 Yan Huang1,2,3†
Yan Huang1,2,3† Xin Ni
Xin Ni
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol., 31 May 2021
Sec. Translational Pharmacology
Volume 12 - 2021 | https://doi.org/10.3389/fphar.2021.677110
This article is part of the Research TopicHydrogen sulfide: Physiology, Pharmacology and Toxicology, Volume IIView all 8 articles
The gasotransmitter H2S is involved in various physiological and pathophysiological processes. The aim of this study was to investigate the physiological functions of H2S in the lungs. In the model of mouse with genetic deficiency in a H2S natural synthesis enzyme cystathionine-γ-lyase (CSE), we found that arterial oxygen saturation (SaO2) was decreased compared with wild type mice. Hypoxyprobe test showed that mild hypoxia occurred in the tissues of heart, lungs and kidneys in Cse-/- mice. H2S donor GYY4137 treatment increased SaO2 and ameliorated hypoxia state in cardiac and renal tissues. Further, we revealed that lung blood perfusion and airway responsiveness were not linked to reduced SaO2 level. Lung injury was found in Cse-/- mice as evidenced by alveolar wall thickening, diffuse interstitial edema and leukocyte infiltration in pulmonary tissues. IL-8, IL-1β, and TNF-α levels were markedly increased and oxidative stress levels were also significantly higher with increased levels of the pro-oxidative biomarker, MDA, decreased levels of the anti-oxidative biomarkers, T-AOC and GSH/GSSG, and reduced superoxide dismutase (SOD) activity in lung tissues of Cse-/- mice compared with those of wild type mice. GYY4137 treatment ameliorated lung injury and suppressed inflammatory state and oxidative stress in lung tissues of Cse-/- mice. A decrease in SaO2 was found in normal mice under hypoxia. These mice displayed lung injury as evidenced by alveolar wall thickening, interstitial edema and leukocyte infiltration. Increased levels of inflammatory cytokines and oxidative stress were also found in lung tissues of the mice with hypoxia insult. GYY4137 treatment increased SaO2 and ameliorated lung injury, inflammation and oxidative stress. Our data indicate that endogenous H2S is an important factor in maintaining normal SaO2 by preventing oxidative stress and inflammation in the lungs.
The endogenous gaseous signaling molecule, H2S, is mainly generated from l-cysteine through the activity of enzymes, including cystathionine-γ-lyase (CSE) and cystathionine-β-synthase (CBS) in a broad spectrum of tissues. Endogenous H2S is involved in many physiological and pathophysiological processes, such as angiogenesis (Kanagy et al., 2017), glucose homeostasis (Pichette and Gagnon, 2016), neural activity (Tabassum and Jeong, 2019), vascular tone (Sun et al., 2011), and ischemia-reperfusion (I/R) injury in the brain, liver, lungs, kidneys and heart (Wu et al., 2015a). Emerging evidence indicates that H2S plays critical roles in inflammatory responses (Sun et al., 2019), oxidative stress (Tabassum and Jeong, 2019), endoplasmic reticulum stress (Wang et al., 2020), and mitochondrial biogenesis (Murphy et al., 2019).
Many studies have shown that H2S is benefit to lung injury upon to various insults such as over-ventilation (Ge et al., 2019), hyperoxia (Faller et al., 2013; Li et al., 2013), lipopolysaccharide (LPS) (Zhang et al., 2016), oleic acid-(Wang et al., 2011a), smoke (Sun et al., 2015; Guan et al., 2020), and ischemia/reperfusion (Qi et al., 2014). H2S-donating compounds remarkably alleviates acute lung injury (ALI) induced by I/R and LPS by inhibiting the inflammatory responses (Qi et al., 2014; Faller et al., 2018). H2S also inhibits production of reactive oxygen species in pulmonary tissues in hyperoxia-induced ALI (HALI) (Faller et al., 2013). However, the physiological functions of H2S in the lungs remain largely unknown to date. Madurga et al. (2015) have demonstrated that H2S plays a key role in pulmonary vascular development and lung alveolarization in mouse. We questioned whether a deficiency of H2S in adult mice leads to dysfunction in lungs. As oxygen saturation in circulation is an important parameter in evaluating pulmonary function, we investigated the arterial blood oxygen saturation (SaO2) in Cse-/- knockout mice. It was found that SaO2 was reduced in these mice, which was reversed by administration of the H2S-donating compound GYY4137. Further, we revealed that reduced SaO2 was associated with increased levels of inflammation and oxidative stress in the lungs. Moreover, we demonstrated that GYY4137 ameliorated the decreased SaO2 induced by 10% hypoxia in wild type (WT) mice and inhibited inflammatory responses and oxidative stress in the lung tissues.
Adult male C57BL/6J (8–10week-old) mice were obtained from Shanghai SLAC Laboratory Animal Co. (Shanghai, China). CSE-deficient (Cse-/-) mice with a C57BL/6J background were provided by Shanghai Biomodel Organism Co., Ltd. (Shanghai, China) and genotyped as described previously (Tan et al., 2017). All animal procedures were carried out in accordance with the guidelines for the use of laboratory animals published by the People’s Republic of China Ministry of Health (May, 2006) and with the approval of the Ethical Committee of Experimental Animals of Central South University and the Ethical Committee of Experimental Animals of Second Military Medical University. Mice were housed under controlled temperature (22 ± 2°C) and humid (50 ± 10%) conditions with regular light–dark cycles (12h) and were given a standard diet and water ad libitum.
Three sets of experiments were performed in the present study. In the first set of experiments, 12 adult male WT and 12 Cse-/- mice (8–10week-old) were randomly divided into two groups, respectively, i. e, control and GYY4137 group (6/group), Mice of GYY4137 group were administered (i.p.) with 50mg/kg GYY4137, while control mice were injected with the same volume of saline. Animals were sacrificed 24h after treatment and blood and tissues were collected. In the second set of experiments, 12 adult male WT and 12 Cse-/- mice were randomly divided into two groups (6/group), respectively. WT and Cse-/- mice were administered (i.p.) 2.8µmol/kg pinacidil or the same volume of saline as the vehicle control. Animals were sacrificed 24h after treatment and blood and tissues were collected. In the third experiment, WT mice were randomly divided into three groups (12/group); two groups of mice were treated with vehicle or GYY4137 (50mg/kg) and immediately subjected to normobaric hypoxia for 24h. The third group was placed under normoxic conditions and treated with vehicle. For hypoxia treatment, mice were placed in a hypoxia box (10% O2, 90% N2) with a standard diet and water ad libitum. GYY4137 and pinacidil were purchased from Sigma-Aldrich (St Louis, MO, United States) and dissolved in saline. The dosages of GYY4137 and pinacidil were based on previous reports (Nagai et al., 1991; Liu et al., 2014).
Mouse SaO2 was noninvasively measured using a MouseOx Plus system (STARR Life Sciences Corp. Oakmont, PA, United States), as described preciously (Sun et al., 2016). Briefly, the mice were anesthetized with pentobarbital sodium (30mg/kg, i.p.). The tail hair was shaved, and a collar clip light sensor was attached to the tail. Then, SaO2 was measured using pulse oximetry.
Tissue hypoxia was detected using the Hypoxyprobe-1 Omni Kit (Hypoxyprobe Inc. Burlington, MA, United States) as described previously (Liu et al., 2015; Sun et al., 2016). Briefly, mice were injected (i.p) with pimonidazole (60mg/kg). After 30min, tissues were collected, fixed overnight in 4% PFA, and embedded in paraffin. Paraffin sections (5μm) of the heart, lungs and kidneys were deparaffinized, rehydrated, and endogenous peroxidase was blocked with 0.3% H2O2. After heat-induced antigen retrieval, the sections were incubated with anti-hypoxyprobe antibody (1:200) overnight at 4°C. The bound antibodies were detected with 1) fluorophore-labeled secondary antibodies (1:1,000, Jackson ImmunoResearch Lab). Nuclei were counterstained with 4′6-diamidino-2-phenylindole (DAPI) (sigma-Aldrich); or 2) the biotin–streptavidin–peroxidase system (UltraSensitive-SP-kit, MaiXin Biotechnology, Fuzhou, China) using diaminobenzidine (Sigma-Aldrich) as the chromogen. Counterstaining was performed with hematoxylin.
