
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol., 20 April 2021
Sec. Cardiovascular and Smooth Muscle Pharmacology
Volume 12 - 2021 | https://doi.org/10.3389/fphar.2021.667254
This article is part of the Research TopicNew Trends in the Renin-Angiotensin-Aldosterone System in Cardiovascular DiseaseView all 9 articles
 Annamaria Mascolo1,2*†
Annamaria Mascolo1,2*† Cristina Scavone1,2†
Cristina Scavone1,2† Concetta Rafaniello1,2
Concetta Rafaniello1,2 Antonella De Angelis2
Antonella De Angelis2 Konrad Urbanek2,3Gabriella di Mauro1,2Donato Cappetta2
Konrad Urbanek2,3Gabriella di Mauro1,2Donato Cappetta2 Liberato Berrino2
Liberato Berrino2 Francesco Rossi1,2‡
Francesco Rossi1,2‡ Annalisa Capuano1,2‡
Annalisa Capuano1,2‡The renin-angiotensin-aldosterone system (RAAS) firstly considered as a cardiovascular circulating hormonal system, it is now accepted as a local tissue system that works synergistically or independently with the circulating one. Evidence states that tissue RAAS locally generates mediators with regulatory homeostatic functions, thus contributing, at some extent, to organ dysfunction or disease. Specifically, RAAS can be divided into the traditional RAAS pathway (or classic RAAS) mediated by angiotensin II (AII), and the non-classic RAAS pathway mediated by angiotensin 1–7. Both pathways operate in the heart and lung. In the heart, the classic RAAS plays a role in both hemodynamics and tissue remodeling associated with cardiomyocyte and endothelial dysfunction, leading to progressive functional impairment. Moreover, the local classic RAAS may predispose the onset of atrial fibrillation through different biological mechanisms involving inflammation, accumulation of epicardial adipose tissue, and electrical cardiac remodeling. In the lung, the classic RAAS regulates cell proliferation, immune-inflammatory response, hypoxia, and angiogenesis, contributing to lung injury and different pulmonary diseases (including COVID-19). Instead, the local non-classic RAAS counteracts the classic RAAS effects exerting a protective action on both heart and lung. Moreover, the non-classic RAAS, through the angiotensin-converting enzyme 2 (ACE2), mediates the entry of the etiological agent of COVID-19 (SARS-CoV-2) into cells. This may cause a reduction in ACE2 and an imbalance between angiotensins in favor of AII that may be responsible for the lung and heart damage. Drugs blocking the classic RAAS (angiotensin-converting enzyme inhibitors and angiotensin receptor blockers) are well known to exert a cardiovascular benefit. They are recently under evaluation for COVID-19 for their ability to block AII-induced lung injury altogether with drugs stimulating the non-classic RAAS. Herein, we discuss the available evidence on the role of RAAS in the heart and lung, summarizing all clinical data related to the use of drugs acting either by blocking the classic RAAS or stimulating the non-classic RAAS.
The renin-angiotensin-aldosterone system (RAAS) is first considered a cardiovascular circulating hormonal system. it is now accepted also as a local tissue system that works synergistically or independently with the circulating one (Labandeira-Garcia et al., 2014; Mascolo et al., 2017). Evidence states that tissue RAAS locally generates mediators with homeostatic regulatory functions, thus contributing, to some extent, to organ dysfunction or disease (Rossi et al., 2016; Mascolo et al., 2017; Mascolo et al., 2020a; Mascolo et al., 202b). The RAAS can be divided into the traditional RAAS pathway (or classic RAAS) mediated by angiotensin II (AII), and the non-classic RAAS pathway mediated by angiotensin 1–7 (A1-7). Both pathways are locally present in the heart and lung. In the heart, an enhancement of classic RAAS, at the expense of non-classic RAAS, can induce cardiac hypertrophy, fibrosis, and dysfunction leading to heart failure (HF) and atrial fibrillation (AF) (Rossi et al., 2016; Mascolo et al., 2020b). In the lung, the classic RAAS also regulates cell proliferation, immune-inflammatory response, hypoxia, and angiogenesis, contributing to lung injury and different pulmonary diseases (Mascolo et al., 2020a; Catarata et al., 2020). Instead, the local non-classic RAAS counteracts the classic RAAS effects exerting a protective action on both heart and lung. However, it is essential to notice that a component of the non-classic RAAS, the transmembrane angiotensin-converting-enzyme 2 (ACE2), localized on the lung alveolar epithelial cells, is a receptor mediating the viral entry of the severe acute respiratory syndrome coronavirus 1 (SARS-COV-1) and SARS-COV-2, responsible for the SARS and the coronavirus disease 2019 (COVID-19), respectively (Li et al., 2003; Turner et al., 2004; Hoffmann et al., 2020). Despite the main symptoms of COVID-19 are respiratory and flu-like symptoms, which can be complicated by lymphopenia and high levels of pro-inflammatory cytokines leading to acute respiratory distress syndrome (ARDS), organ failure, and disseminated coagulopathy (Guo et al., 2020); some patients also develop cardiovascular symptoms (Huang et al., 2020). In this view, it seems pertinent to summarize the evidence on the role of RAAS in cardiac diseases (such as HF and AF) and pulmonary diseases with a focus on COVID-19. Notably, drugs blocking the classic RAAS, well known to exert a cardiovascular benefit, are under evaluation for blocking AII-induced lung injury together with drugs stimulating the non-classic RAAS. Herein, we discuss the evidence on the role of RAAS in the heart and lung, summarizing all clinical data related to the use of drugs acting either by blocking the classic RAAS or stimulating the non-classic RAAS.
The main effector peptide of classic RAAS is the AII, whose synthesis starts with the cleavage of angiotensinogen into angiotensin I (AI) by the renin and then its conversion into AII by the angiotensin-converting enzyme (ACE) (Figure 1) (Unger, 2002). However, AII can also be synthesized through pathways that involve other enzymes like chymase, chymostatin-sensitive angiotensin II-generating enzyme (CAGE), and cathepsin G (Mascolo et al., 2020b). These alternative pathways play a role in the local production of AII. In fact, in the heart, angiotensin 1–12 can be converted by chymase into AII, and this synthesis is significant in inducing adverse left ventricular remodeling post-myocardial infarction (Ahmad et al., 2014). Once synthesized, AII can interact with three receptors (AT1, AT2, and nonAT1nonAT2). AT1 and AT2 are G protein-coupled receptors (Unger, 2002), while nonAT1nonAT2 is an angiotensinase or an angiotensin clearance receptor (Karamyan et al., 2010). The stimulation of the AT1 receptor induces vasoconstriction, increases the release of catecholamines and the synthesis of aldosterone (Unger, 2002), stimulates fibrosis and inflammation, and reduces the activity of collagenase and the expression of mitogen-activated protein kinase (MAPK) (Mascolo et al., 2017; Mascolo et al., 2020b). The pro-inflammatory action of AT1 receptors involves the down-regulation of the NADPH oxidase expression in smooth muscle cells, the production of reactive oxygen species (ROS), and the activity of pro-inflammatory transcription nuclear factors like nuclear factor-kappaB (NF-κB) and E26 transformation-specific sequence (Ets) (Marchesi et al., 2008; Porreca et al., 2017). Moreover, these receptors induce the release of tumor necrosis factor-α (TNF-α), the interleukin-6 (IL-6), and the monocyte chemoattractant protein-1 (MCP-1) (Dandona et al., 2007) and shift the macrophage phenotype toward the pro-inflammatory M1 polarization state (Yamamoto et al., 2011). On the contrary, the stimulation of AT2 receptors exerts a protective role by inducing anti-inflammatory, anti-oxidative, and anti-fibrotic effects (Unger, 2002). Instead, the primary mediator of non-classic RAAS is the A1-7, whose synthesis can involve two pathways. One pathway starts with the cleavage of AII into A1-7 by the carboxypeptidase ACE2. A second pathway begins with the cleavage of AI into angiotensin 1–9 (A1–9) by ACE2 and its consecutive conversion into A1–7 by ACE (Figure 1) (Mascolo et al., 2020b). ACE2 is classified into the soluble form present in the plasma and a transmembrane form existing locally in both the heart and lung (Mascolo et al., 2020a). Both forms contribute to the generation of A1-7, which can interact with the G protein-coupled receptor MAS1, promoting the nitric oxide release (Fraga-Silva et al., 2008), Akt phosphorylation (Dias-Peixoto et al., 2008), and anti-inflammatory effects (da Silveira et al., 2010). Moreover, ACE and ACE2 participate in the inflammation as components of a local RAAS at sites infiltrated by monocytes/macrophages. Both enzymes are expressed by human monocytes where metabolize AI to multiple angiotensin peptides. In particular, classical monocytes (CD14++CD16−) produce both AII and A1–9/A1–7, whereas the non-classical subtype (CD14+CD16++) produces mainly A1–7 (Rutkowska-Zapała et al., 2015).
