 Shuang Wei1,2†
Shuang Wei1,2† Wang-Ping Hu
Wang-Ping Hu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol., 24 May 2021
Sec. Neuropharmacology
Volume 12 - 2021 | https://doi.org/10.3389/fphar.2021.685460
This article is part of the Research TopicPharmacological Aspects of Ligand-gated Ion Channels as Targets of Natural and Synthetic AgentsView all 18 articles
Dexmedetomidine (DEX), a selective α2 adrenergic receptor (α2-AR) agonist, has been shown to have peripheral analgesic effects in a variety of pain conditions. However, the precise molecular mechanisms have not yet been fully elucidated. Acid sensing ion channels (ASICs) are the major player in pain associated with tissue acidosis. Given that both α2-ARs and ASICs exist in dorsal root ganglia (DRG) neurons, we therefore investigated the effects of DEX on the functional activity of ASICs. Herein, whole-cell patch-clamp recordings demonstrated that DEX suppressed ASIC-mediated and acid-evoked currents and action potentials in dissociated rat DRG neurons. DEX shifted downwards concentration-response curve to protons, with a decrease of 35.83 ± 3.91% in the maximal current response to pH 4.5. DEX-induced inhibition of ASIC currents was blocked by the α2A-AR antagonist BRL44408 in DRG neurons. DEX also inhibited ASIC3 currents in CHO cells co-expressing ASIC3 and α2A-ARs, but not in ASIC3 transfected CHO cells without α2A-ARs expression. DEX-induced inhibition of ASIC currents was mimicked by the protein kinase A inhibitor H-89, and blocked by intracellular application of the Gi/o protein inhibitor pertussis toxin and the cAMP analog 8-Br-cAMP. In addition, peripherally administration of DEX dose-dependently relieved nociceptive responses to intraplantar injection of acetic acid in rats through local α2A-ARs. Our results indicated that DEX inhibited the functional activity of ASICs via α2A-ARs and intracellular Gi/o proteins and cAMP/protein kinase A signaling pathway in rat DRG neurons, which was a novel potential mechanism that probably mediated peripheral analgesia of DEX.
Dexmedetomidine (DEX) is a potent highly selective α2 adrenergic receptor (α2-AR) agonist. It is approved as an analgesic agent since the early 1970s (Kamibayashi and Maze, 2000). DEX’ action is related to the wide distribution of α2-ARs in the pain signaling pathway, including in primary afferents and spinal dorsal horn (Gold et al., 1997; Stone et al., 1998; Shi et al., 2000; Nicholson et al., 2005). DEX has shown potential analgesic effects at the supraspinal, spinal and peripheral levels in various pain conditions. Intrathecal or systemic administration of DEX produces analgesic effects in animals and humans (Aho et al., 1991; Kingery et al., 2000; Kimura et al., 2012). Peripheral DEX also shows anti-nociceptive effects since peripherally restricted α2-AR antagonist could block the effect of systemic DEX on neuropathic pain (Poree et al., 1998; Lee et al., 2013). Moreover, DEX has stronger analgesic effects following peripheral nerve injury via α2-ARs (Poree et al., 1998; Malmberg et al., 2001). Studies with α2-AR knock-out animal models suggest that α2-AR plays a major role in DEX analgesia (Hunter et al., 1997; Stone et al., 1997; Malmberg et al., 2001). In addition to neuropathic pain, studies have also confirmed that DEX has analgesic effects on pain under postoperative, acute and chronic inflammatory conditions (Mahmoud and Mason, 2015; Zhao et al., 2020). Clinically, DEX has been used to relieve acute postoperative pain, ischemic pain, refractory cancer pain (Hoy and Keating, 2011).
Apart from α2-ARs, a number of ligand-gated and voltage-gated ion channels are also expressed in primary sensory neurons, including dorsal root ganglion (DRG) neurons, which can convert noxious stimulation into ascending nociceptive signals (Mccleskey and Gold, 1999). Thus, modulation of these ion channels may be a potential mechanism underlying peripheral anti-nociceptive effects of DEX. For example, DEX has been shown to inhibit voltage-gated sodium channels via α2-ARs in DRG and trigeminal ganglion neurons (Gu et al., 2015; Im et al., 2018). DEX also inhibits the activity of TRPV1 in DRG neurons depending on the activation of α2-ARs followed by the inhibition of the adenylate cyclase/cAMP/protein kinase A (PKA) pathway (Lee et al., 2020).
To detect changes in pH, primary sensory neurons also express several acid sensors, such as acid sensitive ion channels (ASICs), TRPV1, proton-sensing GPCRs, and certain K2P channels (Holzer, 2009; Pattison et al., 2019). Among these, ASICs and TRPV1 have been most thoroughly studied. ASICs, as pH sensors, are expressed in both DRG cell bodies and sensory terminals, where they contribute to proton-evoked nociceptive signaling (Alvarez De La Rosa et al., 2002; Benson et al., 2002). To date, there are seven different ASIC subunits encoded by five different genes (Wemmie et al., 2013). Among these ASIC subunits, ASIC3 subunit is the most abundant in DRG and has emerged as a critical pH sensor (Deval et al., 2008; Lee et al., 2013). Proton, a canonical ligand for ASICs, is released and causes tissue acidosis under multiple pathological conditions such as inflammation, tissue injury, ischemic stroke and cancer (Deval et al., 2011; Deval and Lingueglia, 2015). Amiloride, a non-selective ASIC inhibitor, can significantly alleviate the pain caused by moderate (up to pH 6.0) pH, suggesting that ASICs, rather than TRPV1 or other acid sensors, mainly mediate the pain sensation (Ugawa et al., 2002; Deval et al., 2008). ASICs, especially ASIC3, are the major player in pain associated with tissue acidosis and represent novel potential targets for development of analgesics (Reeh and Steen, 1996; Wemmie et al., 2013; Li and Xu, 2015; Dulai et al., 2020).
Given that both α2-ARs and ASICs exist in DRG neurons, the aim of this study was to investigate whether ASICs are also modulatory targets of DEX. We observed that DEX inhibited the electrophysiological activity of ASICs through activation of α2-ARs and an intracellular cAMP and PKA signaling pathway in rat DRG neurons. DEX also relieved ASIC-mediated nociceptive behaviors in rats by activating peripheral α2-ARs.
All experimental protocols were approved by the animal research ethics committee of Hubei University of Science and Technology. Sprague-Dawley male rats (5–6 weeks old) were anesthetized and then killed. The DRGs were removed and chopped with thin spring scissors. The minced ganglia were transferred to a test tube containing Dulbecco’s modified Eagle’s medium (DMEM, Sigma) and incubated in a shaking for 25–30 min at 35°C. Incubation solution contained 1.0 mg/ml collagenase (type I-A, Sigma), 0.5 mg/ml trypsin (type II-S, Sigma) and 0.1 mg/ml DNase (type IV, Sigma). Trypsin digestion was terminated by adding1.25 mg/ml Soybean trypsin inhibitor (type II-S, Sigma). The cells were cultured in DMEM supplemented with 10% fetal bovine serum and 100 ng/ml never growth factor (NGF) for 12–24 h at 37°C in a water saturated atmosphere with 95% O2 and 5% CO2.
