 Jianwei Liao
Jianwei Liao Xin Wang3
Xin Wang3 Dongsheng Ouyang
Dongsheng Ouyang- 1Department of Clinical Pharmacology, Xiangya Hospital, Central South University, Changsha, China
- 2Institute of Clinical Pharmacology, Central South University, Changsha, China
- 3Zhejiang Hisun Pharmaceutical Co., Ltd, Taizhou, China
- 4Department of Geriatric Medicine, Xiangya Hospital, Central South University, Changsha, China
- 5Hunan Key Laboratory for Bioanalysis of Complex Matrix Samples, Changsha, China
Background and objectives: Hyzetimibe is a candidate drug being investigated as the second-in-class cholesterol absorption inhibitor; it lowers plasma levels of low-density lipoprotein cholesterol (LDL-C) by blocking the Niemann-Pick C1-like 1 protein, a transporter mainly expressed in the intestine that allows dietary cholesterol to enter the body from the intestinal lumen. Previous studies on the metabolism of hyzetimibe in healthy volunteers were not enough to show the biotransformation and excretion pathway; in particular, whether hyzetimibe maintains pharmacological action for duration sufficient to pass through the hepatic-intestinal circulation remains unknown. Furthermore, it remains unclear whether the differences between the chemical structures of ezetimibe and hyzetimibe would result in different pharmacokinetic characteristics. Given that the molecular target is in the intestine and the substantial hepatic-intestinal circulation is a metabolic characteristic of the drug, a study of hyzetimibe as an oral 14C-radiolabeled drug, compared with routinely metabolized drugs, would play an important role in uncovering pharmacokinetic details.
Methods: After an overnight fast and before taking medication, six healthy male volunteers swallowed an investigational product suspension containing 20 mg/∼100 μCi of 14C-labeled hyzetimibe as a single dose. Whole-blood, plasma, urine, and fecal samples were collected, and hyzetimibe and its metabolites were measured. Pharmacokinetic variables of hyzetimibe and its metabolites were calculated and statistically analyzed according to obtained concentration data. Safety data were collected throughout the study.
Results: The major metabolite detected in plasma was hyzetimibe-glucuronide, which accounted for 97.2% of the total plasma radioactivity. The mean cumulative excretion of total radioactivity of the dose was 16.39% in urine and 76.90% in feces. Unchanged drug and hyzetimibe-glucuronide were identified as the major components in the feces and the urine, respectively. The main metabolic conversions of hyzetimibe were glucuronidation (M1), mono-oxidation (M4), and mono-oxidation with additional sulfonation (M7). Hyzetimibe was considered generally safe and well tolerated.
Conclusion: This study of 14C-radiolabeled hyzetimibe provides a full profile of the biotransformation and excretion routes of hyzetimibe to improve the understanding of the pharmacokinetic characteristics of hyzetimibe. The changed hydroxyl group in the hyzetimibe structure made it easier for that drug, compared with ezetimibe, to combine with glucuronic acid and subsequently increased the urinary excretion of hyzetimibe vs. ezetimibe. These differences highlight the need to investigate in more detail the different pharmacokinetic impacts on the efficacy and safety of hyzetimibe and ezetimibe.
Introduction
Hyzetimibe (HS-25) is a second-in-class, potent, orally administered lipid-lowering compound that is currently under development. It can selectively inhibit the absorption of cholesterol and related phytosterols in the small intestine. Clinical trials of hyzetimibe have been carried out in America and China since 2012 (NCT03413462, NCT03433196, NCT03464682, NCT02087917), and a new drug application had been submitted to the China Center for Drug Evaluation of National Medical Products Administration. The chemical structure of hyzetimibe is similar to that of ezetimibe, a first-in-class cholesterol absorption inhibitor.
Both drugs can maintain their pharmacological action for a long time through hepatic-intestinal circulation, and the special metabolic route makes them have little clinical interaction with the combined drugs such as Atorvastatin.
The results of preclinical and clinical studies of hyzetimibe have shown that hyzetimibe notably lowers the levels of low-density lipoprotein cholesterol (LDL-C), total cholesterol (TC), non-high-density lipoprotein cholesterol (non-HDL-C), and apolipoprotein B (apo-B) with fast and stable efficacy. Long-term use of hyzetimibe is necessary, so it is particularly important to obtain information about the excretion, mass balance, and metabolism in humans. A previous study in animals and in healthy adult volunteers (Ruan et al., 2014) determined the metabolic characteristics of hyzetimibe in animals according to preclinical pharmacokinetic studies and discovered that hyzetimibe was metabolized to its active metabolite by different isoforms of UDP-glucuronosyltransferase in the intestine and liver; however, the metabolite profiles, excretion pathways, and mass balance of hyzetimibe (the excretion of hyzetimibe and all metabolites in urine and feces) in humans remained unknown. In that study, it was not clear whether the small differences in the chemical structures between ezetimibe and hyzetimibe would result in different pharmacokinetic characteristics. Ruan et al. (2014) also examined the metabolic characteristics of the hepatic-intestinal circulation of hyzetimibe, because its molecular targets were in the intestine. Those authors determined that a study of orally administered 14C-radiolabeled hyzetimibe would offer substantial details about the drug metabolism. Therefore, this study was developed to assess the distribution of hyzetimibe and its metabolites in whole blood and plasma; to explore the pharmacokinetics of total radioactive hyzetimibe and hyzetimibe-M1; to examine the mass balance, excretion pathways, and biotransformation pathways; and to report on the safety and tolerability of a single oral dose of 14C-labeled hyzetimibe in healthy male volunteers to provide a reference for the rational use of the drug.
Methods
Overall Study Design
This was an open-label, single-dose study. The screening/baseline period of the study was approximately seven days, and the observation period was approximately seven-to-ten days, for a total study time of approximately two weeks. Whole-blood, plasma, and pooled urine and fecal samples were obtained on a regular basis to recover the radioactive dose. Safety data were collected throughout the study. The length of the observation period might have been shortened or prolonged if deemed appropriate according to emerging data. After the mass balance cumulative recovery of >85% had been achieved or <1% of dose had been collected in urine and feces within two consecutive days, volunteers were permitted to leave the study center. If emerging data showed that adequate dose recovery was not achieved after ten days (i.e., day 11), those volunteers were asked to continue providing urine and/or fecal samples at twenty-four-hour intervals. When the plasma drug radioactivity concentration was less than two times the plasma background value within two consecutive time points, the collection of radioactive blood samples was stopped. All adverse events (AEs) were collected for each volunteer from the time of informed consent until the follow-up visit or the last day of urine/fecal collection.