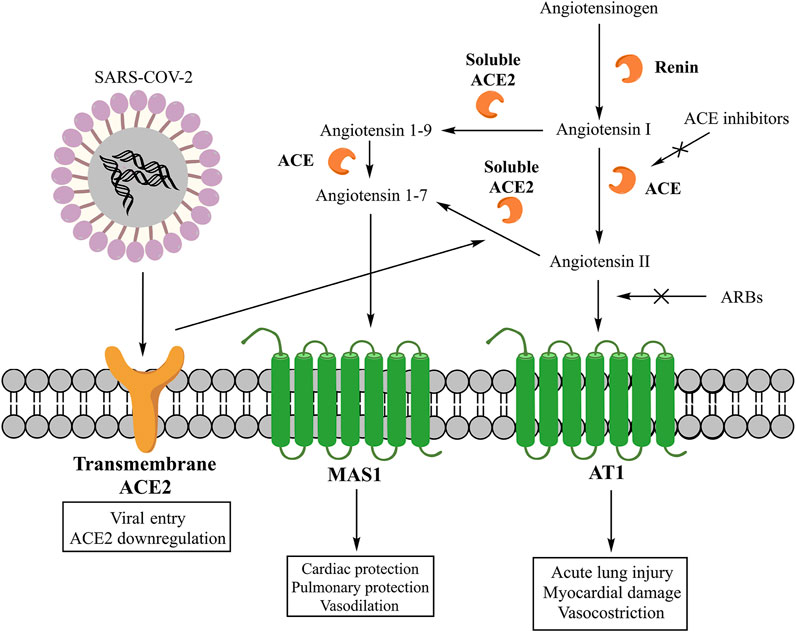
FIGURE 1. Classic and non-classic RAAS and its interaction with SARS-COV-2. From Mascolo A, Scavone C, Rafaniello C, Ferrajolo C, Racagni G, Berrino L, Paolisso G, Rossi F, Capuano A. Renin-Angiotensin System and Coronavirus disease 2019: A Narrative Review. Front Cardiovasc Med. 2020 Aug 11;7:143. doi: 10.3389/fcvm.2020.00143.
Finally, the stimulation of MAS1 receptors on macrophages can inhibit their polarization to inflammatory phenotype and the release of pro-inflammatory cytokines (Mascolo et al., 2020b). Thus, A1-7 can be considered a beneficial axis component that exerts opposite effects to the classic RAAS (Santos et al., 2013).
It is recognized that the classic RAAS is involved in developing cardiac diseases such as HF and AF, which are closely interconnected. Atrial fibrillation’s key component is the structural remodeling that breaks tissue microarchitecture and disturbs ion currents and physiological cell-to-cell interconnections, but its importance extends beyond this arrhythmia. Atrial remodeling frequently corresponds with the ventricular remodeling in HF, increasing the complexity of the problem. Moreover, neurohormonal and structural alterations of HF can increase the probability of developing and advancing AF, and AF can favor incident HF development (Stewart et al., 2002; Maisel and Stevenson, 2003). The pathophysiological mechanisms of RAAS in these diseases are reported below. The AII stimulates collagen synthesis and fibroblast proliferation in the heart, inducing cardiac hypertrophy and fibrosis, which are critical elements of the adverse ventricular remodeling (Rossi et al., 2016; Mascolo et al., 2020b). Specifically, the local cardiac production of AII has been associated with an increase in myocardial mRNA expression of collagen I/III and fibronectin (Fielitz et al., 2001). Moreover, AII can stimulate the myocardial generation of aldosterone, which can also contribute to the synthesis of collagen and to the local production of AII. These effects drive the characteristics hemodynamics alterations (Rossi et al., 2016). Additionally, other than inducing fibrosis, AII can stimulate inflammatory processes and change the heart’s electrophysiological properties (electrical cardiac remodeling) (Li et al., 2001; Novo et al., 2008). These processes can influence the onset of AF. Of note, up-regulation of AT1 receptors was found in left atrial tissue of patients with lone AF or AF with underlying mitral valve disease compared to patients in sinus rhythm. In contrast, no difference was observed in the expression of AT2 receptors (Boldt et al., 2003). AII exerts electrical cardiac remodeling effects by shortening the atrial effective refractory period and the action potential duration potentiating the slow component of delayed rectifier K+ channels in guinea pig atrial myocytes (Zankov et al., 2006). Finally, a more recent hypothesis on the role of AII in inducing AF suggests that the classic RAAS may mediate epicardial fat accumulation and inflammation, which can, in turn, cause AF (Patel et al., 2016a). Epicardial fat accumulation can induce AF through direct and indirect pathophysiological mechanisms (Wong et al., 2017). The direct mechanism consists of epicardial adipocytes’ infiltration into the underlying atrial myocardium (Hatem and Sanders, 2014), while indirect mechanisms are: the release of inflammatory adipokines (such as TNF-α, IL-6, and MCP-1), ROS, and secrete matrix metalloproteinases 2 and 7, which can stimulate atrial remodeling and fibrosis (Carnes et al., 2001; Boixel et al., 2003; Conway et al., 2004; Malavazos et al., 2007; Kourliouros et al., 2011; Smit et al., 2012; Venteclef et al., 2015); the switch of macrophages from an anti-inflammatory M2 to a pro-inflammatory M1 polarization state (Jung and Choi, 2014); the activation of ganglionated plexi located in the epicardial fat (Wong et al., 2017); the stimulation of AF triggers (Nagashima et al., 2012; Nakahara et al., 2014).
On the contrary, the non-classic RAAS exerts a protective role in the heart by reducing inflammation, fibrosis, and cardiac electrical remodeling along with vasodilation and the reduction of hypertrophy and thrombosis (Esposito et al., 2018; Santos et al., 2013). As anti-fibrotic effects, A1–7 has shown the ability to increase the mRNA expression of extracellular signal-regulated kinase-1 (ERK)1/ERK2 (Liu et al., 2010). Moreover, the overexpression of ACE2 has been associated with a reduction in the expression of transient receptor potential melastatin 7, which is a Ca2+ channel expressed on fibroblasts that can contribute to the fibrogenesis mediated by the transforming growth factor (TGF) (Zhou et al., 2017). In opposition, ACE2 knockout animal models showed a worse left ventricular remodeling in response to the AII-induced acute injury, suggesting a protective role of non-classic RAAS in the myocardium recovery (Kassiri et al., 2009). As mentioned above, A1-7 inhibits the pro-inflammatory macrophage polarization state and the release of pro-inflammatory cytokines (Souza and Costa-Neto, 2012). Moreover, the non-classic RAAS can reduce the inflammation of the epicardial adipose tissue. An increase of adipose tissue macrophages, pro-inflammatory cytokines (TNF-α, IL-1β, IL-6), and iNOS was observed in ACE2 knockout mice (Patel et al., 2016b). Finally, A1-7 has shown the ability to prevent the ionic remodeling of AF in preclinical models (Liu et al., 2011). Based on the mechanisms mentioned above, a stimulation of the non-classic RAAS can benefit both AF and HF.
Classic RAAS blockers are renin inhibitors, ACE inhibitors, and Angiotensin Receptor Blockers (ARBs). Among them, ACE inhibitors and ARBs are widely used to treat cardiovascular diseases. Clinical evidence has also shown their potential for the prevention of AF (Novo et al., 2008; Mascolo et al., 2020b). Specifically, RAAS blockers effectively prevented primary AF in patients with early stage of HF and/or not severe hypertension. This is in accordance with their effect of blocking local inflammation and cardiac remodeling, which are expected to be at a maximum in patients in patients at an early stage of the disease. Therefore, it is not surprising to find a lower efficacy of these drugs for the secondary prevention of AF and in populations of patients at a more advanced stage of the disease (Mascolo et al., 2020b).
