
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 20 April 2021
Sec. Experimental Pharmacology and Drug Discovery
Volume 12 - 2021 | https://doi.org/10.3389/fphar.2021.643857
This article is part of the Research Topic Flavonoids: From Biosynthesis and Metabolism to Health Benefits View all 16 articles
 Ines L. Paraiso1,2
Ines L. Paraiso1,2 Thai Q. Tran2
Thai Q. Tran2 Armando Alcazar Magana1,2,3
Armando Alcazar Magana1,2,3 Payel Kundu4
Payel Kundu4 Jaewoo Choi1
Jaewoo Choi1 Claudia S. Maier3
Claudia S. Maier3 Gerd Bobe1,5
Gerd Bobe1,5 Jacob Raber2,4,6
Jacob Raber2,4,6 Chrissa Kioussi2*
Chrissa Kioussi2* Jan F. Stevens1,2*
Jan F. Stevens1,2*The farnesoid X receptor (FXR) plays a critical role in the regulation of lipid and bile acid (BA) homeostasis. Hepatic FXR loss results in lipid and BA accumulation, and progression from hepatic steatosis to nonalcoholic steatohepatitis (NASH). This study aimed to evaluate the effects of xanthohumol (XN), a hop-derived compound mitigating metabolic syndrome, on liver damage induced by diet and FXR deficiency in mice. Wild-type (WT) and liver-specific FXR-null mice (FXRLiver−/−) were fed a high-fat diet (HFD) containing XN or the vehicle formation followed by histological characterization, lipid, BA and gene profiling. HFD supplemented with XN resulted in amelioration of hepatic steatosis and decreased BA concentrations in FXRLiver−/− mice, the effect being stronger in male mice. XN induced the constitutive androstane receptor (CAR), pregnane X receptor (PXR) and glucocorticoid receptor (GR) gene expression in the liver of FXRLiver−/− mice. These findings suggest that activation of BA detoxification pathways represents the predominant mechanism for controlling hydrophobic BA concentrations in FXRLiver−/− mice. Collectively, these data indicated sex-dependent relationship between FXR, lipids and BAs, and suggest that XN ameliorates HFD-induced liver dysfunction via FXR-dependent and independent signaling.
Dyslipidemia coincides with other metabolic disorders such as obesity, hypertension, and glucose intolerance, defined as metabolic syndrome (MetS), which increase the risk to develop type 2 diabetes (T2D) and cardiovascular diseases (Porez et al., 2012). Obesity and T2D are also associated with nonalcoholic fatty liver disease (NAFLD), a spectrum of chronic liver abnormalities from simple steatosis to nonalcoholic steatohepatitis (NASH) to liver cirrhosis (Larter et al., 2010; Chiang, 2013). The growing prevalence of obesity and high-fat diet (HFD)-induced dyslipidemia represent a public health problem worldwide and the development of drugs with a combined effect on different risk factors may be more effective than the use of combinatorial therapy to manage patients' global risks.
Before the discovery of statins, hypercholesterolemia was primarily treated with bile acid (BA) sequestrants, which bind BAs in the intestine and prevent their reabsorption, thereby promoting the hepatic synthesis of BAs from cholesterol (Staels and Kuipers, 2007; Porez et al., 2012). BA synthesis in hepatocytes occurs largely through the classical pathway initiated by the rate-limiting enzyme cholesterol 7α-hydroxylase (CYP7A1). The classical pathway forms the primary BAs, cholic acid (CA) and chenodeoxycholic acid (CDCA), following a multistep enzymatic process. In complement, CYP27A1 initiates an alternative pathway of BA synthesis that also leads to CDCA synthesis (Garcia et al., 2018). Shortly after their synthesis, BAs are conjugated to glycine or taurine and stored into the gallbladder (Chiang, 2013; Garcia et al., 2018). Besides their involvement in transcriptional regulation of cholesterol metabolism (Chiang, 2002; Trauner et al., 2010), BAs regulate hepatic gluconeogenesis, glycogen synthesis and insulin sensitivity (Ma et al., 2006; Trauner et al., 2010). BAs also modulate neurotransmission, neuroendocrine responses, and neurogenesis indicating their importance in neurological functions (Schubring et al., 2012; McMillin and DeMorrow, 2016). However, BA accumulation causes inflammation, hepatic injury (Chiang, 2017) and is associated with motor and cognitive impairments (Huang et al., 2016; McMillin et al., 2016). A key regulator of maintaining lipid and BA homeostasis is the farnesoid X receptor (FXR, NR1H4), which upon activation by BAs, polyunsaturated fatty acids and farnesylated proteins (Forman et al., 1995; Makishima et al., 1999; Zhao et al., 2004), regulates the expression of target genes involved in various physiological processes (Chawla et al., 2001; Sun et al., 2021). An increase of intracellular BAs also activates the constitutive androstane receptor (CAR) and pregnane X receptor (PXR). They modulate transcriptional regulation of their targets including genes encoding hepatic BA metabolizing enzymes and BA/organic anion transporters (Guo et al., 2003; Uppal et al., 2005; Lee et al., 2006). Subsequently, FXR, CAR and PXR have emerged as promising targets for the treatment of metabolic disorders associated with MetS (Gao and Xie, 2012; Porez et al., 2012).
Xanthohumol (XN) is a hop-derived flavonoid, which mitigates obesity-related metabolic impairments by improving dysfunctional glucose and lipid metabolism in HFD-fed animals (Miranda et al., 2016; Miranda et al., 2018). Treatment of HFD-fed C57BL/6J mice with a diet containing XN decreases their plasma low-density lipoprotein cholesterol (LDL-c), IL-6, Homeostatic Model Assessment of Insulin Resistance (HOMA-IR) and leptin concentrations (Miranda et al., 2016). XN enhances fatty acid oxidation as a result of mild mitochondrial uncoupling (Kirkwood et al., 2013) and decreases adipocyte markers such as PPARγ, C/EBPα and DGAT1 (Yang et al., 2007). This effect might be at least partly mediated by FXR, since XN is a ligand of FXR (Yang et al., 2016) that modulates FXR downstream gene expression in a manner similar to selective bile acid receptor modulators (SBARM) (Nozawa, 2005; Paraiso et al., 2020). However, the extent to which FXR signaling mediates the in vivo effects of XN is unknown. Both activation of hepatic FXR and inhibition of intestinal FXR have beneficial effects in obesity-related metabolic diseases (Sun et al., 2021) due to differential effects on metabolic regulation (Kim et al., 2007; Schmitt et al., 2015). These effects are further emphasized by the observation that intestine-specific FXR knockout mice are resistant to HFD-induced obesity, while HFD-fed liver-specific FXR knockout mice develop NAFLD (Li et al., 2013; Schmitt et al., 2015). Therefore, tissue-specific mouse models are necessary to dissect the complex effects of FXR on dyslipidemia. In the current study, we used liver-specific FXR-null mice (FXRLiver−/−) to investigate the effect of XN on dyslipidemia and BA accumulation. Our findings demonstrate that XN ameliorate HFD-induced hepatic injury and dysfunctional lipid and BA metabolism in WT and FXRLiver−/− mice. We also provide evidence that XN induces expression of nuclear receptors (NRs) including CAR, PXR and the glucocorticoid receptor (GR) involved in the metabolism of BAs and lipids. These findings have potentially important implications in the treatment of metabolic and cholestatic diseases.
All animal experiments were performed in accordance with institutional and National Health and Medical Research Council guidelines. The experimental protocol was approved by the Institutional Animal Care and Use Committee at Oregon State University and the studies were carried out in accordance with the approved protocol (IACUC 2019-0001). Nine-week-old WT male and female C57BL/6J mice were obtained from Jackson Laboratory (Bar Harbor, ME, United States). FXRLiver−/− mice were generated by crossing FXRFL/FL mice with mice harboring the Cre recombinase under the control of the albumin promoter (AlbCre) to produce the AlbCre:FXRFL/FL or FXRLiver−/− mice (Kong et al., 2016). All mice were in C57BL/6J genetic background for over 12 generations. Mice were housed in groups of two–3 in ventilated cages under a 12–12-h light-dark cycle and fed a HFD (Dyets Inc. Bethlehem, PA, United States) containing 60, 20 and 20% total calories from fat, carbohydrate and protein, respectively. XN (purity >99%) from Hopsteiner Inc (New York, NY, United States) was mixed into the diet as previously described (Miranda et al., 2018) to deliver a dose of 60 mg/kg body weight/day. The control diet contained an identical amount of the vehicle. 15 WT mice (8 females, 7 males) and 18 FXRLiver−/− mice (10 females, 8 males) were fed a control HFD, while 15 WT mice and 18 FXRLiver−/− mice were treated with XN for a duration of 12 weeks. Food intake and body weights were recorded weekly. At week 10, fasting glucose was measured after 6 h of fasting by using the One Touch UltraMini glucometer (LifeScan Inc. Milpitas, CA, United States). At the end of 12 weeks of feeding, fed-state mice were euthanized by cervical dislocation, their blood collected, and their liver and hippocampus were dissected for further analyses. Deletion of FXR in the liver of FXRLiver−/− mice was confirmed by genotyping at weaning (Kong et al., 2016). Quantitative PCR after the feeding experiment. FXR mRNA levels were ∼ 3-fold lower in the liver of mutant compared to WT mice (Supplementary Figure S1).
Liver biopsies from n = 3 male mice per genotype-diet group were fixed in 4% paraformaldehyde, embedded in OCT and 10 µm-thick sections were used for histology. Hematoxylin and Eosin (H&E) and Sudan black staining were performed as previously described (Chang et al., 2019).
To measure ALT and AST enzymatic activities, liver samples (n = 6 per genotype-diet group) were homogenized in 10 ml of 100 mM Tris (pH = 7.8) per Gram of tissue. The homogenates were centrifuged at 10,000 ×g for 15 min at 4°C. The supernatants were analyzed for ALT and AST activity using colorimetric assay kits purchased from Cayman Chemical (Ann Arbor, MI, United States). Plasma leptin concentrations (n = 5–7 per genotype-diet group) were measured using the Enzyme Immunoassay kit from SPI Bio Inc.. (Sherbrooke, QC, Canada) as per manufacturer’s instructions.
Mouse liver samples (50 mg, n = 15–18 per genotype-diet group) were spiked with SPLASH® Lipidomix® internal standards from Avanti Lipids (Alabaster, AL, United States) and homogenized with zirconium oxide beads and 1 ml of cold methylene chloride: isopropanol: methanol (25:10:65, v/v/v) + 0.1% butylated hydroxytoluene (BHT). The mixture was incubated at –20°C for 1 h and centrifuged at 13,000 rpm for 10 min 20 µL of the supernatant was diluted 1/10 in extraction solvent before MS analysis. UPLC was performed using a 1.7 μm particle, 2.1 mm × 100, CSH C18 Column (Waters, Milford, MA, United States) coupled to a quadrupole TOF mass spectrometer (AB SCIEX, TripleTOF 5600) operated in information-dependent MS/MS acquisition mode. LC and MS conditions were developed by our group and described previously by Choi et al. (Choi et al., 2015) with some adjustments. For positive ion mode LC-QToF-MS/MS, the mobile phases consisted of (A) 60:40 (v/v) acetonitrile: water with ammonium formate (10 mM) and formic acid (0.1%) and (B) 90:10 (v/v) isopropanol: acetonitrile with ammonium formate (10 mM) and formic acid (0.1% formic acid). For analyses run in the negative ion mode, ammonium acetate (10 mM) was used as the modifier. Quantification of lipid species was performed using MultiQuant Software version 3.0.2 (SCIEX), after annotation in PeakView Software Version 1.2 (SCIEX) based on accurate masses and retention times for each lipid. The library of lipid profiling for identification was introduced by Cajka et al. (Cajka et al., 2017).