Lung blood perfusion was evaluated using a Laser Doppler image (LDI) analyzer (Moor Instruments, Cambridge, United Kingdom) as described previously (Ono et al., 2002; Shigemura et al., 2005). Before measurement, mice were anesthetized with pentobarbital sodium (30mg/kg, i.p.) and maintained under a rodent respirator. After midline thoracotomy, the chest was opened to expose the lungs. The mice were placed under the instrument, and the lung surface was scanned with a laser. The lung blood perfusion values were recorded and then calculated using the moorFLPI-2 measurement software (Ver. 1.0).
Lung inspiratory resistance (Ri), expiratory resistance (Re), and dynamic compliance (Cdyn) were measured using the AniRes2005 lung function analysis system (SYNOL High-Tech, Beijing China). As described by Ni et al. (Ni et al., 2011) and Wang et al. (Wang et al., 2011b), mice were anesthetized with pentobarbital sodium (30mg/kg, i.p.), tracheotomized, and connected to a mechanical ventilator. The mice were then placed in a whole-body plethysmograph to measure airway pressure and compliance. Ri, Re, and Cdyn were calculated using AniRes 2005 software from the digitized signals of dynamic airway pressure (ΔP) and volume of chamber (ΔV).
Mice were sacrificed and lung tissues were collected, fixed overnight in 4% PFA, and embedded in paraffin (4mm). Sections were stained with hematoxylin and eosin (H&E) and scored by two blinded pathologists according to previously described criteria (Yang et al., 2010). Briefly, the criteria were as follows: 0, normal tissue; 1, minimal inflammatory change; 2, no obvious damage to the lung architecture; 3, thickening of the alveolar septae; 4, formation of nodules or areas of pneumonitis that distorted the normal architecture; 5, total obliteration of the field.
We determined the real-time kinetics of H2S production in pulmonary tissues using a miniaturized H2S micro-respiration sensor (Model H2S-MRCh, Unisense, Aarhus, Denmark) coupled to Unisense PA 2000 amplifier was used as previously described (Zhu et al., 2010). The pulmonary tissues were homogenized, and then homogenate was centrifuged for 5min at 5,000rpm at 4°C to remove any remaining tissue chunks. One ml analysis buffer (1mM L-cysteine and 2mM pyridoxal-5’-phosphate in PBS) at 37°C was added into a temperature-controlled microrespiration chamber (Unisense) inside a well-grounded Faraday cage. To avoid the spontaneous H2S oxidation, nitrogen was used to deoxygenate the analysis buffer in the respiratory chamber. After the sensor signals were stabilized, a volume of 50μl pulmonary protein solution (10–20mg) was injected into the chamber, real-time H2S production trace was recorded. H2S production rates were determined at the initial steepest slopes of each trace. The H2S sensor was calibrated after each experiment with freshly prepared anoxic sodium sulfide stock solution (0–100μmol/L) according to the manufacturer's manual, using the same buffer and conditions.
Pulmonary tissues were fixed in buffered formalin prior to processing the paraffin sections. Paraffin sections (5µm) were cut, rehydrated and microwaved in citric acid buffer to retrieve antigens. The specific antibodies for CSE were purchased from Santa Cruz Biotechnology (Santa Cruz Biotechnology, Inc. Santa Cruz, CA). Sections were incubated with 3% H2O2 to inhibit endogenous peroxidases, and then with 10% rabbit serum for 30min to block the unspecific antibody binding. The sections were incubated with CSE antibody (1:200; Santa Cruz Biotech.; Cat# sc-365381) in PBS containing 1% BSA for 24h at 4°C. The bound antibodies were detected with the biotin–streptavidin–peroxidase system (UltraSensitive-SP-kit, MaiXin Biotechnology, Fuzhou, China) using diaminobenzidine (Sigma-Aldrich) as chromogen. Counterstaining was performed with hemalum. Negative controls were performed by substituting primary antibody with a normal serum in same dilution.
The concentrations of IL-1β, IL-8, and TNF-α were determined using an ELISA kit (Westang Biotech Co., Ltd., Shanghai, China) according to the manufacturer’s instructions.
Lung tissues (150mg) were homogenized in cold saline and centrifuged at 1,000 × g for 15min and the supernatant was collected. The ratio of reduced/oxidized glutathione (GSH/GSSG), catalase (CAT), superoxide dismutase (SOD) activity, H2O2 concentration, and total antioxidant capacity (T-AOC) in the lung tissues were measured using a GSH/GSSG colorimetric assay kit, CAT activity assay kit, total SOD activity assay kit, H2O2 detection kit, and T-AOC detection kit, respectively. All kits were purchased from Westang Biotech Co., Ltd.
For measurement of malondialdehyde (MDA) levels, 100°mg of lung tissue was homogenized in 1ml of 1.15% KCl solution containing 0.85% NaCl. Homogenates were then centrifuged at 1,500 × g for 15min, and the supernatant was collected. The levels of MDA were determined using a MDA assay (Westang Biotech Co., Ltd.).
Isolation of mitochondria was performed using the Mitochondria Fractionation Kit (Beyotime, China). Briefly, mouse lung tissues were quickly removed and placed in beakers containing chilled (4°C) isolation media (0.25M sucrose, 10mM Tris–HCl buffer, pH 7.4, 1mM EDTA and 250μg BSA/ml. The tissues were minced and washed three times with the isolation media to remove adhering blood and 10% (w/v) homogenates were prepared using homogenizer. The nuclei and cell debris were sedimented by centrifugation at 600g for 10min and discarded. The supernatant was subjected to a further centrifugation at 10000g for 10min. Mitochondrial pellets were suspended in the isolation medium. Respiratory control ratio (RCR) was used to assess the quality of isolated mitochondria.
Mitochondrial superoxide was measured by using a MitoSOX™ Red mitochondrial superoxide indicator (Invitrogen, United States) as described previously (Wang et al., 2014). Briefly, the 5mM stock solution was prepared by adding 13μl DMSO to each tube of MitoSOX™, and then the stock solution was diluted with HBSS to a 5μM working solution. Next, 190μl MitoSOX™ working solution was added to each well of a 96-well cell culture plate, then 10μl of fresh prepared mitochondrial was added and incubated for 10min at 37°C in dark. The plates were measured by the microplate reader with the excitation light of 510nm and emission light of 580nm.
JC-1 probe (Beyotime, China) was employed to measure mitochondrial depolarization. Briefly, 180μl of diluted JC-1 working solution was added to each well of a 96-well cell plate, and then 20μl of mitochondrial solution was added. Mitochondrial membrane potentials were monitored by determining the relative amounts of dual emissions from mitochondrial JC-1 monomers and polymers. The fluorescence value was detected by a microplate reader: for JC-1 monomer with the excitation light of 490nm and emission light of 530nm; for JC-1 polymer with the excitation light of 525nm and emission light of 590nm. The ratio of fluorescence at 590 vs. 530nm emission was used for measuring the mitochondrial membrane potential.
The ATP concentrations were measured with enhanced ATP assay kit (Beyotime, China) according to the manufacture’s protocol. Briefly, 100µl of ATP working solution was added to 1.5ml EP tubes and incubated for 5min at room temperature. Next 10µl fresh prepared mitochondrial solution was transferred to the ATP working solution. And the amount of luminescence emitted was measured with a luminometer (Promega, Madison, WI, United States) immediately. The luminescence data were normalized against sample protein amounts.
Tissue samples were lyzed with cold RIPA lysis buffer (Beyotime, China) and followed by centrifuging at 12,000 × g for 15min at 4°C. The supernatants were then collected. The protein concentration was determined by Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific, MA, United States). Then, tissue extracts were mixed with 4× loading buffer containing 250mmol/L Tris-HCl, 10%SDS, 0.5% bromophenol blue, 50% glycerol and 7.5% DTT at pH 6.8. Samples were heated to 99°C for 10min before loading on a gel. The samples were separated by 10% SDS-PAGE and subsequently transferred to nitrocellulose membranes (Millipore Corp, Bedford, MA). The membranes were incubated with blocking buffer (Tris-buffered saline containing 0.1% Tween-20 and 5% skimmed milk powder) for 2h at room temperature, and then incubated with primary antibodies against CSE (Santa Cruz Biotech.; Cat# sc-365381), CBS (Santa Cruz Biotech.; Cat# sc-133208) or β-actin (Abcam; Cat# ab-8226) at 4°C overnight. After incubation with a secondary horseradish peroxidase-conjugated IgG (Santa Cruz) for 1h at room temperature, immunoblots were visualized using the enhanced chemiluminescence Western blotting detection system (Millipore). The chemiluminiscent signal from the membranes was quantified by a GeneGnome HR scanner using GeneTools software (SynGene).
