 Kenji Rowel Q. Lim
Kenji Rowel Q. Lim Toshifumi Yokota
Toshifumi Yokota- 1Department of Medical Genetics, Faculty of Medicine and Dentistry, University of Alberta, Edmonton, AB, Canada
- 2The Friends of Garrett Cumming Research and Muscular Dystrophy Canada, HM Toupin Neurological Science Research Chair, Edmonton, AB, Canada
Facioscapulohumeral muscular dystrophy (FSHD) is an autosomal dominant disorder characterized by progressive, asymmetric muscle weakness at the face, shoulders, and upper limbs, which spreads to the lower body with age. It is the third most common inherited muscular disorder worldwide. Around 20% of patients are wheelchair-bound, and some present with extramuscular manifestations. FSHD is caused by aberrant expression of the double homeobox protein 4 (DUX4) gene in muscle. DUX4 codes for a transcription factor which, in skeletal muscle, dysregulates numerous signaling activities that culminate in cytotoxicity. Potential treatments for FSHD therefore aim to reduce the expression of DUX4 or the activity of its toxic protein product. In this article, we review how genetic approaches such as those based on oligonucleotide and genome editing technologies have been developed to achieve these goals. We also outline the challenges these therapies are facing on the road to translation, and discuss possible solutions and future directions
Introduction
Facioscapulohumeral muscular dystrophy (FSHD) is an autosomal dominant disorder that affects 1 in 8,000–22,000 people in the world and is the third most common inherited muscular dystrophy (Wang and Tawil, 2016). It presents as a progressive, distinctively asymmetric weakening of muscles in the face, shoulders, and upper limbs. Muscles in other regions may become affected with age; around 20% of patients become wheelchair-bound (Richards et al., 2012). Extramuscular symptoms are uncommon, with a few patients experiencing restrictive lung disease, cardiac conduction abnormalities, hearing loss, or retinal vasculopathy (Fitzsimons et al., 1987; Padberg et al., 1995; Laforêt et al., 1998; Lutz et al., 2013; Scully et al., 2014; Lim et al., 2020c). Age of onset and disease severity are both widely variable in FSHD (Tawil et al., 2014; Wang and Tawil, 2016). Intriguingly though, 4–21% of patients who manifest symptoms before the age of five almost all follow a more severe and rapid course of the disease (Klinge et al., 2006; Goselink et al., 2017). There is no available cure for FSHD. Patients are currently managed for their symptoms at best.
While the genetic mechanisms leading to FSHD are diverse and complex, these all result in aberrant expression of the double homeobox protein 4 (DUX4) gene in skeletal muscle. DUX4 has roles in early embryonic development, where it appears to be essential for zygotic gene activation (De Iaco et al., 2017; Hendrickson et al., 2017). Under healthy conditions, DUX4 is epigenetically silenced after the 4-cell stage in humans and kept as such in all developed tissues but the testis (Snider et al., 2010) and thymus (Das and Chadwick, 2016). This repression is compromised in patients with FSHD, allowing for the synthesis of the DUX4 transcription factor whose activities in skeletal muscle induce potent cytotoxicity by dysregulating pathways involved in cell death, oxidative stress, and muscle development, among others (Dixit et al., 2007; Lim et al., 2020c).
Various approaches are being explored to treat FSHD. Pharmacological treatments have been evaluated mostly with the aim of improving muscular symptoms, and include the use of prednisone, β2 receptor agonists, myostatin inhibitors, and antioxidants, among others. Unfortunately, these generally offered little to no therapeutic benefit based on results from clinical trials (Hamel and Tawil, 2018; Le Gall et al., 2020). Intramuscular transplantation of myoblasts or mesoangioblasts (perivascular myogenic stem cells) from unaffected muscles of FSHD patients into immunodeficient mice revealed that these could integrate with recipient muscle fibers fairly well (Vilquin et al., 2005; Morosetti et al., 2011). However, follow-up studies examining the benefits of such cell-based therapies on FSHD muscle pathology or function are currently unavailable and so their potential for treating FSHD remains uncertain.
