
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 13 April 2021
Sec. Ethnopharmacology
Volume 12 - 2021 | https://doi.org/10.3389/fphar.2021.654489
This article is part of the Research Topic Combating Redox Imbalance-Associated Complications with Natural Products View all 6 articles
 Shashank Kumar Maurya1
Shashank Kumar Maurya1 Neetu Bhattacharya2
Neetu Bhattacharya2 Suman Mishra3
Suman Mishra3 Amit Bhattacharya1
Amit Bhattacharya1 Pratibha Banerjee4
Pratibha Banerjee4 Sabyasachi Senapati4
Sabyasachi Senapati4 Rajnikant Mishra5*
Rajnikant Mishra5*Microglia, a type of innate immune cell of the brain, regulates neurogenesis, immunological surveillance, redox imbalance, cognitive and behavioral changes under normal and pathological conditions like Alzheimer’s, Parkinson’s, Multiple sclerosis and traumatic brain injury. Microglia produces a wide variety of cytokines to maintain homeostasis. It also participates in synaptic pruning and regulation of neurons overproduction by phagocytosis of neural precursor cells. The phenotypes of microglia are regulated by the local microenvironment of neurons and astrocytes via interaction with both soluble and membrane-bound mediators. In case of neuron degeneration as observed in acute or chronic neurodegenerative diseases, microglia gets released from the inhibitory effect of neurons and astrocytes, showing activated phenotype either of its dual function. Microglia shows neuroprotective effect by secreting growths factors to heal neurons and clears cell debris through phagocytosis in case of a moderate stimulus. But the same microglia starts releasing pro-inflammatory cytokines like TNF-α, IFN-γ, reactive oxygen species (ROS), and nitric oxide (NO), increasing neuroinflammation and redox imbalance in the brain under chronic signals. Therefore, pharmacological targeting of microglia would be a promising strategy in the regulation of neuroinflammation, redox imbalance and oxidative stress in neurodegenerative diseases. Some studies present potentials of natural products like curcumin, resveratrol, cannabidiol, ginsenosides, flavonoids and sulforaphane to suppress activation of microglia. These natural products have also been proposed as effective therapeutics to regulate the progression of neurodegenerative diseases. The present review article intends to explain the molecular mechanisms and functions of microglia and molecular dynamics of microglia specific genes and proteins like Iba1 and Tmem119 in neurodegeneration. The possible interventions by curcumin, resveratrol, cannabidiol, ginsenosides, flavonoids and sulforaphane on microglia specific protein Iba1 suggest possibility of natural products mediated regulation of microglia phenotypes and its functions to control redox imbalance and neuroinflammation in management of Alzheimer’s, Parkinson’s and Multiple Sclerosis for microglia-mediated therapeutics.
The brain is one of the immune-privileged organs whose mechanisms of the immune response are poorly understood. However, immune-responsive lymphocytes, microglia, and transcription factors have been observed in the brain. Pathological conditions in the brain have been observed associated with increased oxidative stress which leads to redox imbalance. The microglia responds to pathogens and injury. They accumulate in the region of inflammation or deterioration. It produces cytokines to maintain brain homeostasis, during development, adulthood and aging (Bitzer-Quintero and Gonzalez-Burgos, 2012; London et al., 2013; Burda and Sofroniew, 2014). They serve to communicate between the central nervous system and the peripheral immune system (Perry and Teeling, 2013). The microglia gets primed to release pro-inflammatory cytokines during chronic systemic inflammation, neuronal dysfunction, viral or bacterial infection, healthy aging, and dementia (Perry and Teeling, 2013). During postnatal brain development, microglia regulate the over-production of neurons via phagocytosing neural precursor cells, shape synaptic fields through synaptic pruning (Kim and Joh, 2006; Paolicelli et al., 2011; Li et al., 2012; London et al., 2013; Nayak et al., 2014). Studies also suggest that microglia support the laminar structure of the cortex, outgrowth of dopaminergic axons, and neurite formation (Pont-Lezica et al., 2011; Squarzoni et al., 2014).
In an adult healthy brain, microglia remain in a “resting” or “quiescent” state with a ramified morphology (Hernangomez et al., 2014; Andreasson et al., 2016; Hristovska and Pascual, 2016; Reemst et al., 2016; Arcuri et al., 2017). The microglia of aged organisms show activated state, morphological changes, alterations in telomere length, increased expression of MHC-II, and CD11b. They limit neurogenesis during aging and induce neuroinflammatory reactivity in astrocytes, reduce synapse and pre-synaptic function. Since aged astrocytes are induced by microglia and become neuroinflammatory, the targeting of astrocytes may protect synapses in aging-related diseases (Boisvert et al., 2018). The malfunctioning of microglia cause redox imbalance and increased oxidative stress leading to the recruitment of lymphocytes from blood to damaged sites (London et al., 2013).
Microglia was identified as a small population of phagocytic, migratory cells of mesodermal origin by (del Rio-Hortega, 1932). In 1939, del Rio-Hortega coined the term “microglial cell.” The two statements from del Rio-Hortega while describing microglia raised the controversy over microglia origin. In one statement, he proposed that microglia may arise from mesodermal cells of the innermost layer of meninges i.e., pia mater. In another statement, he proposed that microglia may of myeloid origin because of phenotypic similarities in morphology and phagocytic activity like mononuclear cells of the blood. Despite some controversies (Ginhoux and Prinz, 2015) on the mesodermal or ectodermal origin of microglia, several investigators presented data in support of the mesodermal origin of microglia. Through a series of experiments, it was further revealed that unique embryonic precursors, the yolk sac macrophages, are precursors of microglia. They are not found in the bone marrow. They reached the brain rudiment and appear to persist there into adulthood (Ginhoux and Prinz, 2015).
Microglia cells serve as innate immune cells in the brain and accumulate in regions of degeneration and produce a wide variety of cytokines to maintain homeostasis. The local microenvironment of neurons and astrocytes also affects phenotypes of microglia by interaction with both soluble and membrane-bound mediators (Wohleb, 2016). When neurons degenerate, either due to acute or chronic neurodegenerative diseases, the microglia get released from neurons and show activated phenotype (Perry and Teeling, 2013; Chen and Trapp, 2016). They exhibit dual functions. When the signal is short and moderate, microglia shows a neuro-protective phenotype to clear cell debris by phagocytosis and release growth factors for the healing of neurons. In acute and chronic signals, microglia release pro-inflammatory cytokines like TNF-α, IFN-γ, reactive oxygen species (ROS), nitric oxide (NO), all of which endanger neuronal damage. The malfunctioning of microglia results in the recruitment of lymphocytes from blood to damage sites, which further maintain brain homeostasis (London et al., 2013; ElAli and Rivest, 2016; Donat et al., 2017; Skaper et al., 2018). The microglia-mediated secretion pro-inflammatory cytokines, ROS and NO helps in restoring brain homeostasis as described. However, same redox imbalance or elevated oxidative stress also acts on microglia and affects its function. Increased inflammatory cytokines also induces microglia to secrete high levels of glutamate which in turn suppresses astrocytes glutamate transporters essential for maintaining extracellular physiological glutamate levels leads to excitotoxicity mediated neuronal damage. Gap junctions and hemichannels also play critical role in communication between different cells and are involved in development and progression of various neurological diseases. Literature suggest that activated microglia release excess of glutamate via Cx32 hemichannels. Excessive release of glutamate and proinflammatory cytokines from microglia also disrupt gap junctional communication and increased activity of hemichannels in astrocytes, adversely affect brain homeostasis and increase the severity of disease (Orellana et al., 2009; Takeuchi and Suzumura, 2014). Therefore, identification of microglia specific genes and its targeting with natural products may help in bringing back microglia to resting state, restoring its immunological surveillance function and in regulation of redox balance in the brain to mitigate disease conditions.
In activated microglia, Ionized binding protein1 (Iba1), a 17 kDa calcium-binding EF-hand protein, participates in actin-bundling, membrane ruffling, cell migration, and phagocytosis (Ito et al., 1998; Ito et al., 2001; Ohsawa et al., 2004). The Iba1-positive cells have small nuclei, scanty cytoplasm, and branched processes (Ito et al., 1998). In facial nerve axotomy, ischemia, and several other brain diseases, microglia show activated phenotype with up-regulated expression of Iba1 (Ito et al., 1998; Ito et al., 2001). The Iba1 immunostaining was also considered useful to study the pathophysiological role of activated microglia (Ito et al., 2001). It gets up-regulated by the pro-inflammatory cytokine IFN-γ and forms complex with L-fimbrin and co-localizes with F-actin and small GTPase Rac participates in membrane ruffling and phagocytosis activity in activated microglia, whereas Iba1 mutant mice completely lack membrane ruffling and phagocytosis (Oshawa et al., 2000; Kanazawa et al., 2002). In the hippocampus, the morphology of Iba1 positive microglial cells exhibits small somata with thin tortious and ramified processes. Iba1 has been shown to localize in the cytoplasm and nucleus of microglia (Jinno and Kosaka, 2008). In the dentate gyrus, Iba1 positive microglia were seen in the inner molecular layer, sub-granular zone and also found in the border between granule cell layer and molecular layer. The Iba1 has also been described as more suitable markers than trans-membrane and plasma membrane specific markers for studies on the complex organization and structural analysis of microglia (Korzhevskii and Kirik, 2016). Genome-wide transcriptome and epigenome studies have shown that microglia is different from other tissue macrophages and glial cells. It was also found that Cx3cr1, Trem2, Tyrobp, Cd33, Sall1, Tmem119, Siglec-H, Iba1, and P2ry12 specifically expressed in mouse microglia (Chiu et al., 2013; Galatro et al., 2017). Recently, another microglia specific marker Tmem119 has been identified as a marker of mature microglia in the brain (Bennett et al., 2016; Satoh et al., 2016; Bohnert et al., 2020) and Siglec-H expression specific to activated microglia where it cause phagocytosis of glioma cells of brain tumor (Kopatz et al., 2013; Konishi et al., 2017) and Olfml3 as a new Tgf-β1/Smad2 target gene in microglia (Neidert et al., 2018) has been reported. However, detailed studies on targeting these identified genes to understand microglia function in the regulation of oxidative stress and neurodegeneration are still elusive.
Microglia act as the first responders to infection and inflammation in the CNS followed by migration of the peripheral macrophages into the CNS during neuroinflammation. Activation of microglia by intensive acute or chronic stress cause loss of essential function of microglia and they start showing more of deleterious effect, which in turn cause immune compromised and alteration in CNS protection (London et al., 2013). Understanding the disease-associated-microglia (DAM), its heterogeneity and key regulators, and emerging “microgliome” will help to distinguish transcriptionally distinct and neurodegeneration-specific microglial profiles for clinical research and drug discovery (Sousa et al., 2017). The innate immune receptor and its adaptor protein, such as triggering receptor expressed on myeloid cells 2 (TREM2) and DNAX-activating protein of 12 kDa (DAP12), has been associated with a range of CNS disease phenotypes (Painter et al., 2015). Impaired signalling in disease pathogenesis is caused by mutations in TREM2-DAP12 pathway leading to altered microglial survival and proliferation which in turn affect phagocytosis, cytokine production, and immune responses (Konishi and Kiyama, 2018). In the TREM2-DAP12 signaling, the TREM2 signal via DAP12 receptor activates the protein tyrosine kinase ERK (ERK1/2), regulating actin cytoskeleton polarization and cytoskeleton organization and stimulates microglial phagocytosis. TREM2 also upregulates the expression of the CCR7 chemokine receptor (Mecca et al., 2018). Secondly, TREM2 interaction with DAP12 promotes microglial survival by activating the Wnt/β-catenin signalling pathway. TREM2-DAP12 interaction activates PI3K/Akt signaling, leading to blockage of GSK3β and stabilizing β-catenin that moves into the nucleus, thereby triggering pro-mitotic and anti-apoptotic genes. TREM2-DAP12 signaling activates PI3K/Akt pathway through the regulation of NF-κB (inflammatory genes) and inhibition of RAF (MAPK signaling) causing TLRs signaling inhibition (Zheng et al., 2017; Mecca et al., 2018). Gene ontology analyses coupled with bioinformatics approaches (such as weighted co-expression network analysis, WGCNA) and confirmation in AD mouse models (adult WT and 5xFAD mice) have revealed distinct pro-inflammatory and anti-inflammatory phenotypes within DAM (Rangaraju et al., 2018). Results proposed that the pro-inflammatory DAM was characterized by higher expression of CD44, CD45, and Kv1.3 channels and are controlled by NFkB, Stat1, and RelA signaling pathways, while the anti-inflammatory DAM profile was characterized by CXCR4 expression and regulated by LXRα/β (Rangaraju et al., 2018). Further, the findings in human AD suggested an increase in the expression of pro-inflammatory DAM proteins at pre-clinical stages which showed a positive link with tau and neurofibrillary tangle pathology (Rangaraju et al., 2018). A recent report highlighted the potential therapeutic targets in modulating microglial functions for Alzheimer’s disease (AD) as NLRP3 inflammasome activation (VRAC, P2X7, NLRP3), complement production and signalling (C1, C1q, C3, CR1, Factor B, Factor D, and Properdin), and TREM2/DAP12 signalling (TREM2, SHIP-1, PLCγ2, and Apolipoprotein E) (Nizami et al., 2019). Alterations in TREM receptors (TREM-1/2) are emerging as key components associated with inflammatory bowel disease (IBD) and neurodegenerative disorders spreading inflammation through the gut-brain-axis namely microbiota dysbiosis, leaky gut, and inflammation (Natale et al., 2019). Over the years, the novel molecular and functional signature for brain-resident microglia has been proposed. Neurodegeneration models have proposed cell markers like P2ry12, Fcrls, Siglec-H, Olfml3, and Tmem119 (Chiu et al., 2013; Butovsky et al., 2014; Bennett et al., 2016), and several of them need to be validated further. An anatomically complete atlas of the adult human brain transcriptome critically analyzed transmembrane protein 119 (TMEM119) in bulk human brain tissue data from the Allen Brain Institute (Hawrylycz et al., 2012). The Tmem119 is a stable and robustly expressed microglial marker of brain-derived microglia in both mice and humans (Bennett et al., 2016). The cell-type expression profiling tool (CellMapper) identified 30 additional genes along with TMEM119 i.e., ACY3, ADAM28, ADORA3, ALOX5AP, C1QB, C3, CD33, CD84, CIITA, CPED1, CSF2RA, DHRS9, FCER1G, FYB, GPR34, HPGDS, IGSF6, LAPTM5, LY86, P2RY13, RASAL3, SASH3, SELPLG, SPN, SUCNR1, SUSD3, SYK, TBXAS1, TLR7, and TREM2 (Bonham Luke et al., 2019). Interestingly, scientists found that microglial module genes are strongly expressed in numerous healthy human brain regions known to be vulnerable in AD. Recent studies using weighted gene co-expression network analysis (WGCNA) strongly characterized a gene network that relates to both aging and neurodegenerative diseases (Mukherjee et al., 2019). Since research on the human brain transcriptome is restricted to post-mortem tissue analyses, thus mouse genetic experimentation mouse is now widely characterized as the model organism of choice for studying the diseases of humans and brain aging, as they share 99% of their genes with humans (Rosenthal and Brown, 2007). Thus, the discovery of similarly preserved networks and cell type signature genes in animal models will help to get mechanistic insights and analyze differential expression in Alzheimer’s disease, Parkinson’s disease, and related disorders.