Study Population Selection
Healthy male volunteers between 18 and 45 years of age with a body mass index between 19 and 26 kg/m2 were enrolled in the study. Participants with irregular bowel movements or with long-term exposure to radioactivity—from radioactive working conditions, substantial radiation exposure (more than two chest/abdominal computed tomography scans or more than three other X-ray examinations), or those who participated in radiopharmaceutical labeling trial within one year of this study—were excluded from the study. The approval and implementation of this study strictly followed the ethical principles of the Declaration of Helsinki, the International Conference on Harmonization (ICH)/Good Clinical Practice (GCP), and applicable regulatory requirements. The Medical Ethics Committee of the First Affiliated Hospital of Suzhou University approved this study (Approval No. 091-1). All volunteers gave written informed consent and satisfied the study inclusion/exclusion criteria of the clinical study protocol before they were administered the investigational product.
Doses and Treatment Regimens
Each bottle of hyzetimibe solid powder for oral administration contained 20 mg/∼100 μCi of 14C-labeled hyzetimibe. The radiochemical purity was 100.00%, and the specific activity was 5.033 μCi/mg (equivalent to 2.13 mCi/mmoL).
In the phase II clinical study on the lipid-lowering effect and efficacy of hyzetimibe, observation of the 20-mg dose group confirmed good safety and effectiveness. The 20-mg dose was proposed for daily clinical administration and so was used as the oral dose in this study.
On day 1, after an overnight fast for at least 10 h and no water for at least 1 h before medication administration, volunteers swallowed the investigational suspension containing 20 mg/∼100 μCi of 14C-labeled hyzetimibe as a single dose. The planned dose of 100 μCi represented a small risk level of human radiation exposure (International Commission on Radiological Protection, 1991; International Commission on Radiological Protection, 2007; Code of Federal Regulations, 2020). According to our preliminary estimation, the effective dose of the labeled drug used in this study would not exceed 0.0297 mSv, which aligns with the radiation dose limit recommended by the International Commission on Radiologic Protection (International Commission on Radiological Protection, 1991; International Commission on Radiological Protection, 2007; Code of Federal Regulations, 2020). After ingestion of the administered dose, the vial used for administration was rinsed multiple times with purified water. Volunteers ingested the investigational suspension, the rinses, and additional purified water for a total fluid ingestion of 240 ml. After the volunteers received the medicine, the residual radioactive dose in the drug container and related articles were determined.
The use of radioactive compounds was in accordance with the requirements of the radiation safety license of the study site (Jiangsu environmental radiation license A0666). The treatment of radioactive waste generated during the study was in accordance with urban radioactive waste management measures and with the basic national standards of ionizing radiation protection and radiation source safety (GB18871-2002) issued by General Administration of Quality Supervision, Inspection and Quarantine of China.
Sample Collection
To determine the plasma concentrations of hyzetimibe and its metabolites and to measure the total radioactivity, blood samples were collected before the dose and at 0.25, 0.5, 1, 1.5, 2, 2.5, 3, 4, 5, 6, 8, 10, 12, 24, 36, 48, 72, 96,120, 168, and 240 h after the dose. Whole-blood samples were collected before and at 0.5, 1, 2, 6, 12, and 24 h after the dose to obtain the ratio of total radioactivity in whole blood to plasma. Urine samples were collected before and at 0–4, 4–8, 8–12, 12–24, 24–48, 48–72, 72–96, 96–120, 120–144, 144–168, 168–192, 192–216, and 216–240 h after the dose, and the sample weights were recorded. Fecal samples were collected before and at 0–24, 24–48, 48–72, 72–96, 96–120, 120–144, 144–168, 168–192, 192–216, and 216–240 h after the dose, and the sample weights were recorded. To determine metabolic conversion and the metabolic rate, plasma samples were collected before and at 0.25, 1, 2.5, and 8 h after the dose. Four mixed urine samples and four mixed fecal samples at different time periods were also used for metabolic conversion and metabolic rate assessments.
Sample Preparation and Bioanalysis
Blood samples were vortexed, weighed, and combusted in a Sample Oxidizer 501 (Harvey, United States) to detect total radioactivity. Plasma was well vortexed and weighed to detect total radioactivity. Urine was well mixed and weighed to detect total radioactivity. Pooled fecal homogenates (in a 50% isopropanol solution) were weighed, dried, and combusted in a Sample Oxidizer 501 (Harvey, United States) to detect total radioactivity. Plasma samples were processed by protein precipitation using acetonitrile (1/4, v/v) to determine the plasma concentrations of hyzetimibe and hyzetimibe-M1.
For the pharmacokinetic analysis, total radioactivity levels in whole-blood and plasma samples were quantified using liquid scintillation counting in a Tri-Carb 4910 TR liquid scintillation spectrometer (PerkinElmer, United States). Plasma concentrations of hyzetimibe and its metabolites in the volunteers were determined at different time points using a validated liquid chromatography/tandem mass spectrometry (MS/MS) method (Chen et al., 2015). The plasma samples were analyzed by ultra-performance liquid chromatography (LC-30AD; Shimadzu, Japan combined with mass spectrometry (UPLC-MS/MS). The high performance liquid chromatography (HPLC) column was a Hypersil GOLD C18 system (50 × 2.1 mm, 1.9 μm; kept at 40°C, mobile phase flow rate of 0.4 ml/min; Thermo, United States with Waters Critical Clean PER 735001035 tubing (Waters, United States). The mass spectrometer (QTRAP®5500, AB SCIEX, United States) was equipped with a heated electrospray ionization interface. The mobile phase consisted of mobile phase A (water) and mobile phase B (methanol). From 0.00 to 0.80 min, the gradient solvent composition was 95% mobile phase A and 5% mobile phase B. From 0.80 to 2.80 min, mobile phase A gradually decreased to 5%, and mobile phase B gradually increased to 95%. From 2.80 to 3.50 min, the gradient solvent composition was 5% mobile phase A and 95% mobile phase B. From 3.51 to 4.50 min, the gradient solvent composition was 95% mobile phase A and 5% mobile phase B. The data processing systems were Analyst®1.6.3 and MultiQuant™ 3.0.2 (AB SCIEX, United States. The sample sizes were 5 µl each. The linear range of determination was 0.05–20 ng/ml for hyzetimibe and was 1–400 ng/ml for hyzetimibe-M1.