In response to developing a more targeted form of treatment, reducing muscle-specific DUX4 expression and DUX4-mediated toxicity have become attractive goals for FSHD therapy (Bao et al., 2016; Bouwman et al., 2020; Cohen et al., 2020). Indeed, a number of genetic methods have been employed to achieve one or both of these, including oligonucleotide-based strategies to knockdown DUX4 transcript levels or reduce DUX4 protein activity, and genome editing to correct FSHD-associated mutations. The pre-clinical development of these strategies and others has shown much promise, and identifies possible candidates for clinical trials. Compared to pharmaceutical and cell-based interventions, genetic treatments target the root cause of the disease (i.e., DUX4) and are thus expected to lead to more effective or far-reaching therapeutic effects. In this article, we review the various genetic approaches that have been developed for FSHD therapy, discuss the challenges they may be facing on their way to the clinic, and offer some potential solutions as well as directions for future research.
DUX4 Expression and FSHD
Much of the complexity associated with FSHD genetics comes from the curious location of DUX4 in the genome. The DUX4 gene is part of the D4Z4 macrosatellite repeat array at chromosome 4q35, which is typically 11–100 repeats long in healthy individuals (Gabriëls et al., 1999; Lemmers et al., 2010). There is a homologous D4Z4 repeat array at chromosome 10q26, but mutations in this region have not been linked to FSHD (Bakker et al., 1995; Deidda et al., 1995; Lemmers et al., 2010). Each D4Z4 repeat contains the first two exons of DUX4, with the entire open reading frame of the gene in exon 1 (Gabriëls et al., 1999) (Figure 1). DUX4 has other exons downstream of the array; the full-length isoform that contributes to FSHD pathology ends at exon 3 (Snider et al., 2010; Himeda and Jones, 2019). Only exons from the last D4Z4 repeat contribute to the DUX4 mRNA, and a polyadenylation signal (PAS) at exon 3 is required to stabilize the pathogenic DUX4 transcript, a feature that is only present in the disease-permissive 4qA haplotype (Lemmers et al., 2002; Lemmers et al., 2004; Lemmers et al., 2010). Finally, the 4q35 D4Z4 repeat array is normally hypermethylated, which keeps the DUX4 gene repressed in most adult tissues (Hewitt et al., 1994). Two mechanisms activate DUX4 expression in FSHD: D4Z4 repeat array contraction, and mutations in genes coding for epigenetic regulators (Figure 1). These cause approximately 95% (FSHD1) and 5% (FSHD2) of cases, respectively (Wang and Tawil, 2016). Despite vast differences in their underlying genetics, FSHD1 and FSHD2 are clinically indistinguishable, implying that aberrant DUX4 expression is the key genetic event leading to FSHD pathogenesis.
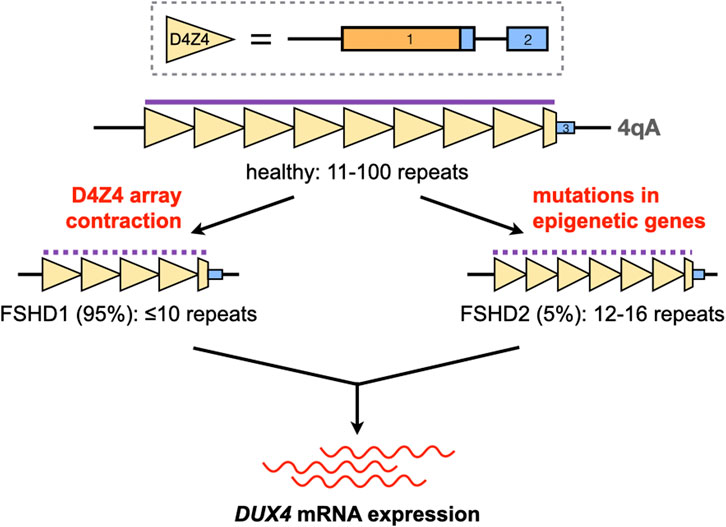
FIGURE 1. Activation of DUX4 expression in FSHD. The DUX4 gene is located in the D4Z4 macrosatellite repeat array at chromosome 4q35. Each D4Z4 repeat (yellow triangles) contains DUX4 exons 1 and 2 (solid boxes; orange, open reading frame); exon three is found downstream of the last repeat in the array. The D4Z4 array is normally 11–100 repeats long and hypermethylated (purple line) in healthy individuals. Contractions of this array or mutations in genes coding for epigenetic regulators, in the 4qA haplotype, disrupt the silencing of DUX4 (dotted purple line) and lead to its aberrant expression in skeletal muscle.
