
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 06 October 2021
Sec. Pharmacogenetics and Pharmacogenomics
Volume 12 - 2021 | https://doi.org/10.3389/fphar.2021.669516
This article is part of the Research Topic Emerging Technologies Powering Rare and Neglected Disease Diagnosis and Therapy Development View all 21 articles
 Hua Li1†
Hua Li1† Wei Wang2,3
Wei Wang2,3 Xiaodi Han1
Xiaodi Han1 Yujia Zhang1
Yujia Zhang1 Lifang Dai1
Lifang Dai1 Manting Xu1
Manting Xu1 Jie Deng1Changhong Ding1Xiaohui Wang1
Jie Deng1Changhong Ding1Xiaohui Wang1 Chunhong Chen1
Chunhong Chen1 Xiaofeng Yang3*†
Xiaofeng Yang3*† Fang Fang1*†
Fang Fang1*†Alpers’ syndrome is an early inceptive neurodegenerative disorder with a poor prognosis, characterized by developmental regression, intractable epilepsy, and hepatic dysfunction. Candidate genes, such as POLG, PARS2, CARS2, FARS2, NARS2, and GABRB2 are distinguished and registered following research on large cohorts that portray the clinical phenotype in such patients using expanded access to whole-exome sequencing (WES). In this study, we aimed to better understand the electroencephalogram (EEG) characteristics and clinical phenotype of different genotypes of the Alpers’ syndrome, which are currently insufficiently studied. We conducted a study on seven patients with Alpers’ syndrome who received treatment in Beijing Children’s Hospital and had a detailed clinical EEG. Furthermore, a substantial literature search of the Chinese Biomedical Literature Database, PubMed, and Cochrane Central Register of Controlled Trials EMBASE was also conducted, which revealed a total of 22 reported cases between January 2008 to January 2021. We analyzed 29 cases of Alpers’ syndrome caused by different gene variants, of which 22 cases were related to POLG gene mutation and 7 cases were related to PARS2, CARS2, FARS2, NARS2, and GABRB2 gene mutation, and found that patients with distinctive pathogenic variants exhibited comparable phenotypes and similar EEG patterns. And we defined EEG characteristics found specifically in Alpers’ syndrome. Rhythmic high-amplitude delta with superimposed (poly) spikes (RHADS) is a characteristic EEG finding in the early stages of Alpers’ syndrome and is a kind of epileptic phenomenon, which can provide clues for the early diagnosis of the disease.
Alpers’ syndrome is an early inceptive neurodegenerative disorder with a poor prognosis, characterized by the triad of developmental regression, intractable epilepsy, and hepatic dysfunction (Sofou et al., 2012; Tarka et al., 2020). The patients with Alpers’ syndrome are generally less than 4 years old after the initial presentation of symptoms (Hannah and Yasir, 2021). It usually occurs in infancy or early childhood, and rarely in juveniles. In the early stages of the disease, epileptic seizures, as an early symptom, are the most common. The epileptic seizures develop in the form of focal, multifocal or myoclonic seizures evolving into epilepsia partialis continua (EPC) or myoclonic status epilepticus (MSE). The sudden outbreak of therapeutically refractory seizures in infancy or adolescence is considered an average clinical indicator of Alpers’ syndrome. The electroencephalograms (EEG) finds have shown a specific EEG phenomenon, known as rhythmic high-amplitude delta with superimposed (poly) spikes (RHADS) (Wolf et al., 2009; van Westrhenen et al., 2018). The epileptic seizures may be have an evolution of EEG in the development of Alpers’ syndrome (Hannah and Yasir, 2021). EEG might provide in the potential diagnostic clue (Wolf et al., 2009; van Westrhenen et al., 2018).
Recent studies have identified molecular genetic causes of Alpers’ syndrome, including pathogenic mutations in the gene encoding the catalytic subunit of polymerase gamma (POLG), PARS2 which encodes prolyl-tRNA synthetase, FARS2 which encodes phenylalanyl-tRNA synthetase, and NARS2 which encodes asparaginyl-tRNA synthetase, etc, POLG gene variation is the most common (Sofou et al., 2015; Walker et al., 2016; Samanta et al., 2018; Sofou et al., 2021).
Alpers’ syndrome is one of the most serious phenotypes of mitochondrial disease caused by POLG gene mutation. More than 90% of Alpers’ syndrome cases happen due to autosomal recessive mutations in the nuclear-encoded catalytic subunit (Bicknese et al., 1992; Hance et al., 2005; Chrysostomou et al., 2016). The results of POLG flaws are mtDNA depletion/accumulation of mtDNA deletions. The depletion of mtDNA causes consequent cellular dysfunction, mitochondrial respiratory chain impairments, and apoptosis in the brain and liver (Anagnostou et al., 2016; Naviaux RK et al., 1999; Tzoulis C et al., 2014).
With the development of genetic detection and analysis techniques, the pathogenic genes of Alpers’ syndrome have been discovered. The candidate genes such as POLG, PARS2, CARS2, FARS2, NARS2, and GABRB2 have been distinguished and registered using large cohorts that portray the clinical phenotype in such patients using expanded access to WES. However, at present, no systematic studies have been performed on the clinical phenotypes and the EEG findings that occur in patients with different genetic pathogenic variation mutations. In this study, we aimed to characterize the EEG features and clinical phenotypes of patients with different genotypes of the Alpers’ syndrome and assist in future studies.
This study was conducted on seven patients with Alpers’ syndrome who underwent a thorough medical treatment from May 2013 to December 2020 in the Beijing Children’s Hospital. They were examined for detailed clinical, EEG, and pathogenic information.
In addition to these, a substantial literature search was carried out in EMBASE, Cochrane Central Register of Controlled Trials, and PubMed. Prior to an extensive literature search, an eligibility criterion was taken into consideration. It included EEG case studies based on patient’s medical history with Alpers’ syndrome. Publications in any language other than English, anthropoidal studies, limited resources of literature review, and unauthenticated abstracts were considered as criteria for exclusion, and comprehensive analysis was reported. After applying the inclusion and exclusion criteria, we shortlisted 22 cases that were reported in the various databases from January 2008 to January 2021. Thus, we examined a total of 29 patients whose details may be found in Supplementary Table S1.
The clinical features that were evaluated for these 29 patients with Alpers’ syndrome were as follows: sex of the patient, age at disease onset, age at seizure onset, categories of seizures, seizure types at disease onset, development, EEG, pathogenic information, and other clinical features. Seizures and epilepsy syndromes were categorized as per the rules and regulations of the International League Against Epilepsy (ILAE) (Belousova et al., 2017).