Electrophysiological experiments were carried out as described previously (Wei et al., 2021). Whole-cell patch clamp recordings were carried out at room temperature (22–25°C) using EPC-10 patch clamp amplifier and PULSE software (HEKA Electronic, Lambrecht, Germany). The isolated DRG neurons were transferred to a 35 mm culture dish and kept in normal external solution for at least 60 min before electrophysiological recordings. The external solution contained the following (in mM): 150 NaCl, 5 KCl, 2 MgCl2, 2.5 CaCl2, 10 HEPES, 10 d-glucose. Its pH and osmolarity was adjusted to 7.4 with NaOH and 330 mOsm/L with sucrose, separately. Recording pipettes were pulled using a Sutter P-97 puller (Sutter Instruments, CA, United States) and its resistance was in the range of 3–6 MΩ. The micropipettes solution contained (in mM): 140 KCl, 2 MgCl2, 11 EGTA, 10 HEPES, 4 ATP, and 0.3 Na2GTP. Its pH and osmolarity was adjusted to 7.2 with KOH and 310 mOsm/L with sucrose, separately. After whole-cell configuration established, 70–80% series resistance and membrane capacitance current were compensated. The recording currents were sampled at 10 kHz and filtered at 2 kHz. Data detection and analysis were performed using the pCLAMP 10 software (Axon Instruments, CA, United States). The neurons selected for electrophysiological recordings were 15–35 μm in diameter, which are thought to be nociceptive neurons. The membrane voltage was maintained at −60 mV in all voltage-clamp experiments. Current-clamp recordings were obtained by switching to current-clamp mode after a stable whole-cell configuration was formed in voltage-clamp mode. Only cells with a stable resting membrane potential (more negative than −50 mV) were used in the study.
Rat ASIC3 and human α2A-AR cDNAs were used for heterologous expression in Chinese hamster ovary (CHO) cells as described previously (Wu et al., 2017). In brief, CHO cells were cultured at 37°C in a humidified atmosphere of 5% CO2 and 95% O2 and passaged twice a week. Transient transfection of CHO cells was performed using a HilyMax liposome transfection reagent (Dojindo Laboratories). CHO cells were maintained in F-12 nutrient mixture (added 1.176 g of NaHCO3/L medium) supplemented with 10% fetal bovine serum and 1% gluta-MAXTM-1 (×100; Invitrogen). When ASIC3 and α2A-AR cDNA were co-transfected, the ratio was maintained at 1:1. All plasmids contained, in addition to the desired ASIC3 cDNA, the coding sequence for enhanced green fluorescent protein to aid in the identification of transfected cells. Electrophysiological measurements were performed 24–48 h after transfection.
In the experiment, drugs included hydrochloric acid, dexmedetomidine, clonidine, yohimbine, BRL44408, 8-Br-cAMP, H-89, amiloride, APETx2, capsaicin, and AMG 9810. They were obtained from Sigma Chemical Co. (St. Louis, MO, United States). Different pH values were configured with hydrochloric acid and external solution. The working concentration of drugs was freshly prepared in normal external solution and their pH was adjusted to7.4 with NaOH. Each working drug was stored in a series of independent reservoirs and applied by gravity. The distance was ∼30 μm between drug exit and recorded neurons. To block G-protein and intracellular signal, some antagonists or blockers were dissolved in the internal solution and applied for intracellular dialysis through recording patch pipettes as described previously (Liu et al., 2021). To ensure that dialysis drugs are infused into the cell interior, there is at least a 30 min interval between the establishment of the whole cell pathway and the current measurement. To functionally characterize ASIC activity, we used AMG9810 (5 μM) to block TRPV1 in the extracellular solution.
Behavior experiments were carried out as described previously (Wei et al., 2021). Male rats were allowed to habituate for at least 30 min before nociceptive behavior experiments in a Plexiglas chamber. Separate groups of rats were coded and pretreated with 50 μl AMG 9810 (10 μM) together with vehicle, different dose (10, 30 and 100 ng) of DEX, 150 ng BRL44408 + 100 ng DEX in ipsilateral hindpaw using a 30-gauge needle connected to a 100 μl Hamilton syringe. After 5 min, another experimenter subcutaneously administered acetic acid solution (1%, 50 μl) into the hindpaw and tested nociceptive behavior. In one group, acetic acid was injected into one hindpaw and DEX (100 ng) was injected into contralateral hindpaw. Nociceptive behavior (that is, number of flinches) was counted over a 5 min period starting immediately after the injection (Deval et al., 2008; Omori et al., 2008).
Data were statistically compared using the Student's t-test or one-way analysis of variance (ANOVA), followed by Bonferroni’s post hoc test. Statistical analysis of concentration–response data was performed using nonlinear curve-fitting program ALLFIT. Data are expressed as mean ± S.E.M.
In the majority of DRG neurons tested (76.9%, 10/13), a perfusion of pH 5.0 acid solution to DRG neurons for 5 s evoked a rapid inward current (IpH5.0), even though blockade of proton-induced TRPV1 activation by addition of AMG9810 (5 μM) in external solution (Figure 1A). Most (70.0%, 7/10) of IpH5.0 were characterized by a fast inactivated inward current, followed by a smaller and non-desensitizing sustained current in ten DRG neurons sensitive to pH 5.0 acid stimuli. All seven IpH5.0 currents with this characteristic could be completely blocked by the broad-spectrum ASIC channel blocker amiloride (10 μM) and ASIC3 blocker APETx2 (2 μM). In the presence of AMG9810, capsaicin (100 nM) failed to evoke any membrane currents in all DRG neurons tested. However, capsaicin (100 nM) evoked an inward current in the majority of DRG neurons sensitive to pH 5.0 acid stimuli (71.4%, 5/7) after washout of AMG9810. Thus, these proton-induced currents were considered to be ASIC currents or ASIC3 currents after TRPV1 activation was blocked by AMG9810. In the following study, we mainly observed the ASIC3-like currents with the above characteristics.
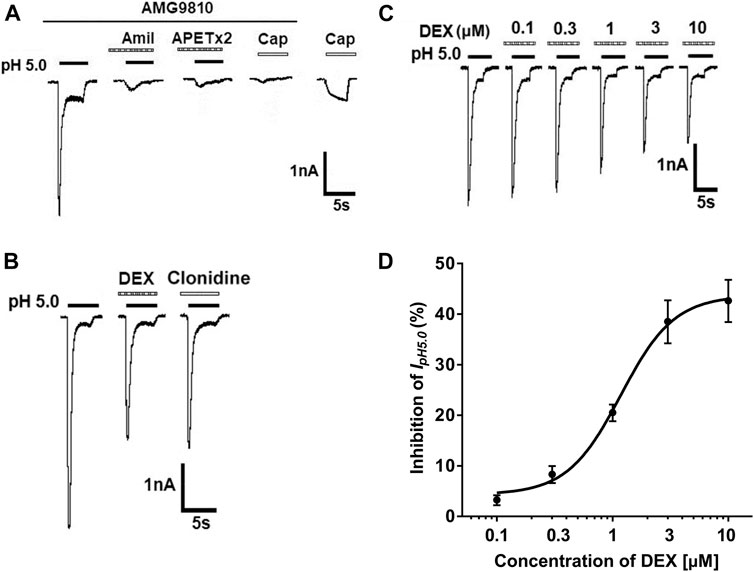
FIGURE 1. DEX concentration-dependently inhibits proton-gated currents in rat DRG neurons. (A). Representative current traces were evoked by application of a pH 5.0 acidic solution for 5 s in a tested DRG neuron in the presence of AMG9810 (5-μM). The proton-gated current (IpH5.0) could be blocked by broad-spectrum ASIC channel blocker amiloride (Amil, 10 μM) and ASIC3 blocker APETx2 (2 μM). Capsaicin (Cap, 100 nM) failed to evoke any membrane currents in the presence of AMG9810 (5-μM). However, capsaicin (100 nM) evoked an inward current after washout of AMG9810 in the same neuron. All membrane potentials were clamped at −60 mV. (B). In the same DRG cell, pre-application of α2-ARagonist DEX or clonidine caused an inhibitory effect on IpH5.0. (C). The sequential current traces illustrate that IpH5.0 amplitude was inhibited by pre-application of different concentration of DEX (for 2 min) in a representative DRG neuron. (D). The graph shows concentration-effect curve of DEX on IpH5.0 with an IC50 of 1.18 ± 0.11 μM. Each point represents the mean ± SEM of 6–10 cells.
