
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 14 January 2022
Sec. Pharmacology of Anti-Cancer Drugs
Volume 12 - 2021 | https://doi.org/10.3389/fphar.2021.724192
 Mengyuan Niu1,2,3†
Mengyuan Niu1,2,3† Bin Zhang1,4†
Bin Zhang1,4† Li Li1,2†
Li Li1,2† Zhonglan Su5
Zhonglan Su5 Wenyuan Pu1,2
Wenyuan Pu1,2 Chen Zhao1,2
Chen Zhao1,2 Lulu Wei1,2
Lulu Wei1,2 Panpan Lian1,2
Panpan Lian1,2 Renwei Lu1,2
Renwei Lu1,2 Ranran Wang1,2
Ranran Wang1,2 Junaid Wazir1,2
Junaid Wazir1,2 Qian Gao1,2*
Qian Gao1,2* Shiyu Song1,2*
Shiyu Song1,2* Hongwei Wang1,2*
Hongwei Wang1,2*Lung cancer is one of the most common malignant cancers worldwide. Searching for specific cancer targets and developing efficient therapies with lower toxicity is urgently needed. HPS90 is a key chaperon protein that has multiple client proteins involved in the development of cancer. In this study, we investigated the transcriptional levels of HSP90 isoforms in cancerous and normal tissues of lung cancer patients in multiple datasets. The higher expression of HSP90AA1 in cancer tissues correlated with poorer overall survival was observed. The higher levels of transcription and expression of HSP90AA1 and the activity of AKT1/ERK pathways were confirmed in lung cancer patient tissues. In both human and mouse lung cancer cell lines, knocking down HSP90AA1 promoted cell apoptosis through the inhibition of the pro-survival effect of AKT1 by decreasing the phosphorylation of itself and its downstream factors of mTOR and BAD, as well as downregulating Mcl1, Bcl-xl, and Survivin. The knockdown also suppressed lung cancer cell proliferation by inhibiting ERK activation and downregulating CyclinD1 expression. The treatment of 17-DMAG, an HSP90 inhibitor, recaptured these effects in vitro and inhibited tumor cell growth, and induced apoptosis without obvious side effects in lung tumor xenograft mouse models. This study suggests that targeting HSP90 by 17-DMAG could be a potential therapy for the treatment of lung cancer.
Lung cancer is one of the most commonly diagnosed cancers and the leading cause of cancer-related deaths worldwide (Torre et al., 2015; Wood et al., 2015). The non-small cell lung cancer (NSCLC), including squamous cell carcinoma (SCC), adenocarcinoma (AC), and large cell carcinoma (LCC) accounts for approximately 85% of all cases of lung cancer (Hussain et al., 2001). Despite various therapeutic approaches, including chemotherapy, immune checkpoint inhibitors, targeted protein kinase inhibitors et al., the 5-years survival rate of late-stage tumors remains poor with approximately 15% (Novello et al., 2013; Park et al., 2014).
Heat shock protein 90 (HSP90) is a chaperon protein that is highly conserved in various species across the different kingdoms (Chen et al., 2006). There are two major cytosolic isoforms of HSP90AA1, HSP90AB1, and one paralog HSP90B1, also known as GRP94, located in the endoplasmic reticulum in human cells (Chen et al., 2006). HSP90 plays a crucial role in maintaining cellular protein homeostasis by assisting other proteins in folding properly, stabilizing proteins against heat stress, and promoting protein degradation (Schopf et al., 2017).
HSP90 has been shown to regulate a variety of biological processes, including cell growth, differentiation, and survival, particularly in tumor cells (Schopf et al., 2017). Because of the presence of mutant proteins and their rapid proliferation, tumor cells place a greater emphasis on controlling proteostasis (Kamal et al., 2003; Schopf et al., 2017). As a result, HSP90 may be a promising therapeutic target, and several clinical trials are currently underway to assess the efficacy of HSP90 inhibitors alone or in combination with other drugs in the treatment of various types of cancer (Eiseman et al., 2005; Yuno et al., 2018). Geldanamycin (GA) and its analogue 17-allylamino-17-demethoxygeldanamycin (17-AAG or anespimycin) and 17-demethylaminoethylamino-17-demethoxygeldanamycin (17-DMAG or alvespimycin) were discovered as inhibitors of HSP90 (Trepel et al., 2010). They were able to bind to HSP90 at the N-terminal ATP pocket, thus prevents the interaction of the chaperone and the clients (Bucci et al., 2000). When compared with GA or 17-AAG, 17-DMAG exhibits increased water solubility, higher oral bioavailability, longer plasma half-life, less toxicity to normal cells and superior antitumor activity (Kaur et al., 2004; Smith et al., 2005; Trepel et al., 2010). 17-DMAG has been tested in clinical trials and has highly attractive pharmaceutical properties (Egorin et al., 2002). In addition, it is reported that in vivo administration of 17-DMAG enhances EphA2+ tumor cell recognition by specific CD8+ T cells. And the treatment outcomes were also improved in sarcoma and renal cell carcinoma (Kawabe et al., 2009; Rao et al., 2012). Whether 17-DMAG participated in immune regulation in lung cancer was investigated in our assay.
However, the clinical significance of HSP90 isoforms and the pathways regulated by HSP90 in lung cancer are still unknown. In this study, we compared the transcriptional levels of HSP90 isoforms in cancerous and normal tissue and analyzed the correlation between HSP90 expression and overall survival in multiple datasets. Furthermore, the higher transcription and expression levels of HSP90AA1 and AKT1/ERK pathways were validated in lung cancer patient tissues. In addition, the anti-tumor effect and regulatory pathways of 17-DMAG were tested in vitro and in vivo.
The transcription data of the lung cancer and normal tissue was downloaded from the GEO database (GSE numbers: GSE10072, GSE32863, GSE19188, and GSE40419) with the “GEOquery” R package (Davis and Meltzer, 2007). The Level 3 mRNA expression data of LUAD was downloaded from the TCGA database. The expression data of normal lungs was downloaded from the GTEX database. The expression values not in log form were log2-transformed. In processing the dataset based on a microarray, the highest probe of the same gene was selected. The correlation between the expression of the indicated genes was analyzed by Spearman’s ranked correlation test. The survival test of the genes was analyzed by the “Survival” R package (Therneau and Grambsch, 2015). Spearman’s rank correlation test was used to determine the correlation of each gene’s expression with HSP90AA1. A positive correlation was defined as rho value greater than >0.3 and p < 0.05. And the enrichment of the genes was performed by the “clusterProfiler” (Yu et al., 2012). The protein-protein interaction network was constructed by the String website (STRING, http://www.string-db.org) (Szklarczyk et al., 2019), and the PPI was, analyzed, and visualized by Cytoscape v3.8.2 software (Shannon et al., 2003). The GSEA analysis was performed by GSEA software (Version 4.1.0) (Subramanian et al., 2005), according to the Pearson correlation of HSP90AA1 expression.
The study was approved by the Ethics Committee in the Medical School of Nanjing University (IRB no.: 20200115003). All patients provided written informed consent. The information details of patients were listed in Supplementary Table S1.
LLC cells were cultured in DMEM (Gibco, Carlsbad, CA) containing 10% fetal bovine serum (FBS, Gibco, Carlsbad, CA) supplemented with penicillin (100 U ml−1) and streptomycin (100 µg ml−1) in 5% CO2 at 37°C. Cells were obtained from American Type Culture Collection.
Cancer cell lines (A549, Solarbio, China) originating from human lung tumors were cultured in RPMI-1640 medium (Gibco, CA, USA) with 10% fetal bovine serum (FBS; Biological Industries, Israel) and antibiotics (100 μg/ml penicillin-streptomycin, Beyotime, China). The cell cultures were incubated in an environment containing 5% CO2 at 37°C.
Animal experiments were performed as described previously (Yang et al., 2019). Male Balb/c mice, aged 6–8 weeks were maintained under standard conditions and cared for in accordance with the institutional guidelines for animal care. All the animal experiments were approved by the Institutional Animal Care and Use Committee of Nanjing University.
LLC cells in equal volumes of PBS were subcutaneously injected into the right flanks of mice to establish tumor xenografts (5 × 105 cells per mouse). When the tumor xenografts reached approximately 5 mm in diameter, the mice were randomly divided into treatment and control groups. The treatment group received an intraperitoneal injection of 17-DMAG (MCE, Cat#HY-12024, NJ, USA) (15 mg/kg, daily, dissolved in 1% DMSO +30% PEG300 + 1% Tween 80) every other day. Whereas the control group received a vehicle solvent.
The tumor size was measured with calipers every 2 days. The tumor volume was calculated using the following equation: The tumor volumes were calculated as 1/2 (length × width2) (Faustino-Rocha et al., 2013). The inhibition rates of 17-DMAG on the tumor were calculated as (average tumor weights of the control group-17-DMAG treated group)/average tumor weights of the control group. At the end of the experiment, the tumors were collected for western blot and histological analysis.