Neurodegenerative diseases are a complex group of disorders characterized by progressive decline in structure and function of central and/or peripheral nervous system. The most common neurodegenerative diseases are Alzheimer’s disease, Parkinson’s disease and Multiple Sclerosis. These diseases lead to loss of neurons and thus affect the brain homeostasis. The disease pathogenesis very often caused by factors like oxidative stress, neuroinflammation, altered clearance mechanism, deposition of proteins like amyloid-beta, alpha-synuclein etc., malfunctioning of brain-specific immune cells (microglia), disturbed blood-brain barrier, and infiltration of lymphocytes. Many mechanisms and key genes have been identified which play a central role in neurodegenerative disease where involvement of microglia specific proteins showed the importance in regulating healthy immune system of brain to combat the effects of neurodegeneration (Figure 1). In this review diseases like Alzheimer’s, Parkinson’s and Multiple Sclerosis disease were discussed to highlight the microglial proteins which have a key role in brain and neuronal homeostasis.
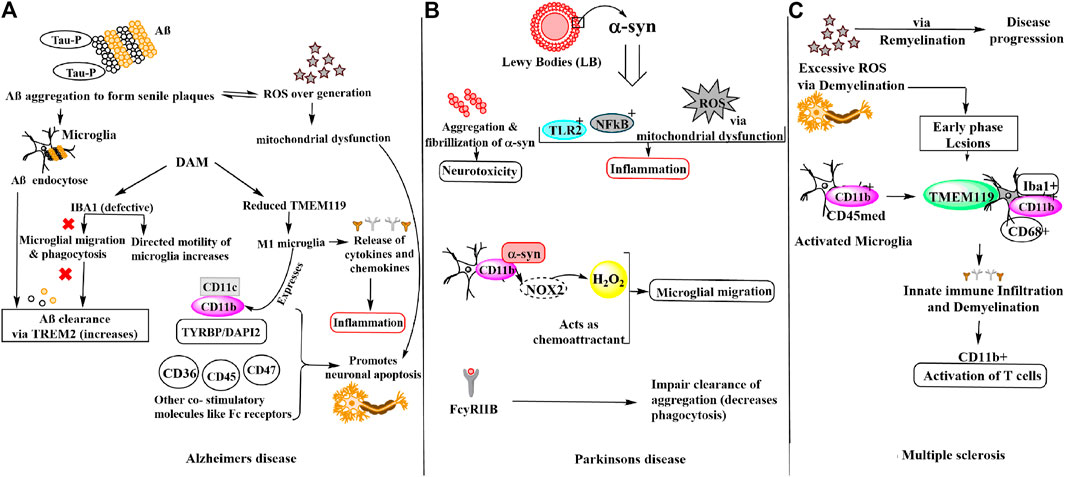
FIGURE 1. Schematic representation of microglial involvement in disease pathogenesis of (A) Alzheimer’s disease (B) Parkinson’s disease (C) Multiple sclerosis.
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder affecting majorly the elderly population. Specific molecular signatures of AD include extracellular amyloid plaques deposits of amyloid-beta (Aβ) peptide, neurofibrillary tangles of tau (microtubule-binding protein), dystrophic neurites, loss of neurons and synapses and fibrous glia (Murphy and LeVine, 2010; Hansen et al., 2018). The tau protein hyper-phosphorylation observed at Ser202/Thr205, Thr212, Ser214, Thr217, Ser262, and Ser422 amino acid residues in AD due to oxidative stress results in neurofibrillary tangles via self-assembly of paired helical filaments admixed with straight filaments (Miao et al., 2019). Further, the aggregates of AD-P (AD-hyperphosphorylated) tau are seen in dystrophic neuritis around the Aβ plaque (Vickers et al., 2016). Tau pathology has also been found to be linked with reactive microglia, astrocytes and increased levels of pro-inflammatory molecules including cytokines and complement proteins (Vogels et al., 2019). It is noteworthy that in per mole of tau protein, 2–3 mol of phosphate is present in normal condition. However, during anesthesia and hypothermia a transient rise is observed while in AD pathology, the phosphate concentration increases drastically i.e., >6 mol/1 mol of tau (Iqbal et al., 2010; Simic et al., 2016; Miao et al., 2019). Another hallmark of AD pathology is the oxidative stress due to redox imbalance caused by overproduction of ROS. Excess of ROS or free radicals lead to nucleic oxidation, protein oxidation and/or lipid peroxidation, metal homeostasis disturbance and mitochondrial dysfunction. Notably, there is a vicious cycle that modulates ROS by Aβ secretion or vice versa. It is also reported that generation of Aβ via APP processing leads to enhanced ROS generation (Moneim 2015; Wojsiat et al., 2018). Aβ40 and Aβ42 monomers produced from APP processing by β-secretases, γ-secretases, oligomerize and aggregate into senile plaques that lead to activation of microglia. Microglia then endocytose Aβ aggregates contributing toward their clearance via TREM2 surface receptors, simultaneously triggering an innate immune response against the aggregation (Tiwari et al., 2019). Earlier, this microglia reaction was thought to be concomitant and triggered by amyloid deposits and dystrophic neurites in AD. However, recent GWAS (genome-wide association studies) have identified that the majority of AD risk loci are located within or near to the genes that are highly and sometimes uniquely expressed in microglia. Thus, microglia being critically involved in the early steps of the disease and identified as important potential therapeutic targets (Hemonnot et al., 2019). Notably, in both physiological and disease states the microglia expresses Iba1 which participates in both baselines and directed motility. In AD pathogenesis, microglia shifts from baseline to directed-motility, involving proteins including Iba1, P2ry12, Cfl1, and Coro1a. Aβ immunotherapy suggested to result in increased microglial baseline motility (suggested by increased IBA1 and P2RY12 expression, which are considered “homeostatic” microglial markers), potentially due to decreased Aβ and/or tau, but without restoration of the physiological status of microglia (Franco-Bocanegra et al., 2019). To identify microglia in humans and experimental models IBA1 has been widely used as a marker as it is distributed along the cell body and processes. However, there is experimental evidence that microglial motility is impaired in AD, for instance in a post-mortem study this was observed by the changes seen in the interactions of motility-related proteins with Aβ as well as with each other. Iba1 is crucial for the formation of actin bundles (Ohsawa et al., 2000), which in turn help in the formation of lamellipodia, filopodia, and membrane ruffles essential for microglial migration and phagocytosis, important for Aβ clearance (Ohsawa et al., 2000; Zotova et al., 2013). In disease-associated microglia (DAM) state, expression of some homeostatic gene, like TMEM119 (transmembrane protein 119) and other genes (e.g., CX3CR1 and P2RY12) is reduced but their major role in AD is unknown (Hansen et al., 2018). Although another molecule CD11b (αM integrin), along with the increased expression of APOE, TREM2, CD68, CLEC7a, and ITGAX (CD11c) referred to as DAM, microglial neurodegenerative phenotype (MGnD), or activated response microglia (ARM) are also observed under neurodegenerative conditions (Keren-Shaul et al., 2017; Krasemann et al., 2017; Sala Frigerio et al., 2019). Remarkably in AD, M1 (proinflammatory) microglia express MHC-II integrins (CD11b, CD11c) and other co-stimulatory (CD36, CD45, CD47) and Fc receptors. Thus, it is found that in microglia, CD11b is required with TYROBP/DAP-12 to control the production of microglial superoxide ions that promotes neuronal apoptosis during brain development. Additionally, M1 microglia produces pro-inflammatory cytokines and chemokines, ROS and NOS. While, M2 microglia produce anti-inflammatory cytokines (IL-10, TGF-β), growth factors (IGF1, FGF, CSF1), and neurotrophic growth factors (nerve-derived growth factor, brain-derived neurotrophic factor (Bdnf), neurotrophins, glial cell-derived neurotrophic factor (Gdnf), which downregulate, protect, or repair in response to oxidative stress and inflammation (Sarlus and Heneka, 2017).
Parkinson’s disease (PD) is the second most common neurodegenerative disorder after AD with a sharp increase in incidence after 60 years (de Lau and Breteler, 2006). PD is characterized by four cardinal signs (resting tremor, bradykinesia, rigidity, and postural instability). Notably, the hallmark of PD is the presence of Lewy bodies (LBs) whose primary structural component is filamentous α-synuclein (α-syn) (Eriksen et al., 2005; Kouli et al., 2018) and the degeneration of dopaminergic neurons in the substantia nigra pars compacta responsible for the motor impairments (Moore et al., 2005). α-syn was found to increase ROS levels in dopaminergic cells. Thus, it was reported that oxidative damage could be modulated by α-syn perhaps via modulation of mitochondrial function because mutant α-syn over-expression leads to significant enhanced sensitivity of dopaminergic neurons to mitochondrial toxins including mitochondrial processing peptidases (MPP)+ and 6-hydroxydopamine, ensuing an augmented protein carbonylation and lipid peroxidation (Chinta and Andersen 2008). Although, identification of SNCA genes that encodes α-syn and many other genetic variants, susceptibility for PD plays a crucial role in the disease pathogenesis. Interestingly, reactive microglia and neuroinflammation have been well known in fibrillary formation in PD (McGeer et al., 1988; Polymeropoulos et al., 1997). Microglia and the innate immune system are essential for synaptic pruning to impart changes to the neural world around them and suggested their contribution to both neurological and psychiatric illnesses (Stevens et al., 2007; Hong et al., 2016; Sekar et al., 2016; Vainchtein et al., 2018). In the same manner, microglia-derived inflammation might stimulate astrocytes to adopt a neurotoxic role or to lose neurotrophic or synaptoptrophic functionality (Liddelow et al., 2017). However, the microglial response to excess or mutant α-syn is still under investigation. The innate immune surface receptors of microglia including toll-like receptor 2 (TLR2) get activated by extracellular α-syn oligomers that function as damage-associated molecular patterns (DAMPs) (Kim et al., 2013; Kam et al., 2020). Under physiological conditions, α-syn regulates the trafficking of synaptic vesicles and the formation of the SNARE complex in the presynaptic terminals, but in pathologic states, α-syn undergoes aggregation and fibrillization that results in neurotoxicity in PD (Burre et al., 2015). Microglia targets highly active neurons by sensing and modulating neuronal activity in the brain (York et al., 2018). The neurons release α-syn in CSF and extracellular space, as well as conditioned media of neurons in a calcium-dependent manner (Emmanouilidou et al., 2010) and crucial for the so-called spreading of α-syn and dependent on neuronal firing (Emmanouilidou et al., 2016; Yamada and Iwatsubo, 2018). Furthermore, microglial migration is being directed by Α-syn as a chemo-attractant. Interaction CD11b with α-syn and subsequent NOX2 activation lead to increase H2O2 (hydrogen peroxide), which can also act as a final direct signal for migration (Wang et al., 2015a). This is evident with the post-mortem study of PD patients where the activated microglia is found in close contact with neurons presenting Α-syn pathological accumulation (Croisier et al., 2005). However, it should be noted that microglia can and will encounter α-syn by the trogocytosis of neuronal structures as well (Weinhard et al., 2018). α-syn interacts with microglial surface receptors including Fc gamma receptor IIB (FcγRIIB) to reduce microglial phagocytosis, which in turn could impair the clearance of aggregated species or other parenchymal debris (Choi et al., 2015). Inflammatory microglial response activated by fibrillary α-syn via NF-κB pathway (Yun et al., 2018). Microglial uptake of α -syn fibrils is regulated by Fyn kinase and class B scavenger receptor CD36 which was genetically linked to PD as per recent GWAS (Nalls et al., 2019; Panicker et al., 2019). Microglia releases IL-1β through a signalling mechanism that led to NLRP3 inflammasome priming and activation. Thus, aggregated α-syn induces pro-inflammatory microglial behaviors via both classic innate immune receptors and interactions with intracellular signaling cascades. (Kam et al., 2020). During high levels of inflammation by chemokines such as CX3C ligand 1 (CX3CL1) produced by neurons or C-C motif chemokine ligand 2 (CCL2) and CXC ligand 10 (CXCL10) produced by microglia, astrocytes, and other inflammatory cells, peripheral NK cells are recruited to the CNS. Other important inhibitory receptors found on Natural Killer (NK) cells are NKG2A for Qa-1b and CD244 for CD48 in mice and CD85 for HLA-A, CD94 for HLA-E in humans. CD48 found on the surface of lymphocytes and other immune cells, endothelial cells, and dendritic cells is a member of the CD2 subfamily of immunoglobulin-like receptors which include signalling lymphocyte activation molecules and functions in activation and differentiation pathways of these cells. CD48 does not have a transmembrane domain but is held at the cell surface by a GPI anchor via a C-terminal domain which may be cleaved to yield a soluble form of the receptor. Microglia-NK cell interaction exerts protective function which can induce decreased expression of the MHC class I molecule Qa1 on activated microglia, which in turn triggers NK cell-mediated cytotoxicity towards hyperactive microglia. As microglia are known to become aberrantly activated in the presence of sustained α-syn burden, targeting microglial activation states by suppressing their deleterious pro-inflammatory neurotoxicity may be a valid therapeutic approach for PD treatment. Furthermore, NK cell-deficient animals displayed increased inflammation in the CNS, as shown through glial fibrillary acidic protein (GFAP) and Iba1 (Mihara et al., 2008).
