 Livio Luongo
Livio Luongo Francesca Guida
Francesca Guida Sabatino Maione
Sabatino Maione Kenneth A. Jacobson
Kenneth A. Jacobson Daniela Salvemini4
Daniela Salvemini4
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
OPINION article
Front. Pharmacol. , 16 April 2021
Sec. Neuropharmacology
Volume 12 - 2021 | https://doi.org/10.3389/fphar.2021.651038
This article is part of the Research Topic Purinergic Signaling 2020: the State-of-The-Art Commented by the Members of the Italian Purine Club View all 41 articles
The pharmacological treatment of chronic pain is still unsatisfactory. The cellular and molecular mechanisms at the basis of pain chronification are still poorly understood.
The commercially available drugs for treating chronic pain, including neuropathic pain, are effective in few patients and own several side effects that often limit the compliance of the patients. In addition, the opioids, which are the most potent analgesics, often fail in chronic pain conditions, especially in neuropathic pain syndromes. Moreover, it is well known that opiates can lead to addiction, tolerance and hyperalgesia. Therefore, new targets for treating neuropathic pain are needed. Purines, including ATP, ADP and adenosine and their receptors are deeply involved in the pathophysiology of neuropathic pain, particularly in the immune-mediated reactions that are responsible for the induction of the tactile allodynia, that represents the main symptom of neuropathic pain. The first response to the insult is mediated by the production of ATP and the activation of P2X receptors (Jacobson et al., 2020). In particular, the P2X4 receptors on microglia have been identified to be essential for the involvement of microglia cells in the pain pathophysiology (Trang et al., 2012). Beside the P2X, the P2Y receptors tend to boost the immune cells in response to nucleotides, with their ligands acting as immediate danger signals (Cekic and Linden, 2016). The subsequent stimulation of the metabotropic P1 adenosine receptors (ARs), of which are known four subtypes (A1, A2A, A2B, and A3), is overall associated with the reduction of both immunoinflammatory response and pain (Vincenzi et al., 2020).
In the present opinion paper we will focus on the role of the P1 adenosine receptors in the pathophysiology of chronic neuropathic pain. The involvement of P2X and P2Y receptors in chronic pain has been discussed elsewhere (Jacobson et al., 2020).
The role of the A1AR in pain and nociception has been well described in both preclinical and clinical studies. Preclinical evidence showed a potent beneficial effect of several A1AR agonists in different animal models of chronic pain (Sowa et al., 2010; Luongo et al., 2012; Kan et al., 2018). It has been suggested that A1AR stimulation in peripheral nerves, might represents the molecular mechanism through which acupuncture exerts an antinociceptive effect (Goldman et al., 2010). A1AR knock out (KO) mice were used to evaluate the role of the A1AR in nociception. Under normal conditions, as well as during inflammatory or neuropathic pain, A1AR KO animals showed a lower thermal threshold as compared to the wild-type (WT) mice. A1AR KO mice also showed a reduced antinociceptive response to morphine given intrathecally, but not systemically (Wu et al., 2005). This is in agreement with other data showing that intrathecal morphine generates an antiallodynic effect through A1AR activation (Zhang et al., 2005). A1AR is widely expressed in the nervous system. A1AR activation induces presynaptic inhibition of primary afferent fibers at the dorsal horn level. This inhibition is associated with decreased release of glutamate, substance P, and other proinflammatory mediators from primary afferents fibers to the spinal cord. A1AR also hyperpolarizes dorsal horn neurons by increasing K+ conductance and reducing Ca2+ influx (Bai et al., 2017).
Moreover, recent evidence also highlights the expression of the A1AR in primary microglia cell cultures (Luongo et al., 2014), assuming its potential role also in neuroinflammatory processes at the basis of the induction of tactile allodynia. Clinical studies also have been carried out for A1AR agonists. However, several side effects, especially at the cardiovascular level were associated to the use of those compounds (Zylka, 2011).
The role of the A2AAR in pain is still a matter of debate since both pronociceptive and antinociceptive effects have been reported in animal models of inflammatory and neuropathic pain. Several reports highlighted the long lasting antiallodynic effect of the spinally-injected A2AAR agonists (Loram et al., 2009; Kwilasz et al., 2018). CGS21680, a selective A2AAR agonist, reduced the formalin-induced nocifensive behavior in both the early (0–15°minutes) and late (15–60°minutes) phases in a mouse model of formalin-induced inflammatory pain (Nalepa et al., 2010).