Balb/c Nude (6–8 weeks of age) mice were obtained from the Model Animal Research Center of Nanjing University for this study. The mice were housed under specific pathogen-free conditions at 23°C and given free access to food and water. To prepare A549 tumor xenografts, the left flank of the mice was subcutaneously inoculated with A549 tumor cell suspension (5 × 106 cells/100 μL) (Zhu et al., 2020). Three days after tumor-cell inoculation, the mice were divided into two groups (n = 10): Con group (control group, no treatment), 17-DMAG-treated group (15 mg/kg/day), which were administered intraperitoneal injection daily until the end of the experiments. Mice were killed when their minor axis of tumors was longer than 20 mm. All experiments with mice were approved by the ethics committee.
Cells were seeded in 96-well plates at 5,000 cells per well in a final culture volume of 100 μL for 24 h before the addition of increasing concentrations of 17-DMAG that were incubated for 24 h. Viable cell number was determined using the Celltiter 96 AQueous Nonradioactive Cell Proliferation Assay (Promega, WI, United States). The value of the background absorbance at 490 nm (A490) of wells not containing cells was subtracted. Percentage of viable cells was calculated as following: A490 of 17-DMAG treated sample/A490 untreated cells × 100. The IC50 was defined as the concentration that gave rise to a 50% viable cell number.
For apoptosis detection, the LLC cells or A549 cells were cultured in a six-well plate. Then LLC cells were treated with 17-DMAG or vehicle (DMSO 0.1%) for 24 h; A549 cells were treated with 17-DMAG or vehicle (DMSO 0.1%) for 24 h. Next, the cells were washed once with a complete medium, resuspended in binding buffer, and then stained with Annexin V-fluorescein isothiocyanate and propidium iodide (PI). After 20 min of incubation in the dark at room temperature, stained A549 cells were examined by flow cytometry analysis (BD LSRII SORP, Franklin Lakes, United States). The analysis was repeated three times.
TUNEL assay was performed by the ApoBrdU DNA Fragmentation Assay Kit (Biovision, San Francisco, CA, United States) following the manufacturer’s instructions. Briefly, the tumorsphere was removed from the implanted region and fixed with 4% paraformaldehyde, and embedded in paraffin. And then, remove the paraffin by immersing the slides in fresh xylene twice. After rehydration, the slides were fixed with 4% paraformaldehyde and washed. Proteinase K was added to remove the remained protein on the slide; then the slides were washed and incubated with a DNA labeling solution. FITC labeled anti-BrdU antibody was added after two washes and then incubated on the slides at RT for 30 min. Then the slides were washed, and PI was adopted to reveal the nuclei of the cells. And the images were captured by the FV10i Laser Scanning Confocal Microscope (Olympus, Center Valley, PA, United States).
Immunohistochemistry was carried out according to a standard protocol. Briefly, the tumor spheres were removed from the implanted region and fixed with 4% paraformaldehyde, and embedded in paraffin. After hydrolysis and antigen retrieval, the slides of both tumor-bearing mice and human patients were blocked and washed with PBS. Immunostaining was carried out with a rabbit monoclonal antibody to CD8 at 4°C overnight. Hematoxylin & eosin (H & E) staining was performed for conventional morphological evaluation under a light microscope (Olympus, Tokyo, Japan).
The tumor samples were lysed on ice using RIPA buffer containing a cocktail of protease inhibitors and phosphatase inhibitors (Thermo fisher, Cat#78440, CA, United States) with constant shaking for 30 min; cellular debris was pelleted by centrifugation at 10,000 × g for 5 min at 4°C, and supernatants were harvested. Protein concentrations were measured by the BCA protein assay kit. Target proteins (30 µg per lane) were separated via SDS-PAGE. Following separation with different concentrations of acrylamide gel (6% for mTOR; 10% for AKT1; 12% for caspase-3 and cleaved caspase-3), the proteins were transferred onto PVDF membranes. The membrane containing the proteins was successively incubated in a blocking buffer (overnight at 4°C), with a primary antibody (37°C for 1 h) and with a secondary antibody (37°C for 1 h). Primary antibodies against HSP-90, CyclinD, pBAD, AKT1, phosphor-AKT1 (Ser473), mTOR, phosphor-mTOR (Ser2448), caspase-3, cleaved caspase-3, ERK and Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) were purchased from Cell Signaling Technology (Boston, MA, United States). Antibodies against Bcl-xl, Survivin, Mcl-1, and GAPDH were purchased from Abcam (Burlingame, CA, United States). The BCA protein assay kit was purchased from Pierce (Rockford, IL, United States).
Total mRNA was extracted from cultured cells and tumorspheres using the RNeasy Micro Kit (Qiagen, Hilden, Germany), mRNA was reverse transcripted into cDNA with the PrimeScript RT Master Mix (TaKaRa, Otsu, Japan). SYBR green quantitative real-time PCR was performed, using PCR Master Mix (Life Technologies). The relative expression of target genes was determined to beta-actin and was calculated by the ΔΔCt method. Primers: Human AKT1, forward: 5′-GCTTCTTTGCCGGTATCGTG-3′, reverse: 5′-GGCCGTGAACTCCTCATCAA-3′; Human CyclinD1, forward: 5′-GCTGCGAAGTGGAAACCATC-3′; reverse: 5′-CCTCCTTCTGCACACATTTGAA-3′; Human Mcl-1, forward: 5′-GGCTAAACACTTGAAGACCATAA-3′, reverse: 5′-GAAGAACTCCACAAACCCATC-3′; Human Bcl-xl, forward: 5′-AAAGCGTAGACAAGGAGATGC-3′, reverse: 5′-TCCCATAGAGTTCCACAAAAGT-3′; Human Survivin, forward: 5′-TTACGCCTGTAATACCAGCAC-3′, reverse: 5′-TCACCAAGGGTTAATTCTTCA-3′; Human beta-actin, forward: 5′-GGACGACATGGAGAAAATCTG-3′, reverse: 5′-GGTCTCAAACATGATCTGGGT-3′.
Mouse AKT1, forward: 5′-ATCTGAGTCCACAGCAAGGTC-3′, reverse: 5′-GAGTCTCTTCTCGGTAGGCTG-3′; Mouse CyclinD1, forward: 5′-CAACTTCCTCTCCTGCTACCG-3′, reverse: 5′-CCTTGTTTAGCCAGAGGCCG-3′; Mouse Mcl1, forward: 5′-AAAGGCGGCTGCATAAGTC-3′, reverse: 5′-TGGCGGTATAGGTCGTCCTC-3′; Mouse Bcl-xl, forward: 5′-ACATCCCAGCTTCACATAACCC-3′, reverse: 5′-CCATCCCGAAAGAGTTCATTCAC-3′; Mouse Survivin, forward: 5′-GAGGCTGGCTTCATCCACTG-3′, reverse: 5′-ATGCTCCTCTATCGGGTTGTC-3′; Mouse GzmA, forward: 5′-GGGGGCCATCTCTTGCTACT-3′, reverse: 5′-TGAGTGAGGAACAACCGTGTC-3′; Mouse GzmB, forward: 5′-CAGGCCAATGGAACACCTCT-3′, reverse: 5′-GTGGAGAGGGCAAACTTCCA-3′; Mouse Ifng, forward: 5′-AGCAAGGCGAAAAAGGATGC-3′, reverse: 5′-TCATTGAATGCTTGGCGCTG-3′; Mouse beta-actin, forward: 5′- GTGACGTTGACATCCGTAAAGA-3′, reverse: 5′-GCCGGACTCATCGTACTCC-3′.
SiRNA transfection was performed using Lipofectamine RNAimax reagent (Thermo, #13778150, CA, United States), according to the manufacture’s instruction. Briefly, tumor cells were seeded in 6-well plates at 60–80% confluence, 10,000 cells per well in 96-well plates. 25 pmol per well in 6-well plates, and 1 pmol per well in 96-well plates of siRNA was diluted in opt-MEM medium, and mixed with transfection reagent and added in each well. After 48 h of transfection, the efficiency of the transfection and the viability of the cell were evaluated. siRNA sequences: human HSP90AA1 (CUUCACAGACUUGUCGUUCUU, scramble: GCGGCACUAAAAUUGUACAGG); mouse Hsp90aa1 (CUUCACAGAUUUGUCAUUCUU, scramble: CAAAGUUCUCACUUGUUUCUU).
Each experiment was performed at least three times, and representative data are shown. Data in bar graphs are given as the means ± S.D. Means were checked for statistical difference using the t-test and p-values less than 0.05 were considered significant (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
To investigate the role of HSP90 in lung cancer, the transcription levels of 3 HSP90 isoforms (AA1, AB1, B1) were compared between lung cancer and normal lung tissue in three microarray and one RNA sequencing datasets. As shown in Figure 1A, these three isoforms of HSP90 are all significantly up-regulated in cancer tissues compared with normal lung tissues. Although they were positively correlated with each other (Figure 1B), only the higher expression of HSP90AA1 was significantly correlated with shortened overall survival time in three datasets, including TCGA, in which survival data was available (Figure 1C).