The mechanisms mediated by ACE-inhibitors and ARBs for cardiac protection are the inhibition of atrial fibrosis and inflammation, the prevention of electrical cardiac remodeling, and the epicardial adipose tissue’s modulation. Concerning inflammation, many studies have demonstrated that ARBs and ACE-inhibitors are associated with anti-oxidative and anti-inflammatory effects. Specifically, these drugs can reduce pro-inflammatory mediators such as C-reactive protein, IL-6, MCP-1, intercellular adhesion molecule-1, vascular cell adhesion molecule-1, NF-κB, and ROS, and increase anti-inflammatory mediators such as the inhibitor of κB and IL-10 (Dandona et al., 2007). Some ARBs exert anti-inflammatory effects because they are agonists of the peroxisome proliferator-activated receptor γ (PPARγ). This intracellular nuclear hormone receptor controls the expression of pro-inflammatory genes through the inhibition of the AP-1 and NF-κB transcription factors. Among ARBs, telmisartan (with a biphenyl tetrazole group) has a higher affinity to PPARγ, followed by candesartan and losartan (Saavedra, 2012).
Regarding the prevention of electrical cardiac remodeling, RAAS blockers have shown the ability to prevent the shortening of the atrial effective refractory period (Nakashima et al., 2000), improve intra-atrial conduction (Wang and Li, 2018), and prolong the action potential duration (Zankov et al., 2006). Moreover, a preclinical study of dogs with ventricular tachypacing-induced congestive HF found that enalapril, an ACE-inhibitor, can reduce conduction abnormalities, atrial fibrosis, and ERK activation (Li et al., 2001; Moccia et al., 2015). Finally, ACE-inhibitors and ARBs may exert cardiac protection by inhibiting epicardial fat accumulation and downsizing epicardial adipocytes (Mori et al., 2007).
Drugs stimulating the non-classic RAAS, such as the human recombinant ACE2 and agonists of MAS1 receptors, are under investigation for cardiovascular diseases (Mascolo et al., 2017). Preclinical evidence in wild-type mice showed that human recombinant ACE2 reduced AII-induced cardiac remodeling and myocardial fibrosis. ACE2 reduced the transcription of fibronectin, TGF-β1, procollagen type I α 1, and procollagen type III α 1, the phosphorylation of the Janus kinase 2, extracellular signal-regulated 1/2, and the levels of protein kinase C-α and protein kinase C-β1 (Zhong et al., 2010). Moreover, the human recombinant ACE2 showed the ability to attenuate diabetic kidney injury, reduced blood pressure and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity in Akita mouse models (Oudit et al., 2010). Finally, its administration showed a protective effect in murine models of AII-induced HF with preserved ejection fraction and pressure-overload mediated HF with reduced ejection fraction (Patel et al., 2017). Regarding clinical evidence, the human recombinant ACE2 has completed phase I (NCT00886353) and phase II (NCT01597635) clinical trials, and its administration was well tolerated with no evident cardiovascular effect in healthy subjects (Haschke et al., 2013). Among MAS1 receptor agonists investigated for treating cardiovascular diseases, there is the non-peptide compound AVE 0991 and the A1-7. AVE 0991 was studied in combination with a renin inhibitor (aliskiren) in rats with experimental hypertension and showed a synergistic effect in lowering the blood pressure (Singh et al., 2013). A1-7 has been investigated in a vector of hydroxypropyl-β-cyclodextrin. With this new formulation, designed to protect A1-7 from degradation and to increase its half-life through a slow-release, A1-7 lowered the blood pressure in animal models (Bertagnolli et al., 2014). Moreover, A1–7 has shown a beneficial cardioprotective effect in various murine models of HF with reduced or preserved ejection fraction (Patel et al., 2017). Finally, clinical data on A1-7 in HF are lacking.
The RAAS seems involved in the development of multiple lung diseases, such as idiopathic pulmonary fibrosis, sarcoidosis, pulmonary hypertension, acute respiratory distress syndrome, lung cancer, and COVID-19 (Catarata et al., 2020; Mascolo et al., 2020a). An increased expression of ACE was observed in lung interstitium in several diseases, supporting the notion of a pulmonary local RAAS and a role for the AII in lung injury and fibrosis (Marshall, 2005). Both AT1 and AT2 receptors are present in the normal and pathological human lung (Catarata et al., 2020). The AT1 receptors were found on vascular smooth muscle cells, alveolar macrophages and in the stroma underneath the airway epithelium, while AT2 receptors were detected in bronchial epithelium and endothelial cells (Bullock et al., 2001). Physiological and pathophysiological effects of AII are mainly mediated through the activation of AT1 receptors (Chung et al., 1996). These receptors mediate vasoconstriction, cell proliferation, angiogenesis, and inflammation with increased pro-inflammatory cytokines, oxidative stress and fibrosis, inflammatory cell chemotaxis, and epithelial cell apoptosis (Kaparianos and Argyropoulou, 2011). Moreover, in vitro studies have demonstrated that the epithelial to mesenchymal transition (EMT) induced by TGF-β1 was associated with an increased expression of angiotensinogen and AT1 receptor in human lung fibroblasts (Abdul-Hafez et al., 2009; Renzoni et al., 2004; Uhal et al., 2007). Finally, the expression of TGF-β1 in human lung myofibroblasts was reduced by AT1 receptor blockade and associated with collagen synthesis inhibition (Uhal et al., 2007). In contrast, AT2 receptors were associated with opposite effects, although some pro-inflammatory effects were observed through the NF-kB pathway activation (Kaparianos and Argyropoulou, 2011). The impact of the classic RAAS in lung pathophysiology was also evident in studies that found inhibition of bleomycin-, γ irradiation-, amiodarone- and paraquat-induced pulmonary fibrosis with the administration of ACE inhibitors (captopril, enalapril, lisinopril, and perindopril) in rats (Mohammadi-Karakani et al., 2006; Molteni et al., 2007; Wang et al., 2000). Moreover, a post hoc analysis of data from a phase 3, placebo-controlled, clinical trial showed a slower disease progression in patients with idiopathic pulmonary fibrosis treated with ACE inhibitors (Kreuter et al., 2019). Because AII and TGF-β1 may influence each other’s activity or act in synergy, the inhibition of both local mediators could delay the progression of lung fibrosis.
Regarding the non-classic RAAS, ACE2 was found in endothelial and smooth muscle cells, alveolar epithelial type I and II cells, and bronchial epithelial cells (Catarata et al., 2020). In the lung, ACE2 has multiple physiological roles: it exerts opposing effects to the classic RAAS as a negative regulator, and it is the receptor for SARS-COV-1 and SARS-COV-2 entry (Figure 1) (Gheblawi et al., 2020). As the negative regulator, the non-classic RAAS can reduce lung injury and prevent acute respiratory distress (Wösten-Van Asperen et al., 2011; Chen et al., 2013; Meng et al., 2015). As the SARS-COV-2 receptor, ACE2 binds the SARS-COV-2’s glycosylated spike (S) protein. This bond is mediated by the human androgen-sensitive transmembrane serine protease type 2 (TMPRSS211) (Mascolo et al., 2020a; Hoffmann et al., 2020) that cleaves the S protein into S1 and S2 subunits (South et al., 2020). The S1 subunit binds the ACE2 and facilitates the viral attachment, whereas the S2 subunit drives the membrane fusion and viral internalization in the pulmonary epithelium (Hoffmann et al., 2020). An important consideration that needs to be done for the pathophysiology of COVID-19 is related to the ACE2 internalization mediated by SARS-COV-2 that could potentially induce a reduction of ACE2 on cell surface and then determine the absence of a key factor important for the local pulmonary synthesis of A1-7. Indeed, an imbalance between AII and A1-7 levels may exacerbate the lung injury caused by SARS-COV-2, contributing to the reduction of the pulmonary function and the increase of fibrosis and inflammation (Triassi et al., 2019; South et al., 2020).
In conclusion, a complete understanding of the role of RAAS in the pulmonary inflammation and fibrosis is fundamental and may open new therapeutic possibilities for the treatment of respiratory diseases, including COVID-19.
The use of RAAS blockers (ACE-inhibitors and ARBs) in COVID-19 patients has been object of discussion during the last year. First, evidence suggested that RAAS blockers may contribute to more adverse health outcomes by increasing the expression of ACE2 mRNA and then potentiating the virulence of SARS-COV-2 (Vaduganathan et al., 2020; Zheng et al., 2020). However, today, there is no study suggesting this association. Even if there was such association, there is no evidence demonstrating a causal relationship between the ACE2 activity and the SARS-COV-2 associated mortality (Kuster et al., 2020).
Another hypothesis considers the ability of SARS-COV-2 to enter any tissue expressing the ACE2, including the heart or other cardiovascular tissues (South et al., 2020). By this mechanism, SARS-COV-2 can induce a reduction of ACE2 in favor of the classic RAAS (increase in AII) that can cause heart damage, which might be even worse in patients with underlying cardiovascular diseases (South et al., 2020; Yousif et al., 2012). However, in this scenario, the RAAS blocker could be protective and beneficial for preventing AII-induced cardiac damage. As RAAS blocker are known to determine clinical benefits, another vital aspect to be considered is the potential damage when a RAAS blocker therapy is stopped in a patient with a stable cardiovascular condition (Mascolo et al., 2020a).