In the present study, all samples were captured by whole-exome reagent hybridization and using Illumina instruments for next-generation sequencing. Sequence data aligned with the human genome reference (hg19) and variants-calling were carried out with NextGene V2.3.4 software (Softgenetics LLC., State College, PA, United States). Variants were screened as follows:1) preference to the disease-related variants, small insertions and deletions (INDEL), canonical splice sites, and nonsense variants; 2) minor allele frequency (MAF) in normal populations <5% (except for known MAF ≥5% pathogenicity); 3) preference to variants in the Human Gene Mutation Database (HGMD), ClinVar; and 4) preference to variants in the Online Mendelian Inheritance in Man database. Pathogenic variants were defined according to the standards and guidelines for the interpretation of sequence variants published by the American College of Medical Genetics in 2015 with Human Genome Variation Society nomenclature (Richards et al., 2015). We performed Sanger sequencing of all samples to validate the identified POLG mutations and test the parental origin of available family members. Of these, we also used quantitative PCR technology to verify the deletion of large fragments of the genome in ample No. 6.
Video-EEG was documented and digitized at the sample rate of 1000 Hz using the standard international 10–20 system (Natus Medical Incorporated, Pleasanton, CA, United States). Impedances were kept below 10 KΩ. A low-cut filter at 1 Hz was used before digital sampling. At least a prolonged continuous EEG monitoring (more than 24-h-long video-EEG recording). All EEG data were interpreted and reviewed by qualified neurophysiologists adopting the following criteria for RHADS: 1) occipital strength; 2) a moderate movement (<1 Hz) of high amplitude (200–1,000 uV); 3) frequent occurrence; and 4) a set of superimposed polyspikes. EEG data that showed results as ‘inexplicit’ were excluded for additional analysis.
In EEGs with RHADS, gamma oscillations (30–80 Hz) were marked visually via time-frequency analyses for EEG. First, the EEG data were reviewed visually in bipolar 10–20-Montage, and stage-Ⅱ NREM sleep data (the delta band was estimated higher than 25% of all delta bands in a 30-s epoch by visual inspection) was selected (Bagshaw et al., 2009; Jacobs et al., 2016). Then, after band-pass filtering the raw data (30–80 Hz), time-frequency analysis techniques were used in each segment of data. Finally, the gamma oscillations were identified and compared with the original EEG data. The EEG data were visually examined by two EEG professional specialists, and channels with significant artifacts were excluded from the analysis.
A total of 29 data samples were collected in this study, including 22 cases in the POLG group and 7 cases in the NON-POLG group. The differences of various indicators in the two groups were compared. For continuous variables that did not conform to the normal distribution, the median description was adopted, and a non-parametric test was used to compare the differences. For the comparison of classification variables, the composition ratio description was adopted, and the differences were compared by Fisher exact test was used to compare proportions of patients with microcephaly, Infantile spasm, Hepatic dysfunction, and EEG feature. For survival analysis, the endpoint was time to death which was defined as the time in months from the date of disease onset to the date of death. The survival rate of the two groups was compared by Kaplan-Meier survival curve analysis. The software selected for this analysis was SPSS26 and Graphpad7.0, and the significance level was 0.05.
The present study has been approved by the Ethics Committee of Beijing Children’s Hospital and written informed consent was obtained from the guardians/families of the patients for their participation in the study.
Our study revealed a total of 12 different variations within the POLG gene in seven patients (patient 1 to 5, previously reported (Dai et al., 2019) (Figure 1). 10 mutations were reported and two mutations were novel (Table 1, patient 6: ex.2-23del and patient 7: p. H569N). Patient 6 was compound heterozygous for the c.3218C>T mutation and a novel variation, a deletion in exon 2–23 (chr15:89859622–89876990). Quantitative PCR technology was used to verified the deletion, the proband and his mother were heterozygous deletions.
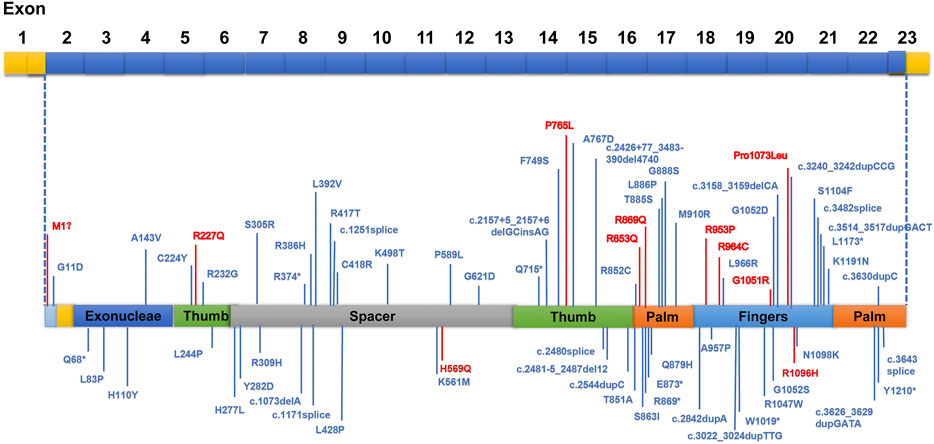
FIGURE 1. Areas of distinguished variants in POLG mutation in Alpers’ syndrome. Mutations presented in red were identified in this current study, whereas those in blue have been reported by past investigations.