Plasma samples collected post-euthanasia (20 μL, n = 15–18 per genotype-diet group) were spiked with 0.1 ng of cholic acid-d4 internal standard (Cayman Chemical, Ann Harbor, MI, United States) per µL of plasma. 1 ml of ice-cold acetonitrile was added, and the mixture was vortexed and centrifuged at 13,000 rpm for 10 min. The supernatant was evaporated under vacuum and reconstituted in 50% MeOH.
Liver samples without gallbladder (25 mg, n = 15–18 per genotype-diet group) were homogenized in 1 ml of solvent (isopropanol/water, 2:1, v/v with 0.1% formic acid) containing 1.6 ng/ml of cholic acid-d4 internal standard. Samples were homogenized using a counter-top bullet blender for 5 min and centrifuged at 13,000 rpm for 5 min. The supernatants were filtered with OSTRO phospholipid removal plate (Waters, Milford, MA, United States), evaporated under vacuum and reconstituted in 50% MeOH.
Left and right hippocampus were pooled, ground in liquid nitrogen and freeze-dried. The samples were weighed and spiked with 1 pg of cholic acid-d4 internal standard per mg of hippocampus. Approximately 8 mg of hippocampus (dry weight, n = 13–17 per genotype-diet group) were homogenized with 1:30 μL (m/v) of 50% MeOH using a counter-top bullet blender for 10 min and centrifuged at 15,000 rpm for 20 min and supernatants used for HPLC analysis.
UPLC was performed using a 1.7 μm particle, 2.1 mm × 100, CSH C18 column (Waters, Milford, MA, United States) coupled to a hybrid triple quadrupole linear ion trap mass spectrometer (AB SCIEX, 4000 QTRAP). LC and MS conditions were developed by our group and described in the Supplemental data. BAs were identified by matching their retention time, isotopic pattern, exact mass of the [M-H]- ion and fragmentation pattern with those of authentic standards (IROA Technologies, Sea Girt, NJ, United States). SRM transitions used for quantification are listed in (Supplementary Table S1) and additional parameters such as collision energy are listed in (Supplementary Table S2).
100% of the mice had hepatic and plasma BA above the detection limit and 82% (52 out of 63 mice) had hippocampal BA above the detection limit, i.e. 74% of the WT mice vs. 89% of the FXRLiver−/− mice.
Liver and plasma extracts (n = 15–17 per genotype-diet group) were analyzed for XN and metabolites by LC-MS/MS using a hybrid triple quadrupole linear ion trap mass spectrometer (AB SCIEX, 4000 QTRAP). Analytes were separated by UPLC carried out using a 2.1 × 50 mm Agilent Zorbax 300 SB-C8 3.5 μm column (Agilent, Santa Clara, CA, United States). Each run lasted 6 min at a flow rate of 0.4 ml/min. The elution gradient started at 30% solvent B (0.1% formic acid in acetonitrile) in solvent A (0.1% formic acid in water) and was increased to 60% solvent over the initial 1.5 min. The gradient was held at 60% for 1 min, increased to 100% B for 0.5 min, held at 100% B from 3.0 to 3.8 min, then dropped to 30% B in 0.1 min. The column was equilibrated for 2.1 min until 6.0 min. SRM transitions for quantification were m/z 353 → 119 for XN and isoxanthohumol (IX), m/z 339 → 219 for 8-prenylnaringenin (8 PN), m/z 355 → 249 for α,β-dihydroxanthohumol (DXN), and m/z 341 → 235 for O-desmethyl-α,β-dihydroxanthohumol (DDXN).
RNA was prepared from liver samples (n = 4-5 per genotype-diet group) and sequenced as previously described (Singh et al., 2018). All samples were processed and analyzed in parallel. Sequence quality was assessed by FastQC. Reads were aligned by Hisat2 (Kim et al., 2019) and Samtools (Li et al., 2009). A gene count matrix was generated byStringtie (Pertea et al., 2015). Two data sets, HFD (control) and HFD-XN (treatment) were derived from the gene count matrix. Each set was analyzed in parallel by DESeq2 (Love et al., 2014) for differential expression (DE) calculation. DE was calculated for FXRLiver−/− mutant over wildtype. PCA plots were generated using the DESeq2 package. Benjamini-Hochberg multiple-test correction was applied to control for the number of false positives with an adjusted 5% statistical significance threshold (Benjamini and Hochberg, 1995). A heatmap was created using the pheatmap package in R (version 3.6). Functional annotation clustering was achieved in Network Analyst v3.0 using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database.
RNA samples from mouse liver (n = 4-5 per genotype-diet group) were reverse-transcribed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Waltham, MA, United States). universal SYBR® Green Supermix (Bio-Rad, Hercules, CA, United States) was used following the manufacturer’s protocol and amplifications were performed using the ABI Prism 7300 Real-Time PCR System (Applied Biosystems, Waltham, MA, United States). Each sample had two technical replicates. Gene expression was normalized to levels of Polymerase-II. Relative gene expression was calculated using the 2−ddCt method. All primers were purchased from IDT technologies (Coralville, IA, United States) and are listed in (Supplementary Table S3).
Statistical data were analyzed in SAS version 9.4 (SAS Ins. Inc., Cary, NC). Plasma leptin, AST, and ALT concentrations, intestinal gene expression, and liver receptor data were not normally distributed and could not be normalized through transformation. Therefore, these parameters were analyzed using the non-parametric Wilcoxon rank sum test after checking for interactions. We categorized values into elevated and normal and used Fisher’s exact test to compare treatment groups. BAs and XN concentrations were not normally distributed but rather distributed logarithmically to the base 10, where 1 is equal to 10, two is equal to 100, and 3 is equal to 1000, and were analyzed on that scale. In addition, we categorized BA values into elevated and normal and compared treatment groups using Fisher’s exact test. The remaining lipid data were analyzed without transformation. The effect of XN-treatment was evaluated separately for WT and FXRLiver−/− mice using a generalized linear model in PROC GLM with XN-treatment, sex, and their interaction, because FXRLiver−/− mice had larger variance estimates than WT mice. The effects of genotype and sex were evaluated in untreated mice using a generalized linear model in PROC GLM with genotype, sex, and their interactions, as XN modified the effect of genotype and sex. All statistical tests were two-sided. Significance was declared at p ≤ 0.05. Correlations were tested by calculating non-parametric Spearman’s correlation coefficient, r.
During the course of the study, weight gain in HFD-fed WT and FXRLiver−/− mice were comparable (Table 1). HFD-fed males gained more body weight than females (Supplementary Figure S2); this effect was significant in WT mice (p < 0.0001) but not in FXRLiver−/− mice (p = 0.1). To ensure oral bioavailability of XN in WT and FXRLiver−/− mice, we measured liver and plasma concentrations of XN and its metabolites in XN-treated mice. Oral bioavailability of XN was comparable in both genotypes (Supplementary Table S4), but IX, a product of XN isomerization, reached higher concentrations in the liver of WT mice compared to FXRLiver−/− mice. Moreover, we observed sex-related differences as females had significantly higher concentrations of XN and IX than males (Table 2). Since there was no difference in food intake, this observation is likely a result of the lower body weight in females compared to males. α,β-Dihydroxanthohumol (DXN), a bacterial metabolite of XN (Paraiso et al., 2019) was not affected by sex or genotype, while 8-prenylnaringenin (8PN) hepatic concentrations were elevated in male FXRLiver−/− mice. These observations suggest an influence of sex on XN pharmacokinetics, likely due to the differences in weight and volume of distribution between males and females.
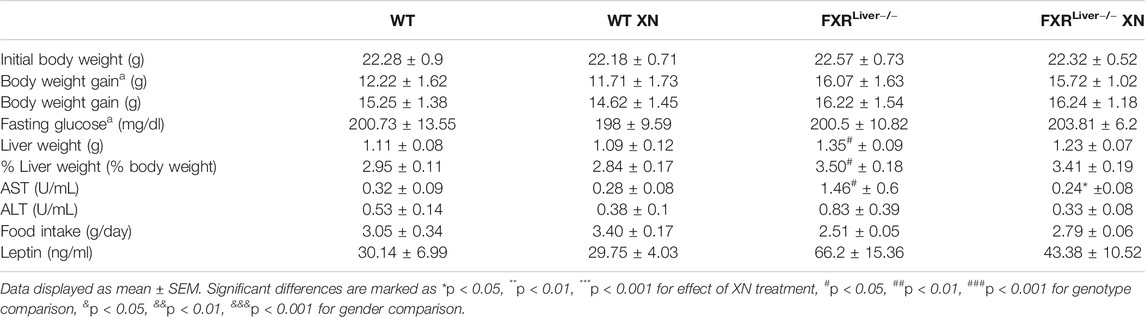
TABLE 1. A list of metabolic parameters measured in WT and FXRLiver−/− mice upon 10 weeks (a) or 12°weeks of HFD ± XN.
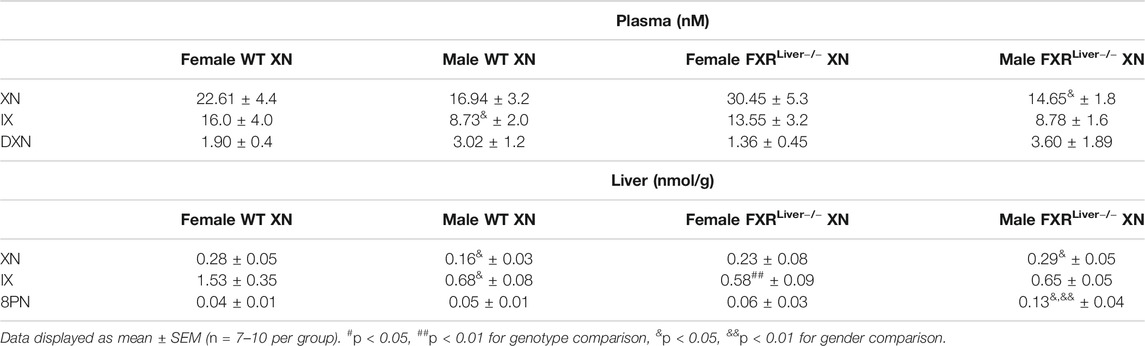
TABLE 2. Concentrations of XN and metabolites (IX, 8PN, DXN) in the plasma and liver of females vs. males HFD-fed WT and FXRLiver−/− mice.
To assess if the HFD successfully induced NAFLD, we examined liver sections from three representative male mice per treatment group. Hematoxylin and eosin (H&E) stained liver sections showed hepatic steatosis in the form of vacuoles with a clear appearance in HFD-fed WT and FXRLiver−/− mice (Figures 1A,B). We observed a reduction in number and size of these vacuoles in both genotypes in XN-treated mice (Figures 1C,D). Sections stained with Sudan black confirmed an increase in lipid vacuoles in FXRLiver−/− mice compared to WT (Figures 1E,F), which was reversed in XN-treated mice (Figures 1G,H, Supplementary Figure S3). Another marker of NAFLD is the proportion of liver weight (LW) over total body weight (LW%). After 12 weeks on the HFD, untreated FXRLiver−/− mice exhibited increased LW (p = 0.02) and LW% (p = 0.03) than WT mice (Table 1). In addition, males had increased LW than females in WT mice (p = 0.001) and FXRLiver−/− mice (p < 0.0001, Supplementary Figure S4A). Fasting glucose was also elevated in males compared to females in both genotypes (Supplementary Figure S4B).