Another chronic inflammatory neurodegenerative disease of the central nervous system (CNS) is multiple sclerosis (MS) which is characterized by demyelination and neuroaxonal damage that leads to the formation of lesions throughout the central nervous system (CNS). Pathological conditions of MS also include generation of excessive ROS, reactive nitrosative species (RNS) responsible for oxidative stress and redox imbalance. ROS plays dual role in causing axonal damage that subsequently leads to neurodegeneration in MS either by affecting demyelination causing apoptosis of oligodendrocytes or remyelination process by oligodendrocyte precursor cells maturation disruption. ROS can also activate NF-kB which alleviates the expression of TNFα and many other genes involved in MS pathogenesis (French et al., 2009; Cai and Xiao, 2016; Cunniffe and Coles, 2021). Gray matter (GM) demonstrated a low number of activated microglia and little infiltration of lymphocytes compared to white matter (WM) lesions (Deng and Sriram, 2005). The early stage of MS is characterized by relapses and the later stage, by progressive disability. Microglia and macrophages play a crucial role in the disease course by actively initiating immune infiltration and the demyelination cascade during the early phase of the disease while promoting remyelination involved in disease progression in later stages. A controversial distinction of microglia from monocyte-derived macrophages depends on expression profiles of CD11b and CD45 (Koeniger and Kuerten, 2017; Martin et al., 2017). Other, microglial valuable markers are TMEM119 (Bennett et al., 2016; Satoh et al., 2016; Li et al., 2019), P2RY12 (Mildner et al., 2017), SALL1 (Buttgereit et al., 2016), and Siglec-H (Konishi et al., 2017) which distinguishes them from other macrophages. Studies suggested that TMEM119 is expressed exclusively on Iba1+CD68+ microglia and not on infiltrated Iba1+CD68+ macrophages within demyelinating lesions of MS that govern the edge of active lesions in MS (Zrzavy et al., 2017), indicating that microglial activation is related to disease development while its expression is absent in immature microglia (Satoh et al., 2016). In experimental autoimmune encephalomyelitis (EAE) mice the appearance of inflammatory T cells in the CNS is consistent with the activation of CD11b + microglia (Murphy and LeVine, 2010; Wang et al., 2019). Microglial immune activation is present in MS lesions, and microglia play a role as phagocytes in demyelination, which is not well understood. Even though, cell membrane receptors present, at low levels than that observed in circulating antigen-presenting cells, still include those that are responsible for activation of T cells [MHC class I and II, CD40, and B7.1 and B7.2 adhesion molecules (LFA, CD11a, CD11b)], along with complement, cytokine, and chemokine receptors causing disease pathogenicity (Deng and Sriram, 2005). The pathogenesis of three neurodegenerative diseases discussed above involves majorly IBA1, CD11b, and TMEM119 molecules in microglial migration, motility and phagocytosis in response to disease pathogenesis.
Based on the microglia functions in regulating neuroinflammation, drug targeting key homeostatic proteins of microglia has been proposed as a potential approach to deal with inflammation in the brain which ultimately leads to neuronal damage. This can be achieved by regulation or modification of receptors present on microglia, signaling pathways like JAK/STAT and NF-κB, using drugs.
Receptors such as TLRs and CB2 has been reported as a novel pharmacological target for neurodegenerative disorders like PD. Candesartan cilexetil, is a synthetic, benzimidazole-derived angiotensin II receptor antagonist prodrug with antihypertensive activity also observed to inhibit TLR2 and TLR4 expression in PD which in turn may reverse microglial phenotype from pro-inflammatory to anti-inflammatory in the oligomeric α-synuclein microenvironment (Daniele et al., 2015; Liu et al., 2019). Another drug, rifampin, and its autoxidation product rifampicin quinone, an antibiotic regulates α-synuclein induced TLR2 and P2X7 dependent microglia inflammatory response under in vitro condition (Acuña et al., 2019). In vitro studies have shown that antagonist of TLR4 such as TAK-242 or RSLA reduces oxidative stress and microglia-mediated TNF-α production and neuronal death (Hughes et al., 2019).
JAK/STAT and NF-κB signaling pathways play important role in the polarization of microglia to the M1 state (Yan et al., 2018). Therefore, targeting this signaling pathway may provide an alternative strategy for the supression of the microglia-mediated neuroinflammation. Phytochemical from a-asarone from Annonaceae and Araceae species has been shown to inhibit the NF-κB signaling which suppresses microglia polarization to M1 state and reduce the production of pro-inflammatory cytokines in MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) induced PD mouse model (Kim et al., 2015). Another drug, basically a bioactive flavonoid named Tanshinone I prevent nigrostriatal dopaminergic neurodegeneration by selectively inhibiting NF-κB signaling activation, and reverses microglia form M1 pro-inflammatory to M2 anti-inflammatory state (Wang et al., 2015b).
Apart from this, several drugs that are currently in use for a disease like lenalidomide for multiple myeloma, zonisamide for epilepsy, minocycline for infectious diseases, dimethyl fumarate for multiple sclerosis are capable of inhibiting activation of microglia and attenuating production of pro-inflammatory cytokines in a mouse model of PD (Liu et al., 2019). However, the mechanism of action of these drugs on microglia is still elusive.
CD70 derived adenosine A2A signaling, Histamine-4 receptor and NLRP3 mediated inflammasome in microglia has also been reported as a promising target in modulating microglial activation for PD (Rocha et al., 2016; Rajasundaram, 2018). Similarly, IL10, cyclic AMP, and vitamin D have been shown to enhance anti-inflammatory phenotype by converting microglia from M1 to M2 state in PD mice model (Subramaniam and Federoff, 2017). Intracerebral injection of IL-10 has been shown to decrease the expression of iNOS and increase the production of TGF-β in the MPTP mouse model (Schwenkgrub et al., 2013). Cyclic AMP in combination with IL-4 causes conversion of microglia from M1 to M2 state decreases the production of oxidative molecules and pro-inflammatory cytokines and improves the phagocytic activity in in vitro (Ghosh et al., 2016). Cyclic adenosine monophosphate (AMP) is a major microglia homeostasis regulator which in turn gets modulated by phosphodiesterase. PDE inhibitors such as rolipram, sildenafil, yonkenafil, and ibudilast have been proposed as a potential therapeutic drug for the neurodegenerative disorder, including MS (Liu et al., 2019). Vitamin D has been shown to inhibit microglial activation; a shift from M1 to M2 polarizes states which in turn protect dopaminergic neurons against neuroinflammation and oxidative stress in OHDA/MPTP induced mouse PD model (Calvello et al., 2017). Another molecular target PPARs has been shown to modulate microglial polarization. Pioglitazone, rosiglitazone is agonists of PPAR-g have shown anti-inflammatory and neuroprotective role (Omeragic et al., 2019). Kca3.1 inhibitors charybdotoxin, clotrimazole, senicapoc, and TRAM-34 reduce microglial neurotoxicity in AD pathology. Among them, Senicapoc has been used for the treatment of sickle cell anemia. Repurposing senicapoc has been shown to reduce the amyloid load, neuroinflammation, and improves neuronal plasticity in 5xFAD mice (Jin L. W. et al., 2019). Another pathway known as Kynurenine has been proposed to contribute to CNS disease by producing several s neuroprotective products during its metabolism. In microglia, an enzyme kyurenine 3-monooxygenase (KMO) expression converts KYN to neurotoxic metabolites such as 3-hydroxykynurenine (3-HK) and quinolinic acid (Quin). Studies have shown region-specific increased production of KYN as well as decreased KYM metabolism toward the production of neurotoxic microglia metabolites in post-mortem AD brain (Biber et al., 2019; Sorgdrager et al., 2019). However, understanding of the role of KYN metabolic products in the development and progression of AD, neurotoxicity, and its association with microglia-mediated neuroinflammation need to be addressed for identification of specific KYN metabolite inhibitor.
Pharmacological antibody-mediated targeting of TREM2 showed to activate TREM2 signaling which further increases microglial survival and migration (Zheng et al., 2017; Schlepckow et al., 2020). However, detail study about TREM2 protein normal function in microglia regulation and pathophysiology of the brain need to be addressed so that it can be investigated further for a promising drug targeting. S100A4 as a marker of microglial reactivity, suggesting the contribution of S100A4 regulated pathways to neuroinflammation, and identify niclosamide as a possible drug in the control and attenuation of reactive phenotypes of microglia, thus opening the way to further investigation for a new application in neurodegenerative conditions (Serrano et al., 2019). HuR, an RNA binding protein is a key regulator of many genes that make up the molecular signature of activated microglia, including increased production of proinflammatory cytokines, chemokines, and migration-associated factors. HuR may be a therapeutic target depending on the disease and stage of the disease. HuR expression is not limited to microglia, and thus it remains to be seen whether other cells could be adversely affected by its inhibition (Matsye et al., 2017). In the Intracerebral hemorrhage (ICH) brain, TLR4 is upregulated mainly in microglia (Lively and Schlichter, 2018). In the blood injection ICH model, deletion of TLR4 inhibits NF-κB signaling pathway and improves ICH outcomes (Lively and Schlichter, 2018). The in vivo and in vitro studies on glioma stem cells and BV-2 cells respectively showed that TLR4 activation is essential for M1 microglial polarization (Dzaye et al., 2016; Gong et al., 2016). Pinocembrin (5, 7-dihydroxyflavanone), a natural product extracted from Propolis, decreases M1 phenotype microglia, as evidenced by decreases in the number of the CD16/32 + microglia and iNOS expression in MACS-sorted CD11b + microglia. However, this effect of pinocembrin was abolished in TLR4lps-del mice with ICH, indicating that pinocembrin reduced microglial M1 polarization mainly by TLR4 inhibition (Lan et al., 2017). Cocaine self-administration also increases microglial activation. The current results also highlight a distinct molecular pathway in microglia through which cocaine increases BDNF, involving the phosphorylation of MeCP2 and subsequent translocation from the nucleus to the cytosol, which frees the BDNF promoter and permits its transcriptional activation. This leads to a significant release of BDNF by microglia (Cotto et al., 2018). Metformin, an anti-diabetic drug decreased Iba-1 staining in the dorsal horn and inhibits development of neuropathic pain in spared nerve injury (SNI) mice model. But the effect of metformin on microglia and in improving positive behavior was only observed in male mice indicating that the neuropathic pain modifying effects of metformin are sex-specific supporting a differential role for microglial activation in male and female mice (Inyang et al., 2019).
Neurodegenerative diseases (NDs), mainly Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), and multiple sclerosis (MS), onset and progression are characterized by oxidative stress, redox imbalance and neuroinflammation caused by microglia. The activated microglia is polarized into M1 (pro-inflammatory) and M2 (anti-inflammatory) functional phenotypes. One of the proposed therapeutic approaches for the treatment of NDs and related diseases is inhibition of the M1 phenotype while stimulating the M2 phenotype. Several natural products have been screened for their therapeutic modulatory role of microglial polarization in NDs (Jin X. et al., 2019). The major advantages of using these natural products are their novel structure (Figure 2), multi-target molecular mechanism, potent efficiency, and safe pharmacodynamics. In this section, we primarily summarized the therapeutic potential of natural products, such as curcumin, resveratrol, cannabidiol, ginsenosides, flavonoids, and sulforaphane, for their molecular mechanisms in the treatment of NDs by targeting microglial proteins.
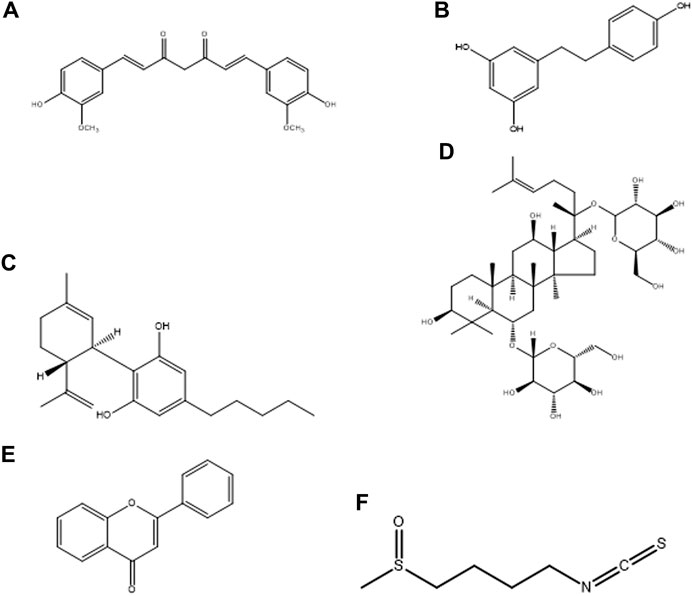
FIGURE 2. Chemical structure of natural products, (A) Curcumin, (B) Resveratrol, (C) Cannabidiol, (D) Ginsenosides, (E) Flavonoids, and (F) Sulforaphane.
Curcuma longa L., a member of the ginger family (Zingiberaceae), is a rhizomatous herbaceous perennial herb and is cultivated in countries with a tropical climate in Asia like India, China, etc. The rhizomes of turmeric (Curcuma longa L.) grow below the ground and curcumin is a major polyphenol isolated from them. Curcuma longa L. and curcumin has been used for thousands of years as a remedy and cited in the ancient Indian medical system, Ayurveda, for treatment of several illnesses, such as inflammation, gastric, hepatic, infectious diseases, and blood disorders. (Goel et al., 2008; Hatcher et al., 2008; Ghasemi et al., 2019). The phytochemical curcumin is a free radical scavenger and hydrogen donor and exhibits both pro-oxidant and antioxidant activity. In the experimental diabetic rats induced by streptozotocin (STZ), the effect of curcumin on liver anti-oxidative stress showed inhibition of polyol pathway associated with down-regulated expression of protein kinase C (PKC) and poly ADP ribose polymerase (PARP) in the curcumin-treated group compared with the diabetic model (Xie et al., 2017). Curcumin directly reacts and scavenge reactive oxygen species (such as superoxide anion, hydroxyl radicals, hydrogen peroxide, singlet oxygen, nitric oxide, peroxynitrite and peroxyl radicals). It further indirectly induces the expression of cytoprotective proteins as an antioxidant response, these includes superoxide dismutase (SOD), catalase (CAT), heme oxygenase 1 (HO-1), glutathione-S-transferase (GST), glutathione reductase (GR), glutathione peroxidase (GPx), NAD(P)H: quinone oxidoreductase 1 (NQO1) and γ-glutamylcysteine ligase (γGCL) (Trujillo et al., 2013). Recently the experimental model of the Saccharomyces cerevisiae budding yeast demonstrated that curcumin significantly increases oxidative stress and accelerates replicative and chronological aging of yeast cells devoid of anti-oxidative protection (with SOD1 and SOD2 gene deletion) and deprived of DNA repair mechanisms (RAD52). Curcuminoids decrease hepatic fat accumulation (hypertriglyceridemia) and avert accumulation of lipid droplets within hepatocytes (steatosis) by downregulating lipogenic factors and activating AMPK in liver (Sahebkar, 2014; He et al., 2017).