Data available on this topic come from observational studies that found no association between the use of ARBs or ACE-inhibitors with COVID-19 diagnosis (Gnavi et al., 2020; Mancia et al., 2020), admission to hospital for COVID-19 (de Abajo et al., 2020), or COVID-19 severity (Reynolds et al., 2020). Moreover, another large observational study that compared the use of ACE-inhibitors and ARBs with active control (calcium channel blockers, and thiazide or thiazide-like diuretics) found no association between COVID-19 diagnosis and ACE-inhibitor or ARB use, and no significant difference between drug classes for the risk of hospital admission with COVID-19, hospital admission with pneumonia, acute respiratory distress syndrome, acute kidney injury, or sepsis across all comparisons (Reynolds et al., 2020). Finally, a cross-sectional, observational, multicenter, nationwide Italian study found that ACE inhibitors or other antihypertensive agents did not affect the outcome of COVID-19 (Iaccarino et al., 2020).
Regarding mortality, two observational studies found similar mortality rates between the use of RAAS blockers and non-RAAS blockers in COVID-19 patients (Gao et al., 2020; Jung et al., 2020). One retrospective study showed a lower risk of COVID-19 mortality in hospitalized patients with COVID-19 and hypertension who received ACE inhibitor/ARB than those who did not receive this treatment (Zhang et al., 2020). However, as recently reported in the preliminary results of a randomized trial (BRACE CORONA, NCT04364893), presented at the European Society of Cardiology Congress, the use of RAAS blockers was not associated with a beneficial effect, but considering that mortality was very low (2.7–2.8%) in the trial its validity is under question (de Abajo, 2020).
Scientific Societies recommend continuing the treatment with the usual anti-hypertensive agent in patients with COVID-19 and not stopping the RAAS inhibitor therapy as no evidence suggests so (American Heart Association, 2020; European Society of Cardiology, 2020; Italian Society of Cardiology, 2020; Italian Society of Hypertension, 2020; Italian Society of Pharmacology, 2020).
Several clinical trials (ClinicalTrials.gov identifier, NCT04351581, NCT04353596, NCT04329195, NCT04351724) are ongoing to evaluate the clinical benefit of continuing or not the treatment with RAAS blockers in patients with COVID-19. Besides, based on the organ protective effects of RAAS blockers, many studies are ongoing to investigate their efficacy in patients with COVID-19. The beneficial effects of ACE inhibitors and ARBs is hypothesized to be related to the block of the classic RAAS in favor of the ACE2/A1-7 pathway as demonstrated in experimental studies (Chappell, 2016; Santos et al., 2019). In this regard, several clinical trials are ongoing to investigate the role of losartan (NCT04312009, NCT04311177, NCT04328012, NCT04428268, NCT04643691, NCT04606563, NCT04447235, NCT04340557, NCT04335123), valsartan (NCT04335786), and telmisartan (NCT04360551, NCT04355936, NCT04359953, NCT04510662, NCT04466241, NCT04356495) for the treatment of COVID-19 (Table 1).
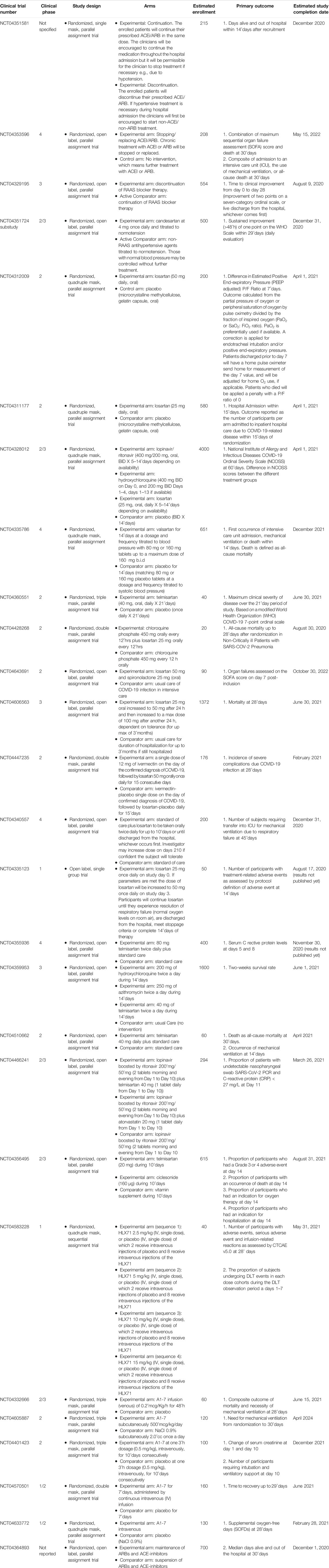
TABLE 1. Characteristics of ongoing clinical trials on the continuation or suspension of RAAS blockers in patients with COVID-19, and on the efficacy of ACE inhibitors, ARBs, angiotensin 1-7, and ACE2 for COVID-19.
Considering the beneficial effects of the non-classic RAAS in the heart and lung, which seems in part lacking in patients with COVID-19, hypotheses were advanced on the potential therapeutic approach of restoring the ACE2/A1-7 pathway. Preclinical evidence showed that the infusion of A1-7 improved oxygenation, and reduced inflammation and fibrosis in two ARDS models (Wösten-Van Asperen et al., 2011; Zambelli et al., 2015; Cuomo et al., 2017). Moreover, the therapy with the soluble human recombinant ACE2 reversed the lung-injury process induced by other viral infections (Zou et al., 2014; Gu et al., 2016). It is crucial to notice that by administering the soluble ACE2, it is possible to stimulate the protective non-classic RAAS without increasing the transmembrane ACE2, avoiding potentiating the viral entry into cells.
Clinical evidence on the role of the non-classic RAAS in COVID-19 is scarce. A phase 2 clinical trial showed that the infusion of ACE2 safely reduced the level of AII in patients with ARDS. However, this trial had no enough power to show efficacy in pulmonary function improvement (Khan et al., 2017). There is an ongoing phase 1 clinical trial (NCT04583228) aiming to evaluate safety, tolerability, pharmacodynamics, pharmacokinetics, and immunogenicity of the human recombinant ACE2-Fc fusion protein (HLX71) in healthy subjects. Finally, several clinical trials are ongoing to assess efficacy and safety of A1-7 infusion in COVID-19 patients (NCT04332666, NCT04605887, NCT04401423, NCT04570501, and NCT04633772). Characteristics of the aforementioned ongoing studies are shown in Table 1.
The classic RAAS plays an important role in the pathophysiology of cardiac diseases, while the non-classic RAAS exerts cardioprotective effects. Classic RAAS blockers are widely used for their efficacy in cardiovascular diseases and benefit from preventing primary AF. These drugs are also under consideration for preventing AII-induced lung injury. Indeed, many clinical trials are ongoing to evaluate their use in COVID-19. The rationale for using such drugs in COVID-19 is related to the imbalance between AII and A1-7 in favor of AII that can be caused by SARS-COV-2 internalization. A reduction in ACE2 can indeed further contribute to pulmonary function deterioration and myocardial damage. Moreover, for patients with COVID-19 already in treatments with RAAS blockers, Scientific Societies recommend not to suspend this treatment. Finally, clinical trials are ongoing to evaluate the beneficial pulmonary effect of restoring the ACE2/A1-7 pathway in COVID-19 patients.
Drafting the work and revising it for important intellectual content: AM, CS, CR, AD, KU, GD, DC, LB, FR, AC. Final approval of the version to be published: AM, CS, CR, AD, KU, GD, DC, LB, FR, AC. Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately discussed: AM, CS, CR, AD, KU, GD, DC, LB, FR, AC. Developed the concept and designed the study: FR and AC. Wrote the paper: AM and CS.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abdul-Hafez, A., Shu, R., and Uhal, B. D. (2009). JunD and HIF‐1α Mediate Transcriptional Activation of Angiotensinogen by TGF‐β1 in Human Lung Fibroblasts. FASEB j. 23, 1655–1662. doi:10.1096/fj.08-114611
Ahmad, S., Varagic, J., Groban, L., Dell’Italia, L. J., Nagata, S., Kon, N. D., et al. (2014). Angiotensin-(1-12): A Chymase-Mediated Cellular Angiotensin II Substrate. Curr. Hypertens. Rep. 16, 429. doi:10.1007/s11906-014-0429-9
American Heart Association (2020). HFSA/ACC/AHA statement addresses concerns re: using RAAS antagonists in COVID-19. Available at: https://www.acc.org/latest-in-cardiology/articles/2020/03/17/08/59/hfsa-acc-aha-statement-addresses-concerns-re-using-raas-antagonists-in-covid-19 (Accessed January 5, 2021).