In some DRG neurons sensitive to pH 5.0 acid stimuli (52.9%, 9/17), pre-application of α2-AR agonist DEX (3 μM) for 2 min decreased the peak amplitude of the ASIC currents (Figure 1B). In all nine cells response to DEX, pre-application of clonidine (10 μM) for 2 min had also similar inhibitory effects on the ASIC currents (Figure 1B). In addition, DEX shortened the inactivation time constant of ASICs from 1,413.81 ± 184.64 to 1,094.44 ± 146.38 msec (p < 0.05, paired t-test, n = 9). The inhibition of IpH5.0 was dependent upon the concentration of DEX (Figures 1C,D). In a representative DRG neuron, the peak amplitude of IpH5.0 progressively decreased as concentration of pre-treated DEX increased from 0.1 to 10 μM (Figure 1B). Figure 1C shows concentration-effect curve of DEX on IpH5.0 with an IC50 (half-maximal effective concentration) value of 1.18 ± 0.11 μM. The results indicated that DEX concentration-dependently inhibited ASIC currents in rat DRG neurons.
We then investigated the effect of DEX on concentration-response curve to protons. Currents were measured by applying a range of different low pH values before and after treatment with DEX. Figure 2A shows that pre-application of DEX (3 μM for 2 min) decreased all peak amplitudes of IpH6.5, IpH5.5 and IpH4.5. Figure 2B shows concentration-response to protons in the absence and presence of DEX (3 μM). First, the maximal current response (IpH4.5) of curve decreased 35.83 ± 3.91% after DEX was treated to DRG neurons. Second, the Hill coefficient or slope of two curves had not significant difference (pH: n = 1.23 ± 0.16; DEX + pH: n = 1.16 ± 0.21; p > 0.1, post hoc Bonferroni’s test). Third, the pH0.5 (pH for half-maximal activation) values of two curves had also no statistical difference (pH: pH0.5 = 5.97 ± 0.17; DEX + pH: pH0.5 = 5.89 ± 0.20; p > 0.1, post hoc Bonferroni’s test). These results indicated that the maximum response to protons was inhibited by DEX, but that no shift in proton sensitivity.
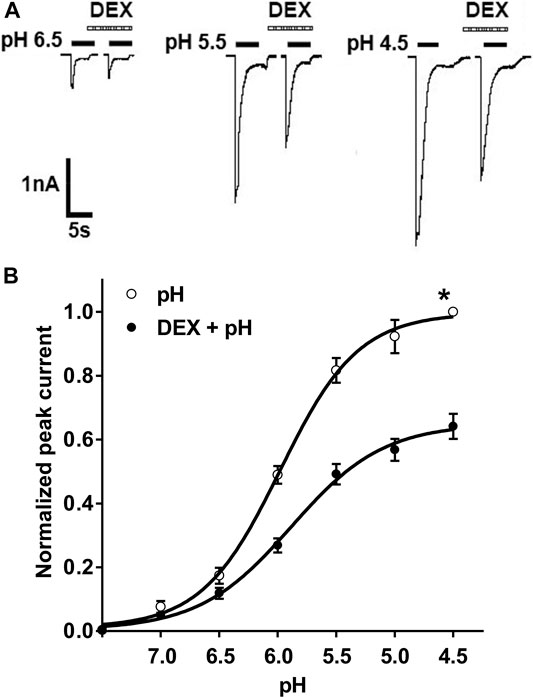
FIGURE 2. DEX shifts downwards the concentration–response curve for protons. (A). Sequential currents were evoked by different low pH values in the absence and presence of DEX (3 μM) pretreatment. (B). Proton concentration-response curves for ASIC activation in the absence (○) and presence (●) of extracellular 3 μM DEX. Pre-application of DEX shifted downwards the concentration-response curve for proton. Each point represents the mean ± S.E.M. of 6–10 neurons. All peak current values were normalized to the peak current maximally activated by pH 4.5 applied alone in the absence of DEX (marked with asterisk). The figure shows averaged data fitted with the Hill equation.
DEX is a selective α2-AR agonist. To address whether inhibition of ASIC currents by DEX application was mediated by α2-ARs, we examined the effect of yohimbine, an α2-AR antagonist, on inhibitory effects of DEX on ASIC currents. As shown in Figures 3A,B, DEX-induced suppression of IpH5.0 was significantly blocked by yohimbine. The amplitude of IpH5.0 decreased 38.48 ± 4.25% by 3 μM DEX pre-treatment alone. In contrast, the amplitude of IpH5.0 decreased only 3.73 ± 3.23% in DRG neurons pre-treated with both 3 μM yohimbine and 3 μM DEX (p < 0.01, compared with DEX pretreatment alone, one-way ANOVA followed by post hoc Bonferroni’s test, n = 7). Moreover, we also examined the effect of BRL44408, an α2A-AR antagonist, on DEX-induced inhibition of ASIC currents. Pre-incubation of BRL44408 (3 μM) alone did not affect the amplitude of IpH5.0, but significantly blocked DEX-induced suppression of IpH5.0 (Figures 3A,B). The results indicated that DEX inhibited ASIC currents mainly through α2A-ARs in DRG neurons.
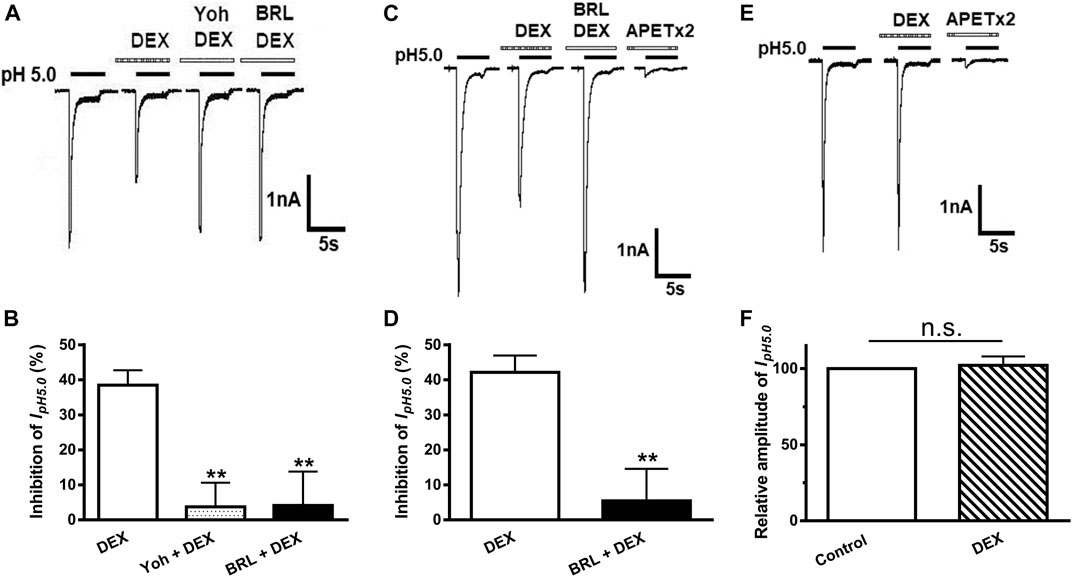
FIGURE 3. α2-ARs mediate DEX-induced inhibition of ASIC currents in DRG and CHO cells. The current traces in (A) and the bar graph in (B) show that IpH5.0 was inhibited by DEX (3 μM) pre-applied alone for 2 min in DRG neurons, and DEX inhibition of IpH5.0 was blocked by the co-application of α2-AR antagonist yohimbine (3 μM) or α2A-AR antagonist BRL44408 (3 μM). The current traces in (C) and the bar graph in (D) show that IpH5.0 was inhibited by DEX (3 μM) pre-applied alone for 2 min in CHO cells co-expressing ASIC3 and α2A-ARs. This inhibiting effect was blocked by the coapplication of α2A-AR antagonist BRL44408 (3 μM). The current traces in (E) and the bar graph in (F) show that DEX (3 μM) had no effect on the IpH5.0 in CHO cells expressing ASIC3 alone, but not expressing α2A-ARs. Currents were normalized to the control (100%, whitecolumn). Statistical tests were performed using one-way ANOVA followed by post hoc Bonferroni’s test, and significance is shown **p < 0.01, n. s, not significant. n = 6 in each column.