The total radioactivity levels in the homogenates of urine and feces were measured by liquid scintillation counting in a Tri-Carb 4910 TR liquid scintillation spectrometer (PerkinElmer, United States).
Selected plasma, urine, and fecal samples were mixed, extracted and analyzed by reversed-phase HPLC (LC-2030C, Shimadzu, Japan) with online/offline radioactivity detection (vARC Radio-Assay System, Model 3, AIM, United States) for metabolic profiling. The main radioactive metabolites in samples of mixed plasma, urine, and feces were identified by HPLC combined with low-energy radioactivity monitor and mass spectrometry (LC/RAM/MS). The RP-HPLC column was an ACE C18 (ACE, British) (150 × 4.6 mm, 3.0 μm, kept at 40°C, mobile phase flow rate of 0.7 ml/min. The mass spectrometer (Thermo LTQ Orbitrap XL LC-MS/MS System, Thermo Scientific, United States) was equipped with a heated electrospray ionization interface. The sample spray was delivered through a formate-amine solution. The data processing system was Xcalibur (Thermo Scientific, United States).
Pharmacokinetics and Safety Measurements
Pharmacokinetic outcome measures included the following: maximum plasma concentration (Cmax); time to maximum plasma concentration (Tmax); area under concentration-time curve from zero (predose) extrapolated to infinity (AUC0-∞); area under the concentration-time curve from zero (predose) to the time of the last quantifiable concentration (AUC0-t); apparent terminal elimination half-life (t1/2); and mean residence time (MRT).
To evaluate the safety and tolerability of a single oral dose of hyzetimibe in healthy male volunteers, adverse events (AEs), vital signs, physical examination, 12-lead electrocardiogram (ECG), and laboratory variables (hematology, chemistry, urinalysis, and hemagglutination) were assessed. AEs were recorded from the time of informed consent to the end of the study or to the early withdrawal assessment. AEs were graded using the National Cancer Institute Common Terminology Criteria for Adverse Events grading system, version 4.03.
Statistical Methods
The sample size of the study was not based on hypothesis testing. Rather, on the basis of similar previously conducted studies of mass balance, a sample size of six volunteers was considered sufficient to gain adequate absorption, distribution, metabolism, and excretion information while exposing as few volunteers as possible to the investigational product and procedures. The pharmacokinetic analysis set included six volunteers who received one dose of hyzetimibe. These six volunteers were included in the safety population. Because this was an exploratory study, the data analysis relied mainly on descriptive statistics and did not involve formal power calculations or model-based statistical analyses. Pharmacokinetic variables, plasma concentrations, urine and fecal samples of 14C-labeled radioactivity data, and safety/tolerability were summarized using appropriate descriptive statistics [e.g., mean, (SD), coefficient of variation (CV%), geometric mean, geometric coefficient of variation (GCV%), minimum, median, and maximum]. Figures of the mean concentration-time data were developed to present plasma 14C-labeled radioactivity concentrations, plasma hyzetimibe concentrations, and plasma hyzetimibe-M1 concentrations.
Results
Demographic Characteristics
The enrolled participants were healthy Chinese men. The mean age of the participants was 30.2 years, and the mean height was 169.25 cm. The mean body mass index was 21.88 kg/m2, within the protocol-specified range of 19–26 kg/m2.
Pharmacokinetic Results
Mean concentration-time profiles in plasma after a single dose of 14C-labeled hyzetimibe are shown in Figure 1. The mean hyzetimibe-M1 concentrations were close to the total radioactivity, but the mean hyzetimibe concentrations were notably lower than the total radioactivity. Pharmacokinetic variables are shown in Table 1. The total radioactivity in plasma was determined by liquid scintillation counter. The median Tmax of total radioactivity was 1.00 h (minimum, maximum: 0.250, 2.50 h). The mean maximum concentration (Cmax) of total radioactivity was 139 ± 43.8 ng Eq/ml. The mean concentration of radioactivity was 14.2 ± 6.01 ng Eq/ml at 24 h after administration. Only a small amount of radioactivity was detected in the plasma of three volunteers at 48 h after administration. In the other three volunteers, plasma levels were below the lower limit of radioactivity quantification (3.76 ng Eq/g). The mean AUC0-t of the total radioactivity in plasma was 961 ± 307 h × ng Eq/ml. The mean MRT0-t of total radioactivity was 10.5 ± 2.22 h. The mean t1/2 of total radioactivity was 13.2 ± 5.81 h.
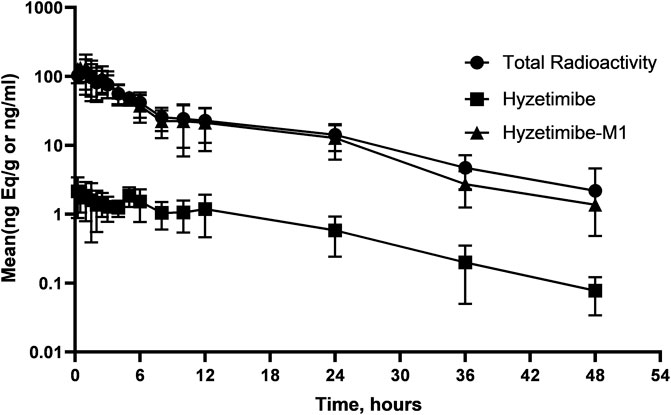
FIGURE 1. Mean (SD) hyzetimibe and hyzetimibe-M1 concentration-time and total radioactivity-time profiles in plasma.
