 Ziyu Yu
Ziyu Yu Nan Jiang
Nan Jiang Wenru Su
Wenru Su Yehong Zhuo
Yehong Zhuo
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 12 July 2021
Sec. Translational Pharmacology
Volume 12 - 2021 | https://doi.org/10.3389/fphar.2021.701564
This article is part of the Research Topic Advanced Perspectives in Cell Therapy and Correlated Immunopharmacology View all 15 articles
Neuroinflammation is a complex inflammatory process in the nervous system that is expected to play a significant role in neurological diseases. Necroptosis is a kind of necrosis that triggers innate immune responses by rupturing dead cells and releasing intracellular components; it can be caused by Toll-like receptor (TLR)-3 and TLR-4 agonists, tumor necrosis factor (TNF), certain microbial infections, and T cell receptors. Necroptosis signaling is modulated by receptor-interacting protein kinase (RIPK) 1 when the activity of caspase-8 becomes compromised. Activated death receptors (DRs) cause the activation of RIPK1 and the RIPK1 kinase activity-dependent formation of an RIPK1-RIPK3-mixed lineage kinase domain-like protein (MLKL), which is complex II. RIPK3 phosphorylates MLKL, ultimately leading to necrosis through plasma membrane disruption and cell lysis. Current studies suggest that necroptosis is associated with the pathogenesis of neuroinflammatory diseases, such as Alzheimer’s disease, Parkinson’s disease, and traumatic brain injury. Inhibitors of necroptosis, such as necrostatin-1 (Nec-1) and stable variant of Nec (Nec-1s), have been proven to be effective in many neurological diseases. The purpose of this article is to illuminate the mechanism underlying necroptosis and the important role that necroptosis plays in neuroinflammatory diseases. Overall, this article shows a potential therapeutic strategy in which targeting necroptotic factors may improve the pathological changes and clinical symptoms of neuroinflammatory disorders.
Inflammation, which is usually caused by injury or infection (Andersson and Tracey, 2011), are fundamentally distinguished by pathology (Kearney and Martin, 2017) between acute and chronic forms of inflammation. Acute inflammation responds to irritants in the early stages, which is an essential response that prepares the body to repair damaged areas during acute inflammation and neuroinflammation, including traumatic brain injury (TBI), stroke, and encephalitis (Huang et al., 2018a; Zhang et al., 2018; Venkatesan et al., 2019). Chronic inflammation is caused by persistent stimuli, which leads to injury of the nerve tissue, resulting in neurodegeneration and inducing neuroinflammation into a vicious cycle (Nasef et al., 2017), including Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS) (Dong et al., 2018; Ren et al., 2018; Lee et al., 2019). A variety of immunocytes are involved in inflammation (Mack, 2018; Tsai et al., 2018). Acute inflammation is dominated by neutrophil infiltration (Pinho-Ribeiro et al., 2018; Kapur et al., 2019), whereas chronic inflammation is often accompanied by macrophage and lymphocyte infiltration (Cao et al., 2019; Faissner et al., 2019; Lodygin et al., 2019; Na et al., 2019).
Neuroinflammation is a complex inflammatory response of the nervous system that may be triggered by various pathogens or toxins and induce immunocyte infiltration and activation (Stephenson et al., 2018). Eventually, the effects lead to neuronal and/or axonal degeneration or death (Estes and McAllister, 2016). A feature essential to maintain neuroplasticity is neuroinflammatory homeostasis. Homeostasis is regulated by the interaction between neurons, glial cells and vascular endothelial cells. Homeostatic imbalance caused by different reasons (such as injury, infection or stress) may have similar pathological manifestations (DiSabato et al., 2016).
However, the mechanisms of neuroinflammation remain unclear in different situations. To better understand the inflammatory disease neuritis, acute and chronic neuroinflammation will be discussed separately in the following sections.
Historically, two forms of cell death, apoptosis and necrosis, have been recognized because of their important roles in homeostasis, development, and pathogenesis (Pasparakis and Vandenabeele, 2015). In the past, necrosis was thought to be accidental death due to excessive cytotoxic damage, carried out via conventional molecular events (Vanden Berghe et al., 2014). In contrast, apoptosis is defined as programmed cell death. Under a microscope, apoptosis is characterized by apoptotic bodies, nuclear pyknosis and fragmentation, and an intact cell membrane (Kerr et al., 1972; Choo et al., 2019). However, an increasing number of studies have described another form of necrosis that performs as programmed and regulated cell death, named necroptosis.
Morphologically, necroptosis has the following characteristics: (1) it resembles necrosis–dying cells cluster together, with disrupted membranes, swollen cell bodies and organelles, and fragmented chromatin; (2) a large quantity of inflammasomes; (3) autophagy (Vanden et al., 2013). Compared with apoptosis, necroptotic cells passively pass through the damaged membrane into the extracellular matrix (Zhang et al., 2017a).
At the molecular level, intracellular and extracellular stimuli and corresponding ligands of the death receptor family trigger necrosis (Zhou and Yuan, 2014). The currently known key components of the necroptotic signaling pathway are receptor-interacting protein kinase 3 (RIPK3) and its substrate, mixed lineage kinase domain-like protein (MLKL), which is a pseudokinase (Yuan et al., 2019). Moreover, small molecules, such as necrostatin-1 (Nec-1), Nec-5, and Nec-7, are thought to inhibit the necroptotic signaling pathway (Gonzalez-Juarbe et al., 2015; Strilic et al., 2016; Fuji et al., 2018). Nec-1 is an ATP-competitive allosteric inhibitor of RIPK1. In a mouse stroke model, Nec-1 was able to reduce brain damage caused by reperfusion (Chu et al., 2018). MicroRNA-155 is also considered an inhibitor of RIPK1 (Liu et al., 2011).
Necroptosis is involved in many pathological processes, such as trauma, cerebral ischemia-reperfusion injury, and inflammatory diseases (Wang et al., 2012; Li et al., 2019; Ni et al., 2019; Zhang et al., 2019). Necroptosis has been reported to play a crucial role in the pathogenesis of certain neuroinflammatory disorders (Ito et al., 2016; Chia et al., 2018; Zhang et al., 2019). Because necroptosis shows the potential to be a target for intervention in neuroinflammatory disorders, it has garnered increasing focus from researchers. In our review, the molecular mechanisms of necroptosis and the role of necroptosis in the development and progression of acute and chronic neuroinflammatory disorders will be described, which may be helpful for finding treatments for neuroinflammatory diseases.
Tumor necrosis factor receptor 1 (TNFR1), which has been reported by studies of TNF signaling in necroptosis, induces the expression of many genes that regulate inflammation (Liu et al., 2014). However, under some conditions, TNF-⍺ also induces cell death (Annibaldi and Meier, 2018; Lafont et al., 2018). TNF-induced cell death requires TNF-⍺ to bind to TNFR1 on the cell membrane and recruit a series of proteins in the cell to form different complexes (Micheau and Tschopp, 2003; Amin et al., 2018). Among them, complex I includes TNFR-associated death domain (TRADD), RIPK1, TNFR-associated factor 2 (TRAF2), the cellular inhibitor of apoptosis protein 1 (cIAP1), cylindromatosis, and the ubiquitin complex (Amin et al., 2018) (Figure 1).
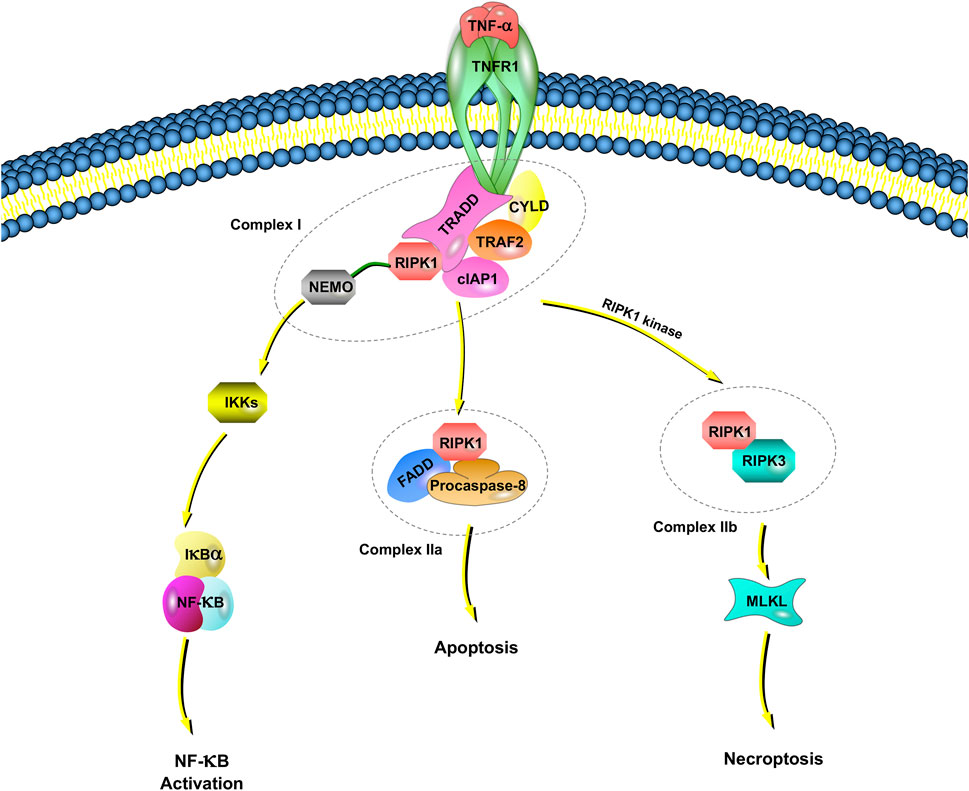
FIGURE 1. TNFR1-dependent necroptosis pathways. TNF-⍺ binds to TNFR1 and recruits a series of proteins, including TRADD, RIPK1, TRAF2, cIAP1, CYLD, and NEMO, which is called complex I. NEMO recruits IKK⍺/IKKβ, resulting in the IKK-mediated phosphorylation of IkB⍺. Once IkB⍺ is phosphorylated, NF-kB signaling pathways are activated. When NF-κB activation is inhibited, deubiquitinated RIPK1, FADD and procaspase-8 are assembled as complex IIa. Complex IIa is involved in apoptosis by activated caspase-8 and cleavage RIPK1. When there is a lack of caspase-8, the cIAP1 in complex I rapidly ubiquitinates RIPK1, leading to the combination of RIPK1 and RIPK3, which is called complex IIb. Complex IIb leads to MLKL-mediated necroptosis.