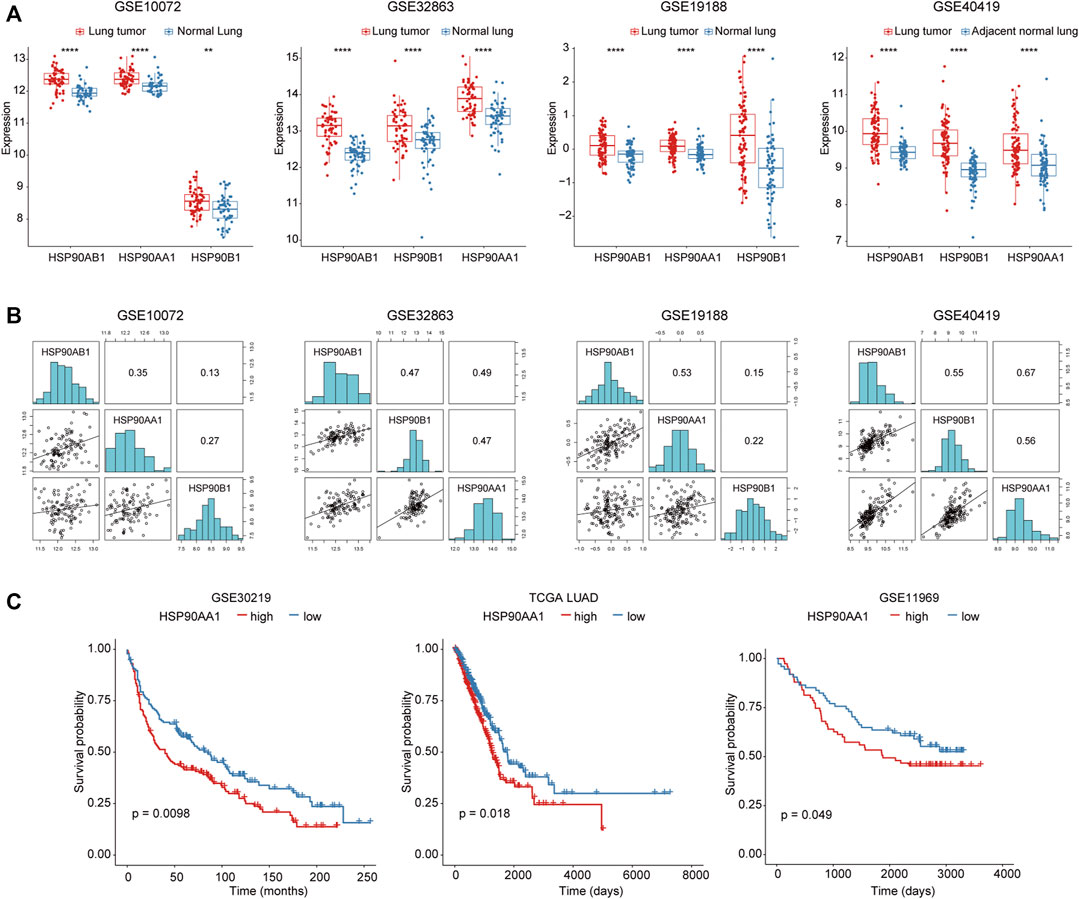
FIGURE 1. Increased HSP90AA1 is correlated with decreased overall survival time in lung cancer patients. (A) HSP90AA1, HSP90AB1, HSP90B1 are all significantly up-regulated in lung cancer tissues in four different high throughput datasets. **p < 0.01, ****p < 0.0001. (B) Three HSP90 isoforms (AA1, AB1, B1) are positively correlated with each other. (C) HSP90AA1 is significantly correlated with a shortened overall survival time in three datasets.
To further explore the mechanism of HSP90AA1 regulation in cancer development, we tried to compare the differences between the HSP90AA1 correlated genes in normal (GTEX normal lung tissue) and cancer lung (TCGA LUAD) tissues. According to the criteria in the method section (Spearman rho >0.3, p < 0.05), 1428 genes in the GTEX database and 556 genes in the TCGA LUAD database were considered as significantly positively correlated with HSP90AA1 expression (Supplementary Figure S1). Interestingly, only 83 genes were shared between normal and lung cancer patients (Figure 2A). There are 473 genes uniquely positively correlated with HSP90AA1 in cancer patients. Next, we enriched the KEGG pathway of these 473 genes in cancer patients. As shown in Figures 2B,C, the KEGG pathways of hsa04110: Cell cycle, hsa03013: RNA transport, hsa03030: DNA replication, and hsa03050: Proteasome, et al., were significantly enriched by these genes. And the heatmap of the enriched genes in the pathways indicated that key genes involving tumor proliferation such as BRCA1, CDK1, and CCNB1 et, al (Figure 2D). The results were further validated in the GTEX database; the correlation between HSP90AA1 and BRCA1 and CDK1 was only observed in cancer patients (Figure 2E). The enrichment of Gene Ontology showed that the molecular function of GO:0008094 DNA-dependent ATPase activity, GO:0003678 DNA helicase activity, and GO:0061575 cyclin-dependent protein serine/threonine kinase activator activity were significantly enriched in these genes. Furthermore, the molecular functions of the biological process, such as GO:0007059 chromosome segregation, GO:0000086 mitotic cell cycle G2/M transition, and GO:0006260 DNA replication, were significantly enriched in these genes. (Figure 2F), implying that HSP90AA1 contributes to tumor cell hyperproliferation.
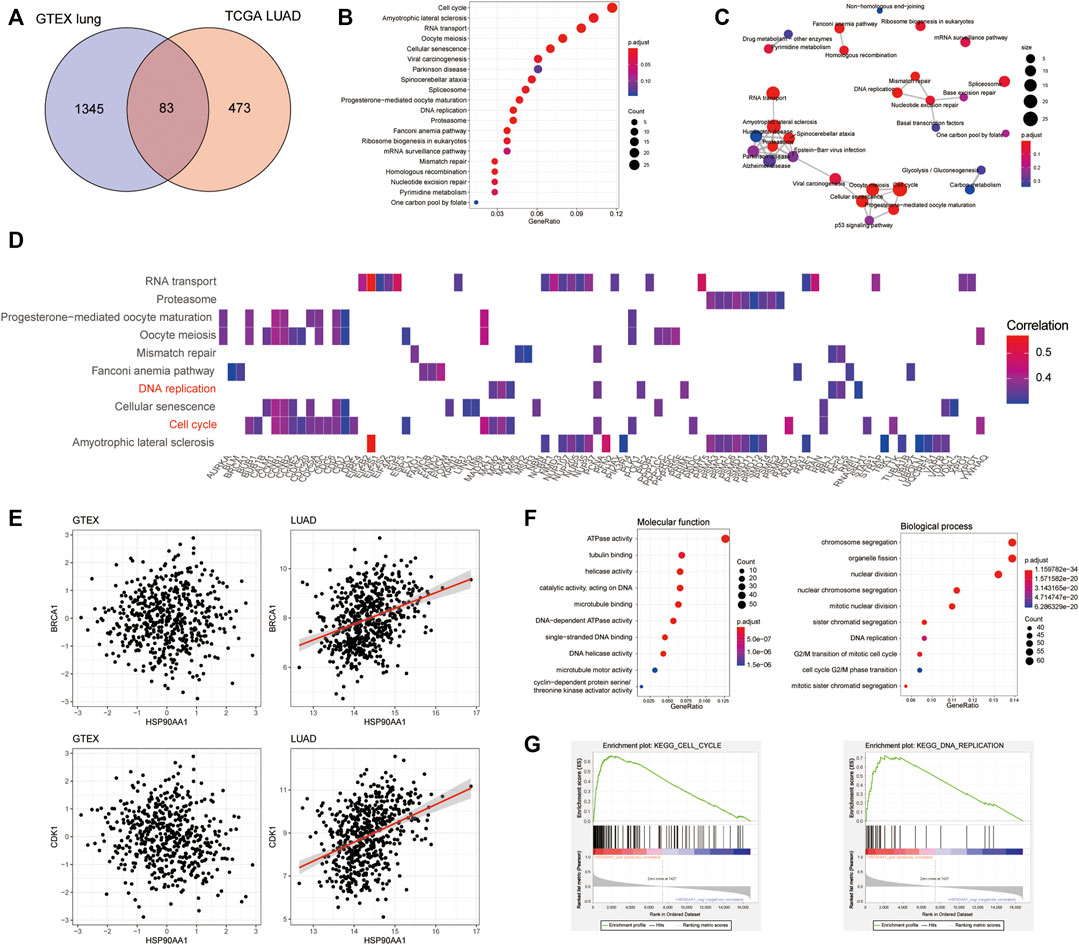
FIGURE 2. The transcription profile of HSP90AA1 correlated genes was different between lung cancer and normal tissues. (A) The Venn plot indicated the overlapping and differences of the genes positively correlated with HSP90AA1 expression in normal (GTEX) and cancerous (TCGA LUAD) lungs. (B & C) Enrichment and correlations of HSP90 positively correlated genes in KEGG pathways. (D) The heatmap of indicated genes enriched in the corresponding pathways and their correlation with HSP90AA1 expression. (E) The correlation of BRCA1 and CDK1 with HSP90AA1 in the GTEX and TCGA databases. (F) The enrichment of GO terms of molecular function and biological process in HSP90AA1 correlated genes. (G) GSEA analysis of HSP90AA1 indicated the possible correlated genes enriched in KEGG pathways of cell cycle and DNA replication.
Besides that, the GSEA assay of the KEGG pathways validated these findings, with the expression pattern of HSP90AA1 positive correlated genes significantly enriched in cell cycle and DNA replication pathways (Figure 2G).