To further verify whether α2A-ARs mediate DEX-induced inhibition of acid-evoked currents, α2A-ARs were co-expressed with ASIC3 in CHO cells. We recorded ASIC3-mediated acid currents, since they were completely blocked by APETx2 (2 μM). Similar to that observed in DRG neurons, the ASIC3 currents were inhibited by the pre-application of DEX (3 μM) in CHO cells co-expressing ASIC3 and α2A-ARs (Figures 3C,D). The DEX-induced inhibition of ASIC3 currents was also blocked by pre-incubation of 3 μM BRL44408, an α2A-AR antagonist (Figures 3C,D). In contrast, DEX had no effect on ASIC3 currents at a concentration of 3 μM in CHO cells expressing ASIC3 alone, but not expressing α2A-ARs (Figures 3E,F). The pH 5.0 induced ASIC3 channel activation were completely blocked by APETx2 (2 μM) in CHO cells expressing ASIC3 alone. Guanabenz, another α2A-AR agonist, is recently found to activate ASIC3 at neutral pH (Callejo et al., 2020). However, we did not observe that DEX had such an effect when applied in CHO cells expressing ASIC3 alone. DEX (3 or 500 μM) failed to induce any membrane currents at neutral pH (n = 8, data not shown). These results further indicated that α2A-ARs mediated DEX-induced inhibition of ASIC3 currents.
α2A-ARs belong to Gi/o protein-coupled receptor family, which leads to a cascade of events, such as inhibition of adenylate cyclase (AC) activity and the cAMP/PKA pathway (Wu et al., 1988). Therefore, we first examined whether DEX-induced inhibition of ASIC currents occurs via Gi/o proteins. Pertussis toxin (PTX, 1 μg/ml), an inhibitor of Gi/o-proteins, was applied internally to DRG neurons and significantly prevented the decrease of IpH5.0 amplitude induced by DEX (Figures 4A,B). To further explore intracellular signal transduction mechanisms underlying suppression of ASIC currents by DEX, we observed the effect of 8-Br-cAMP, a membrane permeable cAMP analog, on DEX suppression of ASIC currents. When seven DRG neurons were pre-treated with 8-Br-cAMP (1 mM) for 5 min, the relative amplitudes of IpH5.0 were increased to 142.46 ± 13.64% of control. In the presence of 8-Br-cAMP (1 mM), pre-application of DEX (3 μM for 2 min) produced only decreases of 6.25 ± 4.38% on IpH5.0 (p > 0.1, compared with 8-Br-cAMP treatment alone, one-way ANOVA followed by post hoc Bonferroni’s test, n = 7; Figures 4C,D). We then observed the effect of H-89, a membrane-permeable inhibitor of PKA, on DEX suppression of ASIC currents. When DRG neurons were pre-treated with H-89 (0.3 μM) for 5 min, the relative amplitudes of IpH5.0 were reduced to 51.21 ± 7.95% of control (n = 7), suggesting that H-89 mimicked the action of DEX on IpH5.0 (Figures 4E,F). In the presence of H-89 (0.3 μM), pre-application of DEX (3 μM for 2 min) no longer further inhibited IpH5.0 (p > 0.1, compared with H-89 treatment alone, one-way ANOVA followed by post hoc Bonferroni’s test, n = 7; Figures 4E,F). Together, the results indicated that the suppression of ASIC currents by DEX was dependent upon Gi/o proteins and intracellular cAMP/PKA signaling pathway.
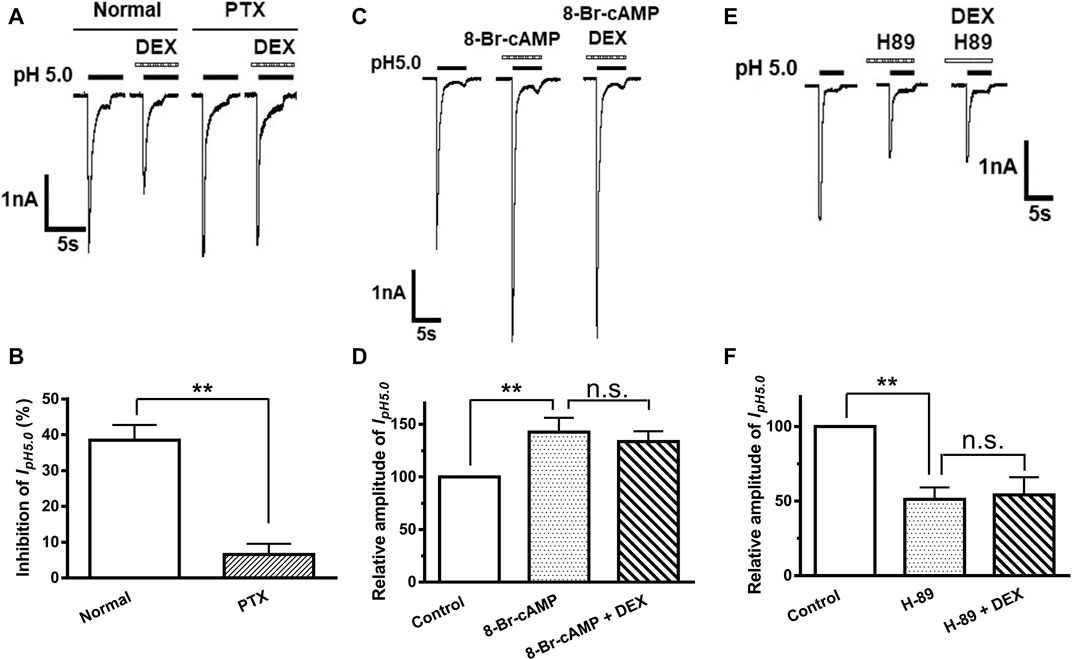
FIGURE 4. Gi/o-protein and PKA signaling participate in DEX-induced inhibition of ASIC currents. The current traces in (A) and the bar graph in (B) show the effects of DEX (3 μM) on IpH5.0 in recording pipettes filled with normal and PTX (1 μg/ml) containing internal solution conditions. The current traces in (C) and the bar graph in (D) show the enhancement of IpH5.0 by pre-incubation 8-Br-cAMP (1 mM), a membrane permeable cAMP analog. DEX (3 μM) failed to cause inhibition of IpH5.0 in the presence of 8-Br-cAMP (1 mM). The current traces in (E) and the bar graph in (F) show inhibition of IpH5.0 by pre-incubation PKA inhibitor H-89 (0.3 μM). DEX (3 μM) did not cause a further inhibition of IpH5.0 in the presence of H-89 (0.3 μM). Statistical tests were performed using one-way ANOVA followed by post hoc Bonferroni’s test, and significance is shown **p < 0.01, n. s, not significant. n = 7 in each column.