Statistical analyses were performed using SPSS Ver. 20. All data are expressed as mean ± standard error of the means (SEM) and normal distribution was assessed using the Shapiro–Wilk test. Statistical significance was determined according to sample distribution and homogeneity of variance. Statistical comparisons between two groups were determined by two-tailed Student’s t-test. One-way analysis of variation (ANOVA) using Bonferroni’s post hoc test and the Kruskal–Wallis test with Dunn’s post hoc test was performed for multi-group analysis. p < 0.05 was considered statistically significant.
We previously demonstrated that H2S plays an important role in oxygen transport (Wang et al., 2021). In the present study, we found that SaO2 was notably decreased in Cse-/- mice compared with that in WT mice (Figure 1A). We next assessed tissue hypoxia in the kidney, heart and lung using the Hypoxyprobe method. Compared with WT mice, immunofluorescence analysis of Hypoxyprobe signals showed slightly elevated staining in cardiac and renal but not in pulmonary tissues of Cse-/- mice (Figure 1B). Cse-/- mice showed a remarkably reduced H2S level, as demonstrated by our and other studies (Yang et al., 2008; Liu et al., 2014; Wang et al., 2021). We then investigated whether decreased SaO2 and tissue hypoxia are attributed to H2S deficiency in Cse-/- mice. As shown in Figures 1A,B, treatment of Cse-/- mice with a slow-releasing H2S donor, GYY4137 led to an increase in SaO2 and a decrease in hypoxyprobe signals in heart and kidney of Cse-/- mice.
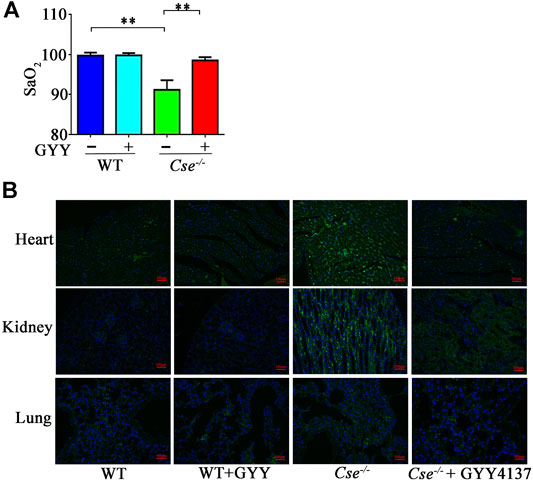
FIGURE 1. Reduced H2S levels contribute to decreased SaO2 and tissue hypoxia in Cse-/- mice. WT and Cse-/- mice were administered with saline or GYY4137 (50mg/kg). Twenty-four hours after injection, the mice were anesthetized and SaO2 level was measured. Some mice were then injected (i.p) with Hypoxyprobe solution. The animals were sacrificed at 30min after injection of Hypoxyprobe solution. Biopsies of kidneys, lungs and heart were fixed. Hypoxyprobe signals were analysis by using fluorescent analysis. (A), SaO2 level in WT and Cse-/- mice with saline or GYY4137 treatment. (B), representative images of Hypoxyprobe signals in the heart, lungs, and kidneys in WT and Cse-/- mice with saline or GYY4137 administration. Data were expressed as mean ± SEM; n = 8–12 per group; **p < 0.05.
Given that airway resistance and lung blood perfusion can affect SaO2 level, we examined these parameters. Airway responsiveness was evaluated using pulmonary ventilation. As shown in Figures 2A–C, there were no differences in Ri, Re, and Cdyn between WT and Cse-/- mice, and GYY4137 treatment had no effect on airway responsiveness.
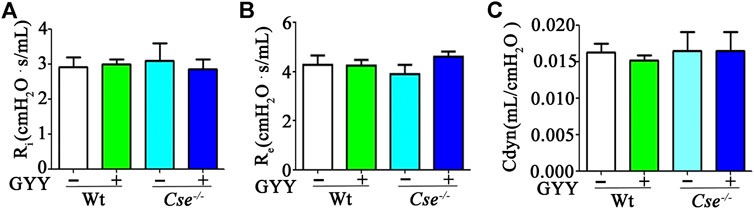
FIGURE 2. H2S deficiency does not affect lung airway resistance. WT and Cse-/- mice were administered with saline or GYY4137 (50mg/kg). Twenty-four hours later, the mice were anesthetized and then lung inspiratory resistance (Ri) (A), expiratory resistance (Re) (B), and dynamic compliance (Cdyn) (C) were analyzed by using AniRes2005 lung function analysis system. The data were expressed as Mean ± SEM; n = 8–11 per group.
H2S is an important vasodilator by controlling KATP channel activity. Cse-/- mice therefore display hypertension because increased contractility of arterials (Yang et al., 2008). H2S can increase lung blood perfusion by relaxing blood vessels in lungs. Thus, pinacidil, a KATP channel activator, was used to determine whether reduced SaO2 and tissue hypoxia in Cse-/- mice are linked to hypertension. In a previous study, we demonstrated that H2S and pinacidil could both reduce the blood pressure in Cse-/- mice (Wang et al., 2021). Here, H2S and pinacidil treatment increased lung blood perfusion in WT and Cse-/- mice (Figures 3A,B). However, pinacidil treatment had no effect on SaO2 and hypoxia in heart and kidney (Figures 3C,D), indicating that H2S maintenance of SaO2 is not associated with lung blood perfusion.
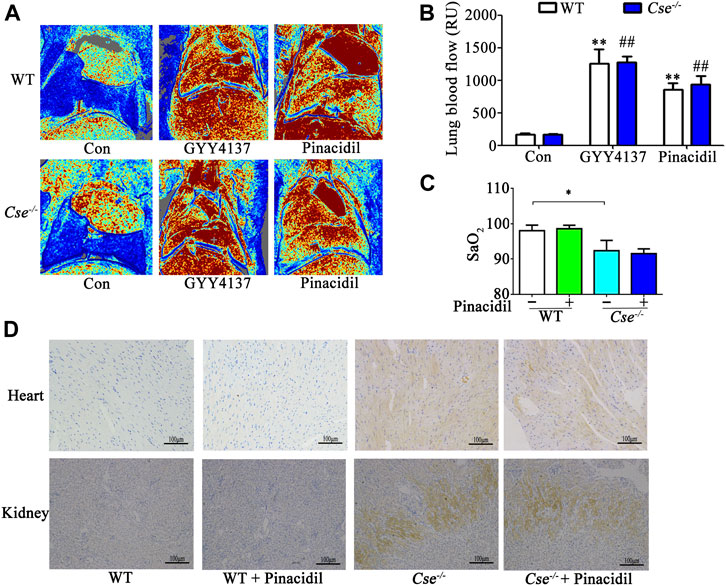
FIGURE 3. Increased lung blood flow does not affect SaO2 and tissue hypoxia in kidney and heart. WT and Cse-/- mice were administered with saline, pinacidil (2.8μmol/kg), or GYY4137 (50mg/kg). Twenty-four hours later, the mice were anesthetized and then lung blood flow and SaO2 was evaluated. Some mice were injected with Hypoxyprobe solution, then the mice were sacrificed at 30min after injection. Biopsies of kidneys and heart were fixed. Hypoxyprobe signals were detected by using immunocytochemistry analysis. (A), representative LDI analysis of lung blood flow. (B), cumulative data of lung blood flow in the mice. Data were expressed as Mean ± SEM; n = 6–10 per group; **p < 0.01 vs. WT, ##p < 0.01 vs. Cse-/-. (C), SaO2 level in WT and Cse-/- mice with saline or pinacidil treatment. Data were expressed as Mean ± SEM; n = 8–9 per group; *p < 0.05. (D), representative images of Hypoxyprobe signals in the heart and kidneys in WT and Cse-/- mice with saline or pinacidil treatment.
