 Satoshi Saito
Satoshi Saito Masashi Tanaka
Masashi Tanaka Noriko Satoh-Asahara4
Noriko Satoh-Asahara4 Roxana Octavia Carare
Roxana Octavia Carare Masafumi Ihara
Masafumi Ihara
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Pharmacol., 12 February 2021
Sec. Neuropharmacology
Volume 12 - 2021 | https://doi.org/10.3389/fphar.2021.643357
This article is part of the Research TopicCurrent Trends in Medicinal Plant Research and Neurodegenerative DisordersView all 16 articles
Cerebral amyloid angiopathy (CAA) is characterized by the accumulation of β-amyloid (Aβ) in the walls of cerebral vessels, leading to complications such as intracerebral hemorrhage, convexity subarachnoid hemorrhage and cerebral microinfarcts. Patients with CAA-related intracerebral hemorrhage are more likely to develop dementia and strokes. Several pathological investigations have demonstrated that more than 90% of Alzheimer’s disease patients have concomitant CAA, suggesting common pathogenic mechanisms. Potential causes of CAA include impaired Aβ clearance from the brain through the intramural periarterial drainage (IPAD) system. Conversely, CAA causes restriction of IPAD, limiting clearance. Early intervention in CAA could thus prevent Alzheimer’s disease progression. Growing evidence has suggested Taxifolin (dihydroquercetin) could be used as an effective therapy for CAA. Taxifolin is a plant flavonoid, widely available as a health supplement product, which has been demonstrated to exhibit anti-oxidative and anti-inflammatory effects, and provide protection against advanced glycation end products and mitochondrial damage. It has also been shown to facilitate disassembly, prevent oligomer formation and increase clearance of Aβ in a mouse model of CAA. Disturbed cerebrovascular reactivity and spatial reference memory impairment in CAA are completely prevented by Taxifolin treatment. These results highlight the need for clinical trials on the efficacy and safety of Taxifolin in patients with CAA
Cerebral amyloid angiopathy (CAA) refers to the abnormal accumulation of amyloid proteins in the walls of cerebral vasculature (Love et al., 2014; Saito et al., 2020b). Seven amyloid proteins have so far been reported in CAA including β-amyloid (Aβ), cystatin C, transthyretin, gelsolin, prion protein, ABri/ADan and immunoglobulin light chain amyloid (Yamada, 2015). The most common form is Aβ-type CAA, which is present in over 90% cases of sporadic, non-familial age-related Alzheimer’s disease (AD) (Saito and Ihara, 2016). The shared role of Aβ deposition in AD and CAA implies interaction between neurodegenerative and cerebrovascular processes (Saito et al., 2015). In this review, we discuss the pathophysiological basis of such interactions and how Taxifolin could act as a potential therapeutic agent for CAA.
CAA induces smooth muscle cell degeneration, vessel wall thickening, luminal narrowing and concentric wall splitting, resulting in varying degrees of intracerebral hemorrhage (ICH) (Love et al., 2014). Lobar, but not deep, ICH is associated with CAA (Saito et al., 2020a). Finger-like projections and subarachnoid hemorrhage extension of lobar ICH, together with the ApoE4 genotype, are reliable diagnostic markers for CAA (Rodrigues et al., 2018; Renard et al., 2019) (Figure 1A). Cerebral microbleeds (CMBs) are commonly observed in patients with CAA. Strictly lobar CMBs are highly specific for CAA, while CMBs in deep brain may indicate hypertensive arteriopathy (Greenberg and Charidimou, 2018; Jung et al., 2020). The estimated annual incidence of CAA-related ICH is 5.3 per 100,000 people in the United Kingdom and 5.8 per 100,000 people in Japan; however, the incidence of ICH not resulting from CAA, and mainly associated with hypertensive arteriopathy, is 2.5-fold higher in Japan than the United Kingdom (Yakushiji et al., 2020). Early diagnosis of CAA is clinically important for guiding prognosis and treatment decisions. A recent prospective study, with a median follow-up time of 2.5 years, showed progression to dementia in more than 25% of patients with CAA-related ICH, even if no dementia had presented after the acute phase of ICH (Xiong et al., 2019). ICH recurrence was more frequent in patients with CAA than other potential causes (Pasi et al., 2018).
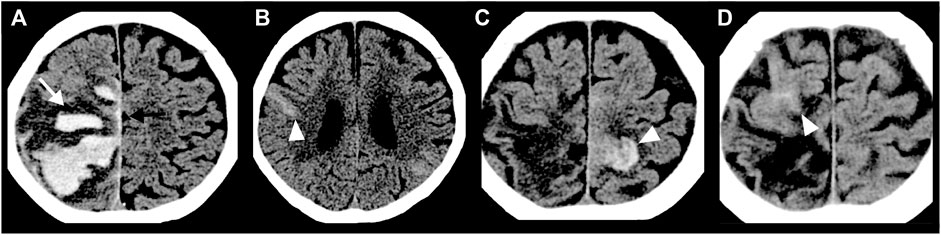
FIGURE 1. Head CT images of an 84-year-old woman with CAA showing repeated non-traumatic intracranial hemorrhage over four months. (A) ICH presenting finger-like projections (white arrow) with SAH extension (black arrow). (B) Acute convexity SAH (arrowhead) at two months later than A. (C) Acute convexity SAH (arrowhead) at three months later than A. (D) Acute convexity SAH (arrowhead) at four months later than A.