ASICs are trimeric cation-permeable channels. Once activation of ASICs by protons causes cation influx (largely Na+) and membrane potential depolarization, resulting in bursts of action potentials if the proton-induced depolarization is large enough to activate Nav subunits (Pattison et al., 2019). We further observed whether DEX had effects on acid-evoked action potentials of rat DRG neurons. Although proton-induced TRPV1 activation was blocked in the presence of 5-μM AMG9810, we observed that an acid stimulus of pH 5.0 could trigger bursts of action potentials (APs) in DRG neurons under current-clamp conditions (Figures 5A,C). Consistent with that observed under voltage-clamp conditions, DEX also decreased the number of APs evoked by acidic stimuli of pH 5.0 in DRG neurons (Figures 5A,B). In seven DRG neurons with DEX (3 μM for 2 min) pre-treatment, the number of APs evoked by acidic stimuli of pH 5.0 significantly decreased (p < 0.01, paired t-test, n = 7, Figure 5B). However, the number of APs had not changed in other seven DRG cells co-treated with both BRL44408(BRL, 3 μM) and DEX (3 μM) (p > 0. 1, paired t-test; n = 7, Figures 5C,D). These results indicated that DEX also suppressed acid-evoked action potentials in rat DRG neurons through α2A-ARs.
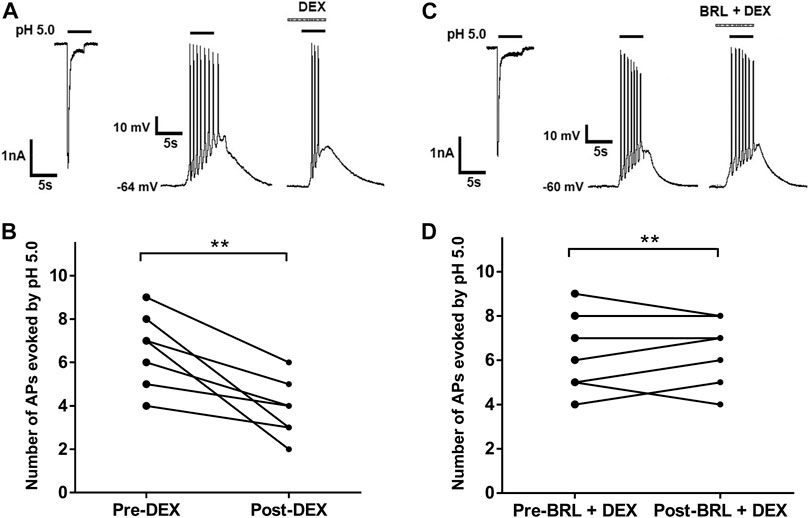
FIGURE 5. DEX suppresses acid-evoked action potentials in rat DRG neurons. (A,C). In the same DRG neuron, pH 5.0 induced not only an inward current with voltage-clamp recording, but also action potentials with current-clamp recording. Original action potentials were recorded in two DRG neurons before and after application of DEX (3 μM) alone (A) or coapplication of both DEX (3 μM) and BRL44408(BRL, 3 μM) for 2min (C). AMG9810 (5 μM) was used to block proton-induced TRPV1 activation. (B,D). The graphs show the number of pH 5.0 acid-evoked action potentials decreased by pre-application of DEX alone, but not by coapplication of both DEX and BRL44408. **p < 0.01, paired t-test, n = 7 cells.
Above results demonstrated that DEX inhibited ASIC activity in vitro. We further ascertained whether DEX had effect on ASIC-mediated nociceptive behaviors through interacting with ASICs in vivo. After acetic acid was injected into rat hind paws, rats displayed an intense flinch/shaking response even if AMG 9810 (10 μM) blocked the activation of TRPV1. We found that pretreatment with DEX (10, 30, and 100 ng) dose-dependently relieved the acid-induced nociceptive behaviors (p < 0.05 and 0.01, one-way ANOVA followed by post hoc Bonferroni’s test, n = 10; Figure 6). However, Co-injection of BRL44408 (BRL, 150 ng) blocked the relieving effect of 100 ng DEX on acid-induced nociceptive behaviors. The mean number of flinches in these rats significantly increased, compared with that observed in rats pretreated with 100 ng DEX alone (p < 0.01, one-way ANOVA followed by post hoc Bonferroni’s test, n = 10; Figure 6), and were no different from control rats without DEX pretreatment. In addition, injection of 100 ng DEX into the contralateral paws did not relieved acid-induced nociceptive behaviors. These results indicated that DEX relieved acid-induced nociceptive behaviors in rats through activation of α2A-ARs localized in the injected hindpaw.
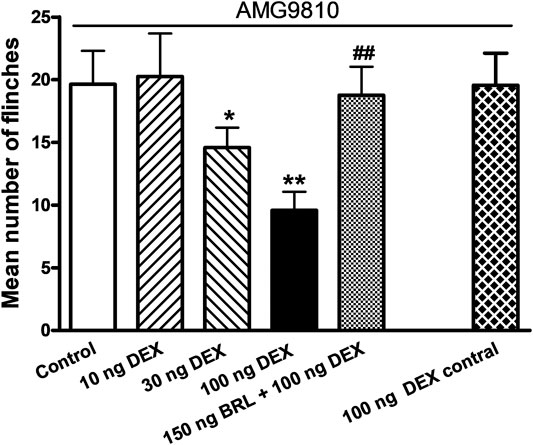
FIGURE 6. DEX relieves acid-induced nociceptive behaviors in rats. In the presence of the TRPV1 inhibitor AMG9810 (10 μM), nociceptive responses were evoked by intraplantar injection of acetic acid (1%, 50 μl) in each rat. Intraplantar pretreatment of DEX (10, 30 and 100 ng) dose-dependently reduced the number of acid-induced flinching. DEX (100 ng) inhibition of acid-induced nociceptive behaviors is reversed by co-injection of α2A-AR antagonist BRL44408 (BRL, 150 ng) so that behaviors in these rats were no different from the control rats treated with acetic acid alone. Rats injected with acetic acid in one hindpaw and DEX into the contralateral hindpaw (100 ng contral) had nociceptive behaviors similar to those seen in control rats. Each bar represents the number of flinches that animals spent licking/lifting the injected paw during first 5 min observation period. **p < 0.01, one-way ANOVA followed by post hoc Bonferroni’s test, compared with control column; ##p < 0.01, one-way ANOVA followed by post hoc Bonferroni’s test, compared with 100 ng DEX column. Each group represents the mean ± S.E.M. of 10 rats.
The present data demonstrated that selective α2-AR agonist DEX inhibited the electrophysiological activity of ASICs through α2A-ARs. DEX reduced ASIC-mediated and acid-evoked currents and action potentials in dissociated rat DRG neurons. PTX-sensitive Gi/o proteins and cAMP/PKA signaling cascade was involved in the inhibition of ASICs by DEX. Behaviorally, DEX also relieved acid-induced nociceptive responses in rats by activating peripheral α2A-ARs.