Gas exchange occurs at the alveolocapillary barrier. We hypothesized that CSE deficiency causes histological alternation in the lungs, thereby leading to reduction of SaO2. Since H2S produced locally in the lungs might be critical for maintenance of normal morphology, we firstly examined CSE expression distribution and capacity of H2S production in WT mice and Cse-/- mice. As shown in Figure 4A, CSE positive staining was identified in alveolar epithelial cells, vascular endothelial cells, and smooth muscle cells in WT mice. There was no obvious staining of CSE in the lungs of Cse-/- mice. The lung tissues were able to produce H2S upon to L-cysteine supply. H2S production rate was remarkably reduced in the lung tissues of Cse-/- mice (Figure 4B).
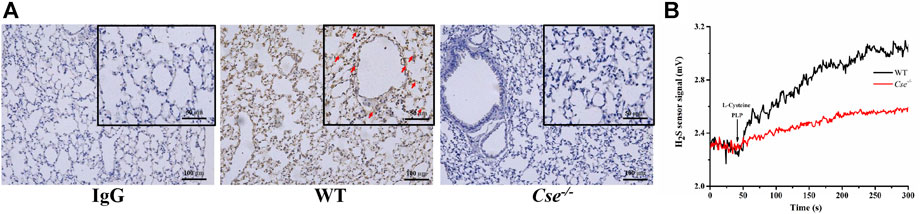
FIGURE 4. CSE expression localization and H2S production in the lung tissue of WT and Cse-/- mice. (A), CSE expression in lung tissues of WT and Cse-/- mice was analyzed by immunocytochemistry. Representative images of CSE expression in the lung tissue of WT and Cse-/- mice. Arrows indicate CSE positive staining. (B), H2S production rate in the lung tissues of WT and Cse-/- mice. Black trace, representative trace of H2S production in the homogenate of lung tissue in WT mice. Red trace, representative trace of H2S production in the homogenate of lung tissue in Cse-/- mice.
Histological analysis showed that alveolar was thickened, alveolar air space was decreased, and diffuse interstitial edema and leukocyte infiltration were found in Cse-/- mice compared with WT mice (Figure 5A). Assessment of histopathologic score showed that Cse-/- mice obtained higher histopathologic score in pulmonary tissues than WT mice (Figure 5B). GYY4137 treatment ameliorated alveolar thickening, diffuse interstitial edema, and leukocyte infiltration, and decreased histopathologic score in Cse-/- mice.
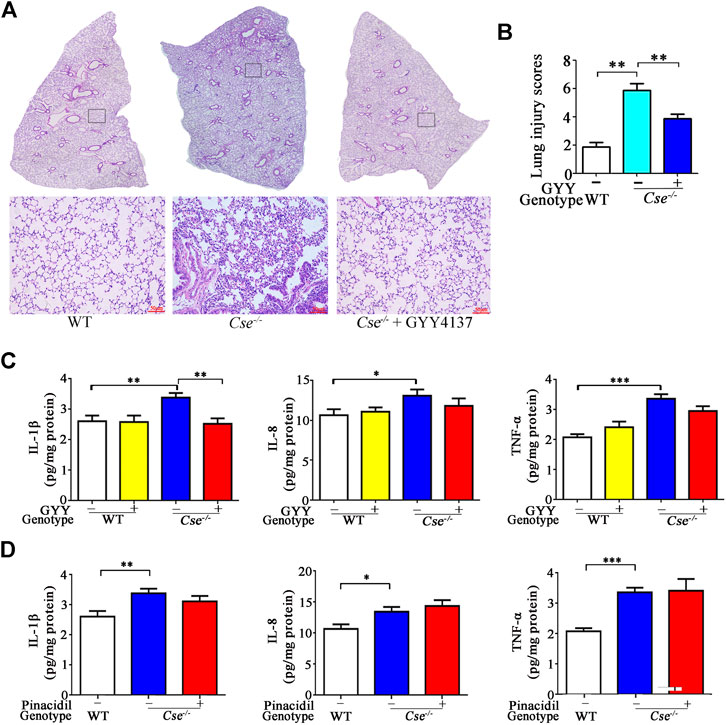
FIGURE 5. GYY4137 treatment ameliorates pathological alternation and inhibits inflammation in lung tissues in Cse-/- mice. WT and Cse-/- mice were administered with saline, GYY4137 (50mg/kg) or pinacidil (2.8μmol/kg). Twenty-four hours after injection, the mice were sacrificed, and the lung tissues were then collected for morphological analysis and determination of proinflammatory cytokines. (A), representative images of H&E staining of lung tissues of WT and Cse-/- mice with saline or GYY4137 treatment. (B), cumulative data of lung injury scores of WT and Cse-/- mice with saline or GYY4137 treatment. Data were expressed as mean ± SEM; n = 8 per group. (C), IL-8, IL-1β and TNF-α levels in lung tissues of WT and Cse-/- mice with saline or GYY4137 treatment. Data were expressed as Mean ± SEM; n = 8 per group. (D), IL-8, IL-1β, and TNF-α levels in lung tissues of WT and Cse-/- mice with saline or pinacidil treatment. Data are expressed as mean ± SEM; n = 8–11 per group; *p < 0.05, **p < 0.01, ***p < 0.001.
As histological analysis showed inflammation in the lungs of Cse-/- mice, we therefore determined inflammatory cytokines in the lungs of Cse-/- mice. As shown in Figure 5C, the pro-inflammatory cytokine IL-8, IL-1β, and TNF-α levels were elevated in lung tissues of Cse-/- mice compared with that of WT mice. Administration of GYY4137 reversed the levels of these pro-inflammatory cytokines. However, pinacidil treatment had no effect on IL-8, IL-1β, or TNF-α levels (Figure 5D).
H2S deficiency is associated with oxidative stress in various tissues (Yang et al., 2015; Bai et al., 2018; Liu et al., 2020). Our previous study had shown that CSE deficiency leads to mitochondrial dysfunction in adrenal gland and subsequently results in oxidative stress (Wang et al., 2014, Wang et al., 2018). We therefore examined mitochondrial function in the lungs of Cse-/- mice. As shown in Figure 6A, mitochondrial ATP production and membrane potential were significantly reduced whilst ROS level was significantly increased in the pulmonary tissues of Cse-/- mice compared with those of WT mice.
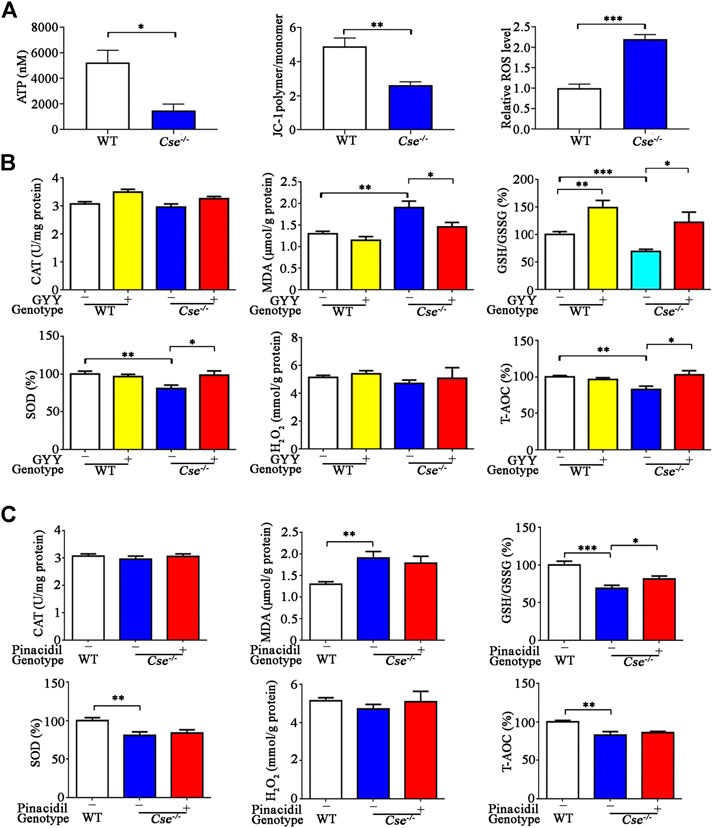
FIGURE 6. Cse-/- mice exhibit mitochondria dysfunction and oxidative stress in lung tissues and GYY4137 treatment ameliorated oxidative stress in lung tissues. (A), mitochondrial function in the lungs of WT and Cse-/- mice. Mitochondria were isolated from lung tissues. ATP production, membrane potential and ROS production were determined as described in Materials and Methods. (B), parameters of oxidative status in lung tissues of WT and Cse-/- mice with GYY4137 treatment. WT and Cse−/- mice were administered with saline or GYY4137 (50mg/kg). Mice were sacrificed at 24h after injection. The lung tissues were collected for determination of GSH/GSSG, CAT, SOD, H2O2 and T-AOC. (C), parameters of oxidative status in lung tissues of WT and Cse-/- mice with pinacidil treatment. WT and Cse-/- mice were administered with saline or pinacidil (2.8μmol//kg). After 24h, the mice were sacrificed at 24h after injection. The lung tissues were collected for determination of GSH/GSSG, CAT, SOD, H2O2, and T-AOC. Data are expressed as mean ± SEM; n = 8–11 per group; *p < 0.05, **p < 0.01, ***p < 0.001.