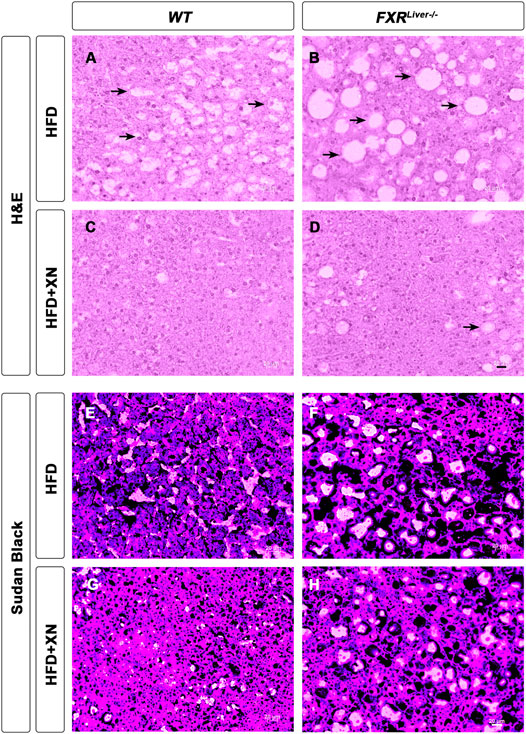
FIGURE 1. XN prevents HFD-induced hepatic steatosis. Representative liver histology by H&E (A-D) and Sudan Black (E-H) of male WT and FXRLiver−/− mice fed HFD ± XN. Arrows indicate vacuoles, a characteristic structure of hepatic steatosis.
To measure the extent to which the steatosis had resulted in liver tissue damage, we measured aspartate aminotransferase (AST) and alanine aminotransferase (ALT) enzymatic activities in liver homogenates. Absence of hepatic FXR might promote liver tissue damage as AST levels were increased in untreated FXRLiver−/− compared to WT mice (p = 0.02, Table 1). XN reduced AST levels in treated FXRLiver−/− mice (p = 0.03). Plasma leptin concentrations were elevated in FXRLiver−/− mice but differences in leptin and food intake among groups were not significant. These results suggest that HFD-induced NAFLD is accentuated in absence of hepatic FXR and the severity of the hepatic steatosis is attenuated by XN supplementation.
We annotated and measured the relative abundances of 116 individual hepatic lipids including triglycerides (TG), free cholesterol, esterified cholesterol (CE), ceramides and sphingomyelins (SM; Figure 2A). We observed sex, genotype and XN-dependent effects on lipid composition (Figures 2B–H).
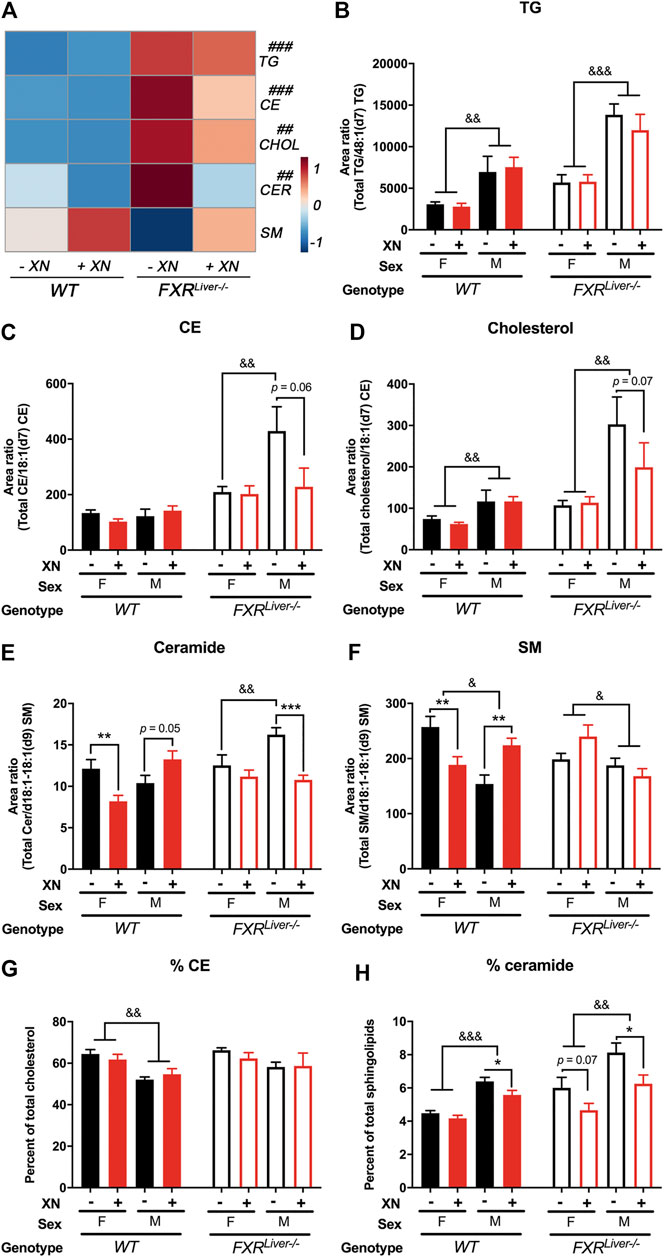
FIGURE 2. XN prevents HFD-induced ceramide accumulation (A) Heatmap of hepatic triglycerides (TG), cholesterol esters (CE), free cholesterol (CHOL), ceramides (CER) and sphingomyelins (SM) in HFD-fed WT and FXRLiver−/− mice untreated or treated with XN. Total relative abundance of (B) TG (C) CE (D) free cholesterol (E) ceramide and (F) SM in the liver of WT and FXRLiver−/− mice untreated or treated with XN. Proportion of (G) CE and (H) ceramide in the liver of WT and FXRLiver−/− mice untreated or treated with XN. Values are mean ± SEM (n = 7–10 per group). *p < 0.05, **p < 0.01, ***p < 0.001 for effect of XN treatment; #p < 0.05, ##p < 0.01, ###p < 0.001 for genotype comparison; &p < 0.05, &p < 0.01, &p < 0.001 for gender comparison.
Hepatic TG were increased in female FXRLiver−/− compared to their WT counterparts, but male FXRLiver−/− mice displayed the most severe hepatic steatosis. Male FXRLiver−/− mice had elevated hepatic TG, cholesterol, CE and ceramides compared to female FXRLiver−/− mice and compared to male WT mice (Figures 2B–E). The proportion of CE over total cholesterol (%CE) and the proportion of ceramide over total sphingolipids (%ceramide) was higher in FXRLiver−/− males compared to WT males (Figures 2G,H). These data indicate that male mice are more responsive to diet-induced hepatic steatosis in the absence of FXR signaling in the liver.
In FXRLiver−/− mice, XN had a predominant effect in male mice, which exhibited the highest hepatic lipid accumulation. Total CE (p = 0.06), cholesterol (p = 0.07) and ceramides (p = 0.0005) were decreased in XN-treated male FXRLiver−/− mice compared to the untreated mice. The %ceramide, a measure of the proportion of ceramides among other sphingolipids, correlated better with histological improvements than ceramide abundances. XN treatment decreased %ceramide in male WT (p = 0.01) and male FXRLiver−/− mice (p = 0.02); both groups had the most elevated %ceramide among the untreated groups. SM abundances followed a trend opposite to that of other lipids and were increased in XN-treated WT males (Figure 2F). These data suggest that XN regulates lipid metabolism via pathways independent of hepatic FXR signaling.
We screened for 34 individual BAs in the plasma, liver and hippocampus of WT and FXRLiver−/− mice using UPLC-MS/MS. Hippocampal BAs were measured to investigate BA retention in tissues deficient in BA detoxification and export mechanisms. Fifteen BAs were detected and quantified in the liver, 12 BAs in the plasma and 7 BAs in the hippocampus.
FXRLiver−/− had higher BA concentrations in plasma (p < 0.0001, Figure 3A) and liver (p = 0.01, Figure 3C) than WT mice, while an increase in hippocampal BAs was observed in FXRLiver−/− males only (p = 0.04, Supplementary Figure S5). The most severe BA accumulation occurred in the plasma of FXRLiver−/− mice, with a 7-fold increase in total BAs vs. 2-fold increase in the liver. The change was driven by an increase in primary conjugated BAs (Figures 3A–D). Hippocampal BA retention was more pronounced than hepatic BA retention as FXRLiver−/− mice exhibited 9-fold increase in total hippocampal BAs compared to WT (Figures 3E,F) indicating passage of BAs through a possibly altered blood brain barrier (BBB). These data suggest that hepatic mechanisms of BA efflux remained more efficient than cerebral mechanisms of BA efflux in FXRLiver−/− mice. This is further supported by our observation that, in FXRLiver−/− mice, plasma BAs were more strongly correlated to hippocampal BAs (r = 0.90, p < 0.0001) than to hepatic BAs (r = 0.49, p = 0.005, Figures 3G,H). BA pool composition was also modified in FXRLiver−/− mice. The percentage of plasma primary conjugated BAs over total BAs was 50% in WT mice vs. 85% in FXRLiver−/− mice (Figure 3B). Hepatic conjugation of BAs improves their hydrophilicity and reduces their toxicity suggesting that FXRLiver−/− mice developed a metabolic mechanism to counter BA-mediated toxicity.
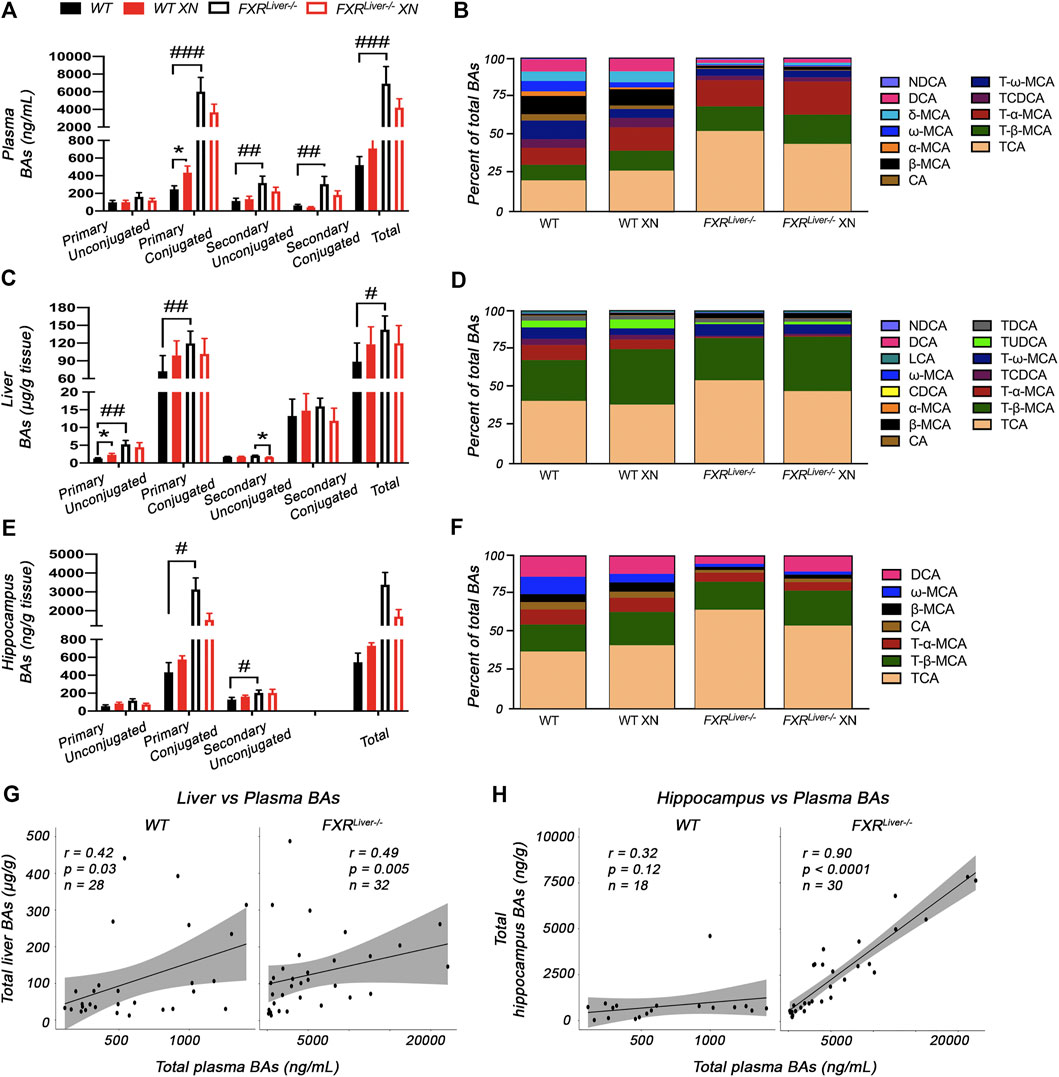
FIGURE 3. XN modulates BA composition (A) Total BAs and (B) composition of the BA pool in the plasma of HFD-fed WT and FXRLiver−/− mice (C) Total BAs and (D) composition of BAs in the liver of HFD-fed WT and FXRLiver−/− mice (E) Total BAs and (F) composition of BAs in the hippocampus of HFD-fed WT and FXRLiver−/− mice (G) Correlations between liver and plasma BAs in WT and FXRLiver−/− mice (H) Correlations between hippocampus and plasma BAs in WT and FXRLiver−/− mice. Bar graphs values are mean ± SEM (n = 15–18 per group). *p < 0.05, **p < 0.01, ***p < 0.001 for effect of XN treatment; #p < 0.05, ##p < 0.01, ###p < 0.001 for genotype comparison. Abbreviations: chenodeoxycholic acid (CDCA), cholic acid (CA), deoxycholic acid (DCA), lithocholic acid (LCA), muricholic acid (MCA), nordeoxycholic acid (NDCA), taurochenodeoxycholic acid (TCDCA), taurocholic acid (TCA), tauromuricholic acid (T-MCA), taurodeoxycholic acid (TDCA), tauroursodeoxycholic acid (TUDCA).