In brain, curcumin is a pleiotropic molecule showing therapeutic properties on microglia, such as inhibits microglia transformation, inflammatory mediators, and consequent NDs. CNB-001, a synthetic pyrazole derivative of curcumin, at a potent concentration (1–10 µM) suppressed the production of lipopolysaccharide (LPS)-induced nitric oxide (NO) and expression of inducible NO synthase (iNOS) in primary cultured rat microglia (Akaishi and Abe 2018). Further, this study suggested that the CNB-001 exerts its anti-inflammatory effects through inhibition of NF-κB and NO production by blockade of the p38 mitogen-activated protein kinase (MAPK) signaling pathways in microglia culture (Akaishi and Abe, 2018). Eun et al., 2017 investigated the neuroprotective effect of fermented Curcuma longa L (FCL) and its efficacy for the regulation of memory dysfunction in brain cell lines (C6, rat glioma cells, and BV2, murine microglia) and scopolamine-treated mice. The result indicated FCL inhibited the production of NO and prostaglandin E2 (PGE2) by the blockage of iNOS and cyclooxygenase-2 (COX-2) expression in BV2 cells. In the FCL, the curcuminoids content was 1.44%. (Eun et al., 2017). In another study, curcumin significantly inhibited LPS-mediated induction of COX-2 expression in both mRNA and protein levels in microglial cells. Additionally, curcumin distinctly inhibited LPS-induced NF-kB and activator protein-1 (AP-1) DNA bindings in BV2 microglial cells (Kang et al., 2004). Studies on AD pathology have shown numerous pharmacologic effects and mechanisms of action of curcumin, such as lessened β-amyloid plaques, deferred degradation of neurons, metal-chelation, anti-inflammatory, antioxidant, and reduced microglia formation, leading to the overall memory enhancement in patients with AD (Mishra and Palanivelu, 2008).
In recent years, a type of natural phenol resveratrol (RES) has fascinated scientists especially for its potent protective effect in NDs. The phenolic resveratrol acts as a commanding antioxidant in the presence of the ascorbate or glutathione system through both classical, hydroxyl-radical scavenging and a new, glutathione-sparing mechanism (Burkitt and Duncan, 2000). Resveratrol was found to act as an antioxidant, anti-inflammatory and anti-mutagen effects in leukemic HL-60 cells and acts on the major stages of carcinogenesis. It mediated anti-initiation activity by inducing phase II drug-metabolizing enzymes, anti-promotion activity by inhibiting cyclooxygenase and hydroperoxidase functions and antiprogression activity by inducing human promyelocytic leukemia cell differentiation (Jang et al., 1997). With regards to brain pathologies, the therapeutic potential of RES is through the modulation of microglial activation and regulation of neuroinflammation. The possible molecular mechanisms are direct antioxidant property of RES, TLR4 and NF-κB signaling pathway, SIRT1 and AMPK signaling, MAPKs signaling pathway, Nrf2/HO-1 signaling, and NLRP3 inflammasome signaling pathway (Huang et al., 2020). In the Yang et al., 2017 suggests RES reduces inflammatory damage and encouraged microglia polarization to the M2 phenotype via PGC-1α measured in in vitro and in vivo. Furthermore, they found RES ameliorated LPS-induced neuroinflammation behavior in mice models (Yang et al., 2017). It has been shown that the potential therapeutic activity of RES is by inhibition of COX-1 activity. Interestingly, RES potently reduced PGE2 synthesis and the formation of 8-iso-prostaglandin F2α (8-iso-PGF2α) in LPS-activated primary rat microglia. Resveratrol is reported as the first known inhibitor that specifically prevents the expression of mPGES-1 in a dose-dependent manner (mRNA and protein) without affecting the expression of COX-2 levels (Candelario-Jalil et al., 2007). mPGES-1 is a key enzyme for the synthesis of PGE2 by activated microglia cells.
Cannabidiol (CBD), one of the important pharmacologically active Phytocannabinoids of Cannabis sativa L., is non-psychoactive, nevertheless has several valuable pharmacological effects including anti-inflammatory and anti-oxidant activity which are well documented (Iffland and Grotenhermen, 2017). In the keratinocytes isolated from the skin of psoriatic patients or healthy volunteers, the cannabidiol treatment has effect on the redox balance and phospholipid metabolism in UV-A/UV-B-irradiated keratinocytes (Jarocka-Karpowicz et al., 2020). The CBD treatment resulted in accumulation of cannabidiol in UV-irradiated psoriasis cells and keratinocyte membranes, lessened the oxidative phospholipid changes and increased the level of the level of palmitoylethanolamide (PEA) that leads to reduced inflammation (Jarocka-Karpowicz et al., 2020). In AD cases, CBD reduces oxidative stress, lessens mitochondrial dysfunction, and diminishes generation of reactive oxygen species. The CBD treated PC12 cells showed inhibition of amyloid beta (Aβ)-induced tau protein hyperphosphorylation linked to the downregulation of an inhibitor of Wnt pathway called as p-GSK-3β (glycogen synthase kinase-3β) (Vallée et al., 2017). They further showed CBD suppression is mainly through initiation of peroxisome proliferator-activated receptor γ (PPARγ), and pro-inflammatory signaling. In regards to oxidative stress management in brain, the rat model of mania showed therapeutic protection by CBD treatment in the brain damage against d-amphetamine-induced oxidative stress (Valvassori et al., 2011). While in human neuroblastoma cells (SHSY5YAPP+), CBD demonstrated neuroprotective properties by promoting ubiquitination of the amyloid precursor protein (APP), which is an indicator of cellular changes in the brain of patients with Alzheimer’s disease (Scuderi et al., 2014). Interestingly, increased endogenous cannabinoids or endocannabinoids (eCB) signaling is associated with an anti-inflammatory, neuroprotective phenotype in microglia (Araujo et al., 2019). The components of the endocannabinoid system (ECS) and signaling controls microglial activity include eCB (small, lipid-derived molecules), enzymes that metabolize these ligands, to cannabinoid receptor 1 (CB1) and cannabinoid receptor 2 (CB2). Microglial activity in CNS development and homeostasis can be modulated by eCB signaling, thus understanding the ECS can provide advanced therapeutics for autism spectrum disorder (ASD) and its associated conditions (Araujo et al., 2019).
Panax ginseng (Genus: Panax; Family: Araliaceae) is a group of slowly growing plants with fleshy roots and Ginseng is the most valuable herbal medicine consumed globally (Tyler 1995). The unique triterpenoid saponins known as Ginsenosides are found exclusively in Panax species and have been isolated from roots, leaves, stems, fruits, and/or flower heads of ginseng. Ginsenoside Rd, a dammarane-type steroid glycoside, has showed encouraging neuroprotective efficacy and diminishes redox imbalance to improve stroke outcome in laboratory and clinical studies. Ginsenoside Rd has exhibited improved stroke outcome after focal cerebral ischemia in aged mice brain, attenuates redox imbalance and neuroprotection against ischemic cerebral damage both in vitro and in vivo. In 24 h post-ischemia, Ginsenoside Rd significantly repressed the build-ups of DNA, protein and lipid peroxidation products (Ye et al., 2011a). At 4 and 24 h after reperfusion, it showed to protect mitochondria as observed by preserved respiratory or electron chain complex activities and aconitase activity, hyperpolarized mitochondrial membrane potential and decreased hydrogen peroxide formation (Ye et al., 2011a). Ye et al., 2011b study showed in rats that the Ginsenoside Rd crossed the intact blood-brain barrier and employs neuroprotection in both transient and permanent middle cerebral artery occlusion (MCAO) (Ye et al., 2011b). Apart from this, Zhao et al., 2009 detected in hippocampus of 12 months old C57BL/6 mice, that many ginsenosides including Ra, Rb1, Rb2, Rc, Rd, Re, Rf, Rg1, Rg2, Rg3, and Ro were effective to prevent memory damage by upregulating plasticity-related proteins and modulation of synaptic plasticity. Some of the important ginsenosides-modulated CNS targets associated with potentiating of brain functions are Rg1 targets iNOS, NMDA receptor (of the hippocampus in ischemic gerbils), Synapses, L-type Ca2+ channels, P38 (modulation of COX-2 expression), DMT1 and FPI, IGF-1, GR CDK4, pRB, and E2F1; Rg3 targets voltage-dependent Ca2+ channels, glycine binding site of NMDA receptor, neprilysin gene, and MSRA; Rg2 targets α1β1δɛ or α3β4 subunits of NMDA receptor, V291, F292, and I295 residues of 5-HT (3A) receptor channels; Rb1 targets (cAMP)-dependent PKA, Choline acetyltransferase, NMDA receptor, α3β4 subunits of nicotinic receptors and P35; Rb2 targets transcription factor AP2 binding site; Rf targets unidentified G protein-coupled receptor; Rh2 targets Na+ channels, Polyamine binding site of NMDA receptor, PACAP and NF-κB and MAP kinase; Re targets Bcl2, Bax, iNOS. And caspase-3; Rd targets iNOS and COX-2, and GR; PNS targets ICAM-1 (Radad et al., 2011). The protopanaxadiol ginsenosides from Panax ginseng showed ginsenosides are positive modulators of P2X4 receptors. Further, in silico docking studies with a homology model of human P2X4 showed the possible ginsenoside binding site in the central vestibule region of the large ectodomain of P2X4 (Dhuna et al., 2019).
Flavonoids are a diverse group of plant metabolites or phytonutrients found in almost all fruits and vegetables. Flavonoids are thought to provide health benefits through their anti-oxidative, anti-inflammatory, anti-mutagenic and anti-carcinogenic properties. They are now considered a crucial constituent in a variety of nutraceutical, pharmaceutical, medicinal, and cosmetic products (Panche et al., 2016). The potential interventions of dietary flavonoids to improve redox imbalance in obesity and related co-morbidities (such as type 2 diabetes mellitus, CVD, gastrointestinal disorders and cognitive impairment) are well-researched and are also effective in reducing levels of oxidative stress and related inflammatory conditions (Gentile et al., 2018). The reduction of oxidative stress by flavonoids is coordinated through mechanisms regulated by the human body’s antioxidant system i.e., glyoxalase pathway, ability to scavenge ROS and regulate antioxidant defense mechanisms and cell signaling pathways (Frandsen and Narayanasamy, 2018). Fisetin, a plant flavonol, inhibited microglial cell migration, and ROS production in BV-2 microglial cells (Chuang et al., 2014). The regulatory molecular mechanism of fisetin-induced HO-1 expression works through the PI-3k/AKT and p38 signaling pathways in microglia cells. They also reported substantially reduced inflammation-related microglial activation and coordination deficit in in vivo mice model (Chuang et al., 2014). Several studies on flavonoids have shown that they exert their neuroprotection (Jang and Johnson, 2010) through impeding microglia activation and subsequent release of many inflammatory molecules such as Luteolin reduced IL-1β, TNFα, NO, PGE2 and attenuated neuronal cell death (Jang et al., 2010); Apigenin inhibited NO and PGE2 (Ha et al., 2008); Blueberry extract inhibited LPS-induced NO, TNFα and IL-1b in BV-2 experimental model (Lau et al., 2007). Isoflavone metabolites decreased LPS-induced NO and TNFα in primary microglia BV-2 (Park et al., 2007).
Sulforaphane (SFN) is a potent activator of Nuclear Factor Erythroid 2-related factor 2 (Nrf2) and a natural dietary isothiocyanate, present in the species of the Brassicaceae such as broccoli and cabbage vegetables. In the cell, the master regulator of cellular detoxification responses and redox balance is known as Nuclear factor E2 factor-related factor (Nrf 2) which kindles the cellular defense mechanism and detoxification process. In our cellular redox system, sulforaphane treatment modulates redox status by activating redox regulator Nrf 2 (Ruhee and Suzuki, 2020). Further, reports suggest sulforaphane alters the cellular Keap1/Nrf2/ARE pathway that is useful for improving health and diseased conditions. Pyridoxamine (PM) and Sulforaphane (SFN) monotherapy in the Goto-kakizaki (GK) rats (an animal model of non-obese type 2 diabetes), triggered significant improvement in endothelial dysfunction in aorta and mesenteric arteries associated with type 2 diabetes, reducing vascular oxidative damage, advanced glycation end products (AGE) and HbA1c (hemoglobin A1c) levels (Pereira et al., 2017). SFN has potent anti-inflammatory activity and may have neuroprotective properties in diverse neurodegenerative diseases related to inflammation (Qin et al., 2018). The underlying mechanisms of SFN against microglia-mediated neuronal damage remain elusive. Qin et al., 2018 have shown that SFN significantly blocked the phosphorylation of both MAPKs (p38, JNK, and ERK1/2) and NF-κB p65, and with mitogen-activated protein kinase (MAPK) inhibitors (SB203580, SP 600125, and U0126) or an NF-κB inhibitor (PDTC) in BV-2 microglia cells. Their results on microglia-mediated neuronal necroptosis demonstrated that the SFN reduces LPS-induced pro-inflammatory processes through down-regulation of MAPK/NF-κB signaling pathway in BV-2 cells (Qin et al., 2018). Another study (Subedi et al., 2019) showed SFN exerts an anti-inflammatory effect on microglia by decreasing c-Jun N-terminal kinase (JNK) phosphorylation levels, subsequently causing a reduction in NF-κB and AP-1 signaling, and suppress the expression of the inflammatory mediators (iNOS, COX-2, NO, and PGE2) and proinflammatory cytokines (TNF-α, IL-6, and IL-1β). SFN also increased the expression of Nrf2 and HO-1 and the manufacture of the anti-inflammatory cytokines IL-10 and IL-4 (Subedi et al., 2019). Thus, the use of various natural products having anti-inflammatory and immunomodulatory effects has opened new avenues in the treatment of various neurological disorders.