Bertagnolli, M., Casali, K. R., De Sousa, F. B., Rigatto, K., Becker, L., Santos, S. H. S., et al. (2014). An Orally Active Angiotensin-(1-7) Inclusion Compound and Exercise Training Produce Similar Cardiovascular Effects in Spontaneously Hypertensive Rats. Peptides. 51, 65–73. doi:10.1016/j.peptides.2013.11.006
Boixel, C., Fontaine, V., Rücker-Martin, C., Milliez, P., Louedec, L., Michel, J.-B., et al. (2003). Fibrosis of the Left Atria during Progression of Heart Failure Is Associated with Increased Matrix Metalloproteinases in the Rat. J. Am. Coll. Cardiol. 42, 336–344. doi:10.1016/S0735-1097(03)00578-3
Boldt, A., Wetzel, U., Weigl, J., Garbade, J., Lauschke, J., Hindricks, G., et al. (2003). Expression of Angiotensin II Receptors in Human Left and Right Atrial Tissue in Atrial Fibrillation with and without Underlying Mitral Valve Disease. J. Am. Coll. Cardiol. 42, 1785–1792. doi:10.1016/j.jacc.2003.07.014
Bullock, G. R., Steyaert, I., Bilbe, G., Carey, R. M., Kips, J., De Paepe, B., et al. (2001). Distribution of Type-1 and Type-2 Angiotensin Receptors in the Normal Human Lung and in Lungs from Patients with Chronic Obstructive Pulmonary Disease. Histochem. Cel Biol. 115, 117–124. doi:10.1007/s004180000235
Carnes, C. A., Chung, M. K., Nakayama, T., Nakayama, H., Baliga, R. S., Piao, S., et al. (2001). Ascorbate Attenuates Atrial Pacing-Induced Peroxynitrite Formation and Electrical Remodeling and Decreases the Incidence of Postoperative Atrial Fibrillation. Circ. Res. 89, E32-8. doi:10.1161/hh1801.097644
Catarata, M. J., Ribeiro, R., Oliveira, M. J., Robalo Cordeiro, C., and Medeiros, R. (2020). Renin-angiotensin System in Lung Tumor and Microenvironment Interactions. Cancers. 12, 1457–1524. doi:10.3390/cancers12061457
Chappell, M. C. (2016). Biochemical Evaluation of the Renin-Angiotensin System: The Good, Bad, and Absolute? Am J Physiol Heart Circ Physiol. 310, H137–H152. doi:10.1152/ajpheart.00618.2015
Chen, L.-N., Yang, X.-H., Nissen, D. H., Chen, Y.-Y., Wang, L.-J., Wang, J.-H., et al. (2013). Dysregulated Renin-AngioteNsin System Contributes to Acute Lung Injury Caused by Hind-Limb Ischemia-Reperfusion in Mice. Shock. 40, 420–429. doi:10.1097/SHK.0b013e3182a6953e
Chung, O., Kühl, H., Stoll, M., and Unger, T. (1998). Physiological and Pharmacological Implications of AT1 versus AT2 Receptors. Kidney Int. Suppl. 67, S95–S99. doi:10.1046/j.1523-1755.1998.06719.x
Conway, D. S. G., Buggins, P., Hughes, E., and Lip, G. Y. H. (2004). Prognostic Significance of Raised Plasma Levels of Interleukin-6 and C-Reactive Protein in Atrial Fibrillation. Am. Heart J. 148, 462–466. doi:10.1016/j.ahj.2004.01.026
Cuomo, D., Porreca, I., Cobellis, G., Tarallo, R., Nassa, G., Falco, G., et al. (2017). Carcinogenic Risk and Bisphenol A Exposure: A Focus on Molecular Aspects in Endoderm Derived Glands. Mol. Cell Endocrinol. 457, 20–34. doi:10.1016/j.mce.2017.01.027
da Silveira, K. D., Coelho, F. M., Vieira, A. T., Sachs, D., Barroso, L. C., Costa, V. V., et al. (2010). Anti-inflammatory Effects of the Activation of the Angiotensin-(1-7) Receptor, MAS, in Experimental Models of Arthritis. J.I. 185, 5569–5576. doi:10.4049/jimmunol.1000314
Dandona, P., Dhindsa, S., Ghanim, H., and Chaudhuri, A. (2007). Angiotensin II and Inflammation: The Effect of Angiotensin-Converting Enzyme Inhibition and Angiotensin II Receptor Blockade. J. Hum. Hypertens. 21, 20–27. doi:10.1038/sj.jhh.1002101
de Abajo, F. J. (2020). Renin–angiotensin System Inhibitors and COVID-19: Overwhelming Evidence against an Association. Lancet Digit. Heal. 3, e70-e71. doi:10.1016/S2589-7500(20)30294-6
de Abajo, F. J., Rodríguez-Martín, S., Lerma, V., Mejía-Abril, G., Aguilar, M., García-Luque, A., et al. (2020). Use of Renin-Angiotensin-Aldosterone System Inhibitors and Risk of COVID-19 Requiring Admission to Hospital: a Case-Population Study. Lancet. 395, 1705–1714. doi:10.1016/S0140-6736(20)31030-8
Dias-Peixoto, M. F., Santos, R. A. S., Gomes, E. R. M., Alves, M. N. M., Almeida, P. W. M., Greco, L., et al. (2008). Molecular Mechanisms Involved in the angiotensin-(1-7)/Mas Signaling Pathway in Cardiomyocytes. Hypertension. 52, 542–548. doi:10.1161/HYPERTENSIONAHA.108.114280
Esposito, F., Nardone, A., Fasano, E., Scognamiglio, G., Esposito, D., Agrelli, D., et al. (2018). A Systematic Risk Characterization Related to the Dietary Exposure of the Population to Potentially Toxic Elements through the Ingestion of Fruit and Vegetables from a Potentially Contaminated Area. A Case Study: The Issue of the “Land of Fires” Area in Campania Region, Italy. Environ. Pollut. 243, 1781–1790. doi:10.1016/j.envpol.2018.09.058
European Society of Cardiology (2020). Position Statement of the ESC Council on Hypertension on ACE-Inhibitors and Angiotensin Receptor Blockers. Available at: https://www.escardio.org/Councils/Council-on-Hypertension-(CHT)/News/position-statement-of-the-esc-council-on-hypertension-on-ace-inhibitors-and-ang (Accessed January 5, 2021).
