doi:10.1016/j.jaad.2019.11.038
Riedl, J., Pickett‐Leonard, M., Eide, C., Kluth, M. A., Ganss, C., Frank, N. Y., et al. (2021). ABCB5+ Dermal Mesenchymal Stromal Cells with Favorable Skin Homing and Local Immunomodulation for Recessive Dystrophic Epidermolysis Bullosa Treatment. Stem Cells 39 (7), 897–903. doi:10.1002/stem.3356
Roda, Â., Mendonça-Sanches, M., Travassos, A. R., Soares-de-Almeida, L., and Metze, D. (2017). Infliximab Therapy for Netherton Syndrome: A Case Report. JAAD Case Rep. 3 (6), 550–552. doi:10.1016/j.jdcr.2017.07.019
Schneider, H., Faschingbauer, F., Schuepbach-Mallepell, S., Körber, I., Wohlfart, S., Dick, A., et al. (2018). Prenatal Correction of X-Linked Hypohidrotic Ectodermal Dysplasia. N. Engl. J. Med. 378 (17), 1604–1610. doi:10.1056/NEJMoa1714322
Schräder, N. H. B., Duipmans, J. C., Molenbuur, B., Wolff, A., and Jonkman, M. F. (2019). Combined THC and CBD to Treat Pain in EB. Brit. J. Dermatol. 180, 922–924.
Sebastiano, V., Zhen, H. H., Haddad, B., Derafshi, B. H., Bashkirova, E., Melo, S. P., et al. (2014). Human COL7A1-Corrected Induced Pluripotent Stem Cells for the Treatment of Recessive Dystrophic Epidermolysis Bullosa. Sci. Transl Med. 6 (264), 264ra163. doi:10.1126/scitranslmed.3009540
Serena Tommasini-Ghelfi, W. A. (1975). Cancer-Associated Mutation and Beyond: The Emerging Biology of Isocitrate Dehydrogenases in Human Disease. Biochem. Biophys. Res. Commun. 66 (4), 1349–1356.
Shabbir, A., Cox, A., Rodriguez-Menocal, L., Salgado, M., and Van Badiavas, E. (2015). Mesenchymal Stem Cell Exosomes Induce Proliferation and Migration of Normal and Chronic Wound Fibroblasts, and Enhance Angiogenesis In Vitro. Stem Cell Dev 24 (14), 1635–1647. doi:10.1089/scd.2014.0316
Shehadeh, W., Sarig, O., Bar, J., Sprecher, E., and Samuelov, L. (2020). Treatment of Epidermolysis Bullosa Pruriginosa-Associated Pruritus with Dupilumab. Br. J. Dermatol. 182 (6), 1495–1497. doi:10.1111/bjd.18855
Shinkuma, S., Guo, Z., and Christiano, A. M. (2016). Site-specific Genome Editing for Correction of Induced Pluripotent Stem Cells Derived from Dominant Dystrophic Epidermolysis Bullosa. Proc. Natl. Acad. Sci. U S A. 113 (20), 5676–5681. doi:10.1073/pnas.1512028113
Signa, S., Campione, E., Rusmini, M., Chiesa, S., Grossi, A., Omenetti, A., et al. (2019). Whole Exome Sequencing Approach to Childhood Onset Familial Erythrodermic Psoriasis Unravels a Novel Mutation of CARD14 Requiring Unusual High Doses of Ustekinumab. Pediatr. Rheumatol. Online J. 17 (1), 38. doi:10.1186/s12969-019-0336-3
Steuer, A. B., and Cohen, D. E. (2020). Treatment of Netherton Syndrome with Dupilumab. JAMA Dermatol. 156 (3), 350–351. doi:10.1001/jamadermatol.2019.4608
Supp, D. M., Hahn, J. M., Combs, K. A., McFarland, K. L., Schwentker, A., Boissy, R. E., et al. (2019). Collagen VII Expression Is Required in Both Keratinocytes and Fibroblasts for Anchoring Fibril Formation in Bilayer Engineered Skin Substitutes. Cel Transpl. 28 (9–10), 1242–1256. doi:10.1177/0963689719857657
Suzuki, S., Nomura, T., Miyauchi, T., Takeda, M., Fujita, Y., Nishie, W., et al. (2019). Somatic Recombination Underlies Frequent Revertant Mosaicism in Loricrin Keratoderma. Life Sci. Alliance 2 (1). doi:10.26508/lsa.201800284
Süßmuth, K., Traupe, H., Loser, K., Ständer, S., Kessel, C., Wittkowski, H., et al. (2021). Response to Dupilumab in Two Children with Netherton Syndrome: Improvement of Pruritus and Scaling. J. Eur. Acad. Dermatol. Venereol. 35 (2), e152–5.