To validate this finding, the expression of HSP90AA1 was tested by qPCR and immunoblotting in 40 paired lung cancer and adjacent tissues. HSP90AA1 was consistently up-regulated in lung cancer at both transcriptional and expression levels (Figures 3A,B). The overactivation of AKT and ERK is a common molecular characteristic in lung cancer patients (Riquelme et al., 2016). And in turn, they will regulate a variety of downstream protein substrates, including gGSK3β, mTOR, BAD and CyclinD1, et al., which are critical in the deregulation of apoptosis, proliferation, and cell motility in cancer cells (Liu et al., 2012; Chen et al., 2019). We then found that AKT1/ERK pathways were also significantly activated in these lung cancer tissues, suggesting a possible contribution of HSP90AA1 to their activation.
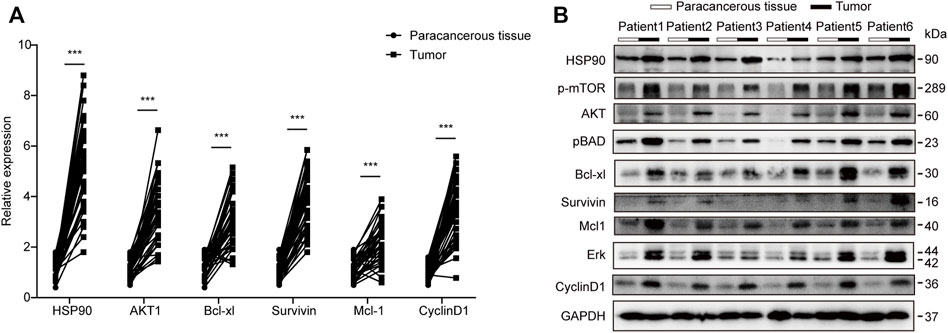
FIGURE 3. Transcription and western blot validation of AKT1/ERK signaling in lung cancer patients. (A) qPCR analysis of HSP90, AKT1, Bcl-xl, Survivin, Mcl1, and CyclinD1 in lung cancer patients’ tumors compared with Para cancerous tissue. Patients number, N = 40 The data represents the mean ± standard deviation (SD). ***p < 0.001 (B) Western blot analysis of HSP90, AKT1, Bcl-xl, Survivin, Mcl1, and CyclinD1 in lung cancer patient’s tumor compared with Para cancerous tissue. We randomly chose 6 patients and repeated the experiment three times.
17-DMAG, a geldanamycin analog that has been shown to inhibit HSP90 in multiple models, was used to investigate the potential of HSP90 targeting (Ambade et al., 2012; Trepel et al., 2010; Wang Y. L. et al., 2016). Figure 4A depicts the molecular structure of 17-DMAG. In a time-course assay, 17-DMAG significantly inhibited both mouse lung cancer LLC (IC50: 0.171 μM) and human lung cancer A549 (IC50: 1.096 μM) growth in a dose-dependent manner (Figures 4B,C). However, it had no discernible effect on viability in BEAS-2B (a normal human lung epithelial cell line) and LO-2 (a normal human liver cell line) at concentrations more than ten-fold higher than its IC50, indicating its safety (Figure 4D). To see if 17-DMAG inhibited cell proliferation by inducing apoptosis, an AnnexinV-PI cytometry assay was performed. As shown in Figure 4E, 17-DMAG treatment significantly induced massive cell apoptosis in both LLC and A549 cells, implying that its anti-cancer activity is dependent on its pro-apoptotic effect.
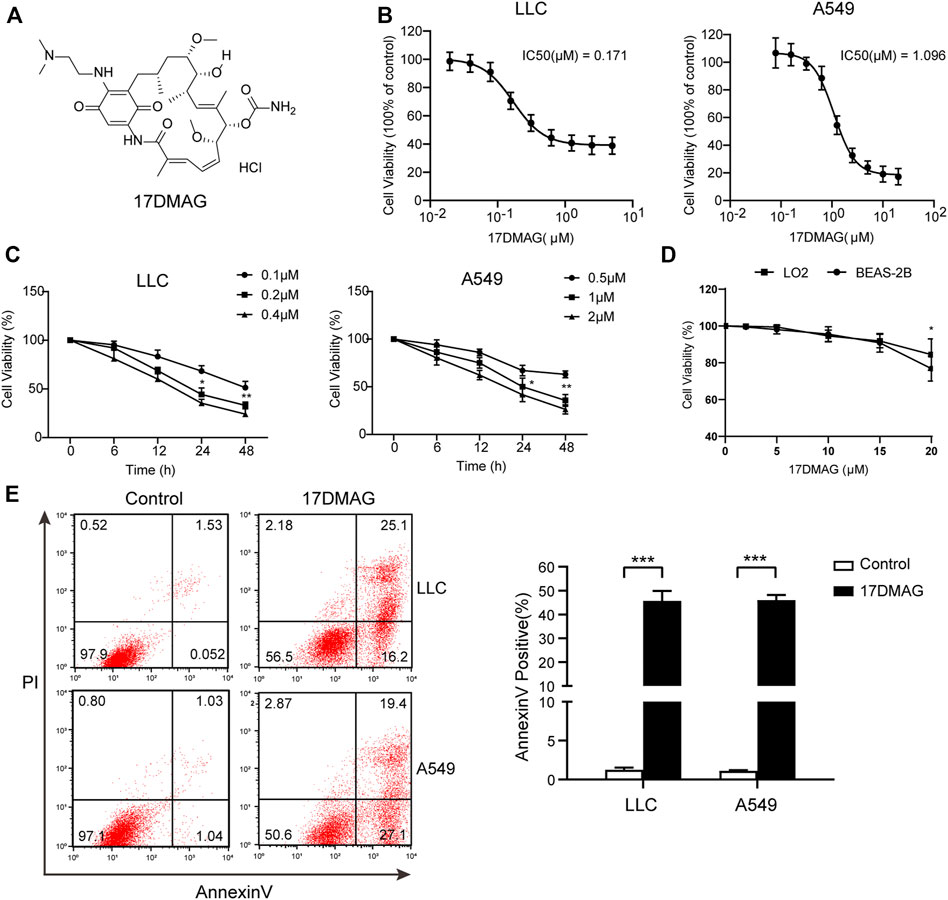
FIGURE 4. 17-DMAG inhibited lung cancer proliferation and induced cell apoptosis. (A) Molecular structural formula of 17-DMAG. (B) LLC and A549 lung cancer cells were treated with folded diluted 17-DMAG. Cell viability was determined by the MTS method. The IC50 value was determined to evaluate the inhibition of tumor cell growth by 17-DMAG. The value of each cell was the mean value of three independent experiments. (C) Time-course analysis of the growth inhibition effect of 17-DMAG in LLC and A549 cells. (D) BEAS-2B cells were treated with 17-DMAG at indicated concentrations for 24 h. The cell viability was determined by an MTS assay (n = 5). (E) LLC and A549 cells were treated with 0.2 or 1 μM 17-DMAG and then stained with PI and AnnexinV-FITC to detect apoptosis by flow cytometry. The columns showed the statistical FACS results of three independent experiments. ***p < 0.001.
Following that, we attempted to investigate the molecular mechanism of HSP90’s anti-cancer effect in lung cancer. As previously stated, high levels of AKT1/ERK pathways were commonly observed in lung cancers (Li et al., 2012; Wu et al., 2021), which was confirmed in our study (Figure 1D). HSP90 has been shown to regulate the activation of AKT1 and ERK in leukemia and esophageal SCC (Rahmani et al., 2003; Friedman et al., 2013). As a result, we attempted to investigate the possible regulation of HSP90 on AKT1/ERK activation in lung cancer cells. 17-DMAG inhibited AKT1 and ERK phosphorylation in a dose-dependent manner, as shown in Figures 5A,B. The treatment also reduced the phosphorylation of the key pro-apoptotic protein BAD, which was directly phosphorylated by AKT1 (Datta et al., 1997), implying that AKT1 kinase activity was inhibited. In addition, 17-DMAG down-regulated the expressions of several pro-survival proteins, including Mcl-1, Survivin, and Bcl-xl, while up-regulated the levels of cleaved-caspase3. Furthermore, 17-DMAG inhibited the protein and transcriptional levels of CyclinD1, which is important in cell cycle regulation (Figure 5C). We next knocked down HPS90AA1 in these two cell lines to confirm that the pivotal effect HSP90AA1 in lung cancer. In both LLC and A549 cells, knocking down (KD) of HSP90AA1 mimicked the effect of 17-DMAG at protein level and transcriptional levels (Figures 6A,B). It also inhibited the growth of LLC and A549 in a time-course assay (Figure 6C). The AnnexinV-PI cytometry assay and the TUNEL assay demonstrated that HSP90AA1 KD significantly induced cell apoptosis in both LLC and A549 cells, which mimicked the effect of 17-DMAG in vitro (Figures 6D,E).