In FSHD1, contraction of the 4qA D4Z4 array to ≤10 repeats activates DUX4 expression by increasing chromatin accessibility and promoting DNA hypomethylation in the region (van Overveld et al., 2003; Hewitt, 2015). It was previously thought that individuals with ≤10 D4Z4 repeats in one 4qA chromosome form a homogeneous FSHD1 population, but it is now known that this is not the case. Clinical variability is high in individuals with 7–10 D4Z4 repeats, with most cases ranging from mild to asymptomatic (Ricci et al., 2013; Lemmers et al., 2015). This spread of phenotypes is attributed to inter-individual differences in D4Z4 methylation, indicating that factors other than array contraction may be more important in determining disease penetrance within this repeat range (Van Overveld et al., 2005; Lemmers et al., 2015). Conversely, penetrance is more complete in individuals with 1-6 D4Z4 repeats. Disease severity is also roughly inversely correlated with repeat count in these patients, e.g. those with the severe early-onset form of FSHD typically have 1-3 D4Z4 repeats (Lunt et al., 1995; Ricci et al., 2013; Nikolic et al., 2016). Considering the 1–10 D4Z4 repeat range, it appears that the lower the number of repeats present, the less contribution factors other than contraction size have in influencing the FSHD1 phenotype.
Unlike in FSHD1, moderately-sized D4Z4 arrays are observed in FSHD2. On average, FSHD2 patients have 12–16 D4Z4 repeat units on at least one 4qA chromosome, which is at the shorter end of the range that characterizes the general population (de Greef et al., 2010; Himeda and Jones, 2019). However, at our current level of understanding, D4Z4 array size has little to do with FSHD2 development. Instead, the majority of FSHD2 patients (∼80%) carry mutations in SMCHD1, which codes for a protein involved in maintaining repressive chromatin architecture (Lemmers et al., 2012); others have mutations in DNMT3B or LRIF1, which code for a DNA methyltransferase or an interactor of SMCHD1, respectively (van den Boogaard et al., 2016; Hamanaka et al., 2020). These mutations lead to D4Z4 hypomethylation independent of D4Z4 array size, creating a permissive environment for DUX4 expression on the 4qA chromosome. One study showed that the extent of D4Z4 hypomethylation correlated with disease severity in FSHD2, at least for SMCHD1 mutation carriers (Lemmers et al., 2015). Because of their role in D4Z4 methylation, SMCHD1 and DNMT3B are also genetic modifiers for FSHD1, leading to cases with characteristics of both FSHD1 and FSHD2 (Sacconi et al., 2013; van den Boogaard et al., 2016; de Greef et al., 2018; Sacconi et al., 2019).
Genetic Therapies for FSHD
Figure 2 summarizes the genetic therapies that have been developed for FSHD, which are covered in the following sections. Briefly, we have potential genetic therapies targeting DUX4 at the DNA, RNA, and protein levels. There are also therapies that focus on inhibiting the effects of DUX4-mediated toxicity, thereby modifying the disease phenotype.