Swartling, C., Karlqvist, M., Hymnelius, K., Weis, J., and Vahlquist, A. (2010). Botulinum Toxin in the Treatment of Sweat-Worsened Foot Problems in Patients With Epidermolysis Bullosa Simplex and Pachyonychia Congenita. Br. J. Dermatol. 163 (5), 1072–1076. doi:10.1111/j.1365-2133.2010.09927.x
Takashima, S., Shinkuma, S., Fujita, Y., Nomura, T., Ujiie, H., Natsuga, K., et al. (2019). Efficient Gene Reframing Therapy for Recessive Dystrophic Epidermolysis Bullosa with CRISPR/Cas9. J. Invest. Dermatol. 139 (8), 1711–e4. doi:10.1016/j.jid.2019.02.015
Tamai, K., and Uitto, J. (2016). Stem Cell Therapy for Epidermolysis Bullosa-Does it Work. J. Invest. Dermatol. 136 (11), 2119–2121. doi:10.1016/j.jid.2016.07.004
Tang, J. Y., Mackay-Wiggan, J. M., Aszterbaum, M., Yauch, R. L., Lindgren, J., Chang, K., et al. (2012). Inhibiting the Hedgehog Pathway in Patients With the Basal-Cell Nevus Syndrome. N. Engl. J. Med. 366 (23), 2180–2188. doi:10.1056/NEJMoa1113538
Teng, J. M. C., Bartholomew, F. B., Patel, V., and Sun, G. (2018). Novel Treatment of Painful Plantar Keratoderma in Pachyonychia Congenita Using Topical Sirolimus. Clin. Exp. Dermatol. 43 (8), 968–971. doi:10.1111/ced.13686
Teramae, A., Kobayashi, Y., Kunimoto, H., Nakajima, K., Suzuki, T., Tsuruta, D., et al. (2019). The Molecular Basis of Chemical Chaperone Therapy for Oculocutaneous Albinism Type 1A. J. Invest. Dermatol. 139 (5), 1143–1149. doi:10.1016/j.jid.2018.10.033
Thomas, B. R., and Sahota, A. (2020). Pachyonychia Congenita and Botulinum Toxin. Br. J. Dermatol. 182 (3), 531–532. doi:10.1111/bjd.18784
Tolar, J., McGrath, J. A., Xia, L., Riddle, M. J., Lees, C. J., Eide, C., et al. (2014). Patient-specific Naturally Gene-Reverted Induced Pluripotent Stem Cells in Recessive Dystrophic Epidermolysis Bullosa. J. Invest. Dermatol. 134 (5), 1246–1254. doi:10.1038/jid.2013.523
Tolar, J., and Wagner, J. E. (2013). Allogeneic Blood and Bone Marrow Cells for the Treatment of Severe Epidermolysis Bullosa: Repair of the Extracellular Matrix. Lancet 382 (9899), 1214–1223. doi:10.1016/S0140-6736(13)61897-8
Uitto, J., Christiano, A. M., McLean, W. H., and McGrath, J. A. (2012). Novel Molecular Therapies for Heritable Skin Disorders. J. Invest. Dermatol. 132 (3), 820–828. doi:10.1038/jid.2011.389
Valentin, F., Wiegmann, H., Tarinski, T., Nikolenko, H., Traupe, H., Liebau, E., et al. (2020). Development of a Pathogenesis-Based Therapy for Peeling Skin Syndrome Type 1. Br. J. Dermatol.