We then determined the oxidative status in pulmonary tissues by measuring the pro-oxidative biomarker MDA and several anti-oxidative biomarkers including H2O2, GSH/GSSG, and T-AOC, as well as the antioxidant enzymes SOD and CAT. As shown in Figure 6B, oxidative status was remarkably elevated in lung tissues of Cse-/- mice compared with those of WT mice as evidenced by an increase in MDA and a decrease in GSH/GSSG, T-AOC, and SOD activity. GYY4137 treatment reduced MDA and increased GSH/GSSG, T-AOC, and SOD level in lung tissues of Cse-/- mice. As blood perfusion in the lung might be associated with oxidative stress, we examined whether pinacidil affected these responses. As shown in Figure 6C, pinacidil treatment did not affect the levels of MDA, T-AOC, and SOD, but increased the GSH/GSSG level in Cse-/- mice.
Prior studies have demonstrated that H2S production in tissues and H2S levels in circulation were significantly reduced in response to hypoxia (Wu et al., 2015b). We hypothesized that decreased H2S production in the lungs could aggravate tissue damage and hypoxia. At first, we examined the CSE and CBS expression levels in pulmonary in the mice exposed to 10% O2 for 1–3days. As shown in Figure 7A, CSE, but not CBS expression was remarkably reduced in the lung tissues after hypoxia for 1 and 3days, suggesting that H2S production is reduced in the lungs upon to hypoxia insult.
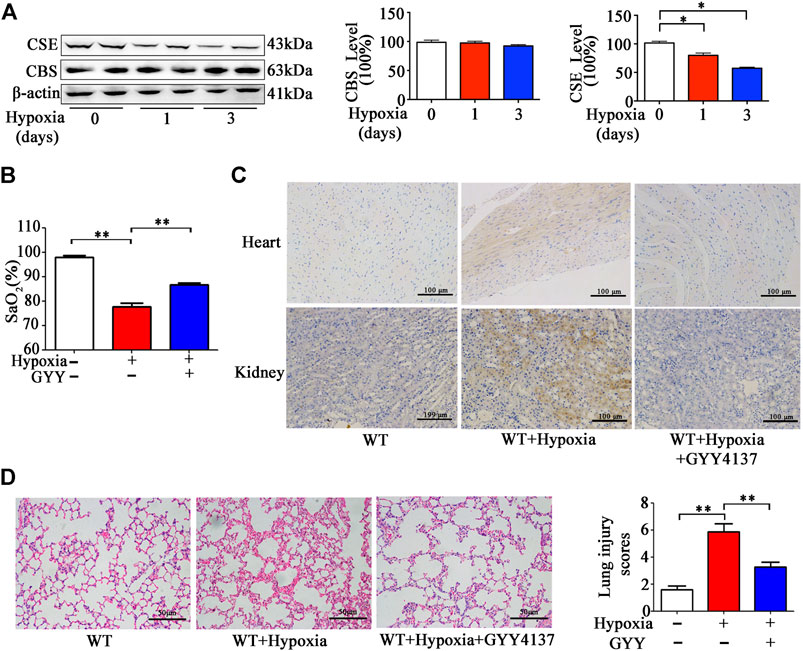
FIGURE 7. GYY4137 treatment increases SaO2 level and attenuates pathological alternation in the lungs and Hypoxyprobe signals in heart and kidneys of the mice with hypoxia insult. (A), CBS and CSE levels in lung tissues of the mice with hypoxia insult. Mice were exposed to normobaric hypoxia (10% oxygen) for the indicated times. Levels of CSE and CBS in lung tissues were determined by western blotting analysis. Representative protein bands are shown to the left of the cumulative histograms. (B–D), the effects of GYY4137 on SaO2 and lung morphology in the mice with hypoxia insult. Mice were treated with GYY4137 (50mg/kg) and immediately subjected to normobaric hypoxia for 24h. Then, the mice were anesthetized for determination of SaO2 level. (B), Some mice were sacrificed at 30min after injection of Hypoxyprobe solution. Biopsies of kidneys, and heart were fixed for Hypoxyprobe signals detection by using immunocytochemistry. (C), panel: representative images of Hypoxyprobe signals in the hearts and kidneys. The lung tissues were collected and fixed for H&E staining and lung injury scores were then calculated. (D), Data are expressed as mean ± SEM; n = 8–11 per group; *p < 0.05, **p < 0.01.
Then we investigated the effects of H2S donor treatment on SaO2, histological injury, inflammation and oxidative status in pulmonary tissues of the mice upon to hypoxia insult. As expected, SaO2 level was decreased in the mice under hypoxia (10% O2) for 24h (Figure 7B). SaO2 level was higher in the mice with GYY4137 treatment compared those with vehicle treatment. Hypoxyprobe signals in lungs, heart and kidneys were ameliorated by GYY4137 treatment (Figure 7C). Histological analysis showed that leukocyte infiltration and interstitial edema occurred, and alveolar space was diminished in pulmonary tissues in the mice exposed to 10% O2 for 24h. GYY4137 treatment significantly attenuated these pathological changes and reduced the lung pathological injury scores (Figure 7D).
In consistence with prior studies (Li et al., 2020), inflammatory cytokines IL-1β, IL-8, and TNF-α levels were significantly increased in lung tissues of the mice 10% O2 for 24h. GYY4137 treatment decreased the levels of these proinflammatory cytokines (Figure 8A). As expected, oxidative stress occurred in lung tissues in the mice under hypoxia. The pro-oxidative biomarker MDA was increased and the anti-oxidative biomarkers H2O2 and SOD were significantly decreased in the mice exposed to hypoxia (Figure 8B). GYY4137 administration reversed the above effects. However, GSH/GSSG level was not significantly changed in hypoxia group compared with normoxia group. GYY4137 administration increased GSH/GSSG level.
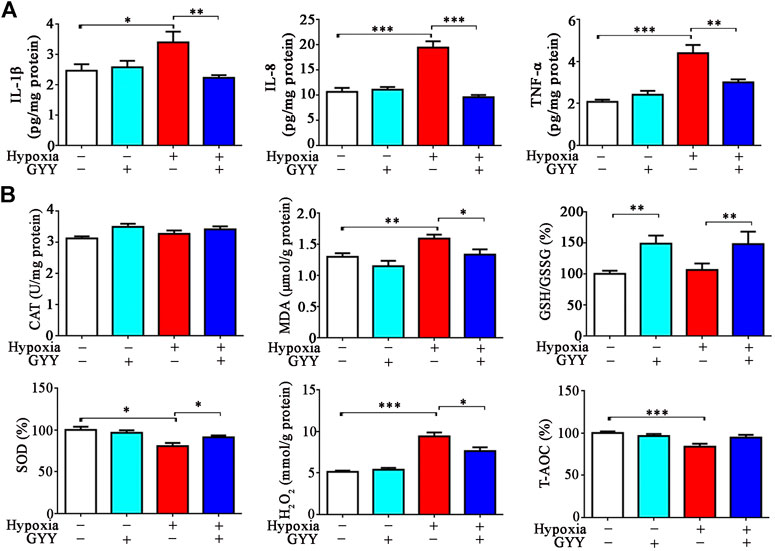
FIGURE 8. GYY4137 treatment reduces the levels of proinflammatory cytokines and improves oxidative stress in lung tissues of the mice with hypoxia insult. H2S alleviates hypoxia-induced inflammation and oxidative stress in the lungs. Mice were treated with GYY4137 (50mg/kg) and immediately subjected to normobaric hypoxia for 24h. Mice were then sacrificed, and lungs tissues were collected for determination of proinflammatory cytokine levels and oxidative stress parameters. (A), IL-8, IL-1β, and TNF-α levels in lung tissues. (B), GSH/GSSG, CAT, SOD, H2O2 and T-AOC in lung tissues. Data are expressed as mean ± SEM; n = 8–11 per group; *p < 0.05, **p < 0.01, ***p < 0.001.