An IkB kinase (IKK) complex consisting of a subunit essential regulator (nuclear factor-kappa B essential modulator, NEMO, also named IKKγ) and two catalytic subunits (IKK⍺ and IKKβ) plays an important role in mediating immunoinflammatory responses and promoting cell survival and oncogenesis (Silke and Brink, 2010). NEMO recruits IKK⍺/IKKβ, resulting in the rapid and selective IKK-mediated phosphorylation of IkB⍺. IkB⍺ activates NF-kB and upregulates genes encoding prosurvival and proinflammatory molecules.
In complex I, E3 ligase (e.g., cIAP1) rapidly ubiquitinates RIPK1. The ubiquitination of RIPK1 is important for regulating its kinase activity. Inhibiting RIPK1 ubiquitination by antagonizing E3 ligase leads to an increased sensitivity of cells to TNF-induced necroptosis (Feoktistova et al., 2011). When NF-κB activation is inhibited, deubiquitinated RIPK1, Fas-associated death domain (FADD) and procaspase-8 are assembled into the death-inducing signaling complex, and the complex finally dissociates from the plasma membrane, now referred to as Complex IIa. Complex IIa is involved in apoptosis by affecting the activation of caspase-8 and the subsequent cleavage of RIPK1 (Lin et al., 1999). When RIPK1 is deubiquitinated, the formation of complex IIb, which is also called the necrosome, is facilitated by RIPK1, RIPK3, MLKL, FADD and procaspase-8.
Recently, cellular FADD-like IL-1β-converting enzyme-inhibitory protein (c-FLIP), a noncatalytic inactive homolog of caspase-8, was reported to take part in the regulation of necroptosis (Tsuchiya et al., 2015). If c-FLIP long is involved in the composition of the heterodimer, caspase-8 maintains high proteolytic activity to inhibit the association of RIPK1, RIPK3 and FADD, thus suppressing necroptosis (Tsuchiya et al., 2015). However, caspase-8 has no proteolytic activity if it is formed by c-FLIP short; RIPK1 and RIPK3 can therefore be assembled to promote necroptosis. Inhibitors of the necroptotic signaling pathway, such as Nec-1, Nec-1s (a stable variant of Nec-1) and other small molecules, have been widely applied to elucidate the molecular mechanisms of necroptosis (Takahashi et al., 2012). Necroptosis is also regulated by the pseudokinase MLKL, which is a functional RIP3 substrate. The subsequent conformational change in MLKL causes rapid cell membrane breakage (a morphological sign of necrosis) by the formation of disulfide bond-dependent amyloid-like polymers (Wang et al., 2014).
Unlike TNF-induced necroptosis, the molecular mechanisms of microbial-triggered necroptosis are more elusive (Figure 2). Innate immunocytes and macrophages, for example, detect microbial activities and initiate antimicrobial responses though pattern-recognition receptors (Kawai and Akira, 2010). Some of the most well-characterized members of the pattern-recognition receptor family are the TLRs (Fitzgerald and Kagan, 2020). TLRs respond to many pathogen-associated molecular patterns (bacteria, viruses, fungi, parasites, etc.) (Tartey and Takeuchi, 2017). TLR3 detects viral double-stranded RNA or artificial analogs, whereas TLR4 responds to lipopolysaccharide (LPS) (Alexopoulou et al., 2001; Fitzgerald et al., 2003). Toll/IL-1 receptor domain-containing adaptors are recruited after the binding of TLR3 and TLR4 to their corresponding ligands (Netea et al., 2009). Then, inflammatory cytokines are released, leading to type I interferon (IFN) responses. There is only one adaptor protein for TLR3, Toll/IL-1 receptor domain-containing adaptor inducing IFN-β/TIR domain-containing adaptor molecule 1 (TRIF/TICAM-1), whereas the TLR4 pathway can be activated by either TRIF or myeloid differentiation factor 88 (MyD88) (Fitzgerald et al., 2001; Akira and Hoshino, 2003; Kagan et al., 2008).
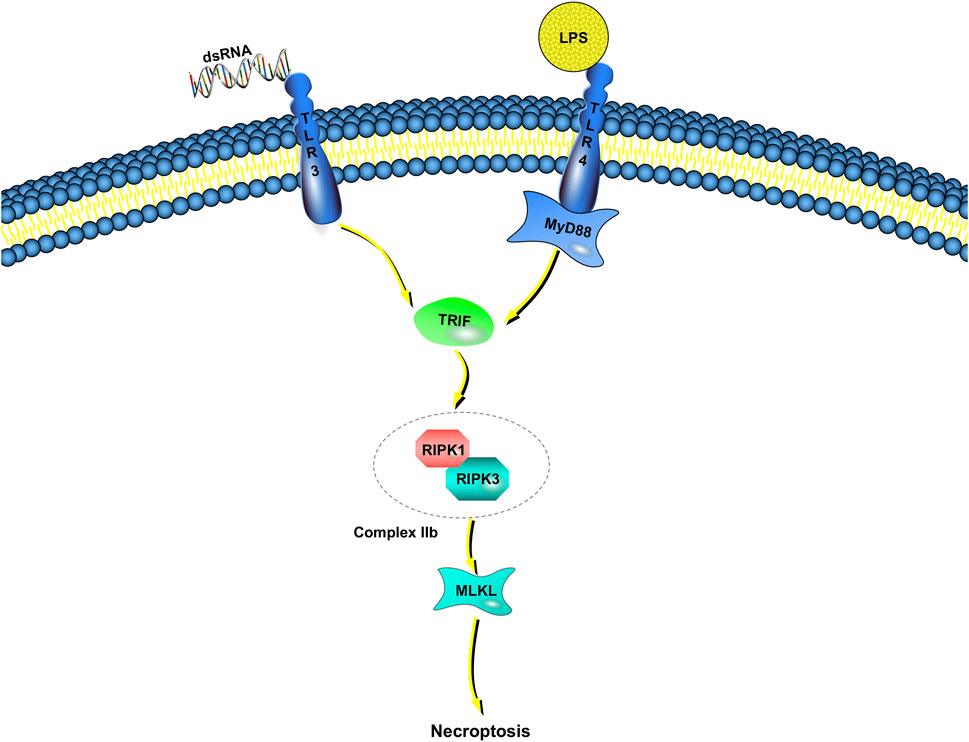
FIGURE 2. TLR-dependent necroptosis pathways. Engagement of TLR3/TLR4 with dsDNA or LPS induces the interaction between TRIF and complex IIb, which is combined RIPK1 with RIPK3. If caspase-8 catalytic activity is impaired, complex IIb triggers MLKL-dependent necroptosis. TRIF is the only adaptor protein of TLR3, whereas the TLR4 pathway can be activated by either TRIF or myeloid MyD88.
In addition to these immune responses, LPS induces necroptosis instead of apoptosis in human macrophages when inhibitors (such as zVAD-fmk) suppress the activation of caspase-8 (Lawlor et al., 2015). Poly (I:C) leads to Jurkat cell (human leukemia T cell) apoptosis when combined with IFN-γ, but the absence of caspase-8 or FADD results in necroptosis rather than apoptosis (Alvarez-Diaz et al., 2016; Boyd-Tressler et al., 2017). Although these results show that necroptosis may be achieved through TLR3 and TLR4 pathways, how to proceed is still unclear (He et al., 2011).
TBI is a disease caused by external forces or other pathological changes in the brain, accompanied by changes in brain function (Maas et al., 2008; McDonald et al., 2016). The mortality rate of severe TBI is high and is estimated at 30–40% in unselected populations in observational studies (Rosenfeld et al., 2012). Some TBI patients lose their lives, and even survivors suffer from enormous physical, mental, emotional and cognitive impairments that undermine the lives of patients and their families and cause enormous losses to society (Limb, 2014; Jenkins et al., 2016).

TABLE 1. Necroptosis in acute neurodegenerative diseases.
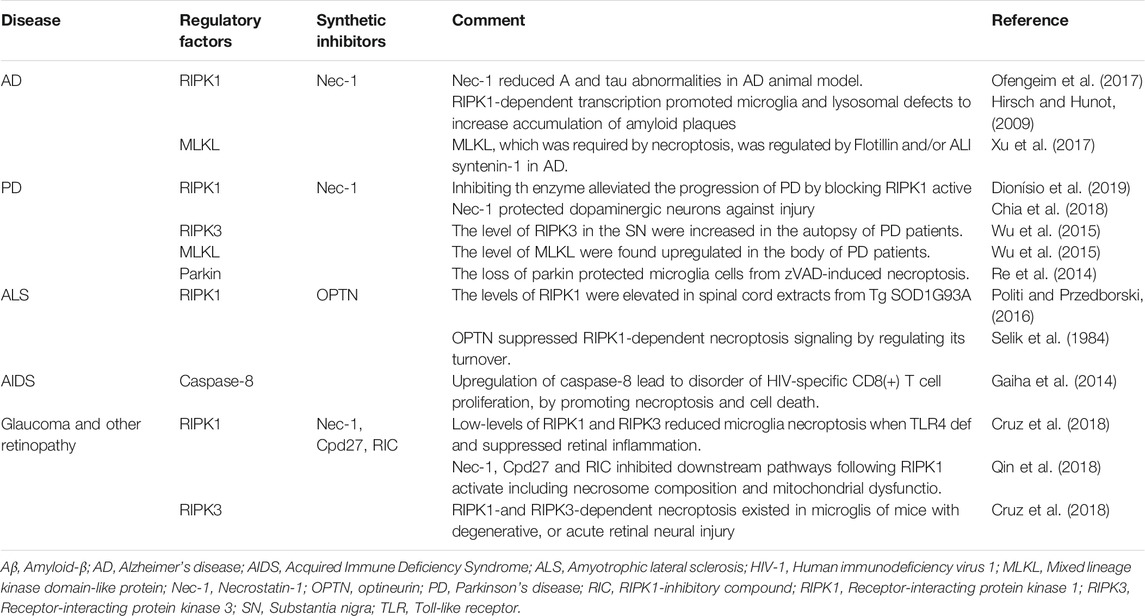
TABLE 2. Necroptosis in chronic neurodegenerative diseases.