Van Damme, A., Seront, E., Dekeuleneer, V., Boon, L. M., and Vikkula, M. (2020). New and Emerging Targeted Therapies for Vascular Malformations. Am. J. Clin. Dermatol. 21 (5), 657–668. doi:10.1007/s40257-020-00528-w
Vanden Oever, M., Muldoon, D., Mathews, W., McElmurry, R., and Tolar, J. (2016). miR-29 Regulates Type VII Collagen in Recessive Dystrophic Epidermolysis Bullosa. J. Invest. Dermatol. 136 (10), 2013–2021. doi:10.1016/j.jid.2016.05.115
Vasher, M., Smithberger, E., Lien, M. H., and Fenske, N. A. (2010). Familial Pityriasis Rubra Pilaris: Report of a Family and Therapeutic Response to Etanercept. J. Drugs Dermatol. 9 (7), 844–850.
Vélez-Cruz, R., Zadorin, A. S., Coin, F., and Egly, J. M. (2013). Sirt1 Suppresses RNA Synthesis after UV Irradiation in Combined Xeroderma Pigmentosum Group D/Cockayne Syndrome (XP-D/CS) Cells. Proc. Natl. Acad. Sci. U S A. 110 (3), E212–E220. doi:10.1073/pnas.1213076110
Venot, Q., Blanc, T., Rabia, S. H., Berteloot, L., Ladraa, S., Duong, J. P., et al. (2018). Targeted Therapy in Patients With PIK3CA-related Overgrowth Syndrome. Nature. 558 (7711), 540–546. doi:10.1038/s41586-018-0217-9
Venugopal, S. S., Yan, W., Frew, J. W., Cohn, H. I., Rhodes, L. M., Tran, K., et al. (2013). A Phase II Randomized Vehicle-Controlled Trial of Intradermal Allogeneic Fibroblasts for Recessive Dystrophic Epidermolysis Bullosa. J. Am. Acad. Dermatol. 69 (6), 898–e7. doi:10.1016/j.jaad.2013.08.014
Vermeesch, J. R., Voet, T., and Devriendt, K. (2016). Prenatal and Pre-implantation Genetic Diagnosis. Nat. Rev. Genet. 17 (10), 643–656. doi:10.1038/nrg.2016.97
Volc, S., Maier, L., Gritsch, A., Aichelburg, M. C., and Volc-Platzer, B. (2020). Successful Treatment of Netherton Syndrome with Ustekinumab in a 15-Year-Old Girl. Br. J. Dermatol. 183 (1), 165–167. doi:10.1111/bjd.18892
Wagner, J. E., Ishida-Yamamoto, A., McGrath, J. A., Hordinsky, M., Keene, D. R., Woodley, D. T., et al. (2010). Bone Marrow Transplantation for Recessive Dystrophic Epidermolysis Bullosa. N. Engl. J. Med. 363 (7), 629–639. doi:10.1056/NEJMoa0910501
Wagner, R. N., Hofbauer, J. P., Wally, V., Kofler, B., Schmuth, M., Rosa, L. D., et al. (2021). Epigenetic and Metabolic Regulation of Epidermal Homeostasis. Exp. Dermatol. 30 (8), 1009–1022. doi:10.1111/exd.14305
Walker, J. A., and Upadhyaya, M. (2018). Emerging Therapeutic Targets for Neurofibromatosis Type 1. Expert Opin. Ther. Targets 22 (5), 419–437. doi:10.1080/14728222.2018.1465931
Wally, V., Hovnanian, A., Ly, J., Buckova, H., Brunner, V., Lettner, T., et al. (2018). Diacerein Orphan Drug Development for Epidermolysis Bullosa Simplex: A Phase 2/3 Randomized, Placebo-Controlled, Double-Blind Clinical Trial. J. Am. Acad. Dermatol. 78 (5), 892–e7. doi:10.1016/j.jaad.2018.01.019
Wally, V., Klausegger, A., Koller, U., Lochmüller, H., Krause, S., Wiche, G., et al. (2008). 5' Trans-splicing Repair of the PLEC1 Gene. J. Invest. Dermatol. 128 (3), 568–574. doi:10.1038/sj.jid.5701152