XN treatment promoted BA synthesis in WT mice but attenuated BA accumulation in FXRLiver−/− mice. XN effect on BA concentrations was independent of sex in WT mice. XN supplementation of WT mice resulted in increased plasma primary conjugated BAs (p = 0.03, Figure 3A) and increased hepatic primary unconjugated BAs (p = 0.02, Figure 3C). These observations are in accordance with previous reports that XN induces CYP7A1 and hepatic BA synthesis in WT mice resulting in increased BA concentrations (Paraiso et al., 2020). DCA, TCA, β-MCA and FXR antagonists, T-α-MCA and T-β-MCA, were increased in the liver and/or plasma of XN-treated WT mice (Figures 4A,B). By contrast, XN treatment resulted in decreased BA concentrations in FXRLiver−/− mice. CA and CA-derived BAs including DCA, TCA and TDCA were decreased in the liver of FXRLiver−/− mice in both sexes, with males exhibiting more significant changes (Figures 4A–C). CA, DCA and ω-MCA were decreased in the plasma of XN-treated male FXRLiver−/− mice.

FIGURE 4. XN differentially modulates classical and alternative pathways of synthesis in WT vs. FXRLiver−/− mice. Heatmaps of individual BA concentrations in the plasma (A), liver (B) and hippocampus (C) of HFD-fed WT and FXRLiver−/− mice. *p < 0.05, **p < 0.01, ***p < 0.001 for effect of XN treatment (n = 7–10 per group). Abbreviations: chenodeoxycholic acid (CDCA), cholic acid (CA), deoxycholic acid (DCA), lithocholic acid (LCA), muricholic acid (MCA), nordeoxycholic acid (NDCA), taurochenodeoxycholic acid (TCDCA), taurocholic acid (TCA), tauromuricholic acid (T-MCA), taurodeoxycholic acid (TDCA), tauroursodeoxycholic acid (TUDCA).
In summary, XN reduced most individual BAs of the classical pathway in FXRLiver−/− mice and increased BAs from the alternative pathway of synthesis in WT mice (Figures 4A,B; Supplementary Figure S6). Since BAs did not reach pathological concentrations in WT mice, these results suggest an adaptation of XN mechanism of action to the pathophysiological conditions and the possible activation of BA receptors independent of FXR. These observations further support a genotype-specific differential modulation of metabolism by XN.
We analyzed changes in global gene expression profiles in hepatic tissue of HFD-fed WT and FXRLiver−/− mice. Since a smaller subset of samples was sequenced, male and female mice RNA sequencing data were pooled to increase the power of the analysis. Functional annotation clustering revealed that genes differentially affected by HFD in WT and FXRLiver−/− mice can be classified into two main functional groups: genes involved in metabolic processes vs. genes involved in inflammation and carcinogenic processes (Supplementary Table S5). The comparison between XN-treated and untreated FXRLiver−/− mice revealed 243 shared genes, with 759 features unique to untreated mice and 170 features unique to XN-treated mice (Figure 5A). Within the shared features, XN supplementation impacted several gene networks including lipid metabolism (Mgat2, Sptlc2, Smpd3), ABC transporters involved in BA transport (Abcc4, Abcc3, Abcb11), metabolism of xenobiotics with genes involved in phase I and II metabolism (Gsta1, Gstm3, Ugt1a7c, Sult2a7), PI3K-Akt signaling pathway (Tnc, Tlr2, Spp1, Thbs1, Lamb3), cytokine-cytokine receptor interactions (Ccl2, Cxcl9/10, Cd9, Tnfrsf1a) and amino acid metabolism (Sardh, Aadat, Kyat3) (Figure 5B).

FIGURE 5. XN alters hepatic gene profiles (A) Venn diagram comparing genes in HFD-fed mice vs. HFD-fed mice treated with XN (B) Relative expression of 106 shared genes classified according to KEGG pathway (Log2FC of FXRLiver−/− ± XN/WT) (C) Genes regulated by XN in WT mice (D) Genes regulated by XN in FXRLiver−/− mice. Values are mean of n = 4-5 mice (males and females) per group.
In WT mice, presence of XN was associated with increased expression of genes involved in lipid and xenobiotic metabolism such as Mgat1, Cyp1a1, Ugt1a7c, and decreased expression of genes involved in energy metabolism (Clock, Rnf146) and inflammation (Cebpg, Saa1, Saa2) (Figure 5C; Table 3). CCAAT-enhancer binding proteins (C/EBP) interact with the proximal promoter of the Saa genes and regulate hepatic expression of SAA (Ray et al., 1995). The concurrent decrease in Cebpg and Saa expression suggests that XN-mediated repression of Saa1 and Saa2 is mediated by inhibition of Cebpg expression.
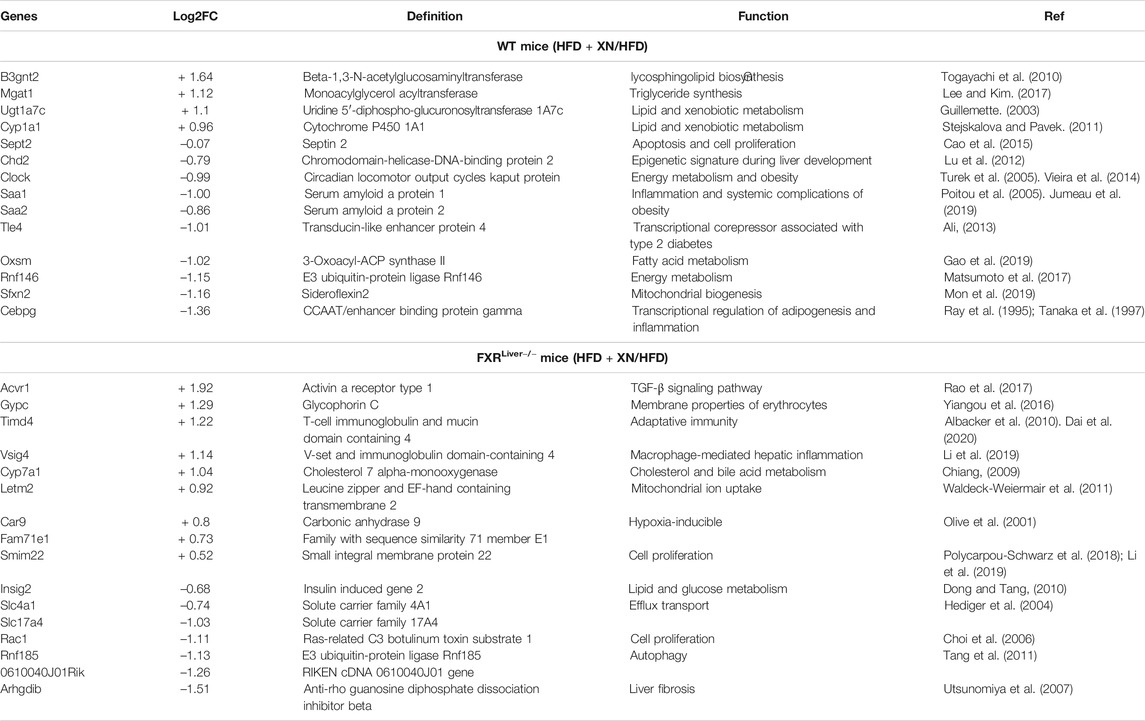
TABLE 3. Hepatic genes regulated by XN in WT and FXRLiver−/− mice and their roles in metabolic function.
In FXRLiver−/− mice, XN induced Vsig4, which attenuates macrophage-mediated hepatic inflammation (Li et al., 2019), Acvr1, which is involved in activin signaling (Rao et al., 2017), and Timd4, that controls adaptive immunity by clearing antigen-specific T-cells (Albacker et al., 2010) (Figure 5D; Table 3). While XN induced increased expression of Cyp7a1, which is involved in the classical pathway of BA synthesis, there were no changes in the expression of genes involved in the alternative pathway of BA synthesis. RhoGDI2, encoded by Arhgdib, is involved in the molecular pathogenesis of liver fibrosis (Utsunomiya et al., 2007) and acts as a positive regulator of Rac1 (Kardol-Hoefnagel et al., 2020). The decreased expression of both genes indicated that XN inhibition of Rac1 transcription might be mediated by repression of Arhgdib in FXRLiver−/− mice (Table 3). These data show that XN promotes lipid and BA metabolism and decreases acute inflammation in WT mice, while XN attenuates inflammation by controlling immune response, inhibits cell proliferation and liver fibrosis in FXRLiver−/− mice.
RNA sequencing revealed several genes involved in phase II reactions such as glucuronidation (UGTs), sulfation (SULTs) and glutathione conjugation (GSTs) were regulated by XN. Therefore, we performed a quantitative analysis of the hepatic expression of a panel of NRs known to regulate phase II BA metabolism including CAR, PXR and GR. BAs interact with CAR (Moore et al., 2002), PXR (Staudinger et al., 2001), as well as GR (Tanaka and Makino, 1992), which also regulates biosynthesis and transport of bile salts (Xiao et al., 2016). XN treatment resulted in higher CAR expression in all sex and genotype groups (Figure 6A), although XN effect was stronger in males. XN also induced gene expression of PXR in both WT and FXRLiver−/− mice, while an increase in GR transcript levels was observed in XN-treated FXRLiver−/− mice only (p = 0.009, Figure 6A).

FIGURE 6. XN induces hepatic expression of FXR-independent NRs (A) Quantitative relative expression of CAR, PXR and GR in the liver of HFD-fed WT and FXRLiver−/− mice (B) Correlations between relative mRNA expression of CAR, PXR and GR vs. hepatic unconjugated BAs concentrations in FXRLiver−/− mice (C) Correlations between relative mRNA expression of GR vs. relative abundances of hepatic CE, cholesterol and ceramide in FXRLiver−/− mice. Bar graphs values are mean ± SEM, n = 4-5 mice (males and females) per group. *p < 0.05, **p < 0.01, ***p < 0.001 for effect of XN treatment.