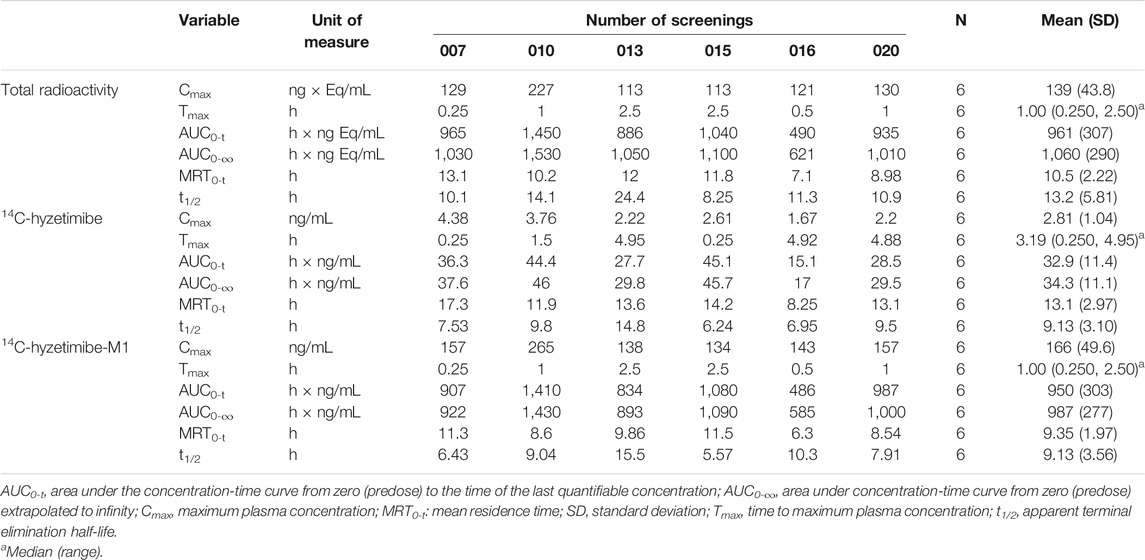
TABLE 1. Summary of pharmacokinetic parameters and total radioactivity of hyzetimibe and hyzetimibe-M1.
A validated LC-MS/MS method was used to determine the plasma concentrations of hyzetimibe and hyzetimibe-M1. The median Tmax in plasma was 3.19 h (minimum, maximum: 0.250, 4.95 h) for hyzetimibe and was 1.00 h (minimum, maximum: 0.250, 2.50 h) for hyzetimibe-M1. The mean maximum concentration (i.e., Cmax) was 2.81 ± 1.04 ng/ml for hyzetimibe and was 166 ± 49.6 ng/ml for hyzetimibe-M1. One volunteer had a plasma concentration at 48 h after administration that was below the lower limit of quantification (hyzetimibe: 0.0500 ng/ml, hyzetimibe-M1: 1.00 ng/ml). The mean plasma concentration of the other five volunteers was 0.0779 ± 0.0438 ng/ml for hyzetimibe and was 1.37 ± 0.888 ng/ml for hyzetimibe-M1 at 48 h after administration. The mean AUC0-t in the plasma was 32.9 ± 11.4 h × ng/ml for hyzetimibe and was 950 ± 303 h × ng/ml for hyzetimibe-M1. The mean ± SD MRT0-t was 13.1 ± 2.97 h for hyzetimibe and was 9.35 ± 1.97 h for hyzetimibe-M1. The t1/2 was 9.13 ± 3.10 h for hyzetimibe and was 9.13 ± 3.56 h for hyzetimibe-M1. The exposure level of total radioactivity in the plasma (Cmax and AUC0-t) was similar to the sum of the exposure levels of hyzetimibe and hyzetimibe-M1, indicating that no other major products of metabolism existed in the blood circulation.
Total Radioactivity Ratio of Whole Blood to Plasma
The total radioactivity ratio of whole blood to plasma was less than 1 and ranged from 0.409 to 0.764. This result suggests that total radioactivity did not bind to blood cells and that the radioactive substances in circulation were mainly distributed in the plasma.
Urine and Fecal Recovery of Radioactive Dose (Mass Balance)
Recovery of the total cumulative radioactivity in the urine and feces from 0 to 168 h after the dose is presented in Figure 2 and Table 2. The mean cumulative recovery of the total radioactivity in urine and feces from 0 to 168 h period after the dose amounted to 93.29 ± 1.82% of the dose. The mean cumulative recovery of the total radioactivity in feces accounted for 76.90 ± 6.17%; the mean cumulative recovery in the urine accounted for 16.39 ± 5.25%. Excretion of total radioactivity mainly occurred within 72 h; accounted for 90.91 ± 3.18%.
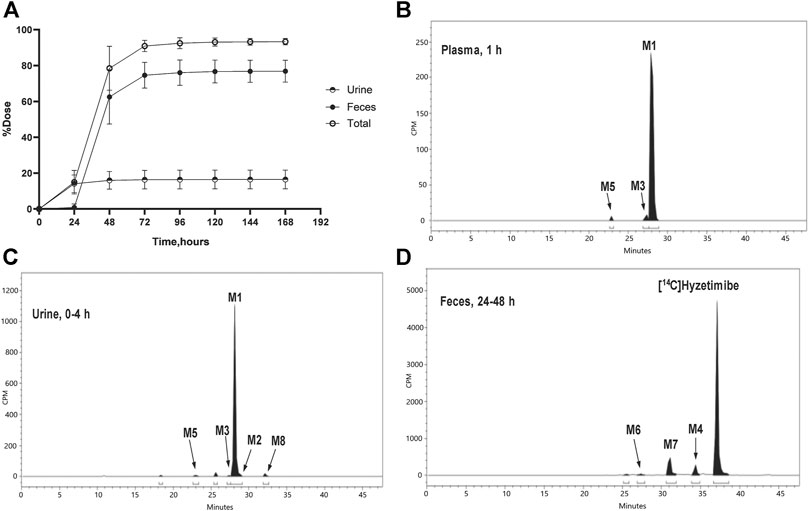
FIGURE 2. (A) Mean (SD) cumulative recoveries of total radioactivity in urine, feces, and combined (n = 6, mean ± SD). Radio-chromatograms of (B) 1 h plasma (C) 0–4 h urine, and (D) 24–48 h feces of six healthy male volunteers. CPM, count per minute.