CAA is likely clinically underdiagnosed, due to the various clinical presentations outside of lobar ICH (Sakai et al., 2019; Fakan et al., 2020). Subarachnoid hemorrhage (SAH), resulting from bleeding into the subarachnoid space, known as “convexity SAH” in the acute phase (Figures 1B–D) and “superficial siderosis” in the chronic phase, can be induced by CAA (Saito et al., 2020a). Most CAA-induced bleeding into the subarachnoid space is limited without the involvement of the adjoining brain parenchyma (Kumar et al., 2010). Many convexity SAH are asymptomatic, though the risk of future intracranial hemorrhage and death of patients with CAA-convexity SAH is very high (Calviere et al., 2019; Saito et al., 2020a).
CAA also induces ischemic strokes consisting of both macro and microinfarcts (Saito et al., 2015; Saito and Ihara, 2016). Cerebral microinfarcts were originally defined as infarcts only visible by microscopy (Okamoto et al., 2012). However, technological advances in imaging modalities, such as ultra-high-field MRI, have enabled cerebral microinfarct observation (Ishikawa et al., 2020; Ter Telgte et al., 2020). AD and CAA patients frequently possess cortical cerebral microinfarcts near Aβ-laden vessels (Okamoto et al., 2009; Okamoto et al., 2012). Cerebral microinfarcts were replicated in CAA model mice following chronic cerebral hypoperfusion by bilateral common carotid artery stenosis (Okamoto et al., 2012). Impaired vasodilation due to vascular Aβ accumulation may contribute to cerebral microinfarct pathogenesis. AD and CAA patients have numerous, sometimes exceeding 1,000 (Westover et al., 2013), cerebral microinfarcts (van Rooden et al., 2014), which are likely to contribute to cognitive impairment (Saito et al., 2015).
CAA plays a pivotal role in the pathogenesis of dementia and is independently associated with cognitive decline (Boyle et al., 2015; Banerjee et al., 2018). Since there is little evidence for overproduction, the failure of clearance of Aβ peptides is a likely key factor in the pathological development of AD and CAA (Mawuenyega et al., 2010; Iturria-Medina et al., 2014). There is therefore increasing interest in developing agents that promote the safe elimination of Aβ from the brains of aged people (Saito and Ihara, 2014). The necessity of promoting Aβ clearance has been demonstrated in clinical trials using Aβ immunization. In AN-1792-vaccinated AD patients, the number and extent of parenchymal Aβ plaques diminished, while cerebrovascular Aβ accumulation and CAA increased (Nicoll et al., 2003; Patton et al., 2006). This finding was also observed in patients treated with solanezumab, a monoclonal anti-Aβ antibody (Roher et al., 2016). Antibody-solubilized Aβ appears to be removed from the cortex and re-deposited in the walls of the cerebral blood vessels via intramural periarterial clearance pathways (Carare et al., 2020).
Intramural periarterial drainage (IPAD), is a mechanism for the drainage of fluid and solutes from the brain along the walls of cerebral arteries (Tarasoff-Conway et al., 2015; Saito et al., 2019) (Figure 2). The central nervous system is devoid of lymph vessels. Instead, interstitial fluid and solutes within the extracellular matrix, including soluble Aβ, enter the IPAD pathways within the basement membranes of capillaries and continue to the basement membranes surrounding smooth muscle cells (SMCs) of the intracerebral and leptomeningeal arteries (Carare et al., 2020), which lead to the cervical lymph nodes (Piotrowska et al., 2020). This process has been examined in detail by several imaging methods including electron (Morris et al., 2016), confocal (Carare et al., 2008; MacGregor Sharp et al., 2020) and two-photon (Arbel-Ornath et al., 2013; Kim et al., 2020), microscopy. IPAD flow rapidly moves toward the leptomeningeal arteries where the deposition of Aβ is prominent in CAA (Keable et al., 2016). Aβ levels in the cerebrospinal fluid are decreased in CAA (Verbeek et al., 2009; van Etten et al., 2017), suggesting that Aβ transport is impeded in the IPAD pathways.
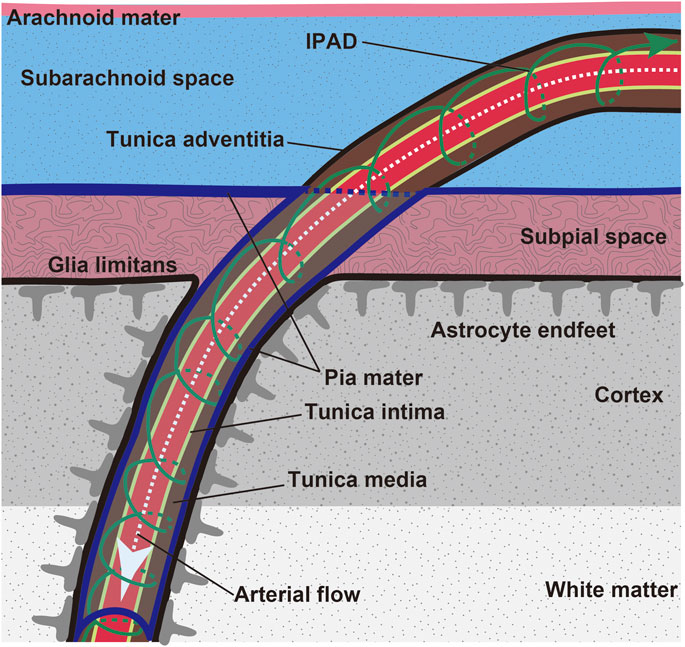
FIGURE 2. Scheme of leptomeninges and penetration of leptomeningeal artery into the brain parenchyma. Brain waste is cleared through the IPAD route (green arrow). Aβ is preferentially deposited in the tunica media of the leptomeningeal and cortical arteries in CAA.