In this study, we have demonstrated that H2S is an important factor in maintenance of SaO2 under physiological condition. These effects are associated with its protection of the lungs against oxidative stress and inflammatory responses.
SaO2, the ratio of oxygenated hemoglobin (HbO2) and total hemoglobin, in the range of 96–100% is considered normal. In this study, we found that SaO2 in Cse-/- mice was around 90.48%, indicating that the mice were underwent mild hypoxia. Hypoxyprobe test confirmed that hypoxia occurred in tissues such as heart and kidney in Cse-/- mice. Abnormal SaO2 is associated with many pathophysiological processes, in particular, pulmonary disorders, such as abnormal lung blood perfusion, impaired pulmonary ventilation function and acute and chronic lung injury. In humans, reduced SaO2 is likely to be due to smoking or pollution-related chronic obstructive airway pulmonary disease (COPD) or early interstitial lung disease and pulmonary fibrosis. Congenital heart disease can change the pathway of blood through the heart and decrease blood perfusion in the lungs to reduce gas exchange (Rhodes et al., 2008). In the present study, we revealed that lung airway resistance was not changed in Cse-/- mice, and increased blood perfusion did not affect SaO2 level in Cse-/- mice. However, lung injury including thickened alveolar wall, diffuse interstitial edema and leukocyte infiltration occurred in Cse-/- mice, which suggests that histological alternation in the lungs contributes to reduced SaO2 level in these mice.
The dysregulation of redox homeostasis and chronic inflammatory processes represent interdependent factors implicated in the pathogenesis of multiple diseases, including cardiovascular diseases, neurodegenerative diseases, chronic lung disease, and diabetes (Biswas, 2016). Both oxidative stress and inflammation are orchestrated to accentuate each other and induce progressive damage. Currently, H2S generation has been reported to be involved in a variety of acute and chronic inflammatory lung diseases, such as ALI (Ali et al., 2018) and COPD (Sun et al., 2015). The protective role of H2S in lung disease is associated with anti-inflammatory and antioxidant activities. H2S treatment can alleviate lung injury induced by LPS (Zhang et al., 2016; Ali et al., 2018), ventilation (Ge et al., 2019) and smoking (Sun et al., 2015) by regulating inflammation and oxidative stress. Our previous study has demonstrated that circulatory H2S level is decreased in Cse-/- mice (Wang et al., 2021), which has been supported elsewhere (Liu et al., 2014; Wang et al., 2021). In the present study, we revealed that pathological changes in the lung tissues including interstitial edema, leukocyte infiltration, inflammation, and redox imbalance were reversed by administration of GYY4137 in Cse-/- mice. These data suggest that H2S is an important factor to suppress inflammation and oxidative stress in the lungs, thereby maintaining physiological homeostasis of the lungs.
CSE utilizes L-cysteine as a substrate to generate H2S. Bibli et al. (Bibli et al., 2019) demonstrated that CSE deficiency in endothelial cells was associated with endothelial dysfunction, which was reversed by treatment with H2S donor. Zhang et al.(2013) showed that deletion of CSE promotes ovalbumin-induced airway hyper-responsiveness and aggravates airway inflammation, which is alleviated by treatment with the H2S donor NaHS. In a cell model, CSE knockdown and administration of the CSE inhibitor PAG significantly enhances ox-LDL-induced TNF-α generation, which is inhibited by exogenous H2S (Wang et al., 2013). Our study showed that CSE was mainly expressed alveolar epithelial cells and vascular endothelial cells in the lungs. As mentioned, H2S donor could inhibit inflammation, and oxidative stress in the lungs of Cse-/- mice. Taken together, it might suggest that H2S contributes to the physiological functions of CSE in the lungs.
It is known that mitochondria dysfunction can lead to oxidative stress and vice versa. Our previous studies have shown that CSE and CBS dysregulation and reduced H2S generation result in mitochondria dysfunction and subsequently lead to oxidative stress in adrenal glands in mice (Wang et al., 2014; 2018). Some studies also demonstrated that H2S metabolism contributes to maintenance of mitochondria function in various tissues (Guo et al., 2012; Módis et al., 2016; Meng et al., 2018; Murphy et al., 2019). Consistently, it was found that mitochondria dysfunction occurred in the pulmonary tissues in Cse-/- mice as evidenced by reduced membrane potential and ATP production and increased ROS level. Thus, mitochondria injury and oxidative stress aggravate lung injury in Cse-/- mice.
We previously demonstrated that circulatory H2S levels are significantly decreased in the mice exposure to hypoxia (Wang et al., 2021). Here, we showed that CSE but not CBS level was reduced in pulmonary tissues under hypoxia, and H2S donor ameliorated lung injury, suppressed levels of proinflammatory cytokines and oxidative stress in the lungs of the mice under hypoxia. These data suggest that decreased H2S level in lung tissue disrupts the balance of pro- and anti-oxidants, enhances oxidative stress and inflammatory responses, and aggravates lung injury. Notably, H2S donor treatment could increase SaO2 level in the mice with hypoxia insult. Together, it indicates that supplementation of H2S might be a potential strategy for improving lung injury and SaO2 under hypoxia.
S-sulfhydration has been implicated to be responsible for H2S biological function (Ju et al., 2017). Protein sulfhydration of ion channels, transcription factors, and enzymes, has been shown to have protective roles. For instance, H2S can inhibit inflammation through sulfhydration of the p65 subunit of nuclear factor-kappa B (NF-κB) at cysteine-38 (Sen et al., 2012). Several studies have demonstrated that H2S could sulfhydrate Keap1 at cysteine-151, inducing the dissociation of Nrf2 from Keap1 and enhancing the nuclear translocation of Nrf2. Nrf2 binds to the antioxidant response element (ARE) to promote antioxidant gene transcription (Yang et al., 2013; Guo et al., 2014; Xie et al., 2016). Thus, it is possible that the sulfhydration of NF-κB and Keap1 may be linked to the roles of H2S in pulmonary physiological homeostasis. Nevertheless, it requires to be confirmed in the future studies.
In conclusion, our study has demonstrated that CSE deficiency leads to reduced SaO2 level and tissue hypoxia in many organs and H2S supplementation reverses SaO2 level and improves tissue hypoxia. Reduced SaO2 level is associated with lung injury but not with airway resistance and blood perfusion in the lungs in CSE deficiency mice. Endogenous H2S is involved in the physiological balance between pro- and antioxidants in pulmonary tissues, thereby protecting the lungs against oxidative stress and inflammatory responses. Our findings shed light on the role of H2S as a therapeutic agent for hypoxia.
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by The Ethical Committee of Experimental Animals of Central South University and the Ethical Committee of Experimental Animals of Second Military Medical University.
XN designed the experiments, analyzed data and supervised the study; YH, GW, and ZZ performed the most of experiments, analyzed data and wrote manuscript; ZT, NZ, and XZ assisted data analysis.