A series of studies in 2012 revealed that multiple cell death pathways participated in the development of TBI and that Nec-1 simultaneously inhibited apoptosis and autophagy (Wang et al., 2012). Pathological and biochemical changes related to necroptosis in a rat model of fluid percussion injury (FPI) were observed by Liu (Liu et al., 2016). In an early phase (6 h) after TBI, RIPK 1 and 3, MLKL, HMGB1 and proinflammatory factors (such as TNF-α, IL-6 and IL-18) were increased in the cortex (Liu et al., 2016). Posttraumatic hypothermia (33 °C) led to decreases in necroptosis regulators, proinflammatory cytokines and brain injury in TBI rats compared to treatment with normal temperature (You et al., 2008). Notably, by targeting necroptosis signaling after TBI, the injured central nervous system (CNS) can be protected from tissue damage and inflammation (You et al., 2008). The following year, hypothermia was reported to significantly reduce RIPK-1 upregulation in moderate TBI rat models, which may inhibit the necroptosis pathway after moderate TBI (Zhang et al., 2017b). Moreover, cognitive dysfunction and glial activation could be observed in TBI mice, and these changes were attenuated in RIPK3-knockout (KO) mice (Liu et al., 2018). Notably, in vitro studies have shown that RIPK3-knockdown of astrocytes can reduce oxidative stress, inflammation and apoptosis, which is dependent on the activation of adenosine 5‘-monophosphate-activated protein kinase-alpha (AMPKα) (The, 2018). In conclusion, inhibition of RIPK3 may be a therapeutic target against cerebral damage by suppressing immunoinflammatory responses, oxidative stress and apoptosis.
Stroke is a broad term that includes diseases caused by blockage or bleeding of blood vessels that supply the brain (Lakhan et al., 2009; The, 2018). Its incidence remains high, while the number of approved treatment methods is low (Ceulemans et al., 2010). As society ages, the number of stroke patients continues to increase and will become an important socioeconomic burden, as 80% of stroke patients remain disabled (Durukan and Tatlisumak, 2007; Candelario-Jalil, 2009).
Ischemia-reperfusion injury (IRI) is a common feature when the blood supply is restored after a period of ischemia (Wu et al., 2018). Several studies have suggested that different mechanisms, including oxidative stress, leukocyte infiltration, platelet adhesion and aggregation, blood-brain barrier disruption, complement activation, and mitochondria-mediated mechanisms, are involved in the pathogenesis of IRI in the nervous system (Bavarsad et al., 2019). In 2005, scientists demonstrated that delayed ischemic brain injury in a mouse model was due to necroptosis, a mechanism different from apoptosis (Degterev et al., 2005). This finding shows a new therapeutic target for stroke with an extended period of time for neuroprotection. The researchers also identified a specific and potent small-molecule inhibitor of necroptosis, Nec-1 (Chu et al., 2018).
Studies in 2018 indicated that necroptosis is probably involved in intracerebral hemorrhage (ICH) (Chu et al., 2018). In an in vivo mouse ICH model, pretreatment with Nec-1 protected astrocytes (Zille et al., 2017). Intracerebroventricular treatment with Nec-1 was helpful in reducing cell death, cerebral edema, hematoma volume and neurological score insufficiency (Su et al., 2015; Shen et al., 2017). In addition, the expression level of RIPK3 was increased after ICH, and pretreatment with Nec-1 reduced the interaction between RIPK3 and RIPK1 and promoted cell survival after ICH (Shen et al., 2017). The nucleotide-binding oligomerization domain (NOD)-like receptor (NLR) family pyrin domain-containing 3 (NLRP3) inflammasome was reported to participate in necroptosis, accompanied by changes in inflammatory factors such as IL-1β. The study authors also found that NLRP3 is expressed not only in immune cells, such as microglia, but also in necroptotic neurons when caspase is inhibited by Q-VD-OPH (Teng et al., 2018). Furthermore, in an IRI rat brain model, NLRP3 inflammasome deficiency protects cerebral tissue from injury, suggesting that the NLRP3 inflammasome plays an important role in neuronal necroptosis.
Some acute neuroinflammation is caused by viruses, such as Japanese encephalitis virus (JEV) and West Nile virus (WNV) (Turtle and Solomon, 2018; Bai et al., 2019). In Asia and the Western Pacific, the most common pathogen of viral encephalitis is JEV (Erlanger et al., 2009). JEV infection and inflammation lead to neuronal death, and subsequent cytotoxicity induces deterioration of Japanese encephalitis (JE) (Bai et al., 2019). In 2017, for the first time, necroptosis was discovered to be a reason for neuronal loss in the JE brain (Bian et al., 2017). When JEV infected neurons, the expression of MLKL was upregulated in vitro and in vivo. The loss of MLKL attenuates the progression of JE and the level of inflammatory factors in a rodent model (Bai et al., 2019).
The same year, RIPK3 was found to restrict WNV pathogenesis independently of cell death in a mouse model of WNV encephalitis (Daniels et al., 2017). Ripk3−/− mice showed higher mortality rates than wild-type (WT) mice, whereas mice with a deficiency of MLKL or MLKL and caspase-8 were little affected (Daniels et al., 2017). The expression of neuronal chemokines was inhibited in Ripk3−/− mice, and recruitment of T cells and other immunocytes in the CNS was reduced, which led to an enhanced susceptibility to death in Ripk3−/− mice. These studies demonstrate the multiple functions of RIPK3 in the pathogenesis of viral diseases. Thus, RIPK3 may be a key regulatory factor during CNS immune processes.
AD is recognized by gradual memory worsening, personality disorders and a decline in general cognition (Scheltens et al., 2016; Barnett, 2019). It is the sixth leading cause of death in the United States, causing more than five million deaths (Alzheimer’s Association., 2016). Neuropathologically, the main characteristic of AD is severe neuronal loss, paraprotein [tau, amyloid-β (Aβ)] accumulation, and significant neuroinflammation (Parbo et al., 2018; Whitwell et al., 2018). There is growing evidence that the pathogenesis of AD is not limited to the neuronal compartment but is closely related to immune mechanisms in the brain. However, the mechanisms of neuronal death remain unclear (Whitwell et al., 2018; Dionisio-Santos et al., 2019).
In 2013, Zhang et al., observed that Nec-1 prevents neural cells from degenerating in a mouse model of AD (Qinli et al., 2013). Next, Yang SH et al. researched Nec-1, which reduces Aβ and tau abnormalities to mitigate memory loss in an AD animal model (Yang et al., 2017). Caccamo and others observed necroptosis in postmortem brains of AD patients, which was closely related to brain weight and cognitive levels. In addition, they found that RIPK1 plays a crucial role at the transcriptome level in AD. Moreover, they observed that inhibition of necroptosis reduced cell death in a mouse model of AD (Caccamo et al., 2017).
Etiologically, dysfunction of microglia plays a fundamental role in AD, and in 2017 it was found that RIPK1-dependent transcription promotes disease-associated microglia and lysosomal defects to mediate the accumulation of amyloid plaques in AD. The pseudokinase MLKL is moved from the cytosol to the plasma membrane after it is phosphorylated by the kinase RIPK3, which is required for necroptosis (Ofengeim et al., 2017). Fan et al., also found that phosphorylated MLKL was translocated from membranes through ALIX–syntenin-1–mediated exocytosis or flotillin-mediated endocytic lysosomal degradation (Fan et al., 2019). Thus, targeting RIPK1 and/or RIPK3 may provide an important therapeutic blueprint for the treatment of AD.
PD is the second most common neurodegenerative disorder. Pathologically, dopaminergic neurons in the substantia nigra (SN) pars compacta degenerate slowly and progressively (Hirsch and Hunot, 2009). The underlying causes of PD remain uncertain, but existing data support the significance of noncellular autonomic pathological mechanisms in PD, most of which are activated by glial cells and/or peripheral immune cells (Xu et al., 2017; Seo et al., 2020). This cell response to neurodegenerative changes can trigger harmful events (such as oxidative stress and cytokine receptor-mediated apoptosis), ultimately resulting in the death of dopamine cells and leading to disease progression (Liu et al., 2019; Trist et al., 2019).
Clinical studies report significant increases in dopaminergic neuron degeneration in the SN of PD patients. In contrast to the control group, RIPK1, RIPK3 and MLKL in the SN were increased at the autopsy of PD patients (Oñate et al., 2020). Downregulation of transforming growth factor β-activated kinase-1 (TAK1) may promote age-dependent PD in the CNS of aging humans. Mitochondrial and lysosomal dysfunction may promote intracellular RIPK1 activation and are prone to necroptosis. Inhibiting the enzyme can slow the progression of PD (Iannielli et al., 2018). Furthermore, scientists have performed some research on PC12 cells. Nec-1, which is associated with the apoptosis signaling pathway in this process, exerted a protective response against injury on dopaminergic neurons (Wu et al., 2015). In 2019, parkin was found to be an E3 ubiquitin ligase involved in PD, suggesting that parkin may alter inflammation and necroptosis by participating in ubiquitination events. The loss of parkin protected microglial cells from zVAD-induced necroptosis, thus accelerating primary neuronal injury caused by inflammation (Dionísio et al., 2019). However, further studies are needed for a detailed understanding of the mechanisms underlying these effects of necroptosis in PD.
ALS is an incurable, adult-onset paralytic disease that manifests as a sporadic disease. It is caused by a decrease in motor neurons in the spinal cord, brainstem, and brain, and 10% of all ALS cases are due to genetic mutations (Chia et al., 2018). For example, mutations in the gene superoxide dismutase 1 (SOD1) lead to familial amyotrophic lateral sclerosis (fALS).