Transcytosis is another vascular-mediated Aβ clearance system closely associated with AD and CAA. The brain parenchyma is separated from capillary lumen by the blood brain barrier (BBB), which prevents passive exchange between the brain and blood, allowing controlled carrier-mediated bidirectional transport of nutrients and waste products (Sweeney et al., 2018; Sweeney et al., 2019). Several molecules, such as low-density lipoprotein receptor related protein-1 (LRP-1), are thought to be involved in Aβ efflux from brain to blood (Shibata et al., 2000; Deane et al., 2004). Aβ binds to LRP-1 at the abluminal side of the vascular endothelium, either as a free peptide or bound to ApoE2 and ApoE3. Aβ-ApoE2 and Aβ-ApoE3 complexes are rapidly cleared across the BBB into the blood, while Aβ bound to ApoE4 interacts poorly with LRP-1 and is less efficiently removed from brain (Deane et al., 2008). Aβ deposition is frequently found in the cerebral capillaries in subjects possessing the ApoE4 allele (Thal et al., 2008).
CAA is not merely a consequence of impaired IPAD or transcytosis but also an important contributor to these processes (Kim et al., 2020; Rosas-Hernandez et al., 2020). CAA damages arterial structure and function, leading to worsening of cerebrovascular function and cognition. Therefore, early intervention strategies against CAA could be key to preventing progression of AD.
Development of novel treatments for CAA has proved challenging, with no pharmaceutical agents currently available (Smith and Markus, 2020). While more than 100 trials are in progress for AD (Cummings et al., 2020), to our knowledge, there are no ongoing clinical trials for agents targeting CAA (The U.S. National Institutes of Health, 2020), though a clinical trial of minocycline, a tetracycline derivative with anti-inflammatory properties is being planned in the Netherlands. Previous clinical trials on agents targeting CAA have reported mixed findings. Tramiprosate (3-amino-1-propanesulfonic acid), a low-molecular-weight ionic compound with preferential binding to soluble form of Aβ, has been shown to effectively block the deposition and facilitate the clearance of Aβ from the brains of transgenic mice expressing a double mutant (K670N/M671L and V717F) human APP gene (Gervais et al., 2007) but does not bind to insoluble fibrillar Aβ (Gervais et al., 2007). However, a phase-II trial of tramiprosate demonstrated no beneficial effects on CMBs despite causing no major safety issues (Greenberg et al., 2006; Gauthier et al., 2009; Aisen et al., 2011; Smith and Markus, 2020). In another trial, the anti-Aβ-monoclonal antibody, ponezumab was investigated in patients with CAA (Leurent et al., 2019). Ponezumab was well tolerated and plasma levels of Aβ40 were increased in the ponezumab-treated group, suggesting effective removal from the brain. However, ponezumab did not improve visual task-related functional MRI activation, a marker for cerebrovascular reactivity (Leurent et al., 2019).
Taxifolin is emerging as a viable safe therapeutic agent for the prevention and treatment of CAA. Taxifolin, also known as dihydroquercetin, is a bioactive flavanonol commonly found in grapes, citrus fruits, onions, green tea, olive oil, wine and several herbs such as milk thistle, French maritime bark, Douglas fir bark, and Smilacis Glabrae Rhizoma (Yang et al., 2016). Taxifolin is also widely used as a food additive and can be found in health supplement products including silymarin (Yang et al., 2016). Taxifolin has received increasing attention as a potential treatment for various diseases such as cancer, cardiovascular diseases, viral hepatitis, dyslipidemia and neurodegenerative disorders (Weidmann, 2012). It exhibits various pharmacological effects (Sunil and Xu, 2019), including anti-oxidant (Guo et al., 2015), advanced glycation end product suppressing (Harris et al., 2011), and mitochondrial protecting (Haraguchi et al., 1996) properties. Inhibition of Aβ fibril formation by Taxifolin has been demonstrated by using transmission electron microscopy imaging (Sato et al., 2013a; Sato et al., 2013b; Saito et al., 2017). Thioflavin T fluorescence assays have also shown that aggregated Aβ fibrils can be disaggregated by Taxifolin (Sato et al., 2013a), seemingly due to its chemical structure properties. Taxifolin is oxidized to form o-quinone on its B-ring. Since Lys16 and Lys28 are involved in the formation of β-sheets of Aβ, oxidized Taxifolin prevents the aggregation of Aβ as it covalently binds to Aβ at Lys16 and Lys28 residues (Sato et al., 2013b; Tanaka et al., 2019).
Aβ disassembly by Taxifolin treatment was confirmed in vivo. We administered Taxifolin or vehicle to a mouse model of CAA expressing the human APP gene with Swedish/Dutch/Iowa triple mutations (Saito et al., 2017). Filter trap assays showed a significant decrease in the concentration of Aβ oligomer in the soluble fraction of brain of Taxifolin-treated mice (Saito et al., 2017). However, the amount of total Aβ in the soluble fraction was similar between the Taxifolin-treated and vehicle-treated CAA mice, suggesting Taxifolin prevented the formation of Aβ oligomers from monomers (Saito et al., 2017). Furthermore, Taxifolin treatment prevented spatial memory deficits induced by injection of oligomeric Aβ into the hippocampus of wild-type mice (Wang et al., 2018). Decreased levels of Aβ oligomers by Taxifolin treatment were seen even in advanced stages of CAA (Saito et al., 2017). Higher blood Aβ levels were found in Taxifolin-treated CAA mice, suggesting facilitation of Aβ clearance from brain to blood. Taxifolin also fully restored both cerebrovascular reactivity and spatial reference memory in CAA mice (Saito et al., 2017). Higher expression of triggering receptor expressed on myeloid cell 2 (TREM2) is associated with the inflammation in the brain (Tanaka et al., 2020). We reported that Taxifolin suppressed inflammation, alleviating the accumulation of TREM2-expressing cells in the brains of CAA model mice (Inoue et al., 2019). Furthermore, Taxifolin suppressed glutamate levels and oxidative tissue damage, resulting in the amelioration of apoptotic cell death. In short, Taxifolin exhibits pleiotropic neuroprotective effects against CAA (Inoue et al., 2019).