Weon, J. L., and Glass, D. A. (2019). Novel Therapeutic Approaches to Xeroderma Pigmentosum. Br. J. Dermatol. 181 (2), 249–255. doi:10.1111/bjd.17253
Witting, M., Molina, M., Obst, K., Plank, R., Eckl, K. M., Hennies, H. C., et al. (2015). Thermosensitive Dendritic Polyglycerol-Based Nanogels for Cutaneous Delivery of Biomacromolecules. Nanomedicine 11 (5), 1179–1187. doi:10.1016/j.nano.2015.02.017
Wiegmann, H., Valentin, F., Tarinski, T., Liebau, E., Loser, K., Traupe, H., et al. (2019). LEKTI Domains D6, D7 and D8+9 Serve as Substrates for Transglutaminase 1: Implications for Targeted Therapy of Netherton Syndrome. Br. J. Dermatol. 181 (5), 999–1008. doi:10.1111/bjd.17820
Wong, T., Gammon, L., Liu, L., Mellerio, J. E., Dopping-Hepenstal, P. J., Pacy, J., et al. (2008). Potential of Fibroblast Cell Therapy for Recessive Dystrophic Epidermolysis Bullosa. J. Invest. Dermatol. 128 (9), 2179–2189. doi:10.1038/jid.2008.78
Woodley, D. T., Cogan, J., Hou, Y., Lyu, C., Marinkovich, M. P., Keene, D., et al. (2017). Gentamicin Induces Functional Type VII Collagen in Recessive Dystrophic Epidermolysis Bullosa Patients. J. Clin. Invest. 127 (8), 3028–3038. doi:10.1172/JCI92707
Woodley, D. T., Remington, J., Huang, Y., Hou, Y., Li, W., Keene, D. R., et al. (2007). Intravenously Injected Human Fibroblasts Home to Skin Wounds, Deliver Type VII Collagen, and Promote Wound Healing. Mol. Ther. 15 (3), 628–635. doi:10.1038/sj.mt.6300041
Woodley, D. T., Wang, X., Amir, M., Hwang, B., Remington, J., Hou, Y., et al. (2013). Intravenously Injected Recombinant Human Type VII Collagen Homes to Skin Wounds and Restores Skin Integrity of Dystrophic Epidermolysis Bullosa. J. Invest. Dermatol. 133 (7), 1910–1913. doi:10.1038/jid.2013.10
Wu, W., Lu, Z., Li, F., Wang, W., Qian, N., Duan, J., et al. (2017). Efficient In Vivo Gene Editing Using Ribonucleoproteins in Skin Stem Cells of Recessive Dystrophic Epidermolysis Bullosa Mouse Model. Proc. Natl. Acad. Sci. U S A. 114 (7), 1660–1665. doi:10.1073/pnas.1614775114
Yalcin, A. D. (2016). A Case of Netherton Syndrome: Successful Treatment with Omalizumab and Pulse Prednisolone and its Effects on Cytokines and Immunoglobulin Levels. Immunopharmacol Immunotoxicol 38 (2), 162–166. doi:10.3109/08923973.2015.1115518
Yarosh, D., Klein, J., O'Connor, A., Hawk, J., Rafal, E., and Wolf, P. (2001). Effect of Topically Applied T4 Endonuclease V in Liposomes on Skin Cancer in Xeroderma Pigmentosum: a Randomised Study. Xeroderma Pigmentosum Study Group. Lancet. 357 (9260), 926–929. doi:10.1016/s0140-6736(00)04214-8
Yehia, L., Ngeow, J., and Eng, C. (2019). PTEN-opathies: from Biological Insights to Evidence-Based Precision Medicine. J. Clin. Invest. 129 (2), 452–464. doi:10.1172/JCI121277
Zhao, Y., Gartner, U., Smith, F. J., and McLean, W. H. (2011). Statins Downregulate K6a Promoter Activity: A Possible Therapeutic Avenue for Pachyonychia Congenita. J. Invest. Dermatol. 131 (5), 1045–1052. doi:10.1038/jid.2011.41