Sate Key Research and Development Program of China (2018YFC1002802), Natural Science Foundation of China (No. 8162010801) Hunan Provincial Science and Technology Department (2018RS3030).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ali, F. F., Abdel-Hamid, H. A., and M., N. D. (2018). H2S Attenuates Acute Lung Inflammation Induced by Administration of Lipopolysaccharide in Adult Male Rats. Gen. Physicol. Biophys. 37, 421–431. doi:10.4149/gpb_2018002
Bai, Y. D., Yang, Y. R., Mu, X. P., Lin, G., Wang, Y. P., Jin, S., et al. (2018). Hydrogen Sulfide Alleviates Acute Myocardial Ischemia Injury by Modulating Autophagy and Inflammation Response under Oxidative Stress. Oxid Med. Cel Longev 2018, 3402809. doi:10.1155/2018/3402809
Bibli, S.-I., Hu, J., Sigala, F., Wittig, I., Heidler, J., Zukunft, S., et al. (2019). Cystathionine γ Lyase Sulfhydrates the RNA Binding Protein Human Antigen R to Preserve Endothelial Cell Function and Delay Atherogenesis. Circulation 139, 101–114. doi:10.1161/circulationaha.118.034757
Biswas, S. K. (2016). Does the Interdependence between Oxidative Stress and Inflammation Explain the Antioxidant Paradox?. Oxid Med. Cel Longev 2016, 5698931. doi:10.1155/2016/5698931
Faller, S., G. Spassov, S., K. Zimmermann, K., W. Ryter, S., Buerkle, H., Loop, T., et al. (2013). Hydrogen Sulfide Prevents Hyperoxia-Induced Lung Injury by Downregulating Reactive Oxygen Species Formation and Angiopoietin-2 Release. Curr. Pharm. Des. 19, 2715–2721. doi:10.2174/1381612811319150006
Faller, S., Hausler, F., Goeft, A., Von Itter, M. A., Gyllenram, V., Hoetzel, A., et al. (2018). Hydrogen Sulfide Limits Neutrophil Transmigration, Inflammation, and Oxidative Burst in Lipopolysaccharide-Induced Acute Lung Injury. Sci. Rep. 8, 14676. doi:10.1038/s41598-018-33101-x
Ge, X., Sun, J., Fei, A., Gao, C., Pan, S., and Wu, Z. (2019). Hydrogen Sulfide Treatment Alleviated Ventilator-Induced Lung Injury through Regulation of Autophagy and Endoplasmic Reticulum Stress. Int. J. Biol. Sci. 15, 2872–2884. doi:10.7150/ijbs.38315
Guan, R., Wang, J., Li, D., Li, Z., Liu, H., Ding, M., et al. (2020). Hydrogen Sulfide Inhibits Cigarette Smoke-Induced Inflammation and Injury in Alveolar Epithelial Cells by Suppressing PHD2/HIF-1α/MAPK Signaling Pathway. Int. Immunopharmacology 81, 105979. doi:10.1016/j.intimp.2019.105979
Guo, C., Liang, F., Shah Masood, W., and Yan, X. (2014). Hydrogen Sulfide Protected Gastric Epithelial Cell from Ischemia/reperfusion Injury by Keap1 S-Sulfhydration, MAPK Dependent Anti-apoptosis and NF-Κb Dependent Anti-inflammation Pathway. Eur. J. Pharmacol. 725, 70–78. doi:10.1016/j.ejphar.2014.01.009
Guo, W., Kan, J. T., Cheng, Z. Y., Chen, J. F., Shen, Y. Q., Xu, J., et al. (2012). Hydrogen Sulfide as an Endogenous Modulator in Mitochondria and Mitochondria Dysfunction. Oxid Med. Cel Longev 2012, 878052. doi:10.1155/2012/878052
Ju, Y., Fu, M., Stokes, E., Wu, L., and Yang, G. (2017). H₂S-Mediated Protein S-Sulfhydration: A Prediction for its Formation and Regulation. Molecules 22. doi:10.3390/molecules22081334
Kanagy, N. L., Szabo, C., and Papapetropoulos, A. (2017). Vascular Biology of Hydrogen Sulfide. Am. J. Physiology-Cell Physiol. 312, C537–C549. doi:10.1152/ajpcell.00329.2016
Li, H.-D., Zhang, Z.-R., Zhang, Q.-X., Qin, Z.-C., He, D.-M., and Chen, J.-S. (2013). Treatment with Exogenous Hydrogen Sulfide Attenuates Hyperoxia-Induced Acute Lung Injury in Mice. Eur. J. Appl. Physiol. 113, 1555–1563. doi:10.1007/s00421-012-2584-5
Li, X., Berg, N. K., Mills, T., Zhang, K., Eltzschig, H. K., and Yuan, X. (2020). Adenosine at the Interphase of Hypoxia and Inflammation in Lung Injury. Front. Immunol. 11, 604944. doi:10.3389/fimmu.2020.604944
Liu, Q., Hu, T., He, L., Huang, X., Tian, X., Zhang, H., et al. (2015). Genetic Targeting of Sprouting Angiogenesis Using Apln-CreER. Nat. Commun. 6, 6020. doi:10.1038/ncomms7020
Liu, Y., Yang, R., Liu, X., Zhou, Y., Qu, C., Kikuiri, T., et al. (2014). Hydrogen Sulfide Maintains Mesenchymal Stem Cell Function and Bone Homeostasis via Regulation of Ca2+ Channel Sulfhydration. Cell Stem Cell 15, 66–78. doi:10.1016/j.stem.2014.03.005
Liu, Z., Wang, X., Li, L., Wei, G., and Zhao, M. (2020). Hydrogen Sulfide Protects against Paraquat-Induced Acute Liver Injury in Rats by Regulating Oxidative Stress, Mitochondrial Function, and Inflammation. Oxid Med. Cel Longev 2020, 6325378. doi:10.1155/2020/6325378
Madurga, A., Golec, A., Pozarska, A., Ishii, I., Mižíková, I., Nardiello, C., et al. (2015). The H2S-Generating Enzymes Cystathionine β-synthase and Cystathionine γ-lyase Play a Role in Vascular Development during normal Lung Alveolarization. Am. J. Physiology-Lung Cell Mol. Physiol. 309, L710–L724. doi:10.1152/ajplung.00134.2015
Meng, G., Liu, J., Liu, S., Song, Q., Liu, L., Xie, L., et al. (2018). Hydrogen Sulfide Pretreatment Improves Mitochondrial Function in Myocardial Hypertrophy via a SIRT3-dependent Manner. Br. J. Pharmacol. 175, 1126–1145. doi:10.1111/bph.13861
Módis, K., Ju, Y., Ahmad, A., Untereiner, A. A., Altaany, Z., Wu, L., et al. (2016). S- Sulfhydration of ATP Synthase by Hydrogen Sulfide Stimulates Mitochondrial Bioenergetics. Pharmacol. Res. 113 (Pt A), 116–124. doi:10.1016/j.phrs.2016.08.023
Murphy, B., Bhattacharya, R., and Mukherjee, P. (2019). Hydrogen Sulfide Signaling in Mitochondria and Disease. FASEB J. 33, 13098–13125. doi:10.1096/fj.201901304r
Nagai, H., Kitagaki, K., Goto, S., Suda, H., and Koda, A. (1991). Effect of Three Novel K+ Channel Openers, Cromakalim, Pinacidil and Nicorandil on Allergic Reaction and Experimental Asthma. Jpn. J. Pharmacol. 56, 13–21. doi:10.1016/s0021-5198(19)39892-0
Ni, X., Li, X., Fang, X., Li, N., Cui, W., Zhang, B., et al. (2011). Kidins220/ARMS Contributes to Airway Inflammation and Hyper-Responsiveness in OVA-Sensitized Mice. Respir. Physiol. Neurobiol. 175, 97–103. doi:10.1016/j.resp.2010.09.012
Ono, M., Sawa, Y., Matsumoto, K., Nakamura, T., Kaneda, Y., and Matsuda, H. (2002). In Vivo gene Transfection with Hepatocyte Growth Factor via the Pulmonary Artery Induces Angiogenesis in the Rat Lung. Circulation 106, I264–I269. doi:10.1161/01.cir.0000032879.55215.f4
Pichette, J., and Gagnon, J. (2016). Implications of Hydrogen Sulfide in Glucose Regulation: How H2S Can Alter Glucose Homeostasis through Metabolic Hormones. Oxid Med. Cel Longev 2016, 3285074. doi:10.1155/2016/3285074
