Commentary: Dapagliflozin Mediates Plin5/PPARα Signaling Axis to Attenuate Cardiac Hypertrophy
 Jing Yu
Jing Yu Huanhuan Zhao
Huanhuan Zhao Xin Qi2,3
Xin Qi2,3 Liping Wei
Liping Wei Hao Wu
Hao Wu- 1Department of Physiology and Pathophysiology, Tianjin Medical University, Tianjin, China
- 2Department of Cardiology, Tianjin Union Medical Center, Nankai University Affiliated Hospital, Tianjin, China
- 3Nankai University School of Medicine, Tianjin, China
Objective: The purpose of this study was to investigate the effect of dapagliflozin (DAPA), a sodium-glucose cotransporter 2 inhibitor, on relieving cardiac hypertrophy and its potential molecular mechanism.
Methods: Cardiac hypertrophy induced by abdominal aortic constriction (AAC) in mice, dapagliflozin were administered in the drinking water at a dose of 25 mg/kg/d for 12 weeks was observed. Echocardiography was used to detect the changes of cardiac function, including LVEF, LVFS, LVEDd, LVEDs, HR and LV mass. Histological morphological changes were evaluated by Masson trichrome staining and wheat germ agglutinin (WGA) staining. The enrichment of differential genes and signal pathways after treatment was analyzed by gene microarray cardiomyocyte hypertrophy was induced by AngII (2 μM) and the protective effect of dapagliflozin (1 μM) was observed in vitro. The morphological changes of myocardial cells were detected by cTnI immunofluorescence staining. ELISA and qRT-PCR assays were performed to detect the expressions levels of cardiac hypertrophy related molecules.
Results: After 12 weeks of treatment, DAPA significantly ameliorated cardiac function and inhibited cardiac hypertrophy in AAC-induced mice. In vitro, DAPA significantly inhibited abnormal hypertrophy in AngII-induced cardiacmyocytes. Both in vivo and in vitro experiments have confirmed that DAPA could mediate the Plin5/PPARα signaling axis to play a protective role in inhibiting cardiac hypertrophy.
Conclusion: Dapagliflozin activated the Plin5/PPARα signaling axis and exerts a protective effect against cardiac hypertrophy.
Introduction
Cardiovascular diseases, especially acute myocardial infarction and atherosclerosis, remain the leading cause of death worldwide. In China, the prevalence of cardiovascular disease is rising, causing nearly 4 million deaths in 2016 alone (Liu et al., 2019). Epidemiological studies showed the incidence of cardiac hypertrophy is high worldwide, with the prevalence of about 1/500 (Wasserstrum et al., 2019). Cardiac hypertrophy is the basis of abnormal ventricular remodeling and plays a vital role in the development of most cardiovascular diseases (Pichler et al., 2020). To some extent, elucidating the pathological mechanism of cardiac hypertrophy provides theoretical support for targeted treatment strategies for cardiovascular diseases. Thus, it is urgent to explore the molecular mechanisms underlying cardiac hypertrophy and to seek the effective strategies. Pathological cardiac hypertrophy that occurs in cardiovascular disease is often accompanied by inflammation and myocardial fibrosis, ultimately leading to heart failure (Tham et al., 2015). In addition, the typical pathological basis of cardiac hypertrophy includes the accumulation and deposition of excessive extracellular matrix (ECM) and abnormal cardiomyocyte hypertrophy. At present, the drugs used in clinical treatment of cardiac hypertrophy mainly include angiotensin-converting enzyme inhibitors (e.g., Captopril) (Zhang et al., 2019), angiotensin II receptor blockers (e.g., Losartan) (Gonzalez et al., 2021) and calcium channel blocker (e.g., Amlodipine) (Lu et al., 2016).
As a novel class of hypoglycemic agent, sodium-glucose co-transporter 2 (SGLT-2) inhibitors (e.g., empagliflozin, dapagliflozin) prevented the reabsorption of glucose and sodium from the proximal convoluted tubules, resulting in glycourine and sodium-rich properties. Dapagliflozin increased the amount of glucose excreted in the urine and improved fasting and postprandial blood glucose levels in patients with T2D (Dhillon, 2019). In addition, it has been reported that dapagliflozin has been widely used in the clinical treatment of diabetes and cardiovascular disease. Empagliflozin (EMPA) has been reported to improve cardiac remodeling and cardiac metabolism and ATP homeostasis in rats with myocardial infarction (Yurista et al., 2019). Excitingly, Ye et al. (2017) found that DAPA has a potential cardiovascular protective effect in the treatment of diabetes. Numerous studies (Aimo et al., 2020; Zannad et al., 2020) have shown the importance of DAPA in reducing hospitalization for heart failure in patients with HFrEF and suggest that DAPA may also reduce the risk of cardiovascular events and cardiovascular mortality associated with diabetes. Besides, Lahnwong et al. (2020) reported that acute administration of DAPA could play a protective role by reducing myocardial infarction size and improving left ventricular function in myocardial ischemia/reperfusion rats. It was reported (Zhang et al., 2021) that DAPA ameliorated Ang II-induced cardiac remodeling by regulating TGF-β1/Smad signaling pathway. Despite the potential cardioprotective effects of DAPA has been widely proven, its mechanisms against cardiac hypertrophy remain unclear.
Modern pharmacology reported (Sharma et al., 2004; Chen et al., 2015) that with the development of the pathological process of cardiac hypertrophy, cardiac energy metabolism is disturbed, fatty acid utilization is limited, and the accumulation of lipids will aggravate the occurrence of cardiac hypertrophy and heart failure. Recent studies (Martin and Parton, 2006; Wang et al., 2015) have shown that as dynamic lipid storage organelles, lipid droplets (LDs) is regarded as a key regulator of lipid metabolism in various kinds of cells including adipocytes and cardiomyocytes. The perilipin family of proteins is a classic LDs related protein, among which perilipin 5 (Plin5) is highly expressed in heart, liver and other tissues (Wolins et al., 2006). Wang et al. (2015) confirmed that Plin5 deficiency increased liver lipid metabolism, promoted mitochondrial proliferation, and led to ROS burst. Plin5 deficiency also increased the oxidation of fatty acids in the heart, leading to cardiac dysfunction. Besides, Plin5 deficiency has been reported to exacerbate TAC-induced cardiac hypertrophy and heart failure. Peroxisome proliferator-activated receptor-α (PPARα) is a ligand-activated transcription factor, a member of the nuclear hormone receptor superfamily, which is known for its critical role in the transcriptional regulation of lipid metabolism (Smeets et al., 2008). Peroxisome proliferator-activated receptors (PPARs) are members of the ligand-activated nuclear receptor superfamily and are the key transcriptional regulators that control the capacity for myocardial mitochondrial fatty acid oxidation (FAO) (Stanley et al., 2005). PPARs consists of three member subfamilies: PPAR-α, PPAR-β and PPAR-γ (Montaigne et al., 2021). PPAR-α is highly expressed in cardiomyocytes, and it regulates the expression of key components in fatty acid uptake, esterification, and oxidation through transcriptional activation of genes encoding key proteins in the signaling pathway, and maintains metabolic homeostasis in cardiomyocytes (Kar and Bandyopadhyay, 2018). Loss of PPAR-α has been shown to lead to more pronounced hypertrophic growth and cardiac dysfunction, suggesting a key regulatory role for PPAR-α in cardiac hypertrophy (Smeets et al., 2008). PPARα activator attenuated cardiac hypertrophy by negatively regulating the binding activity of activated protein-1 (AP-1) (Jia et al., 2014; Shimojo et al., 2014) confirmed that Fenofibrate protected against cardiac hypertrophy by activating the PPAR-α signaling and ameliorating myocardial energy metabolism.
Considering the molecular mechanism of Plin5 and PPARα in cardiac hypertrophy, we hypothesized that DAPA might mediate the protective effect of Plin5/PPARα signaling axis. Therefore, we performed the present study to confirm the effect of DAPA towards cardiac hypertrophy in AAC-induced mice and to explore the regulatory effect on Plin5/PPARα signal axis. Our data revealed that the molecular mechanisms by which DAPA attenuated cardiac hypertrophy, which is of great significance for clinical use.
Methods
Animals and Groups
Eight-week-old male C57BL/6 mice (23 ± 2 g) were obtained from Beijing Vital River Laboratory Animal Technology Co., Ltd. The mice were kept in wages and given free access to food and water. Mice were housed under suitable conditions, including 12-h light/dark cycle, temperature at 25 ± 2°C and relative humidity 50–60%. Follow-up experiments were started after 1 week of adaptive feeding. Twenty-four mice were randomly divided into three groups: sham group (n = 8), AAC group (n = 8) and dapagliflozin treatment group (n = 8). Sham group and AAC group were treated with saline for 12 weeks. In the treatment group, after the establishment of AAC model, dapagliflozin was administered in the drinking water at a dose of 25 mg/kg/d for 12 weeks (Kraakman et al., 2017). All the animal testing procedures covered in this study complied with the guidelines of the Care and Use of Laboratory Animals and were approved by the Laboratory Animal Center of Tianjin Medical University. All procedures and care methods minimize the suffering of the mice.