Zhou, A. G., Little, A. J., and Antaya, R. J. (2021). Epidermolysis Bullosa Pruriginosa Treated with Dupilumab. Pediatr. Dermatol. 38 (2), 526–527. doi:10.1111/pde.14493
ABEs adenine base editors
AEC ankyloblepharon ectodermal defects cleft lip palate syndrome
AED anhidrotic ectodermal dysplasia/HED hypohidrotic ectodermal dysplasia
ARCI autosomal recessive congenital ichthyosis
BCC basal cell carcinoma
BM bone marrow
C7 type VII collagen
CARD 14
CBEs cytosine base editors
CRISPR/Cas9 clustered regularly interspaced short palindromic repeats
CS Cockayne Syndrome
CXCR4 C-X-C Motif Chemokine Receptor 4
DEJ dermo-epidermal junction
DSB double strand bond
EB epidermolysis bullosa (JEB junctional EB; DEB dystrophic EB)
EBS epidermolysis bullosa simplex
EDA ectodysplasin A
EGFR epidermal growth factor receptor
GLA
GOF gain of function
HB-EGF heparin-binding epidermal growth factor-like growth factor
HDR homology directed repair
HHD Hailey-Hailey disease
HMGB1 high mobility group box 1 protein
HSV human herpes virus
LOF loss of function loss of function
HI Harlequin ichthyosis
HPAE highly branched polyβ‐amino esters
ID intradermal
Indels insertions or deletions
IV intravenous
JAK
LAMB3:
LNPs lipid based nanoparticles
LOF loss of function loss of function
MAPK mitogen-activated protein kinase
MSC mesenchymal stem cells
NHEJ non-homologous end joining
NF1 neurofibromatosis type 1
NF-κB nuclear factor-κB
NIPAL4
NMD non sense meditated m-RNA decay
NOS2 nitric oxide synthase
NS Netherton syndrome
OCA oculocutaneous albinism
PDGFRα platelet-derived growth factor receptor-α
PPK palmoplantar keratoderma
PRP pityriasis rubra pilaris
PTC premature termination codons
SDF1a stromal cell derived factor 1a
SERPINB7
SMO smoothened oncogene
SCC squamous cell carcinoma
STAT
TALEN transcription-activator like effector nucleases
TGF-α (−β) transforming growth factor α (b)
TGM-1 Transglutaminase 1
tNG thermoresponsive nanogels
tRNA transfer RNA
TRPV3 Transient Receptor Potential Vanniloid-3
XP xeroderma pigmentosum (XPC type C XP)
ZFNs zinc finger nucleases
Keywords: genodermatoses, unmet medical needs, reoriented drugs, cell therapy, genetic engeneering, personalised medicine
Citation: Morren M-A, Legius E, Giuliano F, Hadj-Rabia S, Hohl D and Bodemer C (2022) Challenges in Treating Genodermatoses: New Therapies at the Horizon. Front. Pharmacol. 12:746664. doi: 10.3389/fphar.2021.746664
Received: 24 July 2021; Accepted: 29 November 2021;
Published: 05 January 2022.
Edited by:
Timothy Martin Cox, University of Cambridge, United KingdomReviewed by:
Robert Sarkany, Guy’s and St Thomas’ NHS Foundation Trust, United KingdomCopyright © 2022 Morren, Legius, Giuliano, Hadj-Rabia, Hohl and Bodemer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marie-Anne Morren, bWFyaWUtYW5uZS5tb3JyZW5AY2h1di5jaA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.