Changes in these NR expression profiles were linked to hepatic BA concentrations. Correlation analyses revealed that relative expression levels of these receptors were negatively correlated with unconjugated BAs in the liver absence of FXR (Figure 6B). Additionally, relative expression of GR was negatively correlated with relative abundances of lipids regulated by XN, i.e. CE (r = -0.78, p = 0.01), cholesterol (r = -0.67, p = 0.04) and ceramide (r = -0.68, p = 0.03) in absence of FXR. Collectively, these data suggest that, in absence of hepatic FXR, induction of CAR, PXR and GR is involved in XN-mediated decrease of lipid and BA concentrations.
NRs regulate ligand-activated transcriptional activation of a myriad of genes for the elimination and detoxification of potentially toxic biliary constituents accumulating in cholestasis (Halilbasic et al., 2013). FXR controls the transcriptional activation of several genes involved in the regulation of glucose and lipid metabolism and maintenance of BA homeostasis, thereby protecting the host against liver damage associated with lipid and BA accumulation. BAs act as signaling molecules through BA receptors such as FXR, TGR5, PXR and VDR to regulate TG, cholesterol, glucose, and energy homeostasis (Schaap et al., 2014; Fiorucci and Distrutti, 2015). BAs inhibit their own synthesis mainly via FXR-mediated negative feedback of CYP7A1, the rate-limiting enzyme in the catabolism of cholesterol into BAs (Goodwin et al., 2000). As a result, FXR knockout mice exhibit dyslipidemia (Sinal et al., 2000; Kok et al., 2003) and hepatic steatosis that progresses to NASH (Armstrong and Guo, 2017).
Liver histology revealed lipid vacuoles characteristic of fatty liver disease in FXRLiver−/− mice and elevated hepatic levels of triglycerides, free cholesterol, cholesterol esters and ceramides. The increased liver enzymes indicate liver injury associated with inflammation and NASH. HFD-induced dyslipidemia was aggravated by FXR deficiency with sex differences. Risk factors associated with HFD-induced obesity including fasting glucose and dyslipidemia were more pronounced in males than females. This is in accordance with previous report that male western diet-fed FXR−/− mice had higher hepatic and serum lipids than their female counterparts (Sheng et al., 2017). In fact, NRs play a crucial role in the calibration of sex-specific metabolic pathways and androsterone (Wang et al., 2006) as well as estrogen (Song et al., 2014) were reported to modulate FXR activity. This suggests that interactions between the receptor and gonadal hormones warrant further investigation. The severity of the FXRLiver−/− phenotype was further aggravated by the accumulation of BAs, which regulate several signaling pathways independent from FXR. In our study, elevated BA concentrations in the plasma of FXRLiver−/− mice were accompanied with higher concentrations of BAs in the hippocampus indicating passage of BAs through the BBB. The concentrations measured in HFD-fed FXRLiver−/− mice are comparable to BA concentrations in the blood and brain of FXR−/− mice with hepatic enceph-alopathy (Huang et al., 2015). In fact, in pathological conditions such as acute liver failure and cholestasis, elevated plasma BAs were reported to increase permeability of the BBB (Quinn et al., 2014; McMillin et al., 2016), warranting the investigation of therapeutic alternatives regulating BA concentrations.
XN anti-hyperlipidemic effect was more accentuated in male FXRLiver−/− mice that developed severe dyslipidemia. Our observation that XN and metabolites reached higher concentrations in female mice suggests that pharmacodynamic effect of XN in vivo depends more on the severity of the phenotype and less on XN concentrations in biological tissues. Ceramide abundances were heavily influenced by their precursor, SM. The lipotoxicity of ceramides is well-documented (Summers, 2006; Chaurasia and Summers, 2015), but the role of SM in NAFLD and NASH is controversial. Several studies report lower SM levels in NASH patients (Kartsoli et al., 2020). In fact, SM are important components of biological cell membranes (Slotte and Ramstedt, 2007) with no demonstrated intrinsic lipotoxicity. The increase in hepatic SM in XN-treated WT males was accompanied by an increase in hepatic ceramides that did not correlate with metabolic improvements. Therefore, we used the ratio of ceramides over total sphingolipids to estimate ceramide relative abundances more precisely. This ratio was decreased in XN-treated WT and FXRLiver−/− mice.
Taken together, XN protected FXRLiver−/− mice from liver damage, as evaluated by liver transaminase activity, liver histopathology and hepatic expression levels of anti-inflammatory genes. These observations indicate that XN effect is not exclusively mediated by hepatic FXR. We hypothesized that the observed XN effect might also derive from its potential as SBARM and from XN-dependent modulation of BA composition because BAs regulate energy expenditure in mice (Chiang, 2002). In WT mice, XN treatment led to an FXR-dependent increase in the most hydrophilic BAs among which are T-α-MCA and T-β-MCA. FXR antagonists, including T-α-MCA and T-β-MCA, have been reported to improve HFD-induced metabolic dysfunction by inducing thermogenesis and repressing intestinal FXR-FGF15 signaling (Sayin et al., 2013; Jiang et al., 2015). Intestinal FXR-FGF15 signaling regulates CYP7A1 gene expression (Kim et al., 2007). In turn, CYP7A1 regulates T-α-MCA and T-β-MCA synthesis, while CYP8B1 is required for TCA synthesis (Jiang et al., 2015; Chiang, 2009; Qi et al., 2015). In accordance with our results, previous studies have demonstrated that CYP7A1 mRNA levels are induced after XN treatment (Nozawa, 2005; Paraiso et al., 2020). This resulted in the increase of T-α-MCA and T-β-MCA, which repress intestinal FXR-FGF15 signaling to increase hepatic BA synthesis and prevent HFD-induced insulin resistance and obesity (Li et al., 2013; Sayin et al., 2013; Jiang et al., 2015; Gonzalez et al., 2016). Contrary to WT mice, XN decreased hydrophobic BAs such as DCA, CA and their taurine conjugates in FXRLiver−/− mice. Moreover, XN promoted genes that attenuate macrophage-mediated inflammation suggesting a shift toward detoxification in absence of hepatic FXR (Figure 7). Increased concentrations of hydrophobic BAs impairs phagocytosis activity of tissue-resident macrophages called Kupffer cells (KCs), induce neutrophil-mediated inflammation and alter hepatic T-cell immunity (Zhu et al., 2016). The selective depletion of liver-resident KCs restores hepatic insulin sensitivity and improves whole-body and hepatic fat accumulation (Neyrinck et al., 2009; Huang et al., 2010).
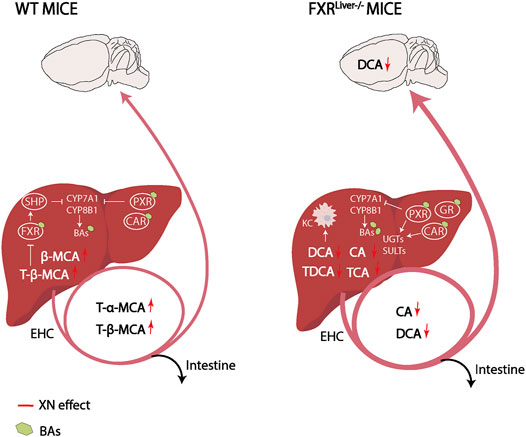
FIGURE 7. Working hypothesis on XN mechanism of control of BA synthesis and detoxification in WT and FXRLiver−/− mice. XN promotes BA synthesis in WT mice and increases FXR antagonists in the liver and plasma. This results in downstream expression of enzymes involved in BA synthesis, CYP7A1 and CYP8B1. Negative feedback on BA synthesis is exerted by BAs agonists of FXR, PXR and CAR. Compared to WT mice, FXRLiver−/− mice had increased BA synthesis, bigger BA pool sizes and increased passage of BAs through the BBB. In FXRLiver−/− mice, BAs such as CA and DCA impair hepatic Kupffer cells (KCs) activity, which XN might attenuate by decreasing CA and DCA concentrations. XN-mediated activation of PXR and CAR, and GR slows down de novo BA synthesis by inhibition of CYP7A1 and induces metabolizing enzymes to stimulate BA excretion. Abbreviations: EHC (Enterohepatic circulation), KC (Kupffer cell).
The major mechanisms underlying XN-mediated attenuation of liver damage in FXRLiver−/− mice are the reduction of BA concentrations and the mitigation of hepatic inflammation due the activation of NRs CAR/PXR/GR. FXR, PXR and CAR have complementary roles in the protection against BA toxicity (Guo et al., 2003). RNA sequencing revealed that several genes involved in metabolism of xenobiotics such as CYPs, UGTs, SULTs and GSTs were regulated by XN in FXRLiver−/− mice. Conjugation of hydrophilic groups by UGTs, SULTs, and GSTs increases the water solubility of BAs and xenobiotics to facilitate their renal elimination (Garcia et al., 2018). Hepatic xenobiotic-sensing receptors CAR and PXR mediate phase I and II BA metabolism by regulating CYP450s, UGTs, SULTs and GSTs that catalyze synthesis, oxidation, sulfonation and glucuronidation of BAs (Keppler, 2011; Garcia et al., 2018; Lv and Huang, 2020). Phase III clearance of BA is also regulated by FXR, PXR and CAR. Once BAs are transformed into more hydrophilic metabolites in the liver, they are pumped into the bile via efflux transporters BSEP and MRP2 as a route for fecal elimination (Wagner et al., 2005; Garcia et al., 2018). Moreover, GR enhances CAR/PXR-mediated transcriptional regulation of target genes such as UGT1A1 (Sugatani et al., 2005). CAR, PXR and GR are activated by unconjugated BAs such as LCA, CA, DCA and UDCA (Tanaka and Makino, 1992; Staudinger et al., 2001; Moore et al., 2002; Carazo et al., 2017). The strong correlations between these NRs and hepatic unconjugated BA concentrations suggest that BA metabolism in XN-treated FXRLiver−/− mice was primarily regulated by the activation of CAR, PXR and GR. Since the expression levels of these receptors were exclusively correlated with unconjugated BAs, it is conceivable that endogenous BAs might also play a role in the activation of these NRs in vivo. Consistent with our observations in FXRLiver−/− mice, taurine conjugated BA species such as TCA are increased in the liver of NASH patients (Lake et al., 2013). The decreased concentrations of hepatic TCA by XN also indicated a normalization of BA metabolism independent from FXR. Due to the affinity of TCA to FXR, it has been hypothesized that TCA is elevated as a compensatory effect to activate the receptor and normalize metabolism (Sheng et al., 2017). GR activation by XN in FXRLiver−/− mice is involved in the lipid-lowering effect of the flavonoid. Partial agonism of GR reverses NAFLD by preventing hepatic TG and cholesterol accumulation (Koorneef et al., 2018). Moreover, the anti-inflammatory effects of GR activation have a positive impact on hepatic lipid accumulation (Rando and Wahli, 2011; Scheschowitsch et al., 2017).
Our results support the hypothesis of a compensatory interaction between FXR, CAR and PXR. XN-mediated induction of CAR and PXR was not FXR-dependent. In the absence of FXR, the complementary regulation by CAR, PXR and GR might be involved in XN-mediated decrease of BAs concentrations. Chronic BA overload and prolonged activation of detoxification pathways may lead to the desensitization of BA-sensing receptors, which would contribute to the chronicity of BA-mediated damage. By improving the efficiency of phase II metabolism and phase III hepatic clearance of BAs, XN alleviated the sustained activation of detoxification pathways, improved BA signaling, lipid metabolism and relieved inflammation. Future studies assessing the pharmacodynamic activity of XN metabolites and quantifying glucuronidated and sulfated BA metabolites are necessary to evaluate XN effect more accurately.
