 Ekaterina Yu Podyacheva
Ekaterina Yu Podyacheva Ekaterina A. Kushnareva
Ekaterina A. Kushnareva Andrei A. Karpov
Andrei A. Karpov Yana G. Toropova
Yana G. Toropova
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol., 03 June 2021
Sec. Cardiovascular and Smooth Muscle Pharmacology
Volume 12 - 2021 | https://doi.org/10.3389/fphar.2021.670479
Today the pharmacological possibilities of treating cancer are expanding and as a result, life expectancy is increasing against the background of chemotherapy and supportive treatment. In the conditions of successful antitumor treatment, complications associated with its toxic effect on healthy tissues and organs began to come to the fore. Anthracycline cardiomyopathy was the first serious cardiovascular complication to draw the attention of oncologists and cardiologists around the world. Anthracycline drugs such as doxorubicin, epirubicin, idarubicin are still widely used in oncological practice to treat a wide range of solid and hematological malignancies. Doxorubicin-induced cardiomyopathy is closely associated with an increase in oxidative stress, as evidenced by reactive oxygen species (ROS) nduced damage such as lipid peroxidation, and decreased levels of antioxidants. Myofibrillar destruction and dysregulation of intracellular calcium are also important mechanisms, usually associated with doxorubicin-induced cardiotoxicity. Despite the abundance of data on various mechanisms involved in the implementation of doxorubicin-induced cardiotoxicity, a final understanding of the mechanism of the development of doxorubicin cardiomyopathy has not yet been formed. It poses the most significant challenges to the development of new methods of prevention and treatment, as well as to the unambiguous choice of a specific treatment regimen using the existing pharmacological tools. In order to resolve these issues new models that could reflect the development of the chemotherapy drugs effects are needed. In this review we have summarized and analyzed information on the main existing models of doxorubicin cardiomyopathy using small laboratory animals. In addition, this paper discusses further areas of research devoted to the development and validation of new improved models of doxorubicin cardiomyopathy suitable both for studying the mechanisms of its implementation and for the preclinical drugs effectiveness assessment.
According to the global statistics, the neoplasm is one of the main reasons for morbidity and mortality worldwide. Frequently encountered issues are seeking medical care in late stages of the disease and inaccessibility of diagnosis. Despite the active development of various approaches to cancer treatment, systemic chemotherapy remains the mainstay cancer treatment. The pharmacological development in the second half of the 20th century led to a stable annual increase in the life expectancy of cancer patients (Markham et al., 2020). However, within the conditions of successful cancer treatment, complications associated with its toxic effect on healthy tissues and organs began to come to the fore. Anthracycline cardiomyopathy was the first serious cardiovascular complication to have been noticed by oncologists and cardiologists worldwide.
Anthracyclines [doxorubicin (DOX), epirubicin, idarubicin] are still widely used as a chemotherapeutic agent in the treatment of solid and hematological malignancies. During the study of anthracyclines toxic effects, a dose-dependent nature of cardiovascular complications was discovered. Thus, the incidence of heart failure is approximately 3–5% with a cumulative dose of 400 mg/m2 of DOX, and the incidence of complications reaches 48% with an increase in the total dose of the resulting drug to 700 mg/m2 (Zamorano et al., 2016).
From a prognostic point of view, it is important to separate cardiovascular complications during treatment with anthracycline drugs according to the timing of the development. To date, acute cardiotoxicity has been distinguished, which developed at the time of chemotherapeutic treatment and manifested in the first year after completion of the anthracyclines administration. At the same time, chronic cardiotoxicity develops after the anticancer treatment (Hole et al., 2013). Сhronic cardiovascular complications are of great importance in the prognosis of cancer patients as they can lead to a significant deterioration in the quality and expectancy of life of those who have been successfully treated for malignant neoplasms.
Today the mechanisms of the damaging effect of anthracyclines on the myocardium are being actively studied in various experimental models. There is evidence supporting the role of oxidative stress, systemic inflammation, calcium metabolism disorders in the development of anthracycline cardiomyopathy (Zhang et al., 2012; Varela-López et al., 2019; Wallace et al., 2020). Despite a number of articles on this issue, there is no review devoted to the analysis of these models. Nonetheless, it is of vital importance when developing a model, which would comply with the modern approaches to the anticancer treatment.
It is assumed that the main mechanisms of action of anthracyclines are as follows: they are easily captured by cells and localized in the nucleus. DOX has an ability to inhibit DNA biosynthesis by DNA intercalation. This drug has a high affinity for sites containing GC base pairs and forming hydrogen bonds between DOX and guanine; it leads to the appearance of ternary DOX-DNA-topoisomerase II complexes, which activate DNA damage responses and, ultimately, cause cell death (Santos and Goldenberg, 2018). Anthracyclines prevent the re-ligation of double-stranded DNA breaks, thus “poisoning” the enzyme. A disruption of this process, ATP-dependent cleavage of DNA strands, followed by a transfer of strands through the break and its ligation, leads to the DNA damage, which, in its turn, leads to mitotic and apoptotic cell death. Topoisomerase II (Top2) falls into two subtypes: Top2α and Top2β. The key mechanism of antitumor treatment with DOX is that it targets Top2α highly expressed in rapidly proliferating malignant cells. This leads to the suppression of the formation of the Top2-DNA cleavage complex, followed by the transcription arrest, which then results in the DNA damage and, ultimately, cell death (Minotti et al., 2004; Octavia et al., 2012; Santos and Goldenberg, 2018). Top2β is more common in resting cells such as cardiomyocytes and is associated with the development of cardiomyopathy. Top2β dysfunction results in the activation of the altered P53 tumor suppressor pathway, impaired calcium leads to the suppression processing, mitochondrial dysfunction, and increased apoptosis (McGowan et al., 2017).
Mechanisms that activate external and internal pathways of apoptosis are of key importance in the modern understanding of the pathogenesis of DOX cardiomyopathy. The quinone fragment of anthracyclines can undergo redox reactions with formation of excess reactive oxygen species (ROS) in the presence of oxidoreductive enzymes such as cytochrome P450 reductase, NADH dehydrogenase, and xanthine oxidase (May et al., 1980). Moreover, the ROS formation can be induced by the release of calcium ions from the sarcoplasmic reticulum of cardiomyocytes, which increases under the influence of DOX. In its turn, oxidative stress provokes the activation of the apoptosis regulators, such as BCl-2 associated X protein (BAX), which activates the release of cytochrome C through the pores in the mitochondrial membrane. This process induces the caspase cascade of activation of the external and internal pathways of apoptosis, which ultimately leads to the death of cardiomyocytes and the impairment of the contractile function of the heart. DOX-induced apoptosis targets not only cardiomyocytes, but also endothelial cells, as indicated by the activation of caspase and the degradation of the internucleosomal DNA (Lüscher and Barton, 2000; Octavia et al., 2012). In addition, cardiotoxicity associated with DOX administration is partially mediated by changes in the high-energy phosphate pool, endothelin-1 levels, and impaired adrenergic myocardial signaling (Renu et al., 2018). The current understanding of the role of oxidative stress in the development of DOX cardiomyopathy is presented in Figure 1.
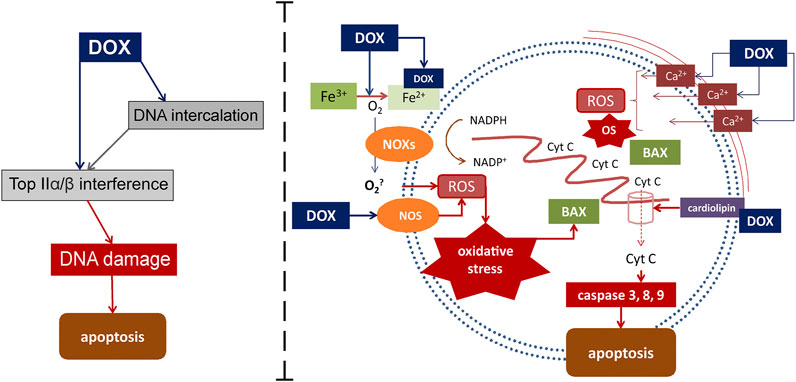
FIGURE 1. Mechanisms of DOX-induced cardiomyocyte damage. DOX targets Top2α/β. This leads to the suppression of the formation of the Top2-DNA cleavage complex, followed by the transcription arrest, which then results in the DNA damage and cell death. DOX also mediates apoptosis through interaction with Fe with subsequent active oxygen releasing and oxidative stress activation. Another way for oxidative stress activation is DOX-induced increasing of calcium ions releasing from the sarcoplasmic reticulum. Binding to cardiolipin DOX leads to mitochondrial dysfunction. Active cytochrome C releasing from mitochondrias to cytoplasm through the pores activates inner ways of apoptosis. BAX, Bcl-2-associated X protein; Cyt C-cytochrome C; DOX-doxorubicin; NOS-nitric oxide synthases; eNOS-endothelial NOS; NOXs-nicotinamide adenine dinucleotide phosphate oxidases; NADPH-nicotinamide adenine dinucleotide phosphate; ROS-reactive oxygen species.