Among all the know microglial genes, Iba1 has been consistently in use as a pan marker of microglia. It is a calcium binding protein, found in the cytoplasm of microglia and its expression has been observed upregulated in activated microglia. It regulated switching of microglia phenotypes during disease conditions. Iba1 participates in membrane ruffling and phagocytic activity of activated microglia via Rac signaling, by interacting with small G protein Rac. In microglia, Rac is a key molecule involved in proliferation, migration, regulation of actin cytoskeleton, microglia activation, phagocytosis and bioactive agents production. In vitro study suggest M-CSF, IL-6 and IFN-γ cause morphological changes to activated microglia and enhance Iba1 expression (Imai and Kohsaka, 2002). Recent study suggest that siRNA based silencing of Iba1 leads to reduced proliferation, cell adhesion whereas increased phagocytic activity of BV2 microglia cells indicating a new microglia target to modulate microglia and its functions (Gheorghe et al., 2020). Natural products are widely used in reducing redox imbalance and inflammation in different organ system including brain. However, molecular targets of these natural products are still not know. Therefore, we aimed to target microglia specific Iba1 protein for molecular docking studies using natural products. The human Iba1 3D structure was retrieved from Protein Data Bank (PDB ID: 2D58) and was used to perform molecular docking with natural products such as Curcumin, Cannabidiol, Ginsenosides, Resveratrol and Sulforane using AutoDock 4.2 (Morris et al., 2009). Further, visualization of drug pockets on Iba1 protein, protein-ligand interaction and analysis of hydrogen bond formation was analyzed by ProteinsPlus server (Schöning-Stierand et al., 2020) and LigPlot2. The DoGSiteScorer analysis of ProteinsPlus server for predicting potential drug binding sites (pockets) showed presence of 5 drug pocket sites on Iba1 which can be used for drug binding. The protein-ligand interaction using Curcumin as ligand showed its binding to drug pocket 2 of Iba1 with volume of 214.4 Å3 and surface area of 426.42 Å2 and drug score of 0.46 (Figure 3A; Table 1). Similar observation was also obtained for Cannabidiol, Ginsenosides, Resveratrol and Sulforane binding on Iba1 protein drug pockets. Further, visualization of Iba1 interaction with Curcumin showed 3 hydrogen bond between Gln20, Leu59 and Arg72 residue of Iba1 with Curcumin (Figures 3B,C) has a distance of 2.60 and 3.10 and 2.99 Å, respectively (Figure 3D) and binding energy of −4.79 Kcal/Mol and inhibition constant of value 306.23 µM (Table 1). Similarly, Cannabidiol, Ginsenosides and Resveratrol also showed lower binding energy with Iba1 with values of −4.84, −6.83 and −6.29 Kcal/Mol respectively, whereas number of hydrogen bonds between ligand and protein observed least in Ginsenosides as compared to Curcumin, Cannabidiol and Resveratrol (Table 2). Lower the binding energy between protein-ligand interactions, higher the affinity of ligand for the protein where presence of hydrogen bond and other hydrophobic interactions helps in forming good binding affinity (Azam and Abbasi, 2013). Drug targeting of any protein requires interaction between them. Our analysis for binding energy and interactions among selected natural products and Iba1 indicates promising therapeutic approach by targeting microglia specific genes in altered redox imbalance and neuroinflammation of brain during pathological conditions. Widespread screening of natural products for targeting microglia specific genes could be one of the approach in combating redox imbalance and neuroinflammation in brain pathologies. However, further analysis of microglia specific proteins like Tmem119, Siglec H, CD40 etc. with natural products would help us in identifying more details on promising drug target for microglia.
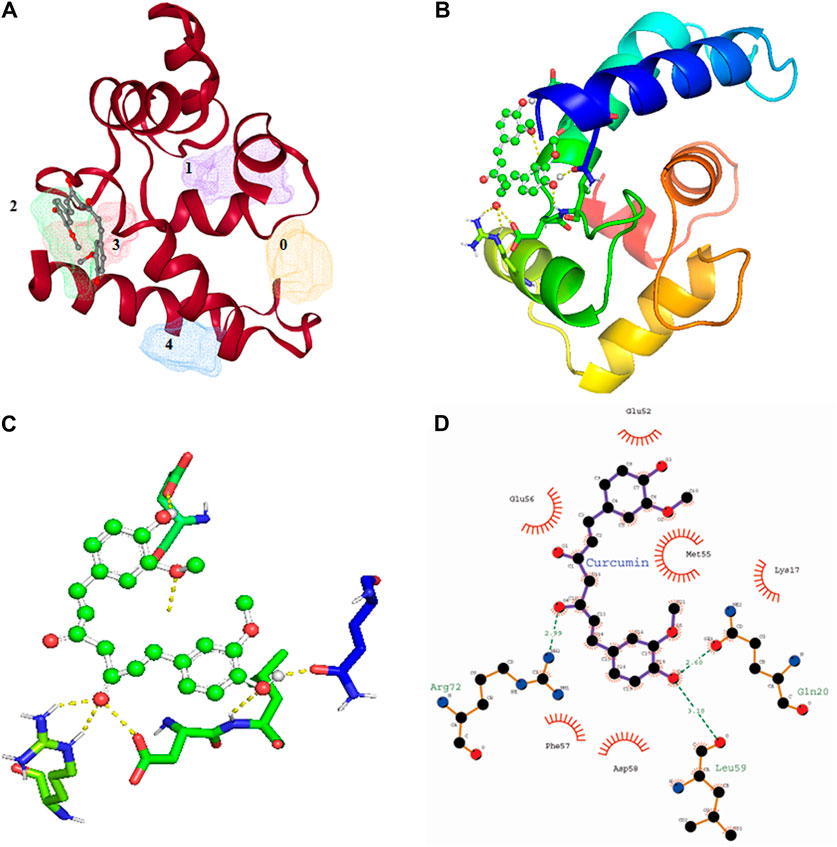
FIGURE 3. Molecular docking showing the interaction of Iba1 protein with Curcumin. (A) DoGSiteScorer analysis for predicting potential drug binding sites (pockets) on protein 3D structure. Out of 5 pockets for drug binding detected, Curcumin has been observed to fall in pocket 2 (green). (B, C) Pymol analysis of protein-ligand interaction after molecular docking using Autodock. The dotted yellow line indicates hydrogen bonding between Iba1 and Curcumin. (D) LigPlot Plus analysis of Autodock result generated 2D ligand-protein interaction diagram depicting hydrogen bonds between Iba1 Gln20, Leu59 and Arg72 amino acids with Curcumin. The hydrogen bond has been shown in green dotted line with a distance of protein and ligand hydrogen bond mentioned in Angstroms (Å).
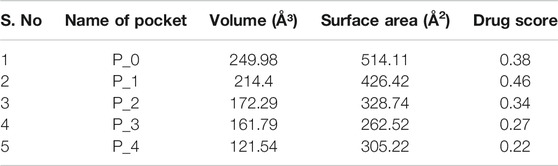
TABLE 1. Binding site prediction granularity: Properties and druggability for Iba1.
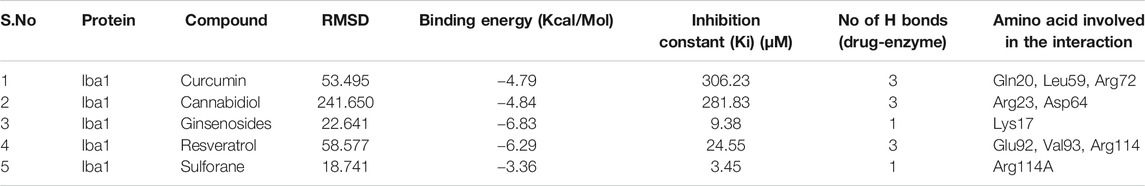
TABLE 2. Protein-ligand interaction analysis of Iba1 with natural compounds showed the lowest binding energy with Ginsenosides and Resveratrol.
Redox imbalance in microglia plays a critical role in neuroinflammation and neurodegeneration, where several promising druggable targets are being identified in recent studies. Molecular docking results from microglia specific proteins and diverse range of natural products like Curcumin, Cannabidiol, and Resveratrol support targeting microglia using natural products as a possible intervention to regulate redox imbalance and inflammation in neurological diseases. Precise drug designing and in vivo studies are warranted to further validate the drug molecules in mitigating the microglia induced neurodegeneration implicated in several neurodegenerative diseases.
SKM and RM designed the study. SKM, NB, SM, AB, PB, and SS did literature survey, wrote the part of manuscript. SKM performed in silico analysis, compiled the manuscript. RM reviewed and edited the manuscript. All authors read and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
RM acknowledges financial support from Department of Science & Technology-Science and Engineering Research Board, India (EMR/2016/006913). SKM, and AB, acknowledge Ramjas College, the University of Delhi for its continuous support and motivation. NB acknowledges Dyal Singh College, the University of Delhi for its continuous support and motivation. SM acknowledges the Department of Molecular Medicine and Biotechnology, SGPGI, Lucknow. PB acknowledges ICMR JRF (33/6/2019TF/Rare/BMS) and DST INSPIRE Fellowship (DST/INSPIRE Fellowship/2019/IF190501). SS acknowledges financial support from ICMR (33/6/2019TF/Rare/BMS) and DST-FIST support (SR/FST/LS-I/2017/49-C).
Acuña, L., Hamadat, S., Corbalán, N. S., González-Lizárraga, F., Dos-Santos-Pereira, M., Rocca, J., et al. (2019). Rifampicin and its derivative rifampicin quinone reduce microglial inflammatory responses and neurodegeneration induced in vitro by α-Synuclein fibrillary aggregates. Cells 8, 776. doi:10.3390/cells8080776
Akaishi, T., and Abe, K. (2018). CNB-001, a synthetic pyrazole derivative of curcumin, suppresses lipopolysaccharide-induced nitric oxide production through the inhibition of NF-κB and p38 MAPK pathways in microglia. Eur. J. Pharmacol. 819, 190–197. doi:10.1016/j.ejphar.2017.12.008
Andreasson, K. I., Bachstetter, A. D., Colonna, M., Ginhoux, F., Holmes, C., Lamb, B., et al. (2016). Targeting innate immunity for neurodegenerative disorders of the central nervous system. J. Neurochem. 138, 653–693. doi:10.1111/jnc.13667
Araujo, D. J., Tjoa, K., and Saijo, K. (2019). The endocannabinoid system as a window into microglial biology and its relationship to autism. Front Cell Neurosci. 13, 424. doi:10.3389/fncel.2019.00424
Arcuri, C., Mecca, C., Bianchi, R., Giambanco, I., and Donato, R. (2017). The pathophysiological role of microglia in dynamic surveillance, phagocytosis and structural remodeling of the developing CNS. Front. Mol. Neurosci. 10, 191. doi:10.3389/fnmol.2017.00191
Azam, S. S., and Abbasi, S. W. (2013). Molecular docking studies for the identification of novel melatoninergic inhibitors for acetylserotonin-o-methyltransferase using different docking routines. Theor. Biol. Med. Model. 10, 63. doi:10.1186/1742-4682-10-63
Bennett, M. L., Bennett, F. C., Liddelow, S. A., Ajami, B., Zamanian, J. L., Fernhoff, N. B., et al. (2016). New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. USA 113, E1738–E1746. doi:10.1073/pnas.1525528113
Biber, K., Bhattacharya, A., Campbell, B. M., Piro, J. R., Rohe, M., Staal, R., et al. (2019). Microglial drug targets in AD: opportunities and challenges in drug discovery and development. Front. Pharmacol. 10, 840. doi:10.3389/fphar.2019.00840
Bitzer-Quintero, O. K., and González-Burgos, I. (2012). Immune system in the brain: a modulatory role on dendritic spine morphophysiology? Neural plast. 2012, 348642. doi:10.1155/2012/348642
Bohnert, S., Seiffert, A., Trella, S., Bohnert, M., Distel, L., Ondruschka, B., et al. (2020). TMEM119 as a specific marker of microglia reaction in traumatic brain injury in postmortem examination. Int. J. Leg. Med. 134, 2167–2176. doi:10.1007/s00414-020-02384-z
Boisvert, M. M., Erikson, G. A., Shokhirev, M. N., and Allen, N. J. (2018). The aging astrocyte transcriptome from multiple regions of the mouse brain. Cel Rep. 22, 269–285. doi:10.1016/j.celrep.2017.12.039
Bonham Luke, W., Sirkis Daniel, W., and Yokoyama Jennifer, S. (2019). The transcriptional landscape of microglial genes in aging and neurodegenerative disease. Front. Immunol. 10, 1170. doi:10.3389/fimmu.2019.01170
Burda, J. E., and Sofroniew, M. V. (2014). Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 81, 229–248. doi:10.1016/j.neuron.2013.12.034
Burkitt, M. J., and Duncan, J. (2000). Effects of trans-resveratrol on copper-dependent hydroxyl-radical formation and DNA damage: evidence for hydroxyl-radical scavenging and a novel, glutathione-sparing mechanism of action. Arch. Biochem. Biophys. 381 (2), 253–263. doi:10.1006/abbi.2000.1973
Burré, J., Sharma, M., and Südhof, T. C. (2015). Definition of a molecular pathway mediating α-synuclein neurotoxicity. J. Soc. Neurosci. 35 (13), 5221–5232. doi:10.1523/JNEUROSCI.4650-14.2015
Butovsky, O., Jedrychowski, M. P., Moore, C. S., Cialic, R., Lanser, A. J., Gabriely, G., et al. (2014). Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat. Neurosci. 17, 131–143. doi:10.1038/nn.3599
Buttgereit, A., Lelios, I., Yu, X., Vrohlings, M., Krakoski, N. R., Gautier, E. L., et al. (2016). Sall1 is a transcriptional regulator defining microglia identity and function. Nat. Immunol. 17, 1397–1406. doi:10.1038/ni.3585
Cai, Z., and Xiao, M. (2016). Oligodendrocytes and Alzheimer's disease. Int. J. Neurosci. 126, 97–104. doi:10.3109/00207454.2015.1025778
Calvello, R., Cianciulli, A., Nicolardi, G., De Nuccio, F., Giannotti, L., Salvatore, R., et al. (2017). Vitamin D treatment attenuates neuroinflammation and dopaminergic neurodegeneration in an animal model of Parkinson’s disease, shifting M1 to M2 microglia responses. J. Neuroimmune Pharmacol. 12, 327–339. doi:10.1007/s11481-016-9720-7
Candelario-Jalil, E., de Oliveira, A. C. P., Gräf, S., Bhatia, H. S., Hüll, M., Muñoz, E., et al. (2007). Resveratrol potently reduces prostaglandin E2production and free radical formation in lipopolysaccharide-activated primary rat microglia. J. Neuroinflammation 4, 25. doi:10.1186/1742-2094-4-25
Chen, Z., and Trapp, B. D. (2016). Microglia and neuroprotection. J. Neurochem. 136, 10–17. doi:10.1111/jnc.13062
Chinta, S. J., and Andersen, J. K. (2008). Redox imbalance in Parkinson's disease. Biochim. Biophys. Acta 1780, 1362–1367. doi:10.1016/j.bbagen.2008.02.005
Chiu, I. M., Morimoto, E. T. A., Goodarzi, H., Liao, J. T., O'Keeffe, S., Phatnani, H. P., et al. (2013). A neurodegeneration-specific gene-expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Rep. 4, 385–401. doi:10.1016/j.celrep.2013.06.018
Choi, Y. R., Kang, S. J., Kim, J. M., Lee, S. J., Jou, I., Joe, E. H., et al. (2015). FcγRIIB mediates the inhibitory effect of aggregatedα-synuclein on microglial phagocytosis. Neurobiol. Dis. 83, 90–99. doi:10.1016/j.nbd.2015.08.025
Chuang, J. Y., Chang, P. C., Shen, Y. C., Lin, C., Tsai, C. F., Chen, J. H., et al. (2014). Regulatory effects of fisetin on microglial activation. Molecules 19, 8820–8839. doi:10.3390/molecules1907882
Cotto, B., Li, H., Tuma, R. F., Ward, S. J., and Langford, D. (2018). Cocaine-mediated activation of microglia and microglial MeCP2 and BDNF production. Neurobiol. Dis. 117, 28–41. doi:10.1016/j.nbd.2018.05.017
Croisier, E., Moran, L. B., Dexter, D. T., Pearce, R. K., and Graeber, M. B. (2005). Microglial inflammation in the parkinsonian substantia nigra: relationship to alpha-synuclein deposition. J. neuroinflammation 2, 14. doi:10.1186/1742-2094-2-14
Cunniffe, N., and Coles, A. (2021). Promoting remyelination in multiple sclerosis. J. Neurol. 268, 30–44. doi:10.1007/s00415-019-09421-x
Daniele, S. G., Béraud, D., Davenport, C., Cheng, K., Yin, H., and Maguire-Zeiss, K. A. (2015). Activation of MyD88-dependent TLR1/2 signaling by misfolded α-synuclein, a protein linked to neurodegenerative disorders. Sci. Signaling 8, ra45. doi:10.1126/scisignal.2005965
de Lau, L. M., and Breteler, M. M. (2006). Epidemiology of Parkinson's disease. Lancet Neurol. 5, 525–535. doi:10.1016/S1474-4422(06)70471-9
del Río-Hortega, P. (1932). “Microglia,” in Cytology and cellular pathology of the nervous system. Editors W Penfield, and P. B. Hoeber (New York, 2, 483–534.