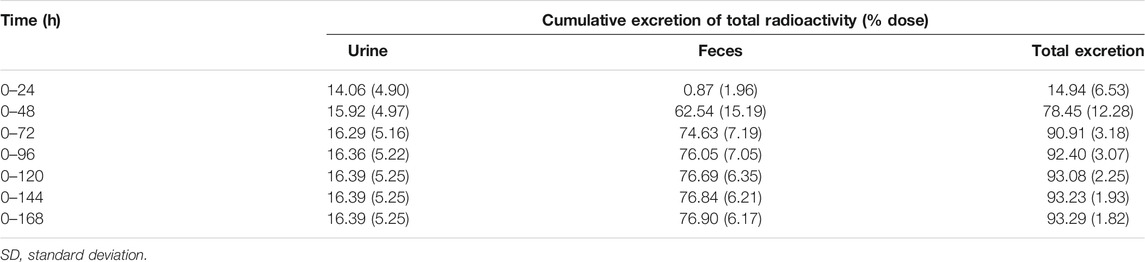
TABLE 2. Mean (SD) cumulative excretion of total radioactivity in urine and feces (n = 6).
Metabolite Profiles
As shown in Table 3, eight main metabolites were identified in addition to unchanged drug: glucuronidation of phenolic hydroxyl (M1), glucuronidation of aliphatic hydroxyl (M2), mono-oxidation (M4), glucuronidation of M4 (M3), glucuronidation of phenolic hydroxyl and aliphatic hydroxyl (M5), sulfonation of M4 (M7), oxidation and hydrogenation of M7 (M6), and formylation of M1 (M8). The percentages of unchanged drug and products of metabolism in the total radioactivity exposure in the plasma (% AUC), urine (% dose), and feces (% dose) of healthy male volunteers are summarized in Table 4.
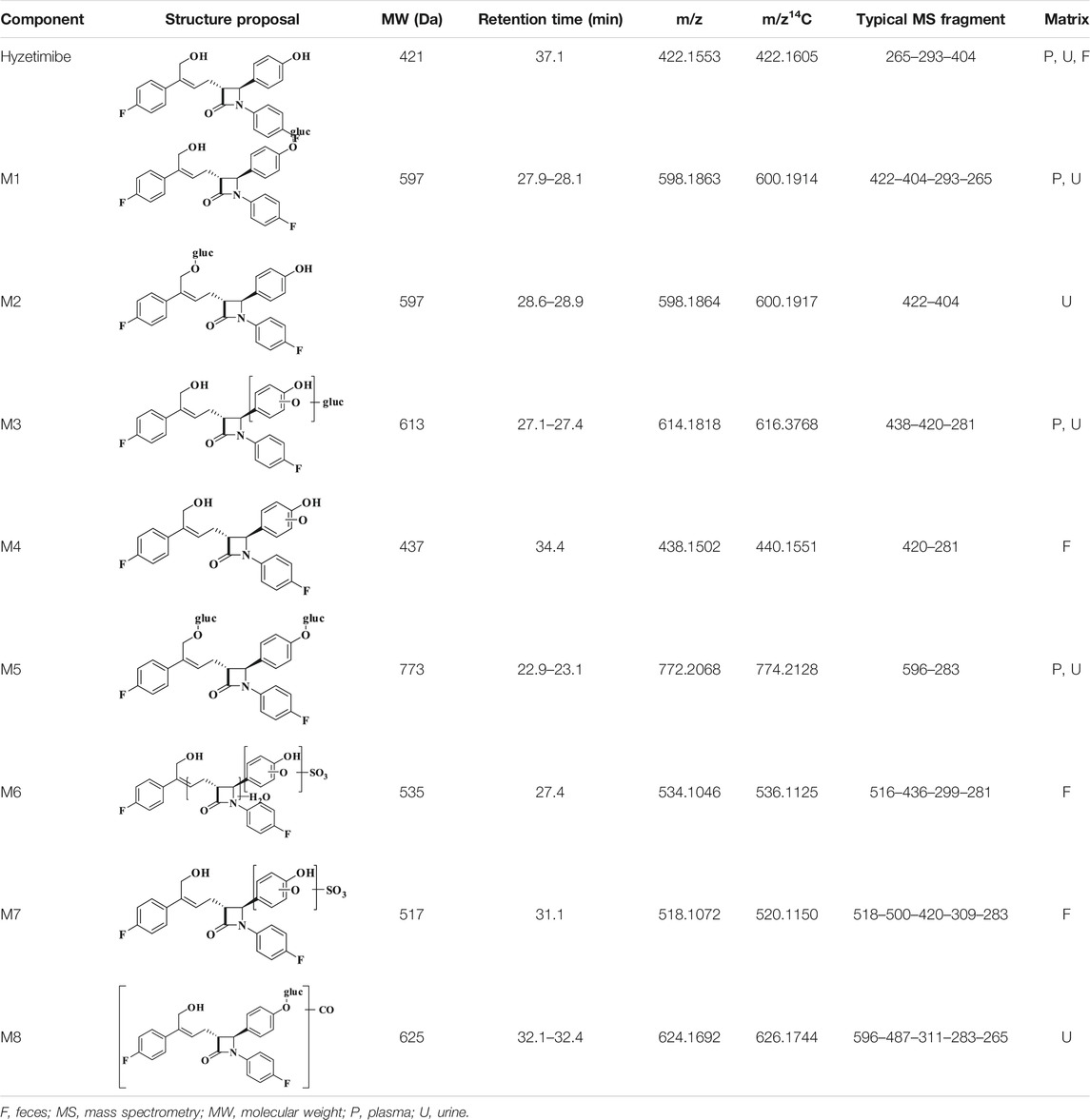
TABLE 3. Identification of in vivo metabolites of hyzetimibe.
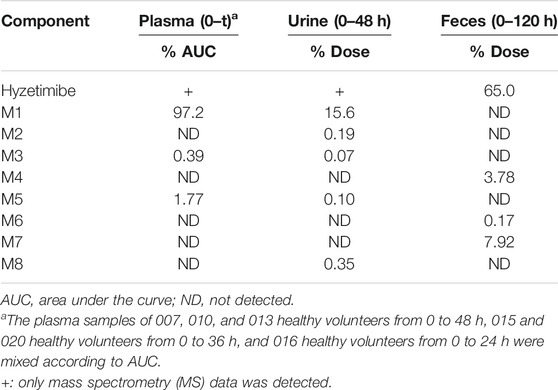
TABLE 4. Mean percentages of hyzetimibe and metabolites excreted in plasma, urine, and feces after a single oral dose of 20 mg of 14C-hyzetimibe suspension.