Abdominal Aortic Constriction Model
AAC mice were performed as previously described. (Duan et al., 2007). Briefly, after anesthesia with 2% isoflurane, the mice were placed in the supine position on the platform and cut along the midline of the abdomen to expose the abdominal cavity. Subsequently, the abdominal aorta was ligated with 7-0 silk sutures with a 27G needle. The degree of AAC was controlled by needle removal under suture to ensure the stability and uniformity of the operation. Close the abdominal cavity, carefully suture the skin, and clean the wound. In the sham operation, the silk was removed without ligation. After the operation, the mice were injected with penicillin and put back into the cage when they woke up.
Echocardiography
After 12 weeks of continuous administration, the mice were anesthetized with 2% isoflurane, and cardiac function was detected by echocardiography. As previously reported (Chai et al., 2020), Doppler images of the lateral and septal mitral annulus were recorded. Heart rate (HR) was recorded by synchronized electrocardiography. Left ventricular ejection fraction (LVEF), fraction shortening (LVFS), left ventricular end-diastolic volume (LVEDV), left ventricular end-systolic volume (LVESV) were calculated.
Microarray Profiling
After 12 weeks of treatment, the total RNA was isolated from the heart tissue using Trizol reagent (Invitrogen, CA, United States). Determine the concentration of total RNA by fluorescence-based quantitation using an RNA RiboGreen® dye assay (e.g., Quant-iT™ RiboGreen® RNA Reagent and Kit) and the NanoDrop Fluorospectrometer for initial RNA concentration of 5 pg/μL to 1 ng/μL (www.nanodrop.com). RNA integrity was tested using the Agilent 2,100 Bioanalyzer with an RNA LabChip Kit. Global transcriptome analysis of total RNA (50 ng/μl) was performed using the Affymetrix GeneChip Primeview™, according to the manufacturer’s instructions. Hybridization, staining, normalization and data analysis were performed following the standard protocol established by Agilent Technologies, Inc.
Morphological and Histological Analysis
The mice were anesthetized, the hearts were immediately removed, washed with PBS, and fixed with 4% paraformaldehyde for 24 h. Then paraffin-embedded sections with a thickness of 3–5 μm were stained according to Masson trichrome staining, and wheat germ agglutinin (WGA) staining (Zhu et al., 2018). Masson trichrome staining was performed to evaluate cardiac fibrosis. WGA staining was used to assess the cell size. For WGA staining, briefly, the slides were stained with WGA-FITC labeled antibodies for 30 min at room temperature. The nuclei were stained with DAPI for 5 min. For each analysis, six fields were randomly selected for each sample under the microscope. All histomorphological analyses were performed by pathologists who were unaware of the experimental grouping design.
Cell Culture and Treatment
Neonatal mouse cardiac myocytes (NMCMs) were isolated from hearts of 1–3 days-old C57BL/6 mice according to the previous report, then the heart was cut into small pieces (Bei et al., 2019). Briefly, the heart was immediately removed and washed with ice-cold PBS. Next the cardiac myocytes were cultured in DMEM (Dulbecco’s modified Eagle’s Medium, Gibco) containing 10% FBS (fetal bovine serum). Then, NMCMs were inoculated in two 25 cm2 culture flasks and incubated in 5% CO2 for 1 h to isolate cardiomyocytes and cardiomyocytes. The purified mouse primary cardiomyocytes (NMCMs) were cultured for 3 days, which was used for the subsequent experiment. The cells were divided into three groups: control group, AngII group and dapagliflozin group. After the starvation for 4 h, the control group was given a normal medium for 48 h. The cells in AngII group were treated with AngII (2 μM) for 48 h, the medium was changed every 24 h. In the dapagliflozin group, NMCMs were treated with AngII (2 μM) and dapagliflozin (1 μM) for 48 h, the medium was changed every 24 h (Uthman et al., 2018).
RNA Interference and Groups
After 24 h of cell culture, the purified cardiomyocytes were replaced with the fresh complete medium containing a cardiomyocyte growth supplement. After 24 h, the NMCMs were washed with PBS, and cells were transfected with 1 μM of Lipofectamine 2000 (Invitrogen, CA, United States) and siRNA (si-NC, si-negative control or si-Plin5, Guangzhou RiboBioCo., Ltd., China) in Opti-MEM® I Reduced-Serum Medium according to the manufacturer instructions. 6 h later, cells were treated with AngII (2 μM) and dapagliflozin (1 μM), DMSO or GW6471 (10 μM) for 24 h. The cells were then washed with PBS, harvested, lysed with TRIzol and stored in an −80°C refrigerator for subsequent protein and RNA detection. This part of the experiment is divided into the following groups:
A: AngII + DAPA (dapagliflozin)
B: AngII + DAPA (dapagliflozin) + DMSO
C: AngII + DAPA (dapagliflozin) + DMSO + GW6471 (PPARα inhibitor)
D: AngII + DAPA (dapagliflozin) + si-NC
E: AngII + DAPA (dapagliflozin) + si-Plin5
Immunofluorescence Staining
Cultured NMCM cells with a density of 1 × 105/mL were harvested and allowed to air dry naturally on the slides. Then the cells were washed with PBS three times and fixed for 15 min with 4% paraformaldehyde at room temperature. 2% bovine serum albumin (BSA) was added to block nonspecific binding at room temperature for 2 h. Then, cTnI antibody was incubated at 37°C for 3 h. After PBS washing, the cells were incubated with biotinylated goat anti-mouse IgG as the secondary antibody, incubated at 37°C for 1 h. Subsequently, the slides were washed with PBS for three times. And the nucleus was counterstained with DAPI. Finally, the cells were observed and photographed under a fluorescence microscope (Olympus, Tokyo, Japan).
Western Blotting Assay
Heart tissue or NMCMs were lysed in RIPA buffer with PMSF, protease and phosphatase inhibitors. Protein samples were isolated after centrifugation, and the protein concentration was determined by the BCA kit. Protein samples were diluted to 10 μg/μL by PBS, and then stored in −80°C refrigerator for later use. The equal amount of protein was separated in 10% SDS-PAGE gel, transferred to PVDF membranes and blocked with 5% BSA. PVDF membranes were incubated with primary antibodies (STAT1, ab109461; PPARα, ab126285; HMGCS2, ab137043; PDK4, ab214938; GAPDH, ab181602, Abcam, United Kingdom; Plin5, A20418, Abclonal, Wuhan, China) at 4°C overnight. After washing 3 times with TBST and incubated with secondary antibodies at room temperature for 2 h. The bands were visualized with enhanced chemiluminescence (ECL kit) in dark. Then image J software was used to measure the gray value of the target protein bands. GAPDH was used as the loading control. This section of the experiment was repeated independently at least three times.
Quantitative Real-Time Polymerase Chain Reaction Analysis
Total RNA was isolated from NMCMs and cardiac tissue using Trizol according to standard protocols. The RNA was then determined for purity and concerntration, and cDNAs were synthesized with 500 ng RNA using a reverse transcription kit. qRT-PCR analysis was performed using Takara SYBR Premix Ex Taq™ on Roche LightCycler480 PCR System. The mRNA expression was calculated by −2ΔΔCT method, and GAPDH was used as the loading control.
Enzyme-Linked Immunosorbent Assay
The contents of ANP, BNP, β-MHC and cTnI in the serum were detected by ELISA kits (Zhang et al., 2019). The test was conducted according to the manufacturers’ instructions.
Statistical Analysis
The data in the study are presented as the means ± SD, and the data was analyzed using one-way ANOVA followed by the Student t test for multiple comparisons by SPSS 22.0 software. The p value of less than 0.05 was considered statistically significant.
Results
Protective Effects of Dapagliflozin on AAC-Induced Cardiac Hypertrophy in Mice
After 12 weeks of treatment with dapagliflozin, the protective effect of dapagliflozin on AAC-induced cardiac hypertrophy was observed. As shown in Figures 1A–C, we found compared with sham group, the left ventricular ejection fraction (LVEF) and fraction shortening (LVFS) in AAC model group were significantly inhibited, indicating a significant decrease in cardiac function in AAC-induced mice. After dapagliflozin treatment, the LVEF and LVFS in the treatment group were improved significantly compared with AAC group. Furthermore, LV mass in DAPA group was reduced markedly compared with AAC group (Figure 1D). According to the echocardiography results (Figures 1E–G), we also obtained HR, LVEDV and LVESV in different groups. There was no significant difference in HR between AAC group and treatment group. While, LVEDV and LVESV in AAC group were significantly increased compared with those in sham group, which was consistent with previous reports of cardiac hypertrophy (Ye et al., 2020). After treatment, LVEDV and LVESV were decreased significantly compared with AAC group. The results suggested that dapagliflozin can significantly improve cardiac function in AAC-induced mice. Masson staining results (Figure 1H) showed that dapagliflozin significantly blunted the myocardial fibrosis. WGA staining results (Figure 1I) were used to detect the changes of cell size. Consistent with the H&E results, dapagliflozin significantly inhibited abnormal hypertrophy of cardiomyocytes after treatment. We observed the changes of the heart as a whole (Figures 1J,K), and we found that the abnormal enlargement of the heart induced by AAC was significantly improved in the treatment group. The ratio of HW to BW was increased in AAC-induced mice, which could be reversed by dapagliflozin. Taken together, we demonstrated that dapagliflozin attenuated cardiac hypertrophy and improved cardiac function.