On the other hand, several papers showed a facilitative role of the A2AAR on nociceptive threshold. In particular, it has been suggested that mice lacking the A2AAR are less responsive to the noxious stimuli and, in these mice, the spinal cord neurons seems to be less active (Hussey et al., 2007; Hussey et al., 2010). Moreover, A2AAR KO mice showed reduced tactile allodynia as compared to the wild type animals in a mouse model of neuropathic pain due to the sciatic nerve injury (Bura et al., 2008).
The role of the A3AR emerged only recently in the pain field. The analgesic effect of adenosine was, in fact, believed to be mediated mainly by A1AR stimulation and, at least in part, by A2AAR (Sawynok, 2016; Kwilasz et al., 2018). In addition, A1AR and A2AAR modulating drugs, although effective in the preclinical models, did not reach clinical experimentation for their important cardiovascular side effect (Zylka, 2011; Jacobson et al., 2020). Contrary to what emerged from the early studies showing A3AR levels in the brain that were difficult to detect (Rivkees et al., 2000), the A3AR is expressed in different areas of the central nervous system (CNS) of both rodents and humans (Yaar et al., 2002; Jacobson et al., 2018). Besides the A3AR expression in neurons, which is lower as compared to the A1AR and A2AAR in physiological conditions, there is a high expression of this receptor in immune cells in the periphery and CNS. Indeed, it has been demonstrated that A3AR is expressed by astrocytes, oligodendrocytes, microglia, and endothelial cells, including in human tissue (Zhang et al., 2016). This is important since recent evidence highlighted the key role of the microglia and astrocytes in the induction and maintenance of tactile allodynia, which represents a major symptom associated with neuropathic pain.
After early confusing reports about the involvement of the A3AR in chronic pain, recent evidence supports a pivotal role of this receptor in the reduction of tactile allodynia in different preclinical models of neuropathic pain. The recent synthesis of highly selective agonists for A3AR (>10,000-fold in comparison to other AR subtypes) and the possibility to use A3AR KO animals paved the way for more definitively investigating these receptors in the pain axis. In fact, the selective pharmacological stimulation of the A3AR induced pronounced and prolonged antiallodynic effects in traumatic nerve-injury, chemotherapy-induced and other models of neuropathic pain (Bar-Yehuda et al., 2011; Chen et al., 2012; Fishman et al., 2012; Janes et al., 2015; Little et al., 2015; Tosh et al., 2015; Terayama et al., 2018; Wahlman et al., 2018; Stockstill et al., 2020). Moreover, A3AR-selective agonists reduced the formalin-induced nocifensive behaviour and diabetic neuropathy (Yan et al., 2016; Petrelli et al., 2017). The A3AR seems to exert its beneficial effect at different levels of the pain axis. Indeed, the receptors can be activated at the spinal cord (SC) and rostral ventromedial medulla (RVM) levels, where the mRNA has been found (Little et al., 2015). Moreover, the selective activation of the A3AR can recruit several downstream pathways (Cohen and Fishman, 2019; Kim et al., 2019). These pleiotropic mechanisms might be the reason for the high efficacy of the A3AR agonists. Among several downstream mechanisms, the A3AR stimulation has been shown capable of reducing the expression of proinflammatory cytokines including tumor necrosis factor (TNFα) and interleukin-1β (IL-1β), and enhancing the expression of antiinflammatory cytokines, such as interleukin-10 (IL-10) and interleukin-4 (IL-4) in the SC (Janes et al., 2015; Wahlman et al., 2018). Intriguingly, it has also been observed that the A3AR signaling is associated with a spinal mechanism of action that modulates the chloride potassium symporter 5 (KCC2 transporter), which has been shown to be downregulated and, in turn, responsible for the GABAergic gradient shift from inhibitory to excitatory signaling in neuropathic pain (Ford et al., 2015). This latter mechanisms is very important for the establishment of tactile allodynia, which also involves microglia cells (Coull et al., 2005). Coppi and coworkers also highlighted the capability of A3AR agonists to inhibit the pronociceptive N-type Ca2+ currents and cell excitability in dorsal root ganglion (DRG) neurons (Coppi et al., 2019). This latter mechanism has also been suggested by Lucarini and colleagues in a model of visceral pain in rats (Lucarini et al., 2020).