It has been found that six ASIC subunits are expressed in neuron cell bodies in DRG, and ASIC3 subunit is the most abundant (Deval et al., 2008). In the presence of AMG9810, the low pH-evoked currents were mediated by ASICs in rat DRG cells, since they were completely blocked by amiloride and APETx2. The present study showed that DEX, a selective α2-AR agonist, inhibited ASIC currents and the number of action potentials evoked by low pH stimulation. Moreover, DEX accelerated the inactivation of ASICs, which also helps to reduce the number of action potentials evoked by acidic stimuli. DEX inhibited the electrophysiological activity of ASICs in rat DRG neurons through α2-ARs. First, another α2-AR agonist clonidine has also been shown to effectively inhibit ASIC currents. Second, DEX-induced inhibition was completely blocked by the α2A-AR antagonist BRL44408. Last, DEX could also inhibit acid-evoked currents mediated by ASIC3 in CHO cells co-expressing α2A-ARs and ASIC3, but not in CHO cells only expressed ASIC3 and lacked α2A-AR expression, further suggesting that the inhibition of ASIC3 currents by DEX required the co-expression of α2A-ARs. In addition, only some, but not all, ASIC-like responses were inhibited by DEX/clonidine, which may be related to the degree of the co-expression of α2A-ARs and ASIC3 in rat DRG neurons. Thus, we speculated that DEX-induced inhibition of ASIC currents maybe also occur only in DRG neurons co-expressing α2A-ARs and ASICs, although the direct evidence needs to further determine. It has been shown that three subtypes of the α2-ARs (α2A-, α2B- and α2C-ARs) are expressed in the rat DRGs (Shi et al., 2000). But α2B-AR subtype is found only in a very small population (Chung et al., 1993; Shi et al., 2000). The expression of α2A and α2C-AR subtypes is regulated by peripheral nerve injury. Proteins and mRNA levels of α2A and α2C-AR subtypes are increased and decreased, respectively, in DRG neurons after chronic constriction injury of sciatic nerve (Cho et al., 1997; Shi et al., 2000). In addition to ASICs, other cation channels, such as Nav1.8, TRPV1 and TRPM8 are also inhibited via α2A-AR subtype in DRG neurons (Bavencoffe et al., 2010; Gu et al., 2015; Lee et al., 2020). In addition, α2A-AR agonist is recently found to activate ASIC3 at neutral pH and potentiate acid-gated currents (Callejo et al., 2020). However, we observed that DEX failed to induce any membrane currents at neutral pH when applied in CHO cells expressing ASIC3 alone. The lack of such a role of DEX may be due to the difference of the molecular backbone between DEX and guanabenz, although both of them are α2A-AR agonists.
α2A-AR is a Gi/o protein-coupled receptor that inhibits AC and reduces cAMP production upon activation (Wu et al., 1988). The present data demonstrated that Gi/o proteins and intracellular cAMP/PKA signal transduction were involved in DEX-induced inhibition of ASIC currents in rat DRG neurons. First, preventing Gi/o recruitment by PTX treatment blocked DEX-induced inhibition of ASIC currents. Second, DEX-induced inhibition was lack after DRG neurons treated with the cAMP analog 8Br-cAMP. Last, inhibition of intracellular PKA with H-89 mimicked the inhibiting effect of DEX on ASIC currents. Then on the base of these, DEX no longer further inhibited ASIC currents. These findings were consistent with the general notion that stimulation of α2A-ARs by DEX brings about Gi/o-mediated inhibition of AC and reduction of intracellular cAMP levels, suggesting that DEX inhibited ASIC currents through PTX-sensitive Gi/o proteins and classical AC/cAMP/PKA signaling pathway. Previous studies show that ASICs are modulated by intracellular cAMP/PKA signaling. For example, inhibition of PKA phosphorylation of ASICs decreases the amplitude of ASIC currents in cortical neurons (Chai et al., 2007). It has been shown that cAMP-dependent signaling pathway involves in the inhibition of ASIC-mediated functional activity by sumatriptan, a Gi/o protein-coupled 5-HT1D receptor agonist, in rat trigeminal ganglion neurons (Guo et al., 2018). Our previous studies found that activation of μ-opioid receptors or CB1 cannabinoid receptors inhibits ASICs in DRG neurons in a cAMP/PKA-dependent manner (Liu et al., 2012; Cai et al., 2014). These studies provide further support that the suppression of ASIC currents by DEX was dependent upon intracellular cAMP and PKA signaling pathway.
Intraplantar injection of low pH solution produces an intense flinch/shaking response in rats by activating ASICs (Deval et al., 2008; Omori et al., 2008). In the presence of AMG9810, ASICs are activated by injected protons, causing cation influx (largely Na+) and membrane depolarization in nociceptors, where nociception will occur if the depolarization is large enough to activate Nav subunits, resulting in generation of action potential that transmit nociceptive signals (Pattison et al., 2019). The present data show that peripheral pretreatment of the DEX relieved the ASIC-mediated nociceptive behaviors in a dose-dependent manner. The DEX′ actions occurred locally rather than systematically, since the low pH- evoked nociceptive behaviors did not changed when DEX was injected into contralateral hindpaws. Local application of BRL44408, a specific α2A-AR antagonist, significantly blocked the effect of DEX on ASIC-mediated nociceptive behaviors, suggesting that the DEX exerted its analgesic effect by acting directly on α2A-AR subtypes localized on nociceptors. It has been shown that anti-noceptive effects of DEX are mainly mediated via α2A-AR subtype, since the effects are absent in α2A-AR mutant mice, but intact in α2B- and α2C-AR mutant mice (Malmberg et al., 2001). Obviously, the behavioral data corroborated the electrophysiological studies and vice versa. In this work, we used cell bodies of DRG neurons as a simple and accessible model to examine the characteristics of the membrane of peripheral terminals. Inhibition of ASICs by DEX may also occur in peripheral terminals. So it's also possible that DEX relieved low pH- evoked nociceptive behaviors by inhibiting ASICs localized in nociceptors, at least partially. DEX activated α2A-ARs and then decreased ASIC-mediated currents and action potentials in primary sensory neurons, resulting in reduction of ASIC-mediated pain.
Protons are released and result in a decrease in pH value in some pathological pain conditions, such as postoperative pain. The extracellular decreased pH is sustained for a few days after incision (Woo et al., 2004). Peripheral ASIC3-containing channels have been found to detect local changes in pH and contribute to postoperative pain (Deval et al., 2011). Intra-operative application of an ASIC3 inhibitor has a potent analgesic effect on postoperative pain (Deval et al., 2011). Clinically, intra-operative DEX decreases requirements for postoperative analgesics of various surgical patients, improves pain management in post-anesthesia care unit, and has no obvious side effects (Alhashemi and Kaki, 2004; Arain et al., 2004). The present results that DEX-induced inhibition of ASICs in primary sensory neurons provided a clue that ASICs may be therapeutic targets for peripheral DEX to relieve some types of pain, such as postoperative pain.
In summary, our results suggested DEX-induced inhibition of ASIC-mediated the electrophysiological activity and pain, revealing a novel peripheral mechanism underlying DEX analgesia. It is clinically important that local application of DEX can not only effectively relieve pain by inhibiting ASICs in primary sensory neurons, but also this route of administration would avoid side-effects that accompany systemic activation of α2A-ARs.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation
The animal study was reviewed and approved by the animal research ethics committee of Hubei University of Science and Technology. Written informed consent was obtained from the owners for the participation of their animals in this study.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
This work was supported by the National Natural Science Foundation of China (No. 81671101), and Science and Technology Research Project of Hubei Provincial Department of Education (No. Q20202804).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
α2-AR, α2 adrenergic receptor; AC, adenylate cyclase; ANOVA, analysis of variance; APs, action potentials; ASIC, acid-sensing ion channels; CHO, Chinese hamster ovary; DEX, dexmedetomidine; DRG, dorsal root ganglion; IpH, proton-gated current; PKA, protein kinase A; PTX, pertussis toxin; TRPV1, transient receptor potential vanilloid receptor-1.