Mitochondria are subcellular organelles which are most affected by DOX and show severe damage. This is conditioned by the fact that DOX, being a cationic drug, is retained in the inner mitochondrial membrane. Thus, an almost irreversible complex with cardiolipin is formed (Goormaghtigh et al., 1980). For the proteins in the electron transport chain to function properly, they must be bound to cardiolipin. However, it has been shown that as DOX disrupts the cardiolipin-protein interface, more superoxide (O2-) is produced (Kashfi et al., 1990; Schlame et al., 2000). These events obstruct mitochondrial (and therefore cellular) metabolism, since mitochondria generate more than 90% of the ATP used by cardiomyocytes (Ventura-Clapier et al., 2004). This functional disorder leads to ultrastructural pathological changes. In a rat model of chronic cardiomyopathy induced by DOX a general transition from erobic to anaerobic metabolism of the heart muscle was shown. This is directly related to a decrease in the oxidation of long-chain-fatty acids in the cardiac mitochondria and an increase in glucose metabolism. This shift in metabolism is a common feature of heart failure.
Starting from 1980, the first studies have showed that DOX has a strong affinity for iron (May et al., 1980). The doxorubicin-iron complex can induce lipid peroxidation by interacting with negatively charged membranes. Reduction of DOX in the presence of free iron starts the cycle of free radical formation (redox cycle). The interaction of the doxorubicinol metabolite with thiol groups of proteins exacerbates cell damage (Kappus et al., 1980; Xu et al., 2005). Under physiological conditions, free iron is insufficient to combine with DOX to such extent that it causes cardiomyopathy because the free iron content in most cells, including cardiomyocytes, is very low. More recent studies have shown that the effect of DOX on iron metabolism is mediated through proteins that sequester and bind intracellular iron. One of those mechanisms involves a formation of complexes of the doxorubicinol metabolite with the Fe-S group of cytoplasmic aconitase/IRE-BP (the iron-responsive element-binding proteins). This increases the stability of transferrin mRNA and prevents the translation of iron sequestration proteins (Minotti et al., 1998; Minotti et al., 2004). The subsequent decrease in IRE-BP leads to an increase in free iron, which can maintain the cycle of free radical generation. Thus, impaired iron sequestration remains a critical component of the DOX-induced cardiotoxicity (Nitobe et al., 2003).
NADPH is another enzyme that promotes formation of free radicals in the redox cycle (Santos and Goldenberg, 2018). It is a group of enzymes associated with the plasma membrane that serves as a source of ROS. Nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOX) help convert the quinone moiety to a semi-quinone radical that can react with oxygen to form hydrogen peroxide. NOX can be activated by many stimuli, including TNF-α, and plays a key role in cardio remodeling (Santos and Goldenberg, 2018). In addition, it has been found that Nox 2 is a major mediator of NOX-derived ROS.
Nitric oxide (NO) is also a major contributor to oxidative stress caused by DOX. NO is a vasodilator that mediates heartbeats under normal homeostatic conditions. An increased level of NO is observed in DOX-induced cardiotoxicity due to NOS (NO-synthase) isoforms, namely endothelial NOS, inducible NOS, and neuronal NOS (Nebigil and Désaubry, 2018). DOX captures electrons from NADPH by direct binding of the eNOS reductase domain, and thatcontributes to the superoxide formation.
It is generally accepted that DOX-induced oxidative stress activates apoptotic signaling leading to apoptosis of cardiomyocytes (Nitobe et al., 2003). This involves both external and internal pathways of apoptosis (Papadopoulou et al., 1999). In the DOX model, oxidative stress activates HSF-1, which produces more Hsp25. Hsp25 stabilizes p53 and increases the production of pro-apoptotic proteins (Vedam et al., 2010; Octavia et al., 2012). These mechanisms play a significant role in the development of both acute and chronic cardiotoxicity. Liu et al. demonstrated that overexpression of Hsp27 also prevents DOX-induced apoptosis and myocardial dysfunction due to the protective role of Hsp27 in regulating oxidative stress responses and maintaining mitochondrial function (Liu et al., 2007). Hsp27 is known for its cardioprotective action against ischemia/reperfusion injury. Overexpression of Hsp10 and Hsp60 similarly leads to an increase in post-translational modification of Bcl-2 proteins, shifting the balance towards the transmission of antiapoptotic signals.
DOX-induced cardiotoxicity is also associated with increased intracellular calcium levels. Dysregulation of intracellular calcium concentrations results in the ROS formation. DOX causes a release of calcium from the sarcoplasmic reticulum by increasing the likelihood of the channel transition to an open state (Holmberg and Williams, 1990). Saeki et al. suggested that the ryanodine channel has multiple binding sites for DOX, and that binding occurs regardless of whether the channel is open or closed (Keung et al., 1991; Saeki et al., 2002). Most of the intracellular calcium in cardiomyocytes is present in the sarcoplasmic reticulum. Oxidative stress can cause calcium leakage, activation of calpain, and cleavage of caspase-12.
Caspase-12 activates apoptotic pathways, and its activation depends on the dysregulation of calpain, which sends distress signals from the sarcoplasmic reticulum (Octavia et al., 2012). Calpains are proteases activated by calcium. Calpains have been found to degrade titin, which is a large protein and a key component of the cardiac sarcomere that helps maintain cardiac contractility. Therefore, prevention of calpain activity helps maintain contractility after exposure to DOX (Santos and Goldenberg, 2018). It has also been found that DOX increases the sensitivity of mitochondria to intracellular calcium. A study in rats has shown that the mitochondria of heart cells treated with DOX have a reduced ability to retain calcium (Octavia et al., 2012).
Endothelin-1 (ET-1) signaling increases cell survival signaling in cardiomyocytes, which explains its activation in heart failure (Suzuki and Miyauchi, 2001). The expression of ET-1 and the expression of its receptors are increased in the myocardium of rats with congestive heart failure, in patients with idiopathic dilated cardiomyopathy and in patients treated with DOX (Picard et al., 1998; Pieske et al., 1999; Sayed-Ahmed et al., 2001). In the vasculature the activity of ET-1 is balanced by the action of NO. However, in patients treated with DOX, NO production is impaired (May et al., 1980; Miller et al., 1993; Li et al., 2006; Nakahara et al., 2018).
Despite the abundance of data on various mechanisms involved in the implementation of DOX-induced cardiotoxicity, a final understanding of the mechanism of the DOX cardiomyopathy development has not yet been formed. This is what causes difficulties in working out new methods of prevention and treatment, as well as in the unambiguous choice of a specific treatment regimen using the existing pharmacological tools.
The review is based on the results of studies from articles and reviews published for the last 40 years. We have conducted a literature search using publicly available databases (PubMed, Google Scholar, Europe PubMed Central). All publications used while preparing this review are written in English, published in peer-reviewed journals, and contain original data on the experimental models including basic animal characteristics.
Both large and small laboratory animals are used to simulate the damaging effect of anthracycline antibiotics. Rabbits were the first animals to experience the chronic cardiotoxic effects of DOX. Maral et al. and later Jaenke have described histological and ultrastructural changes in the myocardium of rabbits treated with DOX for 3–4 months (Maral et al., 1967; Jaenke, 1974). These researchers have reported that the incidence and severity of cardiomyopathic lesions were the highest in the left ventricular wall and interventricular septum, intermediate in the atria, and the lowest in the right ventricular wall. Nephropathy (glomerular vacuolation, tubular injury, deposition of protein casts in the tubular lumen, and clinical and laboratory evidence of anephrotic syndrome with hypercholesterolemia) is also a common and important side effect of chronic DOX-induced toxicity in rabbits and other rodents. Such lesions do not develop in people treated with these DOX (Herman et al., 1985; Herman and Ferrans, 1997).
The main advantages of large laboratory animals, such as pigs, monkeys, dogs, include the similarity of their cardiovascular system anatomical structure with that of humans, similar pathological changes in the myocardium that occur during the administration of anthracyclines, as well as the possibility of conducting a wide range of functional studies characterizing the work of the heart (Herman and Ferrans, 1997). However, the large animal model has significant drawbacks, including the amount of medication needed and the technical difficulties that arise while blood sampling and performing multiple intravenous injections of DOX (anterior vena cava) (Herman and Ferrans, 1983; Herman and Ferrans, 1997). In addition, achieving a cardiotoxic effect requires a larger amount of anticancer drug and a longer period of time from the beginning of exposure to the development of signs of cardiotoxicity (Robert, 2007). These disadvantages make experiments on large models less productive and significantly more expensive.
The need for small laboratory animal’s models arose when it became necessary to study toxic properties of a number of anthracycline analogs. A model that would allow to study drugs on a fairly large sample of animals for a relatively short period of time was required, which, at the same time, would let assess the degree of myocardial damage and the effectiveness of potentially cardioprotective therapy. The most commonly used models that meet the above criteria are laboratory mice and rats.
Mettler et al. first reported in 1977 that CDF rats (Fischer) treated with DOX 1–2 mg/kg per week for 10–14 weeks showed signs of congestive heart failure. Histological changes in these animals included vacuolization and degeneration of myocytes, interstitial edema and mild fibrosis (Mettler et al., 1977). In search of the best rat model Herman et al. in 1985 compared the severity of chronic DOX cardiotoxicity in adult male rats with spontaneous hypertension and in genetically related normotensive Wistar-Kyoto rats (Herman et al., 1985). Heart lesions were noted in both strains of rats. However, at a dose level of 0.25–1 mg/kg per week for 12 weeks, spontaneously hypertensive rats were significantly more sensitive to the cardiotoxic effects of DOX than normotensive rats. The mechanism of this hypersensitivity has not been determined. Because of their sensitivity and high degree of reproducibility of heart lesions, rats with spontaneous hypertension are more suitable than other rat strains as small animal models for studying anthracycline cadiotoxicity (Herman and Ferrans, 1997). However, this model reflects only a narrow proportion of patients with severe arterial hypertension who have undergone chemotherapy.