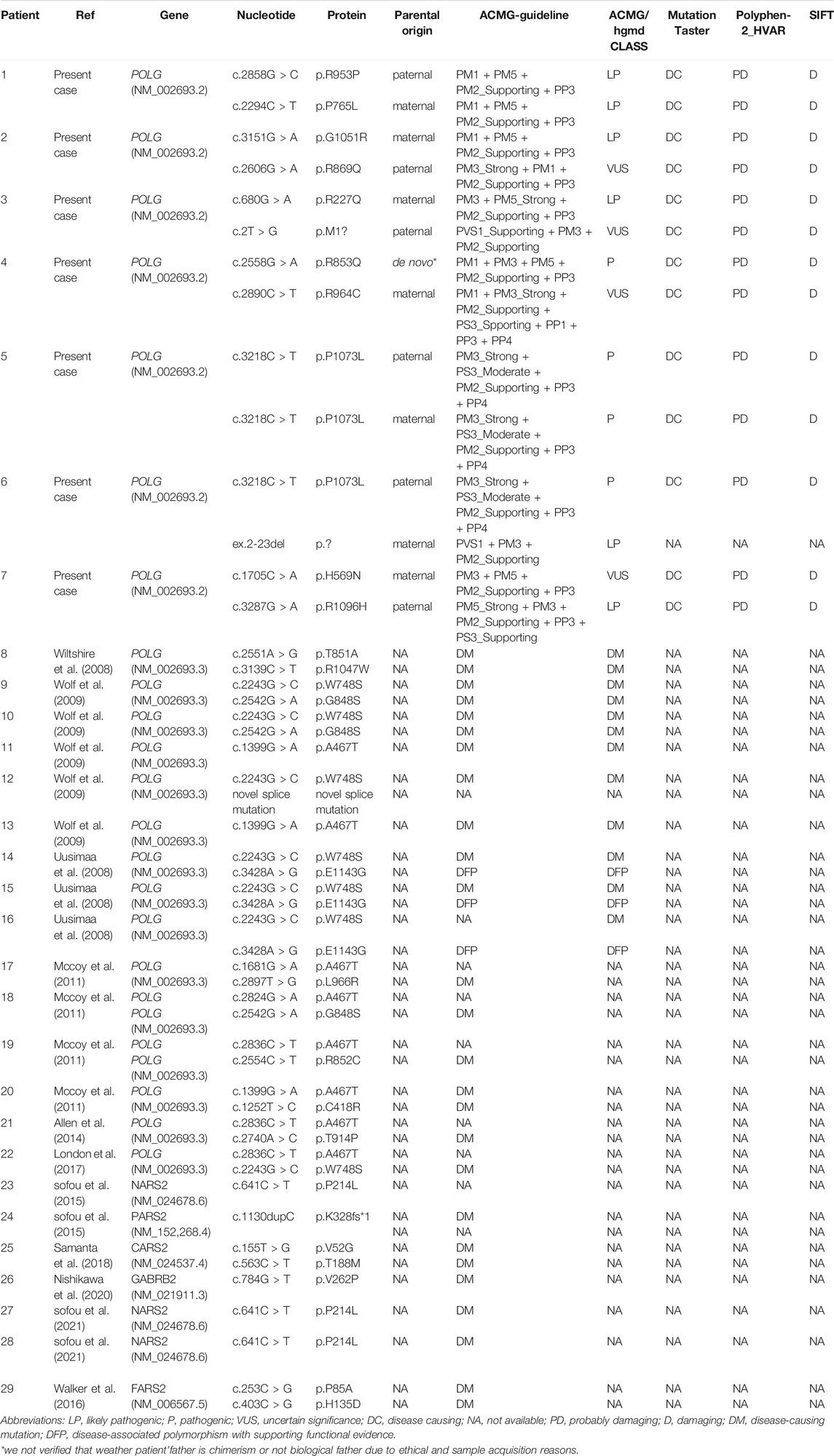
TABLE 1. Summary of the Genetic mutation variants of 29 cases with Alpers’ syndrome.
We identified pathogenic/likely pathogenic/VUS variants in seven patients in our cohort. One patient (patient5) in our study shared the same homozygous variant c.3218C>T (p.P1073L), which was also reported in another two Chinese studies (Qian et al., 2015). Three different recurrent variants (p.W748S,p.E1143G, and p. A467T) were reported (Uusimaa et al., 2008; Wolf et al., 2009). Two unrelated patients shared the same recurrent homozygous variant c.3218C>T (p.P1073L), this recommends a foundation effect in the Chinese population. The other five patients harbored compound heterozygous variants, including 2 variants (Exon 2–23 del and c.1705C>A,p.H569N) newly reported in this study (Table 1).
Genetic (the classification of the variants according to the American College of Medical Genetics and Genomics (ACMG), parental origin, etc) details of these patients are shown in Table 1. Among the 29 Alpers’ syndrome patients, 79.31% (23/29) carried at least 1 missense variant. Only 1 truncation variant in POLG was identified (Wolf et al., 2009). Variants in PARS2, CARS2, GABRB2, FARS2, and NARS2 were also reported in Alpers’ syndrome patients (Sofou et al., 2015; Walker et al., 2016; Samanta et al., 2018; Sofou et al., 2021). All the variations were consistent with the autosomal recessive inheritance pattern, except for that in GABRB2. Aiko et al. reported a de novo missense variant in GABRB2 (c.784G>T (p.Val262Phe)). A recurrent missense variant, c.641C>T (p.P214L), was identified in NARS2 and 1 truncation variant was identified in PARS2 (Sofou et al., 2015). All together 5 recurrent missense variants in 2 genes were identified in Alpers’ syndrome patients, and only 2 truncation variants were observed in 2 genes.
Our data showed that patients with pathogenic POLG variations showed the characteristics of mitochondrial encephalopathy, with refractory epilepsy, progressive liver function impairment, as the main clinical symptoms, acute liver failure occured after valproic acid application. For the analysis of POLG-related Alpers’ syndrome, 22 out of 29 patients were included (8 females, 14 males, The median age at onset was 2.33 years, the range of age-onset was from 2 months to 17 years). The clinical characteristics of POLG mutation-related Alpers’ syndrome were analyzed as follows.
All 22 patients (100%) with POLG mutation-related Alpers’ syndrome experienced epileptic seizures. A variety of refractory epileptic seizures were exhibited in 21 patients (95.5%). Seizures were the most common initial symptom (63.6%), and began at a median age of 2.67 years (ranging from 3 months to 17 years). The main type of seizures were focal/myoclonic seizures that lead to focal motor status or epileptic status. A total of 14 (63.6%) patients showed the symptoms of focal seizures, 12 (54.5%) patients had focal motor status or epileptic status, whereas myoclonic seizures occurred in 3 of 22 patients (13.6%) and generalized tonic-clonic seizures occurred in 2 of 22 patients (9.1%). Only 1 (4.5%) patient presented with the symptoms of generalized seizure. Eight of the 22 patients presented with the symptoms of epilepsia partialis continua (EPC; 36.4%) (Supplementary Table S1).
A notable developmental delay with the signs of POLG mutation was found in all patients who suffered from Alpers’ syndrome in life. A total of 6 out of 22 patients (27.3%) showed the symptoms of developmental delay that were evident before the onset of seizures. Nine patients exhibited normal development until the beginning of intractable seizures. One patient who was mentioned in the literature review was having the symptoms of severe-to-profound intellectual disability.
A series of features such as visual symptoms and visual disturbance, ataxia (including congenital ataxia), hypotonia, vomiting, language delayed/lost speech, and different levels of liver failure were found in all patients. Four patients had severe liver failure after valproic acid application and one patient showed multiorgan failure.
In addition, Our data showed that patients with pathogenic NON-POLG (NARS2, FARS2, PARS2, CARS2, and GABRB2) variations showed the characteristics of age at onset was earlier, mostly in the early infantile period, with prominent infantile spasms, less liver damage, and microcephaly, most of the survival time after treatment is longer. The characteristics are significantly different from POLG-related Alpers’ syndrome. For the analysis of NON-POLG mutation-related Alpers’ syndrome, 7 out of 29 patients were included (2 females, 5 males, The age at onset was the early infantile period). The clinical characteristics of NON-POLG mutation-related Alpers’ syndrome were analyzed as follows.