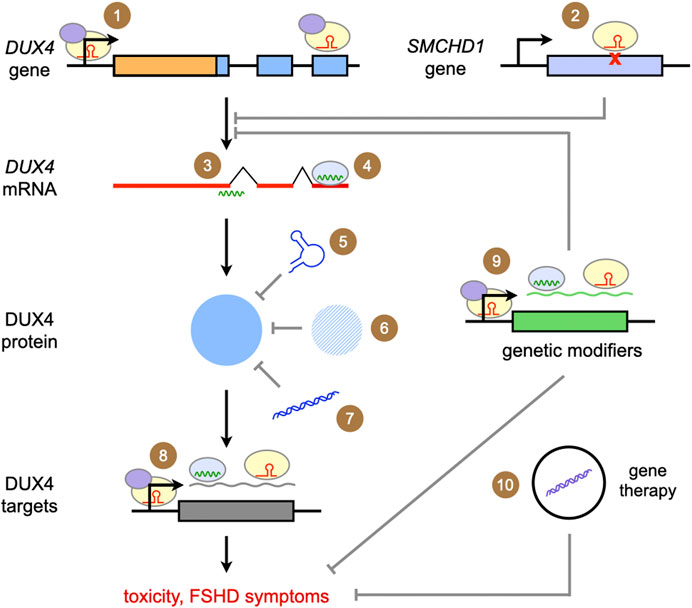
FIGURE 2. Summary of genetic approaches for the treatment of FSHD. The approaches covered in this review are depicted above. Ultimately, all focus on reducing DUX4 expression, DUX4 protein activity, or the effects of DUX4-mediated toxicity. (1) Targeted gene repression with CRISPR/dCas9-KRAB; (2) correction of a SMCHD1 mutation with CRISPR/Cas9; (3) gene knockdown with antisense oligonucleotides; (4) gene knockdown/silencing with RNA interference; competing with DUX4 activity through (5) DNA aptamers, (6) introduction of proteins homologous or similar to DUX4, and (7) DNA decoys; (8) gene knockdown/silencing of DUX4 downstream targets; (9) gene knockdown/silencing of genetic modifiers of DUX4 expression or DUX4-mediated toxicity (10) delivery of genes coding for proteins that ameliorate DUX4-mediated toxicity or secondary features of FSHD pathology.
Oligonucleotide Therapies
Depending on their structure and chemistry, oligonucleotides can inhibit DUX4 expression in a variety of ways. One of the most extensively tested for FSHD are antisense oligonucleotides (AOs), single-stranded nucleic acid analogues that can bind target mRNA sequences by Watson-Crick base-pairing. There are two kinds of AOs. The first are those that reduce gene expression by interfering with mRNA splicing and processing (Lim and Yokota, 2018). These AOs act as steric blockers, preventing factors from accessing critical sequences in the mRNA such as splice sites, and are usually phosphorodiamidate morpholino oligomers (PMOs) or phosphorothioated 2′-O-methyl RNAs (2′-OMePS). The second are those that reduce gene expression by inducing target mRNA degradation (Lim and Yokota, 2020). The AOs in this group are gapmers, fully phosphorothioated oligonucleotides that have a central DNA stretch flanked by bases of modified chemistry, e.g. locked nucleic acids (LNA) or 2′-O-methoxyethyl RNAs (2′-MOE). When a gapmer binds its target mRNA, a DNA/RNA hybrid is created in the middle of the AO that is recognized by ribonuclease H, which proceeds to bind the hybrid and cleave its RNA portion.