Functional annotation clustering revealed that hepatic genes involved in inflammation and neoplastic processes were altered in FXRLiver−/− mice. Hydrophobic BAs are well-known for their cancer-promoting effects and promote carcinogenesis in several tumor models, including hepatocellular carcinoma, colon cancer, and breast cancer (Nagengast et al., 1995; Debruyne et al., 2001; Kim et al., 2006). Our research sheds new light on the chemopreventive potential of XN and highlights the potential of XN as adjuvant therapy in cancers associated with accumulation of BAs such as bile duct cancer and hepatocellular carcinoma.
BA synthesis and transport are tightly regulated by BA and xenobiotic-sensing NRs, which regulate genes in synthesis, metabolism and clearance of BAs and play a critical role in BA detoxification. Our current study extends previous research and shows the novel findings that 1) XN ameliorates HFD-induced inflammation and tissue damage in FXRLiver−/− mice; 2) XN improves HFD-induced dysfunctional lipid and BA metabolism via FXR-dependent and independent signaling, including the induction of CAR, PXR and GR. To the best of our knowledge, the potential of XN as adjuvant therapy in the management of cholestatic diseases has not been reported and merits further investigation.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA687670.
The animal study was reviewed and approved by the Institutional Animal Care Use Committee, Oregon State University, Corvallis, Oregon (USA), IACUC-2019-0001. The animal studies were carried out in accordance with the approved protocol.
IP, CK, JS designed the experiments. IP, CK, AM performed the experiments. IP, TT, GB. PK. JR. CK. JS. analyzed the data and performed statistical analyses. All the authors provided scientific support, wrote and reviewed the manuscript.
This work was funded by the Linus Pauling Institute, the National Institutes of Health (NIH grants S10RR022589, S10RR027878, and R01AT009168-04S1), the OSU College of Pharmacy and the OSU Foundation Buhler-Wang Research Fund.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank Grace L. Guo for providing the FXRLiver−/− mice and Cristobal Miranda for providing technical guidance.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2021.643857/full#supplementary-material.
Albacker, L. A., Karisola, P., Chang, Y. J., Umetsu, S. E., Zhou, M., Akbari, O., et al. (2010). TIM-4, a receptor for phosphatidylserine, controls adaptive immunity by regulating the removal of antigen-specific T cells. J. Immunol. 185, 6839–6849. doi:10.4049/jimmunol.1001360
Armstrong, L. E., and Guo, G. L. (2017). Role of FXR in liver inflammation during nonalcoholic steatohepatitis. Curr. Pharmacol. Rep. 3 (2), 92–100. doi:10.1007/s40495-017-0085-2
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodological) 57 (1), 289–300. doi:10.1111/j.2517-6161.1995.tb02031.x
Cajka, T., Smilowitz, J. T., and Fiehn, O. (2017). Validating quantitative untargeted lipidomics across nine liquid chromatography-high-resolution mass spectrometry platforms. Anal. Chem. 89 (22), 12360–12368. doi:10.1021/acs.analchem.7b03404
Cao, L. Q., Shao, Z. L., Liang, H. H., Zhang, D. W., Yang, X. W., Jiang, X. F., et al. (2015). Activation of peroxisome proliferator-activated receptor-γ (PPARγ) inhibits hepatoma cell growth via downregulation of SEPT2 expression. Cancer Lett. 359 (1), 127–135. doi:10.1016/j.canlet.2015.01.004
Carazo, A., Hyrsova, L., Dusek, J., Chodounska, H., Horvatova, A., Berka, K., et al. (2017). Acetylated deoxycholic (DCA) and cholic (CA) acids are potent ligands of pregnane X (PXR) receptor. Toxicol. Lett. 265, 86–96. doi:10.1016/j.toxlet.2016.11.013
Chang, C. N., Singh, A. J., Gross, M. K., and Kioussi, C. (2019). Requirement of Pitx2 for skeletal muscle homeostasis. Dev. Biol. 445 (1), 90–102. doi:10.1016/j.ydbio.2018.11.001
Chaurasia, B., and Summers, S. A. (2015). Ceramides - lipotoxic inducers of metabolic disorders. Trends Endocrinol. Metab. 26 (10), 538–550. doi:10.1016/j.tem.2015.07.006
Chawla, A., Repa, J. J., Evans, R. M., and Mangelsdorf, D. J. (2001). Nuclear receptors and lipid physiology: opening the X-files. Science 294 (5548), 1866. doi:10.1126/science.294.5548.1866
Chiang, J. Y. L. (2013). Bile acid metabolism and signaling. Compr. Physiol. 3 (3), 1191–1212. doi:10.1002/cphy.c120023
Chiang, J. Y. L. (2017). Bile acid metabolism and signaling in liver disease and therapy. Liver Res. 1 (1), 3–9. doi:10.1016/j.livres.2017.05.001
Chiang, J. Y. L. (2002). Bile acid regulation of gene expression: roles of nuclear hormone receptors. Endocr. Rev. 23 (4), 443–463. doi:10.1210/er.2000-0035
Chiang, J. Y. L. (2009). Bile acids: regulation of synthesis. J. lipid Res. 50 (10), 1955–1966. doi:10.1194/jlr.r900010-jlr200
Choi, J., Leonard, S. W., Kasper, K., McDougall, M., Stevens, J. F., Tanguay, R. L., et al. (2015). Novel function of vitamin E in regulation of zebrafish (Danio rerio) brain lysophospholipids discovered using lipidomics. J. Lipid Res. 56 (6), 1182–1190. doi:10.1194/jlr.m058941
Choi, S. S., Sicklick, J. K., Ma, Q., Yang, L., Huang, J., Qi, Y., et al. (2006). Sustained activation of Rac1 in hepatic stellate cells promotes liver injury and fibrosis in mice. Hepatology 44 (5), 1267–1277. doi:10.1002/hep.21375
Dai, W., Zhang, B., Jiang, X. M., Su, H., Li, J., Zhao, Y., et al. (2020). Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 368 (6497), 1331. doi:10.1126/science.abb4489
Debruyne, P. R., Bruyneel, E. A., Li, X., Zimber, A., Gespach, C., and Mareel, M. M. (2001). The role of bile acids in carcinogenesis. Mutat. Research/Fundamental Mol. Mech. Mutagenesis 480-481, 359–369. doi:10.1016/s0027-5107(01)00195-6
Dong, X.-Y., and Tang, S.-Q. (2010). Insulin-induced gene: a new regulator in lipid metabolism. Peptides 31 (11), 2145–2150. doi:10.1016/j.peptides.2010.07.020
Fiorucci, S., and Distrutti, E. (2015). Bile acid-activated receptors, intestinal microbiota, and the treatment of metabolic disorders. Trends Mol. Med. 21 (11), 702–714. doi:10.1016/j.molmed.2015.09.001
Forman, B. M., Goode, E., Chen, J., Oro, A. E., Bradley, D. J., Perlmann, T., et al. (1995). Identification of a nuclear receptor that is activated by farnesol metabolites. Cell 81 (5), 687–693. doi:10.1016/0092-8674(95)90530-8
Gao, J., and Xie, W. (2012). Targeting xenobiotic receptors PXR and CAR for metabolic diseases. Trends Pharmacol. Sci. 33 (10), 552–558. doi:10.1016/j.tips.2012.07.003
Gao, T., Qian, S., Shen, S., Zhang, X., Liu, J., Jia, W., et al. (2019). Reduction of mitochondrial 3-oxoacyl-ACP synthase (OXSM) by hyperglycemia is associated with deficiency of α-lipoic acid synthetic pathway in kidney of diabetic mice. Biochem. Biophysical Res. Commun. 512 (1), 106–111. doi:10.1016/j.bbrc.2019.02.155
Garcia, M., Thirouard, L., Sedès, L., Monrose, M., Holota, H., Caira, F., et al. (2018). Nuclear receptor metabolism of bile acids and xenobiotics: a coordinated detoxification System with impact on health and diseases. Ijms 19 (11), 3630. doi:10.3390/ijms19113630
Gonzalez, F. J., Jiang, C., and Patterson, A. D. (2016). An intestinal microbiota-farnesoid X receptor Axis modulates metabolic disease. Gastroenterology 151 (5), 845–859. doi:10.1053/j.gastro.2016.08.057
Goodwin, B., Jones, S. A., Price, R. R., Watson, M. A., McKee, D. D., Moore, L. B., et al. (2000). A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol. Cel 6 (3), 517–526. doi:10.1016/s1097-2765(00)00051-4
Guillemette, C. (2003). Pharmacogenomics of human UDP-glucuronosyltransferase enzymes. Pharmacogenomics J. 3 (3), 136–158. doi:10.1038/sj.tpj.6500171
Guo, G. L., Lambert, G., Negishi, M., Ward, J. M., Brewer, H. B., Kliewer, S. A., et al. (2003). Complementary roles of farnesoid X receptor, pregnane X receptor, and constitutive androstane receptor in protection against bile acid toxicity. J. Biol. Chem. 278 (46), 45062–45071. doi:10.1074/jbc.m307145200
Halilbasic, E., Baghdasaryan, A., and Trauner, M. (2013). Nuclear receptors as drug targets in cholestatic liver diseases. Clin. Liver Dis. 17 (2), 161–189. doi:10.1016/j.cld.2012.12.001
Hediger, M. A., Romero, M. F., Peng, J.-B., Rolfs, A., Takanaga, H., and Bruford, E. A. (2004). The ABCs of solute carriers: physiological, pathological and therapeutic implications of human membrane transport proteins. Pflugers Archiv Eur. J. Physiol. 447 (5), 465–468. doi:10.1007/s00424-003-1192-y
Huang, C., Wang, J., Hu, W., Wang, C., Lu, X., Tong, L., et al. (2016). Identification of functional farnesoid X receptors in brain neurons. FEBS Lett. 590 (18), 3233–3242. doi:10.1002/1873-3468.12373