Aho, M., Lehtinen, A.-M., Erkola, O., Kallio, A., and Korttila, K. (1991). The Effect of Intravenously Administered Dexmedetomidine on Perioperative Hemodynamics and Isoflurane Requirements in Patients Undergoing Abdominal Hysterectomy. Anesthesiology 74, 997–1002. doi:10.1097/00000542-199106000-00005
Alhashemi, J. A., and Kaki, A. M. (2004). Dexmedetomidine in Combination with Morphine PCA Provides superior Analgesia for Shockwave Lithotripsy. Can. J. Anesth. 51, 342–347. doi:10.1007/bf03018237
Alvarez De La Rosa, D., Zhang, P., Shao, D., White, F., and Canessa, C. M. (2002). Functional Implications of the Localization and Activity of Acid-Sensitive Channels in Rat Peripheral Nervous System. Proc. Natl. Acad. Sci. 99, 2326–2331. doi:10.1073/pnas.042688199
Arain, S. R., Ruehlow, R. e. M., Uhrich, T. D., and Ebert, T. J. (2004). The Efficacy of Dexmedetomidine versus Morphine for Postoperative Analgesia after Major Inpatient Surgery. Anesth. Analgesia 98, 153–158. table of contents. doi:10.1213/01.ane.0000093225.39866.75
Bavencoffe, A., Gkika, D., Kondratskyi, A., Beck, B., Borowiec, A.-S., Bidaux, G., et al. (2010). The Transient Receptor Potential Channel TRPM8 Is Inhibited via the α2A Adrenoreceptor Signaling Pathway. J. Biol. Chem. 285, 9410–9419. doi:10.1074/jbc.m109.069377
Benson, C. J., Xie, J., Wemmie, J. A., Price, M. P., Henss, J. M., Welsh, M. J., et al. (2002). Heteromultimers of DEG/ENaC Subunits Form H+-gated Channels in Mouse Sensory Neurons. Proc. Natl. Acad. Sci. 99, 2338–2343. doi:10.1073/pnas.032678399
Cai, Q., Qiu, C.-Y., Qiu, F., Liu, T.-T., Qu, Z.-W., Liu, Y.-M., et al. (2014). Morphine Inhibits Acid-Sensing Ion Channel Currents in Rat Dorsal Root Ganglion Neurons. Brain Res. 1554, 12–20. doi:10.1016/j.brainres.2014.01.042
Callejo, G., Pattison, L. A., Greenhalgh, J. C., Chakrabarti, S., Andreopoulou, E., Hockley, J. R. F., et al. (2020). In Silico screening of GMQ-like Compounds Reveals Guanabenz and Sephin1 as New Allosteric Modulators of Acid-Sensing Ion Channel 3. Biochem. Pharmacol. 174, 113834. doi:10.1016/j.bcp.2020.113834
Chai, S., Li, M., Lan, J., Xiong, Z.-G., Saugstad, J. A., and Simon, R. P. (2007). A Kinase-Anchoring Protein 150 and Calcineurin Are Involved in Regulation of Acid-Sensing Ion Channels ASIC1a and ASIC2a. J. Biol. Chem. 282, 22668–22677. doi:10.1074/jbc.m703624200
Cho, H.-J., Kim, D.-S., Lee, N.-H., Kim, J.-K., Lee, K.-M., Han, K.-S., et al. (1997). Changes in the α2-adrenergic Receptor Subtypes Gene Expression in Rat Dorsal Root Ganglion in an Experimental Model of Neuropathic Pain. Neuroreport 8, 3119–3122. doi:10.1097/00001756-199709290-00022
Chung, K., Kim, H. J., Na, H. S., Park, M. J., and Chung, J. M. (1993). Abnormalities of Sympathetic Innervation in the Area of an Injured Peripheral Nerve in a Rat Model of Neuropathic Pain. Neurosci. Lett. 162, 85–88. doi:10.1016/0304-3940(93)90566-4
Deval, E., and Lingueglia, E. (2015). Acid-Sensing Ion Channels and Nociception in the Peripheral and central Nervous Systems. Neuropharmacology 94, 49–57. doi:10.1016/j.neuropharm.2015.02.009
Deval, E., Noel, J., Gasull, X., Delaunay, A., Alloui, A., Friend, V., et al. (2011). Acid-sensing Ion Channels in Postoperative Pain. J. Neurosci. 31, 6059–6066. doi:10.1523/jneurosci.5266-10.2011
Deval, E., Noël, J., Lay, N., Alloui, A., Diochot, S., Friend, V., et al. (2008). ASIC3, a Sensor of Acidic and Primary Inflammatory Pain. EMBO J. 27, 3047–3055. doi:10.1038/emboj.2008.213
Dulai, J. S., Smith, E. S. J., and Rahman, T. (2020). Acid-Sensing Ion Channel 3: An Analgesic Target. Channels (Austin) 15, 94–127. doi:10.1080/19336950.2020.1852831
Gold, M. S., Dastmalchi, S., and Levine, J. D. (1997). α 2-Adrenergic Receptor Subtypes in Rat Dorsal Root and superior Cervical Ganglion Neurons. Pain 69, 179–190. doi:10.1016/s0304-3959(96)03218-6
Gu, X.-Y., Liu, B.-L., Zang, K.-K., Yang, L., Xu, H., Pan, H.-L., et al. (2015). Dexmedetomidine Inhibits Tetrodotoxin-Resistant Nav1.8 Sodium Channel Activity through Gi/o-dependent Pathway in Rat Dorsal Root Ganglion Neurons. Mol. Brain 8, 15. doi:10.1186/s13041-015-0105-2
Guo, L., Zhao, L., Ming, P., Hong, L., Liu, A., and Li, R. (2018). Sumatriptan Inhibits the Electrophysiological Activity of ASICs in Rat Trigeminal Ganglion Neurons. Eur. J. Pharmacol. 841, 98–103. doi:10.1016/j.ejphar.2018.10.013
Holzer, P. (2009). Acid-sensitive Ion Channels and Receptors. Handb Exp. Pharmacol. 194, 283–332. doi:10.1007/978-3-540-79090-7_9
Hoy, S. M., and Keating, G. M. (2011). Dexmedetomidine. Drugs 71, 1481–1501. doi:10.2165/11207190-000000000-00000
Hunter, J. C., Fontana, D. J., Hedley, L. R., Jasper, J. R., Lewis, R., Link, R. E., et al. (1997). Assessment of the Role of α2 -adrenoceptor Subtypes in the Antinociceptive, Sedative and Hypothermic Action of Dexmedetomidine in Transgenic Mice. Br. J. Pharmacol. 122, 1339–1344. doi:10.1038/sj.bjp.0701520
Im, S. T., Jo, Y. Y., Han, G., Jo, H. J., Kim, Y. H., and Park, C. K. (2018). Dexmedetomidine Inhibits Voltage-Gated Sodium Channels via Alpha2-Adrenoceptors in Trigeminal Ganglion Neurons. Mediators Inflamm. 2018, 1782719. doi:10.1155/2018/1782719
Kamibayashi, T., Maze, M., Weiskopf, R. B., Weiskopf, R. B., and Todd, M. M. (2000). Clinical Uses of α2-Adrenergic Agonists. Anesthesiology 93, 1345–1349. doi:10.1097/00000542-200011000-00030
Kimura, M., Saito, S., and Obata, H. (2012). Dexmedetomidine Decreases Hyperalgesia in Neuropathic Pain by Increasing Acetylcholine in the Spinal Cord. Neurosci. Lett. 529, 70–74. doi:10.1016/j.neulet.2012.08.008
Kingery, W. S., Guo, T. Z., Davies, F. M., Limbird, L., and Maze, M. (2000). The α2A Adrenoceptor and the Sympathetic Postganglionic Neuron Contribute to the Development of Neuropathic Heat Hyperalgesia in Mice. Pain 85, 345–358. doi:10.1016/s0304-3959(99)00286-9