In light of the promising preclinical data outlined in this review, we are currently preparing a clinical trial of Taxifolin in CAA patients. Nevertheless, several caveats on the use of this exciting potential treatment option should be addressed. Firstly, preclinical studies have not demonstrated that Taxifolin mitigates or prevents ICH, suggesting this may represent an inappropriate efficacy outcome in a clinical trial. Considering that Taxifolin restores cerebrovascular reactivity in mice (Saito et al., 2017), improvement of the cerebrovascular reserve capacity may be a more suitable in evaluating efficacy in CAA patients; indeed, impaired vascular reactivity is an early manifestation of CAA (van Opstal et al., 2017). However, the use of a surrogate, instead of a clinical, endpoint as a primary outcome of the efficacy of a drug in a clinical trial of a common disease such as CAA remains controversial (Broich et al., 2011). Secondly, the optimal dose and usage of Taxifolin in humans should be assessed. We reported the inhibition of Aβ oligomer formation in mice using a high dose of Taxifolin (Saito et al., 2017). In our experiments, 3% Taxifolin was administered orally to mice weighing approximately 30 g and consuming 3–5 g chow a day. It is still unknown whether smaller doses of Taxifolin initiate Aβ disassembly in vivo. Daily doses of 100 mg per day of Taxifolin are frequently administered as a health supplement product. However, whether high doses of Taxifolin are safe and tolerated in humans has yet been established and the elimination half-life period of Taxifolin is short at less than 1 h (Saito et al., 2017). Thirdly, as many as 191 metabolites of Taxifolin were reported in rats (Yang et al., 2016). Given that some of the metabolites could exhibit anti-CAA effects as well as Taxifolin, individual differences in the metabolism of Taxifolin may affect the response on Aβ disassembly in each patient, meaning the safety of such derivatives should be also evaluated. Finally, the identification of predictive indicators of favorable response of Taxifolin on CAA should be prioritized. Heterogeneity and multimorbidity are common in the elderly (Barnett et al., 2012), meaning the pharmacokinetic and pharmacodynamic effects of Taxifolin may vary in different individuals. However, the grouping of the patients based on the results of predictive indicators may facilitate more targeted, stratified or precision medicine treatments (Hampel et al., 2018).
Although numerous agents derived from natural plants now play pivotal roles in the prevention and treatment of various diseases, the importance of medicinal plant research may be underestimated in the field of AD and CAA. The beneficial effects demonstrated in preclinical studies suggest more promise for the clinical use of Taxifolin than other drug candidates for CAA. Future basic and clinical studies of this commonly used bioactive flavonoid could open new avenues for preemptive medicine for AD and CAA.
All the authors contributed to conceptualization and writing.
This work was supported by Grant-in-Aid for Japan Society for the Promotion of Science Fellows and Shimadzu Science Foundation (SS) as well as the Collaborative Research Project of Brain Research Institute, Niigata University (MI).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the late Yoko Okamoto for useful discussions, and Ahmad Khundakar at Newcastle University for editorial assistance.
Aisen, P. S., Gauthier, S., Ferris, S. H., Saumier, D., Haine, D., Garceau, D., et al. (2011). Tramiprosate in mild-to-moderate Alzheimer’s disease—a randomized, double-blind, placebo-controlled, multi-centre study (the Alphase Study). Arch. Med. Sci. 7 (1), 102–111. doi:10.5114/aoms.2011.20612
Arbel-Ornath, M., Hudry, E., Eikermann-Haerter, K., Hou, S., Gregory, J. L., Zhao, L., et al. (2013). Interstitial fluid drainage is impaired in ischemic stroke and Alzheimer’s disease mouse models. Acta Neuropathol. 126 (3), 353–364. doi:10.1007/s00401-013-1145-2
Banerjee, G., Wilson, D., Ambler, G., Osei-Bonsu Appiah, K., Shakeshaft, C., Lunawat, S., et al. (2018). Cognitive impairment before intracerebral hemorrhage is associated with cerebral amyloid angiopathy. Stroke 49 (1), 40–45. doi:10.1161/STROKEAHA.117.019409
Barnett, K., Mercer, S. W., Norbury, M., Watt, G., Wyke, S., and Guthrie, B. (2012). Epidemiology of multimorbidity and implications for health care, research, and medical education: a cross-sectional study. Lancet 380 (9836), 37–43. doi:10.1016/S0140-6736(12)60240-2
Boyle, P. A., Yu, L., Nag, S., Leurgans, S., Wilson, R. S., Bennett, D. A., et al. (2015). Cerebral amyloid angiopathy and cognitive outcomes in community-based older persons. Neurology 85 (22), 1930–1936. doi:10.1212/WNL.0000000000002175