In general, models can be divided into short-term and long-term ones, depending on the injection protocols (Robert, 2007; Nakahara et al., 2018). In short-term rat models animals receive a single dose of DOX (10–30 mg/kg) intravenously (Todorova et al., 2012). Other short-term models use 2–3.4 mg/kg of DOX every other day, six times intraperitoneally (the total dose is 12–20 mg/kg) (Zbinden et al., 1978; Lanza et al., 1989; Carvalho et al., 2010; Razmaraii et al., 2016; Cappetta et al., 2018; Aygun and Gul, 2019; Barış et al., 2019). Long-term rat models typically use 1–5 mg/kg every week for 2–12 weeks with a cumulative dose of 3–25 mg/kg (Villani et al., 1991; Sacco et al., 2001; Sayed-Ahmed et al., 2001; Rahimi Balaei et al., 2010; Hydock et al., 2012; Hole et al., 2013; Toblli et al., 2014; Kang et al., 2017; Medeiros-Lima et al., 2019; Wang et al., 2019; Chakouri et al., 2020; Sharma et al., 2020). Thus, a short-term high-dose injection model would be suitable for assessing acute cardiotoxicity, while a long-term low-dose model would be suitable for assessing chronic cardiotoxicity.
Recent studies often research models that use DOX 6 times every other day at 2.5 mg/kg (Merlet et al., 2013; Lončar-Turukalo et al., 2015; Cappetta et al., 2017a; Ahmad et al., 2017; Cappetta et al., 2017b; Elhadidy et al., 2020). Histological examination showed that this injection protocol results in irregularly spaced myocardial fibers with inflammation, edema, and leukocyte cell infiltration. Other models use 1 mg/kg of DOX for 10 days and 1.25 mg/kg of DOX 4 times a week for 4 weeks (Medeiros-Lima et al., 2019; Aykan et al., 2020). The results show that DOX significantly reduced body, heart and left ventricular weight one week after stopping DOX. This trend persists after 2 and 4 weeks. Interstitial fibrosis of the left ventricle progressively increases after the end of DOX administration. Cardiac troponin I levels increase significantly one and two weeks after the end of DOX administration increase even more after four weeks.
In a study aimed at achieving DOX-induced chronic cardiomyopathy, rats received 2.15 mg/kg of Dox for 3 weeks 7 times (Ivanová et al., 2012). The researchers noted that animals treated with DOX had lower body weight compared to the control animals (treated with saline). However, the total heart weight as well as the left ventricular weight did not change significantly. Eight weeks after the end of the treatment systolic blood pressure increased in DOX-treated rats compared to the control animals, whereas there was no difference in heart rate. Electron microscopic examination of the heart shows that DOX induces heterogeneous subcellular changes characterized by degeneration and/or loss of myofibrils, as well as cytoplasmic vacuolization of cardiomyocytes. These changes are known as structural markers of DOX cardiotoxicity. Structural disorganization of the extracellular space of the heart is also observed, which is manifested in an increase in the density of extracellular matrix proteins. Simultaneously, ultrastructural analysis shows the presence of damaged capillary endothelial cells. The damage is characterized by accidental disruption of plasmalemmal integrity, damage to mitochondria, changes in cytoplasmic density, number of ribosomes, pinocytic vesicles, and random disorganization and accumulation of chromatin, which indicates activation and dysfunction of endothelial cells. Moreover, in the hearts of rats treated with DOX the following has been noted: an increased accumulation of interstitial collagen fibers; presence of macrophages, vacuoles forming large membrane-bound spaces, Cajal cell processes and proliferating fibroblasts. All these changes appear in the fourth week after the end of the DOX administration and worsen by the eighth week (Ivanová et al., 2012).
Thus, there is still no single universal model that creates DOX chronic cardiotoxicity in rats. Table 1 presents data on the use of rat models of DOX cardiomyopathy (Table 1).
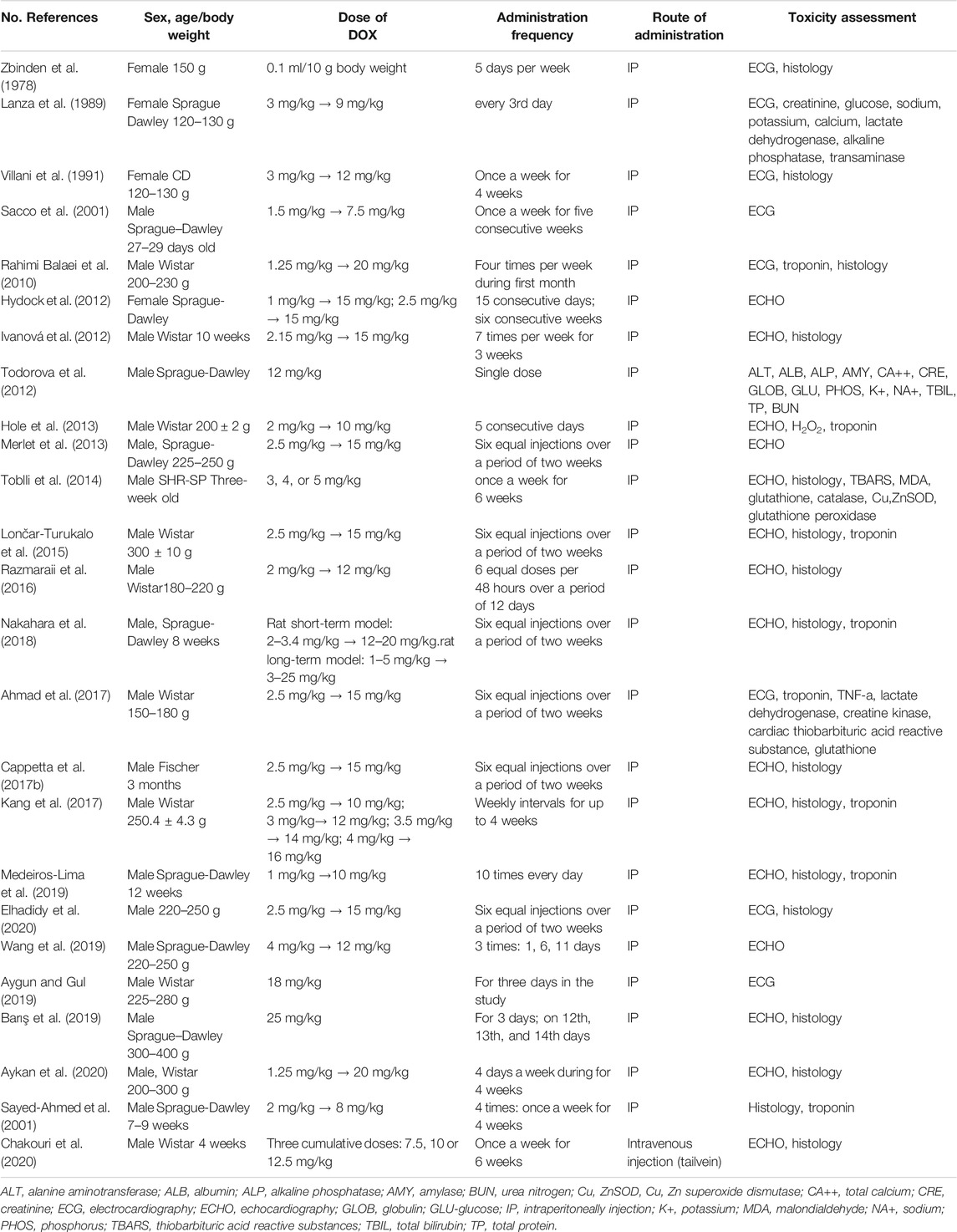
TABLE 1. Data on the modeling of doxorubicin cardiomyopathy in rats.
The first mouse DOX cardiomyopathy model was presented by Bertazzoli et al. (1979). Two administration regimens were studied: 1.4 and 4 mg/kg twice a week up to 10 administrations in total. After a low dose administration cardiomyocyte vacuolization was detected. However, with a higher dose of DOX more severe pathological changes were noted, such as atrophy, myofibrils loss, necrosis with connective tissue proliferation, etc. In addition, significantly higher involvement of other tissues and organs (kidneys, spleen and liver) was discovered. Initial attempts to establish DOX cardiomyopathy models predominantly used intravenous (IV) administration through the tail vein (Forssen and Tokes, 1981; Kanter et al., 1985; van der Vijgh et al., 1988; van Acker et al., 1996). However, this method was not convenient in the context of handling of a large number of animals and it also required specialized technical skills.
Later, intraperitoneal (IP) method of DOX administration was tested and accepted into practice (Mostafa et al., 2000; Abd-Allah et al., 2002; Li et al., 2006; Neilan et al., 2006; Pei et al., 2016). Johansen compared DOX pharmacokinetic properties using two different types of administration: IV and IP. (Johansen, 1981). These experiments showed that plasma drug concentration after 24 hours was similar in the compared groups. He also evaluated DOX concentration in different tissues—that of muscles, kidneys, liver and heart. It was shown that after 24 hours the drug concentration in tissues was also similar. This method of administration has been widely put into practice and is now the most commonly used method for DOX cardiomyopathy modeling. However, inaccurate drug administration still poses risk of damaging organs.