Fielitz, J., Hein, S., Mitrovic, V., Pregla, R., Zurbrügg, H. R., Warnecke, C., et al. (2001). Activation of the Cardiac Renin-Angiotensin System and Increased Myocardial Collagen Expression in Human Aortic Valve Disease. J. Am. Coll. Cardiol. 37, 1443–1449. doi:10.1016/S0735-1097(01)01170-6
Fraga-Silva, R. A., Pinheiro, S. V. B., Gonçalves, A. C. C., Alenina, N., Bader, M., Souza Santos, R. A., et al. (2008). The Antithrombotic Effect of Angiotensin-(1-7) Involves Mas-Mediated NO Release from Platelets. Mol. Med. 14, 28–35. doi:10.2119/2007-00073.Fraga-Silva
Gao, C., Cai, Y., Zhang, K., Zhou, L., Zhang, Y., Zhang, X., et al. (2020). Association of Hypertension and Antihypertensive Treatment with COVID-19 Mortality: a Retrospective Observational Study. Eur. Heart J. 41, 2058–2066. doi:10.1093/eurheartj/ehaa433
Gheblawi, M., Wang, K., Viveiros, A., Nguyen, Q., Zhong, J.-C., Turner, A. J., et al. (2020). Angiotensin-Converting Enzyme 2: SARS-CoV-2 Receptor and Regulator of the Renin-Angiotensin System. Circ. Res. 126, 1456–1474. doi:10.1161/CIRCRESAHA.120.317015
Gnavi, R., Demaria, M., Picariello, R., Dalmasso, M., Ricceri, F., and Costa, G. (2020). Therapy with Agents Acting on the Renin-Angiotensin System and Risk of Severe Acute Respiratory Syndrome Coronavirus 2 Infection. Clin. Infect. Dis. 71, 2291–2293. doi:10.1093/cid/ciaa634
Gu, H., Xie, Z., Li, T., Zhang, S., Lai, C., Zhu, P., et al. (2016). Angiotensin-converting Enzyme 2 Inhibits Lung Injury Induced by Respiratory Syncytial Virus. Sci. Rep. 6, 19840. doi:10.1038/srep19840
Guo, Y.-R., Cao, Q.-D., Hong, Z.-S., Tan, Y.-Y., Chen, S.-D., Jin, H.-J., et al. (2020). The Origin, Transmission and Clinical Therapies on Coronavirus Disease 2019 (COVID-19) Outbreak - an Update on the Status. Mil. Med Res 7, 11. doi:10.1186/s40779-020-00240-0
Haschke, M., Schuster, M., Poglitsch, M., Loibner, H., Salzberg, M., Bruggisser, M., et al. (2013). Pharmacokinetics and Pharmacodynamics of Recombinant Human Angiotensin-Converting Enzyme 2 in Healthy Human Subjects. Clin. Pharmacokinet. 52, 783–792. doi:10.1007/s40262-013-0072-7
Hatem, S. N., and Sanders, P. (2014). Epicardial Adipose Tissue and Atrial Fibrillation. Cardiovasc. Res. 102, 205–213. doi:10.1093/cvr/cvu045
Hoffmann, M., Kleine-Weber, H., Schroeder, S., Krüger, N., Herrler, T., Erichsen, S., et al. (2020). SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 181, 271–280.e8. doi:10.1016/j.cell.2020.02.052
Huang, C., Wang, Y., Li, X., Ren, L., Zhao, J., Hu, Y., et al. (2020). Clinical Features of Patients Infected with 2019 Novel Coronavirus in Wuhan, China. Lancet 395, 497–506. doi:10.1016/S0140-6736(20)30183-5
Iaccarino, G., Grassi, G., Borghi, C., Ferri, C., Salvetti, M., Volpe, M., et al. (2020). Age and Multimorbidity Predict Death Among COVID-19 Patients. Hypertension. 76, 366–372. doi:10.1161/HYPERTENSIONAHA.120.15324
Italian Society of Cardiology (2020). GUIDA CLINICA COVID-19 PER CARDIOLOGI. Available at: https://www.sicardiologia.it/public/Documento-SIC-COVID-19.pdf (Accessed January 5, 2021).
Italian Society of Hypertension Farmaci antiipertensivi e rischio di COVID-19 (2020). Il comunicato della SIIA | SIIA. Available at: https://siia.it/notizie-siia/farmaci-antiipertensivi-e-rischio-di-covid-19-il-comunicato-della-siia/ (Accessed April 4, 2020).
Italian Society of Pharmacology SIF | Documento Informativo della Società Italiana di Farmacologia - Uso di Ace-Inibitori/Sartani ed infezione da COVID-19 (2020). Available at: https://www.sifweb.org/documenti/document_2020-03-13_documento-informativo-della-societa-italiana-di-farmacologia-uso-di-ace-inibitori-sartani-ed-infezione-da-covid-19 (Accessed April 17, 2020).
Jung, S.-Y., Choi, J. C., You, S.-H., and Kim, W.-Y. (2020). Association of Renin-Angiotensin-Aldosterone System Inhibitors with Coronavirus Disease 2019 (COVID-19)- Related Outcomes in Korea: A Nationwide Population-Based Cohort Study. Clin. Infect. Dis. 71, 2121–2128. doi:10.1093/cid/ciaa624
Jung, U., and Choi, M.-S. (2014). Obesity and its Metabolic Complications: The Role of Adipokines and the Relationship between Obesity, Inflammation, Insulin Resistance, Dyslipidemia and Nonalcoholic Fatty Liver Disease. Ijms 15, 6184–6223. doi:10.3390/ijms15046184
Kaparianos, A., and Argyropoulou, E. (2011). Local Renin-Angiotensin II Systems, Angiotensin-Converting Enzyme and its Homologue ACE2: Their Potential Role in the Pathogenesis of Chronic Obstructive Pulmonary Diseases, Pulmonary Hypertension and Acute Respiratory Distress Syndrome. Curr Med Chem. 18, 3506–3515. doi:10.2174/092986711796642562
Karamyan, V. T., Arsenault, J., Escher, E., and Speth, R. C. (2010). Preliminary Biochemical Characterization of the Novel, Non-AT1, Non-AT2 Angiotensin Binding Site from the Rat Brain. Endocr. 37, 442–448. doi:10.1007/s12020-010-9328-2
Kassiri, Z., Zhong, J., Guo, D., Basu, R., Wang, X., Liu, P. P., et al. (2009). Loss of Angiotensin-Converting Enzyme 2 Accelerates Maladaptive Left Ventricular Remodeling in Response to Myocardial Infarction. Circ. Heart Fail. 2, 446–455. doi:10.1161/CIRCHEARTFAILURE.108.840124
Khan, A., Benthin, C., Zeno, B., Albertson, T. E., Boyd, J., Christie, J. D., et al. (2017). A Pilot Clinical Trial of Recombinant Human Angiotensin-Converting Enzyme 2 in Acute Respiratory Distress Syndrome. Crit. Care. 21, 234. doi:10.1186/s13054-017-1823-x
Kourliouros, A., Karastergiou, K., Nowell, J., Gukop, P., Tavakkoli Hosseini, M., Valencia, O., et al. (2011). Protective Effect of Epicardial Adiponectin on Atrial Fibrillation Following Cardiac Surgery. Eur. J. Cardio-Thoracic Surg. 39, 228–232. doi:10.1016/j.ejcts.2010.05.006
Kreuter, M., Lederer, D. J., Molina-Molina, M., Noth, I., Valenzuela, C., Frankenstein, L., et al. (2019). Association of Angiotensin Modulators with the Course of Idiopathic Pulmonary Fibrosis. Chest. 156, 706–714. doi:10.1016/j.chest.2019.04.015
Kuster, G. M., Pfister, O., Burkard, T., Zhou, Q., Twerenbold, R., Haaf, P., et al. (2020). SARS-CoV2: Should Inhibitors of the Renin-Angiotensin System Be Withdrawn in Patients with COVID-19? Eur. Heart J. 41, 1801–1803. doi:10.1093/eurheartj/ehaa235
Labandeira-García, J. L., Garrido-Gil, P., Rodriguez-Pallares, J., Valenzuela, R., Borrajo, A., and Rodríguez-Perez, A. I. (2014). Brain Renin-Angiotensin System and Dopaminergic Cell Vulnerability. Front. Neuroanat. 8, 67. doi:10.3389/fnana.2014.00067
Li, D., Shinagawa, K., Pang, L., Leung, T. K., Cardin, S., Wang, Z., et al. (2001). Effects of Angiotensin-Converting Enzyme Inhibition on the Development of the Atrial Fibrillation Substrate in Dogs with Ventricular Tachypacing-Induced Congestive Heart Failure. Circulation. 104, 2608–2614. doi:10.1161/hc4601.099402
Li, W., Moore, M. J., Vasilieva, N., Sui, J., Wong, S. K., Berne, M. A., et al. (2003). Angiotensin-converting Enzyme 2 Is a Functional Receptor for the SARS Coronavirus. Nature. 426, 450–454. doi:10.1038/nature02145