Broich, K., Weiergräber, M., and Hampel, H. (2011). Biomarkers in clinical trials for neurodegenerative diseases: regulatory perspectives and requirements. Prog. Neurobiol. 95 (4), 498–500. doi:10.1016/j.pneurobio.2011.09.004
Calviere, L., Viguier, A., Patsoura, S., Rousseau, V., Albucher, J. F., Planton, M., et al. (2019). Risk of intracerebral hemorrhage and mortality after convexity subarachnoid hemorrhage in cerebral amyloid angiopathy. Stroke 50 (9), 2562–2564. doi:10.1161/STROKEAHA.119.026244
Carare, R. O., Bernardes-Silva, M., Newman, T. A., Page, A. M., Nicoll, J. A., Perry, V. H., et al. (2008). Solutes, but not cells, drain from the brain parenchyma along basement membranes of capillaries and arteries: significance for cerebral amyloid angiopathy and neuroimmunology. Neuropathol. Appl. Neurobiol. 34 (2), 131–144. doi:10.1111/j.1365-2990.2007.00926.x
Carare, R. O., Aldea, R., Agarwal, N., Bacskai, B. J., Bechman, I., Boche, D., et al. (2020). Clearance of interstitial fluid (ISF) and CSF (CLIC) group—part of vascular professional interest area (PIA). Alzheimers Dement (Amst) 12 (1), e12053. doi:10.1002/dad2.12053
Cummings, J., Lee, G., Ritter, A., Sabbagh, M., and Zhong, K. (2020). Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alzheimer’s Res. Ther. 6(1), e12050. doi:10.1002/trc2.12050
Deane, R., Sagare, A., Hamm, K., Parisi, M., Lane, S., Finn, M. B., et al. (2008). apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J. Clin. Invest. 118 (12), 4002–4013. doi:10.1172/JCI36663
Deane, R., Wu, Z., Sagare, A., Davis, J., Du Yan, S., Hamm, K., et al. (2004). LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron 43 (3), 333–344. doi:10.1016/j.neuron.2004.07.017
Fakan, B., Reisz, Z., Zadori, D., Vecsei, L., Klivenyi, P., and Szalardy, L. (2020). Predictors of localization, outcome, and etiology of spontaneous intracerebral hemorrhages: focus on cerebral amyloid angiopathy. J. Neural. Transm. 127 (6), 963–972. doi:10.1007/s00702-020-02174-2
Gauthier, S., Aisen, P. S., Ferris, S. H., Saumier, D., Duong, A., Haine, D., et al. (2009). Effect of tramiprosate in patients with mild-to-moderate Alzheimer’s disease: exploratory analyses of the MRI sub-group of the Alphase study. J. Nutr. Health Aging 13 (6), 550–557. doi:10.1007/s12603-009-0106-x
Gervais, F., Paquette, J., Morissette, C., Krzywkowski, P., Yu, M., Azzi, M., et al. (2007). Targeting soluble Abeta peptide with Tramiprosate for the treatment of brain amyloidosis. Neurobiol. Aging 28 (4), 537–547. doi:10.1016/j.neurobiolaging.2006.02.015
Greenberg, S. M., and Charidimou, A. (2018). Diagnosis of cerebral amyloid angiopathy: evolution of the boston criteria. Stroke 49 (2), 491–497. doi:10.1161/STROKEAHA.117.016990
Greenberg, S. M., Rosand, J., Schneider, A. T., Creed Pettigrew, L., Gandy, S. E., Rovner, B., et al. (2006). A phase 2 study of tramiprosate for cerebral amyloid angiopathy. Alzheimer Dis. Assoc. Disord. 20 (4), 269–274. doi:10.1097/01.wad.0000213845.28624.f4
Guo, H., Zhang, X., Cui, Y., Zhou, H., Xu, D., Shan, T., et al. (2015). Taxifolin protects against cardiac hypertrophy and fibrosis during biomechanical stress of pressure overload. Toxicol. Appl. Pharmacol. 287 (2), 168–177. doi:10.1016/j.taap.2015.06.002
Hampel, H., Vergallo, A., Giorgi, F. S., Kim, S. H., Depypere, H., Graziani, M., et al. (2018). Precision medicine and drug development in Alzheimer’s disease: the importance of sexual dimorphism and patient stratification. Front. Neuroendocrinol. 50, 31–51. doi:10.1016/j.yfrne.2018.06.001
Haraguchi, H., Mochida, Y., Sakai, S., Masuda, H., Tamura, Y., Mizutani, K., et al. (1996). Protection against oxidative damage by dihydroflavonols in Engelhardtia chrysolepis. Biosci. Biotechnol. Biochem. 60 (6), 945–948. doi:10.1271/bbb.60.945
Harris, C. S., Beaulieu, L. P., Fraser, M. H., McIntyre, K. L., Owen, P. L., Martineau, L. C., et al. (2011). Inhibition of advanced glycation end product formation by medicinal plant extracts correlates with phenolic metabolites and antioxidant activity. Planta Med. 77 (2), 196–204. doi:10.1055/s-0030-1250161
Inoue, T., Saito, S., Tanaka, M., Yamakage, H., Kusakabe, T., Shimatsu, A., et al. (2019). Pleiotropic neuroprotective effects of taxifolin in cerebral amyloid angiopathy. Proc. Natl. Acad. Sci. U. S. A. 116 (20), 10031–10038. doi:10.1073/pnas.1901659116
Ishikawa, H., Ii, Y., Shindo, A., Tabei, K. I., Umino, M., Ito, A. O., et al. (2020). Cortical microinfarcts detected by 3-tesla magnetic resonance imaging: differentiation between cerebral amyloid angiopathy and embolism. Stroke 51 (3), 1010–1013. doi:10.1161/STROKEAHA.119.028202
Iturria-Medina, Y., Sotero, R. C., Toussaint, P. J., Evans, A. C., and Alzheimer’s Disease Neuroimaging, I. (2014). Epidemic spreading model to characterize misfolded proteins propagation in aging and associated neurodegenerative disorders. PLoS Comput. Biol. 10 (11), e1003956. doi:10.1371/journal.pcbi.1003956