Deng, X., and Sriram, S. (2005). Role of microglia in multiple sclerosis. Curr. Neurol. Neurosci. Rep. 5, 239–244. doi:10.1007/s11910-005-0052-x
Dhuna, K., Felgate, M., Bidula, S. M., Walpole, S., Bibic, L., Cromer, B. A., et al. (2019). Ginsenosides act as positive modulators of P2X4 receptors. Mol. Pharmacol. 95, 210–221. doi:10.1124/mol.118.113696
Donat, C. K., Scott, G., Gentleman, S. M., and Sastre, M. (2017). Microglial activation in traumatic brain injury. Front. Aging Neurosci. 9, 208. doi:10.3389/fnagi.2017.00208
Dzaye, O., Hu, F., Derkow, K., Haage, V., Euskirchen, P., Harms, C., et al. (2016). Glioma stem cells but not bulk glioma cells upregulate IL-6 secretion in microglia/brain macrophages via toll-like receptor 4 signaling. J. Neuropathol. Exp. Neurol. 75, 429–440. doi:10.1093/jnen/nlw016
ElAli, A., and Rivest, S. (2016). Microglia ontology and signaling. Front. Cel. Dev. Biol. 4, 72. doi:10.3389/fcell.2016.00072
Emmanouilidou, E., Melachroinou, K., Roumeliotis, T., Garbis, S. D., Ntzouni, M., Margaritis, L. H., et al. (2010). Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J. Soc. Neurosci. 30, 6838–6851. doi:10.1523/JNEUROSCI.5699-09.2010
Emmanouilidou, E., Minakaki, G., Keramioti, M. V., Xylaki, M., Balafas, E., Chrysanthou-Piterou, M., et al. (2016). GABA transmission via ATP-dependent K+ channels regulates α-synuclein secretion in mouse striatum. Brain a J. Neurol. 139, 871–890. doi:10.1093/brain/awv403
Eriksen, J. L., Wszolek, Z., and Petrucelli, L. (2005). Molecular pathogenesis of Parkinson disease. Arch. Neurol. 62, 353–357. doi:10.1001/archneur.62.3.353
Eun, C. S., Lim, J. S., Lee, J., Lee, S. P., and Yang, S. A. (2017). The protective effect of fermented Curcuma longa L. on memory dysfunction in oxidative stress-induced C6 gliomal cells, proinflammatory-activated BV2 microglial cells, and scopolamine-induced amnesia model in mice. BMC Complement. Altern. Med. 17, 367. doi:10.1186/s12906-017-1880-3
Franco-Bocanegra, D. K., George, B., Lau, L. C., Holmes, C., Nicoll, J. A. R., and Boche, D. (2019). Microglial motility in Alzheimer’s disease and after Aβ42 immunotherapy: a human post-mortem study. Acta Neuropathol. Commun. 7, 174. doi:10.1186/s40478-019-0828-x
Frandsen, J. R., and Narayanasamy, P. (2018). Neuroprotection through flavonoid: enhancement of the glyoxalase pathway. Redox Biol. 14, 465–473. doi:10.1016/j.redox.2017.10.015
French, H. M., Reid, M., Mamontov, P., Simmons, R. A., and Grinspan, J. B. (2009). Oxidative stress disrupts oligodendrocyte maturation. J. Neurosci. Res. 87, 3076–3087. doi:10.1002/jnr.22139
Galatro, T. F., Holtman, I. R., Lerario, A. M., Vainchtein, I. D., Brouwer, N., Sola, P. R., et al. (2017). Transcriptomic analysis of purified human cortical microglia reveals age-associated changes. Nat. Neurosci. 20, 1162–1171. doi:10.1038/nn.4597
Gentile, D., Fornai, M., Pellegrini, C., Colucci, R., Blandizzi, C., and Antonioli, L. (2018). Dietary flavonoids as a potential intervention to improve redox balance in obesity and related co-morbidities: a review. Nutr. Res. Rev. 31, 239–247. doi:10.1017/S0954422418000082
Ghasemi, F., Bagheri, H., Barreto, G. E., Read, M. I., and Sahebkar, A. (2019). Effects of curcumin on microglial cells. Neurotox. Res. 36, 12–26. doi:10.1007/s12640-019-00030-0
Gheorghe, R. O., Deftu, A., Filippi, A., Grosu, A., Bica-Popi, M., Chiritoiu, M., et al. (2020). Silencing the cytoskeleton protein Iba1 (ionized calcium binding adapter protein 1) interferes with BV2 microglia functioning. Cell Mol. Neurobiol. 40, 1011–1027. doi:10.1007/s10571-020-00790-w
Ghosh, M., Xu, Y., and Pearse, D. D. (2016). Cyclic AMP is a key regulator of M1 to M2a phenotypic conversion of microglia in the presence of Th2 cytokines. J. Neuroinflammation 13, 9. doi:10.1186/s12974-015-0463-9
Ginhoux, F., and Prinz, M. (2015). Origin of microglia: current concepts and past controversies. Cold Spring Harbor Perspect. Biol. 7, a020537. doi:10.1101/cshperspect.a020537
Goel, A., Kunnumakkara, A. B., and Aggarwal, B. B. (2008). Curcumin as “Curecumin”: from kitchen to clinic. Biochem. Pharmacol. 75, 787–809. doi:10.1016/j.bcp.2007.08.016
Gong, L., Wang, H., Sun, X., Liu, C., Duan, C., Cai, R., et al. (2016). Toll-Interleukin 1 Receptor domain-containing adaptor protein positively regulates BV2 cell M1 polarization. Eur. J. Neurosci. 43, 1674–1682. doi:10.1111/ejn.13257
Ha, S. K., Lee, P., Park, J. A., Oh, H. R., Lee, S. Y., Park, J. H., et al. (2008). Apigenin inhibits the production of NO and PGE2 in microglia and inhibits neuronal cell death in a middle cerebral artery occlusion-induced focal ischemia mice model. Neurochem. Int. 52, 878–886. doi:10.1016/j.neuint.2007.10.005
Hansen, D. V., Hanson, J. E., and Sheng, M. (2018). Microglia in Alzheimer’s disease. J. Cell Biol. 217, 459–472. doi:10.1083/jcb.201709069
Hatcher, H., Planalp, R., Cho, J., Torti, F. M., and Torti, S. V. (2008). Curcumin: from ancient medicine to current clinical trials. Cell Mol. Life Sci. 65, 1631–1652. doi:10.1007/s00018-008-7452-4
Hawrylycz, M. J., Lein, E. S., Guillozet-Bongaarts, A. L., Shen, E. H., Ng, L., Miller, J. A., et al. (2012). An anatomically comprehensive atlas of the adult human brain transcriptome. Nature 489, 391–399. doi:10.1038/nature11405
He, L., He, T., Farrar, S., Ji, L., Liu, T., and Ma, X. (2017). Antioxidants maintain cellular redox homeostasis by elimination of reactive oxygen species. Cell Physiol. Biochem. 44, 532–553. doi:10.1159/000485089
Hemonnot, A. L., Hua, J., Ulmann, L., and Hirbec, H. (2019). Microglia in alzheimer disease: well-known targets and new opportunities. Front. Aging Neurosci. 11, 233. doi:10.3389/fnagi.2019.00233
Hernangómez, M., Carrillo-Salinas, F. J., Mecha, M., Correa, F., Mestre, L., Loría, F., et al. (2014). Brain innate immunity in the regulation of neuroinflammation: therapeutic strategies by modulating CD200-CD200R interaction involve the cannabinoid system. Curr. Pharm. Des. 20, 4707–4722. doi:10.2174/1381612820666140130202911
Hong, S., Beja-Glasser, V. F., Nfonoyim, B. M., Frouin, A., Li, S., Ramakrishnan, S., et al. (2016). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352 (6286), 712–716. doi:10.1126/science.aad8373
Hristovska, I., and Pascual, O. (2016). Deciphering resting microglial morphology and process motility from a synaptic prospect. Front. Integr. Neurosci. 9, 73. doi:10.3389/fnint.2015.00073
Huang, J., Huang, N., Xu, S., Luo, Y., Li, Y., Jin, H., et al. (2020). Signaling mechanisms underlying inhibition of neuroinflammation by resveratrol in neurodegenerative diseases. J. Nutr. Biochem. 88, 108552. doi:10.1016/j.jnutbio.2020.108552
Hughes, C. D., Choi, M. L., Ryten, M., Hopkins, L., Drews, A., Botía, J. A., et al. (2019). Picomolar concentrations of oligomeric alpha-synuclein sensitizes TLR4 to play an initiating role in Parkinson’s disease pathogenesis. Acta Neuropathol. 137, 103–120. doi:10.1007/s00401-018-1907-y
Iffland, K., and Grotenhermen, F. (2017). An update on safety and side effects of Cannabidiol: a review of clinical data and relevant animal studies. Cannabis Cannabinoid Res. 2, 139–154. doi:10.1089/can.2016.0034
Imai, Y., and Kohsaka, S. (2002). Intracellular signaling in M-CSF-induced microglia activation: role of Iba1. Glia 40, 164–174. doi:10.1002/glia.10149
Inyang, K. E., Szabo-Pardi, T., Wentworth, E., McDougal, T. A., Dussor, G., Burton, M. D., et al. (2019). The antidiabetic drug metformin prevents and reverses neuropathic pain and spinal cord microglial activation in male but not female mice. Pharmacol. Res. 139, 1–16. doi:10.1016/j.phrs.2018.10.027
Iqbal, K., Liu, F., Gong, C. X., and Grundke-Iqbal, I. (2010). Tau in Alzheimer disease and related tauopathies. Curr. Alzheimer Res. 7, 656–664. doi:10.2174/156720510793611592
Ito, D., Imai, Y., Ohsawa, K., Nakajima, K., Fukuuchi, Y., and Kohsaka, S. (1998). Microglia-specific localisation of a novel calcium binding protein, Iba1. Brain research. Mol. Brain Res. 57, 1–9. doi:10.1016/s0169-328x(98)00040-0
Ito, D., Tanaka, K., Suzuki, S., Dembo, T., and Fukuuchi, Y. (2001). Enhanced expression of Iba1, ionized calcium-binding adapter molecule 1, after transient focal cerebral ischemia in rat brain. Stroke 32 (5), 1208–1215. doi:10.1161/01.str.32.5.1208
Jang, M., Cai, L., Udeani, G. O., Slowing, K. V., Thomas, C. F., Beecher, C. W., et al. 1997. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science 275(5297), 218–220. doi:10.1126/science.275.5297.218
Jang, S., Dilger, R. N., and Johnson, R. W. (2010). Luteolin inhibits microglia and alters hippocampal-dependent spatial working memory in aged mice. J. Nutr. 140, 1892–1898. doi:10.3945/jn.110.123273
Jang, S., and Johnson, R. W. (2010). Can consuming flavonoids restore old microglia to their youthful state? Nutr. Rev. 68, 719–728. doi:10.1111/j.1753-4887.2010.00336.x
Jarocka-Karpowicz, I., Biernacki, M., Wroński, A., Gęgotek, A., and Skrzydlewska, E. (2020). Cannabidiol effects on phospholipid metabolism in keratinocytes from patients with psoriasis vulgaris. Biomolecules 10, 367. doi:10.3390/biom10030367
Jin, L. W., Lucente, J. D., Nguyen, H. M., Singh, V., Singh, L., Chavez, M., et al. (2019). Repurposing the KCa3.1 inhibitor senicapoc for Alzheimer's disease. Ann. Clin. translational Neurol. 6, 723–738. doi:10.1002/acn3.754
Jinno, S., and Kosaka, T. (2008). Reduction of Iba1-expressing microglial process density in the hippocampus following electroconvulsive shock. Exp. Neurol. 212, 440–447. doi:10.1016/j.expneurol.2008.04.028