Very recently, it has been demonstrated that the morphine-induced tolerance could also be mediated by the A3AR, suggesting a possible use of the A3AR agonists as adjuvant therapy in combination with opioids (Doyle et al., 2020). Coadministration of an A3AR agonist at a low dose also reduced withdrawal behavior following morphine administration in rats, without reducing morphine’s antinociceptive effect. A scheme summarizing the molecules acting on the A3AR in different preclinical models of neuropathic pain is shown in Figure 1.
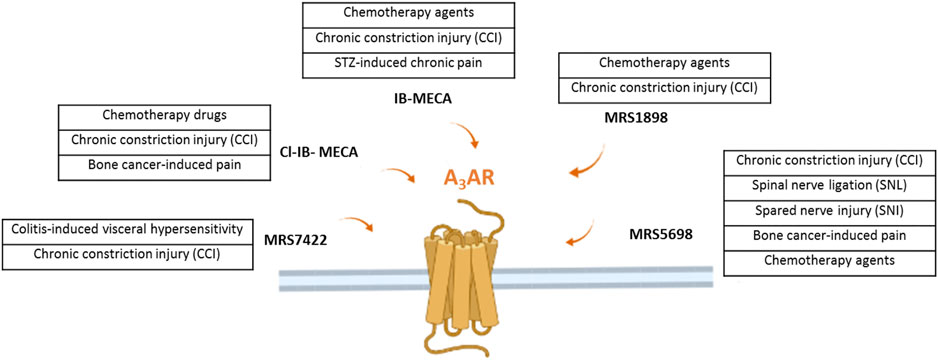
FIGURE 1. The figure illustrates the several A3AR agonists use in different preclinical model of neuropathic pain of various origins.
Purinergic signaling is involved in pain transmission. Data from different laboratories suggested that adenosine metabotropic receptors play key role in chronic pain models. In the previous years, the focus on adenosine in pain was mostly directed to the A1 and A2A receptors and their pharmacological manipulation. Cardiovascular side effects are the most limiting for the use of these compounds clinically.
More recent research has revealed the central role of the A3AR and its pharmacological manipulation for chronic pain of various origins. The efficacy of the A3AR agonists could be due to their pleiotropic mechanism of action, without exerting pronounced cardiovascular side effects. Interestingly, the antinociceptive effect of the A3AR pharmacological stimulation is independent from the recruitment of the opioid or cannabinoid systems, it does not alter physiological pain, thus, avoiding the problems related to the tolerance and abuse (Janes et al., 2016). Worthy of note is also the capability of A3AR to synergize with other drugs commonly used for treating chronic pain including opioids, amitriptyline and gabapentin (Chen et al., 2012).
To conclude, it is clear that the purines deserve further research attention in the pain field offering very promising results indicative of future potential. In particular, A3AR seems to represent a promising candidate for developing safer and more effective drug treatment for neuropathic pain.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
DS is founder of BioIntervene, Inc., a company developing A3AR agonists for clinical use.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Bai, H. H., Liu, J. P., Yang, L., Zhao, J. Y., Suo, Z. W., Yang, X., and Hu, X. D. (2017). Adenosine A1 receptor potentiated glycinergic transmission in spinal cord dorsal horn of rats after peripheral inflammation. Neuropharmacology 126, 158–167. doi:10.1016/j.neuropharm.2017.09.001
Bar-Yehuda, S, Luger, D, Ochaion, A, Cohen, S, Patokaa, R, Zozulya, G, et al. (2011). Inhibition of experimental auto-immune uveitis by the A3 adenosine receptor agonist CF101. Int. J. Mol. Med. 28 (5), 727–31. doi:10.3892/ijmm.2011.753
Bura, A. S., Nadal, X., Ledent, C., Maldonado, R., and Valverde, O. (2008). A2A adenosine receptor regulates glia proliferation and pain after peripheral nerve injury. Pain 140 (1), 95–103. doi:10.1016/j.pain.2008.07.012
Cekic, C, and Linden, J (2016). Purinergic regulation of the immune system. Nat. Rev. Immunol. 16, 177–192. doi:10.1038/nri.2016.4
Chen, Z., Janes, K., Chen, C., Doyle, T., Bryant, L., Tosh, D. K., et al. (2012). Controlling murine and rat chronic pain through A 3 adenosine receptor activation. FASEB J. 26 (5), 1855–1865. doi:10.1096/fj.11-201541