FIGURE 1
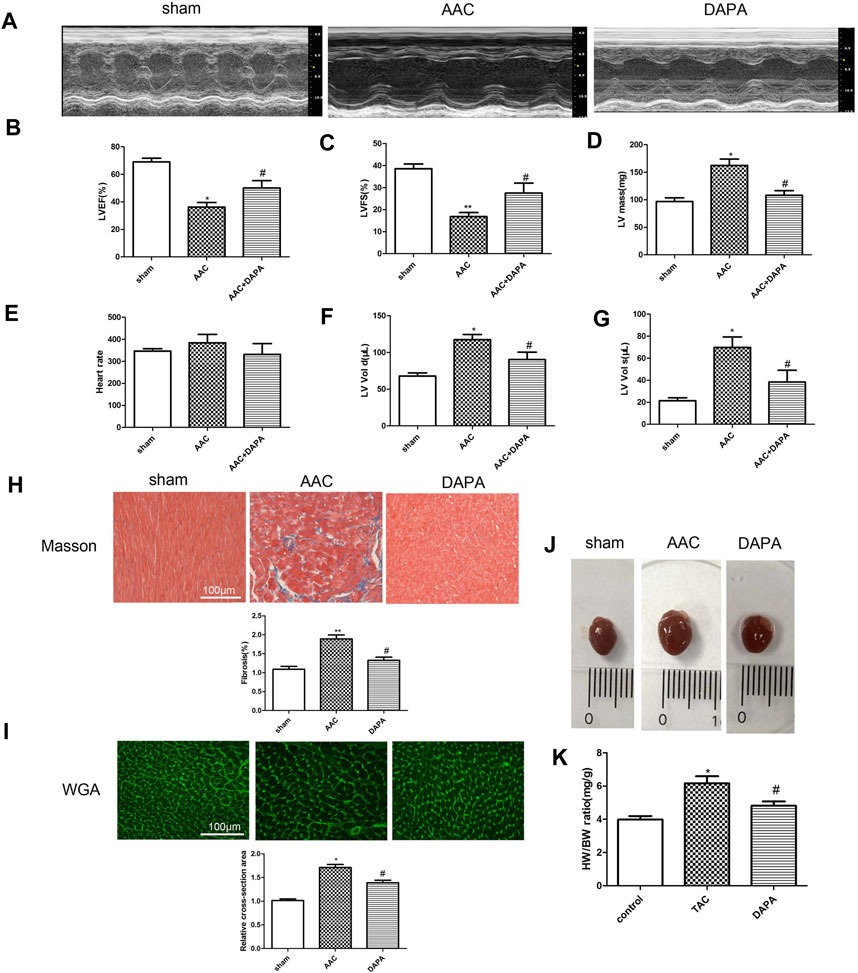
FIGURE 1. Dapagliflozin alleviated cardiac remodeling and improved cardiac function in AAC-induced mice (A) Representative M-mode echocardiography images. In AAC-induced mice, dapagliflozin improved ejection fractions (EF) (B) and fractional shortening (FS) (C), LV mass (D), Heart rate (E), LV end-diastolic volume, LVEDV (F), LV end-systolic volume, LVESV, n = 8 (G). Images of Masson-stained (H) and WGA-stained sections (I) in different groups, n = 3, original magnification, 400× (J) Images of whole heart from mice in different groups after 28 days. (K) The heart weight to body weight ratio was determined, n = 8. The data were expressed as mean ± SD, *p < 0.05, vs. sham group; #p < 0.05, vs. AAC group.
Dapagliflozin Down-Regulates the Levels of Genes Associated With Cardiac Hypertrophy in AAC-Induced Mice
In addition to the decreased cardiac function and the abnormal enlargement of the heart shape, cardiac hypertrophy is characterized by the activation of the fetal gene program, the abnormal up-regulated genes mainly including ANP, BNP, β-MHC and cTNI. In order to further confirm the effect of dapagliflozin on cardiac hypertrophy, ELISA assay was performed to detect the changes in the markers related to cardiac hypertrophy in serum of AAC-induced mice. As shown in Figures 2A–D, compared with sham group, the expression levels of ANP, BNP, β-MHC and cTNI in AAC group were significantly increased. After treatment, the expressions of markers were down-regulated by dapagliflozin. Moreover, qRT-PCR analysis was performed to detect the mRNA expression of ANP, BNP and β-MHC in heart tissues. Similarly, the qRT-PCR results (Figures 2E–G) were consistent with ELISA results. Based on the above results, we hypothesized that dapagliflozin could mitigate cardiac hypertrophy in AAC-induced mice.
FIGURE 2
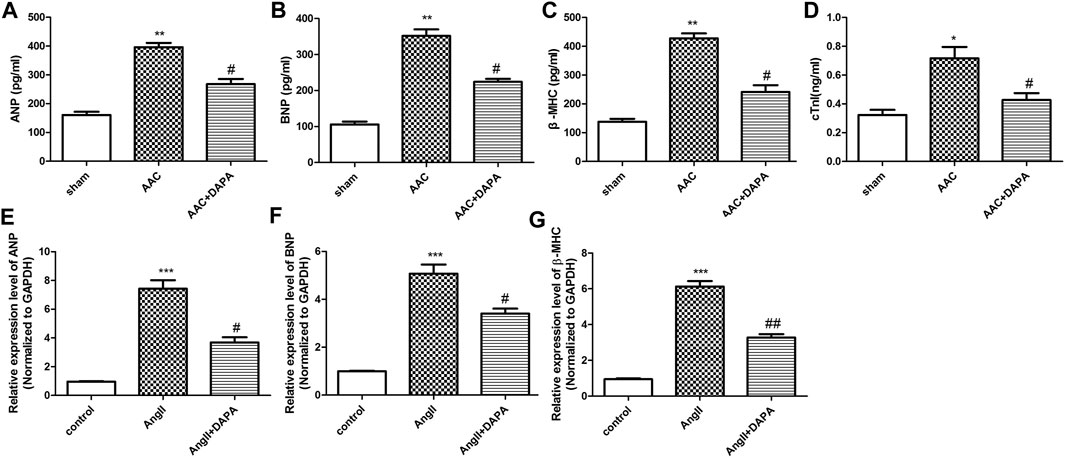
FIGURE 2. Effect of dapagliflozin on the level of cardiac hypertrophy-related genes in AAC-induced mice. The contents of ANP (A), BNP (B), β-MHC (C) and cTnI (D) in cardiac tissues of different groups were detected via ELISA kits. qRT-PCR analysis was performed to detect the mRNA expression of ANP (E), BNP (F) and β-MHC (G). The data were expressed as mean ± SD, *p < 0.05, vs. sham group; #p < 0.05, vs. AAC group.
Dapagliflozin Attenuated AngII-Induced Cardiomyocyte Hypertrophy
Stimulation of NMCMs by AngII could induce cardiac hypertrophy (Mele et al., 2019). Cardiac troponin I (cTnI) is an important structural protein and a cardiomyocyte-specific marker (Luo et al., 2021). Immunofluorescence staining results (Figure 3A) of NMCMs showed obvious hyperplasia of myocardial cells induced by AngII, suggesting that the cardiac hypertrophy model was successful. Cardiomyocyte hypertrophy was inhibited after dapagliflozin treatment, demonstrating that dapagliflozin attenuates cardiac hypertrophy in vitro. And, qRT-PCR analysis was performed to detect the expression level of genes related to cardiac hypertrophy and to evaluate the effect of dapagliflozin on cardiomyocyte hypertrophy in vitro. The results (Figures 3B–E) indicated that compared with control group, the mRNA expressions of Nppa, Nppb, Serca2 and myh7 in AngII group were up-regulated significantly. While the mRNA expressions of Nppa, Nppb, Serca2 and myh7 in treatment group was down-regulated markedly compared to AngII group. Taken together, the results indicated that dapagliflozin attenuated cardiomyocyte hypertrophy in vitro.