Jung, Y. H., Jang, H., Park, S. B., Choe, Y. S., Park, Y., Kang, S. H., et al. (2020). Strictly lobar microbleeds reflect amyloid angiopathy regardless of cerebral and cerebellar compartments. Stroke 51 (12), 3600–3607. doi:10.1161/STROKEAHA.119.028487
Keable, A., Fenna, K., Yuen, H. M., Johnston, D. A., Smyth, N. R., Smith, C., et al. (2016). Deposition of amyloid β in the walls of human leptomeningeal arteries in relation to perivascular drainage pathways in cerebral amyloid angiopathy. Biochim. Biophys. Acta 1862 (5), 1037–1046. doi:10.1016/j.bbadis.2015.08.024
Kim, S. H., Ahn, J. H., Yang, H., Lee, P., Koh, G. Y., and Jeong, Y. (2020). Cerebral amyloid angiopathy aggravates perivascular clearance impairment in an Alzheimer's disease mouse model. Acta Neuropathol Commun 8 (1), 181. doi:10.1186/s40478-020-01042-0
Kumar, S., Goddeau, R. P., Selim, M. H., Thomas, A., Schlaug, G., Alhazzani, A., et al. (2010). Atraumatic convexal subarachnoid hemorrhage: clinical presentation, imaging patterns, and etiologies. Neurology 74 (11), 893–899. doi:10.1212/WNL.0b013e3181d55efa
Leurent, C., Goodman, J. A., Zhang, Y., He, P., Polimeni, J. R., Gurol, M. E., et al. (2019). Immunotherapy with ponezumab for probable cerebral amyloid angiopathy. Ann Clin Transl Neurol 6 (4), 795–806. doi:10.1002/acn3.761
Love, S., Chalmers, K., Ince, P., Esiri, M., Attems, J., Jellinger, K., et al. (2014). Development, appraisal, validation and implementation of a consensus protocol for the assessment of cerebral amyloid angiopathy in post-mortem brain tissue. Am J Neurodegener. Dis. 3 (1), 19–32.
MacGregor Sharp, M., Saito, S., Keable, A., Gatherer, M., Aldea, R., Agarwal, N., et al. (2020). Demonstrating a reduced capacity for removal of fluid from cerebral white matter and hypoxia in areas of white matter hyperintensity associated with age and dementia. Acta Neuropathol Commun 8 (1), 131. doi:10.1186/s40478-020-01009-1
Mawuenyega, K. G., Sigurdson, W., Ovod, V., Munsell, L., Kasten, T., Morris, J. C., et al. (2010). Decreased clearance of CNS beta-amyloid in Alzheimer's disease. Science 330 (6012), 1774. doi:10.1126/science.1197623
Morris, A. W., Sharp, M. M., Albargothy, N. J., Fernandes, R., Hawkes, C. A., Verma, A., et al. (2016). Vascular basement membranes as pathways for the passage of fluid into and out of the brain. Acta Neuropathol. 131 (5), 725–736. doi:10.1007/s00401-016-1555-z
Nicoll, J. A., Wilkinson, D., Holmes, C., Steart, P., Markham, H., and Weller, R. O. (2003). Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat. Med. 9 (4), 448–452. doi:10.1038/nm840
Okamoto, Y., Ihara, M., Fujita, Y., Ito, H., Takahashi, R., and Tomimoto, H. (2009). Cortical microinfarcts in Alzheimer’s disease and subcortical vascular dementia. Neuroreport 20 (11), 990–996. doi:10.1097/WNR.0b013e32832d2e6a
Okamoto, Y., Yamamoto, T., Kalaria, R. N., Senzaki, H., Maki, T., Hase, Y., et al. (2012). Cerebral hypoperfusion accelerates cerebral amyloid angiopathy and promotes cortical microinfarcts. Acta Neuropathol. 123(3), 381–394. doi:10.1007/s00401-011-0925-9
Pasi, M., Charidimou, A., Boulouis, G., Auriel, E., Ayres, A., Schwab, K. M., et al. (2018). Mixed-location cerebral hemorrhage/microbleeds: underlying microangiopathy and recurrence risk. Neurology 90(2), e119–e126. doi:10.1212/WNL.0000000000004797
Patton, R. L., Kalback, W. M., Esh, C. L., Kokjohn, T. A., Van Vickle, G. D., Luehrs, D. C., et al. (2006). Amyloid-beta peptide remnants in AN-1792-immunized Alzheimer’s disease patients: a biochemical analysis. Am. J. Pathol. 169 (3), 1048–1063. doi:10.2353/ajpath.2006.060269
Piotrowska, A., Winter, K., Carare, R. O., and Bechmann, I. (2020). Vital functions contribute to the spread of extracellular fluids in the brain: comparison between life and death. Front. Aging Neurosci. 12, 15. doi:10.3389/fnagi.2020.00015
Renard, D., Parvu, T., and Thouvenot, E. (2019). Finger-like projections in lobar haemorrhage on early magnetic resonance imaging is associated with probable cerebral amyloid angiopathy. Cerebrovasc. Dis. 47(3–4), 121–126. doi:10.1159/000499032
Rodrigues, M. A., Samarasekera, N., Lerpiniere, C., Humphreys, C., McCarron, M. O., White, P. M., et al. (2018). The Edinburgh CT and genetic diagnostic criteria for lobar intracerebral haemorrhage associated with cerebral amyloid angiopathy: model development and diagnostic test accuracy study. Lancet Neurol. 17 (3), 232–240. doi:10.1016/S1474-4422(18)30006-1
Roher, A. E., Maarouf, C. L., Kokjohn, T. A., Belden, C., Serrano, G., Sabbagh, M. S., et al. (2016). Chemical and neuropathological analyses of an Alzheimer’s disease patient treated with solanezumab. Am J Neurodegener Dis. 5 (4), 158–170.