In 2014, scientists used a co-culture model system to study human sporadic astrocyte co-cultured with human embryonic stem cell-derived motor neurons, and necroptosis was found as the central feature of the death of motor neurons (Re et al., 2014). Moreover, RIPK1- and RIPK3-mediated axonal damage has been shown to occur extensively in SOD1 transgenic mice and pathological tissues from ALS patients (Saccon et al., 2013). Mutations of the optineurin (OPTN) gene promoted a marked increase in the secretion of proinflammatory cytokines, as well as neuronal cell death, in both fALS and sporadic ALS (Toth and Atkin, 2018). A lack of OPTN induces the activation of necroptosis in oligodendrocytes, leading to Wallerian-like axonal degeneration (Ito et al., 2016). It was also found that OPTN actively suppressed RIPK1-dependent signaling by regulating its turnover. In contrast to WT oligodendrocytes, OPTN−/− oligodendrocytes are more susceptible to TNF-induced necroptosis, which can be inhibited by Nec-1s (Politi and Przedborski, 2016; Greco et al., 2018). Therefore, the necroptotic pathway is proposed as a novel possible target for the treatment of this incurable disease.
Human immunodeficiency virus 1 (HIV-1) is a 9.7 kb retrovirus found in 1983 that was recognized as the pathogene for an increasing lethal immunodeficiency syndrome named AIDS (Selik et al., 1984). HIV enters and stays in the CNS via myelomonocytic cells, such as monocytes, perivascular cells, and microglia (Yadav and Collman, 2009). HIV-1 infection is described as a progressive decrease in CD4+ T lymphocytes and loss of function of the immune system. AIDS manifests in infected individuals years after the initial infection (Doitsh and Greene, 2016).
Recently, necroptosis has been biologically and pathologically researched in HIV-1-infected cells. Unlike almost all infectious diseases, HIV infection precisely targets microglia in the brain and T lymphocytes in the periphery, which are crucial components of the neuroimmune system, resulting in dysfunction of these cells. One report found that necroptosis exists in both the infected primary CD4+ T lymphocytes and CD4+ T cell lines (Pan et al., 2014). Another article suggested that caspase-8 activity was positively correlated with disease severity and programmed cell death-1 (PD-1) expression but negatively correlated with proliferation in HIV-specific CD8+ T cells (Gaiha et al., 2014). NecroX-5 could inhibit defective HIV-specific CD8+ T cell proliferation by blocking necroptosis (Kim et al., 2010; Gaiha et al., 2014). Therefore, chronic stimulation from HIV contributes to caspase-8 activity and increases the cell death of HIV-specific CD8+ T cells through the activation of necroptosis (Gaiha et al., 2014). Drug therapy combined with the inhibitor of necroptosis may improve HIV treatment.
Oxidative stress, inflammation and neurodegeneration are the main contributors in the most common retinal diseases, such as age-related macular degeneration, glaucoma and diabetic retinopathy (Bapputty et al., 2019; Dieguez et al., 2019). An unbalanced retinal immune reaction involving responses of local microglia and recruited macrophages has been specifically emphasized in retinal degenerative diseases (Akhtar-Schäfer et al., 2018).
Glaucoma is characterized by the loss of retinal ganglion cells and is a leading cause of nonreversible blindness, as well as a deteriorating neurodegenerative disease, with a probable seventy million people suffering worldwide (Tham et al., 2014). Current findings place inflammation and apoptosis as important contributors to retinal cell death under elevated pressure. RIPK1-inhibitory compound (RIC), which performs biochemical functions different from those of previous factors (Nec-1 and Compound 27; Cpd27), inhibits downstream pathways following RIPK1 activation and is mediated by necrosome composition and mitochondrial dysfunction (Do et al., 2017). Microglia play an important role in necroptosis. Necroptotic microglia produce several kinds of proinflammatory cytokines and chemokines, such as TNF-α and chemokine ligand 2 (Chen et al., 2019a), (Qin et al., 2018). RIPK1- and RIPK3-dependent necroptosis existed in microglia of mouse models with degenerative retina or acute retinal neural injury (Huang et al., 2018b). Necrostatin-1 blocked necroptosis, depressing microglia-mediated inflammation, which protected retinal degeneration or reduced neural injury in vivo. In the pathway, knockdown of TLR4 reduces RIPK1 and RIPK3 expression to suppress microglial necroptosis and depress retinal inflammation, which suggests that TLR4 signaling participates in necroptosis-mediated microglial inflammation (Huang et al., 2018b). Thus, microglia in the retina provoke inflammatory activation through TLR4-mediated necroptosis, which aggravates retinal neural damage and retinal degeneration.
In regard to ocular trauma, blast-exposed patients experience subsequent vision loss even after a healthy ophthalmological exam. Increased intraocular RIPK3 suggests that the photoreceptors are depleted because of necroptosis. TNF-a and RIPK3 exacerbate the activation of microglia, indicating that RIPK3 may also result in oxidative stress in the outer retina and lead to progressive cell loss (Thomas et al., 2019). Inhibitors of necroptosis are thought to be a new promising strategy to promote neuroaxonal survival and remyelination, potentially preventing disability in retinal diseases.
Several specific necroptosis inhibitors aimed at RIPK1, RIPK3, or MLKL have been found or developed, such as Nec-1, Nec-1s, cpd27, and GSK872. Studies of these specific necroptosis inhibitors have shown therapeutic effects in various neuroinflammatory diseases. In animal models of TBI, Nec-1 (a kind of RIPK1 inhibitor) inhibits apoptosis and autophagy (Wang et al., 2012). Nec-1 also reduces Aβ and tau abnormalities in an AD animal model (Yang et al., 2017). Dabrafenib (an inhibitor of RIPK3 kinase-dependent necroptosis) reduces ischemic brain damage in mice (Cruz et al., 2018). The novel MLKL inhibitors have been proven to be promising tools for studying the biological function of MLKL and as druggable targets of necroptosis. However, the MLKL inhibitors currently known are few (such as TC13172, necrosulfonamide and GW806742X), and no MLKL inhibitor has reached the clinical trial stage (Chen et al., 2017; Yan et al., 2017). However, most studies have yet to conduct clinical trials, and there are still questions about human safety (Chen et al., 2019b).
Necroptosis is a new type of programmed necrosis that can be activated by multiple kinds of extracellular and intracellular stimuli. Our understanding of the underlying molecular mechanism and biological function of necroptosis has increased in recent years. In general, necroptosis achieves physiological and pathological effects through the TNF or TLR pathway. Depending on the inhibition of caspase-8 activation, RIPK1, RIPK3, MLKL, FADD and procaspase-8 form complex IIb, which leads to necroptosis. The mechanism by which necroptosis is achieved through the TLR3 and TLR4 pathways is unclear. In the rat model of hydraulic shock brain injury, RIPK1 and 3, MLKL and proinflammatory factors were increased in the cortex (Liu et al., 2016). RIPK1 and 3 and the NLRP3 inflammasome were reported to participate in necroptosis of ICH (Shen et al., 2017). In chronic neuroinflammatory diseases such as AD, RIPK3 impacts necroptosis by phosphorylating MLKL (Ofengeim et al., 2017; Fan et al., 2019). Although the role of necroptosis in inflammatory or apoptotic pathologies has been appreciated, our knowledge of the involvement and impact of necroptosis in neuroinflammatory diseases remains limited. Hence, a deeper understanding of necroptosis in neuroinflammatory diseases, such as AD and stroke, could be beneficial for providing insights into the mechanisms of neuronal death and clinical treatments. The precise mechanism of plasma membrane breakage induced by MLKL in necroptosis remains to be uncovered. Whether MLKL plays a role in necroptosis as a carrier protein of some particular proteins, from cytoplasm to nuclei, also needs further study. While some small-molecule inhibitors of RIPK1 and RIPK3 have made some progress in clinical trials, the efficacy of treatment remains to be confirmed by multicenter experiments.
To conclude, we highlighted the increasing evidence about the role of necroptosis in various neuroinflammatory diseases. In the future, improvement of the application of such signaling inhibitors may remove obstacles for replacement therapies for neurological diseases.
ZY drafted and edited the manuscript and prepared the figures. NJ drafted and edited the manuscript and prepared the table. WS and YZ reviewed and edited the manuscript. All authors conceived the idea of this review together. All authors read and approved the final manuscript.