The unchanged drug was not detected by radio-chromatography in mixed plasma. The AUC0-t of the main metabolite, M1, in plasma was 931 ± 291 h × ng Eq/ml, accounting for 97.2 ± 1.81% of the total radioactivity exposure in plasma. The total radioactivity exposure in plasma of M3 and M5 was present in minor quantities of 0.39–1.77%. Other metabolites accounted for 0.70% of the total radioactivity exposure in plasma and were not identified. The excretion of total radioactivity in urine from 0 to 168 h was 16.39 ± 5.25% of the dose. The unchanged drug was only detected by mass spectrometer and could not be quantified by radio-chromatography. The main metabolite identified in the urine was M1 (15.6 ± 4.66% of the dose). M2, M3, M5, and M8 represented a small quantity (0.07–0.35% of the dose). Other metabolites accounted for 0.06% of the dose and were not identified. The excretion of total radioactivity in the feces from 0 to 168 h was 76.90 ± 6.17% of the dose. In the feces, the unchanged drug was identified as the main entity (65.0 ± 7.87% of the dose). The main metabolite identified in the feces was M7 (7.92 ± 5.81% of dose). Two other minor metabolites, M4 and M6, represented 3.78 and 0.17% of the dose, respectively. Radio-chromatograms for human plasma, urine, and feces are illustrated in Figure 2.
According to the results of metabolite identification, the main metabolic routes for hyzetimibe in healthy male volunteers are as follows: glucuronidation (M1), mono-oxidation (M4), and mono-oxidation with additional sulfonation (M7). The proposed metabolic scheme is shown in Figures 3, 4.
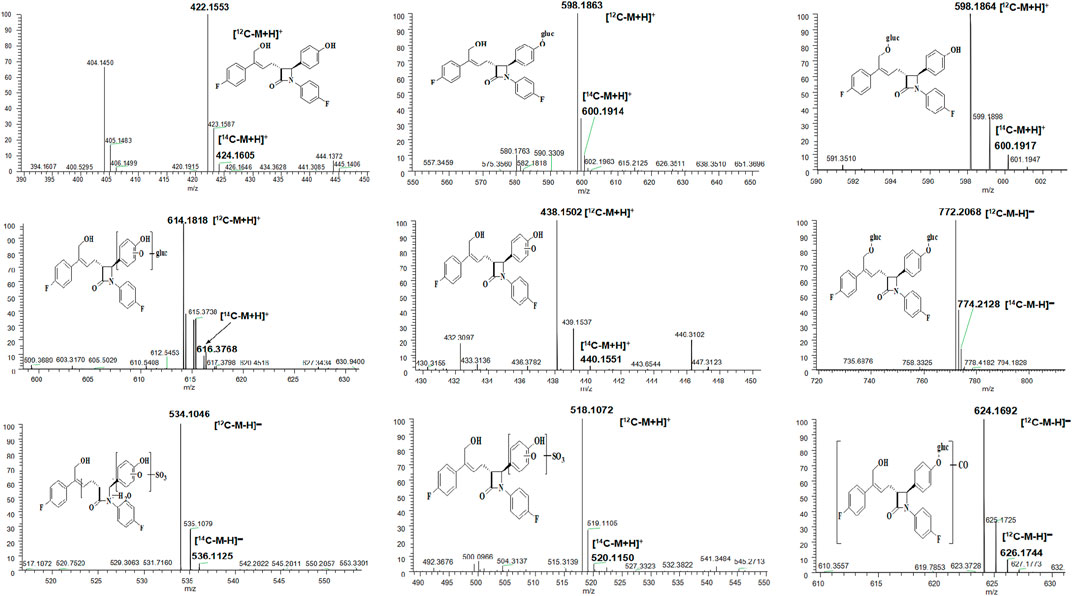
FIGURE 3. Full-scan chromatogram of unchanged drug and metabolites by high performance liquid chromatography combined with low-energy radioactivity monitor/(±) electrospray ionization-Fourier transform mass spectrometry, HPLC-RAM/(±) ESI-FTMS.
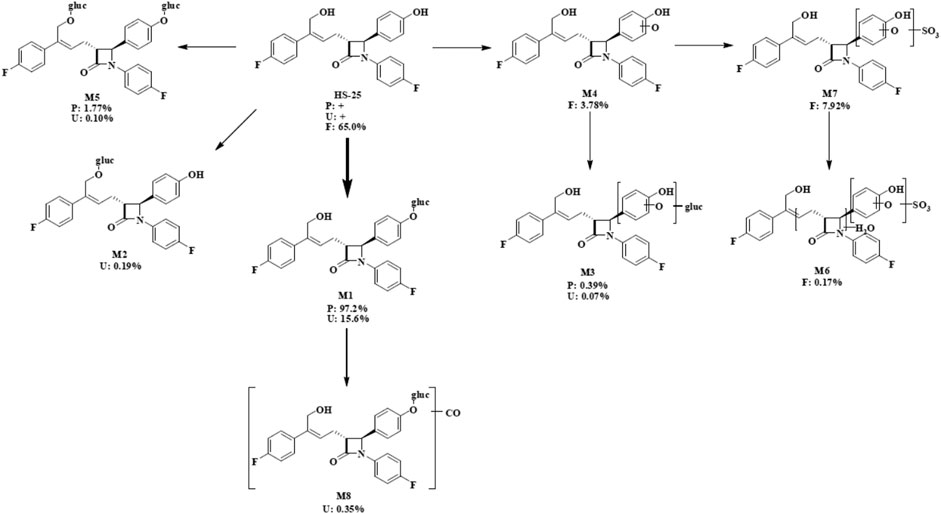
FIGURE 4. Proposed metabolic scheme. F, feces (% dose); gluc, glucuronic acid (C6H8O6); P: plasma [% area under the curve (AUC)]; U, urine (% dose).
Safety Evaluation
Hyzetimibe was generally regarded as safe and well tolerated. No deaths, severe AEs, or AEs leading to discontinuation were reported in the study. Three volunteers reported five AEs. All but one (diarrhea) of the events were considered unrelated to the study drug. No abnormal vital sign measurements, ECG results, and physical examination findings were reported. Two volunteers (volunteer 007 and 016) had uric acid at levels that were considered abnormal and of clinical relevance, and one volunteer (volunteer 015) had abnormal, clinically significant high-sensitivity C-reactive protein (hs-CRP) and fecal occult blood test (FOBT) results. These abnormalities were mild and were not considered related to the drug preparation.