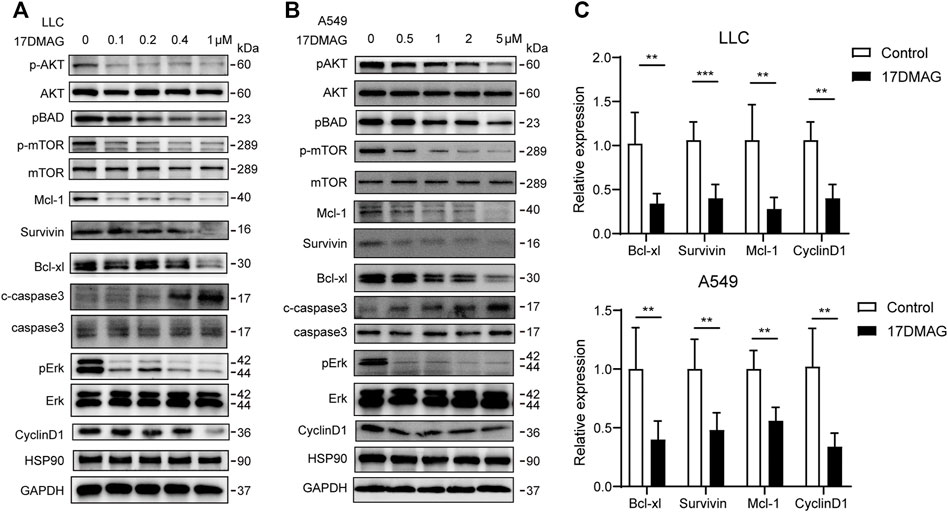
FIGURE 5. Inhibiting HSP90 reversed AKT1/ERK activation in lung cancer cells. (A) LLC cells were treated with indicated concentrations of 17-DMAG and then subjected to western blot for measuring protein levels by indicated antibodies. (B) A549 cells were treated with indicated concentrations of 17-DMAG and then subjected to western blot for measuring protein levels by indicated antibodies. (C) The total RNA of 17-DMAG-treated LLC cells and A549 cells were extracted, and a qPCR method was adopted to test the transcription level of indicated genes. The result was obtained from three independent experiments. **p < 0.01, ***p < 0.001.
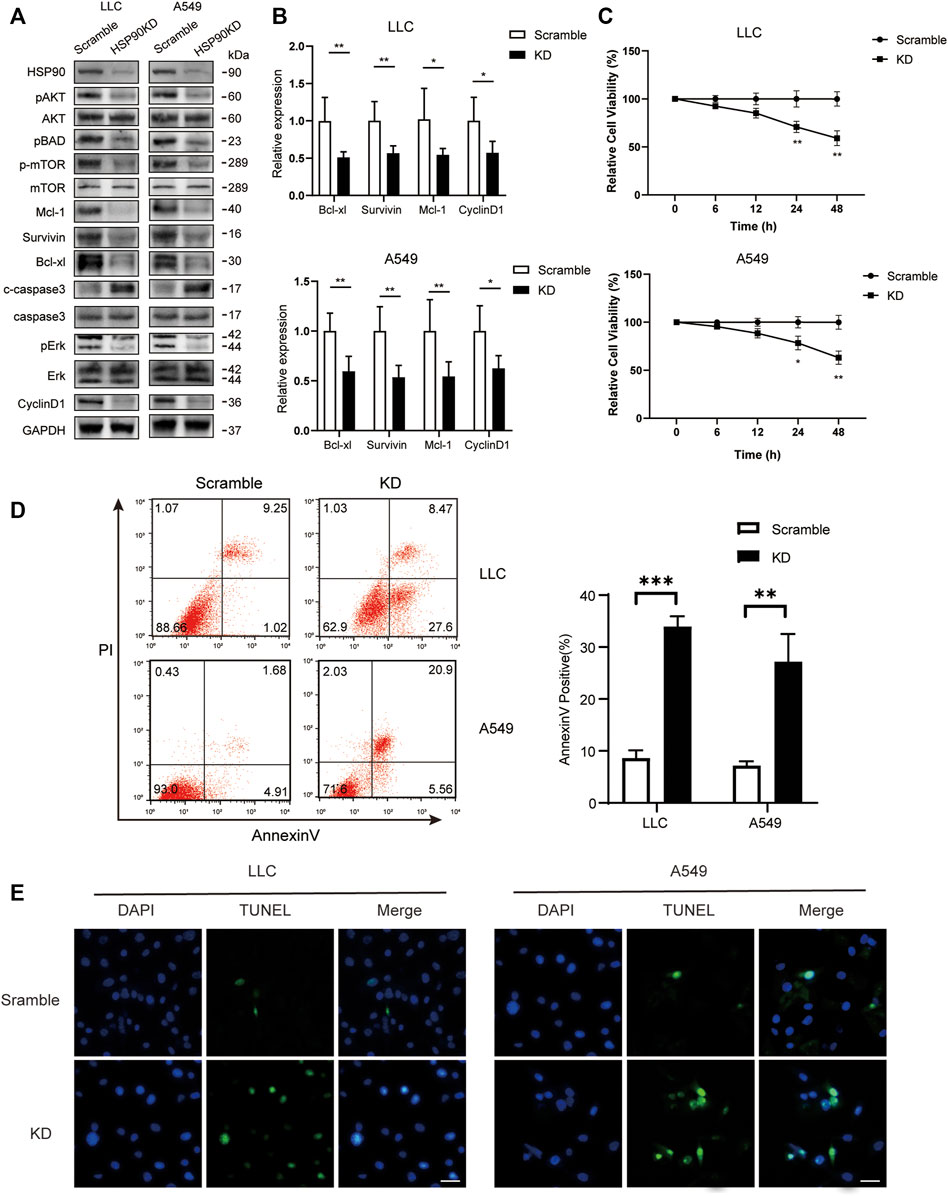
FIGURE 6. Knocking down of HSP90 inhibited lung cancer proliferation and induced cell apoptosis. LLC and A549 cells were transfected with HSP90 siRNA or control siRNA. (A) Western blotting with indicated antibodies was performed. (B) The total RNA of the cells was extracted, and a qPCR method was adopted to test the transcription level of indicated genes. (C) Time-course analysis of the cell growth detected by MTS assay. *p < 0.05, **p < 0.01. (D) The apoptosis level of the cells was evaluated by staining with PI and AnnexinV-FITC antibody and subjected by flow cytometry. The columns showed the statistical FACS results of three independent experiments. **p < 0.01, ***p < 0.001. (E) Analysis of apoptosis by TUNEL staining (blue fluorescence, DAPI staining for nuclei; green fluorescence, TUNEL-positive staining) in LLC and A549 cells. Scale bar = 30 μm.
Finally, we investigated 17-DMAG’s anti-cancer activity in vivo. Subcutaneous injections of LLC and A549 cells were made into the flanks of C57/BL6 and Balb/c nude mice, respectively. The mice were given either vehicle (PBS) or 17-DMAG (15 mg/kg/day) treatment after the cells formed a 100 mm3 sphere. When compared to control groups, both LLC and A549-bearing mice receiving 17-DMAG showed a significant inhibition of tumor growth (Figure 7A). In contrast, the bodyweight of the animals was comparable across groups (Figure 7B), indicating that it was not toxic in vivo. The tumors were removed and weighed at the end of the experiment. Tumors in the 17-DMAG group were significantly lighter than those in the control group, and the inhibition rates of 17-DMAG treatment in LLC and A549 bearing mice were 80.9 and 50.7%, respectively (Figures 7C,D). Furthermore, a TUNEL assay revealed that 17-DMAG induced tumor-cell apoptosis in vivo (Figure 7E).
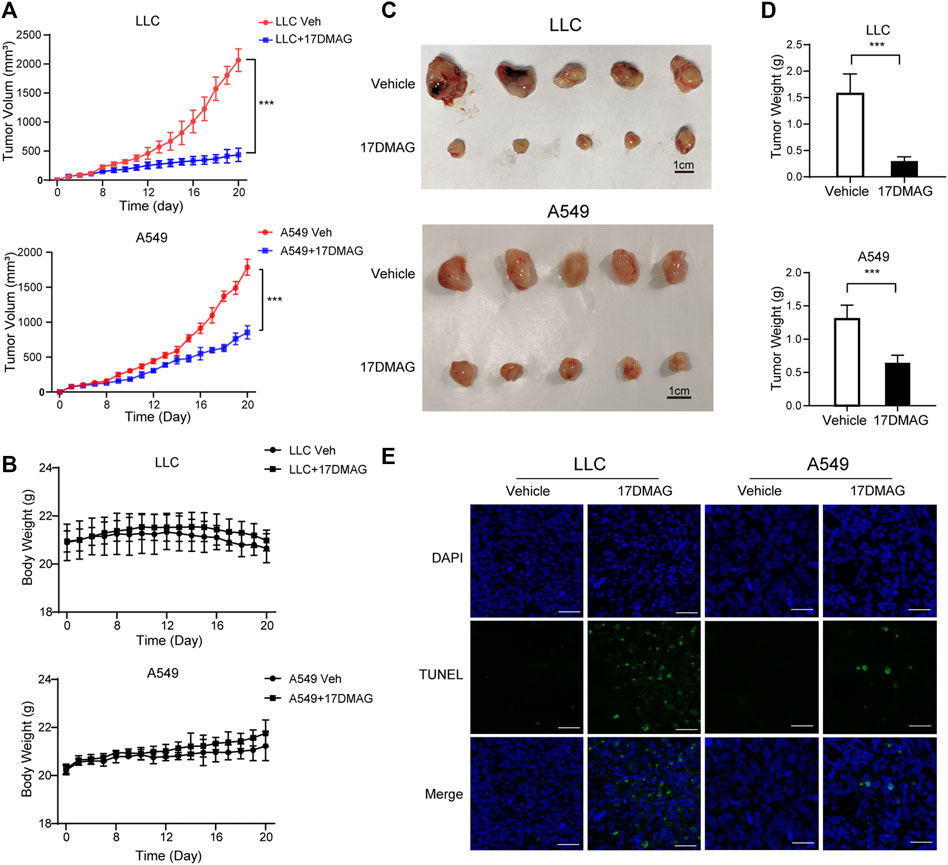
FIGURE 7. 17-DMAG inhibited tumor growth and induced cell death in vivo. Male C57/BL6 and Balb/c nude mice (n = 10) were injected with LLC and A549 cells, respectively (n = 10). After tumors reached approximately 100 mm3, mice were treated with or without 17-DMAG (15 mg/kg/day) intraperitoneally. (A) Tumor volumes of LLC and A549 bearing mice were calculated every day. ***p < 0.001. (B) The weight of each LLC or A549-bearing mouse was measured every other day before the treatment of 17-DMAG (n = 5 per group). (C) Tumor mass images of both LLC and A549 model mice at the end of the experiment (n = 5 per group). (D) LLC and A549 tumors treated with or without 17-DMAG were removed and weighed after the euthanasia of mice. ***p < 0.001. (E) Analysis of apoptosis by TUNEL staining (blue fluorescence, DAPI staining for nuclei; green fluorescence, TUNEL-positive staining) in LLC and A549 cell xenograft mouse-derived tissue from control, 17-DMAG–treated animals. Scale bar = 50 μm.