The year 2019 saw the emerging of a novel method of DOX administration. It involved using slow-released subcutaneous pellets with a 5 mg/kg total cumulative dose, which is released over a period of 21 days (∼0.25 mg/kg/day) (Allen et al., 2019; Naresh et al., 2020). Myocardial changes after the cumulative dose achievement were similar to those after IP administration. Therefore, this method is very innovative for developing a chronic DOX cardiomyopathy model. Nowadays other measures are taken to verify DOX cardiomyopathy besides histological changes, such as evaluation of other cardio-markers, includingtroponin, NT-proBNP and assessment of systolic function by echocardiography (Xujie et al., 2012; Sahu et al., 2016; Pillai et al., 2017; Vandenwijngaert et al., 2017; Ye et al., 2020). Information about features of mouse models is summarized in Table 2.
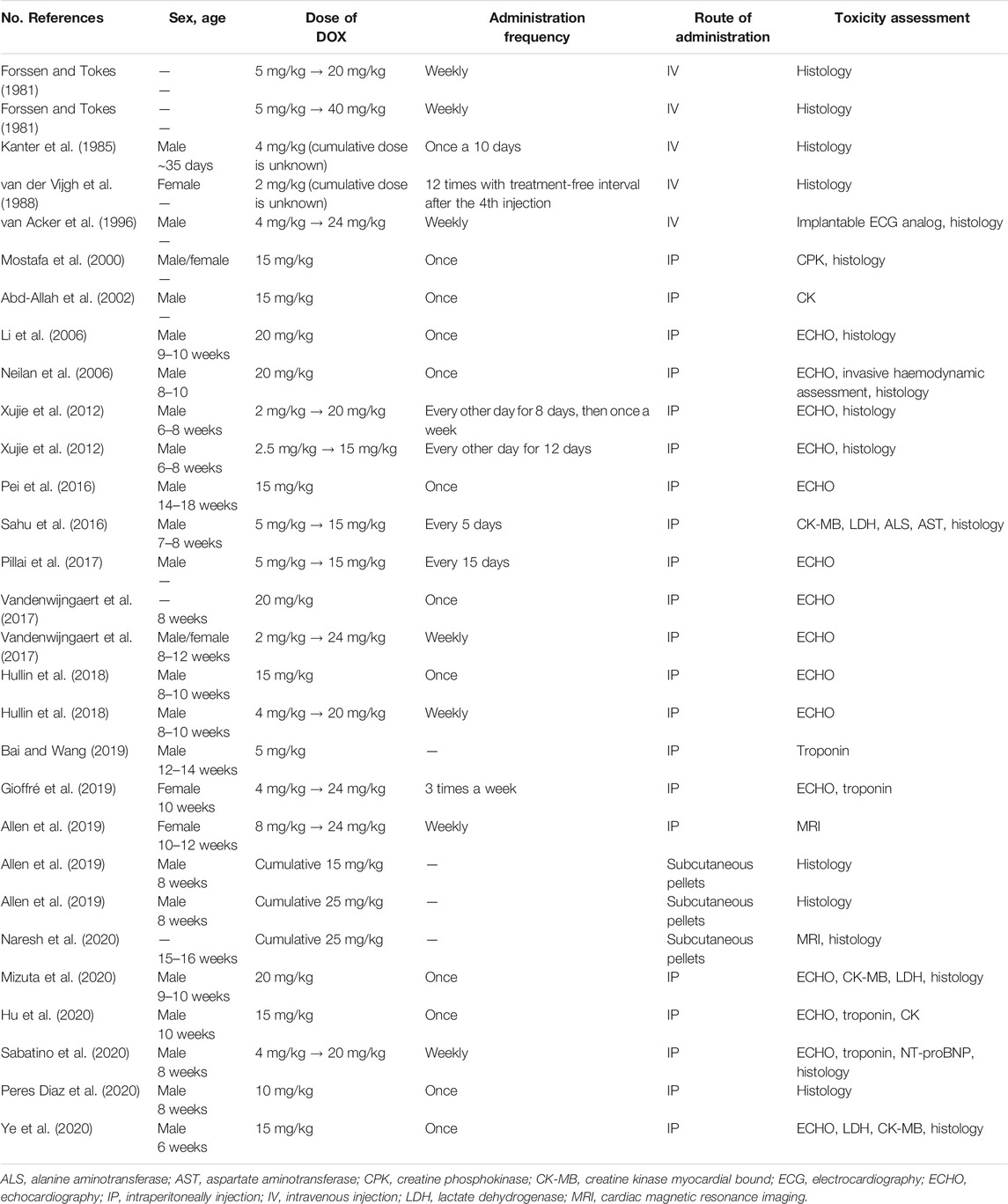
TABLE 2. Data on the modeling of doxorubicin cardiomyopathy in mouse.
In current experimental practice mouse models are used either for acute (with one-time high dose administration) or chronic (with serial administration until the cumulative dose has been achieved) DOX cardiomyopathy modeling (Hullin et al., 2018; Bai and Wang, 2019; Gioffré et al., 2019; Hu et al., 2020; Peres Diaz et al., 2020). It is worth noting that an important criterion for the model quality is the animal survival rate. It is an important factor for the dynamic evaluation and potential cardioprotective drugs examination. Mizuta et al. showed that a single dose administration with 30 mg/kg leads to a high mortality rate during the first 6 days, that is why the dose had to be decreased to 20 mg/kg to continue the dynamic research (Mizuta et al., 2020).
Sabatino et al. revealed that with 4 mg/kg DOX administration until the achievement of the cumulative dose of 20 mg/kg a significant fibrosis, fibrin exhaustion (92%), myocardial fibers disorganization (67%), tortuosity (100%) and myocardial vacuolization (100%) were observed during 5 weeks (Sabatino et al., 2020). As shown in Table 2 there is still no common experimental chronic DOX cardiomyopathy model with good reproducibility. All models vary in antitumor drug dose and administration frequency. Therefore, this fact gives us no opportunity to compare the results of different experiments run in different conditions.
In experimental practice various types of laboratory animals are widely used to create models of DOX cardiomyopathy. However, studies on small laboratory animals (rats and mice) are dominant. At the same time, the criteria for assessing myocardial damage while administering DOX do not differ significantly and include: functional assessment of the myocardium using echocardiography, assessment of markers of myocardial damage (troponin, natriuretic peptide), as well as histological examination with an assessment of the degree of damage and development of fibrosis.
It should be noted that models generated in rats and mice do not show a drastic contrast in the severity of morphological changes in the myocardium. The processes of inflammation, myocardial fibrosis, disorganization and vacuolization of myocardial fibers in both species are widely represented in the myocardium while administering DOX. However, in a number of studies on mice histological examination has not been carried out due to a small amount of myocardial tissue, which was used for other experimental purposes, such as analysis of the expression of proteins, micro-RNA, etc. This circumstance can be considered a significant limitation while selecting mice for modeling DOX cardiomyopathy.
There is a number of studies in which modeling of DOX cardiomyopathy is performed simultaneously with modeling the tumor process. In this case, the choice of mice for research seems to be more rational due to the fact that these animals are genetically more pure lines in comparison with rats. However, the rat is a preferred animal for generating a breast cancer model as simulated tumors in rats provide an adequate similarity to human neoplasms. In particular, models of breast cancer in rats in most cases are hormone sensitive, while in mouse models tumor growth does not depend on estrogen activity (Mollard et al., 2011).
It is also worth noting that in terms of calculating the administered dose of DOX per kilogram of body weight and taking into account the anthropometric characteristics of animals, experiments on mice seem to be more economical in terms of the volume of the drug administered. However, the small size of animals can present difficulties in the selection and dilution of the required single dose of DOX and in its administration. The latter issue has been successfully resolved in recent years by approbation of models with intraperitoneal injection, which demonstrated good bioavailability of the drug and sufficient development of changes in the myocardium to recognize the model of DOX cardiomyopathy as effective (Zbinden et al., 1978).
It should be noted that with the currently available large number of experimental models of DOX cardiomyopathy using small laboratory animals (mice and rats), their prevailing majority is characterized by similar approaches to verification of cardiomyopathy, including histological, biochemical, electron microscopic studies, changes in the functioning of the myocardium. In the above mentioned studies there were no differences in pathological changes in the modeling of DOX cardiomyopathy between female and male rats/mice. Meanwhile, the described diversity of studies is characterized by the use of animals of different genetic status (belonging to the stock, line), as well as the absence of a unified approach to the presentation of data concerning the characteristics of animals. For instance, in some studies, sex and weight of animals are specified as the main characteristics of animals, while in other studies their sex and age are specified. In addition, the presented studies differ in the doses of DOX used. Given the proven role of the genetic background in the formation of phenotypic differences that can influence the results of the experiment (Simon et al., 2013); the complexity of comparing the doses of DOX used with the weight characteristics of animals (hence, the calculation of the cumulative dose of the drug); as well as, in some cases, the lack of the evaluation criteria reflecting the development of pathogenetic links (for example, oxidative stress) and the physical state of animals (reflecting the severity of myocardial damage), we can assume one of the probable reasons for the “failure” of pathogenetically grounded experimental treatment in a clinical setting.