Huang, F., Wang, T., Lan, Y., Yang, L., Pan, W., Zhu, Y., et al. (2015). Deletion of mouse FXR gene disturbs multiple neurotransmitter systems and alters neurobehavior. Front. Behav. Neurosci. 9, 70. doi:10.3389/fnbeh.2015.00070
Huang, W., Metlakunta, A., Dedousis, N., Zhang, P., Sipula, I., Dube, J. J., et al. (2010). Depletion of liver Kupffer cells prevents the development of diet-induced hepatic steatosis and insulin resistance. diabetes 59 (2), 347–357. doi:10.2337/db09-0016
Jiang, C., Xie, C., Lv, Y., Li, J., Krausz, K. W., Shi, J., et al. (2015). Intestine-selective farnesoid X receptor inhibition improves obesity-related metabolic dysfunction. Nat. Commun. 6, 10166. doi:10.1038/ncomms10166
Jumeau, C., Awad, F., Assrawi, E., Cobret, L., Duquesnoy, P., Giurgea, I., et al. (2019). Expression of SAA1, SAA2 and SAA4 genes in human primary monocytes and monocyte-derived macrophages. PLOS ONE 14 (5), e0217005. doi:10.1371/journal.pone.0217005
Kardol-Hoefnagel, T., van Logtestijn, S. A. L. M., and Otten, H. G. (2020). A review on the function and regulation of ARHGDIB/RhoGDI2 expression including the hypothetical role of ARHGDIB/RhoGDI2 autoantibodies in kidney transplantation. Transplant. Direct 6 (5), e548. doi:10.1097/txd.0000000000000993
Kartsoli, S., Kostara, C. E., Tsimihodimos, V., Bairaktari, E. T., and Christodoulou, D. K. (2020). Lipidomics in non-alcoholic fatty liver disease. Wjh 12 (8), 436–450. doi:10.4254/wjh.v12.i8.436
Keppler, D. (2011). “Multidrug resistance proteins (MRPs, ABCCs): importance for pathophysiology and drug therapy,” in Drug transporters. Editors M. F. Fromm, and R. B. Kim (Berlin, Heidelberg: Springer), 299–323. doi:10.1007/978-3-642-14541-4_8
Kim, D., Paggi, J. M., Park, C., Bennett, C., and Salzberg, S. L. (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37 (8), 907–915. doi:10.1038/s41587-019-0201-4
Kim, I., Ahn, S. H., Inagaki, T., Choi, M., Ito, S., Guo, G. L., et al. (2007). Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. J. Lipid Res. 48 (12), 2664–2672. doi:10.1194/jlr.m700330-jlr200
Kim, I., Morimura, K., Shah, Y., Yang, Q., Ward, J. M., and Gonzalez, F. J. (2006). Spontaneous hepatocarcinogenesis in farnesoid X receptor-null mice. Carcinogenesis 28 (5), 940–946. doi:10.1093/carcin/bgl249
Kirkwood, J. S., Legette, L. L., Miranda, C. L., Jiang, Y., and Stevens, J. F. (2013). A metabolomics-driven elucidation of the anti-obesity mechanisms of xanthohumol. J. Biol. Chem. 288 (26), 19000–19013. doi:10.1074/jbc.m112.445452
Kok, T., Hulzebos, C. V., Wolters, H., Havinga, R., Agellon, L. B., Stellaard, F., et al. (2003). Enterohepatic circulation of bile salts in farnesoid X receptor-deficient mice. J. Biol. Chem. 278 (43), 41930–41937. doi:10.1074/jbc.m306309200
Kong, B., Zhu, Y., Li, G., Williams, J. A., Buckley, K., Tawfik, O., et al. (2016). Mice with hepatocyte-specific FXR deficiency are resistant to spontaneous but susceptible to cholic acid-induced hepatocarcinogenesis. Am. J. Physiology-Gastrointestinal Liver Physiol. 310 (5), G295–G302. doi:10.1152/ajpgi.00134.2015
Koorneef, L. L., van den Heuvel, J. K., Kroon, J., Boon, M. R., Hoen, P. A. C., Hettne, K. M., et al. (2018). Selective glucocorticoid receptor modulation prevents and reverses nonalcoholic fatty liver disease in male mice. Endocrinology 159 (12), 3925–3936. doi:10.1210/en.2018-00671
Lake, A. D., Novak, P., Shipkova, P., Aranibar, N., Robertson, D., Reily, M. D., et al. (2013). Decreased hepatotoxic bile acid composition and altered synthesis in progressive human nonalcoholic fatty liver disease. Toxicol. Appl. Pharmacol. 268 (2), 132–140. doi:10.1016/j.taap.2013.01.022
Larter, C. Z., Chitturi, S., Heydet, D., and Farrell, G. C. (2010). A fresh look at NASH pathogenesis. Part 1: the metabolic movers. J. Gastroenterol. Hepatol. 25 (4), 672–690. doi:10.1111/j.1440-1746.2010.06253.x
Lee, F. Y., Lee, H., Hubbert, M. L., Edwards, P. A., and Zhang, Y. (2006). FXR, a multipurpose nuclear receptor. Trends Biochem. Sci. 31 (10), 572–580. doi:10.1016/j.tibs.2006.08.002
Lee, Y. J., and Kim, J. W. (2017). Monoacylglycerol O-acyltransferase 1 (MGAT1) localizes to the ER and lipid droplets promoting triacylglycerol synthesis. BMB Rep. 50 (7), 367–372. doi:10.5483/bmbrep.2017.50.7.036
Li, F., Ji, J. P., Xu, Y., and Liu, R. L. (2019). Identification a novel set of 6 differential expressed genes in prostate cancer that can potentially predict biochemical recurrence after curative surgery. Clin. Transl Oncol. 21 (8), 1067–1075. doi:10.1007/s12094-018-02029-z
Li, F., Jiang, C., Krausz, K. W., Li, Y., Albert, I., Hao, H., et al. (2013). Microbiome remodelling leads to inhibition of intestinal farnesoid X receptor signalling and decreased obesity. Nat. Commun. 4, 2384. doi:10.1038/ncomms3384
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25 (16), 2078–2079. doi:10.1093/bioinformatics/btp352
Li, Y., Sun, J. P., Wang, J., Lu, W. H., Xie, L. Y., Lv, J., et al. (2019). Expression of Vsig4 attenuates macrophage-mediated hepatic inflammation and fibrosis in high fat diet (HFD)-induced mice. Biochem. Biophysical Res. Commun. 516 (3), 858–865. doi:10.1016/j.bbrc.2019.06.045
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15 (12), 550. doi:10.1186/s13059-014-0550-8
Lu, H., Cui, J., Gunewardena, S., Yoo, B., Zhong, X.-b., and Klaassen, C. (2012). Hepatic ontogeny and tissue distribution of mRNAs of epigenetic modifiers in mice using RNA-sequencing. Epigenetics 7 (8), 914–929. doi:10.4161/epi.21113
Lv, C., and Huang, L. (2020). Xenobiotic receptors in mediating the effect of sepsis on drug metabolism. Acta Pharmaceutica Sinica. B 10 (1), 33–41. doi:10.1016/j.apsb.2019.12.003
Ma, K., Saha, P. K., Chan, L., and Moore, D. D. (2006). Farnesoid X receptor is essential for normal glucose homeostasis. J. Clin. Invest. 116 (4), 1102–1109. doi:10.1172/jci25604
Makishima, M., Okamoto, A. Y., Repa, J. J., Tu, H., Learned, R. M., Luk, A., et al. (1999). Identification of a nuclear receptor for bile acids. Science 284 (5418), 1362–1365. doi:10.1126/science.284.5418.1362
Matsumoto, Y., La Rose, J., Lim, M., Adissu, H. A., Law, N., Mao, X., et al. (2017). Ubiquitin ligase RNF146 coordinates bone dynamics and energy metabolism. J. Clin. Invest. 127 (7), 2612–2625. doi:10.1172/jci92233
McMillin, M., and DeMorrow, S. (2016). Effects of bile acids on neurological function and disease. FASEB j. 30 (11), 3658–3668. doi:10.1096/fj.201600275r
McMillin, M., Frampton, G., Quinn, M., Ashfaq, S., de los Santos, M., Grant, S., et al. (2016). Bile acid signaling is involved in the neurological decline in a murine model of acute liver failure. Am. J. Pathol. 186 (2), 312–323. doi:10.1016/j.ajpath.2015.10.005
Miranda, C. L., Elias, V. D., Hay, J. J., Choi, J., Reed, R. L., and Stevens, J. F. (2016). Xanthohumol improves dysfunctional glucose and lipid metabolism in diet-induced obese C57BL/6J mice. Arch. Biochem. Biophys. 599, 22–30. doi:10.1016/j.abb.2016.03.008
Miranda, C. L., Johnson, L. A., de Montgolfier, O., Elias, V. D., Ullrich, L. S., Hay, J. J., et al. (2018). Non-estrogenic xanthohumol derivatives mitigate insulin resistance and cognitive impairment in high-fat diet-induced obese mice. Sci. Rep. 8 (1), 613. doi:10.1038/s41598-017-18992-6
Mon, E. E., Wei, F. Y., Ahmad, R. N. R., Yamamoto, T., Moroishi, T., and Tomizawa, K. (2019). Regulation of mitochondrial iron homeostasis by sideroflexin 2. J. Physiol. Sci. 69 (2), 359–373. doi:10.1007/s12576-018-0652-2
Moore, L. B., Maglich, J. M., McKee, D. D., Wisely, B., Willson, T. M., Kliewer, S. A., et al. (2002). Pregnane X receptor (PXR), constitutive androstane receptor (CAR), and benzoate X receptor (BXR) define three pharmacologically distinct classes of nuclear receptors. Mol. Endocrinol. 16 (5), 977–986. doi:10.1210/mend.16.5.0828
Nagengast, F. M., Grubben, M. J. A. L., and van Munster, I. P. (1995). Role of bile acids in colorectal carcinogenesis. Eur. J. Cancer 31 (7), 1067–1070. doi:10.1016/0959-8049(95)00216-6
Neyrinck, A. M., Cani, P. D., Dewulf, E. M., De Backer, F., Bindels, L. B., and Delzenne, N. M. (2009). Critical role of Kupffer cells in the management of diet-induced diabetes and obesity. Biochem. biophysical Res. Commun. 385 (3), 351–356. doi:10.1016/j.bbrc.2009.05.070
Nozawa, H. (2005). Xanthohumol, the chalcone from beer hops (Humulus lupulus L.), is the ligand for farnesoid X receptor and ameliorates lipid and glucose metabolism in KK-A mice. Biochem. Biophysical Res. Commun. 336 (3), 754–761. doi:10.1016/j.bbrc.2005.08.159
Olive, P. L., Aquino-Parsons, C., MacPhail, S. H., Liao, S. Y., Raleigh, J. A., Lerman, M. I., et al. (2001). Carbonic anhydrase 9 as an endogenous marker for hypoxic cells in cervical cancer. Cancer Res. 61 (24), 8924–8929.