All 7 patients (100%) with NON-POLG mutation-related Alpers’ syndrome experienced epileptic seizures. A variety of refractory epileptic seizures were exhibited in 7 patients (100%). Epileptic seizures were initial symptom in 3 patients. In terms of seizure type, two (43.1%) patients showed the symptoms of focal seizures, two (28.6%) exhibited focal motor status or epileptic status, whereas myoclonic seizures occurred in three of the seven patients (42.9%), generalized tonic-clonic seizures in three of the seven patients (42.9%), myoclonic seizures in 3 of 7 patients (42.9%). Only one (14.3%) patient presented with the symptoms of infantile spasm, one (14.3%) with the symptoms of atypical absence seizure, one (14.3%) with the symptoms of a tonic seizure, and three with the symptoms of EPC.
A notable developmental delay with signs of NON-POLG mutation was found in six of the seven patients who suffered from Alpers’ syndrome in life. Five out of seven patients (71.4%) showed symptoms of developmental delay that were evident before the seizure outbreak. One patient in the literature review showed symptoms of severe intellectual disability. Only one patient exhibited normal development until the beginning of intractable seizures.
The other series of features such as hypotonia (85.7%, 6/7), visual symptoms (42.9%, 3/7), feeding difficulties, vomiting (42.9%, 3/7), progressive microcephaly (42.9%,3/7), language delayed/lost speech (14.3%, 1/7), mild liver failure (28.6%, 2/7), and autism spectrum (14.3%, 1/7) were observed in the patients in this category. A detailed description of the clinical features of each patient is provided in Supplementary Table S1.
Abnormal EEGs with epileptiform discharges and slow activity were detected at the early stage in all patients. A total of 23 out of 29 (79.3%) patients showed a reducing nonspecific background in the EEG. Of the 29 patients, focal epileptiform discharges on EEG were found in 16 patients (55.2%), multifocal epileptiform discharges were found in 2 (6.9%), and hypsarrhythmia occurred only in 1 (3.4%). Focal slowing and epileptiform discharges were seen relatively equally in the early and late stages of the disease. The incidence of RHADS was described in 18 out of 29 patients (62.1%) (Figure 2). RHADS were the most common EEG patterns and were based on focal abnormality along with epileptic discharges, and were described in four patients in our series with the gamma oscillations (Figure 3). RHADS and slowing were the most common factors identified in the occipital region (57.1%), followed by the impairment in the frontal lobe (28.6%) and temporal (14.3%) regions. In the late stage of the disease, six cases of RHADS on EEG performed focal changes (focal slowing, epileptiform discharges), multifocal epileptiform discharges, and burst suppression pattern.
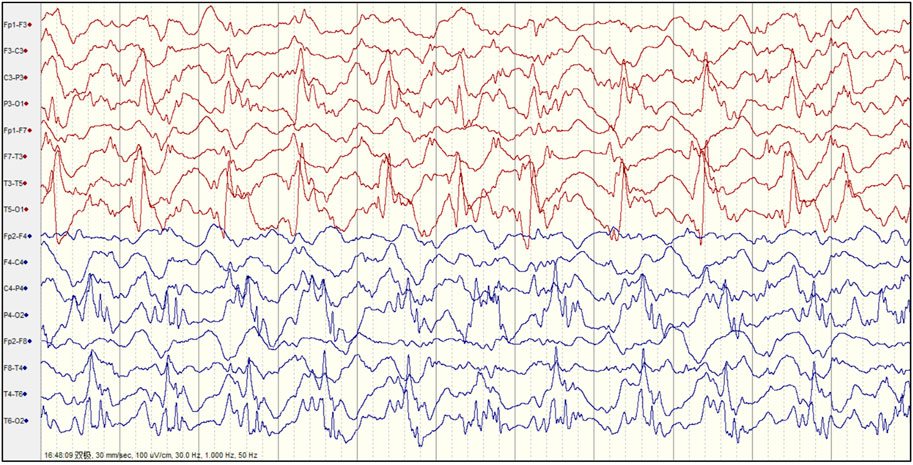
FIGURE 2. The electroencephalogram (EEG) of patient 4 with Alpers’ syndrome in our study showed typical rhythmic high amplitude delta waves with superimposed (poly) spikes (RHADS) in the bilateral posterior head region.
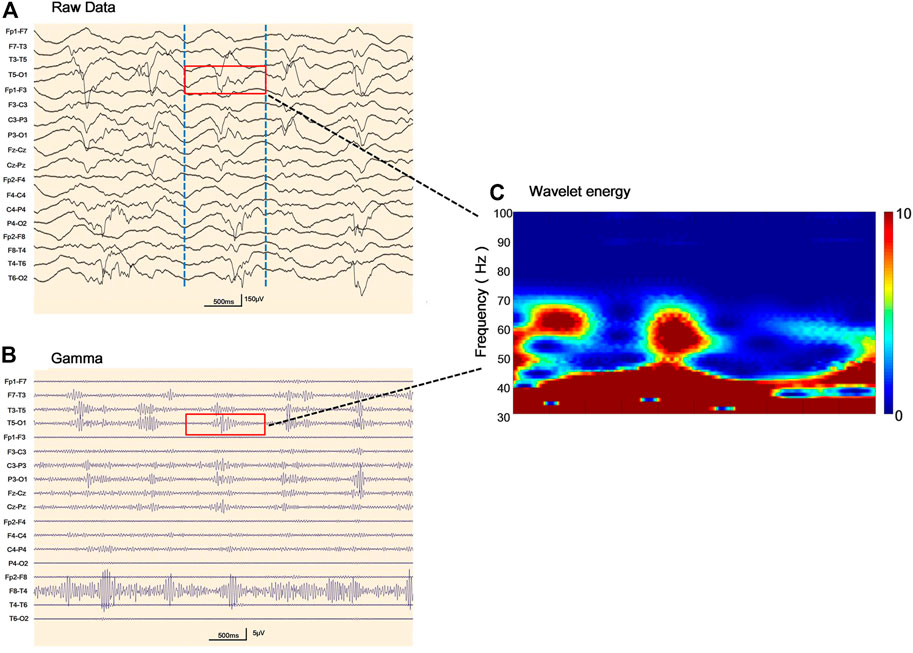
FIGURE 3. Representative electroencephalogram (EEG) follow-ups and corresponding gamma oscillations (A) Raw EEG with the pattern of rhythmic high amplitude delta waves with superimposed (poly) spikes (RHADS) in sleep during stage II non-rapid eye movement period, the red layout marks 1 s data (B) EEG corresponding to Figure 3A after 30–80 Hz filtering, the red line addresses the gamma oscillations visually identified (C) Verification of gamma oscillations. Utilizing the wavelet transform method, the time-frequency spectra exhibit spectral blobs at around 30–80 Hz in the region T5-O1 in association with the corresponding spikes.