FIGURE 3
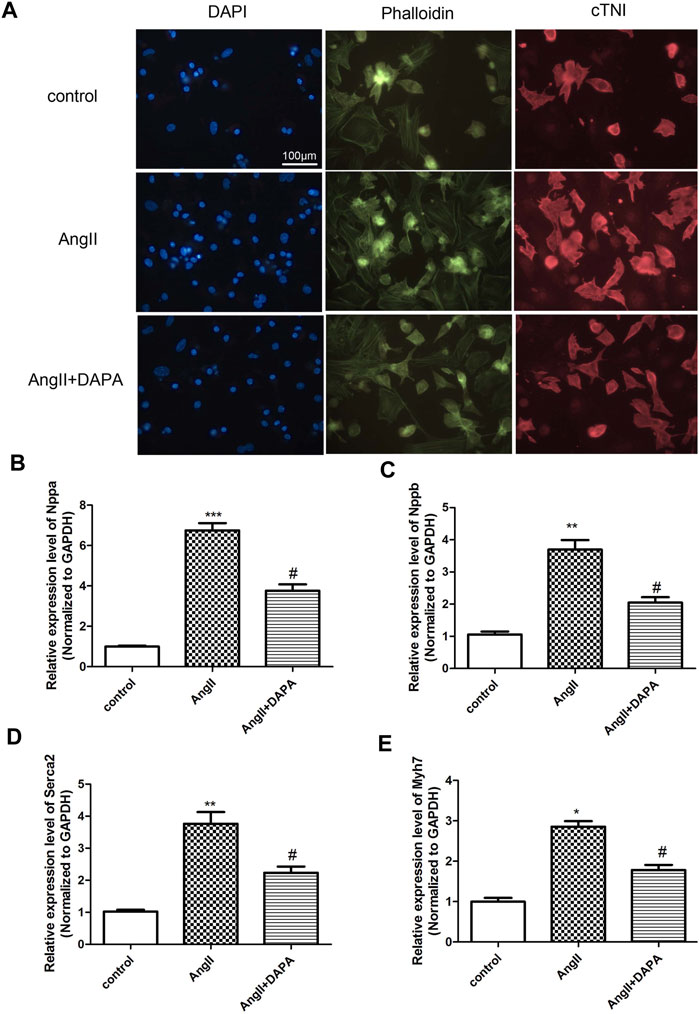
FIGURE 3. Dapagliflozin attenuated AngII-induced cardiomyocyte hypertrophy. (A) The morphological changes of cardiomyocyte cells were evaluated by immunofluorescence staining with anti-cTNI (red), cells were counterstained with DAPI (blue) staining the nucleus, Phalloidin-FITC staining the cytoskeleton (green) qRT-PCR analysis was used to detect the mRNA expression of Nppa (B), Nppb (C), Serca2 (D) and myh7 (E) in different groups. The data were expressed as mean ± SD in three independent experiments, n = 3, *p < 0.05, **p < 0.01, vs. control group; #p < 0.05, ##p < 0.01, vs. AngII group.
Differential Gene Analysis of Dapagliflozin in AAC-Induced Mice Detected by Gene Microarray
To further study the protective mechanism of dapagliflozin in AAC-induced mice, we conducted a microarray analysis. FC > 1.5 or FC < −1.5 represented significant differential expression. As shown in Figure 4A, compared with AAC group, we found 2014 genes changed significantly in the dapagliflozin treatment group, among which 941 genes were up-regulated and 1,073 genes were down-regulated. Besides, signaling pathway enrichment analysis showed the PPAR signal pathway was significantly down-regulated in AAC-induced mice compared with sham group. While PPAR signaling pathway was up-regulated in treatment group compared with AAC group. In addition, we have identified some key differential genes (Table 1), which may be key signaling molecules for the protective effects of dapagliflozin. In subsequent experiments, we explored the mechanism of dapagliflozin by targeting signaling pathways and key molecules.
FIGURE 4
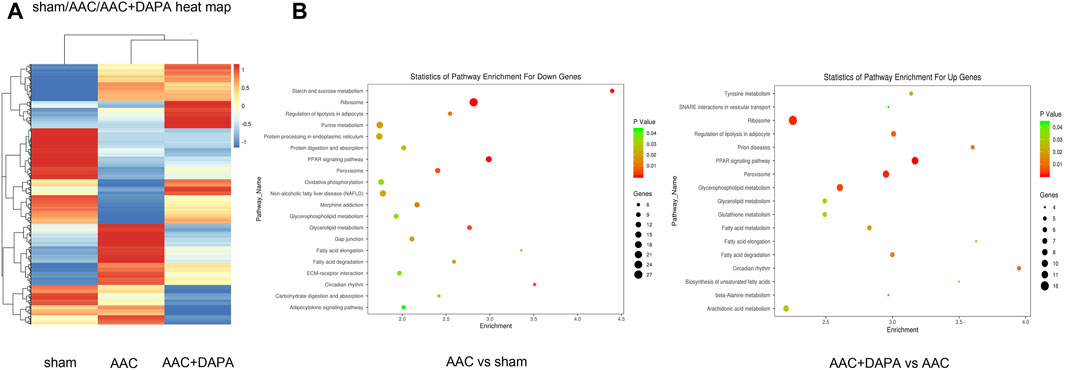
FIGURE 4. Differential gene analysis of dapagliflozin in AAC-induced mice detected by gene microarray (A) Clustered heat map showed the differential gene expression profile after dapagliflozin treatment in AAC-induced mice. (B) Signaling pathway enrichment analysis among different groups, the PPAR signal pathway (red line) is the signal pathway of this research.
TABLE 1
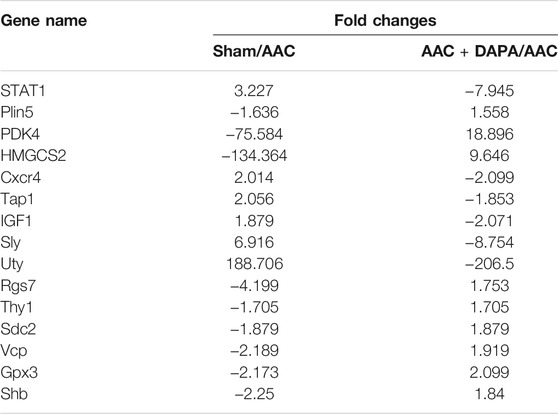
TABLE 1. Representative differential genes after dapagliflozin treatment in AAC-induced mice (Fc < 0, down-regulated; Fc > 0, up-regulated).
Dapagliflozin Mediated the Plin5/PPARα Signaling Axis to Mitigate AngII-Induced Cardiomyocyte Hypertrophy
Combined with the differential genes screened by microarray, qRT-PCR analysis and western blotting assay were used for in vitro validation. We identified the differential gene Plin5, STAT1 and PPARα signaling pathways for verification. As shown in Figures 5A–E, qRT-PCR results showed that compared with control group, the mRNA expression of STAT1 was up-regulated significantly in AngII group, while the expressions of Plin5, PPARα, HMGCS2 and PDK4 were down-regulated. After treatment, the mRNA expressions of STAT1, Plin5, PPARα, HMGCS2 and PDK4 were reversed by dapagliflozin. Interestingly, the qRT-PCR results matched the microarray results. Likewise, western blot results (Figures 5F–K) showed the same trends. To sum up the above results, we hypothesized that dapagliflozin mediated the Plin5/PPARα signaling axis to mitigate cardiac hypertrophy.
FIGURE 5
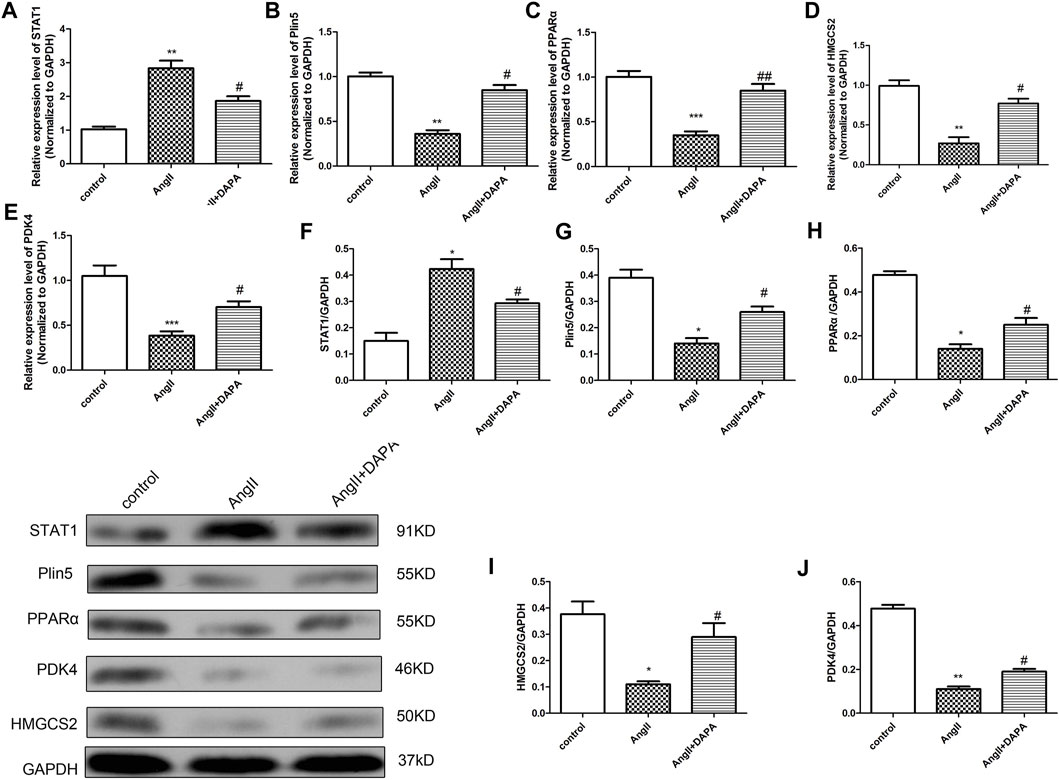
FIGURE 5. Dapagliflozin mediated the Plin5/PPARα signaling axis to mitigate AngII-induced cardiomyocyte hypertrophy. qRT-PCR analysis was used to detect the mRNA expressions of STAT1 (A), Plin5 (B), PPARα (C), HMGCS2 (D) and PDK4 (E) in cardiomyocytes of different groups. (F–J) Western blotting assay was performed to detect the protein expressions of STAT1, Plin5, PPARα, HMGCS2 and PDK4. The data were expressed as mean ± SD, n = 3, *p < 0.05, vs. control group; #p < 0.05, vs. AngII group.