Cohen, S., and Fishman, P. (2019). Targeting the A 3 adenosine receptor to treat cytokine release syndrome in cancer immunotherapy. Drug Des., Dev. Ther. 13, 491–497. doi:10.2147/DDDT.S195294
Coppi, E., Cherchi, F., Fusco, I., Failli, P., Vona, A., Dettori, I., et al. (2019). Adenosine A3 receptor activation inhibits pronociceptive N-type Ca2+ currents and cell excitability in dorsal root ganglion neurons. Pain 160 (5), 1103–1118. doi:10.1097/j.pain.0000000000001488
Coull, J. A. M., Beggs, S., Boudreau, D., Boivin, D., Tsuda, M., Inoue, K., et al. (2005). BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 438 (7070), 1017–1021. doi:10.1038/nature04223
Doyle, T. M., Largent-Milnes, T. M., Chen, Z., Staikopoulos, V., Esposito, E., Dalgarno, R., et al. (2020). Chronic Morphine-Induced Changes in Signaling at the A3 Adenosine Receptor Contribute to Morphine-Induced Hyperalgesia, Tolerance, and Withdrawal. J. Pharmacol. Exp. Ther. 374 (2), 331–341. doi:10.1124/jpet.120.000004
Fishman, P, Bar-Yehuda, S, Liang, BT, and Jacobson, KA (2012). Pharmacological and therapeutic effects of A3 adenosine receptor agonists. Drug Discov. Today 17, 359–366. doi:10.1016/j.drudis.2011.10.007
Ford, A., Castonguay, A., Cottet, M., Little, J. W., Chen, Z., Symons-Liguori, A. M., et al. (2015). Engagement of the GABA to KCC2 signaling pathway contributes to the analgesic effects of A3AR agonists in neuropathic pain. J. Neurosci. 35 (15), 6057–6067. doi:10.1523/jneurosci.4495-14.2015
Goldman, N., Chen, M., Fujita, T., Xu, Q., Peng, W., Liu, W., et al. (2010). Adenosine A1 receptors mediate local anti-nociceptive effects of acupuncture. Nat. Neurosci. 13 (7), 883–888. doi:10.1038/nn.2562
Hussey, M. J., Clarke, G. D., Ledent, C., Hourani, S. M. O., and Kitchen, I. (2007). Reduced response to the formalin test and lowered spinal NMDA glutamate receptor binding in adenosine A2A receptor knockout mice. Pain 129 (3), 287–294. doi:10.1016/j.pain.2006.10.014
Hussey, M. J., Clarke, G. D., Ledent, C., Kitchen, I., and Hourani, S. M. O. (2010). Genetic deletion of the adenosine A2A receptor in mice reduces the changes in spinal cord NMDA receptor binding and glucose uptake caused by a nociceptive stimulus. Neurosci. Lett. 479 (3), 297–301. doi:10.1016/j.neulet.2010.05.084
In, A. J., Ellis, A., Wieseler, J., Loram, L., Favret, J., McFadden, A., Springer, K., Falci, S., Rieger, J., Maier, S. F., and Watkins, L. R. (2018). Sustained reversal of central neuropathic pain induced by a single intrathecal injection of adenosine A 2A receptor agonists. Brain, Behav., Immun. 69, 470–479. doi:10.1016/j.bbi.2018.01.005
Jacobson, K. A., Merighi, S., Varani, K., Borea, P. A., Baraldi, S., Aghazadeh Tabrizi, M., et al. (2018). A3 Adenosine Receptors as Modulators of Inflammation: From Medicinal Chemistry to Therapy. Med. Res. Rev. 38, 1031–1072. doi:10.1002/med.21456
Jacobson, K. A., Giancotti, L. A., Lauro, F., Mufti, F., and Salvemini, D. (2020). Treatment of chronic neuropathic pain: purine receptor modulation. Pain 161 (7), 1425–1441. doi:10.1097/j.pain.0000000000001857
Janes, K., Wahlman, C., Little, J. W., Doyle, T., Tosh, D. K., Jacobson, K. A., et al. (2015). Spinal neuroimmune activation is independent of T-cell infiltration and attenuated by A3 adenosine receptor agonists in a model of oxaliplatin-induced peripheral neuropathy. Brain, Behav. Immun. 44, 91–99. doi:10.1016/j.bbi.2014.08.010