Jin, X., Liu, M. Y., Zhang, D. F., Zhong, X., Du, K., Qian, P., et al. (2019). Natural products as a potential modulator of microglial polarization in neurodegenerative diseases. Pharmacol. Res. 145, 104253. doi:10.1016/j.phrs.2019.104253
Kam, T. I., Hinkle, J. T., Dawson, T. M., and Dawson, V. L. (2020). Microglia and astrocyte dysfunction in Parkinson's disease. Neurobiol. Dis. 144, 105028. doi:10.1016/j.nbd.2020.105028
Kanazawa, H., Ohsawa, K., Sasaki, Y., Kohsaka, S., and Imai, Y. (2002). Macrophage/microglia-specific protein Iba1 enhances membrane ruffling and Rac activation via phospholipase C-gamma -dependent pathway. J. Biol. Chem. 277, 20026–20032. doi:10.1074/jbc.M109218200
Kang, G., Kong, P. J., Yuh, Y. J., Lim, S. Y., Yim, S. V., Chun, W., et al. (2004). Curcumin suppresses lipopolysaccharide-induced cyclooxygenase-2 expression by inhibiting activator protein 1 and nuclear factor kappab bindings in BV2 microglial cells. J. Pharmacol. Sci. 94, 325–328. doi:10.1254/jphs.94.325
Keren-Shaul, H., Spinrad, A., Weiner, A., Matcovitch-Natan, O., Dvir-Szternfeld, R., Ulland, T. K., et al. (2017). A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169, 1276–1290. doi:10.1016/j.cell.2017.05.018
Kim, B. W., Koppula, S., Kumar, H., Park, J. Y., Kim, I. W., More, S. V., et al. (2015). α-Asarone attenuates microglia-mediated neuroinflammation by inhibiting NF kappa B activation and mitigates MPTP-induced behavioral deficits in a mouse model of Parkinson's disease. Neuropharmacology 97, 46–57. doi:10.1016/j.neuropharm.2015.04.037
Kim, C., Ho, D. H., Suk, J. E., You, S., Michael, S., Kang, J., et al. (2013). Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 4, 1562. doi:10.1038/ncomms2534
Kim, Y. S., and Joh, T. H. (2006). Microglia, major player in the brain inflammation: their roles in the pathogenesis of Parkinson's disease. Exp. Mol. Med. 38 (4), 333–347. doi:10.1038/emm.2006.40
Koeniger, T., and Kuerten, S. (2017). Splitting the “Unsplittable”: dissecting resident and infiltrating macrophages in experimental autoimmune encephalomyelitis. Int. J. Mol. Sci. 18, 2072. doi:10.3390/ijms18102072
Konishi, H., and Kiyama, H. (2018). Microglial TREM2/DAP12 signaling: a double-edged sword in neural diseases. Front. Cell. Neurosci. 12, 206. doi:10.3389/fncel.2018.00206
Konishi, H., Kobayashi, M., Kunisawa, T., Imai, K., Sayo, A., Malissen, B., et al. (2017). Siglec-H is a microglia-specific marker that discriminates microglia from CNS-associated macrophages and CNS-infiltrating monocytes. Glia 65, 1927–1943. doi:10.1002/glia.23204
Kopatz, J., Beutner, C., Welle, K., Bodea, L. G., Reinhardt, J., Claude, J., et al. (2013). Siglec-h on activated microglia for recognition and engulfment of glioma cells. Glia 61, 1122–1133. doi:10.1002/glia.22501
Korzhevskii, D. E., and Kirik, O. V. (2016). Brain microglia and microglial markers. Neurosci. Behav. Physi 46, 284–290. doi:10.1007/s11055-016-0231-z
Kouli, A., Torsney, K. M., and Kuan, W. L (2018). “Parkinson’s disease: etiology, neuropathology, and pathogenesis,” in Parkinson’s disease: pathogenesis and clinical aspects [internet]. Editors T. B. Stoker, and J. C. Greenland (Brisbane, AU: Codon Publications), 3–26. doi:10.15586/codonpublications.parkinsonsdisease.2018.ch1
Krasemann, S., Madore, C., Cialic, R., Baufeld, C., Calcagno, N., El Fatimy, R., et al. (2017). The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47, 566–581. doi:10.1016/j.immuni.2017.08.008
Lan, X., Han, X., Li, Q., Li, Q., Gao, Y., Cheng, T., et al. (2017). Pinocembrin protects hemorrhagic brain primarily by inhibiting toll-like receptor 4 and reducing M1 phenotype microglia. Brain Behav. Immun. 61, 326–339. doi:10.1016/j.bbi.2016.12.012
Lau, F. C., Bielinski, D. F., and Joseph, J. A. (2007). Inhibitory effects of blueberry extract on the production of inflammatory mediators in lipopolysaccharide-activated BV2 microglia. J. Neurosci. Res. 85, 1010–1017. doi:10.1002/jnr.21205
Li, Q., Cheng, Z., Zhou, L., Darmanis, S., Neff, N. F., Okamoto, J., et al. (2019). Developmental heterogeneity of microglia and brain myeloid cells revealed by deep single-cell RNA sequencing. Neuron 101, 207–223. doi:10.1016/j.neuron.2018.12.006
Li, Y., Du, X. F., Liu, C. S., Wen, Z. L., and Du, J. L. (2012). Reciprocal regulation between resting microglial dynamics and neuronal activity in vivo. Dev. Cell 23, 1189–1202. doi:10.1016/j.devcel.2012.10.027
Liddelow, S. A., Guttenplan, K. A., Clarke, L. E., Bennett, F. C., Bohlen, C. J., Schirmer, L., et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. doi:10.1038/nature21029
Liu, C. Y., Wang, X., Liu, C., and Zhang, H. L. (2019). Pharmacological targeting of microglial activation: new therapeutic approach. Front. Cell. Neurosci. 13, 514. doi:10.3389/fncel.2019.00514
Lively, S., and Schlichter, L. C. (2018). Microglia responses to pro-inflammatory stimuli (LPS, IFNγ+TNFα) and reprogramming by resolving cytokines (IL-4, IL-10). Front. Cell. Neurosci. 12, 215. doi:10.3389/fncel.2018.00215
London, A., Cohen, M., and Schwartz, M. (2013). Microglia and monocyte-derived macrophages: functionally distinct populations that act in concert in CNS plasticity and repair. Front. Cell. Neurosci. 7, 34. doi:10.3389/fncel.2013.00034
Martin, E., El-Behi, M., Fontaine, B., and Delarasse, C. (2017). Analysis of microglia and monocyte-derived macrophages from the central nervous system by flow cytometry. J. Vis. Exp. 124, 55781. doi:10.3791/55781
Matsye, P., Zheng, L., Si, Y., Kim, S., Luo, W., Crossman, D. K., et al. (2017). HuR promotes the molecular signature and phenotype of activated microglia: implications for amyotrophic lateral sclerosis and other neurodegenerative diseases. Glia 65, 945–963. doi:10.1002/glia.23137
McGeer, P. L., Itagaki, S., Boyes, B. E., and McGeer, E. G. (1988). Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38, 1285–1291. doi:10.1212/wnl.38.8.1285
Mecca, C., Giambanco, I., Donato, R., and Arcuri, C. (2018). Microglia and aging: the role of the TREM2-DAP12 and cx3cl1-cx3cr1 axes. Int. J. Mol. Sci. 19, 318. doi:10.3390/ijms19010318
Miao, J., Shi, R., Li, L., Chen, F., Zhou, Y., Tung, Y. C., et al. (2019). Pathological tau from Alzheimer’s brain induces site-specific hyperphosphorylation and SDS- and reducing agent-resistant aggregation of tau in vivo. Front. Aging Neurosci. 11, 34. doi:10.3389/fnagi.2019.00034
Mihara, T., Nakashima, M., Kuroiwa, A., Akitake, Y., Ono, K., Hosokawa, M., et al. (2008). Natural killer cells of Parkinson’s disease patients are set up for activation: a possible role for innate immunity in the pathogenesis of this disease. Parkinsonism Relat. Disord. 14, 46–51. doi:10.1016/j.parkreldis.2007.05.013
Mildner, A., Huang, H., Radke, J., Stenzel, W., and Priller, J. (2017). P2Y12 receptor is expressed on human microglia under physiological conditions throughout development and is sensitive to neuroinflammatory diseases. Glia 65, 375–387. doi:10.1002/glia.23097
Mishra, S., and Palanivelu, K. (2008). The effect of curcumin (turmeric) on Alzheimer’s disease: an overview. Ann. Indian Acad. Neurol. 11, 13–19. doi:10.4103/0972-2327.40220
Moneim, A. E. (2015). Oxidant/Antioxidant imbalance and the risk of Alzheimer’s disease. Curr. Alzheimer Res. 12, 335–349. doi:10.2174/1567205012666150325182702
Moore, D. J., West, A. B., Dawson, V. L., and Dawson, T. M. (2005). Molecular pathophysiology of Parkinson’s disease. Annu. Rev. Neurosci. 28, 57–87. doi:10.1146/annurev.neuro.28.061604.135718
Morris, G. M., Huey, R., Lindstrom, W., Sanner, M. F., Belew, R. K., Goodsell, D. S., et al. (2009). AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 30 (16), 2785–2791. doi:10.1002/jcc.21256
Mukherjee, S., Klaus, C., Pricop-Jeckstadt, M., Miller, J. A., and Struebing, F. L. (2019). A microglial signature directing human aging and neurodegeneration-related gene networks. Front. Neurosci. 13, 2. doi:10.3389/fnins.2019.00002
Murphy, M. P., and LeVine, H. (2010). Alzheimer’s disease and the amyloid-beta peptide. J. Alzheimers Dis. 19, 311–323. doi:10.3233/JAD-2010-1221
Nalls, M. A., Blauwendraat, C., Vallerga, C. L., Heilbron, K., Bandres-Ciga, S., Chang, D., et al. International Parkinson’s Disease Genomics Consortium (2019). Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurology 18, 1091–1102. doi:10.1016/S1474-4422(19)30320-5
Natale, G., Biagioni, F., Busceti, C. L., Gambardella, S., Limanaqi, F., and Fornai, F. (2019). TREM receptors connecting bowel inflammation to neurodegenerative disorders. Cells 8 (10), 1124. doi:10.3390/cells8101124
Nayak, D., Roth, T. L., and McGavern, D. B. (2014). Microglia development and function. Annu. Rev. Immunol. 32, 367–402. doi:10.1146/annurev-immunol-032713-120240
Neidert, N., von Ehr, A., Zöller, T., and Spittau, B. (2018). Microglia-specific expression of Olfml3 is directly regulated by transforming growth factorβ1-induced smad2 signaling. Front. Immunol. 9, 1728. doi:10.3389/fimmu.2018.01728
Nizami, S., Hall-Roberts, H., Warrier, S., Cowley, S. A., and Di Daniel, E. (2019). Microglial inflammation and phagocytosis in Alzheimer’s disease: potential therapeutic targets. Br. J. Pharmacol. 176, 3515–3532. doi:10.1111/bph.14618
Ohsawa, K., Imai, Y., Kanazawa, H., Sasaki, Y., and Kohsaka, S. (2000). Involvement of Iba1 in membrane ruffling and phagocytosis of macrophages/microglia. J. Cell Sci. 113, 3073–3084.