In cancer tissues, the inhibitory effect of 17-DMAG on AKT1/ERK signaling pathways was then investigated. In both tumor models, 17-DMAG inhibited not only AKT1 and ERK phosphorylation but also proliferative markers such as CyclinD1 and induced cleaved Caspase3 expression, indicating a similar effect on HSP90 regulation of the AKT1/ERK pathway in vitro (Figures 8A–C). As CD8+ T lymphocytes are the major cytotoxic cells that eliminate tumor cells (Prado-Garcia et al., 2012). Immunohistochemical analysis was used to assess the infiltration of CD8+ T lymphocytes in the cancer tissues of the immunocompetent mouse C57BL/6 model. As shown in Figure 8D, 17-DMAG treated mice had a significantly higher number of CD8+ cytotoxic T cells (marked with an arrow) in tumor tissues compared to vehicle-treated controls. Furthermore, the transcripts of genes related to T-cell–cytotoxic granule components (GzmA and GzmB) and the key anti-tumor cytokine (Interferon-γ) were statistically more abundant in 17-DMAG-treated mice tumors (Figure 8E).
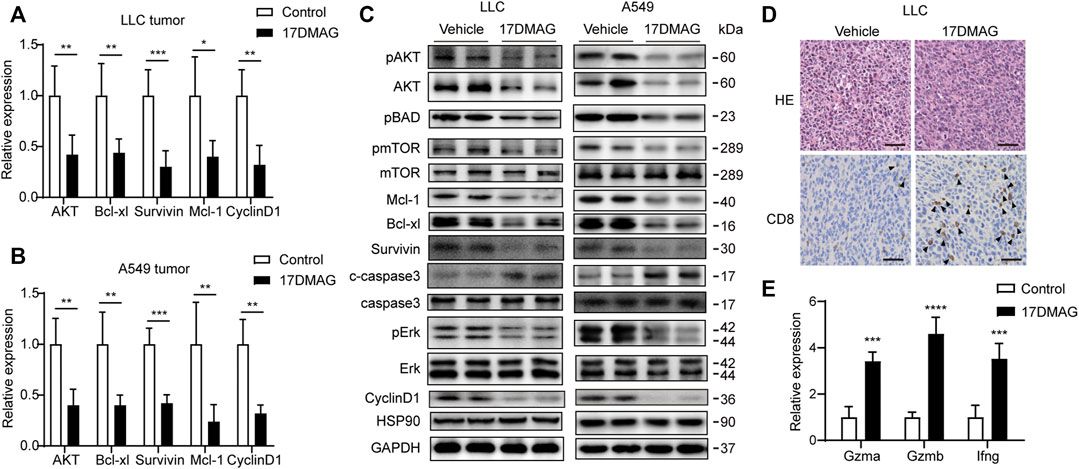
FIGURE 8. Inhibiting HSP90 reversed AKT1/ERK activation in vivo. (A) mRNA levels of indicated genes from LLC tumor tissue were tested by qPCR. *p < 0.05, **p < 0.01, ***p < 0.001. (B) mRNA levels of indicated genes from A549 tumor tissue were tested by qPCR. **p < 0.01, ***p < 0.001. (C) Lysates from LLC and A549 tumor tissue were analyzed by western blotting and probed with indicated antibodies. (D) Representative images of LLC tumor sections stained with CD8 antibody. The positive staining of CD8 was marked with arrows. Scale bar = 50 μm. (E) Expression of genes related to lymphocyte cytotoxicity. Relative mRNA expression levels were adjusted to the level of beta-actin mRNA for each sample. ***p < 0.001, ****p < 0.0001.
Tumor cells face a more severe proteostasis crisis than normal cells due to the presence of a large number of mutant proteins and a disruption in protein synthesis for rapid proliferation (Sherr, 1996). As a result, HSP90 was important in cancer cells for assisting protein folding and enhancing signaling pathways, making it a potential therapeutic target in cancer research (Wang H. et al., 2016). In human cells, there were two cytoplasmic isoforms of HSP90 proteins named HSP90AA1 and Hsp90AB1, one ER-located isoform of HSP90B1, and one mitochondrial paralog TRAP1. We found that the transcription levels of all these isoforms were higher in cancer tissues than normal tissues (Figure 1A) after a systematic screening of their expression in cancer patients. However, only HSP90AA1 showed a significant correlation with a shorter overall survival time in lung cancer patients, indicating that it plays an important role in tumor progression (Figure 1A).
The serine/threonine kinase AKT (or protein kinase B) controls key cellular processes such as glucose metabolism, cell cycle progression, and apoptosis (Lawlor and Alessi, 2001). Active AKT can contribute to tumorigenesis in a large number of human cancers, including brain, gastric, colon, breast, lung, and prostate carcinomas (Brognard et al., 2001; Scheid and Woodgett, 2001). Hyperactivation of ERK promotes the growth and malignant transformation of lung epithelial cells (Qi et al., 2018; Saad et al., 2019). A constitutively active mutation of KRAS (KrasG12D) and epidermal growth factor receptor (EGFR) can both cause abnormal ERK signaling (Rosell and Karachaliou, 2016; Recondo et al., 2018). Other mechanisms, however, contribute to the dysregulation of this signaling. HSP90 is an ATP-dependent molecular chaperone required for the stability of its "client" oncoproteins, many of which are KRAS effectors, such as members of RAF/ERK and PI3K/AKT/mTOR pathways (Chatterjee et al., 2016). The PI3K/AKT signaling pathway is one of the most frequently activated signaling pathways in different tumors, including lung cancer. The link between HSP90 inhibition and PI3K/AKT/mTOR signaling suppression has been previously reported in a number of solid and hematological malignancies, and our results support these findings (Basso et al., 2002; Ohji et al., 2006; Giulino-Roth et al., 2017). In addition, HSP90 has been shown to be critical for B-RAF or RAF-1–induced MEK activation (Grammatikakis et al., 1999). Concurrent pharmacologic targets of the RAF/MEK/ERK and PI3K/AKT/mTOR pathways have been reported in KRAS-driven cancer models, but clinically this strategy has been limited by additive toxicity (Collisson et al., 2012). In our study, the HSP90AA1 knockdown inhibited the AKT1 and ERK pathways, which were over-activated in tumor tissues.
To explore the clinical translational significance of this observation, we tested the anti-cancer activity of an HSP90 specific inhibitor-17-DMAG in both human and mouse lung cancer cell lines. 17-DMAG is a semi-synthetic geldanamycin derivative, which differs from 17-AAG in position 17 side chain of the ansa ring (Smith et al., 2005). Compared to other HSP90 inhibitors, 17-DMAG was more water-soluble than 17-AAG and had lower toxicity to the liver than geldanamycin, making it a more clinically viable agent.
Our findings show that 17-DMAG has a significant anti-cancer effect with minimal toxicity in LLC or A549-bearing mice. 17-DMAG inhibited cell proliferation and induced cell apoptosis in lung cancer cells by inhibiting the AKT1/ERK pathway, and it was well tolerated in normal cells and mouse models. This study conclusively indicates that targeting HSP90 is a promising therapeutic strategy for patients with lung cancer.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by Ethics Committee in the Medical School of Nanjing University. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by Committee of the Care and Use of Laboratory Animals (Nanjing University).
MN, BZ and LL: Conceptualization, Methodology, Data curation, Writing- Original draft preparation. LL and ZS: Visualization, Investigation. WP and CZ: Supervision, Formal analysis. LW, PL, and RL: Software. RW: Validation. JW: Writing-Reviewing and Editing. SS, QG and HW: Resources, Writing-Reviewing and Editing, Project administration, Funding acquisition. All authors read and approved the final manuscript.