Janes, K, Symons-Liguori, AM, Jacobson, KA, and Salvemini, D (2016). Identification of A3 adenosine receptor agonists as novel non-narcotic analgesics. Br. J. Pharmacol. 173, 1253–1267. doi:10.1111/bph.13446
Kan, H. W., Chang, C. H., Lin, C. L., Lee, Y. C., Hsieh, S. T., and Hsieh, Y. L. (2018). Downregulation of adenosine and adenosine A1 receptor contributes to neuropathic pain in resiniferatoxin neuropathy. Pain 159 (8), 1580–1591. doi:10.1097/j.pain.0000000000001246
Kim, Y., Kwon, S. Y., Jung, H. S., Park, Y. J., Kim, Y. S., In, J. H., et al. (2019). Amitriptyline inhibits the MAPK/ERK and CREB pathways and proinflammatory cytokines through A3AR activation in rat neuropathic pain models. Korean J. Anesthesiol. 72 (1), 60–67. doi:10.4097/kja.d.18.00022
Little, J. W., Ford, A., Symons-Liguori, A. M., Chen, Z., Janes, K., Doyle, T., et al. (2015). Endogenous adenosine A3 receptor activation selectively alleviates persistent pain states. Brain 138 (1), 28–35. doi:10.1093/brain/awu330
Loram, L. C., Harrison, J. A., Sloane, E. M., Hutchinson, M. R., Sholar, P., Taylor, F. R., et al. (2009). Enduring reversal of neuropathic pain by a single intrathecal injection of adenosine 2A receptor agonists: A novel therapy for neuropathic pain. J. Neurosci. 29 (44), 14015–14025. doi:10.1523/jneurosci.3447-09.2009
Lucarini, E., Coppi, E., Micheli, L., Parisio, C., Vona, A., Cherchi, F., et al. (2020). Acute visceral pain relief mediated by A3AR agonists in rats: Involvement of N-type voltage-gated calcium channels. Pain 161 (9), 2179–2190. doi:10.1097/j.pain.0000000000001905
Luongo, L., Guida, F., Imperatore, R., Napolitano, F., Gatta, L., Cristino, L., et al. (2014). The A1 adenosine receptor as a new player in microglia physiology. GLIA 62 (1), 122–132. doi:10.1002/glia.22592
Luongo, L., Petrelli, R., Gatta, L., Giordano, C., Guida, F., Vita, P., et al. (2012). 5'-Chloro-5'-deoxy-(±)-ENBA, a Potent and Selective Adenosine A1 Receptor Agonist, Alleviates Neuropathic Pain in Mice Through Functional Glial and Microglial Changes without Affecting Motor or Cardiovascular Functions. Molecules 17 (12), 13712–13726. doi:10.3390/molecules171213712
Nalepa, I., Vetulani, J., Borghi, V., Kowalska, M., Przewłocka, B., Roman, A., et al. (2010). Changes induced by formalin pain in central α1-adrenoceptor density are modulated by adenosine receptor agonists. J. Neural. Transm. 117 (5), 549–558. doi:10.1007/s00702-010-0387-6
Petrelli, R., Scortichini, M., Kachler, S., Boccella, S., Cerchia, C., Torquati, I., et al. (2017). Exploring the Role of N6-Substituents in Potent Dual Acting 5′-C-Ethyltetrazolyladenosine Derivatives: Synthesis, Binding, Functional Assays, and Antinociceptive Effects in Mice. J. Med. Chem. 60 (10), 4327–4341. doi:10.1021/acs.jmedchem.7b00291
Rivkees, S. A., Thevananther, S., and Hao, H. (2000). Are A3 adenosine receptors expressed in the brain? NeuroReport 11 (5), 1025–1030. doi:10.1097/00001756-200004070-00026
Sawynok, J. (2016). Adenosine receptor targets for pain. Neuroscience 338, 1–18. doi:10.1016/j.neuroscience.2015.10.031
Sowa, N. A., Voss, M. K., and Zylka, M. J. (2010). Recombinant ecto-5’-nucleotidase (CD73) has long lasting antinociceptive effects that are dependent on adenosine A1receptor activation. Mol. Pain 6, 20. doi:10.1186/1744-8069-6-20