Ohsawa, K., Imai, Y., Sasaki, Y., and Kohsaka, S. (2004). Microglia/macrophage-specific protein Iba1 binds to fimbrin and enhances its actin-bundling activity. J. Neurochem. 88, 844–856. doi:10.1046/j.1471-4159.2003.02213.x
Omeragic, A., Kara-Yacoubian, N., Kelschenbach, J., Sahin, C., Cummins, C. L., Volsky, D. J., et al. (2019). Peroxisome Proliferator-Activated Receptor-gamma agonists exhibit anti-inflammatory and antiviral effects in an EcoHIV mouse model. Sci. Rep. 9, 9428. doi:10.1038/s41598-019-45878-6
Orellana, J. A., Sáez, P. J., Shoji, K. F., Schalper, K. A., Palacios-Prado, N., Velarde, V., et al. (2009). Modulation of brain hemichannels and gap junction channels by pro-inflammatory agents and their possible role in neurodegeneration. Antioxid. Redox Signal. 11, 369–399. doi:10.1089/ars.2008.2130
Painter, M. M., Atagi, Y., Liu, C. C., Rademakers, R., Xu, H., Fryer, J. D., et al. (2015). TREM2 in CNS homeostasis and neurodegenerative disease. Mol. Neurodegener. 10, 43. doi:10.1186/s13024-015-0040-9
Panche, A. N., Diwan, A. D., and Chandra, S. R. (2016). Flavonoids: an overview. J. Nutr. Sci. 5, e47. doi:10.1017/jns.2016.41
Panicker, N., Sarkar, S., Harischandra, D. S., Neal, M., Kam, T. I., Jin, H., et al. (2019). Fyn kinase regulates misfolded α-synuclein uptake and NLRP3 inflammasome activation in microglia. J. Exp. Med. 216, 1411–1430. doi:10.1084/jem.20182191
Paolicelli, R. C., Bolasco, G., Pagani, F., Maggi, L., Scianni, M., Panzanelli, P., et al. (2011). Synaptic pruning by microglia is necessary for normal brain development. Science 333, 1456–1458. doi:10.1126/science.1202529
Park, J. S., Woo, M. S., Kim, D. H., Hyun, J. W., Kim, W. K., Lee, J. C., et al. (2007). Anti-inflammatory mechanisms of isoflavone metabolites in lipopolysaccharide-stimulated microglial cells. J. Pharmacol. Exp. Ther. 320, 1237–1245. doi:10.1124/jpet.106.114322
Pereira, A., Fernandes, R., Crisóstomo, J., Seiça, R. M., and Sena, C. M. (2017). The Sulforaphane and pyridoxamine supplementation normalize endothelial dysfunction associated with type 2 diabetes. Sci. Rep. 7, 14357. doi:10.1038/s41598-017-14733-x
Perry, V. H., and Teeling, J. (2013). Microglia and macrophages of the central nervous system: the contribution of microglia priming and systemic inflammation to chronic neurodegeneration. Semin. Immunopathol. 35, 601–612. doi:10.1007/s00281-013-0382-8
Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E., Dehejia, A., Dutra, A., et al. (1997). Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 276, 2045–2047. doi:10.1126/science.276.5321.2045
Pont-Lezica, L., Béchade, C., Belarif-Cantaut, Y., Pascual, O., and Bessis, A. (2011). Physiological roles of microglia during development. J. Neurochem. 119, 901–908. doi:10.1111/j.1471-4159.2011.07504.x
Qin, S., Yang, C., Huang, W., Du, S., Mai, H., Xiao, J., et al. (2018). Sulforaphane attenuates microglia-mediated neuronal necroptosis through down-regulation of MAPK/NF-κB signaling pathways in LPS-activated BV-2 microglia. Pharmacol. Res. 133, 218–235. doi:10.1016/j.phrs.2018.01.014
Radad, K., Moldzio, R., and Rausch, W. D. (2011). Ginsenosides and their CNS targets. CNS Neurosci. Ther. 17, 761–768. doi:10.1111/j.1755-5949.2010.00208.x
Rajasundaram, S. (2018). Adenosine A2A receptor signaling in the immunopathogenesis of experimental autoimmune encephalomyelitis. Front. Immunol. 9, 402. doi:10.3389/fimmu.2018.00402
Rangaraju, S., Dammer, E. B., Raza, S. A., Rathakrishnan, P., Xiao, H., Gao, T., et al. (2018). Identification and therapeutic modulation of a pro-inflammatory subset of disease-associated-microglia in Alzheimer’s disease. Mol. Neurodegener. 13, 24. doi:10.1186/s13024-018-0254-8
Reemst, K., Noctor, S. C., Lucassen, P. J., and Hol, E. M. (2016). The indispensable roles of microglia and astrocytes during brain development. Front. Hum. Neurosci. 10, 566. doi:10.3389/fnhum.2016.00566
Rocha, S. M., Saraiva, T., Cristóvão, A. C., Ferreira, R., Santos, T., Esteves, M., et al. (2016). Histamine induces microglia activation and dopaminergic neuronal toxicity via H1 receptor activation. J. Neuroinflammation 13, 137. doi:10.1186/s12974-016-0600-0
Rosenthal, N., and Brown, S. (2007). The mouse ascending: perspectives for human-disease models. Nat. Cell Biol. 9, 993–999. doi:10.1038/ncb437
Ruhee, R. T., and Suzuki, K. (2020). The integrative role of sulforaphane in preventing inflammation, oxidative stress and fatigue: a review of a potential protective phytochemical. Antioxidants (Basel) 9, 521. doi:10.3390/antiox9060521
Sahebkar, A. (2014). Curcuminoids for the management of hypertriglyceridaemia. Nat. Rev. Cardiol. 11, 123. doi:10.1038/nrcardio.2013.140-c1
Sala Frigerio, C., Wolfs, L., Fattorelli, N., Thrupp, N., Voytyuk, I., Schmidt, I., et al. (2019). The major risk factors for Alzheimer’s disease: age, sex, and genes modulate the microglia response to Aβ plaques. Cell Rep. 27, 1293–1306. doi:10.1016/j.celrep.2019.03.099
Sarlus, H., and Heneka, M. T. (2017). Microglia in Alzheimer’s disease. J. Clin. Invest. 127, 3240–3249. doi:10.1172/JCI90606
Satoh, J., Kino, Y., Asahina, N., Takitani, M., Miyoshi, J., Ishida, T., et al. (2016). TMEM119 marks a subset of microglia in the human brain. Neuropathology 36, 39–49. doi:10.1111/neup.12235
Schlepckow, K., Monroe, K. M., Kleinberger, G., Cantuti-Castelvetri, L., Parhizkar, S., Xia, D., et al. (2020). Enhancing protective microglial activities with a dual function TREM2 antibody to the stalk region. EMBO Mol. Med. 12, e11227. doi:10.15252/emmm.201911227
Schöning-Stierand, K., Diedrich, K., Fährrolfes, R., Flachsenberg, F., Meyder, A., Nittinger, E., et al. (2020). ProteinsPlus: interactive analysis of protein-ligand binding interfaces. Nucleic Acids Res. 48, W48–W53. doi:10.1093/nar/gkaa235
Schwenkgrub, J., Joniec-Maciejak, I., Sznejder-Pachołek, A., Wawer, A., Ciesielska, A., Bankiewicz, K., et al. (2013). Effect of human interleukin-10 on the expression of nitric oxide synthases in the MPTP-based model of Parkinson’s disease. Pharmacol. Rep. 65, 44–49. doi:10.1016/s1734-1140(13)70962-9
Scuderi, C., Steardo, L., and Esposito, G. (2014). Cannabidiol promotes amyloid precursor protein ubiquitination and reduction of beta amyloid expression in SHSY5YAPP+ cells through PPARγ involvement. Phytother. Res. 28, 1007–1013. doi:10.1002/ptr.5095
Sekar, A., Bialas, A. R., de Rivera, H., Davis, A., Hammond, T. R., Kamitaki, N., et al. (2016). Schizophrenia risk from complex variation of complement component 4. Nature 530, 177–183. doi:10.1038/nature16549
Serrano, A., Apolloni, S., Rossi, S., Lattante, S., Sabatelli, M., Peric, M., et al. (2019). The S100A4 transcriptional inhibitor niclosamide reduces pro-inflammatory and migratory phenotypes of microglia: implications for amyotrophic lateral sclerosis. Cells 8, 1261. doi:10.3390/cells8101261
Šimić, G., Babić Leko, M., Wray, S., Harrington, C., Delalle, I., Jovanov-Milošević, N., et al. (2016). Tau protein hyperphosphorylation and aggregation in alzheimer's disease and other tauopathies, and possible neuroprotective strategies. Biomolecules 6, 6. doi:10.3390/biom6010006
Skaper, S. D., Facci, L., Zusso, M., and Giusti, P. (2018). An Inflammation-centric view of neurological disease: beyond the neuron. Front. Cell. Neurosci. 12, 72. doi:10.3389/fncel.2018.00072
Sorgdrager, F., Vermeiren, Y., Van Faassen, M., van der Ley, C., Nollen, E., Kema, I. P., et al. (2019). Age- and disease-specific changes of the kynurenine pathway in Parkinson's and Alzheimer's disease. J. Neurochem. 151, 656–668. doi:10.1111/jnc.14843
Sousa, C., Biber, K., and Michelucci, A. (2017). Cellular and molecular characterization of microglia: a unique immune cell population. Front. Immunol. 8, 198. doi:10.3389/fimmu.2017.00198
Squarzoni, P., Oller, G., Hoeffel, G., Pont-Lezica, L., Rostaing, P., Low, D., et al. (2014). Microglia modulate wiring of the embryonic forebrain. Cell Rep. 8, 1271–1279. doi:10.1016/j.celrep.2014.07.042
Stevens, B., Allen, N. J., Vazquez, L. E., Howell, G. R., Christopherson, K. S., Nouri, N., et al. (2007). The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178. doi:10.1016/j.cell.2007.10.036
Subedi, L., Lee, J. H., Yumnam, S., Ji, E., and Kim, S. Y. (2019). Anti-inflammatory effect of sulforaphane on LPS-activated microglia potentially through JNK/AP-1/NF-κB inhibition and Nrf2/HO-1 activation. Cells 8, 194. doi:10.3390/cells8020194
Subramaniam, S. R., and Federoff, H. J. (2017). Targeting microglial activation states as a therapeutic avenue in Parkinson’s disease. Front. Aging Neurosci. 9, 176. doi:10.3389/fnagi.2017.00176
Takeuchi, H., and Suzumura, A. (2014). Gap junctions and hemichannels composed of connexins: potential therapeutic targets for neurodegenerative diseases. Front. Cell. Neurosci. 8, 189. doi:10.3389/fncel.2014.00189
Tiwari, S., Atluri, V., Kaushik, A., Yndart, A., and Nair, M. (2019). Alzheimer's disease: pathogenesis, diagnostics, and therapeutics. Int. J. nanomedicine 14, 5541–5554. doi:10.2147/IJN.S200490
Trujillo, J., Yi, Chirino., Molina-Jijón, E., Andérica-Romero, A. C., Tapia, E., and Pedraza-Chaverrí, J. (2013). Renoprotective effect of the antioxidant curcumin: recent findings. Redox Biol. 1, 448–456. doi:10.1016/j.redox.2013.09.003
Tyler, V. E. (1995). Herbal remedies. J. Pharm. Technol. 11, 214–220. doi:10.1177/875512259501100510
Vainchtein, I. D., Chin, G., Cho, F. S., Kelley, K. W., Miller, J. G., Chien, E. C., et al. (2018). Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science 359, 1269–1273. doi:10.1126/science.aal3589
Vallée, A., Lecarpentier, Y., Guillevin, R., and Vallée, J. N. (2017). Effects of cannabidiol interactions with Wnt/β-catenin pathway and PPARγ on oxidative stress and neuroinflammation in Alzheimer’s disease. Acta Biochim. Biophys. Sin (Shanghai) 49, 853–866. doi:10.1093/abbs/gmx073
Valvassori, S. S., Elias, G., De Souza, B., Petronilho, F., Dal-Pizzol, F., Kapczinski, F., et al. (2011). Effects of cannabidiol on amphetamine-induced oxidative stress generation in an animal model of mania. J. Psychopharmacol. 25, 274–280. doi:10.1177/0269881109106925
Vickers, J. C., Mitew, S., Woodhouse, A., Fernandez-Martos, C. M., Kirkcaldie, M. T., Canty, A. J., et al. (2016). Defining the earliest pathological changes of Alzheimer's disease. Curr. Alzheimer Res. 13, 281–287. doi:10.2174/1567205013666151218150322
Vogels, T., Murgoci, A. N., and Hromádka, T. (2019). Intersection of pathological tau and microglia at the synapse. Acta Neuropathol. Commun. 7, 109. doi:10.1186/s40478-019-0754-y
Wang, J., Wang, J., Wang, J., Yang, B., Weng, Q., and He, Q. (2019). Targeting microglia and macrophages: a potential treatment strategy for multiple sclerosis. Front. Pharmacol. 10, 286. doi:10.3389/fphar.2019.00286
Wang, S., Chu, C. H., Stewart, T., Ginghina, C., Wang, Y., Nie, H., et al. (2015a). α-Synuclein, a chemoattractant, directs microglial migration via H2O2-dependent Lyn phosphorylation. Proc. Nat. Acad. Sci. USA 112, E1926–E1935. doi:10.1073/pnas.1417883112
Wang, S., Jing, H., Yang, H., Liu, Z., Guo, H., Chai, L., et al. (2015b). Tanshinone I selectively suppresses pro-inflammatory genes expression in activated microglia and prevents nigrostriatal dopaminergic neurodegeneration in a mouse model of Parkinson’s disease. J. ethnopharmacology 164, 247–255. doi:10.1016/j.jep.2015.01.042
Weinhard, L., di Bartolomei, G., Bolasco, G., Machado, P., Schieber, N. L., Neniskyte, U., et al. (2018). Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat. Commun. 9, 1228. doi:10.1038/s41467-018-03566-5
Wohleb, E. S. (2016). Neuron-microglia interactions in mental health disorders: "for better, and for worse. Front. Immunol. 7, 544. doi:10.3389/fimmu.2016.00544
Wojsiat, J., Zoltowska, K. M., Laskowska-Kaszub, K., and Wojda, U. (2018). Oxidant/antioxidant imbalance in Alzheimer’s disease: therapeutic and diagnostic prospects. Oxidative Med. Cell. longevity, 6435861. doi:10.1155/2018/6435861
Xie, Z., Zeng, X., Li, X., Wu, B., Shen, G., Wu, Q., et al. (2017). Curcumin attenuates oxidative stress in liver in Type 1 diabetic rats. Open Life Sci. 12, 452–459. doi:10.1515/biol-2017-0053
Yamada, K., and Iwatsubo, T. (2018). Extracellular α-synuclein levels are regulated by neuronal activity. Mol. Neurodegener. 13, 9. doi:10.1186/s13024-018-0241-0
Yan, Z., Gibson, S. A., Buckley, J. A., Qin, H., and Benveniste, E. N. (2018). Role of the JAK/STAT signaling pathway in regulation of innate immunity in neuroinflammatory diseases. Clin. Immunol. (Orlando, Fla.) 189, 4–13. doi:10.1016/j.clim.2016.09.014
Yang, X., Xu, S., Qian, Y., and Xiao, Q. (2017). Resveratrol regulates microglia M1/M2 polarization via PGC-1α in conditions of neuroinflammatory injury. Brain Behav. Immun. 64, 10. doi:10.1016/j.bbi.2017.03.003
Ye, R., Kong, X., Yang, Q., Zhang, Y., Han, J., Li, P., et al. (2011b). Ginsenoside rd in experimental stroke: superior neuroprotective efficacy with a wide therapeutic window. Neurotherapeutics 8, 515–525. doi:10.1007/s13311-011-0051-3
Ye, R., Kong, X., Yang, Q., Zhang, Y., Han, J., and Zhao, G. (2011a). Ginsenoside Rd attenuates redox imbalance and improves stroke outcome after focal cerebral ischemia in aged mice. Neuropharmacology 61, 815–824. doi:10.1016/j.neuropharm.2011.05.029
York, E. M., Bernier, L. P., and MacVicar, B. A. (2018). Microglial modulation of neuronal activity in the healthy brain. Dev. Neurobiol. 78, 593–603. doi:10.1002/dneu.22571
Yun, S. P., Kam, T. I., Panicker, N., Kim, S., Oh, Y., Park, J. S., et al. (2018). Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat. Med. 24, 931–938. doi:10.1038/s41591-018-0051-5
Zhao, H., Li, Q., Pei, X., Zhang, Z., Yang, R., Wang, J., et al. (2009). Long-term ginsenoside administration prevents memory impairment in aged C57BL/6J mice by up-regulating the synaptic plasticity‐related proteins in hippocampus. Behav. Brain Res. 201, 311–317. doi:10.1016/j.bbr.2009.03.002
Zheng, H., Jia, L., Liu, C. C., Rong, Z., Zhong, L., Yang, L., et al. (2017). TREM2 promotes microglial survival by activating Wnt/β-catenin pathway. J. Neurosci. 37, 1772–1784. doi:10.1523/JNEUROSCI.2459-16.2017
Zotova, E., Bharambe, V., Cheaveau, M., Morgan, W., Holmes, C., Harris, S., et al. (2013). Inflammatory components in human Alzheimer’s disease and after active amyloid-β42 immunization. Brain a J. Neurol. 136, 2677–2696. doi:10.1093/brain/awt210
Keywords: Brain, Neuroinflamamation, Natural product, Microglia, Neurodegeneration
Citation: Maurya SK, Bhattacharya N, Mishra S, Bhattacharya A, Banerjee P, Senapati S and Mishra R (2021) Microglia Specific Drug Targeting Using Natural Products for the Regulation of Redox Imbalance in Neurodegeneration. Front. Pharmacol. 12:654489. doi: 10.3389/fphar.2021.654489
Received: 16 January 2021; Accepted: 08 March 2021;
Published: 13 April 2021.
Edited by:
Flávio Reis, University of Coimbra, PortugalReviewed by:
Hideyuki Takeuchi, Yokohama City University, JapanCopyright © 2021 Maurya, Bhattacharya, Mishra, Bhattacharya, Banerjee, Senapati and Mishra. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rajnikant Mishra, cm1pc2hyYWFAYmh1LmFjLmlu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.