Qi, Q. Y., Chen, W., Li, X. L., Wang, Y. W., and Xie, X. H. (2014). H₂S Protecting against Lung Injury Following Limb Ischemia-Reperfusion by Alleviating Inflammation and Water Transport Abnormality in Rats. Biomed. Environ. Sci. 27, 410–418. doi:10.3967/bes2014.070
Sen, N., Paul, B. D., Gadalla, M. M., Mustafa, A. K., Sen, T., Xu, R., et al. (2012). Hydrogen Sulfide-Linked Sulfhydration of NF-Κb Mediates its Antiapoptotic Actions. Mol. Cel 45, 13–24. doi:10.1016/j.molcel.2011.10.021
Shigemura, N., Sawa, Y., Mizuno, S., Ono, M., Ohta, M., Nakamura, T., et al. (2005). Amelioration of Pulmonary Emphysema by In Vivo Gene Transfection with Hepatocyte Growth Factor in Rats. Circulation 111, 1407–1414. doi:10.1161/01.cir.0000158433.89103.85
Sun, H. J., Wu, Z. Y., Nie, X. W., and Bian, J. S. (2019). Role of Endothelial Dysfunction in Cardiovascular Diseases: The Link between Inflammation and Hydrogen Sulfide. Front. Pharmacol. 10, 1568. doi:10.3389/fphar.2019.01568
Sun, K., Zhang, Y., D'alessandro, A., Nemkov, T., Song, A., Wu, H., et al. (2016). Sphingosine-1-phosphate Promotes Erythrocyte Glycolysis and Oxygen Release for Adaptation to High-Altitude Hypoxia. Nat. Commun. 7, 12086. doi:10.1038/ncomms12086
Sun, Y., Tang, C. S., Du, J. B., and Jin, H. F. (2011). Hydrogen Sulfide and Vascular Relaxation. Chin. Med. J. (Engl) 124, 3816–3819. doi:10.3760/cma.j.issn.0366-6999.2011.22.038
Sun, Y., Wang, K., Li, M. X., He, W., Chang, J. R., Liao, C. C., et al. (2015). Metabolic Changes of H2S in Smokers and Patients of COPD Which Might Involve in Inflammation, Oxidative Stress and Steroid Sensitivity. Sci. Rep. 5, 14971. doi:10.1038/srep14971
Tabassum, R., and Jeong, N. Y. (2019). Potential for Therapeutic Use of Hydrogen Sulfide in Oxidative Stress-Induced Neurodegenerative Diseases. Int. J. Med. Sci. 16, 1386–1396. doi:10.7150/ijms.36516
Tan, B., Jin, S., Sun, J., Gu, Z., Sun, X., Zhu, Y., et al. (2017). New Method for Quantification of Gasotransmitter Hydrogen Sulfide in Biological Matrices by LC-MS/MS. Sci. Rep. 7, 46278. doi:10.1038/srep46278
Wang, C., Wang, H. Y., Liu, Z. W., Fu, Y., and Zhao, B. (2011a). Effect of Endogenous Hydrogen Sulfide on Oxidative Stress in Oleic Acid-Induced Acute Lung Injury in Rats. Chin. Med. J. (Engl) 124, 3476–3480. doi:10.3760/cma.j.issn.0366-6999.2011.21.007
Wang, C.-N., Liu, Y.-J., Duan, G.-L., Zhao, W., Li, X.-H., Zhu, X.-Y., et al. (2014). CBS and CSE Are Critical for Maintenance of Mitochondrial Function and Glucocorticoid Production in Adrenal Cortex. Antioxid. Redox Signaling 21, 2192–2207. doi:10.1089/ars.2013.5682
Wang, C., Du, J., Du, S., Liu, Y., Li, D., Zhu, X., et al. (2018). Endogenous H 2 S Resists Mitochondria-Mediated Apoptosis in the Adrenal Glands via ATP5A1 S-Sulfhydration in Male Mice. Mol. Cell Endocrinol. 474, 65–73. doi:10.1016/j.mce.2018.02.011
Wang, G., Huang, Y., Zhang, N., Liu, W., Wang, C., Zhu, X., et al. (2021). Hydrogen Sulfide Is a Regulator of Hemoglobin Oxygen-Carrying Capacity via Controlling 2,3-BPG Production in Erythrocytes. Oxid Med. Cel Longev 2021, 8877691. doi:10.1155/2021/8877691
Wang, H., Shi, X., Qiu, M., Lv, S., and Liu, H. (2020). Hydrogen Sulfide Plays an Important Protective Role through Influencing Endoplasmic Reticulum Stress in Diseases. Int. J. Biol. Sci. 16, 264–271. doi:10.7150/ijbs.38143
Wang, L.-m., Li, W.-h., Xu, Y.-c., Wei, Q., Zhao, H., and Jiang, X.-f. (2011b). Annexin 1-Derived Peptide Ac2-26 Inhibits Eosinophil Recruitment In Vivo via Decreasing Prostaglandin D2. Int. Arch. Allergy Immunol. 154, 137–148. doi:10.1159/000320228
Wang, X.-H., Wang, F., You, S.-J., Cao, Y.-J., Cao, L.-D., Han, Q., et al. (2013). Dysregulation of Cystathionine γ-lyase (CSE)/hydrogen Sulfide Pathway Contributes to Ox-LDL-Induced Inflammation in Macrophage. Cell Signal. 25, 2255–2262. doi:10.1016/j.cellsig.2013.07.010
Wu, B., Teng, H., Zhang, L., Li, H., Li, J., Wang, L., et al. (2015a). Interaction of Hydrogen Sulfide with Oxygen Sensing under Hypoxia. Oxid Med. Cel Longev 2015, 758678. doi:10.1155/2015/758678
Wu, D., Wang, J., Li, H., Xue, M., Ji, A., and Li, Y. (2015b). Role of Hydrogen Sulfide in Ischemia-Reperfusion Injury. Oxid Med. Cel Longev 2015, 186908. doi:10.1155/2015/186908
Xie, L., Gu, Y., Wen, M., Zhao, S., Wang, W., Ma, Y., et al. (2016). Hydrogen Sulfide Induces Keap1 S-Sulfhydration and Suppresses Diabetes-Accelerated Atherosclerosis via Nrf2 Activation. Diabetes 65, 3171–3184. doi:10.2337/db16-0020
Yang, G., An, S. S., Ji, Y., Zhang, W., and Pei, Y. (2015). Hydrogen Sulfide Signaling in Oxidative Stress and Aging Development. Oxid Med. Cel Longev 2015, 357824. doi:10.1155/2015/357824
Yang, G., Wu, L., Jiang, B., Yang, W., Qi, J., Cao, K., et al. (2008). H2S as a Physiologic Vasorelaxant: Hypertension in Mice with Deletion of Cystathionine -Lyase. Science 322, 587–590. doi:10.1126/science.1162667
Yang, G., Zhao, K., Ju, Y., Mani, S., Cao, Q., Puukila, S., et al. (2013). Hydrogen Sulfide Protects against Cellular Senescence via S-Sulfhydration of Keap1 and Activation of Nrf2. Antioxid. Redox Signaling 18, 1906–1919. doi:10.1089/ars.2012.4645
Yang, T., Mao, Y.-F., Liu, S.-Q., Hou, J., Cai, Z.-Y., Hu, J.-Y., et al. (2010). Protective Effects of the Free Radical Scavenger Edaravone on Acute Pancreatitis-Associated Lung Injury. Eur. J. Pharmacol. 630, 152–157. doi:10.1016/j.ejphar.2009.12.025
Zhang, G., Wang, P., Yang, G., Cao, Q., and Wang, R. (2013). The Inhibitory Role of Hydrogen Sulfide in Airway Hyperresponsiveness and Inflammation in a Mouse Model of Asthma. Am. J. Pathol. 182, 1188–1195. doi:10.1016/j.ajpath.2012.12.008
Zhang, H.-X., Liu, S.-J., Tang, X.-L., Duan, G.-L., Ni, X., Zhu, X.-Y., et al. (2016). H2S Attenuates LPS-Induced Acute Lung Injury by Reducing Oxidative/Nitrative Stress and Inflammation. Cell Physiol Biochem 40, 1603–1612. doi:10.1159/000453210
Keywords: hydrogen sulfide, lung, cystathionine-gamma-lyase, SaO2, hypoxia
Citation: Huang Y, Wang G, Zhou Z, Tang Z, Zhang N, Zhu X and Ni X (2021) Endogenous Hydrogen Sulfide Is an Important Factor in Maintaining Arterial Oxygen Saturation. Front. Pharmacol. 12:677110. doi: 10.3389/fphar.2021.677110
Received: 07 March 2021; Accepted: 18 May 2021;
Published: 31 May 2021.
Edited by:
Junbao Du, Peking University First Hospital, ChinaReviewed by:
Yuan Xinxu, Virginia Commonwealth University, United StatesCopyright © 2021 Huang, Wang, Zhou, Tang, Zhang, Zhu and Ni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xin Ni, eGlubmkyMDE4QGNzdS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.