As demonstrated in Table 2, Age at onset, Age at seizure onset, Time of death showed a significant relationship with the POLG or NON-POLG group,p < 0.05; As demonstrated in Table 2, Age at onset, Age at seizure onset, Time of death showed a significant relationship with the POLG or NON-POLG group,p < 0.05; microcephaly, Infantile spasm, Hepaticdys function, EEG feature showed a significant relationship in POLG or NON-POLG group, p < 0.05 (Table 3); As can be seen from the above table, there was a significant difference in survival rate between the NON-POLG group and the POLG group (p < 0.05). From the above table, there are significant differences in survival between NON-POLG group and POLG, p < 0.05. Among them, the median survival of Group POLG was 1.75 years and NON-POLG 14.083, and the survival rate of Group NON-POLG was signnificantly higher than that of Group POLG, with the following KM survival curve (Figure 4).

TABLE 2. Comparison of mutations and clinical features of 29 cases with Alpers’ syndrome by nonparametric test.
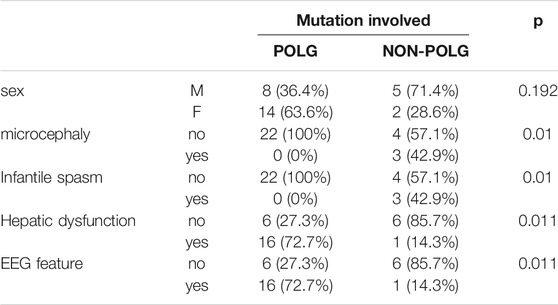
TABLE 3. Comparison of mutations and clinical features of 29 cases with Alpers, syndrome by Fisher exact test.
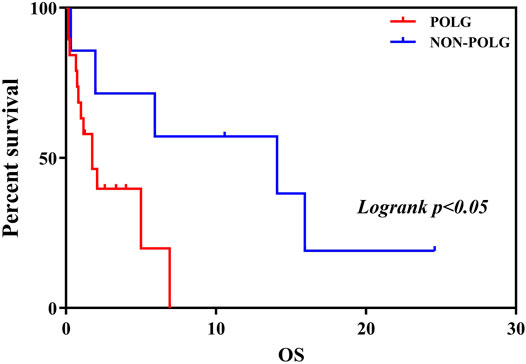
FIGURE 4. Kaplan-Meier curve comparing survival of patients with the POLG or non-POLG group and patients with POLG Group showed carried significantly worse survival.
Alpers’ syndrome is a rare mitochondrial encephalopathy, which is thought to be caused by autosomal recessive mutations in the nuclear-encoded catalytic subunit of POLG (Hannah and Yasir, 2021). Pathogenic variants in POLG are common causes of multiple mtDNA deletions and/or mtDNA depletion. The brain and liver are susceptible to the disease because of their high demand for energy and proportionate need for mitochondria. The loss of mitochondria in these organ systems can lead to various symptoms, the most common being seizures and liver failure. Mitochondrial diseases affect cellular energy production through oxidative phosphorylation (OxPHOS) (Dimauro et al., 2013). The assembly and function of mitochondrial OXPHOS are dependent on a great number of proteins, mainly encoded by nuclear DNA and by mitochondrial DNA. Genetic defects in some of these proteins lead to various phenotypes with different ages of onset and course of the disease. Next-generation sequencing has significantly broadened our understanding of the genetic causes of mitochondrial disease (Jin et al., 2019; Li et al., 2019; Shen and Shi, 2019). In recent years, an increasing number of genes have been related to Alpers’ syndrome, many pathogenic mutations in the mitochondrial aminoacyl-tRNA synthetases (mt-aaRSs), such as FARS2, CARS2, NARS2, and PARS2, etc, are a novel cause of mitochondrial translation disorder, leading to Alpers’ syndrome (Elo et al., 2012; Sofou et al., 2015; Walker et al., 2016; Samanta et al., 2018; Sofou et al., 2021). This study demonstrated the pathogenic mechanism of gene variants resulting in the loss of receptor function (Nishikawa et al., 2020). The patients in this study exhibited different genotypes of Alpers’ syndrome, which suggests different genetic variations resulted in significant changes in mitochondrial function. In 2020, WES revealed a de novo missense variant of GABRB2 (NM_021911.2: c.784G>T, p[Val262Phe]) in a child presenting with daily MSE and RHADS, that was associated with Alpers’ syndrome phenotype. However, the GABRB2 gene encodes γ-GABA receptors. At present, the relationship between γ-GABA receptors and mitochondrial function remains unclear, and further research will be of importance.
The relationship between clinical phenotypes and genotypes has been studied thoroughly based on the collected results of genetic mutation variants. The clinical phenotypes of Alpers’ syndrome are variable, even in those with identical mutations, and this includes different degrees of neurological symptoms and hepatic dysfunction (Wu et al., 2018). The Alpers’ syndrome caused by POLG gene mutations usually affects children aged less than 4 years (Hannah and Yasir, 2021), and inevitably leads to death. Refractory epilepsy is a major phenotypic feature leading to the poor prognostic range and rapidly progressive disease. In the early stages of the disease, epileptic seizures are the most common. The epileptic seizures develop in the form of focal, multifocal or myoclonic seizures evolving into EPC or MSE. In this study, all the patients had exhibited epileptic seizures, most had a focal motor seizure and epileptic status (Supplementary Table S1). In this study, most seizures captured by EEG monitoring originated from the occipital lobe, which appears to be the main site for seizure genesis in Alpers’ syndrome. Numerous clinical and neuropathological studies revealed that the brain lobe is the key spot of the main area of neuronal degeneration and late post-stroke seizures in patients (Montine et al., 1995; Engelsen et al., 2008; Khan et al., 2012; Anagnostou et al., 2016; Wu et al., 2018). Recently, a complete study suggested interneuron and Purkinje cell pathology may be the mechanisms underlying seizure generation and maintenance (Hayhurst et al., 2019). Development is another common neurological feature, this can occur at any time in childhood and may reflect the nature of the process (Supplementary Table S1). In the course of the disease, the individuals with Alpers’ syndrome show different degrees of developmental delay, these findings in this study were consistent with those in previous studies (Supplementary Table S1). Liver involvement is the main characteristic of Alpers’ syndrome, which is rarely the presenting symptom and can rapidly progress to end-stage liver failure. Sodium valproate accelerates liver dysfunction and invariably ends in a fatality (Park et al., 2017). In the current study, four patients presented with progressive liver failure (Supplementary Table S1). In addition, sometimes viral prodrome symptoms, headaches, and vomiting can be observed, which may give rise to clinical suspicion of encephalitis (Rahman and Copeland, 2019), expanding the phenotypic spectrum of Alpers’ syndrome (Lim and Thomas, 2020). In recent years, with the advancement of genetic testing technology, Alpers syndrome caused by NON-POLG genes has also been continuously reported, including mitochondrial aminoacyl transfer RNA synthetase related genes (PARS2, CARS2, FARS2 and NARS2) and γ-GABA receptor related genes Gene (GABRB2). This study retrospectively compared the Alpers’ syndrome caused by NON-POLG and POLG gene mutations, and found that NON-POLG onset was earlier, mostly in the early infantile period, with prominent infantile spasms, less liver damage, and some microcephaly, most of the survival time after treatment is longer (Table 2; Figure 4), the phenotype is significantly different from the Alpers’ syndrome caused by POLG gene mutation (Sofou et al., 2015), so It may be better for these patients with mutations in NON-POLG to be diagnosed as Alpers-like syndrome. Differences in clinical phenotypes between the two groups may be related to the genes themselves, this aspect requires further research for clarification (Bao et al., 2019).