Paraiso, I. L., Plagmann, L. S., Yang, L., Zielke, R., Gombart, A. F., Maier, C. S., et al. (2019). Reductive metabolism of xanthohumol and 8-prenylnaringenin by the intestinal bacterium eubacterium ramulus. Mol. Nutr. Food Res. 63 (2), e1800923. doi:10.1002/mnfr.201970006
Paraiso, I. L., Revel, J. S., Choi, J., Miranda, C. L., Lak, P., Kioussi, C., et al. (2020). Targeting the liver‐brain Axis with hop‐derived flavonoids improves lipid metabolism and cognitive performance in mice. Mol. Nutr. Food Res. 64, 2000341. doi:10.1002/mnfr.202000341
Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T.-C., Mendell, J. T., and Salzberg, S. L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33 (3), 290–295. doi:10.1038/nbt.3122
Poitou, C., Viguerie, N., Cancello, R., De Matteis, R., Cinti, S., Stich, V., et al. (2005). Serum amyloid A: production by human white adipocyte and regulation by obesity and nutrition. Diabetologia 48 (3), 519–528. doi:10.1007/s00125-004-1654-6
Polycarpou-Schwarz, M., Groß, M., Mestdagh, P., Schott, J., Grund, S. E., Hildenbrand, C., et al. (2018). The cancer-associated microprotein CASIMO1 controls cell proliferation and interacts with squalene epoxidase modulating lipid droplet formation. Oncogene 37 (34), 4750–4768. doi:10.1038/s41388-018-0281-5
Porez, G., Prawitt, J., Gross, B., and Staels, B. (2012). Bile acid receptors as targets for the treatment of dyslipidemia and cardiovascular disease. J. lipid Res. 53 (9), 1723–1737. doi:10.1194/jlr.r024794
Qi, Y., Jiang, C., Cheng, J., Krausz, K. W., Li, T., Ferrell, J. M., et al. (2015). Bile acid signaling in lipid metabolism: metabolomic and lipidomic analysis of lipid and bile acid markers linked to anti-obesity and anti-diabetes in mice. Biochim. Biophys. Acta (Bba) - Mol. Cel Biol. Lipids 1851 (1), 19–29. doi:10.1016/j.bbalip.2014.04.008
Quinn, M., McMillin, M., Galindo, C., Frampton, G., Pae, H. Y., and DeMorrow, S. (2014). Bile acids permeabilize the blood brain barrier after bile duct ligation in rats via Rac1-dependent mechanisms. Dig. Liver Dis. 46 (6), 527–534. doi:10.1016/j.dld.2014.01.159
Rando, G., and Wahli, W. (2011). Sex differences in nuclear receptor-regulated liver metabolic pathways. Biochim. Biophys. Acta (Bba) - Mol. Basis Dis. 1812 (8), 964–973. doi:10.1016/j.bbadis.2010.12.023
Rao, S., Zaidi, S., Banerjee, J., Jogunoori, W., Sebastian, R., Mishra, B., et al. (2017). Transforming growth factor-β in liver cancer stem cells and regeneration. Hepatol. Commun. 1 (6), 477–493. doi:10.1002/hep4.1062
Ray, A., Hannink, M., and Ray, B. K. (1995). Concerted participation of NF-κB and C/EBP heteromer in lipopolysaccharide induction of serum amyloid A gene expression in liver. J. Biol. Chem. 270 (13), 7365–7374. doi:10.1074/jbc.270.13.7365
Sayin, S. I., Wahlström, A., Felin, J., Jäntti, S., Marschall, H. U., Bamberg, K., et al. (2013). Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cel Metab. 17 (2), 225–235. doi:10.1016/j.cmet.2013.01.003
Schaap, F. G., Trauner, M., and Jansen, P. L. M. (2014). Bile acid receptors as targets for drug development. Nat. Rev. Gastroenterol. Hepatol. 11 (1), 55–67. doi:10.1038/nrgastro.2013.151
Scheschowitsch, K., Leite, J. A., and Assreuy, J. (2017). New insights in glucocorticoid receptor signaling-more than just a ligand-binding receptor. Front. Endocrinol. 8, 16. doi:10.3389/fendo.2017.00016
Schmitt, J., Kong, B., Stieger, B., Tschopp, O., Schultze, S. M., Rau, M., et al. (2015). Protective effects of farnesoid X receptor (FXR) on hepatic lipid accumulation are mediated by hepatic FXR and independent of intestinal FGF15 signal. Liver Int. 35 (4), 1133–1144. doi:10.1111/liv.12456
Schubring, S. R., Fleischer, W., Lin, J. S., Haas, H. L., and Sergeeva, O. A. (2012). The bile steroid chenodeoxycholate is a potent antagonist at NMDA and GABAA receptors. Neurosci. Lett. 506 (2), 322–326. doi:10.1016/j.neulet.2011.11.036
Sheng, L., Jena, P. K., Liu, H. X., Kalanetra, K. M., Gonzalez, F. J., French, S. W., et al. (2017). Gender differences in bile acids and microbiota in relationship with gender dissimilarity in steatosis induced by diet and FXR inactivation. Scientific Rep. 7 (1), 1748. doi:10.1038/s41598-017-01576-9
Sinal, C. J., Tohkin, M., Miyata, M., Ward, J. M., Lambert, G., and Gonzalez, F. J. (2000). Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell 102 (6), 731–744. doi:10.1016/s0092-8674(00)00062-3
Singh, A. J., Chang, C. N., Ma, H. Y., Ramsey, S. A., Filtz, T. M., and Kioussi, C. (2018). FACS-Seq analysis of Pax3-derived cells identifies non-myogenic lineages in the embryonic forelimb. Scientific Rep. 8 (1), 7670. doi:10.1038/s41598-018-25998-1
Slotte, J. P., and Ramstedt, B. (2007). The functional role of sphingomyelin in cell membranes. Eur. J. Lipid Sci. Technol. 109 (10), 977–981. doi:10.1002/ejlt.200700024
Song, X., Vasilenko, A., Chen, Y., Valanejad, L., Verma, R., Yan, B., et al. (2014). Transcriptional dynamics of bile salt export pump during pregnancy: mechanisms and implications in intrahepatic cholestasis of pregnancy. Hepatology 60 (6), 1993–2007. doi:10.1002/hep.27171
Staels, B., and Kuipers, F. (2007). Bile acid sequestrants and the treatment of type 2 diabetes mellitus. Drugs 67 (10), 1383–1392. doi:10.2165/00003495-200767100-00001
Staudinger, J. L., Goodwin, B., Jones, S. A., Hawkins-Brown, D., MacKenzie, K. I., LaTour, A., et al. (2001). The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc. Natl. Acad. Sci. 98 (6), 3369. doi:10.1073/pnas.051551698
Stejskalova, L., and Pavek, P. (2011). The function of cytochrome P450 1A1 enzyme (CYP1A1) and aryl hydrocarbon receptor (AhR) in the placenta. Cpb 12 (5), 715–730. doi:10.2174/138920111795470994
Sugatani, J., Nishitani, S., Yamakawa, K., Yoshinari, K., Sueyoshi, T., Negishi, M., et al. (2005). Transcriptional regulation of human UGT1A1 gene expression: activated glucocorticoid receptor enhances constitutive androstane receptor/pregnane X receptor-mediated UDP-glucuronosyltransferase 1A1 regulation with glucocorticoid receptor-interacting protein 1. Mol. Pharmacol. 67 (3), 845–855. doi:10.1124/mol.104.007161
Summers, S. (2006). Ceramides in insulin resistance and lipotoxicity. Prog. Lipid Res. 45 (1), 42–72. doi:10.1016/j.plipres.2005.11.002
Sun, L., Cai, J., and Gonzalez, F. J. (2021). The role of farnesoid X receptor in metabolic diseases, and gastrointestinal and liver cancer. Nat. Rev. Gastroenterol. Hepatol. doi:10.1038/s41575-020-00404-2
Tanaka, H., and Makino, I. (1992). Ursodeoxycholic acid-dependent activation of the glucocorticoid receptor. Biochem. Biophysical Res. Commun. 188 (2), 942–948. doi:10.1016/0006-291x(92)91146-h
Tanaka, T., Yoshida, N., Kishimoto, T., and Akira, S. (1997). Defective adipocyte differentiation in mice lacking the C/EBPbeta and/or C/EBPdelta gene. EMBO J. 16 (24), 7432–7443. doi:10.1093/emboj/16.24.7432
Tang, F., Wang, B., Li, N., Wu, Y., Jia, J., Suo, T., et al. (2011). RNF185, a novel mitochondrial ubiquitin E3 ligase, regulates autophagy through interaction with BNIP1.6 (9), e24367. doi:10.1371/journal.pone.0024367
Togayachi, A., Kozono, Y., Kuno, A., Ohkura, T., Sato, T., Hirabayashi, J., et al. (2010). β3GnT2 (B3GNT2), a major polylactosamine synthase: analysis of B3gnt2-deficient mice. Methods Enzymol. 479, 185–204. doi:10.1016/s0076-6879(10)79011-x
Trauner, M., Claudel, T., Fickert, P., Moustafa, T., and Wagner, M. (2010). Bile acids as regulators of hepatic lipid and glucose metabolism. Dig. Dis. 28 (1), 220–224. doi:10.1159/000282091
Turek, F. W., Joshu, C., Kohsaka, A., Lin, E., Ivanova, G., McDearmon, E., et al. (2005). Obesity and metabolic syndrome in circadian clock mutant mice. Science 308 (5724), 1043. doi:10.1126/science.1108750
Uppal, H., Toma, D., Saini, S. P. S., Ren, S., Jones, T. J., and Xie, W. (2005). Combined loss of orphan receptors PXR and CAR heightens sensitivity to toxic bile acids in mice. Hepatology 41 (1), 168–176. doi:10.1002/hep.20512
Utsunomiya, T., Okamoto, M., Wakiyama, S., Hashimoto, M., Fukuzawa, K., Ezaki, T., et al. (2007). A specific gene-expression signature quantifies the degree of hepatic fibrosis in patients with chronic liver disease. Wjg 13 (3), 383–390. doi:10.3748/wjg.v13.i3.383
Vieira, E., Ruano, Eg., Figueroa, A. L., Aranda, G., Momblan, D., Carmona, F., et al. (2014). Altered clock gene expression in obese visceral adipose tissue is associated with metabolic syndrome. PloS one 9 (11), e111678. doi:10.1371/journal.pone.0111678
Wagner, M., Halilbasic, E., Marschall, H.-U., Zollner, G., Fickert, P., Langner, C., et al. (2005). CAR and PXR agonists stimulate hepatic bile acid and bilirubin detoxification and elimination pathways in mice. Hepatology 42 (2), 420–430. doi:10.1002/hep.20784
Waldeck-Weiermair, M., Jean-Quartier, C., Rost, R., Khan, M. J., Vishnu, N., Bondarenko, A. I., et al. (2011). Leucine zipper EF hand-containing transmembrane protein 1 (Letm1) and uncoupling proteins 2 and 3 (UCP2/3) contribute to two distinct mitochondrial Ca2+ uptake pathways. J. Biol. Chem. 286 (32), 28444–28455. doi:10.1074/jbc.m111.244517
Wang, S., Lai, K., Moy, F. J., Bhat, A., Hartman, H. B., and Evans, M. J. (2006). The nuclear hormone receptor farnesoid X receptor (FXR) is activated by androsterone. Endocrinology 147 (9), 4025–4033. doi:10.1210/en.2005-1485
Xiao, Y., Yan, W., Zhou, K., Cao, Y., and Cai, W. (2016). Glucocorticoid treatment alters systemic bile acid homeostasis by regulating the biosynthesis and transport of bile salts. Dig. Liver Dis. 48 (7), 771–779. doi:10.1016/j.dld.2016.03.022
Yang, J. Y., Della-Fera, M. A., Rayalam, S., and Baile, C. A. (2007). Effect of xanthohumol and isoxanthohumol on 3T3-L1 cell apoptosis and adipogenesis. Apoptosis 12 (11), 1953–1963. doi:10.1007/s10495-007-0130-4
Yang, L., Broderick, D., Campbell, Y., Gombart, A. F., Stevens, J. F., Jiang, Y., et al. (2016). Conformational modulation of the farnesoid X receptor by prenylflavonoids: insights from hydrogen deuterium exchange mass spectrometry (HDX-MS), fluorescence titration and molecular docking studies. Biochim. Biophys. Acta (Bba) - Proteins Proteomics 1864 (12), 1667–1677. doi:10.1016/j.bbapap.2016.08.019
Yiangou, L., Montandon, R., Modrzynska, K., Rosen, B., Bushell, W., Hale, C., et al. (2016). A stem cell strategy identifies glycophorin C as a major erythrocyte receptor for the rodent malaria parasite plasmodium berghei 11 (6), e0158238. doi:10.1371/journal.pone.0158238
Zhao, A., Yu, J., Lew, J. L., Huang, L., Wright, S. D., and Cui, J. (2004). Polyunsaturated fatty acids are FXR ligands and differentially regulate expression of FXR targets. DNA Cel Biol. 23 (8), 519–526. doi:10.1089/1044549041562267
Keywords: nonalcoholic fatty liver disease, farnesoid X receptor, bile acids, lipid metabolism, xanthohumol
Citation: Paraiso IL, Tran TQ, Magana AA, Kundu P, Choi J, Maier CS, Bobe G, Raber J, Kioussi C and Stevens JF (2021) Xanthohumol ameliorates Diet-Induced Liver Dysfunction via Farnesoid X Receptor-Dependent and Independent Signaling. Front. Pharmacol. 12:643857. doi: 10.3389/fphar.2021.643857
Received: 18 December 2020; Accepted: 22 March 2021;
Published: 20 April 2021.
Edited by:
M. Carmen González-Mas, University of Valencia, SpainReviewed by:
Saskia Van Mil, University Medical Center Utrecht, NetherlandsCopyright © 2021 Paraiso, Tran, Magana, Kundu, Choi, Maier, Bobe, Raber, Kioussi and Stevens. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chrissa Kioussi, Q2hyaXNzYS5raW91c3NpQG9yZWdvbnN0YXRlLmVkdQ==; Jan F. Stevens, ZnJlZC5zdGV2ZW5zQG9yZWdvbnN0YXRlLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.