Dapagliflozin Mediated the Plin5/PPARα Signaling Axis to Attenuate Cardiac Hypertrophy in vivo
To further clarify our hypothesis, we validated the expression of differential genes and signaling molecules in AAC-induced mice. QRT-PCR analysis and western blotting assay were performed to detect the mRNA and protein expressions of STAT1, Plin5, PPARα, HMGCS2 and PDK4 in heart tissues of different groups. Similarly, we found the same results (Figure 6) as in vitro experiment, confirming that dapagliflozin could attenuate cardiac hypertrophy.
FIGURE 6
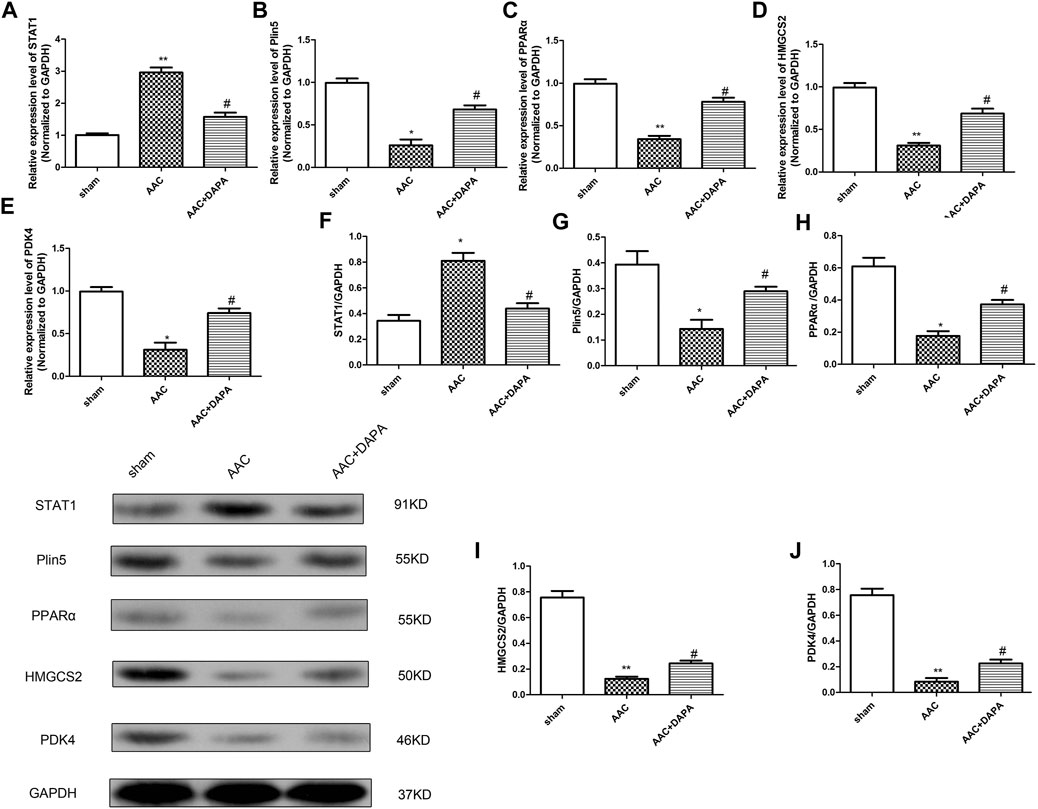
FIGURE 6. Dapagliflozin mediated the Plin5/PPARα signaling axis to attenuate cardiac hypertrophy in vivo. qRT-PCR analysis was used to detect the mRNA expressions of STAT1 (A), Plin5 (B), PPARα (C), HMGCS2 (D) and PDK4 (E) in cardiac tissues. (E) Western blotting assay was performed to detect the protein expressions of STAT1, Plin5, PPARα, HMGCS2 and PDK4. The data were expressed as mean ± SD, n = 3, *p < 0.05, vs. sham group; #p < 0.05, vs. AAC group.
Silence Plin5 or GW6471 Could Reverse the Protective Effect of Dapagliflozin-Mediated PPARα Signaling Axis in AngII-Induced Cardiomyocyte Hypertrophy
Finally, we investigated whether dapagliflozin mediated the Plin5/PPARα signaling axis to exert a protective effect against cardiac hypertrophy. Plin5 silencing and GW6471 (PPARα inhibitors) were used to reduce the expression of Plin5 and PPARα, respectively. The expression levels of Plin5/PPARα signal axis-related proteins were detected by western blotting assay. The results (Figures 7A–D) showed that compared with AngII + DAPA + DMSO group, the expressions of Plin5, PPAR-α, HMGCS2 and PDK4 in AngII + DAPA + GW6471 group were reduced significantly. Similarly, the expressions of Plin5, PPARα, HMGCS2 and PDK4 in AngII + DAPA + si-Plin5 were significantly down-regulated compared with AngII + DAPA + si-NC group. Western blot results suggested that si-Plin5 and GW6471 could reverse the expression levels of Plin5/PPARα signaling axis in AngII-induced cardiomyocyte. Thus, we speculated that dapagliflozin could up-regulate the expressions of Plin5/PPARα and further promote the expression levels of PDK4 and HMGCS2. Subsequently, ELISA and qRT-PCR assays were used to detect the expressions of cardiac hypertrophy-related molecules and verify the effect of si-Plin5 and GW6471 on the protective effect of dapagliflozin. ELISA results (Figures 7E–H) showed that compare to AngII + DAPA + DMSO group, the contents of ANP, BNP, β-MHC and cTNI in AngII + DAPA + GW6471 group were significantly increased. The contents of ANP, BNP, β-MHC and cTNI in AngII + DAPA + si-Plin5 were increased markedly compared with that in AngII + DAPA + si-NC group. Furthermore, qRT-PCR results (Figures 7I–L) showed that compared to AngII + DAPA + DMSO group, the mRNA expressions of Nppa, Nppb, Serca2 and myh7 in AngII + DAPA + GW6471 group were significantly increased. The mRNA expressions of Nppa, Nppb, Serca2 and myh7 in AngII + DAPA + si-Plin5 group were increased significantly compared with those in AngII + DAPA + si-NC group. In conclusion, we found that si-Plin5 and GW6471 could reverse the protective effect of dapagliflozin on cardiac hypertrophy. It also revealed that dapagliflozin regulated the Plin5/PPAR-α signaling axis to inhibit cardiac hypertrophy.
FIGURE 7
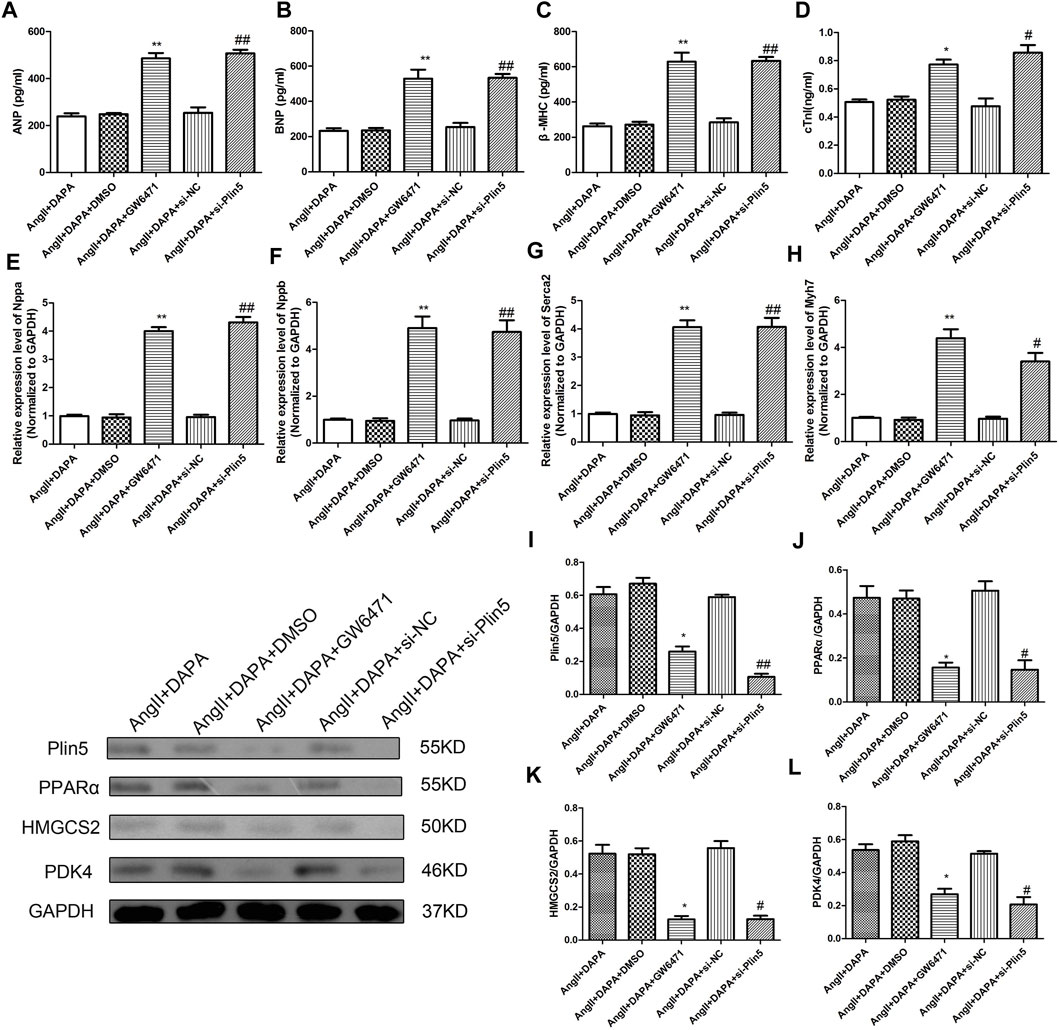
FIGURE 7. Silence Plin5 or GW6471 could reverse the protective effect of dapagliflozin-mediated PPARα signaling axis in AngII-induced cardiomyocyte hypertrophy. The content of ANP (A), BNP (B), β-MHC (C) and cTNI (D) in cell culture supernatant was detected by ELISA kits. qRT-PCR analysis was performed to detect the mRNA expression of Nppa (E), Nppb (F), Serca2 (G) and myh7 (H) in different groups. (I–L) Western blotting assay was used to detect the protein expressions of Plin5, PPARα, HMGCS2 and PDK4 in cardiomyocyte of different groups. The data were expressed as mean ± SD, n = 3, *p < 0.05, vs. AngII + DAPA + DMSO group; #p < 0.05, vs. AngII + DAPA + si-NC group.