Our study systematically describes the EEG findings of various genotypes of Alpers’ syndrome. In this study, background slowing was more commonly seen in Alpers’ syndrome. Eighteen patients exhibited RHADS in the early stage of the disease, although six cases of RHADS on EEG performed nonspecific abnormalities during the late stage of the disease. RHADS may be associated with the staging phase (Boyd et al., 1986; Wolf et al., 2009; van Westrhenen et al., 2018). Our study is in agreement with the previous research that RHADS are highly distinctive in patients with Alpers’ syndrome. RHADS were all in the early stage of the disease with the association of epileptic status and no traces of the influence of drug treatment (Boyd et al., 1986; Wolf et al., 2009; van Westrhenen et al., 2018). In addition, a notable finding was that the patient suffered from RHADS with gamma oscillations (Figure 3). This spatial overlap suggests between the brain areas generating spikes and fast oscillations involve similar ‘‘hyperactive’’ neuronal networks. In this study, RHADS were based on focal abnormality along with epileptic discharges, however, the gamma oscillations were described in four patients in our series, this finding demonstrated that spikes are more sensitive. Yet, it is important to note that gamma activity more specific and accurate to determine the regions of the brain participating in seizure generation, and the occurrence of the fast oscillations directly reflects the degree of epileptic activity of the EEG (Melani et al., 2013). This finding supported the idea that RHADS is a kind of epileptic phenomenon (Zijlmans et al., 2012). We also found that patients with Alpers’ syndrome may have a high frequency of epileptic status. All patients experienced the same episodes of seizures and an indistinguishable EEG pattern during the early phase of the syndrome. EEG is an important tool for the examination of the diagnosis and prognosis of Alpers’ syndrome because the EEG patterns are consistent with fluctuations in the electrical properties of the brain. The EEG correlations at different stages of the disease not only help to recognize the clinical course of the disease, but also provide a platform for better utilization of EEG in the diagnosis of clinical disease development. The explicit EEG pattern remains the foundation for the diagnosis and may help to comprehend the mechanism of Alpers’ syndrome. However, it’s worth noting that Alpers’ syndrome caused by different genotypes showed similar EEG patterns in our study, which may be due to the involvement of a common electrophysiological mechanism. Therefore, future studies may be focused on applying digital video electroencephalography with high sampling rates and series of EEG patterns and the development of an automatic gamma oscillation analysis system.
To sum up, in this article, we the first integrated analysis of the EEG characteristics and clinical phenotype of patients with Alpers’ syndrome of various genotypes. We found that the patients having different pathogenic variants exhibited comparable phenotypes. There are certain differences in the clinical phenotype of Alpers’ syndrome caused by POLG gene mutations and NON-POLG gene mutations. and the differences in clinical phenotypes may be related to the genes themselves. So we believe that the clinical phenotype of NON-POLG gene mutations may be more appropriate to define Alpers-like syndrome. Due to our limited sample size, in the future, we will continue to collect samples and expand the sample size for further research. In addition, we defined EEG characteristics found specifically in Alpers’ syndrome. RHADS is a characteristic EEG finding in the early stage of Alpers’ syndrome, which is a kind of epileptic phenomenon, can provide clues for the early diagnosis of the disease. EEG characteristics of Alpers’ syndrome with various genotypes show similar EEG patterns, the reason may be related to the common electrophysiological mechanism. We only have a few cases and have yet to draw a definitive conclusion in terms of the relationship between pathogenic genes and Alpers syndrome. This is a summative retrospective study, but it will provide useful information for a deeper understanding of the pathophysiology of Alpers’ syndrome.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
The present study has been approved by the Ethics Committee of Beijing Children’s Hospital and written informed consent was obtained from the guardians/families of the patients for their participation in the study.
All authors contributed to the study design, critically reviewed the manuscript, and approved the final version. HL performed literature search and analysis and wrote the manuscript. WW, XH, YZ, LD, MX, JD, CD, XW, and CC performed literature search and analysis. FF and XY revised the manuscript.
This work was supported by the China Association Against Epilepsy Scientific Research Project (CX-B -2021-09), Beijing Children’s Hospital for Children Medication Foundation (YZZD202001), and the Capital Health Development Research Foundation Project (2018-2-2096).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank the patients and their families who participated in this study. We also thank Berry Genomics Co. and Beijing Fulgent Co., for the technical support.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2021.669516/full#supplementary-material
Bg, background; BS, burst suppression pattern; D, damaging; DC, Disease causing; DFP, disease-associated polymorphism with supporting functional evidence; DM, disease-causing mutation; EEG, Electroencephalogram; EPC, epilepsia partialis continua; FED, focal epileptiform discharges; FS, focal seizure; MFCS, Multifocal clonic seizures; MFED, multifocal epileptiform discharges; MS, myoclonic seizure; GTCS, generalized tonic-clonic seizures; MLH, migraine-like headache; GSW, generalised spike-wave or poly spike-wave discharges; Hyps, hypsarrhythmia; GS, generalized seizure; MFMS, multifocal myoclonus seizure; TS, tonic seizure; IS, infantile spasms; LP, likely pathogenic; P, pathogenic; NA, not available; PD, probably damaging; RHADS, rhythmic high-amplitude delta with superimposed (poly) spikes; SE, status epilepticus; VUS, uncertain significance.