Discussion
To our knowledge, this study was the first to report radiolabeled pharmacokinetic details about hyzetimibe, a cholesterol absorption inhibitor, in humans. Preclinical pharmacokinetic studies had suggested that hyzetimibe was metabolized to its active metabolite by different isoforms of UDP-glucuronosyltransferase in the intestine and liver. The data obtained from this radiolabeled study of the absorption, distribution, metabolism, and excretion of 14C-labeled hyzetimibe in healthy male volunteers demonstrated that the main metabolic route of hyzetimibe was glucuronide conjugation (a phase II reaction) in the small intestine and liver. Hyzetimibe and its metabolites are excreted through bile and urine Hyzetimibe was rapidly metabolized into hyzetimibe-glucuronide. The time of maximum concentration (Tmax) for hyzetimibe-M1 occurred from 0.25 to 2.5 h after oral administration. Plasma hyzetimibe-glucuronide accounted for 97.2% of total plasma radioactivity (AUC0-48). A multi-peak phenomenon was observed from plasma concentration-time profiles and indicated that the drug had enterohepatic circulation. By the end of sample collection (168 h), the mean cumulative recovery of the total radioactivity in the urine and feces combined was 93.29% of the dose after a single oral administration of 20 mg/∼100 μCi of 14C-labeled hyzetimibe suspension in six healthy male volunteers. The mean cumulative recovery of the total radioactivity in urine was 16.39% of the dose, and the mean cumulative recovery in feces was 76.90%. These results indicated that the primary route of elimination of radioactivity was by fecal elimination. The excretion of total radioactivity mainly occurred within 72 h, amounting to 90.91% of the dose. The exposure level of the total radioactivity in plasma (Cmax and AUC0-t) was similar to the sum of the exposure level of hyzetimibe and hyzetimibe-M1, indicating that no other major products of metabolism existed in the blood circulation. The major component in the feces was unchanged hyzetimibe, which accounted for 65% of the dose. The major component in the urine was hyzetimibe-glucuronide, which accounted for 15.6% of the dose.
In contrast, Patrick et al. (2002), Zetia (ezetimibe) tablets: pharmacology review(s) and Zetia (ezetimibe) tablets: prescribing information reported a mean Tmax of total radioactivity of 2.63 h after oral administration of 14C-labeled ezetimibe (20 mg) to healthy male participants. The mean maximum concentration (Cmax) of total radioactivity in that study was 75.1 ng Eq/g, and the mean AUC0-t of total radioactivity in plasma was 780 h × ng Eq/g. The mean Tmax in the plasma was 9.88 h for ezetimibe and was 2.31 h for ezetimibe glucuronide (SCH 60663). The mean maximum concentration (Cmax) was 5.21 ng/ml for ezetimibe and was 61.2 ng/ml for ezetimibe glucuronide. The mean AUC0-t in plasma was 86.4 h × ng/ml for ezetimibe and was 636 h × ng/ml for ezetimibe glucuronide. The primary metabolic pathway for ezetimibe was identified as glucuronidation of the phenolic hydroxyl, and minimal oxidative metabolism was observed. The cumulative recovery of radioactivity in the feces was approximately 78% of dose, and cumulative recovery in the urine was 11% after a single oral administration of 20 mg of 14C-labeled ezetimibe in the healthy male volunteers. The major component recovered in the urine was ezetimibe-glucuronide, accounting for 9% of dose (Patrick et al., 2002).
In our study, the change of hydroxyl group in the drug structure made it easier for hyzetimibe to combine with glucuronic acid, which led to more excretion of hyzetimibe (15.6% of dose) than ezetimibe in the urine. A small amount of other oxidative metabolites of ezetimibe were also identified in urine and feces in the study by Patrick et al. (2002). The pharmacokinetic parameter results for oral 14C-radiolabeled hyzetimibe in our study were similar to results with oral hyzetimibe reported by Ruan et al. (2014) with the same dose (Table 5). According to the mean and individual hyzetimibe and hyzetimibe-M1 concentration-time and total radioactivity-time profiles in the plasma, a multi-peak phenomenon appeared for hyzetimibe and hyzetimibe-M1; most of the secondary peaks occurred at 4–6 and 10–12 h. These results indicated that hyzetimibe and hyzetimibe-glucuronide had enterohepatic circulation. Hyzetimibe-glucuronide transformed into hyzetimibe, which was reabsorbed into the blood. The multi-peak times of hyzetimibe, hyzetimibe-glucuronide, and total radioactivity were close to dinner time. Food intake stimulated bile secretion (Fuchs et al., 1998) and then promoted enterohepatic recycling after the transformation of hyzetimibe-glucuronide and hyzetimibe. Because multiple peaks made it was impossible to reliably estimate the terminal-phase rate constant by regression analysis, the shorter mean half-lives of hyzetimibe and hyzetimibe-glucuronide were not consistent with results from a prior clinical study (Ruan et al., 2014), as shown in Table 5. Enterohepatic recycling could affect the efficacy of hyzetimibe, because hyzetimibe acts on the small intestine to lower cholesterol absorption.
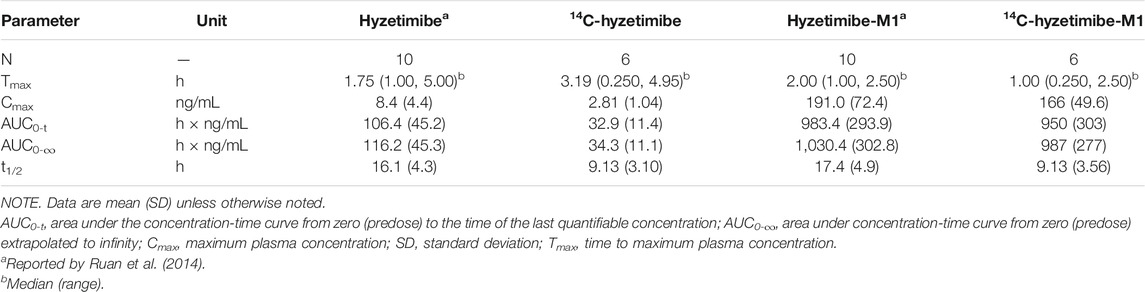
TABLE 5. Pharmacokinetic parameters of hyzetimibe, hyzetimibe-M1,14C-hyzetimibe and 14C-hyzetimibe-M1.