Rosas-Hernandez, H., Cuevas, E., Raymick, J. B., Robinson, B. L., and Sarkar, S. (2020). Impaired amyloid beta clearance and brain microvascular dysfunction are present in the tg-SwDI mouse model of alzheimer’s disease. Neuroscience 440, 48–55. doi:10.1016/j.neuroscience.2020.05.024
Saito, S., and Ihara, M. (2016). Interaction between cerebrovascular disease and Alzheimer pathology. Curr. Opin. Psychiatr. 29 (2), 168–173. doi:10.1097/YCO.0000000000000239
Saito, S., and Ihara, M. (2014). New therapeutic approaches for Alzheimer’s disease and cerebral amyloid angiopathy. Front. Aging Neurosci. 6, 290. doi:10.3389/fnagi.2014.00290
Saito, S., Ikeda, Y., Ando, D., Carare, R. O., Ishibashi-Ueda, H., and Ihara, M. (2020a). Cerebral amyloid angiopathy presenting as massive subarachnoid haemorrhage: a case study and review of literature. Front. Aging Neurosci. 12, 538456. doi:10.3389/fnagi.2020.538456
Saito, S., McLaurin, J., and Carare, R. O. (2020b). Editorial: intramural vascular cells: key therapeutic targets for vascular cognitive impairment. Front. Aging Neurosci. 12, 615780. doi:10.3389/fnagi.2020.615780
Saito, S., Yamamoto, Y., and Ihara, M. (2019). Development of a multicomponent intervention to prevent alzheimer’s disease. Front. Neurol. 10, 490. doi:10.3389/fneur.2019.00490
Saito, S., Yamamoto, Y., and Ihara, M. (2015). Mild cognitive impairment: at the crossroad of neurodegeneration and vascular dysfunction. Curr. Alzheimer Res. 12 (6), 507–512. doi:10.2174/1567205012666150530202508.
Saito, S., Yamamoto, Y., Maki, T., Hattori, Y., Ito, H., Mizuno, K., et al. (2017). Taxifolin inhibits amyloid-β oligomer formation and fully restores vascular integrity and memory in cerebral amyloid angiopathy. Acta Neuropathol. Commun. 5 (1), 26. doi:10.1186/s40478-017-0429-5
Sakai, K., Ueda, M., Fukushima, W., Tamaoka, A., Shoji, M., Ando, Y., et al. (2019). Nationwide survey on cerebral amyloid angiopathy in Japan. Eur. J. Neurol. 26 (12), 1487–1493. doi:10.1111/ene.14031
Sato, M., Murakami, K., Uno, M., Ikubo, H., Nakagawa, Y., Katayama, S., et al. (2013a). Structure-activity relationship for (+)-taxifolin isolated from silymarin as an inhibitor of amyloid β aggregation. Biosci. Biotechnol. Biochem. 77 (5), 1100–1103. doi:10.1271/bbb.120925
Sato, M., Murakami, K., Uno, M., Nakagawa, Y., Katayama, S., Akagi, K., et al. (2013b). Site-specific inhibitory mechanism for amyloid β42 aggregation by catechol-type flavonoids targeting the Lys residues. J. Biol. Chem. 288 (32), 23212–23224. doi:10.1074/jbc.M113.464222
Shibata, M., Yamada, S., Kumar, S. R., Calero, M., Bading, J., Frangione, B., et al. (2000). Clearance of Alzheimer’s amyloid-ss(1-40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J. Clin. Invest. 106 (12), 1489–1499. doi:10.1172/JCI10498
Smith, E. E., and Markus, H. S. (2020). New treatment approaches to modify the course of cerebral small vessel diseases. Stroke 51 (1), 38–46. doi:10.1161/STROKEAHA.119.024150
Sunil, C., and Xu, B. (2019). An insight into the health-promoting effects of taxifolin (dihydroquercetin). Phytochemistry 166, 112066. doi:10.1016/j.phytochem.2019.112066
Sweeney, M. D., Kisler, K., Montagne, A., Toga, A. W., and Zlokovic, B. V. (2018). The role of brain vasculature in neurodegenerative disorders. Nat. Neurosci. 21 (10), 1318–1331. doi:10.1038/s41593-018-0234-x
Sweeney, M. D., Zhao, Z., Montagne, A., Nelson, A. R., and Zlokovic, B. V. (2019). Blood-brain barrier: from physiology to disease and back. Physiol. Rev. 99 (1), 21–78. doi:10.1152/physrev.00050.2017
Tanaka, M., Saito, S., Inoue, T., Satoh-Asahara, N., and Ihara, M. (2019). Novel therapeutic potentials of taxifolin for amyloid-β-associated neurodegenerative diseases and other diseases: recent advances and future perspectives. Int. J. Mol. Sci. 20 (9). doi:10.3390/ijms20092139
Tanaka, M., Saito, S., Inoue, T., Satoh-Asahara, N., and Ihara, M. (2020). Potential therapeutic approaches for cerebral amyloid angiopathy and alzheimer’s disease. Int. J. Mol. Sci. 21 (6). doi:10.3390/ijms21061992
Tarasoff-Conway, J. M., Carare, R. O., Osorio, R. S., Glodzik, L., Butler, T., Fieremans, E., et al. (2015). Clearance systems in the brain-implications for Alzheimer disease. Nat. Rev. Neurol. 11(8), 457–470. doi:10.1038/nrneurol.2015.119
Ter Telgte, A., Scherlek, A. A., Reijmer, Y. D., van der Kouwe, A. J., van Harten, T., Duering, M., et al. (2020). Histopathology of diffusion-weighted imaging-positive lesions in cerebral amyloid angiopathy. Acta Neuropathol. 139 (5), 799–812. doi:10.1007/s00401-020-02140-y
Thal, D. R., Griffin, W. S., de Vos, R. A., and Ghebremedhin, E. (2008). Cerebral amyloid angiopathy and its relationship to Alzheimer's disease. Acta Neuropathol. 115(6), 599–609. doi:10.1007/s00401-008-0366-2
The U.S. National Institutes of Health (2020). ClinicalTrials.gov. Available: https://clinicaltrials.gov/ (Accessed November 1, 2020).