Stockstill, K., Wahlman, C., Braden, K., Chen, Z., Yosten, G. L., Tosh, D. K., et al. (2020). Sexually dimorphic therapeutic response in bortezomib-induced neuropathic pain reveals altered pain physiology in female rodents. Pain 161 (1), 177–184. doi:10.1097/j.pain.0000000000001697
Terayama, R., Tabata, M., Maruhama, K., and Iida, S. (2018). A3 adenosine receptor agonist attenuates neuropathic pain by suppressing activation of microglia and convergence of nociceptive inputs in the spinal dorsal horn. Exp. Brain Res. 236 (12), 3203–3213. doi:10.1007/s00221-018-5377-1
Tosh, D. K., Padia, J., Salvemini, D., and Jacobson, K. A. (2015). Efficient, large-scale synthesis and preclinical studies of MRS5698, a highly selective A3 adenosine receptor agonist that protects against chronic neuropathic pain. Purinergic Signal. 11 (3), 371–387. doi:10.1007/s11302-015-9459-2
Trang, T, Beggs, S, and Salter, M. W. (2012). ATP receptors gate microglia signaling in neuropathic pain. Exp. Neurol. 234, 354–361. doi:10.1016/j.expneurol.2011.11.012
Vincenzi, F., Pasquini, S., Borea, P. A., and Varani, K. (2020). Targeting adenosine receptors: A potential pharmacological avenue for acute and chronic pain. Int. J. Mol. Sci. 21, 8710. doi:10.3390/ijms21228710
Wahlman, C., Doyle, T. M., Little, J. W., Luongo, L., Janes, K., Chen, Z., et al. (2018). Chemotherapy-induced pain is promoted by enhanced spinal adenosine kinase levels through astrocyte-dependent mechanisms. Pain 159 (6), 1025–1034. doi:10.1097/j.pain.0000000000001177
Wu, W. P., Hao, J. X., Halldner, L., Lövdahl, C., DeLander, G. E., Wiesenfeld-Hallin, Z., et al. (2005). Increased nociceptive response in mice lacking the adenosine A1 receptor. Pain 113 (3), 395–404. doi:10.1016/j.pain.2004.11.020
Yaar, R., Lamperti, E. D., Toselli, P. A., and Ravid, K. (2002). Activity of the A3 adenosine receptor gene promoter in transgenic mice: Characterization of previously unidentified sites of expression. FEBS Lett. 532 (3), 267–272. doi:10.1016/s0014-5793(02)03612-8
Yan, H., Zhang, E., Feng, C., and Zhao, X. (2016). Role of A3 adenosine receptor in diabetic neuropathy. J. Neurosci. Res. 94 (10), 936–946. doi:10.1002/jnr.23774
Zhang, Y., Conklin, D. R., Li, X., and Eisenach, J. C. (2005). Intrathecal morphine reduces allodynia after peripheral nerve injury in rats via activation of a spinal A1 adenosine receptor. Anesthesiology 102 (2), 416–420. doi:10.1097/00000542-200502000-00027
Zhang, Y., Sloan, S. A., Clarke, L. E., Caneda, C., Plaza, C. A., Blumenthal, P. D., et al. (2016). Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with mouse. Neuron 89 (1), 37–53. doi:10.1016/j.neuron.2015.11.013
Keywords: metabotropic adenosine receptors, neuropathic pain, new targets, A1AR, A2AR, A3AR
Citation: Luongo L, Guida F, Maione S, Jacobson KA and Salvemini D (2021) Adenosine Metabotropic Receptors in Chronic Pain Management. Front. Pharmacol. 12:651038. doi: 10.3389/fphar.2021.651038
Received: 13 January 2021; Accepted: 22 March 2021;
Published: 16 April 2021.
Edited by:
Francisco Ciruela, University of Barcelona, SpainReviewed by:
Adair Roberto Soares Santos, Federal University of Santa Catarina, BrazilCopyright © 2021 Luongo, Guida, Maione, Jacobson and Salvemini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Livio Luongo, bGl2aW8ubHVvbmdvQGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.