AOs of both kinds have successfully inhibited DUX4 expression in patient-derived cells and FSHD mouse models (Table 1). PMOs and 2′-OMePS AOs targeting splice acceptor sites for DUX4 exons 2 and 3 (Figure 3) gave 30–90% DUX4 mRNA knockdown (at 10 and 50 nM tested doses) in myotubes from treated primary patient myoblasts (Vanderplanck et al., 2011; Ansseau et al., 2017). Corresponding reductions in DUX4 downstream target gene expression and DUX4-positive nuclei, as well as improvements in muscle cell morphology, were observed. AOs targeting the exon 3 splice acceptor site were particularly more effective, one of which was tested in mice as a vivo-PMO (Ansseau et al., 2017; Derenne et al., 2020). Vivo-PMOs are PMOs that have been covalently linked to an octaguanidine dendrimer for improving uptake in tissues (Morcos et al., 2008). Mice transduced with DUX4 constructs at the tibialis anterior (TA) were intramuscularly (i.m.) injected at the same muscle with 10 μg of the vivo-PMO, which led to 30-fold lower DUX4 expression than the control vivo-PMO-treated leg by semi-quantitative RT-PCR, 10 days after treatment (Ansseau et al., 2017). Histopathological improvements were observed in another study using the same AO (Derenne et al., 2020). PMOs have also been used to target the PAS in exon 3 (Figure 3), which knocked down DUX4 transcript expression in immortalized patient-derived myotubes by 25–52% at a 50 nM dose (Marsollier et al., 2016) and in a xenograft FSHD mouse model by nearly 100% with a 20-μg injection (Chen et al., 2016a). Reduced expression of DUX4 downstream target genes, transcriptomic-level restoration, and loss of DUX4-positive nuclei were observed in vitro; treatment showed no significant improvements in muscle cell fusion, however.
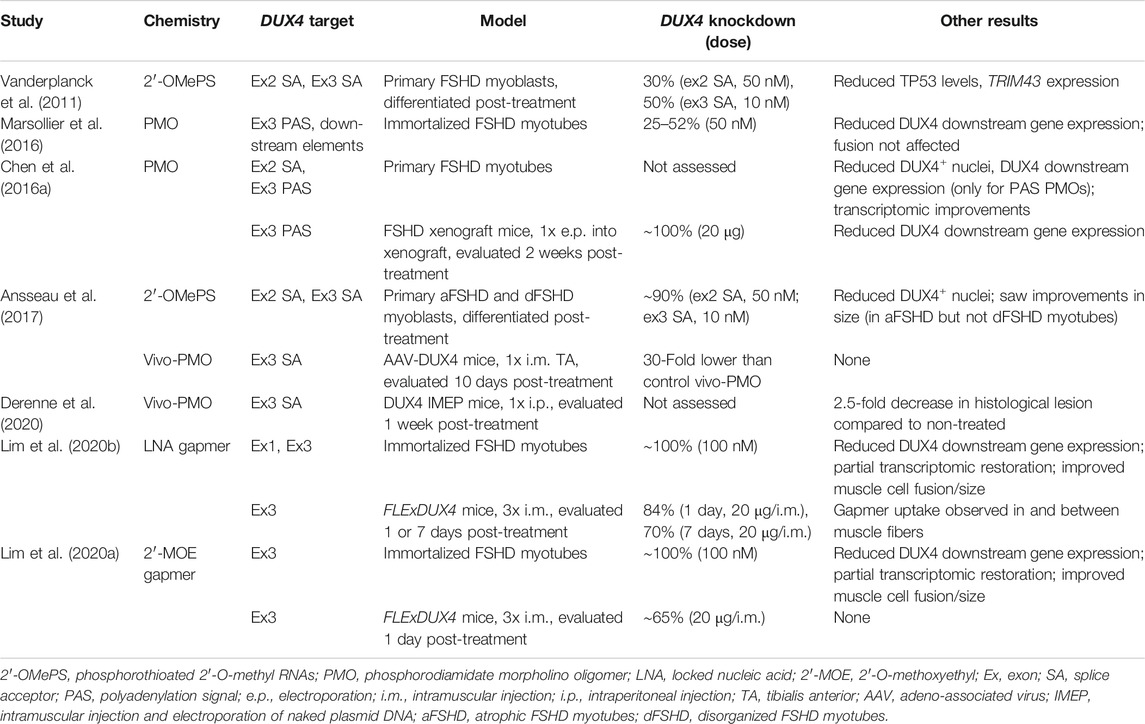
TABLE 1. Summary of results from pre-clinical studies on antisense oligonucleotides for DUX4 knockdown.