It is known that some of the drugs entering clinical development do not confirm their effectiveness previously demonstrated in laboratory conditions (Pound and Ritskes-Hoitinga, 2018; Ferreira et al., 2020). This situation also applies to drugs intended for a preventive or pathogenetic effect on the course of DOX cardiomyopathy (Weidemann and Strotmann, 2006; Vincent et al., 2013; Lipshultz et al., 2014; Abushouk et al., 2017; Kim et al., 2019; Wang et al., 2019; Sharma et al., 2020). Possible reasons for such low transferability can be considered the presence of species differences between humans and laboratory animals, as well as the difficulty in developing an adequate experimental model of the disease. Taking into account the expansion of pharmacological possibilities of cancer treatment, and, as a result, the increase in the life expectancy due to chemotherapy and supportive treatment, it becomes necessary to search for mechanisms that would implement the development of those effects of chemotherapeutic drugs that have not been previously encountered. In order to resolve these issues new models are needed that could reflect these effects.
Ideally, the experimental model should meet certain criteria: the similarity of morphological, biochemical and functional characteristics at various levels of integration of the organism (systemic, organ, cellular, subcellular, molecular); the similarity of the mechanism of development (pathogenesis) of the disease and its phenotypic manifestations (biochemical, functional and morphological signs); the generality of the principles of treatment for humans and the animal model. In order to select the optimal dose of DOX administration to an animal, we must have a good understanding of the clinical symptoms developing in people who received chemotherapy treatment. Subclinical cardiotoxicity in humans is usually defined by cardiac echocardiography as clinically asymptomatic left ventricular systolic dysfunction with a fall in ejection fraction (EF) of more than 10% to an EF value of <50%. If asymptomatic reductions in EF are prospectively detected within the first 12 months of the follow-up after chemotherapy, this indicates early cardiotoxicity (McGowan et al., 2017). The analysis for cardiac troponin also plays an important role in the determination of various heart diseases. In the context of chemotherapy, elevated troponin can quantify both cardiomyocyte apoptosis and myofibril degradation and predict cardiotoxicity, defined as a drop in EF, as well as serious adverse cardiovascular complications. In view of all above mentioned aspects, it can be assumed that when creating an actual model of DOX cardiomyopathy in animals, in this case in rodents, there should be a decrease in the EF by more than 10%, confirmed by echocardiographic studies; an increase in the level of cardiac troponin, which characterizes damage to cardiomyotes; and most importantly, the long-term survival of animals and their satisfactory physical condition.
The most commonly used dose of DOX in rats ranges from 1 to 2.5 mg/kg, which constitutes a cumulative dose of 10–20 mg/kg. The results of most studies demonstrate that DOX significantly reduces body and heart weight, increases cardiac troponin levels, and results in the accumulation of interstitial collagen fibers in the left ventricle, manifested at the end of administration and aggravated by the 4th or 8th week. Of all the studies described in the review concerning the modeling of DOX cardiomyopathy in rats, it can be assumed that the optimal cumulative doses of DOX administration providing clinical manifestations confirmed by echocardiographic and biochemical studies, comparable to people who have undergone chemotherapy and ensuring long-term survival of animals, start from 10 mg/kg and less. It can be assumed that the experimental model characterized by changes that reflect the main pathogenetic links of this disease, as well as the integrated approach to the verification of the model will allow to consider it on the one hand, as a basic model for studying the mechanisms underlying the development of DOX cardiomyopathy, and, on the other hand, as an adequate model for preclinical assessment of pathogenetically based treatment.
In this review, we have summarized and analyzed the main models of DOX cardiomyopathy using small laboratory animals (rats and mice). We have also proposed approaches to improve the model, taking into account the species differences between humans and laboratory animals, biochemical and histological differences, the severity of changes in the morphology and functional characteristics of the myocardium.
EP collected supporting evidence, wrote manuscript and edited manuscript. EK collected supporting evidence and wrote manuscript. AK wrote manuscript. YT provided guidance for the manuscript and wrote manuscript.
The research was carried out within the state assignment of Ministry of Health of the Russian Federation. Study of the influence of intravenous administration of nicothiamide riboside on the development and course of doxorubicin cardiomyopathy, No НИОКТР АААА-А19-119070490037-5.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2021.670479/full#supplementary-material
Abd-Allah, A. R., Al-Majed, A. A., Mostafa, A. M., Al-Shabanah, O. A., Din, A. G. E., and Nagi, M. N. (2002). Protective Effect of Arabic Gum against Cardiotoxicity Induced by Doxorubicin in Mice: A Possible Mechanism of protection. J. Biochem. Mol. Toxicol. 16 (5), 254–259. doi:10.1002/jbt.10046
Abushouk, A. I., Ismail, A., Salem, A. M. A., Afifi, A. M., and Abdel-Daim, M. M. (2017). Cardioprotective Mechanisms of Phytochemicals against Doxorubicin-Induced Cardiotoxicity. Biomed. Pharmacother. 90, 935–946. doi:10.1016/j.biopha.2017.04.033
Ahmad, S., Panda, B. P., Kohli, K., Fahim, M., and Dubey, K. (2017). Folic Acid Ameliorates Celecoxib Cardiotoxicity in a Doxorubicin Heart Failure Rat Model. Pharm. Biol. 55 (1), 1295–1303. doi:10.1080/13880209.2017.1299768
Allen, B. D., Zhang, Z., Naresh, N. K., Misener, S., Procissi, D., and Carr, J. C. (2019). Slow-Release Doxorubicin Pellets Generate Myocardial Cardiotoxic Changes in Mice without Significant Systemic Toxicity. Cardiovasc. Toxicol. 19 (5), 482–484. doi:10.1007/s12012-019-09521-0
Aygun, H., and Gul, S. S. (2019). Cardioprotective Effect of Melatonin and Agomelatine on Doxorubicin-Induced Cardiotoxicity in a Rat Model: an Electrocardiographic, Scintigraphic and Biochemical Study. Bll 120 (4), 249–255. doi:10.4149/bll_2019_045
Aykan, D. A., Yaman, S., Eser, N., Özcan Metin, T., Seyithanoğlu, M., Aykan, A. Ç., et al. (2020). Bisoprolol and Linagliptin Ameliorated Electrical and Mechanical Isometric Myocardial Contractions in Doxorubicin-Induced Cardiomyopathy in Rats. Pharmacol. Rep. 72 (4), 867–876. doi:10.1007/s43440-019-00034-9
Bai, Z., and Wang, Z. (2019). Genistein Protects against Doxorubicin‐induced Cardiotoxicity through Nrf‐2/HO‐1 Signaling in Mice Model. Environ. Toxicol. 34 (5), 645–651. doi:10.1002/tox.22730
Barış, V. Ö., Gedikli, E., Yersal, N., Müftüoğlu, S., and Erdem, A. (2019). Protective Effect of Taurine against Doxorubicin-Induced Cardiotoxicity in Rats: Echocardiographical and Histological Findings. Amino Acids 51 (10-12), 1649–1655. doi:10.1007/s00726-019-02801-7
Bertazzoli, C., Bellini, O., Magrini, U., and Tosana, M. G. (1979). Quantitative Experimental Evaluation of Adriamycin Cardiotoxicity in the Mouse. Cancer Treat. Rep. 63, 1877–1883.
Cappetta, D., De Angelis, A., Sapio, L., Prezioso, L., Illiano, M., Quaini, F., et al. (2017). Oxidative Stress and Cellular Response to Doxorubicin: A Common Factor in the Complex Milieu of Anthracycline Cardiotoxicity. Oxidative Med. Cell Longevity 2017, 1–13. doi:10.1155/2017/15210202017
Cappetta, D., Esposito, G., Coppini, R., Piegari, E., Russo, R., Ciuffreda, L. P., et al. (2017). Effects of Ranolazine in a Model of Doxorubicin-Induced Left Ventricle Diastolic Dysfunction. Br. J. Pharmacol. 174 (21), 3696–3712. doi:10.1111/bph.13791
Cappetta, D., Rossi, F., Piegari, E., Quaini, F., Berrino, L., Urbanek, K., et al. (2018). Doxorubicin Targets Multiple Players: A New View of an Old Problem. Pharmacol. Res. 127, 4–14. doi:10.1016/j.phrs.2017.03.016
Carvalho, R. A., Sousa, R. P. B., Cadete, V. J. J., Lopaschuk, G. D., Palmeira, C. M. M., Bjork, J. A., et al. (2010). Metabolic Remodeling Associated with Subchronic Doxorubicin Cardiomyopathy. Toxicology 270, 92–98. doi:10.1016/j.tox.2010.01.019
Chakouri, N., Farah, C., Matecki, S., Amedro, P., Vincenti, M., Saumet, L., et al. (2020). Screening for In-Vivo Regional Contractile Defaults to Predict the Delayed Doxorubicin Cardiotoxicity in Juvenile Rat. Theranostics 10 (18), 8130–8142. doi:10.7150/thno.47407
Elhadidy, M. G., Elmasry, A., Rabei, M. R., and Eladel, A. E. (2020). Effect of Ghrelin on VEGF-B and Connexin-43 in a Rat Model of Doxorubicin-Induced Cardiomyopathy. J. Basic Clin. Physiol. Pharmacol. 31 (1), 1–11. doi:10.1515/jbcpp-2018-0212
Ferreira, G. S., Veening-Griffioen, D. H., Boon, W. P. C., Moors, E. H. M., and van Meer, P. J. K. (2020). Levelling the Translational gap for Animal to Human Efficacy Data. Animals 10 (7), 1–13. doi:10.3390/ani10071199
Forssen, E. A., and Tokes, Z. A. (1981). Use of Anionic Liposomes for the Reduction of Chronic Doxorubicin-Induced Cardiotoxicity. Proc. Natl. Acad. Sci. 78 (3 I), 1873–1877. doi:10.1073/pnas.78.3.1873
Gioffré, S., Ricci, V., Vavassori, C., Ruggeri, C., Chiesa, M., Alfieri, I., et al. (2019). Plasmatic and Chamber-specific Modulation of Cardiac microRNAs in an Acute Model of DOX-Induced Cardiotoxicity. Biomed. Pharmacother. 110 (October 2018), 1–8. doi:10.1016/j.biopha.2018.11.042
Goormaghtigh, E., Chatelain, P., Caspers, J., and Ruysschaert, J. M. (1980). Evidence of a Specific Complex between Adriamycin and Negatively-Charged Phospholipids. Biochim. Biophys. Acta (Bba)—Biomembranes 597 (1), 1–14. doi:10.1016/0005-2736(80)90145-5
Herman, E., El-Hage, A. N., Ferrans, V. J., and Ardalan, B. (1985). Comparison of the Severity of the Chronic Cardiotoxicity Produced by Doxorubicin in Normotensive and Hypertensive Rats. Toxicol. Appl. Pharmacol. 78 (2), 202–214. doi:10.1016/0041-008X(85)90284-4
Herman, E. H., and Ferrans, V. J. (1983). Influence of Vitamin E and ICRF-187 on Chronic Doxorubicin Cardiotoxicity in Miniature Swinefluence of Vitamin E and ICRF-187 on Chronic Doxorubicin Cardiotoxicity in Miniature Swine. Lab. Invest. 49, 69–77.