Allen, N. M., Winter, T., Shahwan, A., and King, M. D. (2014). Explosive Onset Non-epileptic Jerks and Profound Hypotonia in an Infant with Alpers-Huttenlocher Syndrome. Seizure 23 (3), 237–239. doi:10.1016/j.seizure.2013.07.005
Anagnostou, M. E., Ng, Y. S., Taylor, R. W., and Mcfarland, R. (2016). Epilepsy Due to Mutations in the Mitochondrial Polymerase Gamma (POLG) Gene: A Clinical and Molecular Genetic Review. Epilepsia 57 (10), 1531–1545. doi:10.1111/epi.13508
Bagshaw, A. P., Jacobs, J., Levan, P., Dubeau, F., and Gotman, J. (2009). Effect of Sleep Stage on Interictal High-Frequency Oscillations Recorded from Depth Macroelectrodes in Patients with Focal Epilepsy. Epilepsia 50 (4), 617–628. doi:10.1111/j.1528-1167.2008.01784.x
Bao, Y., Suo, L., Qian, P., Huang, H., Yang, Y., Tang, J., et al. (2019). Clinical and Genetic Analysis of Dent Disease with Nephrotic Range Albuminuria in Shaanxi, China. Sci. China Life Sci. 62 (12), 1590–1593. doi:10.1007/s11427-018-9829-0
Belousova, E. D., Zavadenko, N. N., Kholin, A. A., and Sharkov, A. A. (2017). Psychiatry of the Future: an Overview of Foreign Scientists Opinions of the Position of Psychiatry in the Modern World. Z. Nevrol. Psikhiatr. Im. S.S. Korsakova 117 (7), 99. doi:10.17116/jnevro20171177199-106
Bicknese, A. R., May, W., Hickey, W. F., and Dodson, W. E. (1992). Early Childhood Hepatocerebral Degeneration Misdiagnosed as Valproate Hepatotoxicity. Ann. Neurol. 32 (6), 767–775. doi:10.1002/ana.410320610
Boyd, S. G., Harden, A., Egger, J., and Pampiglione, G. (1986). Progressive Neuronal Degeneration of Childhood with Liver Disease ("Alpers' Disease"): Characteristic Neurophysiological Features. Neuropediatrics 17 (02), 75–80. doi:10.1055/s-2008-1052505
Chrysostomou, A., Grady, J. P., Laude, A., Taylor, R. W., Turnbull, D. M., and Lax, N. Z. (2016). Investigating Complex I Deficiency in Purkinje Cells and Synapses in Patients with Mitochondrial Disease. Neuropathol. Appl. Neurobiol. 42 (5), 477–492. doi:10.1111/nan.12282
Dai, L. F., Fang, F., Liu, Z. M., Shen, D. M., Ding, C. H., Li, J. W., et al. (2019). [Phenotype and Genotype of Twelve Chinese Children with Mitochondrial DNA Depletion Syndromes]. Zhonghua Er Ke Za Zhi 57 (3), 211–216. doi:10.3760/cma.j.issn.0578-1310.2019.03.011
Dimauro, S., Schon, E. A., Carelli, V., and Hirano, M. (2013). The Clinical Maze of Mitochondrial Neurology. Nat. Rev. Neurol. 9 (8), 429–444. doi:10.1038/nrneurol.2013.126
Elo, J. M., Yadavalli, S. S., Euro, L., Isohanni, P., Götz, A., Carroll, C. J., et al. (2012). Mitochondrial Phenylalanyl-tRNA Synthetase Mutations Underlie Fatal Infantile Alpers Encephalopathy. Hum. Mol. Genet. 21 (20), 4521–4529. doi:10.1093/hmg/dds294
Engelsen, B. A., Tzoulis, C., Karlsen, B., Lillebø, A., Laegreid, L. M., Aasly, J., et al. (2008). POLG1 Mutations Cause a Syndromic Epilepsy with Occipital Lobe Predilection. Brain 131 (3), 818–828. doi:10.1093/brain/awn007
Hance, N., Ekstrand, M. I., and Trifunovic, A. (2005). Mitochondrial DNA Polymerase Gamma Is Essential for Mammalian Embryogenesis. Hum. Mol. Genet. 14 (13), 1775–1783. doi:10.1093/hmg/ddi184
Hannah, R. R., and Yasir, A. K. (2021). Alpers-Huttenlochen Syndrome (AHS, Alper Disease). StatPearls 2021, 01.
Hayhurst, H., Anagnostou, M. E., Bogle, H. J., Grady, J. P., Taylor, R. W., Bindoff, L. A., et al. (2019). Dissecting the Neuronal Vulnerability Underpinning Alpers' Syndrome: a Clinical and Neuropathological Study. Brain Pathol. 29 (1), 97–113. doi:10.1111/bpa.12640
Jacobs, J., Vogt, C., LeVan, P., Zelmann, R., Gotman, J., and Kobayashi, K. (2016). The Identification of Distinct High-Frequency Oscillations during Spikes Delineates the Seizure Onset Zone Better Than High-Frequency Spectral Power Changes. Clin. Neurophysiol. 127 (1), 129–142. doi:10.1016/j.clinph.2015.04.053
Jin, Y., Chen, G., Xiao, W., Hong, H., Xu, J., Guo, Y., et al. (2019). Sequencing XMET Genes to Promote Genotype-Guided Risk Assessment and Precision Medicine. Sci. China Life Sci. 62 (7), 895–904. doi:10.1007/s11427-018-9479-5
Khan, A., Trevenen, C., Wei, X. C., Sarnat, H. B., Payne, E., and Kirton, A. (2012). Alpers Syndrome: the Natural History of a Case Highlighting Neuroimaging, Neuropathology, and Fat Metabolism. J. Child. Neurol. 27 (5), 636–640. doi:10.1177/0883073811423973
Li, Z., Zhu, P., Huang, H., Pan, Y., Han, P., Cui, H., et al. (2019). Identification of a Novel COL4A5 Mutation in the Proband Initially Diagnosed as IgAN from a Chinese Family with X-Linked Alport Syndrome. Sci. China Life Sci. 62 (12), 1572–1579. doi:10.1007/s11427-018-9545-3
Lim, A., and Thomas, R. H. (2020). The Mitochondrial Epilepsies. Eur. J. Paediatr. Neurol. 24, 47–52. doi:10.1016/j.ejpn.2019.12.021
London, F., Hadhoum, N., Outteryck, O., Vermersch, P., and Zéphir, H. (2017). Late-onset of Alpers-Huttenlocher Syndrome: an Unusual Cause of Refractory Epilepsy and Liver Failure. Acta Neurol. Belg. 117 (1), 399–401. doi:10.1007/s13760-016-0672-8
Mccoy, B., Owens, C., Howley, R., Ryan, S., King, M., Farrell, M. A., et al. (2011). Partial Status Epilepticus - Rapid Genetic Diagnosis of Alpers' Disease. Eur. J. Paediatr. Neurol. 15 (6), 558–562. doi:10.1016/j.ejpn.2011.05.012
Melani, F., Zelmann, R., Dubeau, F., and Gotman, J. (2013). Occurrence of Scalp-Fast Oscillations Among Patients with Different Spiking Rate and Their Role as Epileptogenicity Marker. Epilepsy Res. 106 (3), 345–356. doi:10.1016/j.eplepsyres.2013.06.003
Montine, T. J., Powers, J. M., Vogel, F. S., and Radtke, R. A. (1995). Alpers' Syndrome Presenting with Seizures and Multiple Stroke-like Episodes in a 17-Year-Old Male. Clin. Neuropathol. 14, 322–326.