Natural Science Foundation of China (81870658) National Key R&D Program of China (2017YA0105804).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Akhtar-Schäfer, I., Wang, L., Krohne, T. U., Xu, H., and Langmann, T. (2018). Modulation of Three Key Innate Immune Pathways for the Most Common Retinal Degenerative Diseases. EMBO Mol. Med. 10 (10). doi:10.15252/emmm.201708259
Akira, S., and Hoshino, K. (2003). Myeloid Differentiation Factor 88-Dependent and -Independent Pathways in Toll-like Receptor Signaling. J. Infect. Dis. 187 (Suppl. 2), S356–S363. doi:10.1086/374749
Alexopoulou, L., Holt, A. C., Medzhitov, R., and Flavell, R. A. (2001). Recognition of Double-Stranded RNA and Activation of NF-Κb by Toll-like Receptor 3. Nature 413 (6857), 732–738. doi:10.1038/35099560
Alvarez-Diaz, S., Dillon, C. P., Lalaoui, N., Tanzer, M. C., Rodriguez, D. A., Lin, A., et al. (2016). The Pseudokinase MLKL and the Kinase RIPK3 Have Distinct Roles in Autoimmune Disease Caused by Loss of Death-Receptor-Induced Apoptosis. Immunity 45 (3), 513–526. doi:10.1016/j.immuni.2016.07.016
Alzheimer's Association. (2016) 2016 Alzheimer's Disease Facts and Figures. Alzheimers Dement 12(4):459–509. doi:10.1016/j.jalz.2016.03.001
Amin, P., Florez, M., Najafov, A., Pan, H., Geng, J., Ofengeim, D., et al. (2018). Regulation of a Distinct Activated RIPK1 Intermediate Bridging Complex I and Complex II in TNFα-Mediated Apoptosis. Proc. Natl. Acad. Sci. USA 115 (26), E5944–E5953. doi:10.1073/pnas.1806973115
Andersson, U., and Tracey, K. J. (2011). HMGB1 Is a Therapeutic Target for Sterile Inflammation and Infection. Annu. Rev. Immunol. 29, 139–162. doi:10.1146/annurev-immunol-030409-101323
Annibaldi, A., and Meier, P. (2018). Checkpoints in TNF-Induced Cell Death: Implications in Inflammation and Cancer. Trends Mol. Med. 24 (1), 49–65. doi:10.1016/j.molmed.2017.11.002
Bai, F., Thompson, E. A., Vig, P., and Leis, A. A. (2019). Current Understanding of West Nile Virus Clinical Manifestations, Immune Responses, Neuroinvasion, and Immunotherapeutic Implications. Pathogens 8 (4). doi:10.3390/pathogens8040193
Bapputty, R., Talahalli, R., Zarini, S., Samuels, I., Murphy, R., and Gubitosi-Klug, R. (2019). Montelukast Prevents Early Diabetic Retinopathy in Mice. Diabetes 68 (10), 2004–2015. doi:10.2337/db19-0026
Barnett, R. (2019). Alzheimer's Disease. The Lancet 393 (10181), 1589. doi:10.1016/s0140-6736(19)30851-7
Bavarsad, K., Barreto, G. E., Hadjzadeh, M.-A. -R., and Sahebkar, A. (2019). Protective Effects of Curcumin against Ischemia-Reperfusion Injury in the Nervous System. Mol. Neurobiol. 56 (2), 1391–1404. doi:10.1007/s12035-018-1169-7
Bian, P., Zheng, X., Wei, L., Ye, C., Fan, H., Cai, Y., et al. (2017). MLKL Mediated Necroptosis Accelerates JEV-Induced Neuroinflammation in Mice. Front. Microbiol. 8, 303. doi:10.3389/fmicb.2017.00303
Boyd-Tressler, A. M., Lane, G. S., and Dubyak, G. R. (2017). Up-regulated Ectonucleotidases in Fas-Associated Death Domain Protein- and Receptor-Interacting Protein Kinase 1-Deficient Jurkat Leukemia Cells Counteract Extracellular ATP/AMP Accumulation via Pannexin-1 Channels during Chemotherapeutic Drug-Induced Apoptosis. Mol. Pharmacol. 92 (1), 30–47. doi:10.1124/mol.116.104000
Caccamo, A., Branca, C., Piras, I. S., Ferreira, E., Huentelman, M. J., Liang, W. S., et al. (2017). Necroptosis Activation in Alzheimer's Disease. Nat. Neurosci. 20 (9), 1236–1246. doi:10.1038/nn.4608
Candelario-Jalil, E. (2009). Injury and Repair Mechanisms in Ischemic Stroke: Considerations for the Development of Novel Neurotherapeutics. Curr. Opin. Investig. Drugs 10 (7), 644–654.
Cao, W., Zhang, T., Feng, R., Xia, T., Huang, H., Liu, C., et al. (2019). Hoxa5 Alleviates Obesity-Induced Chronic Inflammation by Reducing ER Stress and Promoting M2 Macrophage Polarization in Mouse Adipose Tissue. J. Cel Mol Med 23 (10), 7029–7042. doi:10.1111/jcmm.14600
Ceulemans, A.-G., Zgavc, T., Kooijman, R., Hachimi-Idrissi, S., Sarre, S., and Michotte, Y. (2010). The Dual Role of the Neuroinflammatory Response after Ischemic Stroke: Modulatory Effects of Hypothermia. J. Neuroinflammation 7, 74. doi:10.1186/1742-2094-7-74
Chen, A. Q., Fang, Z., Chen, X. L., Yang, S., Zhou, Y. F., Mao, L., et al. (2019). Microglia-derived TNF-Alpha Mediates Endothelial Necroptosis Aggravating Blood Brain-Barrier Disruption after Ischemic Stroke. Cell Death Dis. 10 (7), 487. doi:10.1038/s41419-019-1716-9
Chen, J., Kos, R., Garssen, J., and Redegeld, F. (2019). Molecular Insights into the Mechanism of Necroptosis: The Necrosome as a Potential Therapeutic Target. Cells 8 (12). doi:10.3390/cells8121486
Chen, S., Lv, X., Hu, B., Shao, Z., Wang, B., Ma, K., et al. (2017). RIPK1/RIPK3/MLKL-mediated Necroptosis Contributes to Compression-Induced Rat Nucleus Pulposus Cells Death. Apoptosis 22 (5), 626–638. doi:10.1007/s10495-017-1358-2
Chia, R., Chiò, A., and Traynor, B. J. (2018). Novel Genes Associated with Amyotrophic Lateral Sclerosis: Diagnostic and Clinical Implications. Lancet Neurol. 17 (1), 94–102. doi:10.1016/s1474-4422(17)30401-5
Choo, Z., Loh, A., and Chen, Z. X. (2019). Destined to Die: Apoptosis and Pediatric Cancers. Cancers (Basel) 11 (11). doi:10.3390/cancers11111623
Chu, X., Wu, X., Feng, H., Zhao, H., Tan, Y., Wang, L., et al. (2018). Coupling between Interleukin-1R1 and Necrosome Complex Involves in Hemin-Induced Neuronal Necroptosis after Intracranial Hemorrhage. Stroke 49 (10), 2473–2482. doi:10.1161/strokeaha.117.019253
Cruz, S. A., Qin, Z., Stewart, A. F. R., and Chen, H. H. (2018). Dabrafenib, an Inhibitor of RIP3 Kinase-dependent Necroptosis, Reduces Ischemic Brain Injury. Neural Regen. Res. 13 (2), 252–256. doi:10.4103/1673-5374.226394
Daniels, B. P., Snyder, A. G., Olsen, T. M., Orozco, S., Oguin, T. H., Tait, S. W. G., et al. (2017). RIPK3 Restricts Viral Pathogenesis via Cell Death-independent Neuroinflammation. Cell 169 (2), 301–313. doi:10.1016/j.cell.2017.03.011
Degterev, A., Huang, Z., Boyce, M., Li, Y., Jagtap, P., Mizushima, N., et al. (2005). Chemical Inhibitor of Nonapoptotic Cell Death with Therapeutic Potential for Ischemic Brain Injury. Nat. Chem. Biol. 1 (2), 112–119. doi:10.1038/nchembio711
Dieguez, H. H., Romeo, H. E., Alaimo, A., González Fleitas, M. F., Aranda, M. L., Rosenstein, R. E., et al. (2019). Oxidative Stress Damage Circumscribed to the central Temporal Retinal Pigment Epithelium in Early Experimental Non-exudative Age-Related Macular Degeneration. Free Radic. Biol. Med. 131, 72–80. doi:10.1016/j.freeradbiomed.2018.11.035
Dionísio, P. E. A., Oliveira, S. R., Amaral, J. S. J. D., and Rodrigues, C. M. P. (2019). Loss of Microglial Parkin Inhibits Necroptosis and Contributes to Neuroinflammation. Mol Neurobiol 56 (4), 2990–3004. doi:10.1007/s12035-018-1264-9
Dionisio-Santos, DA, Olschowka, JA, and O'Banion, MK (2019). Exploiting microglial and peripheral immune cell crosstalk to treat Alzheimer's disease. J Neuroinflammation 16 (1), 74. doi:10.1186/s12974-019-1453-0
DiSabato, D. J., Quan, N., and Godbout, J. P. (2016). Neuroinflammation: the Devil Is in the Details. J. Neurochem. 139 (Suppl. 2), 136–153. doi:10.1111/jnc.13607
Do, Y.-J., Sul, J.-W., Jang, K.-H., Kang, N. S., Kim, Y.-H., Kim, Y.-G., et al. (2017). A Novel RIPK1 Inhibitor that Prevents Retinal Degeneration in a Rat Glaucoma Model. Exp. Cel Res. 359 (1), 30–38. doi:10.1016/j.yexcr.2017.08.012
Doitsh, G., and Greene, W. C. (2016). Dissecting How CD4 T Cells Are Lost during HIV Infection. Cell Host & Microbe 19 (3), 280–291. doi:10.1016/j.chom.2016.02.012
Dong, Y., Lagarde, J., Xicota, L., Corne, H., Chantran, Y., Chaigneau, T., et al. (2018). Neutrophil Hyperactivation Correlates with Alzheimer's Disease Progression. Ann. Neurol. 83 (2), 387–405. doi:10.1002/ana.25159
Durukan, A., and Tatlisumak, T. (2007). Acute Ischemic Stroke: Overview of Major Experimental Rodent Models, Pathophysiology, and Therapy of Focal Cerebral Ischemia. Pharmacol. Biochem. Behav. 87 (1), 179–197. doi:10.1016/j.pbb.2007.04.015
Erlanger, T. E., Weiss, S., Keiser, J., Utzinger, J., and Wiedenmayer, K. (2009). Past, Present, and Future of Japanese Encephalitis. Emerg. Infect. Dis. 15 (1), 1–7. doi:10.3201/eid1501.080311
Estes, M. L., and McAllister, A. K. (2016). Maternal Immune Activation: Implications for Neuropsychiatric Disorders. Science 353 (6301), 772–777. doi:10.1126/science.aag3194