Discussion
In the present study, we evaluated the effect of DAPA on AAC-induced cardiac hypertrophy. In vivo, we found that DAPA treatment mitigated AAC-induced myocardial hypertrophy, fibrosis, and cardiac dysfunction. In vitro, we confirmed that DAPA inhibited AngII-induced abnormal cardiomyocytes hypertrophy. Furthermore, we demonstrated an inhibitory role of DAPA on cardiac hypertrophy by activating Plin5/PPARα signaling cascades in the myocardium. These results implied that DAPA could ameliorate cardiac dysfunction in AAC-induced mice, which is consistent with the results of other studies.
DAPA is a new class of oral hypoglycemic agents, SGLT2I, which can enhance renal glucose excretion or glycerine and reduce hyperglycemia (Garcia-Ropero et al., 2018). It has been reported that patients taking DAPA have a lower risk of heart failure and heart disease compared with other glucose-lowering drugs. There is sufficient evidence to suggest that the cardioprotective effects of DAPA are attributable to their systemic effects through glucose and sodium. However, the molecular mechanism of its regulation on cardiac hypertrophy remains unclear.
In the present study, AAC-induced cardiac hypertrophy model in mice was established to further investigate the cardiac function and pathological process after DAPA treatment. We found that DAPA significantly improved cardiac function and increased ejection fraction, which is consistent with Chang et al. reported results (Zhang et al., 2021). After 12 weeks of treatment, we found that the LVEF and LVFS were significantly increased, while LVEDV and LVESV were significantly decreased. In addition, we found that DAPA could reduce cardiac enlargement and inhibit the development of pathological processes, which may contribute to improving cardiac function.
Taking into account the complex pathological factors of cardiac hypertrophy, ventricular hypertrophy and left ventricular weight were considered as the core factors of cardiac hypertrophy aggravated death (Aistrup et al., 2013). In our study, DAPA treatment significantly improved the HW/BW ratio and inhibited myocardial tissue fibrosis. Besides, in vitro experiments also confirmed that DAPA can significantly reduce the abnormal hypertrophy of cardiomyocytes and downregulate the molecular markers related to cardiac hypertrophy.
PPARα is the most abundant PPAR isoform in heart tissue, and the activation of its related signaling pathways directly affected the expressions of key genes in fatty acid oxidation, which is crucial to lipid metabolism and energy metabolism balance in the heart (Campbell et al., 2002). Numerous studies (Zhao et al., 2017) have reported the beneficial effect of activation of the PPARα signal on the development of cardiac hypertrophy, which may be attributed to its regulation of myocardial function and energy metabolism through modulating fatty acid oxidation. Hrvey (Harvey et al., 2020) reported that Nox2 was a key signaling molecule in the pathological reaction of PPARα down-regulation leading to cardiac hypertrophy, which proved that the molecular mechanism of PPARα in cardiac hypertrophy from the negative side. In cardiac hypertrophy, the overexpression of PPARα in cardiac tissue can mediate the p53/GSK3β signaling pathway to significantly improve myocardial energy deficiency and cardiac function, which also supports the important role of PPARα in the pathological process of cardiac hypertrophy (Rana et al., 2019). Interestingly, the absence of PPARα led to a more marked hypertrophic growth response and cardiac dysfunction, which is associated with enhanced expression of inflammatory markers and extracellular matrix remodeling (Smeets et al., 2008). In our study, Plin5 and PPARα signaling pathways were selected as the key signaling molecules in AAC-induced mice according to gene chip detection. Consistent with previous studies (Hou et al., 2021), we found that PPARα was significantly down-regulated in AAC-induced mice. Myocardial hypertrophy was significantly inhibited after treatment, suggesting that DAPA may mediate the PPARα-related signaling axis. It has been reported (Zhao et al., 2019) that microRNA-370 mediated the Plin5-dependent PPAR signaling pathway to protect mice from myocardial ischemia/reperfusion injury. Wang et al. (2019) found that PLIN5 deficiency exacerbated cardiac hypertrophy by promoting oxidative stress.
We demonstrated that DAPA could activate the Plin5/PPARα signaling pathway and further significantly promoted the expressions PDK4 and HMGCS2 in cardiac hypertrophy. It has been reported (Palomer et al., 2009) that PPARs played key roles in the modulation of glucose homeostasis in cardiac cells by regulating PDK4 through PGC-1α. It has been reported (Thapa et al., 2018) that adropin treatment of cardiomyocytes can reduce the inhibition of PDH activity by regulating the expression of PDK4, thus affecting the metabolic balance of glucose in cardiomyocytes, indicating that PDK4 plays a key role in myocardial metabolism. Besides, HMGCS2 could combat metabolic abnormalities by transferring the flux of excess intramitochondrial acetyl-CoA. Upregulation of HMGCS2 can promote myocardial glucose oxidation (Singh et al., 2018). We suggested that DAPA mediated the Plin5/PPARα signal axis, and ultimately affected the expression level of PDK4 and HMGCS2, myocardial cell metabolism, and relieve the pathological progression of cardiac hypertrophy.
At present, dapagliflozin is mainly used in the first-line treatment of diabetes with remarkable effects (Bouter et al., 2020; Shah et al., 2021). Recent clinical studies not only focus on the pathological mechanism of diabetes but also involve the treatment of cardiovascular diseases (Cappetta et al., 2020; Nassif et al., 2021). In the study, we investigated the protective effect of dapagliflozin on AAC-induced cardiac hypertrophy and its mechanism of involving activation of the Plin5/PPARα signaling axis.
Data Availability Statement
The data presented in the study are deposited in the GEO repository, accession number GSE183120.
Ethics Statement
The animal study was reviewed and approved by the Tianjin Medical University.
Author Contributions
JY: Writing, Conceptualization, Methodology, Software HZ: Data curation, Original draft preparation. XQ: Visualization, Investigation, AAC model experiment LW: Supervision, Methodology, AAC model experiment ZL: Software, Validation, Data curation CL: Investigation XZ: Software HW: Design, Reviewing and Editing.
Funding
This study was supported by the project of the Science and Technology Development Fund of Tianjin Education Commission for Higher Education (No. 2020KJ197).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are grateful to all participates for their contributions for the present study.