Naviaux, R. K., Nyhan, W. L., Barshop, B. A., Poulton, J., Markusic, D., Karpinski, N. C., et al. (1999). Mitochondrial DNA Polymerase Gamma Deficiency and mtDNA Depletion in a Child with Alpers' Syndrome. Ann. Neurol. 45 (1), 54–58. doi:10.1002/1531-8249(199901)45:1<54:aid-art10>3.0.co;2-b
Nishikawa, A., Otani, Y., Ito, S., Nagata, S., Shiota, M., Takanashi, J. I., et al. (2020). A De Novo GABRB2 Variant Associated with Myoclonic Status Epilepticus and Rhythmic High-Amplitude delta with Superimposed (Poly) Spikes (RHADS). Epileptic Disord. 22 (4), 476–481. doi:10.1684/epd.2020.1183
Park, S., Kang, H. C., Lee, J. S., Park, Y. N., Kim, S., and Koh, H. (2017). Alpers-Huttenlocher Syndrome First Presented with Hepatic Failure: Can Liver Transplantation Be Considered as Treatment Option? Pediatr. Gastroenterol. Hepatol. Nutr. 20 (4), 259–262. doi:10.5223/pghn.2017.20.4.259
Qian, Y., Ziehr, J. L., and Johnson, K. A. (2015). Alpers Disease Mutations in Human DNA Polymerase Gamma Cause Catalytic Defects in Mitochondrial DNA Replication by Distinct Mechanisms. Front. Genet. 6, 135. doi:10.3389/fgene.2015.00135
Rahman, S., and Copeland, W. C. (2019). POLG-related Disorders and Their Neurological Manifestations. Nat. Rev. Neurol. 15 (1), 40–52. doi:10.1038/s41582-018-0101-0
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and Guidelines for the Interpretation of Sequence Variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Samanta, D., Gokden, M., and Willis, E. (2018). Clinicopathologic Findings of CARS2 Mutation. Pediatr. Neurol. 87, 65–69. doi:10.1016/j.pediatrneurol.2018.06.009
Shen, Y., and Shi, T. (2019). Innovation for Better Health of Children. Sci. China Life Sci. 62 (12), 1561–1562. doi:10.1007/s11427-019-1589-2
Sofou, K., Kollberg, G., Holmström, M., Dávila, M., Darin, N., Gustafsson, C. M., et al. (2015). Whole Exome Sequencing Reveals Mutations in NARS2 and PARS2, Encoding the Mitochondrial Asparaginyl-tRNA Synthetase and Prolyl-tRNA Synthetase, in Patients with Alpers Syndrome. Mol. Genet. Genomic Med. 3 (1), 59–68. doi:10.1002/mgg3.115
Sofou, K., Moslemi, A. R., Kollberg, G., Bjarnadóttir, I., Oldfors, A., Nennesmo, I., et al. (2012). Phenotypic and Genotypic Variability in Alpers Syndrome. Eur. J. Paediatr. Neurol. 16 (4), 379–389. doi:10.1016/j.ejpn.2011.12.006
Sofou, K., Kollberg, G., Hedberg-Oldfors, C., and Oldfors, A. (2021). The Phenotypic Variability and Natural History of NARS2 Associated Disease. Eur. J. Paediatric Neurol. 31, 31–37. doi:10.1016/j.ejpn.2021.01.012
Tarka, S., Laure-Kamionowska, M., Wierzba-Bobrowicz, T., Witulska, K., Ciara, E., Szymańska, K., et al. (2020). POLG Gene Mutation. Clinico-Neuropathological Study. Folia Neuropathol. 58 (4), 386–392. doi:10.5114/fn.2020.102441
Tzoulis, C., Tran, G. T., Coxhead, J., Bertelsen, B., Lilleng, P. K., Balafkan, N., et al. (2014). Molecular Pathogenesis of Polymerase γ-related Neurodegeneration. Ann. Neurol. 76 (1), 66–81. doi:10.1002/ana.24185
Uusimaa, J., Hinttala, R., Rantala, H., Päivärinta, M., Herva, R., Röyttä, M., et al. (2008). Homozygous W748S Mutation in the POLG1 Gene in Patients with Juvenile-Onset Alpers Syndrome and Status Epilepticus. Epilepsia 49 (6), 1038–1045. doi:10.1111/j.1528-1167.2008.01544.x
van Westrhenen, A., Cats, E. A., van den Munckhof, B., van der Salm, S. M. A., Teunissen, N. W., Ferrier, C. H., et al. (2018). Specific EEG Markers in POLG1 Alpers' Syndrome. Clin. Neurophysiol. 129 (10), 2127–2131. doi:10.1016/j.clinph.2018.07.016
Walker, M. A., Mohler, K. P., Hopkins, K. W., Oakley, D. H., Sweetser, D. A., Ibba, M., et al. (2016). Novel Compound Heterozygous Mutations Expand the Recognized Phenotypes of FARS2-Linked Disease. J. Child. Neurol. 31 (9), 1127–1137. doi:10.1177/0883073816643402
Wiltshire, E., Davidzon, G., DiMauro, S., Akman, H. O., Sadleir, L., Haas, L., et al. (2008). Juvenile Alpers Disease. Arch. Neurol. 65 (1), 121–124. doi:10.1001/archneurol.2007.14
Wolf, N. I., Rahman, S., Schmitt, B., Taanman, J. W., Duncan, A. J., Harting, I., et al. (2009). Status Epilepticus in Children with Alpers' Disease Caused by POLG1 Mutations: EEG and MRI Features. Epilepsia 50 (6), 1596–1607. doi:10.1111/j.1528-1167.2008.01877.x
Wu, J., Yang, C., Collins, J., and Ginat, D. T. (2018). Case 250: Alpers-Huttenlocher Syndrome. Radiology 286 (2), 720–725. doi:10.1148/radiol.2017151748
Keywords: Alpers, syndrome, POLG, electroencephalogram, RHADS, diagnosis
Citation: Li H, Wang W, Han X, Zhang Y, Dai L, Xu M, Deng J, Ding C, Wang X, Chen C, Yang X and Fang F (2021) Clinical Attributes and Electroencephalogram Analysis of Patients With Varying Alpers’ Syndrome Genotypes. Front. Pharmacol. 12:669516. doi: 10.3389/fphar.2021.669516
Received: 19 February 2021; Accepted: 30 August 2021;
Published: 06 October 2021.
Edited by:
Tieliu Shi, East China Normal University, ChinaReviewed by:
Bo Liu, Sichuan University, ChinaCopyright © 2021 Li, Wang, Han, Zhang, Dai, Xu, Deng, Ding, Wang, Chen, Yang and Fang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaofeng Yang, eGlhb2Zlbmd5YW5nQHlhaG9vLmNvbQ==; Fang Fang, ZmFuZ2ZhbmdAYmNoLmNvbS5jbg==
†ORCID: Hua Li; orcid.org/0000-0001-7334-534X; , Xiaofeng Yangorcid.org/0000-0002-7847-0665; Fang Fang, orcid.org/0000-0001-6362-7896
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.