Faissner, S., Plemel, J. R., Gold, R., and Yong, V. W. (2019). Progressive Multiple Sclerosis: from Pathophysiology to Therapeutic Strategies. Nat. Rev. Drug Discov. 18, 905–922. doi:10.1038/s41573-019-0035-2
Fan, W., Guo, J., Gao, B., Zhang, W., Ling, L., Xu, T., et al. (2019). Flotillin-mediated Endocytosis and ALIX-Syntenin-1-Mediated Exocytosis Protect the Cell Membrane from Damage Caused by Necroptosis. Sci. Signal. 12 (583). doi:10.1126/scisignal.aaw3423
Feoktistova, M., Geserick, P., Kellert, B., Dimitrova, D. P., Langlais, C., Hupe, M., et al. (2011). cIAPs Block Ripoptosome Formation, a RIP1/caspase-8 Containing Intracellular Cell Death Complex Differentially Regulated by cFLIP Isoforms. Mol. Cel. 43 (3), 449–463. doi:10.1016/j.molcel.2011.06.011
Fitzgerald, K. A., and Kagan, J. C. (2020). Toll-like Receptors and the Control of Immunity. Cell 180 (6), 1044–1066. doi:10.1016/j.cell.2020.02.041
Fitzgerald, K. A., Palsson-McDermott, E. M., Bowie, A. G., Jefferies, C. A., Mansell, A. S., Brady, G., et al. (2001). Mal (MyD88-adapter-like) Is Required for Toll-like Receptor-4 Signal Transduction. Nature 413 (6851), 78–83. doi:10.1038/35092578
Fitzgerald, K. A., Rowe, D. C., Barnes, B. J., Caffrey, D. R., Visintin, A., Latz, E., et al. (2003). LPS-TLR4 Signaling to IRF-3/7 and NF-Κb Involves the Toll Adapters TRAM and TRIF. J. Exp. Med. 198 (7), 1043–1055. doi:10.1084/jem.20031023
Fuji, H., Ohmae, S., Noma, N., Takeiri, M., Yasutomi, H., Izumi, K., et al. (2018). Necrostatin-7 Suppresses RANK-NFATc1 Signaling and Attenuates Macrophage to Osteoclast Differentiation. Biochem. Biophysical Res. Commun. 503 (2), 544–549. doi:10.1016/j.bbrc.2018.05.153
Gaiha, G. D., McKim, K. J., Woods, M., Pertel, T., Rohrbach, J., Barteneva, N., et al. (2014). Dysfunctional HIV-specific CD8+ T Cell Proliferation Is Associated with Increased Caspase-8 Activity and Mediated by Necroptosis. Immunity 41 (6), 1001–1012. doi:10.1016/j.immuni.2014.12.011
Gonzalez-Juarbe, N., Gilley, R. P., Hinojosa, C. A., Bradley, K. M., Kamei, A., Gao, G., et al. (2015). Pore-Forming Toxins Induce Macrophage Necroptosis during Acute Bacterial Pneumonia. Plos Pathog. 11 (12), e1005337. doi:10.1371/journal.ppat.1005337
Greco, V., Spalloni, A., Corasolla, C. V., Pieroni, L., Persichilli, S., Mercuri, N. B., et al. (2018). Proteomics and Toxicity Analysis of Spinal-Cord Primary Cultures upon Hydrogen Sulfide Treatment. Antioxidants (Basel) 7 (7). doi:10.3390/antiox7070087
He, S., Liang, Y., Shao, F., and Wang, X. (2011). Toll-like Receptors Activate Programmed Necrosis in Macrophages through a Receptor-Interacting Kinase-3-Mediated Pathway. Proc. Natl. Acad. Sci. 108 (50), 20054–20059. doi:10.1073/pnas.1116302108
Hirsch, E. C., and Hunot, S. (2009). Neuroinflammation in Parkinson's Disease: a Target for Neuroprotection?. Lancet Neurol. 8 (4), 382–397. doi:10.1016/s1474-4422(09)70062-6
Huang, S., Ge, X., Yu, J., Han, Z., Yin, Z., Li, Y., et al. (2018). Increased miR-124-3p in Microglial Exosomes Following Traumatic Brain Injury Inhibits Neuronal Inflammation and Contributes to Neurite Outgrowthviatheir Transfer into Neurons. FASEB j. 32 (1), 512–528. doi:10.1096/fj.201700673r
Huang, Z., Zhou, T., Sun, X., Zheng, Y., Cheng, B., Li, M., et al. (2018). Necroptosis in Microglia Contributes to Neuroinflammation and Retinal Degeneration through TLR4 Activation. Cell Death Differ 25 (1), 180–189. doi:10.1038/cdd.2017.141
Iannielli, A., Bido, S., Folladori, L., Segnali, A., Cancellieri, C., Maresca, A., et al. (2018). Pharmacological Inhibition of Necroptosis Protects from Dopaminergic Neuronal Cell Death in Parkinson's Disease Models. Cel Rep. 22 (8), 2066–2079. doi:10.1016/j.celrep.2018.01.089
Ito, Y., Ofengeim, D., Najafov, A., Das, S., Saberi, S., Li, Y., et al. (2016). RIPK1 Mediates Axonal Degeneration by Promoting Inflammation and Necroptosis in ALS. Science 353 (6299), 603–608. doi:10.1126/science.aaf6803
Jenkins, P. O., Mehta, M. A., and Sharp, D. J. (2016). Catecholamines and Cognition after Traumatic Brain Injury. Brain 139 (Pt 9), 2345–2371. doi:10.1093/brain/aww128
Kagan, J. C., Su, T., Horng, T., Chow, A., Akira, S., and Medzhitov, R. (2008). TRAM Couples Endocytosis of Toll-like Receptor 4 to the Induction of Interferon-β. Nat. Immunol. 9 (4), 361–368. doi:10.1038/ni1569
Kapur, R., Kasetty, G., Rebetz, J., Egesten, A., and Semple, J. W. (2019). Osteopontin Mediates Murine Transfusion-Related Acute Lung Injury via Stimulation of Pulmonary Neutrophil Accumulation. Blood 134 (1), 74–84. doi:10.1182/blood.2019000972
Kawai, T., and Akira, S. (2010). The Role of Pattern-Recognition Receptors in Innate Immunity: Update on Toll-like Receptors. Nat. Immunol. 11 (5), 373–384. doi:10.1038/ni.1863
Kearney, C. J., and Martin, S. J. (2017). An Inflammatory Perspective on Necroptosis. Mol. Cel 65 (6), 965–973. doi:10.1016/j.molcel.2017.02.024
Kerr, J. F. R., Wyllie, A. H., and Currie, A. R. (1972). Apoptosis: A Basic Biological Phenomenon with Wideranging Implications in Tissue Kinetics. Br. J. Cancer 26 (4), 239–257. doi:10.1038/bjc.1972.33
Kim, H. J., Koo, S. Y., Ahn, B.-H., Park, O., Park, D. H., Seo, D. O., et al. (2010). NecroX as a Novel Class of Mitochondrial Reactive Oxygen Species and ONOO− Scavenger. Arch. Pharm. Res. 33 (11), 1813–1823. doi:10.1007/s12272-010-1114-4
Lafont, E., Draber, P., Rieser, E., Reichert, M., Kupka, S., de Miguel, D., et al. (2018). TBK1 and IKKε Prevent TNF-Induced Cell Death by RIPK1 Phosphorylation. Nat. Cel Biol 20 (12), 1389–1399. doi:10.1038/s41556-018-0229-6
Lakhan, S. E., Kirchgessner, A., and Hofer, M. (2009). Inflammatory Mechanisms in Ischemic Stroke: Therapeutic Approaches. J. Transl Med. 7, 97. doi:10.1186/1479-5876-7-97
Lawlor, K. E., Khan, N., Mildenhall, A., Gerlic, M., Croker, B. A., D'Cruz, A. A., et al. (2015). RIPK3 Promotes Cell Death and NLRP3 Inflammasome Activation in the Absence of MLKL. Nat. Commun. 6, 6282. doi:10.1038/ncomms7282
Lee, J. D., Coulthard, L. G., and Woodruff, T. M. (2019). Complement Dysregulation in the central Nervous System during Development and Disease. Semin. Immunol. 45, 101340. doi:10.1016/j.smim.2019.101340
Li, J., Zhang, J., Zhang, Y., Wang, Z., Song, Y., Wei, S., et al. (2019). TRAF2 Protects against Cerebral Ischemia-Induced Brain Injury by Suppressing Necroptosis. Cel Death Dis 10 (5), 328. doi:10.1038/s41419-019-1558-5
Limb, M. (2014). Traumatic Brain Injury Carries Long Term Health Risks, Finds Study. BMJ 348, g294. doi:10.1136/bmj.g294
Lin, Y., Devin, A., Rodriguez, Y., and Liu, Z.-g. (1999). Cleavage of the Death Domain Kinase RIP by Caspase-8 Prompts TNF-Induced Apoptosis. Genes Dev. 13 (19), 2514–2526. doi:10.1101/gad.13.19.2514
Liu, J.-Q., Chu, S.-F., Zhou, X., Zhang, D.-Y., and Chen, N.-H. (2019). Role of Chemokines in Parkinson's Disease. Brain Res. Bull. 152, 11–18. doi:10.1016/j.brainresbull.2019.05.020
Liu, J., van Mil, A., Vrijsen, K., Zhao, J., Gao, L., Metz, C. H. G., et al. (2011). MicroRNA-155 Prevents Necrotic Cell Death in Human Cardiomyocyte Progenitor Cells via Targeting RIP1. J. Cel Mol Med 15 (7), 1474–1482. doi:10.1111/j.1582-4934.2010.01104.x
Liu, S., Wang, X., Li, Y., Xu, L., Yu, X., Ge, L., et al. (2014). Necroptosis Mediates TNF-Induced Toxicity of Hippocampal Neurons. Biomed. Res. Int. 2014, 1–11. doi:10.1155/2014/290182
Liu, T., Zhao, D. X., Cui, H., Chen, L., Bao, Y. H., Wang, Y., et al. (2016). Therapeutic Hypothermia Attenuates Tissue Damage and Cytokine Expression after Traumatic Brain Injury by Inhibiting Necroptosis in the Rat. Sci. Rep. 6, 24547. doi:10.1038/srep24547
Liu, Z.-M., Chen, Q.-X., Chen, Z.-B., Tian, D.-F., Li, M.-C., Wang, J.-M., et al. (2018). RIP3 Deficiency Protects against Traumatic Brain Injury (TBI) through Suppressing Oxidative Stress, Inflammation and Apoptosis: Dependent on AMPK Pathway. Biochem. Biophysical Res. Commun. 499 (2), 112–119. doi:10.1016/j.bbrc.2018.02.150
Lodygin, D., Hermann, M., Schweingruber, N., Flügel-Koch, C., Watanabe, T., Schlosser, C., et al. (2019). β-Synuclein-reactive T Cells Induce Autoimmune CNS Grey Matter Degeneration. Nature 566 (7745), 503–508. doi:10.1038/s41586-019-0964-2
Maas, A. I., Stocchetti, N., and Bullock, R. (2008). Moderate and Severe Traumatic Brain Injury in Adults. Lancet Neurol. 7 (8), 728–741. doi:10.1016/s1474-4422(08)70164-9
Mack, M. (2018). Inflammation and Fibrosis. Matrix Biol. 68-69, 106–121. doi:10.1016/j.matbio.2017.11.010