Lee, B.-M., Jang, Y., Park, G., Kim, K., Oh, S. H., Shin, T. J., et al. (2020). Dexmedetomidine Modulates Transient Receptor Potential Vanilloid Subtype 1. Biochem. Biophysical Res. Commun. 522, 832–837. doi:10.1016/j.bbrc.2019.11.146
Lee, H. G., Choi, J. I., Kim, Y. O., and Yoon, M. H. (2013). The Role of Alpha-2 Adrenoceptor Subtype in the Antiallodynic Effect of Intraplantar Dexmedetomidine in a Rat Spinal Nerve Ligation Model. Neurosci. Lett. 557, 118–122. doi:10.1016/j.neulet.2013.10.002
Li, W. G., and Xu, T. L. (2015). Acid-sensing Ion Channels: a Novel Therapeutic Target for Pain and Anxiety. Curr. Pharm. Des. 21, 885–894. doi:10.2174/1381612820666141027124506
Liu, T.-T., Wei, S., Jin, Y., Qiu, C.-Y., and Hu, W.-P. (2021). Inhibition of ASIC-Mediated Currents by Activation of Somatostatin 2 Receptors in Rat Dorsal Root Ganglion Neurons. Mol. Neurobiol. 58, 2107–2117. doi:10.1007/s12035-020-02257-x
Liu, Y. Q., Qiu, F., Qiu, C. Y., Cai, Q., Zou, P., Wu, H., et al. (2012). Cannabinoids Inhibit Acid-Sensing Ion Channel Currents in Rat Dorsal Root Ganglion Neurons. PLoS One 7, e45531. doi:10.1371/journal.pone.0045531
Mahmoud, M., and Mason, K. P. (2015). Dexmedetomidine: Review, Update, and Future Considerations of Paediatric Perioperative and Periprocedural Applications and Limitations. Br. J. Anaesth. 115, 171–182. doi:10.1093/bja/aev226
Malmberg, A. B., Hedley, L. R., Jasper, J. R., Hunter, J. C., and Basbaum, A. I. (2001). Contribution of α2 Receptor Subtypes to Nerve Injury-Induced Pain and its Regulation by Dexmedetomidine. Br. J. Pharmacol. 132, 1827–1836. doi:10.1038/sj.bjp.0704032
Mccleskey, E. W., and Gold, M. S. (1999). Ion Channels of Nociception. Annu. Rev. Physiol. 61, 835–856. doi:10.1146/annurev.physiol.61.1.835
Nicholson, R., Dixon, A. K., Spanswick, D., and Lee, K. (2005). Noradrenergic Receptor mRNA Expression in Adult Rat Superficial Dorsal Horn and Dorsal Root Ganglion Neurons. Neurosci. Lett. 380, 316–321. doi:10.1016/j.neulet.2005.01.079
Omori, M., Yokoyama, M., Matsuoka, Y., Kobayashi, H., Mizobuchi, S., Itano, Y., et al. (2008). Effects of Selective Spinal Nerve Ligation on Acetic Acid-Induced Nociceptive Responses and ASIC3 Immunoreactivity in the Rat Dorsal Root Ganglion. Brain Res. 1219, 26–31. doi:10.1016/j.brainres.2008.03.040
Pattison, L. A., Callejo, G., and St John Smith, E. (2019). Evolution of Acid Nociception: Ion Channels and Receptors for Detecting Acid. Phil. Trans. R. Soc. B 374, 20190291. doi:10.1098/rstb.2019.0291
Poree, L. R., Guo, T. Z., Kingery, W. S., and Maze, M. (1998). The Analgesic Potency of Dexmedetomidine Is Enhanced after Nerve Injury. Anesth. Analgesia 87, 941–948. doi:10.1213/00000539-199810000-00037
Reeh, P. W., and Steen, K. H. (1996). Chapter 8. Tissue Acidosis in Nociception and Pain. Prog. Brain Res. 113, 143–151. doi:10.1016/s0079-6123(08)61085-7
Shi, T.-J. S., Winzer-Serhan, U., Leslie, F., and Hökfelt, T. (2000). Distribution and Regulation of α 2-adrenoceptors in Rat Dorsal Root Ganglia. Pain 84, 319–330. doi:10.1016/s0304-3959(99)00224-9
Stone, L. S., Broberger, C., Vulchanova, L., Wilcox, G. L., Hökfelt, T., Riedl, M. S., et al. (1998). Differential Distribution of α2Aand α2CAdrenergic Receptor Immunoreactivity in the Rat Spinal Cord. J. Neurosci. 18, 5928–5937. doi:10.1523/jneurosci.18-15-05928.1998
Stone, L. S., Macmillan, L. B., Kitto, K. F., Limbird, L. E., and Wilcox, G. L. (1997). The α2aAdrenergic Receptor Subtype Mediates Spinal Analgesia Evoked by α2Agonists and Is Necessary for Spinal Adrenergic-Opioid Synergy. J. Neurosci. 17, 7157–7165. doi:10.1523/jneurosci.17-18-07157.1997
Ugawa, S., Ueda, T., Ishida, Y., Nishigaki, M., Shibata, Y., and Shimada, S. (2002). Amiloride-blockable Acid-Sensing Ion Channels Are Leading Acid Sensors Expressed in Human Nociceptors. J. Clin. Invest. 110, 1185–1190. doi:10.1172/jci0215709
Wei, S., Qiu, C. Y., Jin, Y., Liu, T. T., and Hu, W. P. (2021). TNF-alpha Acutely Enhances Acid-Sensing Ion Channel Currents in Rat Dorsal Root Ganglion Neurons via a P38 MAPK Pathway. J. Neuroinflammation 18, 92. doi:10.1186/s12974-021-02151-w
Wemmie, J. A., Taugher, R. J., and Kreple, C. J. (2013). Acid-sensing Ion Channels in Pain and Disease. Nat. Rev. Neurosci. 14, 461–471. doi:10.1038/nrn3529
Woo, Y. C., Park, S. S., Subieta, A. R., and Brennan, T. J. (2004). Changes in Tissue pH and Temperature after Incision Indicate Acidosis May Contribute to Postoperative Pain. Anesthesiology 101, 468–475. doi:10.1097/00000542-200408000-00029
Wu, J., Liu, T. T., Zhou, Y. M., Qiu, C. Y., Ren, P., Jiao, M., et al. (2017). Sensitization of ASIC3 by Proteinase-Activated Receptor 2 Signaling Contributes to Acidosis-Induced Nociception. J. Neuroinflammation 14, 150. doi:10.1186/s12974-017-0916-4
Wu, Y. Y., Goldfien, A., and Roberts, J. M. (1988). Alpha Adrenergic Stimulation Reduces Cyclic Adenosine 3′,5′-Monophosphate Generation in Rabbit Myometrium by Two Mechanisms1. Biol. Reprod. 39, 58–65. doi:10.1095/biolreprod39.1.58
Keywords: dexmedetomidine, α2 adrenergic receptor, acid-sensing ion channel, electrophysiology, dorsal root ganglion neuron, nociceptive behavior
Citation: Wei S, Qiu C-Y, Jin Y, Liu T-T and Hu W-P (2021) Dexmedetomidine Inhibits ASIC Activity via Activation of α2A Adrenergic Receptors in Rat Dorsal Root Ganglion Neurons. Front. Pharmacol. 12:685460. doi: 10.3389/fphar.2021.685460
Received: 25 March 2021; Accepted: 11 May 2021;
Published: 24 May 2021.
Edited by:
Zubair Ahmed, University of Birmingham, United KingdomReviewed by:
Ewan St. John Smith, University of Cambridge, United KingdomCopyright © 2021 Wei, Qiu, Jin, Liu and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wang-Ping Hu, d2FuZ3BpbmdfaHVAMTYzLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.