Herman, E. H., and Ferrans, V. J. (1997). Animal Models of Anthracycline Cardiotoxicity: Basic Mechanisms and Cardioprotective Activity. Prog. Pediatr. Cardiol. 8 (2), 49–58. doi:10.1016/S1058-9813(98)00002-2
Hole, L. D., Larsen, T. H., Fossan, K. O., Limé, F., and Schjøtt, J. (2013). A Short-Time Model to Study Relevant Indices of Cardiotoxicity of Doxorubicin in the Rat. Toxicol. Mech. Methods 23 (6), 412–418. doi:10.3109/15376516.2013.773391
Holmberg, S. R., and Williams, A. J. (1990). Patterns of Interaction between Anthraquinone Drugs and the Calcium-Release Channel from Cardiac Sarcoplasmic Reticulum. Circ. Res. 67 (2), 272–283. doi:10.1161/01.RES.67.2.272
Hu, X., Li, B., Li, L., Li, B., Luo, J., and Shen, B. (2020). Asiatic Acid Protects against Doxorubicin-Induced Cardiotoxicity in Mice. Oxidative Med. Cell Longevity 2020, 1–12. doi:10.1155/2020/5347204
Hullin, R., Métrich, M., Sarre, A., Basquin, D., Maillard, M., Regamey, J., et al. (2018). Diverging Effects of Enalapril or Eplerenone in Primary Prevention against Doxorubicin-Induced Cardiotoxicity. Cardiovasc. Res. 114 (2), 272–281. doi:10.1093/cvr/cvx162
Hydock, D. S., Lien, C.-Y., Jensen, B. T., Parry, T. L., Schneider, C. M., and Hayward, R. (2012). Rehabilitative Exercise in a Rat Model of Doxorubicin Cardiotoxicity. Exp. Biol. Med. (Maywood) 237 (12), 1483–1492. doi:10.1258/ebm.2012.012137
Ivanová, M., Dovinová, I., Okruhlicová, Ľ., Tribulová, N., Šimončíková, P., Barte-ková, M., et al. (2012). Chronic Cardiotoxicity of Doxorubicin Involves Activation of Myocardial and Circulating Matrix Metalloproteinases in Rats. Acta Pharmacol. Sin 33 (4), 459–469. doi:10.1038/aps.2011.194
Jaenke, R. S. (1974). An Anthracycline Antibiotic-Induced Cardiomyopathy in Rabbits. Lab. Invest. 30, 292–304.
Johansen, P. B. (1981). Doxorubicin Pharmacokinetics after Intravenous and Intraperitoneal Administration in the Nude Mouse. Cancer Chemother. Pharmacol. 5 (4), 267–270. doi:10.1007/BF00434396
Kang, Y., Wang, W., Zhao, H., Qiao, Z., Shen, X., and He, B. (2017). Avaliação de cardiotoxicidade subclínica induzida por doxorrubicina em um modelo de rato por speckle-tracking. Arq Bras Cardiol. 109 (2), 132–139. doi:10.5935/abc.20170097
Kanter, M. M., Hamlin, R. L., Unverferth, D. V., Davis, H. W., and Merola, A. J. (1985). Effect of Exercise Training on Antioxidant Enzymes and Cardiotoxicity of Doxorubicin. J. Appl. Physiol. 59 (4), 1298–1303. doi:10.1152/jappl.1985.59.4.1298
Kappus, H., Muliawan, H., and Scheulen, M. E. (1980). In Vivo studies on Adriamycin-Induced Lipid Peroxidation and Effects of Ferrous Ions. Dev. Toxicol. Environ. Sci. 8, 635–638.
Kashfi, K., Israel, M., Sweatman, T. W., Seshadri, R., and Cook, G. A. (1990). Inhibition of Mitochondrial Carnitine Palmitoyltransferases by Adriamycin and Adriamycin Analogues. Biochem. Pharmacol. 40 (7), 1441–1448. doi:10.1016/0006-2952(90)90438-Q
Keung, E. C., Toll, L., Ellis, M., and Jensen, R. A. (1991). L-type Cardiac Calcium Channels in Doxorubicin Cardiomyopathy in Rats Morphological, Biochemical, and Functional Correlations. J. Clin. Invest. 87 (6), 2108–2113. doi:10.1172/JCI115241
Kim, S. J., Kim, H. S., and Seo, Y. R. (2019). Understanding of ROS-Inducing Strategy in Anticancer Therapy. Oxid Med. Cel Longev eCollection, 1–12. doi:10.1155/2019/5381692
Lanza, E., Rozza, A., Favalli, L., Monti, E., Poggi, P., and Villani, F. (1989). The Rat Model in the Comparative Evaluation of Anthracyclines Cardiotoxicity. Tumori 75 (6), 533–536. doi:10.1177/030089168907500603
Li, K., Sung, R. Y. T., Huang, W. Z., Yang, M., Pong, N. H., Lee, S. M., et al. (2006). Thrombopoietin Protects against In Vitro and In Vivo Cardiotoxicity Induced by Doxorubicin. Circulation 113 (18), 2211–2220. doi:10.1161/CIRCULATIONAHA.105.560250
Lipshultz, S. E., Franco, V. I., Sallan, S. E., Adamson, P. C., K. Steiner, R., Swain, S. M., et al. (2014). Dexrazoxane for Reducing Anthracycline-Related Cardiotoxicity in Children with Cancer: An Update of the Evidence. Prog. Pediatr. Cardiol. 36 (1–2), 39–49. doi:10.1016/j.ppedcard.2014.09.007
Liu, B., Bai, Q. X., Chen, X. Q., Gao, G. X., and Gu, H. T. (2007). [Effect of Curcumin on Expression of Survivin, Bcl-2 and Bax in Human Multiple Myeloma Cell Line]. Zhongguo Shi Yan Xue Ye Xue Za Zhi 15, 762–766.
Lončar-Turukalo, T., Vasić, M., Tasić, T., Mijatović, G., Glumac, S., Bajić, D., et al. (2015). Heart Rate Dynamics in Doxorubicin-Induced Cardiomyopathy. Physiol. Meas. 36 (4), 727–739. doi:10.1088/0967-3334/36/4/727
Lüscher, T. F., and Barton, M. (2000). Endothelins and Endothelin Receptor Antagonists: Therapeutic Considerations for a Novel Class of Cardiovascular Drugs. Circulation 102 (19 Suppl. L), 2434–2440. doi:10.1161/01.CIR.102.19.2434
Maral, R., Bourat, G., Ducrot, R., Fournel, J., Ganter, P., Julou, L., et al. (1967). [Toxicologic Study and Experimental Antitumor Activity of Rubidomycin (13,057 R.P.)]. Pathol. Biol. 15, 903–908.
Markham, M. J., Wachter, K., Agarwal, N., Bertagnolli, M. M., Chang, S. M., Dale, W., et al. (2020). Clinical Cancer Advances 2020: Annual Report on Progress against Cancer from the American Society of Clinical Oncology. Jco 38 (10), 1081–1101. doi:10.1200/JCO.19.03141
May, P. M., Williams, G. K., and Williams, D. R. (1980). Solution Chemistry Studies of Adriamycin-Iron Complexes Present In Vivo. Eur. J. Cancer (1965) 16 (9), 1275–1276. doi:10.1016/0014-2964(80)90189-9
McGowan, J. V., Chung, R., Maulik, A., Piotrowska, I., Walker, J. M., and Yellon, D. M. (2017). Anthracycline Chemotherapy and Cardiotoxicity. Cardiovasc. Drugs Ther. 31 (1), 63–75. doi:10.1007/s10557-016-6711-0
Medeiros-Lima, D. J. M., Carvalho, J. J., Tibirica, E., Borges, J. P., and Matsuura, C. (2019). Time Course of Cardiomyopathy Induced by Doxorubicin in Rats. Pharmacol. Rep. 71 (4), 583–590. doi:10.1016/j.pharep.2019.02.013
Merlet, N., Piriou, N., Rozec, B., Grabherr, A., Lauzier, B., Trochu, J.-N., et al. (2013). Increased Beta2-Adrenoceptors in Doxorubicin-Induced Cardiomyopathy in Rat. PLoS One 8 (5), e64711–15. doi:10.1371/journal.pone.0064711
Mettler, F. P., Young, D. M., and Ward, J. M. (1977). Adriamycin-induced Cardiotoxicity (Cardiomyopathy and Congestive Heart Failure) in Rats. Cancer Res. 37 (August), 2705–2713.