McDonald, S. J., Sun, M., Agoston, D. V., and Shultz, S. R. (2016). The Effect of Concomitant Peripheral Injury on Traumatic Brain Injury Pathobiology and Outcome. J. Neuroinflammation 13 (1), 90. doi:10.1186/s12974-016-0555-1
Micheau, O., and Tschopp, J. (2003). Induction of TNF Receptor I-Mediated Apoptosis via Two Sequential Signaling Complexes. Cell 114 (2), 181–190. doi:10.1016/s0092-8674(03)00521-x
Na, Y. R., Stakenborg, M., Seok, S. H., and Matteoli, G. (2019). Macrophages in Intestinal Inflammation and Resolution: a Potential Therapeutic Target in IBD. Nat. Rev. Gastroenterol. Hepatol. 16 (9), 531–543. doi:10.1038/s41575-019-0172-4
Nasef, N. A., Mehta, S., and Ferguson, L. R. (2017). Susceptibility to Chronic Inflammation: an Update. Arch. Toxicol. 91 (3), 1131–1141. doi:10.1007/s00204-016-1914-5
Netea, M. G., Nold-Petry, C. A., Nold, M. F., Joosten, L. A. B., Opitz, B., van der Meer, J. H. M., et al. (2009). Differential Requirement for the Activation of the Inflammasome for Processing and Release of IL-1β in Monocytes and Macrophages. Blood 113 (10), 2324–2335. doi:10.1182/blood-2008-03-146720
Ni, H., Rui, Q., Lin, X., Li, D., Liu, H., and Chen, G. (2019). 2-BFI Provides Neuroprotection against Inflammation and Necroptosis in a Rat Model of Traumatic Brain Injury. Front. Neurosci. 13, 674. doi:10.3389/fnins.2019.00674
Ofengeim, D., Mazzitelli, S., Ito, Y., DeWitt, J. P., Mifflin, L., Zou, C., et al. (2017). RIPK1 Mediates a Disease-Associated Microglial Response in Alzheimer's Disease. Proc. Natl. Acad. Sci. USA 114 (41), E8788–E8797. doi:10.1073/pnas.1714175114
Oñate, M., Catenaccio, A., Salvadores, N., Saquel, C., Martinez, A., Moreno-Gonzalez, I., et al. (2020). The Necroptosis Machinery Mediates Axonal Degeneration in a Model of Parkinson Disease. Cel Death Differ 27 (4), 1169–1185. doi:10.1038/s41418-019-0408-4
Pan, T., Wu, S., He, X., Luo, H., Zhang, Y., Fan, M., et al. (2014). Necroptosis Takes Place in Human Immunodeficiency Virus Type-1 (HIV-1)-Infected CD4+ T Lymphocytes. PLoS One 9 (4), e93944. doi:10.1371/journal.pone.0093944
Parbo, P., Ismail, R., Sommerauer, M., Stokholm, M. G., Hansen, A. K., Hansen, K. V., et al. (2018). Does Inflammation Precede Tau Aggregation in Early Alzheimer's Disease? A PET Study. Neurobiol. Dis. 117, 211–216. doi:10.1016/j.nbd.2018.06.004
Pasparakis, M., and Vandenabeele, P. (2015). Necroptosis and its Role in Inflammation. Nature 517 (7534), 311–320. doi:10.1038/nature14191
Pinho-Ribeiro, F. A., Baddal, B., Haarsma, R., O’Seaghdha, M., Yang, N. J., Blake, K. J., et al. (2018). Blocking Neuronal Signaling to Immune Cells Treats Streptococcal Invasive Infection. Cell 173 (5), 1083–1097. doi:10.1016/j.cell.2018.04.006
Politi, K., and Przedborski, S. (2016). Axonal Degeneration: RIPK1 Multitasking in ALS. Curr. Biol. 26 (20), R932–R934. doi:10.1016/j.cub.2016.08.052
Qin, S., Yang, C., Huang, W., Du, S., Mai, H., Xiao, J., et al. (2018). Sulforaphane Attenuates Microglia-Mediated Neuronal Necroptosis through Down-Regulation of MAPK/NF-κB Signaling Pathways in LPS-Activated BV-2 Microglia. Pharmacol. Res. 133, 218–235. doi:10.1016/j.phrs.2018.01.014
Qinli, Z., Meiqing, L., Xia, J., Li, X., Weili, G., Xiuliang, J., et al. (2013). Necrostatin-1 Inhibits the Degeneration of Neural Cells Induced by Aluminum Exposure. Restor Neurol. Neurosci. 31 (5), 543–555. doi:10.3233/rnn-120304
Re, D. B., Le Verche, V., Yu, C., Amoroso, M. W., Politi, K. A., Phani, S., et al. (2014). Necroptosis Drives Motor Neuron Death in Models of Both Sporadic and Familial ALS. Neuron 81 (5), 1001–1008. doi:10.1016/j.neuron.2014.01.011
Ren, Q., Ma, M., Yang, J., Nonaka, R., Yamaguchi, A., Ishikawa, K.-i., et al. (2018). Soluble Epoxide Hydrolase Plays a Key Role in the Pathogenesis of Parkinson's Disease. Proc. Natl. Acad. Sci. USA 115 (25), E5815–E5823. doi:10.1073/pnas.1802179115
Rosenfeld, J. V., Maas, A. I., Bragge, P., Morganti-Kossmann, M. C., Manley, G. T., and Gruen, R. L. (2012). Early Management of Severe Traumatic Brain Injury. The Lancet 380 (9847), 1088–1098. doi:10.1016/s0140-6736(12)60864-2
Saccon, R. A., Bunton-Stasyshyn, R. K., Fisher, E. M., and Fratta, P. (2013). Is SOD1 Loss of Function Involved in Amyotrophic Lateral Sclerosis?. Brain 136 (Pt 8), 2342–2358. doi:10.1093/brain/awt097
Scheltens, P., Blennow, K., Breteler, M. M. B., de Strooper, B., Frisoni, G. B., Salloway, S., et al. (2016). Alzheimer's Disease. The Lancet 388 (10043), 505–517. doi:10.1016/s0140-6736(15)01124-1
Selik, R. M., Haverkos, H. W., and Curran, J. W. (1984). Acquired Immune Deficiency Syndrome (AIDS) Trends in the United States, 1978-1982. Am. J. Med. 76 (3), 493–500. doi:10.1016/0002-9343(84)90669-7
Seo, J., Park, J., Kim, K., Won, J., Yeo, H.-G., Jin, Y. B., et al. (2020). Chronic Infiltration of T Lymphocytes into the Brain in a Non-human Primate Model of Parkinson's Disease. Neuroscience 431, 73–85. doi:10.1016/j.neuroscience.2020.01.043
Shen, H., Liu, C., Zhang, D., Yao, X., Zhang, K., Li, H., et al. (2017). Role for RIP1 in Mediating Necroptosis in Experimental Intracerebral Hemorrhage Model Both In Vivo and In Vitro. Cel Death Dis 8 (3), e2641. doi:10.1038/cddis.2017.58
Silke, J., and Brink, R. (2010). Regulation of TNFRSF and Innate Immune Signalling Complexes by TRAFs and cIAPs. Cel Death Differ 17 (1), 35–45. doi:10.1038/cdd.2009.114
Stephenson, J., Nutma, E., van der Valk, P., and Amor, S. (2018). Inflammation in CNS Neurodegenerative Diseases. Immunology 154 (2), 204–219. doi:10.1111/imm.12922
Strilic, B., Yang, L., Albarrán-Juárez, J., Wachsmuth, L., Han, K., Müller, U. C., et al. (2016). Tumour-cell-induced Endothelial Cell Necroptosis via Death Receptor 6 Promotes Metastasis. Nature 536 (7615), 215–218. doi:10.1038/nature19076
Su, X., Wang, H., Kang, D., Zhu, J., Sun, Q., Li, T., et al. (2015). Necrostatin-1 Ameliorates Intracerebral Hemorrhage-Induced Brain Injury in Mice through Inhibiting RIP1/RIP3 Pathway. Neurochem. Res. 40 (4), 643–650. doi:10.1007/s11064-014-1510-0
Takahashi, N., Duprez, L., Grootjans, S., Cauwels, A., Nerinckx, W., DuHadaway, J. B., et al. (2012). Necrostatin-1 Analogues: Critical Issues on the Specificity, Activity and In Vivo Use in Experimental Disease Models. Cel Death Dis 3, e437. doi:10.1038/cddis.2012.176
Tartey, S., and Takeuchi, O. (2017). Pathogen Recognition and Toll-like Receptor Targeted Therapeutics in Innate Immune Cells. Int. Rev. Immunol. 36 (2), 57–73. doi:10.1080/08830185.2016.1261318
Teng, X., Chen, W., Liu, Z., Feng, T., Li, H., Ding, S., et al. (2018). NLRP3 Inflammasome Is Involved in Q-VD-OPH Induced Necroptosis Following Cerebral Ischemia-Reperfusion Injury. Neurochem. Res. 43 (6), 1200–1209. doi:10.1007/s11064-018-2537-4
Tham, Y.-C., Li, X., Wong, T. Y., Quigley, H. A., Aung, T., and Cheng, C.-Y. (2014). Global Prevalence of Glaucoma and Projections of Glaucoma Burden through 2040. Ophthalmology 121 (11), 2081–2090. doi:10.1016/j.ophtha.2014.05.013
Thomas, C. N., Thompson, A. M., Ahmed, Z., and Blanch, R. J. (2019). Retinal Ganglion Cells Die by Necroptotic Mechanisms in a Site-specific Manner in a Rat Blunt Ocular Injury Model. Cells 8 (12). doi:10.3390/cells8121517
Toth, R. P., and Atkin, J. D. (2018). Dysfunction of Optineurin in Amyotrophic Lateral Sclerosis and Glaucoma. Front. Immunol. 9, 1017. doi:10.3389/fimmu.2018.01017
Trist, B. G., Hare, D. J., and Double, K. L. (2019). Oxidative Stress in the Aging Substantia Nigra and the Etiology of Parkinson's Disease. Aging Cell 18 (6), e13031. doi:10.1111/acel.13031
Tsai, S., Clemente-Casares, X., Zhou, A. C., Lei, H., Ahn, J. J., Chan, Y. T., et al. (2018). Insulin Receptor-Mediated Stimulation Boosts T Cell Immunity during Inflammation and Infection. Cel Metab. 28 (6), 922–934. doi:10.1016/j.cmet.2018.08.003
Tsuchiya, Y., Nakabayashi, O., and Nakano, H. (2015). FLIP the Switch: Regulation of Apoptosis and Necroptosis by cFLIP. Ijms 16 (12), 30321–30341. doi:10.3390/ijms161226232
Turtle, L., and Solomon, T. (2018). Japanese Encephalitis - the Prospects for New Treatments. Nat. Rev. Neurol. 14 (5), 298–313. doi:10.1038/nrneurol.2018.30
Vanden Berghe, T., Linkermann, A., Jouan-Lanhouet, S., Walczak, H., and Vandenabeele, P. (2014). Regulated Necrosis: the Expanding Network of Non-apoptotic Cell Death Pathways. Nat. Rev. Mol. Cel Biol 15 (2), 135–147. doi:10.1038/nrm3737
Vanden, B. T., Grootjans, S., Goossens, V., Dondelinger, Y., Krysko, D. V., Takahashi, N., et al. (2013). Determination of Apoptotic and Necrotic Cell Death In Vitro and In Vivo. Methods 61 (2), 117–129.