van Etten, E. S., Verbeek, M. M., van der Grond, J., Zielman, R., van Rooden, S., van Zwet, E. W., et al. (2017). β-Amyloid in CSF: biomarker for preclinical cerebral amyloid angiopathy. Neurology 88 (2), 169–176. doi:10.1212/WNL.0000000000003486
van Opstal, A. M., van Rooden, S., van Harten, T., Ghariq, E., Labadie, G., Fotiadis, P., et al. (2017). Cerebrovascular function in presymptomatic and symptomatic individuals with hereditary cerebral amyloid angiopathy: a case-control study. Lancet Neurol. 16 (2), 115–122. doi:10.1016/S1474-4422(16)30346-5
van Rooden, S., Goos, J. D., van Opstal, A. M., Versluis, M. J., Webb, A. G., Blauw, G. J., et al. (2014). Increased number of microinfarcts in Alzheimer disease at 7-T MR imaging. Radiology 270 (1), 205–211. doi:10.1148/radiol.13130743
Verbeek, M. M., Kremer, B. P., Rikkert, M. O., Van Domburg, P. H., Skehan, M. E., and Greenberg, S. M. (2009). Cerebrospinal fluid amyloid beta(40) is decreased in cerebral amyloid angiopathy. Ann. Neurol. 66 (2), 245–249. doi:10.1002/ana.21694
Wang, Y., Wang, Q., Bao, X., Ding, Y., Shentu, J., Cui, W., et al. (2018). Taxifolin prevents β-amyloid-induced impairments of synaptic formation and deficits of memory via the inhibition of cytosolic phospholipase A2/prostaglandin E2 content. Metab. Brain Dis. 33 (4), 1069–1079. doi:10.1007/s11011-018-0207-5
Weidmann, A. E. (2012). Dihydroquercetin: more than just an impurity?. Eur. J. Pharmacol. 684(1-3), 19–26. doi:10.1016/j.ejphar.2012.03.035
Westover, M. B., Bianchi, M. T., Yang, C., Schneider, J. A., and Greenberg, S. M. (2013). Estimating cerebral microinfarct burden from autopsy samples. Neurology 80 (15), 1365–1369. doi:10.1212/WNL.0b013e31828c2f52
Xiong, L., Charidimou, A., Pasi, M., Boulouis, G., Pongpitakmetha, T., Schirmer, M. D., et al. (2019). Predictors for late post-intracerebral hemorrhage dementia in patients with probable cerebral amyloid angiopathy. J. Alzheimers Dis. 71 (2), 435–442. doi:10.3233/JAD-190346
Yakushiji, Y., Tanaka, J., Wilson, D., Charidimou, A., Noguchi, T., Kawashima, M., et al. (2020). Proportion of intracerebral haemorrhage due to cerebral amyloid angiopathy in the East and West: comparison between single hospital centres in Japan and the United Kingdom. J. Neurol. Sci. 416, 117037. doi:10.1016/j.jns.2020.117037
Yamada, M. (2015). Cerebral amyloid angiopathy: emerging concepts. J Stroke 17(1), 17–30. doi:10.5853/jos.2015.17.1.17
Keywords: IPAD, clinical trial, treatment, Alzheimer’s disease, cerebral amyloid angiopathy, Taxifolin
Citation: Saito S, Tanaka M, Satoh-Asahara N, Carare RO and Ihara M (2021) Taxifolin: A Potential Therapeutic Agent for Cerebral Amyloid Angiopathy. Front. Pharmacol. 12:643357. doi: 10.3389/fphar.2021.643357
Received: 18 December 2020; Accepted: 15 January 2021;
Published: 12 February 2021.
Edited by:
Tahir Ali, University of Calgary, CanadaReviewed by:
Gwangho Yoon, Chonnam National University Medical School, South KoreaCopyright © 2021 Saito, Tanaka, Satoh-Asahara, Carare and Ihara. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Satoshi Saito, c2FpdG91LnNhdG9zaGkuNDNtQGt5b3RvLXUuanA=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.