The total radioactivity ratio of whole blood to plasma was less than one. This result suggests that the total radioactivity did not bind to blood cells.
Eight main metabolites and unchanged drug were identified. The main metabolic routes of hyzetimibe in humans were glucuronidation (M1, 15.6% in urine), mono-oxidation with additional sulfonation (M7, 7.92% in feces), and mono-oxidation (M4, 3.78% in feces). Hyzetimibe was considered generally safe and well tolerated in healthy volunteers.
When the metabolism of hyzetimibe and ezetimibe were compared, the following similarities were noted. 1) The primary metabolic pathway was glucuronidation, and the main metabolite was the glucuronide conjugate. 2) The primary route of elimination of drug-derived radioactivity after oral administration was via the feces in the form of unchanged drug. 3) Urine was not the main excretion pathway, and the main metabolite in urine was the glucuronide conjugate of hyzetimibe or ezetimibe. However, some differences existed in their metabolic pathways, such as the following: The amount of elimination of drug-derived radioactivity through the feces and urine differed; the types and contents of secondary metabolites (content <10%) differed; and the metabolic pathways in the human body were not completely consistent.
Conclusion
This pharmacokinetic study of oral 14C-radiolabeled hyzetimibe offers a highly comprehensive profile of the disposition of hyzetimibe and enhances the understanding of hyzetimibe metabolic characteristics. The structural change to the hydroxyl group in hyzetimibe (vs. ezetimibe) improved binding with glucuronic acid and increased urinary excretion compared with ezetimibe. This comparison highlights the need for additional investigation of the pharmacokinetic differences—and the resulting impact on efficacy and safety—between hyzetimibe and ezetimibe.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
Ethics Statement
The studies involving human participants were reviewed and approved by The First Affiliated Hospital of Soochow University. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
DO contributed to the study design; XW participated in the implementation of trials; ZL assisted with data analysis.
Funding
This study was supported by Key R&D Programs of Hunan Province (grant number 2019SK2241), the National Development of Key Novel Drugs for Special Projects of China (grant number 2017ZX09304014), Hunan Key Laboratory for Bioanalysis of Complex Matrix Samples (grant number 2017TP1037), the Innovation and Entrepreneurship Investment Project in Hunan Province (grant number 2019GK5020), the International Scientific and Technological Innovation Cooperation Base for Bioanalysis of Complex Matrix Samples in Hunan Province (grant number 2019CB1014), and the Science and Technology Project of Changsha (grant number kh1902002).
Conflict of Interest
Author XW was employed by Zhejiang Hisun Pharmaceutical Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Liyan Miao, Zheming Gu, and Zhenyu Yang for their help in the writing and support of the study. The authors also are grateful for all of the volunteers; investigators; and medical, nursing, and laboratory staff who participated in this study. They also thank Hisun Pharmaceutical Co., Ltd., which produced and provided hyzetimibe raw materials; the First Affiliated Hospital of Soochow University, which offered the clinical facility; and Value Pharmaceutical Services Co., Ltd., which provided measurement and analysis.
References
Chen, J., Lou, H., Jiang, B., Shao, R., Ruan, Z., and Wang, J. a. (2015). Simultaneous determination of hyzetimibe and its main active metabolite in plasma by LC-MS/MS and its application in PK study. Bioanalysis 7, 1857–1867. doi:10.4155/bio.15.114
Code of Federal Regulations (2020). Title 21 - Food and Drugs. Part 361 Sec. 361.1: Radioactive drugs for certain research uses. Available at: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?FR=361.1 (Accessed November 2, 2020).
Fuchs, WS, von Nieciecki, A, Molz, KH, Popescu, G, Weil, A, Barkworth, MF, et al. (1998). [The effect of bile secretion on the pharmacokinetics of a theophylline sustained-release preparation]. Arzneimittelforschung 48, 597–604.
International Commission on Radiological Protection (1991). Radiological protection in biomedical research. ICRP Publication 62. Ann ICRP 22, 1–28.
International Commission on Radiological Protection (2007). Recommendations of the international commission on radiological protection. ICRP Publication 103. Ann ICRP 37, 1–332.
Patrick, J. E., Kosoglou, T., Stauber, K. L., Alton, K. B., Maxwell, S. E., Zhu, Y., et al. (2002). Disposition of the selective cholesterol absorption inhibitor Ezetimibe in healthy male subjects. Drug Metab. Dispos. 30, 430–437. doi:10.1124/dmd.30.4.430
Ruan, Z., Jiang, B., Chen, J., Zhang, X., Lou, H., Xiang, M., et al. (2014). Pharmacokinetics, pharmacodynamics, safety, and tolerability of hyzetimibe (HS-25) in healthy Chinese subjects. The Journal of Clinical Pharmacology 54, 1144–1152. doi:10.1002/jcph.310
U.S. Food and Drug Administration (2002a). Zetia (ezetimibe) tablets: Clinical Pharmacology Biopharmaceutics Review(s). Available at: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2002/21445_Zetia_biopharmr_P1.pdf (Accessed November 2, 2020).
U.S. Food and Drug Administration (2002b). Zetia (ezetimibe) tablets: prescribing information. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2002/21445lbl.pdf (Accessed November 2, 2020).
Keywords: radiolabel, pharmacokenitics, phase I, healthy men, hyzetimibe, ezetimibe
Citation: Liao J, Wang X, Li Z and Ouyang D (2021) Pharmacokinetic Study of Oral 14C-Radiolabeled Hyzetimibe, A New Cholesterol Absorption Inhibitor. Front. Pharmacol. 12:665372. doi: 10.3389/fphar.2021.665372
Received: 08 February 2021; Accepted: 05 May 2021;
Published: 28 May 2021.
Edited by:
Sabina Passamonti, University of Trieste, ItalyReviewed by:
Cindy Yanfei Li, Amgen, United StatesZhihao Liu, United States Department of Agriculture, United States
Copyright © 2021 Liao, Wang, Li and Ouyang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhenyu Li, liyu1552@csu.edu.cn; Dongsheng Ouyang, ouyangyj@163.com