Liu, E., Xu, Z., Li, J., Yang, S., Yang, W., and Li, G. (2011). Enalapril, Irbesartan, and Angiotensin-(1-7) Prevent Atrial Tachycardia-Induced Ionic Remodeling. Int. J. Cardiol. 146, 364–370. doi:10.1016/j.ijcard.2009.07.015
Liu, E., Yang, S., Xu, Z., Li, J., Yang, W., and Li, G. (2010). Angiotensin-(1-7) Prevents Atrial Fibrosis and Atrial Fibrillation in Long-Term Atrial Tachycardia Dogs. Regul. Peptides 162, 73–78. doi:10.1016/j.regpep.2009.12.020
Maisel, W. H., and Stevenson, L. W. (2003). Atrial Fibrillation in Heart Failure: Epidemiology, Pathophysiology, and Rationale for Therapy. Am. J. Cardiol. 91, 2–8. doi:10.1016/S0002-9149(02)03373-8
Malavazos, A. E., Ermetici, F., Coman, C., Corsi, M. M., Morricone, L., and Ambrosi, B. (2007). Influence of Epicardial Adipose Tissue and Adipocytokine Levels on Cardiac Abnormalities in Visceral Obesity. Int. J. Cardiol. 121, 132–134. doi:10.1016/j.ijcard.2006.08.061
Mancia, G., Rea, F., Ludergnani, M., Apolone, G., and Corrao, G. (2020). Renin-Angiotensin-Aldosterone System Blockers and the Risk of Covid-19. N. Engl. J. Med. 382, 2431–2440. doi:10.1056/nejmoa2006923
Marchesi, C., Paradis, P., and Schiffrin, E. L. (2008). Role of the Renin-Angiotensin System in Vascular Inflammation. Trends Pharmacol. Sci. 29, 367–374. doi:10.1016/j.tips.2008.05.003
Marshall, R. (2003). The Pulmonary Renin-Angiotensin System. Curr Pharm Des. 9, 715–722. doi:10.2174/1381612033455431
Mascolo, A., Scavone, C., Rafaniello, C., Ferrajolo, C., Racagni, G., Berrino, L., et al. (2020a). Renin-angiotensin System and Coronavirus Disease 2019: A Narrative Review. Front. Cardiovasc. Med. 7. doi:10.3389/fcvm.2020.00143
Mascolo, A., Sessa, M., Scavone, C., De Angelis, A., Vitale, C., Berrino, L., et al. (2017). New and Old Roles of the Peripheral and Brain Renin-Angiotensin-Aldosterone System (RAAS): Focus on Cardiovascular and Neurological Diseases. Int. J. Cardiol. 227, 734–742. doi:10.1016/j.ijcard.2016.10.069
Mascolo, A., Urbanek, K., De Angelis, A., Sessa, M., Scavone, C., Berrino, L., et al. (2020b). Angiotensin II and Angiotensin 1-7: Which Is Their Role in Atrial Fibrillation? Heart Fail. Rev. 25, 367–380. doi:10.1007/s10741-019-09837-7
Meng, Y., Li, T., Zhou, G.-s., Chen, Y., Yu, C.-H., Pang, M.-X., et al. (2015). The Angiotensin-Converting Enzyme 2/angiotensin (1-7)/mas axis Protects against Lung Fibroblast Migration and Lung Fibrosis by Inhibiting the NOX4-Derived ROS-Mediated RhoA/Rho Kinase Pathway. Antioxid. Redox Signaling 22, 241–258. doi:10.1089/ars.2013.5818
Moccia, M., Erro, R., Picillo, M., Vassallo, E., Vitale, C., Longo, K., et al. (2015). Quitting Smoking: An Early Non-motor Feature of Parkinson's Disease? Parkinsonism Relat. Disord. 21, 216–220. doi:10.1016/j.parkreldis.2014.12.008
Mohammadi-Karakani, A., Ghazi-Khansari, M., and Sotoudeh, M. (2006). Lisinopril Ameliorates Paraquat-Induced Lung Fibrosis. Clinica Chim. Acta 367, 170–174. doi:10.1016/j.cca.2005.12.012
Molteni, A., Wolfe, L., Ward, W., Hsin Ts’ao, C., Brizio Molteni, L., Veno, P., et al. (2007). Effect of an Angiotensin II Receptor Blocker and Two Angiotensin Converting Enzyme Inhibitors on Transforming Growth Factor-β (TGF-β) and α-Actomyosin (α SMA), Important Mediators of Radiation-Induced Pneumopathy and Lung Fibrosis. Curr. Pharm. Des. 13, 1307–1316. doi:10.2174/138161207780618777
Mori, Y., Itoh, Y., and Tajima, N. (2007). Angiotensin II Receptor Blockers Downsize Adipocytes in Spontaneously Type 2 Diabetic Rats with Visceral Fat Obesity. Am. J. Hypertens. 20, 431–436. doi:10.1016/j.amjhyper.2006.09.016
Nagashima, K., Okumura, Y., Watanabe, I., Nakai, T., Ohkubo, K., Kofune, M., et al. (2012). Does Location of Epicardial Adipose Tissue Correspond to Endocardial High Dominant Frequency or Complex Fractionated Atrial Electrogram Sites during Atrial Fibrillation? Circ. Arrhythm Electrophysiol. 5, 676–683. doi:10.1161/CIRCEP.112.971200
Nakahara, S., Hori, Y., Kobayashi, S., Sakai, Y., Taguchi, I., Takayanagi, K., et al. (2014). Epicardial Adipose Tissue-Based Defragmentation Approach to Persistent Atrial Fibrillation: Its Impact on Complex Fractionated Electrograms and Ablation Outcome. Heart Rhythm. 11, 1343–1351. doi:10.1016/j.hrthm.2014.04.040
Nakashima, H., Kumagai, K., Urata, H., Gondo, N., Ideishi, M., and Arakawa, K. (2000). Angiotensin II Antagonist Prevents Electrical Remodeling in Atrial Fibrillation. Circulation. 101, 2612–2617. doi:10.1161/01.CIR.101.22.2612
Novo, G., Guttilla, D., Fazio, G., Cooper, D., and Novo, S. (2008). The Role of the Renin-Angiotensin System in Atrial Fibrillation and the Therapeutic Effects of ACE-Is and ARBS. Br. J. Clin. Pharmacol. 66, 345–351. doi:10.1111/j.1365-2125.2008.03234.x
Oudit, G. Y., Liu, G. C., Zhong, J., Basu, R., Chow, F. L., Zhou, J., et al. (2010). Human Recombinant ACE2 Reduces the Progression of Diabetic Nephropathy. Diabetes. 59, 529–538. doi:10.2337/db09-1218
Patel, V. B., Basu, R., and Oudit, G. Y. (2016a). ACE2/Ang 1-7 axis: A Critical Regulator of Epicardial Adipose Tissue Inflammation and Cardiac Dysfunction in Obesity. Adipocyte. 5, 306–311. doi:10.1080/21623945.2015.1131881
Patel, V. B., Lezutekong, J. N., Chen, X., and Oudit, G. Y. (2017). Recombinant Human ACE2 and the Angiotensin 1-7 Axis as Potential New Therapies for Heart Failure. Can. J. Cardiol. 33, 943–946. doi:10.1016/j.cjca.2016.12.012
Patel, V. B., Mori, J., McLean, B. A., Basu, R., Das, S. K., Ramprasath, T., et al. (2016b). ACE2 Deficiency Worsens Epicardial Adipose Tissue Inflammation and Cardiac Dysfunction in Response to Diet-Induced Obesity. Diabetes. 65, db150399–95. doi:10.2337/db15-0399
Porreca, I., Ulloa-Severino, L., Almeida, P., Cuomo, D., Nardone, A., Falco, G., et al. (2017). Molecular Targets of Developmental Exposure to Bisphenol A in Diabesity: a Focus on Endoderm-Derived Organs. Obes. Rev. 18, 99–108. doi:10.1111/obr.12471
Renzoni, E. A., Abraham, D. J., Howat, S., Shi-Wen, X., Sestini, P., Bou-Gharios, G., et al. (2004). Gene Expression Profiling Reveals Novel TGFβ Targets in Adult Lung Fibroblasts. Respir. Res. 5, 24. doi:10.1186/1465-9921-5-24
Reynolds, H. R., Adhikari, S., Pulgarin, C., Troxel, A. B., Iturrate, E., Johnson, S. B., et al. (2020). Renin-Angiotensin-Aldosterone System Inhibitors and Risk of Covid-19. N. Engl. J. Med. 382, 2441–2448. doi:10.1056/nejmoa2008975
Rossi, F., Mascolo, A., and Mollace, V. (2017). The Pathophysiological Role of Natriuretic Peptide-RAAS Cross Talk in Heart Failure. Int. J. Cardiol. 226, 121. doi:10.1016/j.ijcard.2016.03.080
Rutkowska-Zapała, M., Suski, M., Szatanek, R., Lenart, M., Węglarczyk, K., Olszanecki, R., et al. (2015). Human Monocyte Subsets Exhibit Divergent Angiotensin I-Converting Activity. Clin. Exp. Immunol. 181, 126–132. doi:10.1111/cei.12612
Saavedra, J. M. (2012). Angiotensin II AT1 Receptor Blockers as Treatments for Inflammatory Brain Disorders. Clin. Sci. 123, 567–590. doi:10.1042/CS20120078
Santos, R. A. S., Ferreira, A. J., Verano-Braga, T., and Bader, M. (2013). Angiotensin-converting Enzyme 2, Angiotensin-(1-7) and Mas: New Players of the Renin-Angiotensin System. J. Endocrinol. 216, R1–R17. doi:10.1530/JOE-12-0341