References
Aimo, A., Pateras, K., Stamatelopoulos, K., Bayes-Genis, A., Lombardi, C. M., Passino, C., et al. (2020). Relative Efficacy of Sacubitril-Valsartan, Vericiguat, and SGLT2 Inhibitors in Heart Failure with Reduced Ejection Fraction: a Systematic Review and Network Meta-Analysis. Cardiovasc. Drugs Ther. 10 (7), 10557–10567. doi:10.1007/s10557-020-07099-2
Aistrup, G. L., Gupta, D. K., Kelly, J. E., O'Toole, M. J., Nahhas, A., Chirayil, N., et al. (2013). Inhibition of the Late Sodium Current Slows T-Tubule Disruption during the Progression of Hypertensive Heart Disease in the Rat. Am. J. Physiol. Heart Circ. Physiol. 305 (7), H1068–H1079. doi:10.1152/ajpheart.00401.2013
Bei, Y., Pan, L. L., Zhou, Q., Zhao, C., Xie, Y., Wu, C., et al. (2019). Cathelicidin-related Antimicrobial Peptide Protects against Myocardial Ischemia/reperfusion Injury. BMC Med. 17 (1), 42. doi:10.1186/s12916-019-1268-y
Bouter, K. E. C., van Bommel, E. J. M., Jansen, H., van Harskamp, D., Schierbeek, H., Ackermans, M. T., et al. (2020). The Effect of Dapagliflozin on Apolipoprotein B and Glucose Fluxes in Patients with Type 2 Diabetes and Well-Controlled Plasma LDL Cholesterol. Diabetes Obes. Metab. 22 (6), 988–996. doi:10.1111/dom.13990
Campbell, F. M., Kozak, R., Wagner, A., Altarejos, J. Y., Dyck, J. R., Belke, D. D., et al. (2002). A Role for Peroxisome Proliferator-Activated Receptor Alpha (PPARalpha ) in the Control of Cardiac Malonyl-CoA Levels: Reduced Fatty Acid Oxidation Rates and Increased Glucose Oxidation Rates in the Hearts of Mice Lacking PPARalpha Are Associated with Higher Concentrations of Malonyl-CoA and Reduced Expression of Malonyl-CoA Decarboxylase. J. Biol. Chem. 277 (6), 4098–4103. doi:10.1074/jbc.M106054200
Cappetta, D., De Angelis, A., Ciuffreda, L. P., Coppini, R., Cozzolino, A., Miccichè, A., et al. (2020). Amelioration of Diastolic Dysfunction by Dapagliflozin in a Non-Diabetic Model Involves Coronary Endothelium. Pharmacol. Res. 157,104781. doi:10.1016/j.phrs.2020.104781
Chai, D., Lin, X., Zheng, Q., Xu, C., Xie, H., Ruan, Q., et al. (2020). Retinoid X Receptor Agonists Attenuates Cardiomyopathy in Streptozotocin-Induced Type 1 Diabetes through LKB1-dependent Anti-fibrosis Effects. Clin. Sci. (Lond) 134 (6), 609–628. doi:10.1042/CS20190985
Chen, T., Liu, J., Li, N., Wang, S., Liu, H., Li, J., et al. (2015). Mouse SIRT3 Attenuates Hypertrophy-Related Lipid Accumulation in the Heart through the Deacetylation of LCAD. PloS one 10 (3),e0118909. doi:10.1371/journal.pone.0118909
Dhillon, S. (2019). Dapagliflozin: A Review in Type 2 Diabetes. Drugs 79 (10), 1135–1146. doi:10.1007/s40265-019-01148-3
Duan, S. Z., Ivashchenko, C. Y., Whitesall, S. E., D'Alecy, L. G., and Mortensen, R. M. (2007). Direct Monitoring Pressure Overload Predicts Cardiac Hypertrophy in Mice. Physiol. Meas. 28 (11), 1329–1339. doi:10.1088/0967-3334/28/11/001
Garcia-Ropero, A., Badimon, J. J., and Santos-Gallego, C. G. (2018). The Pharmacokinetics and Pharmacodynamics of SGLT2 Inhibitors for Type 2 Diabetes Mellitus: the Latest Developments. Expert Opin. Drug Metab. Toxicol. 14 (12), 1287–1302. doi:10.1080/17425255.2018.1551877
Gonzalez, E. A., Tobar Leitão, S. A., Soares, D. D. S., Tavares, A. M. V., Giugliani, R., Baldo, G., et al. (2021). Cardiac Pathology in Mucopolysaccharidosis I Mice: Losartan Modifies ERK1/2 Activation during Cardiac Remodeling. J. Inherit. Metab. Dis. 44 (3), 740–750. doi:10.1002/jimd.12327
Harvey, A., Robinson, E., Edgar, K. S., McMullan, R., O'Neill, K. M., Alderdice, M., et al. (2020). Downregulation of PPARα during Experimental Left Ventricular Hypertrophy Is Critically Dependent on Nox2 NADPH Oxidase Signalling. Int. J. Mol. Sci. 21 (12), 4406. doi:10.3390/ijms21124406
Hou, N., Huang, Y., Cai, S. A., Yuan, W. C., Li, L. R., Liu, X. W., et al. (2021). Puerarin Ameliorated Pressure Overload-Induced Cardiac Hypertrophy in Ovariectomized Rats through Activation of the PPARα/PGC-1 Pathway. Acta Pharmacol. Sin 42 (1), 55–67. doi:10.1038/s41401-020-0401-y
Jia, Z., Xue, R., Liu, G., Li, L., Yang, J., Pi, G., et al. (2014). HMGB1 Is Involved in the Protective Effect of the PPAR α Agonist Fenofibrate against Cardiac Hypertrophy. PPAR Res. 2014,541394 doi:10.1155/2014/541394
Kar, D., and Bandyopadhyay, A. (2018). Targeting Peroxisome Proliferator Activated Receptor α (PPAR α) for the Prevention of Mitochondrial Impairment and Hypertrophy in Cardiomyocytes. Cell Physiol Biochem 49 (1), 245–259. doi:10.1159/000492875
Kraakman, M. J., Lee, M. K., Al-Sharea, A., Dragoljevic, D., Barrett, T. J., Montenont, E., et al. (2017). Neutrophil-derived S100 Calcium-Binding Proteins A8/A9 Promote Reticulated Thrombocytosis and Atherogenesis in Diabetes. J. Clin. Invest. 127 (6), 2133–2147. doi:10.1172/JCI92450
Lahnwong, S., Palee, S., Apaijai, N., Sriwichaiin, S., Kerdphoo, S., Jaiwongkam, T., et al. (2020). Acute Dapagliflozin Administration Exerts Cardioprotective Effects in Rats with Cardiac Ischemia/reperfusion Injury. Cardiovasc. Diabetol. 19 (1), 91. doi:10.1186/s12933-020-01066-9
Liu, S., Li, Y., Zeng, X., Wang, H., Yin, P., Wang, L., et al. (2019). Burden of Cardiovascular Diseases in China, 1990-2016: Findings from the 2016 Global Burden of Disease Study. JAMA Cardiol. 4 (4), 342–352. doi:10.1001/jamacardio.2019.0295
Lu, J., Liu, F., Chen, F., Jin, Y., Chen, H., Liu, D., et al. (2016). Amlodipine and Atorvastatin Improve Ventricular Hypertrophy and Diastolic Function via Inhibiting TNF-α, IL-1β and NF-κB Inflammatory Cytokine Networks in Elderly Spontaneously Hypertensive Rats. Biomed. Pharmacother. 83, 330–339. doi:10.1016/j.biopha.2016.06.034
Luo, J., Zhao, W., Gan, Y., Pan, B., Liu, L., Liu, Z., et al. (2021). Cardiac Troponin I R193H Mutation Is Associated with Mitochondrial Damage in Cardiomyocytes. DNA Cel Biol 40 (2), 184–191. doi:10.1089/dna.2020.5828
Martin, S., and Parton, R. G. (2006). Lipid Droplets: a Unified View of a Dynamic Organelle. Nat. Rev. Mol. Cel Biol 7 (5), 373–378. doi:10.1038/nrm1912
Mele, L., Maskell, L. J., Stuckey, D. J., Clark, J. E., Heads, R. J., and Budhram-Mahadeo, V. S. (2019). The POU4F2/Brn-3b Transcription Factor Is Required for the Hypertrophic Response to Angiotensin II in the Heart. Cell Death Dis 10 (8), 621. doi:10.1038/s41419-019-1848-y
Montaigne, D., Butruille, L., and Staels, B. (2021). PPAR Control of Metabolism and Cardiovascular Functions. Nat. Rev. Cardiol. 10 (3), 569–584. doi:10.1038/s41569-021-00569-6
Nassif, M. E., Windsor, S. L., Tang, F., Husain, M., Inzucchi, S. E., McGuire, D. K., et al. (2021). Dapagliflozin Effects on Lung Fluid Volumes in Patients with Heart Failure and Reduced Ejection Fraction: Results from the DEFINE-HF Trial. Diabetes Obes. Metab. 23 (6), 1426–1430. doi:10.1111/dom.14352
Palomer, X., Alvarez-Guardia, D., Rodríguez-Calvo, R., Coll, T., Laguna, J. C., Davidson, M. M., et al. (2009). TNF-alpha Reduces PGC-1alpha Expression through NF-kappaB and P38 MAPK Leading to Increased Glucose Oxidation in a Human Cardiac Cell Model. Cardiovasc. Res. 81 (4), 703–712. doi:10.1093/cvr/cvn327