Miller, W. L., Cavero, P. G., Aarhus, L. L., Heublein, D. M., and Burnett, J. C. (1993). Endothelin-Mediated Cardiorenal Hemodynamic and Neuroendocrine Effects Are Attenuated by Nitroglycerin In Vivo. Am. J. Hypertens. 6 (2), 156–163. doi:10.1093/ajh/6.2.156
Minotti, G., Menna, P., Salvatorelli, E., Cairo, G., and Gianni, L. (2004). Anthracyclines: Molecular Advances and Pharmacologic Developments in Antitumor Activity and Cardiotoxicity. Pharmacol. Rev. 56 (2), 185–229. doi:10.1124/pr.56.2.6
Minotti, G., Recalcati, S., Mordente, A., Liberi, G., Calafiore, A. M., Mancuso, C., et al. (1998). The Secondary Alcohol Metabolite of Doxorubicin Irreversibly Inactivates Aconitase/iron Regulatory Protein‐1 in Cytosolic Fractions from Human Myocardium. FASEB j. 12 (7), 541–552. doi:10.1096/fasebj.12.7.541
Mizuta, Y., Tokuda, K., Guo, J., Zhang, S., Narahara, S., Kawano, T., et al. (2020). Sodium Thiosulfate Prevents Doxorubicin-Induced DNA Damage and Apoptosis in Cardiomyocytes in Mice. Life Sci. 257 (June), 118074. doi:10.1016/j.lfs.2020.118074
Mollard, S., Mousseau, Y., Baaj, Y., Richard, L., Cook-Moreau, J., Monteil, J., et al. (2011). How Can Grafted Breast Cancer Models Be Optimized? Cancer Biol. Ther. 12 (10), 855–864. doi:10.4161/cbt.12.10.18139
Mostafa, M. G., Mima, T., Ohnishi, S. T., and Mori, K. (2000). S-allylcysteine Ameliorates Doxorubicin Toxicity in the Heart and Liver in Mice. Planta Med. 66 (2), 148–151. doi:10.1055/s-2000-11124
Nakahara, T., Tanimoto, T., Petrov, A. D., Ishikawa, K., Strauss, H. W., and Narula, J. (2018). Rat Model of Cardiotoxic Drug-Induced Cardiomyopathy. Methods Mol. Biol. 1816, 221–232. doi:10.1007/978-1-4939-8597-5_17
Naresh, N. K., Misener, S., Zhang, Z., Yang, C., Ruh, A., Bertolino, N., et al. (2020). Cardiac MRI Myocardial Functional and Tissue Characterization Detects Early Cardiac Dysfunction in a Mouse Model of Chemotherapy‐Induced Cardiotoxicity. NMR Biomed. 33 (9), 1–12. doi:10.1002/nbm.4327
Nebigil, C. G., and Désaubry, L. (2018). Updates in Anthracycline-Mediated Cardiotoxicity. Front. Pharmacol. 9 (NOV), 1–13. doi:10.3389/fphar.2018.01262
Neilan, T. G., Jassal, D. S., Perez-Sanz, T. M., Raher, M. J., Pradhan, A. D., Buys, E. S., et al. (2006). Tissue Doppler Imaging Predicts Left Ventricular Dysfunction and Mortality in a Murine Model of Cardiac Injury. Eur. Heart J. 27 (15), 1868–1875. doi:10.1093/eurheartj/ehl013
Nitobe, J., Yamaguchi, S., Okuyama, M., Nozaki, N., Sata, M., Miyamoto, T., et al. (2003). Reactive Oxygen Species Regulate FLICE Inhibitory Protein (FLIP) and Susceptibility to Fas-Mediated Apoptosis in Cardiac Myocytes. Cardiovasc. Res. 57 (1), 119–128. doi:10.1016/S0008-6363(02)00646-6
Octavia, Y., Tocchetti, C. G., Gabrielson, K. L., Janssens, S., Crijns, H. J., and Moens, A. L. (2012). Doxorubicin-induced Cardiomyopathy: From Molecular Mechanisms to Therapeutic Strategies. J. Mol. Cell Cardiol. 52 (6), 1213–1225. doi:10.1016/j.yjmcc.2012.03.006
Papadopoulou, L. C., Theophilidis, G., Thomopoulos, G. N., and Tsiftsoglou, A. S. (1999). Structural and Functional Impairment of Mitochondria in Adriamycin-Induced Cardiomyopathy in Mice: Suppression of Cytochrome C Oxidase II Gene Expression. Biochem. Pharmacol. 57 (5), 481–489. doi:10.1016/S0006-2952(98)00305-0
Pei, X. M., Tam, B. T., Sin, T. K., Wang, F. F., Yung, B. Y., Chan, L. W., et al. (2016). S100A8 and S100A9 Are Associated with Doxorubicin-Induced Cardiotoxicity in the Heart of Diabetic Mice. Front. Physiol. 7 (AUG), 1–14. doi:10.3389/fphys.2016.00334
Peres Diaz, L. S., Schuman, M. L., Aisicovich, M., Toblli, J. E., Pirola, C. J., Landa, M. S., et al. (2020). Short-term Doxorubicin Cardiotoxic Effects: Involvement of Cardiac Thyrotropin Releasing Hormone System. Life Sci. 261 (July), 118346. doi:10.1016/j.lfs.2020.118346
Picard, P., Smith, P. J. W., Monge, J. C., Rouleau, J. L., Nguyen, Q. T., Calderone, A., et al. (1998). Coordinated Upregulation of the Cardiac Endothelin System in a Rat Model of Heart Failure. J. Cardiovasc. Pharmacol. 31 (Suppl. 1), S294–S297. doi:10.1097/00005344-199800001-00082
Pieske, B., Beyermann, B., Breu, V., Löffler, B. M., Schlotthauer, K., Maier, L. S., et al. (1999). Functional Effects of Endothelin and Regulation of Endothelin Receptors in Isolated Human Nonfailing and Failing Myocardium. Circulation 99 (14), 1802–1809. doi:10.1161/01.CIR.99.14.1802
Pillai, V. B., Kanwal, A., Fang, Y. H., Sharp, W. W., Samant, S., Arbiser, J., et al. (2017). Honokiol, an Activator of Sirtuin-3 (SIRT3) Preserves Mitochondria and Protects the Heart from Doxorubicin-Induced Cardiomyopathy in Mice. Oncotarget 8 (21), 34082–34098. doi:10.18632/oncotarget.16133
Pound, P., and Ritskes-Hoitinga, M. (2018). Is it Possible to Overcome Issues of External Validity in Preclinical Animal Research? Why Most Animal Models Are Bound to Fail. J. Transl Med. 16 (1), 1–8. doi:10.1186/s12967-018-1678-1
Rahimi Balaei, M., Momeny, M., Babaeikelishomi, R., Ejtemaei Mehr, S., Tavangar, S. M., and Dehpour, A. R. (2010). The Modulatory Effect of Lithium on Doxorubicin-Induced Cardiotoxicity in Rat. Eur. J. Pharmacol. 641 (2-3), 193–198. doi:10.1016/j.ejphar.2010.05.046
Razmaraii, N., Babaei, H., Mohajjel Nayebi, A., Asadnasab, G., Ashrafi Helan, J., and Azarmi, Y. (2016). Cardioprotective Effect of Phenytoin on Doxorubicin-Induced Cardiac Toxicity in a Rat Model. J. Cardiovasc. Pharmacol. 67 (3), 237–245. doi:10.1097/FJC.0000000000000339
Renu, K. V. G. A., V.G., A., P.B., T. P., and Arunachalam, S. (2018). Molecular Mechanism of Doxorubicin-Induced Cardiomyopathy - an Update. Eur. J. Pharmacol. 818, 241–253. doi:10.1016/j.ejphar.2017.10.043
Robert, J. (2007). Preclinical Assessment of Anthracycline Cardiotoxicity in Laboratory Animals: Predictiveness and Pitfalls. Cell Biol Toxicol 23 (1), 27–37. doi:10.1007/s10565-006-0142-9
Sabatino, J., De Rosa, S., Tammè, L., Iaconetti, C., Sorrentino, S., Polimeni, A., et al. (2020). Empagliflozin Prevents Doxorubicin-Induced Myocardial Dysfunction. Cardiovasc. Diabetol. 19 (1), 66. doi:10.1186/s12933-020-01040-5
Sacco, G., Bigioni, M., Evangelista, S., Goso, C., Manzini, S., and Maggi, C. A. (2001). Cardioprotective Effects of Zofenopril, a New Angiotensin-Converting Enzyme Inhibitor, on Doxorubicin-Induced Cardiotoxicity in the Rat. Eur. J. Pharmacol. 414 (1), 71–78. doi:10.1016/S0014-2999(01)00782-8
Saeki, K., Obi, I., Ogiku, N., Shigekawa, M., Imagawa, T., and Matsumoto, T. (2002). Doxorubicin Directly Binds to the Cardiac-type Ryanodine Receptor. Life Sci. 70 (20), 2377–2389. doi:10.1016/S0024-3205(02)01524-2
Sahu, B. D., Kumar, J. M., Kuncha, M., Borkar, R. M., Srinivas, R., and Sistla, R. (2016). Baicalein Alleviates Doxorubicin-Induced Cardiotoxicity via Suppression of Myocardial Oxidative Stress and Apoptosis in Mice. Life Sci. 144, 8–18. doi:10.1016/j.lfs.2015.11.018
Santos, D. S. d., and Goldenberg, R. C. d. S. (2018). Doxorubicin-Induced Cardiotoxicity: From Mechanisms to Development of Efficient Therapy. Cardiotoxicity, 3–24. doi:10.5772/intechopen.79588 Published online.