This work was supported by grants from the National Natural Science Foundation of China (Nos. 82070912 and 81773326), the Fundamental Research Funds for the Central Universities (0214-14380509, 0214-14380523), the Natural Science Foundation of Jiangsu Province China (BK20210182, BK20171347 and BE2019676), and grants from the National Key R&D Program of China (2020YFC2005600, 2020YFC2005601, 2020YFC2005100, 2020YFC2005300). Nanjing Medical Science and Technique Development Foundation (YKK17186).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2021.724192/full#supplementary-material
Ambade, A., Catalano, D., Lim, A., and Mandrekar, P. (2012). Inhibition of Heat Shock Protein (Molecular Weight 90 kDa) Attenuates Proinflammatory Cytokines and Prevents Lipopolysaccharide-Induced Liver Injury in Mice. Hepatology 55 (5), 1585–1595. doi:10.1002/hep.24802
Basso, A. D., Solit, D. B., Chiosis, G., Giri, B., Tsichlis, P., and Rosen, N. (2002). Akt Forms an Intracellular Complex with Heat Shock Protein 90 (Hsp90) and Cdc37 and Is Destabilized by Inhibitors of Hsp90 Function. J. Biol. Chem. 277 (42), 39858–39866. doi:10.1074/jbc.M206322200
Brognard, J., Clark, A. S., Ni, Y., and Dennis, P. A. (2001). Akt/protein Kinase B Is Constitutively Active in Non-small Cell Lung Cancer Cells and Promotes Cellular Survival and Resistance to Chemotherapy and Radiation. Cancer Res. 61 (10), 3986–3997.
Bucci, M., Roviezzo, F., Cicala, C., Sessa, W. C., and Cirino, G. (2000). Geldanamycin, an Inhibitor of Heat Shock Protein 90 (Hsp90) Mediated Signal Transduction Has Anti-inflammatory Effects and Interacts with Glucocorticoid Receptor In Vivo. Br. J. Pharmacol. 131 (1), 13–16. doi:10.1038/sj.bjp.0703549
Chatterjee, S., Bhattacharya, S., Socinski, M. A., and Burns, T. F. (2016). HSP90 Inhibitors in Lung Cancer: Promise Still Unfulfilled. Clin. Adv. Hematol. Oncol. 14 (5), 346–356.
Chen, B., Zhong, D., and Monteiro, A. (2006). Comparative Genomics and Evolution of the HSP90 Family of Genes across All Kingdoms of Organisms. BMC Genomics 7 (1), 156. doi:10.1186/1471-2164-7-156
Chen, Y., Tang, Y., Tang, Y., Yang, Z., and Ding, G. (2019). Serine Protease from Nereis Virens Inhibits H1299 Lung Cancer Cell Proliferation via the PI3K/AKT/mTOR Pathway. Mar. Drugs 17 (6), 366. doi:10.3390/md17060366
Collisson, E. A., Trejo, C. L., Silva, J. M., Gu, S., Korkola, J. E., Heiser, L. M., et al. (2012). A central Role for RAF→MEK→ERK Signaling in the Genesis of Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2 (8), 685–693. doi:10.1158/2159-8290.CD-11-0347
Datta, S. R., Dudek, H., Tao, X., Masters, S., Fu, H., Gotoh, Y., et al. (1997). Akt Phosphorylation of BAD Couples Survival Signals to the Cell-Intrinsic Death Machinery. Cell 91 (2), 231–241. doi:10.1016/S0092-8674(00)80405-5
Davis, S., and Meltzer, P. S. (2007). GEOquery: a Bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 23 (14), 1846–1847. doi:10.1093/bioinformatics/btm254
Egorin, M. J., Lagattuta, T. F., Hamburger, D. R., Covey, J. M., White, K. D., Musser, S. M., et al. (2002). Pharmacokinetics, Tissue Distribution, and Metabolism of 17-(dimethylaminoethylamino)-17-Demethoxygeldanamycin (NSC 707545) in CD2F1 Mice and Fischer 344 Rats. Cancer Chemother. Pharmacol. 49 (1), 7–19. doi:10.1007/s00280-001-0380-8
Eiseman, J. L., Lan, J., Lagattuta, T. F., Hamburger, D. R., Joseph, E., Covey, J. M., et al. (2005). Pharmacokinetics and Pharmacodynamics of 17-demethoxy 17-[[(2-dimethylamino)ethyl]amino]geldanamycin (17DMAG, NSC 707545) in C.B-17 SCID Mice Bearing MDA-MB-231 Human Breast Cancer Xenografts. Cancer Chemother. Pharmacol. 55 (1), 21–32. doi:10.1007/s00280-004-0865-3
Faustino-Rocha, A., Oliveira, P. A., Pinho-Oliveira, J., Teixeira-Guedes, C., Soares-Maia, R., da Costa, R. G., et al. (2013). Estimation of Rat Mammary Tumor Volume Using Caliper and Ultrasonography Measurements. Lab. Anim. (NY) 42 (6), 217–224. doi:10.1038/laban.254
Friedman, J. A., Wise, S. C., Hu, M., Gouveia, C., Vander Broek, R., Freudlsperger, C., et al. (2013). HSP90 Inhibitor SNX5422/2112 Targets the Dysregulated Signal and Transcription Factor Network and Malignant Phenotype of Head and Neck Squamous Cell Carcinoma. Transl Oncol. 6 (4), 429–441. doi:10.1593/tlo.13292
Giulino-Roth, L., van Besien, H. J., Dalton, T., Totonchy, J. E., Rodina, A., Taldone, T., et al. (2017). Inhibition of Hsp90 Suppresses PI3K/AKT/mTOR Signaling and Has Antitumor Activity in Burkitt Lymphoma. Mol. Cancer Ther. 16 (9), 1779–1790. doi:10.1158/1535-7163.MCT-16-0848
Grammatikakis, N., Lin, J. H., Grammatikakis, A., Tsichlis, P. N., and Cochran, B. H. (1999). p50(cdc37) Acting in Concert with Hsp90 Is Required for Raf-1 Function. Mol. Cel. Biol. 19 (3), 1661–1672. doi:10.1128/MCB.19.3.1661
Hussain, S. P., Hofseth, L. J., and Harris, C. C. (2001). Tumor Suppressor Genes: at the Crossroads of Molecular Carcinogenesis, Molecular Epidemiology and Human Risk Assessment. Lung Cancer 34 (Suppl. 2), S7–S15. doi:10.1016/s0169-5002(01)00339-7
Kamal, A., Thao, L., Sensintaffar, J., Zhang, L., Boehm, M. F., Fritz, L. C., et al. (2003). A High-Affinity Conformation of Hsp90 Confers Tumour Selectivity on Hsp90 Inhibitors. Nature 425 (6956), 407–410. doi:10.1038/nature01913
Kaur, G., Belotti, D., Burger, A. M., Fisher-Nielson, K., Borsotti, P., Riccardi, E., et al. (2004). Antiangiogenic Properties of 17-(dimethylaminoethylamino)-17-Demethoxygeldanamycin: an Orally Bioavailable Heat Shock Protein 90 Modulator. Clin. Cancer Res. 10 (14), 4813–4821. doi:10.1158/1078-0432.CCR-03-0795
Kawabe, M., Mandic, M., Taylor, J. L., Vasquez, C. A., Wesa, A. K., Neckers, L. M., et al. (2009). Heat Shock Protein 90 Inhibitor 17-Dimethylaminoethylamino-17-Demethoxygeldanamycin Enhances EphA2+ Tumor Cell Recognition by Specific CD8+ T Cells. Cancer Res. 69 (17), 6995–7003. doi:10.1158/0008-5472.CAN-08-4511
Lawlor, M. A., and Alessi, D. R. (2001). PKB/Akt: a Key Mediator of Cell Proliferation, Survival and Insulin Responses? J. Cel Sci. 114 (Pt 16), 2903–2910. doi:10.1242/jcs.114.16.2903
Li, G., Ji, X. D., Gao, H., Zhao, J. S., Xu, J. F., Sun, Z. J., et al. (2012). EphB3 Suppresses Non-small-cell Lung Cancer Metastasis via a PP2A/RACK1/Akt Signalling Complex. Nat. Commun. 3 (1), 667. doi:10.1038/ncomms1675
Liu, Y., Sun, S. Y., Owonikoko, T. K., Sica, G. L., Curran, W. J., Khuri, F. R., et al. (2012). Rapamycin Induces Bad Phosphorylation in Association with its Resistance to Human Lung Cancer Cells. Mol. Cancer Ther. 11 (1), 45–56. doi:10.1158/1535-7163.MCT-11-0578
Novello, S., Vavalà, T., Levra, M. G., Solitro, F., Pelosi, E., Veltri, A., et al. (2013). Early Response to Chemotherapy in Patients with Non-small-cell Lung Cancer Assessed by [18F]-Fluoro-Deoxy-D-Glucose Positron Emission Tomography and Computed Tomography. Clin. Lung Cancer 14 (3), 230–237. doi:10.1016/j.cllc.2012.10.004
Ohji, G., Hidayat, S., Nakashima, A., Tokunaga, C., Oshiro, N., Yoshino, K., et al. (2006). Suppression of the mTOR-Raptor Signaling Pathway by the Inhibitor of Heat Shock Protein 90 Geldanamycin. J. Biochem. 139 (1), 129–135. doi:10.1093/jb/mvj008
Park, M. R., Park, Y. H., Choi, J. W., Park, D. I., Chung, C. U., Moon, J. Y., et al. (2014). Progression-free Survival: an Important Prognostic Marker for Long-Term Survival of Small Cell Lung Cancer. Tuberc. Respir. Dis. (Seoul) 76 (5), 218–225. doi:10.4046/trd.2014.76.5.218
Prado-Garcia, H., Romero-Garcia, S., Morales-Fuentes, J., Aguilar-Cazares, D., and Lopez-Gonzalez, J. S. (2012). Activation-Induced Cell Death of Memory CD8+ T Cells from Pleural Effusion of Lung Cancer Patients is Mediated by the Type II Fas-Induced Apoptotic Pathway. Cancer Immunol. Immunother. 61 (7), 1065–1080. doi:10.1007/s00262-011-1165-5
Qi, M., Tian, Y., Li, W., Li, D., Zhao, T., Yang, Y., et al. (2018). ERK Inhibition Represses Gefitinib Resistance in Non-small Cell Lung Cancer Cells. Oncotarget 9 (15), 12020–12034. doi:10.18632/oncotarget.24147
Rahmani, M., Yu, C., Dai, Y., Reese, E., Ahmed, W., Dent, P., et al. (2003). Coadministration of the Heat Shock Protein 90 Antagonist 17-allylamino- 17-demethoxygeldanamycin with Suberoylanilide Hydroxamic Acid or Sodium Butyrate Synergistically Induces Apoptosis in Human Leukemia Cells. Cancer Res. 63 (23), 8420–8427.