Venkatesan, A., Michael, B. D., Probasco, J. C., Geocadin, R. G., and Solomon, T. (2019). Acute Encephalitis in Immunocompetent Adults. The Lancet 393 (10172), 702–716. doi:10.1016/s0140-6736(18)32526-1
Wang, H., Sun, L., Su, L., Rizo, J., Liu, L., Wang, L.-F., et al. (2014). Mixed Lineage Kinase Domain-like Protein MLKL Causes Necrotic Membrane Disruption upon Phosphorylation by RIP3. Mol. Cel 54 (1), 133–146. doi:10.1016/j.molcel.2014.03.003
Wang, Y.-Q., Wang, L., Zhang, M.-Y., Wang, T., Bao, H.-J., Liu, W.-L., et al. (2012). Necrostatin-1 Suppresses Autophagy and Apoptosis in Mice Traumatic Brain Injury Model. Neurochem. Res. 37 (9), 1849–1858. doi:10.1007/s11064-012-0791-4
Whitwell, J. L., Graff-Radford, J., Tosakulwong, N., Weigand, S. D., Machulda, M. M., Senjem, M. L., et al. (2018). Imaging Correlations of Tau, Amyloid, Metabolism, and Atrophy in Typical and Atypical Alzheimer's Disease. Alzheimer's Demen. 14 (8), 1005–1014. doi:10.1016/j.jalz.2018.02.020
Wu, J. R., Wang, J., Zhou, S. K., Yang, L., Yin, J. L., Cao, J. P., et al. (2015). Necrostatin-1 protection of Dopaminergic Neurons. Neural Regen. Res. 10 (7), 1120–1124. doi:10.4103/1673-5374.160108
Wu, M.-Y., Yiang, G.-T., Liao, W.-T., Tsai, A. P.-Y., Cheng, Y.-L., Cheng, P.-W., et al. (2018). Current Mechanistic Concepts in Ischemia and Reperfusion Injury. Cell Physiol Biochem 46 (4), 1650–1667. doi:10.1159/000489241
Xu, H., Wang, Y., Song, N., Wang, J., Jiang, H., and Xie, J. (2017). New Progress on the Role of Glia in Iron Metabolism and Iron-Induced Degeneration of Dopamine Neurons in Parkinson's Disease. Front. Mol. Neurosci. 10, 455. doi:10.3389/fnmol.2017.00071
Yadav, A., and Collman, R. G. (2009). CNS Inflammation and Macrophage/microglial Biology Associated with HIV-1 Infection. J. Neuroimmune Pharmacol. 4 (4), 430–447. doi:10.1007/s11481-009-9174-2
Yan, B., Liu, L., Huang, S., Ren, Y., Wang, H., Yao, Z., et al. (2017). Discovery of a New Class of Highly Potent Necroptosis Inhibitors Targeting the Mixed Lineage Kinase Domain-like Protein. Chem. Commun. 53 (26), 3637–3640. doi:10.1039/c7cc00667e
Yang, S. H., Lee, D. K., Shin, J., Lee, S., Baek, S., Kim, J., et al. (2017). Nec-1 Alleviates Cognitive Impairment with Reduction of Aβ and Tau Abnormalities in APP/PS 1 Mice. EMBO Mol. Med. 9 (1), 61–77. doi:10.15252/emmm.201606566
You, Z., Savitz, S. I., Yang, J., Degterev, A., Yuan, J., Cuny, G. D., et al. (2008). Necrostatin-1 Reduces Histopathology and Improves Functional Outcome after Controlled Cortical Impact in Mice. J. Cereb. Blood Flow Metab. 28 (9), 1564–1573. doi:10.1038/jcbfm.2008.44
Yuan, J., Amin, P., and Ofengeim, D. (2019). Necroptosis and RIPK1-Mediated Neuroinflammation in CNS Diseases. Nat. Rev. Neurosci. 20 (1), 19–33. doi:10.1038/s41583-018-0093-1
Zhang, F., Yan, C., Wei, C., Yao, Y., Ma, X., Gong, Z., et al. (2018). Vinpocetine Inhibits NF-κb-dependent Inflammation in Acute Ischemic Stroke Patients. Transl. Stroke Res. 9 (2), 174–184. doi:10.1007/s12975-017-0549-z
Zhang, H.-B., Cheng, S.-X., Tu, Y., Zhang, S., Hou, S.-K., and Yang, Z. (2017). Protective Effect of Mild-Induced Hypothermia against Moderate Traumatic Brain Injury in Rats Involved in Necroptotic and Apoptotic Pathways. Brain Inj. 31 (3), 406–415. doi:10.1080/02699052.2016.1225984
Zhang, S., Tang, M. B., Luo, H. Y., Shi, C. H., and Xu, Y. M. (2017). Necroptosis in Neurodegenerative Diseases: a Potential Therapeutic Target. Cel Death Dis 8 (6), e2905. doi:10.1038/cddis.2017.286
Zhang, S., Su, Y., Ying, Z., Guo, D., Pan, C., Guo, J., et al. (2019). RIP1 Kinase Inhibitor Halts the Progression of an Immune-Induced Demyelination Disease at the Stage of Monocyte Elevation. Proc. Natl. Acad. Sci. USA 116 (12), 5675–5680. doi:10.1073/pnas.1819917116
Zhou, W., and Yuan, J. (2014). Necroptosis in Health and Diseases. Semin. Cel Dev. Biol. 35, 14–23. doi:10.1016/j.semcdb.2014.07.013
Zille, M., Karuppagounder, S. S., Chen, Y., Gough, P. J., Bertin, J., Finger, J., et al. (2017). Neuronal Death after Hemorrhagic Stroke In Vitro and In Vivo Shares Features of Ferroptosis and Necroptosis. Stroke 48 (4), 1033–1043. doi:10.1161/strokeaha.116.015609
Aβ Amyloid-β
AD Alzheimer’s disease
AIDS Acquired Immune Deficiency Syndrome
ALS Amyotrophic lateral sclerosis
c-FLIP cellular FADD-like interleukin-1β converting enzyme inhibitory protein
cIAP1 Cellular inhibitor of apoptosis protein 1
CNS Central nervous system
DRs Activated death receptors
FADD Fas-associated death domain
fALS Familial amyotrophic lateral sclerosis
FPI Fluid percussion injury
HIV-1 Human immunodeficiency virus 1
ICH Intracerebral hemorrhage
IKK IκB kinase
IRI Ischemia-reperfusion injury
JE Japanese encephalitis
JEV Japanese encephalitis virus
LPS lipopolysaccharide
MLKL Mixed lineage kinase domain-like protein
Nec-1 Necrostatin-1
NEMO Nuclear factor-Kappa B essential modulator
NLRP3 NLR Family Pyrin Domain Containing 3
OPTN optineurin
PD Parkinson’s disease
PD-1 programmed cell death-1
RIC RIPK1-inhibitory compound
RIPK1 Receptor-interacting protein kinase 1
RIPK3 Receptor-interacting protein kinase 3
sALS sporadic amyotrophic lateral sclerosis
SN Substantia nigra
SOD1 Superoxide dismutase 1
TAK1 Transforming growth factor β-activated kinase-1
TBI Traumatic brain injury
TICAM-1 TIR domain-containing adaptor molecule 1
TLR Toll-like receptor
TNF Tumor necrosis factor
TNFR1 TNF receptor 1
TRADD TNFR-associated death domain
TRIF Toll/IL-1 receptor domain-containing adaptor inducing IFN-β
WNV West Nile virus
WT wild-type
Keywords: neuroinflammation, necroptosis, ripk1, ripk3, mlkl, necrostatin-1
Citation: Yu Z, Jiang N, Su W and Zhuo Y (2021) Necroptosis: A Novel Pathway in Neuroinflammation. Front. Pharmacol. 12:701564. doi: 10.3389/fphar.2021.701564
Received: 28 April 2021; Accepted: 28 June 2021;
Published: 12 July 2021.
Edited by:
Sathya Dev Unudurthi, Masonic Medical Research Institute (MMRI), United StatesReviewed by:
Yusra Saif Al Dhaheri, United Arab Emirates University, United Arab EmiratesCopyright © 2021 Yu, Jiang, Su and Zhuo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenru Su, c3V3cjNAbWFpbC5zeXN1LmVkdS5jbg==; Yehong Zhuo, emh1b3loQG1haWwuc3lzdS5lZHUuY24=
†These authors contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.