Santos, R. A. S., Oudit, G. Y., Verano-Braga, T., Canta, G., Steckelings, U. M., and Bader, M. (2019). The Renin-Angiotensin System: Going beyond the Classical Paradigms. Am J Physiol Heart Circ Physiol. 316, H958–H970. doi:10.1152/ajpheart.00723.2018
Singh, Y., Singh, K., and Sharma, P. L. (2013). Effect of Combination of Renin Inhibitor and Mas-Receptor Agonist in DOCA-Salt-Induced Hypertension in Rats. Mol. Cel. Biochem. 373, 189–194. doi:10.1007/s11010-012-1489-2
Smit, M. D., Maass, A. H., De Jong, A. M., Muller Kobold, A. C., Van Veldhuisen, D. J., and Van Gelder, I. C. (2012). Role of Inflammation in Early Atrial Fibrillation Recurrence. Europace. 14, 810–817. doi:10.1093/europace/eur402
South, A. M., Diz, D. I., and Chappell, M. C. (2020). COVID-19, ACE2, and the Cardiovascular Consequences. Am J Physiol Heart Circ Physiol. 318, H1084–H1090. doi:10.1152/AJPHEART.00217.2020
Souza, L. L., and Costa-Neto, C. M. (2012). Angiotensin-(1-7) Decreases LPS-Induced Inflammatory Response in Macrophages. J. Cel. Physiol. 227, 2117–2122. doi:10.1002/jcp.22940
Stewart, S., Hart, C. L., Hole, D. J., and McMurray, J. J. V. (2002). A Population-Based Study of the Long-Term Risks Associated with Atrial Fibrillation: 20-Year Follow-Up of the Renfrew/Paisley Study. Am. J. Med. 113, 359–364. doi:10.1016/S0002-9343(02)01236-6
Triassi, M., Nardone, A., Giovinetti, M. C., De Rosa, E., Canzanella, S., Sarnacchiaro, P., et al. (2019). Ecological Risk and Estimates of Organophosphate Pesticides Loads into the Central Mediterranean Sea from Volturno River, the River of the “Land of Fires” Area, Southern Italy. Sci. Total Environ. 678, 741–754. doi:10.1016/j.scitotenv.2019.04.202
Turner, A. J., Hiscox, J. A., and Hooper, N. M. (2004). ACE2: From Vasopeptidase to SARS Virus Receptor. Trends Pharmacol. Sci. 25, 291–294. doi:10.1016/j.tips.2004.04.001
Uhal, B., Kyong Kim, J., Li, X., and Molina-Molina, M. (2007). Angiotensin-TGF-β1 Crosstalk in Human Idiopathic Pulmonary Fibrosis:Autocrine Mechanisms in Myofibroblasts and Macrophages. Curr. Pharm. Des. 13, 1247–1256. doi:10.2174/138161207780618885
Unger, T. (2002). The Role of the Renin-Angiotensin System in the Development of Cardiovascular Disease. Am. J. Cardiol. 89, 3–9. doi:10.1016/s0002-9149(01)02321-9
Vaduganathan, M., Vardeny, O., Michel, T., McMurray, J. J. V., Pfeffer, M. A., and Solomon, S. D. (2020). Renin-Angiotensin-Aldosterone System Inhibitors in Patients with Covid-19. N. Engl. J. Med. 382, 1653–1659. doi:10.1056/nejmsr2005760
Venteclef, N., Guglielmi, V., Balse, E., Gaborit, B., Cotillard, A., Atassi, F., et al. (2015). Human Epicardial Adipose Tissue Induces Fibrosis of the Atrial Myocardium through the Secretion of Adipo-Fibrokines. Eur. Heart J. 36, 795–805. doi:10.1093/eurheartj/eht099
Wang, R., Ibarra-Sunga, O., Verlinski, L., Pick, R., and Uhal, B. D. (2000). Abrogation of Bleomycin-Induced Epithelial Apoptosis and Lung Fibrosis by Captopril or by a Caspase Inhibitor. Am. J. Physiology-Lung Cell Mol. Physiol. 279, L143. doi:10.1152/ajplung.2000.279.1.l143
Wang, X., and Li, G. (2018). Irbesartan Prevents Sodium Channel Remodeling in a Canine Model of Atrial Fibrillation. J. Renin Angiotensin Aldosterone Syst. 19, 147032031875526. doi:10.1177/1470320318755269
Wong, C. X., Ganesan, A. N., and Selvanayagam, J. B. (2017). Epicardial Fat and Atrial Fibrillation: Current Evidence, Potential Mechanisms, Clinical Implications, and Future Directions. Eur. Heart J. 38, ehw045–1302. doi:10.1093/eurheartj/ehw045
Wösten-van Asperen, R. M., Lutter, R., Specht, P. A., Moll, G. N., Van Woensel, J. B., Van Der Loos, C. M., et al. (2011). Acute Respiratory Distress Syndrome Leads to Reduced Ratio of ACE/ACE2 Activities and Is Prevented by Angiotensin-(1-7) or an Angiotensin II Receptor Antagonist. J. Pathol. 225, 618–627. doi:10.1002/path.2987
Yamamoto, S., Yancey, P. G., Zuo, Y., Ma, L.-J., Kaseda, R., Fogo, A. B., et al. (2011). Macrophage Polarization by Angiotensin II-type 1 Receptor Aggravates Renal Injury-Acceleration of Atherosclerosis. Arterioscler Thromb. Vasc. Biol. 31, 2856–2864. doi:10.1161/ATVBAHA.111.237198
Yousif, M. H. M., Dhaunsi, G. S., Makki, B. M., Qabazard, B. A., Akhtar, S., and Benter, I. F. (2012). Characterization of Angiotensin-(1-7) Effects on the Cardiovascular System in an Experimental Model of Type-1 Diabetes. Pharmacol. Res. 66, 269–275. doi:10.1016/j.phrs.2012.05.001
Zambelli, V., Bellani, G., Borsa, R., Pozzi, F., Grassi, A., Scanziani, M., et al. (2015). Angiotensin-(1-7) Improves Oxygenation, while Reducing Cellular Infiltrate and Fibrosis in Experimental Acute Respiratory Distress Syndrome. ICMx 3. doi:10.1186/s40635-015-0044-3
Zankov, D. P., Omatsu-Kanbe, M., Isono, T., Toyoda, F., Ding, W.-G., Matsuura, H., et al. (2006). Angiotensin II Potentiates the Slow Component of Delayed Rectifier K + Current via the at 1 Receptor in Guinea Pig Atrial Myocytes. Circulation. 113, 1278–1286. doi:10.1161/CIRCULATIONAHA.104.530592
Zhang, P., Zhu, L., Cai, J., Lei, F., Qin, J. J., Xie, J., et al. (2020). Association of Inpatient Use of Angiotensin-Converting Enzyme Inhibitors and Angiotensin II Receptor Blockers with Mortality Among Patients with Hypertension Hospitalized with COVID-19. Circ. Res. 126, 1671–1681. doi:10.1161/CIRCRESAHA.120.31713410.1161/circresaha.120.317242
Zheng, Y.-Y., Ma, Y.-T., Zhang, J.-Y., and Xie, X. (2020). COVID-19 and the Cardiovascular System. Nat. Rev. Cardiol. 17, 259–260. doi:10.1038/s41569-020-0360-5
Zhong, J., Basu, R., Guo, D., Chow, F. L., Byrns, S., Schuster, M., et al. (2010). Angiotensin-converting Enzyme 2 Suppresses Pathological Hypertrophy, Myocardial Fibrosis, and Cardiac Dysfunction. Circulation 122, 717–728. doi:10.1161/CIRCULATIONAHA.110.955369
Zhou, T., Han, Z., Gu, J., Chen, S., Fan, Y., Zhang, H., et al. (2017). Angiotensin-converting Enzyme-2 Overexpression Improves Atrial Electrical Remodeling through TRPM7 Signaling Pathway. Oncotarget. 8, 78726–78733. doi:10.18632/oncotarget.20221
Keywords: renin-angiotensin-aldosterone system, heart, lung, COVID-19, inflammation
Citation: Mascolo A, Scavone C, Rafaniello C, De Angelis A, Urbanek K, di Mauro G, Cappetta D, Berrino L, Rossi F and Capuano A (2021) The Role of Renin-Angiotensin-Aldosterone System in the Heart and Lung: Focus on COVID-19. Front. Pharmacol. 12:667254. doi: 10.3389/fphar.2021.667254
Received: 12 February 2021; Accepted: 06 April 2021;
Published: 20 April 2021.
Edited by:
Ewa Krystyna Szczepanska-Sadowska, Medical University of Warsaw, PolandReviewed by:
Vincenzo Calderone, University of Pisa, ItalyCopyright © 2021 Mascolo, Scavone, Rafaniello, De Angelis, Urbanek, di Mauro, Cappetta, Berrino, Rossi and Capuano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Annamaria Mascolo, YW5uYW1hcmlhLm1hc2NvbG9AdW5pY2FtcGFuaWEuaXQ=
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work and share last authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.