Sayed-Ahmed, M. M., Khattab, M. M., Gad, M. Z., and Osman, A.-M. M. (2001). Increased Plasma Endothelin-1 and Cardiac Nitric Oxide during Doxorubicin-Induced Cardiomyopathy. Pharmacol. Toxicol. 89 (3), 140–144. doi:10.1111/j.1600-0773.2001.890305.x10.1034/j.1600-0773.2001.d01-148.x
Schlame, M., Rua, D., and Greenberg, M. L. (2000). The Biosynthesis and Functional Role of Cardiolipin. Prog. Lipid Res. 39 (3), 257–288. doi:10.1016/S0163-7827(00)00005-9
Sharma, A., Parikh, M., Shah, H., and Gandhi, T. (2020). Modulation of Nrf2 by Quercetin in Doxorubicin-Treated Rats. Heliyon 6 (4), e03803. doi:10.1016/j.heliyon.2020.e03803
Simon, M. M., Greenaway, S., White, J. K., Fuchs, H., Gailus-Durner, V., Wells, S., et al. (2013). A Comparative Phenotypic and Genomic Analysis of C57BL/6J and C57BL/6N Mouse Strains. Genome Biol. 14 (7), R82–R22. doi:10.1186/gb-2013-14-7-r82
Suzuki, T., and Miyauchi, T. (2001). A Novel Pharmacological Action of ET-1 to Prevent the Cytotoxicity of Doxorubicin in Cardiomyocytes. Am. J. Physiology-Regulatory, Integr. Comp. Physiol. 280 (5 49-5), R1399–R1406. doi:10.1152/ajpregu.2001.280.5.r1399
Toblli, J. E., Rivas, C., Cao, G., Giani, J. F., Funk, F., Mizzen, L., et al. (2014). Ferric Carboxymaltose-Mediated Attenuation of Doxorubicin-Induced Cardiotoxicity in an Iron Deficiency Rat Model. Chemother. Res. Pract. 2014, 1–9. doi:10.1155/2014/570241
Todorova, V. K., Beggs, M. L., Delongchamp, R. R., Dhakal, I., Makhoul, I., Wei, J. Y., et al. (2012). Transcriptome Profiling of Peripheral Blood Cells Identifies Potential Biomarkers for Doxorubicin Cardiotoxicity in a Rat Model. PLoS One 7 (11), e48398–14. doi:10.1371/journal.pone.0048398
van Acker, S. A. B. E., Kramer, K., Voest, E. E., Grimbergen, J. A., Zhang, J., van der Vijgh, W. J. F., et al. (1996). Doxorubicin-induced Cardiotoxicity Monitored by ECG in Freely Moving Mice. Cancer Chemother. Pharmacol. 38 (1), 95–101. doi:10.1007/s002800050453
van der Vijgh, W. J. F., van Velzen, D., van der Poort, J. S. E., Schlüper, H. M. M., Mross, K., Feijen, J., et al. (1988). Morphometric Study of Myocardial Changes during Doxorubicin-Induced Cardiomyopathy in Mice. Eur. J. Cancer Clin. Oncol. 24 (10), 1603–1608. doi:10.1016/0277-5379(88)90052-1
Vandenwijngaert, S., Swinnen, M., Walravens, A.-S., Beerens, M., Gillijns, H., Caluwé, E., et al. (2017). Decreased Soluble Guanylate Cyclase Contributes to Cardiac Dysfunction Induced by Chronic Doxorubicin Treatment in Mice. Antioxid. Redox Signaling 26 (4), 153–164. doi:10.1089/ars.2015.6542
Varela-López, A., Battino, M., Navarro-Hortal, M. D., Giampieri, F., Forbes-Hernández, T. Y., Romero-Márquez, J. M., et al. (2019). An Update on the Mechanisms Related to Cell Death and Toxicity of Doxorubicin and the Protective Role of Nutrients. Food Chem. Toxicol. 134 (July), 110834. doi:10.1016/j.fct.2019.110834
Vedam, K., Nishijima, Y., Druhan, L. J., Khan, M., Moldovan, N. I., Zweier, J. L., et al. (2010). Role of Heat Shock Factor-1 Activation in the Doxorubicin-Induced Heart Failure in Mice. Am. J. Physiology-Heart Circulatory Physiol. 298 (6), H1832–H1841. doi:10.1152/ajpheart.01047.2009
Ventura-Clapier, R., Garnier, A., and Veksler, V. (2004). Energy Metabolism in Heart Failure. J. Physiol. 555, 1–13. doi:10.1113/jphysiol.2003.055095
Villani, F., Galimberti, M., Zunino, F., Monti, E., Rozza, A., Lanza, E., et al. (1991). Prevention of Doxorubicin-Induced Cardiomyopathy by Reduced Glutathione. Cancer Chemother. Pharmacol. 28 (5), 365–369. doi:10.1007/BF00685691
Vincent, D. T., Ibrahim, Y. F., Espey, M. G., and Suzuki, Y. J. (2013). The Role of Antioxidants in the Era of Cardio-Oncology. Cancer Chemother. Pharmacol. 72 (6), 1157–1168. doi:10.1007/s00280-013-2260-4
Wallace, K. B., Sardão, V. A., and Oliveira, P. J. (2020). Mitochondrial Determinants of Doxorubicin-Induced Cardiomyopathy. Circ. Res. 126, 926–941. doi:10.1161/CIRCRESAHA.119.314681
Wang, P., Wang, L., Lu, J., Hu, Y., Wang, Q., Li, Z., et al. (2019). SESN2 Protects against Doxorubicin-Induced Cardiomyopathy via Rescuing Mitophagy and Improving Mitochondrial Function. J. Mol. Cell Cardiol. 133 (June), 125–137. doi:10.1016/j.yjmcc.2019.06.005
Weidemann, F., and Strotmann, J. M. (2006). Detection of Subclinical LV Dysfunction by Tissue Doppler Imaging. Eur. Heart J. 27 (15), 1771–1772. doi:10.1093/eurheartj/ehl144
Xu, X., Persson, H. L., and Richardson, D. R. (2005). Molecular Pharmacology of the Interaction of Anthracyclines with Iron. Mol. Pharmacol. 68 (2), 261–271. doi:10.1124/mol.105.013383
Xujie, L., Xinggang, W., Xian, Z., Yeqing, X., Ruizhen, C., and Haozhu, C. (2012). C57BL/6 Mice Are More Appropriate Than BALB/C Mice in Inducing Dilated Cardiomyopathy with Short-Term Doxorubicin Treatment. Acta Cardiol. Sin 28, 236240.
Ye, D., Wang, Z., Xu, Y., Ye, J., Wang, M., Liu, J., et al. (2020). Interleukin-9 Aggravates Doxorubicin-Induced Cardiotoxicity by Promoting Inflammation and Apoptosis in Mice. Life Sci. 255 (October), 117844. doi:10.1016/j.lfs.2020.117844
Zamorano, J. L., Lancellotti, P., Rodriguez Muñoz, D., Aboyans, V., Asteggiano, R., Galderisi, M., et al. (2016). 2016 ESC Position Paper on Cancer Treatments and Cardiovascular Toxicity Developed under the Auspices of the ESC Committee for Practice Guidelines. Eur. Heart J. 37 (36), 2768–2801. doi:10.1093/eurheartj/ehw211
Zbinden, G., Bachmann, E., and Holderegger, C. (1978). Model Systems for Cardiotoxic Effects of Anthracyclines. Antibiot. Chemother. 23, 255–270. doi:10.1159/000401489
Keywords: anthracycline cardiomyopathy, doxorubicin-induced cardiotoxicity, oxidative stress, anthracycline drugs, doxorubicin
Citation: Podyacheva EY, Kushnareva EA, Karpov AA and Toropova YG (2021) Analysis of Models of Doxorubicin-Induced Cardiomyopathy in Rats and Mice. A Modern View From the Perspective of the Pathophysiologist and the Clinician. Front. Pharmacol. 12:670479. doi: 10.3389/fphar.2021.670479
Received: 22 February 2021; Accepted: 20 May 2021;
Published: 03 June 2021.
Edited by:
Antonella De Angelis, University of Campania Luigi Vanvitelli, ItalyReviewed by:
Tamer. M. A. Mohamed, University of Louisville, United StatesCopyright © 2021 Podyacheva, Kushnareva, Karpov and Toropova. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ekaterina Yu Podyacheva, a2F0cmluc3RhbmZvcmRAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.