Pichler, G., Redon, J., Martínez, F., Solaz, E., Calaforra, O., Andrés, M. S., et al. (2020). Cardiac Magnetic Resonance-Derived Fibrosis, Strain and Molecular Biomarkers of Fibrosis in Hypertensive Heart Disease. J. Hypertens. 38 (10), 2036–2042. doi:10.1097/HJH.0000000000002504
Rana, S., Datta, R., Chaudhuri, R. D., Chatterjee, E., Chawla-Sarkar, M., and Sarkar, S. (2019). Nanotized PPARα Overexpression Targeted to Hypertrophied Myocardium Improves Cardiac Function by Attenuating the P53-Gsk3β-Mediated Mitochondrial Death Pathway. Antioxid. Redox Signal. 30 (5), 713–732. doi:10.1089/ars.2017.7371
Shah, M., Stolbov, L., Yakovleva, T., Tang, W., Sokolov, V., Penland, R. C., et al. (2021). A Model‐based Approach to Investigating the Relationship between Glucose‐insulin Dynamics and Dapagliflozin Treatment Effect in Patients with Type 2 Diabetes. Diabetes Obes. Metab. 23 (4), 991–1000. doi:10.1111/dom.14305
Sharma, S., Adrogue, J. V., Golfman, L., Uray, I., Lemm, J., Youker, K., et al. (2004). Intramyocardial Lipid Accumulation in the Failing Human Heart Resembles the Lipotoxic Rat Heart. FASEB J. 18 (14), 1692–1700. doi:10.1096/fj.04-2263com
Shimojo, N., Jesmin, S., Sakai, S., Maeda, S., Miyauchi, T., Mizutani, T., et al. (2014). Fish Oil Constituent Eicosapentaenoic Acid Inhibits Endothelin-Induced Cardiomyocyte Hypertrophy via PPAR-α. Life Sci. 118 (2), 173–178. doi:10.1016/j.lfs.2014.04.025
Singh, S., McDonough, C. W., Gong, Y., Alghamdi, W. A., Arwood, M. J., Bargal, S. A., et al. (2018). Genome Wide Association Study Identifies the HMGCS2 Locus to Be Associated with Chlorthalidone Induced Glucose Increase in Hypertensive Patients. J. Am. Heart Assoc. 7 (6), e007339. doi:10.1161/JAHA.117.007339
Smeets, P. J., Teunissen, B. E., Willemsen, P. H., van Nieuwenhoven, F. A., Brouns, A. E., Janssen, B. J., et al. (2008). Cardiac Hypertrophy Is Enhanced in PPAR Alpha-/- Mice in Response to Chronic Pressure Overload. Cardiovasc. Res. 78 (1), 79–89. doi:10.1093/cvr/cvn001
Stanley, W. C., Recchia, F. A., and Lopaschuk, G. D. (2005). Myocardial Substrate Metabolism in the normal and Failing Heart. Physiol. Rev. 85 (3), 1093–1129. doi:10.1152/physrev.00006.2004
Tham, Y. K., Bernardo, B. C., Ooi, J. Y., Weeks, K. L., and McMullen, J. R. (2015). Pathophysiology of Cardiac Hypertrophy and Heart Failure: Signaling Pathways and Novel Therapeutic Targets. Arch. Toxicol. 89 (9), 1401–1438. doi:10.1007/s00204-015-1477-x
Thapa, D., Stoner, M. W., Zhang, M., Xie, B., Manning, J. R., Guimaraes, D., et al. (2018). Adropin Regulates Pyruvate Dehydrogenase in Cardiac Cells via a Novel GPCR-MAPK-PDK4 Signaling Pathway. Redox Biol. 18, 25–32. doi:10.1016/j.redox.2018.06.003
Uthman, L., Baartscheer, A., Bleijlevens, B., Schumacher, C. A., Fiolet, J. W. T., Koeman, A., et al. (2018). Class Effects of SGLT2 Inhibitors in Mouse Cardiomyocytes and Hearts: Inhibition of Na+/H+ Exchanger, Lowering of Cytosolic Na+ and Vasodilation. Diabetologia 61 (3), 722–726. doi:10.1007/s00125-017-4509-7
Wang, C., Zhao, Y., Gao, X., Li, L., Yuan, Y., Liu, F., et al. (2015). Perilipin 5 Improves Hepatic Lipotoxicity by Inhibiting Lipolysis. Hepatology 61 (3), 870–882. doi:10.1002/hep.27409
Wang, C., Yuan, Y., Wu, J., Zhao, Y., Gao, X., Chen, Y., et al. (2019). Plin5 Deficiency Exacerbates Pressure Overload-Induced Cardiac Hypertrophy and Heart Failure by Enhancing Myocardial Fatty Acid Oxidation and Oxidative Stress. Free Radic. Biol. Med. 141, 372–382. doi:10.1016/j.freeradbiomed.2019.07.006
Wasserstrum, Y., Barriales-Villa, R., Fernández-Fernández, X., Adler, Y., Lotan, D., Peled, Y., et al. (2019). The Impact of Diabetes Mellitus on the Clinical Phenotype of Hypertrophic Cardiomyopathy. Eur. Heart J. 40 (21), 1671–1677. doi:10.1093/eurheartj/ehy625
Wolins, N. E., Quaynor, B. K., Skinner, J. R., Tzekov, A., Croce, M. A., Gropler, M. C., et al. (2006). OXPAT/PAT-1 Is a PPAR-Induced Lipid Droplet Protein that Promotes Fatty Acid Utilization. Diabetes 55 (12), 3418–3428. doi:10.2337/db06-0399
Ye, Y., Bajaj, M., Yang, H. C., Perez-Polo, J. R., and Birnbaum, Y. (2017). SGLT-2 Inhibition with Dapagliflozin Reduces the Activation of the Nlrp3/ASC Inflammasome and Attenuates the Development of Diabetic Cardiomyopathy in Mice with Type 2 Diabetes. Further Augmentation of the Effects with Saxagliptin, a DPP4 Inhibitor. Cardiovasc. Drugs Ther. 31 (2), 119–132. doi:10.1007/s10557-017-6725-2
Ye, S., Luo, W., Khan, Z. A., Wu, G., Xuan, L., Shan, P., et al. (2020). Celastrol Attenuates Angiotensin II-Induced Cardiac Remodeling by Targeting STAT3. Circ. Res. 126 (8), 1007–1023. doi:10.1161/CIRCRESAHA.119.315861
Yurista, S. R., Silljé, H. H. W., Oberdorf-Maass, S. U., Schouten, E. M., Pavez Giani, M. G., Hillebrands, J. L., et al. (2019). Sodium-glucose Co-transporter 2 Inhibition with Empagliflozin Improves Cardiac Function in Non-diabetic Rats with Left Ventricular Dysfunction after Myocardial Infarction. Eur. J. Heart Fail. 21 (7), 862–873. doi:10.1002/ejhf.1473
Zannad, F., Ferreira, J. P., Pocock, S. J., Anker, S. D., Butler, J., Filippatos, G., et al. (2020). SGLT2 Inhibitors in Patients with Heart Failure with Reduced Ejection Fraction: A Meta-Analysis of the EMPEROR-Reduced and DAPA-HF Trials. Lancet 396 (10254), 819–829. doi:10.1016/S0140-6736(20)31824-9
Zhang, Y., Zhang, L., Fan, X., Yang, W., Yu, B., Kou, J., et al. (2019). Captopril Attenuates TAC-Induced Heart Failure via Inhibiting Wnt3a/β-Catenin and Jak2/Stat3 Pathways. Biomed. Pharmacother. 113,108780. doi:10.1016/j.biopha.2019.108780
Zhang, Y., Lin, X., Chu, Y., Chen, X., Du, H., Zhang, H., et al. (2021). Dapagliflozin: a Sodium-Glucose Cotransporter 2 Inhibitor, Attenuates Angiotensin II-Induced Cardiac Fibrotic Remodeling by Regulating TGFβ1/Smad Signaling. Cardiovasc. Diabetol. 20 (1), 121. doi:10.1186/s12933-021-01312-8
Zhao, M., Jiang, Q., Geng, M., Zhu, L., Xia, Y., Khanal, A., et al. (2017). The Role of PPAR Alpha in Perfluorooctanoic Acid Induced Developmental Cardiotoxicity and L-Carnitine Mediated protection-Results of in Ovo Gene Silencing. Environ. Toxicol. Pharmacol. 56, 136–144. doi:10.1016/j.etap.2017.09.006
Zhao, Y. B., Zhao, J., Zhang, L. J., Shan, R. G., Sun, Z. Z., Wang, K., et al. (2019). MicroRNA-370 Protects against Myocardial Ischemia/reperfusion Injury in Mice Following Sevoflurane Anesthetic Preconditioning through PLIN5-dependent PPAR Signaling Pathway. Biomed. Pharmacother. 113, 108697 doi:10.1016/j.biopha.2019.108697
Keywords: dapagliflozin, cardiac hypertrophy, AngII, Plin5/PPARα signaling, mice
Citation: Yu J, Zhao H, Qi X, Wei L, Li Z, Li C, Zhang X and Wu H (2021) Dapagliflozin Mediates Plin5/PPARα Signaling Axis to Attenuate Cardiac Hypertrophy. Front. Pharmacol. 12:730623. doi: 10.3389/fphar.2021.730623
Received: 25 June 2021; Accepted: 08 September 2021;
Published: 23 September 2021.
Edited by:
Sai Wang Seto, Hong Kong Polytechnic University, Hong Kong, SAR ChinaReviewed by:
Suneet Kaur, Duke University, United StatesCees Korstanje, Scenic Biotech B.V., Netherlands
Copyright © 2021 Yu, Zhao, Qi, Wei, Li, Li, Zhang and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hao Wu, d3VoYW8yMDIwbmtAMTYzLmNvbQ==