Rao, A., Taylor, J. L., Chi-Sabins, N., Kawabe, M., Gooding, W. E., and Storkus, W. J. (2012). Combination Therapy with HSP90 Inhibitor 17-DMAG Reconditions the Tumor Microenvironment to Improve Recruitment of Therapeutic T Cells. Cancer Res. 72 (13), 3196–3206. doi:10.1158/0008-5472.CAN-12-0538
Recondo, G., Facchinetti, F., Olaussen, K. A., Besse, B., and Friboulet, L. (2018). Making the First Move in EGFR-Driven or ALK-Driven NSCLC: First-Generation or Next-Generation TKI? Nat. Rev. Clin. Oncol. 15 (11), 694–708. doi:10.1038/s41571-018-0081-4
Riquelme, E., Behrens, C., Lin, H. Y., Simon, G., Papadimitrakopoulou, V., Izzo, J., et al. (2016). Modulation of EZH2 Expression by MEK-ERK or PI3K-AKT Signaling in Lung Cancer Is Dictated by Different KRAS Oncogene Mutations. Cancer Res. 76 (3), 675–685. doi:10.1158/0008-5472.CAN-15-1141
Rosell, R., and Karachaliou, N. (2016). Lung Cancer: Using ctDNA to Track EGFR and KRAS Mutations in Advanced-Stage Disease. Nat. Rev. Clin. Oncol. 13 (7), 401–402. doi:10.1038/nrclinonc.2016.83
Saad, M. I., Alhayyani, S., Mcleod, L., Yu, L., Alanazi, M., Deswaerte, V., et al. (2019). ADAM17 Selectively Activates the IL-6 Trans-signaling/ERK MAPK axis in KRAS-Addicted Lung Cancer. EMBO Mol. Med. 11 (4), e9976. doi:10.15252/emmm.201809976
Scheid, M. P., and Woodgett, J. R. (2001). PKB/AKT: Functional Insights from Genetic Models. Nat. Rev. Mol. Cel Biol. 2 (10), 760–768. doi:10.1038/35096067
Schopf, F. H., Biebl, M. M., and Buchner, J. (2017). The HSP90 Chaperone Machinery. Nat. Rev. Mol. Cel Biol. 18 (6), 345–360. doi:10.1038/nrm.2017.20
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 13 (11), 2498–2504. doi:10.1101/gr.1239303
Sherr, C. J. (1996). Cancer Cell Cycles. Science 274 (5293), 1672–1677. doi:10.1126/science.274.5293.1672
Smith, V., Sausville, E. A., Camalier, R. F., Fiebig, H. H., and Burger, A. M. (2005). Comparison of 17-Dimethylaminoethylamino-17-Demethoxy-Geldanamycin (17DMAG) and 17-Allylamino-17-Demethoxygeldanamycin (17AAG) In Vitro: Effects on Hsp90 and Client Proteins in Melanoma Models. Cancer Chemother. Pharmacol. 56 (2), 126–137. doi:10.1007/s00280-004-0947-2
Subramanian, A., Tamayo, P., Mootha, V. K., Mukherjee, S., Ebert, B. L., Gillette, M. A., et al. (2005). Gene Set Enrichment Analysis: a Knowledge-Based Approach for Interpreting Genome-wide Expression Profiles. Proc. Natl. Acad. Sci. U S A. 102 (43), 15545–15550. doi:10.1073/pnas.0506580102
Szklarczyk, D., Gable, A. L., Lyon, D., Junge, A., Wyder, S., Huerta-Cepas, J., et al. (2019). STRING V11: Protein-Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-wide Experimental Datasets. Nucleic Acids Res. 47 (D1), D607–D613. doi:10.1093/nar/gky1131
Therneau, T. M., and Grambsch, P. M. (2015). Modeling Survival Data: Extending the Cox Model. New York: Springer.
Torre, L. A., Bray, F., Siegel, R. L., Ferlay, J., Lortet-Tieulent, J., and Jemal, A. (2015). Global Cancer Statistics, 2012. CA Cancer J. Clin. 65 (2), 87–108. doi:10.3322/caac.21262
Trepel, J., Mollapour, M., Giaccone, G., and Neckers, L. (2010). Targeting the Dynamic HSP90 Complex in Cancer. Nat. Rev. Cancer 10 (8), 537–549. doi:10.1038/nrc2887
Wang H, H., Lu, M., Yao, M., and Zhu, W. (2016). Effects of Treatment with an Hsp90 Inhibitor in Tumors Based on 15 Phase II Clinical Trials. Mol. Clin. Oncol. 5 (3), 326–334. doi:10.3892/mco.2016.963
Wang YL, Y. L., Shen, H. H., Cheng, P. Y., Chu, Y. J., Hwang, H. R., Lam, K. K., et al. (2016). 17-DMAG, an HSP90 Inhibitor, Ameliorates Multiple Organ Dysfunction Syndrome via Induction of HSP70 in Endotoxemic Rats. PLoS One 11 (5), e0155583. doi:10.1371/journal.pone.0155583
Wood, S. L., Pernemalm, M., Crosbie, P. A., and Whetton, A. D. (2015). Molecular Histology of Lung Cancer: From Targets to Treatments. Cancer Treat. Rev. 41 (4), 361–375. doi:10.1016/j.ctrv.2015.02.008
Wu, L.-M., Liao, X.-Z., Zhang, Y., He, Z.-R., Nie, S.-Q., Ke, B., et al. (2021). Parthenolide Augments the Chemosensitivity of Non-small-cell Lung Cancer to Cisplatin via the PI3K/AKT Signaling Pathway. Front. Cel Dev. Biol. 8, 610097. doi:10.3389/fcell.2020.610097
Yang, J., Xu, W. W., Hong, P., Ye, F., Huang, X. H., Hu, H. F., et al. (2019). Adefovir Dipivoxil Sensitizes colon Cancer Cells to Vemurafenib by Disrupting the KCTD12-CDK1 Interaction. Cancer Lett. 451, 79–91. doi:10.1016/j.canlet.2019.02.050
Yu, G., Wang, L. G., Han, Y., and He, Q. Y. (2012). clusterProfiler: an R Package for Comparing Biological Themes Among Gene Clusters. OMICS 16 (5), 284–287. doi:10.1089/omi.2011.0118
Yuno, A., Lee, M. J., Lee, S., Tomita, Y., Rekhtman, D., Moore, B., et al. (2018). Clinical Evaluation and Biomarker Profiling of Hsp90 Inhibitors. Methods Mol. Biol. 1709, 423–441. doi:10.1007/978-1-4939-7477-1_29
Keywords: Hsp90, 17-DMAG, lung cancer, AKT1, ERK
Citation: Niu M, Zhang B, Li L, Su Z, Pu W, Zhao C, Wei L, Lian P, Lu R, Wang R, Wazir J, Gao Q, Song S and Wang H (2022) Targeting HSP90 Inhibits Proliferation and Induces Apoptosis Through AKT1/ERK Pathway in Lung Cancer. Front. Pharmacol. 12:724192. doi: 10.3389/fphar.2021.724192
Received: 12 June 2021; Accepted: 28 December 2021;
Published: 14 January 2022.
Edited by:
Feng-Yao Tang, China Medical University, TaiwanCopyright © 2022 Niu, Zhang, Li, Su, Pu, Zhao, Wei, Lian, Lu, Wang, Wazir, Gao, Song and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qian Gao, cWlhbl9nYW9Abmp1LmVkdS5jbg==; Shiyu Song, c29uZ3NoaXl1QG5qdS5lZHUuY24=; Hongwei Wang, aHdhbmdAbmp1LmVkdS5jbg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.