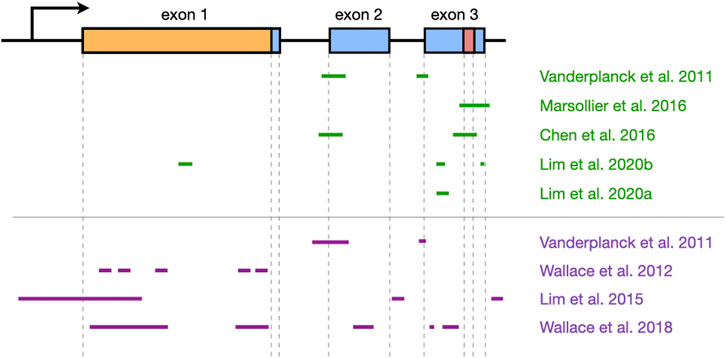
FIGURE 3. Overview of DUX4 regions that have been targeted by oligonucleotide therapies. The structure of the DUX4 gene is shown at the top (arrow indicates promoter region; boxes, exons; lines, introns; orange, open reading frame; red, polyadenylation signal), and the regions that have been targeted by antisense oligonucleotides (green) or RNA interference (purple) are shown at the bottom. Approximate locations are shown, and the figure is not to scale. Note that Ansseau et al. (2017) used the same oligonucleotides as Vanderplanck et al. (2011). DUX4 structure was based on information from Ensembl, transcript ID ENST00000569241.5.
Our group has recently published on the efficacy of using gapmers for inhibiting DUX4 mRNA expression. We designed LNA gapmers to target sites at DUX4 exons 1 and 3 (Lim et al., 2020b), as well as 2′-MOE gapmers to target only sites at exon 3 (Lim et al., 2020a) (Figure 3). The LNA and 2′-MOE gapmers targeted overlapping sequences at exon 3, upstream of the PAS. All gapmers knocked down DUX4 mRNA levels almost completely (∼99%) at 100 nM and by more than 50% at 10 nM regardless of chemistry in immortalized patient-derived myotubes. This demonstrates an increased potency of gapmers compared to steric-blocking AOs, perhaps due to the direct nature of transcript degradation induced by the gapmers. More sustained knockdown of DUX4 downstream target genes was observed for LNA than 2′-MOE gapmers at 10 nM. LNA gapmer treatment also restored more FSHD signature genes upon RNA sequencing analysis, hinting that LNA gapmers may be the more potent of the two in terms of DUX4 knockdown. Focusing on exon 3-targeting gapmers, we saw improvements in muscle cell fusion and size, as well as minimal to no effects on potential off-target genes in vitro. A separate study also found that treatment with one of the 2′-MOE gapmers increased membrane repair in immortalized patient myoblasts (Bittel et al., 2020). One gapmer from each chemistry was also tested in the FLExDUX4 model, which carry a stably integrated, Cre-inducible DUX4 transgene (Jones and Jones, 2018). Non-induced FLExDUX4 mice exhibit low levels of DUX4 expression mimicking what is seen in patient muscle cells, and was used for preliminary studies. Significant DUX4 knockdown was induced in these mice following three 20-μg i.m. TA injections, at 70–84% for the LNA gapmer and 65% for the 2′-MOE gapmer, on average.
Another class of oligonucleotide therapy is RNA interference (RNAi), which makes use of small interfering RNAs (siRNAs) or microRNAs (miRNAs) (Table 2). Unlike AOs, siRNAs and miRNAs require association with effector proteins to reduce target gene expression. siRNAs targeting DUX4 promoter elements or exons (Figure 3) knocked down DUX4 transcript levels by 50–90% in vitro, with corresponding restorative effects on DUX4 downstream targets (Vanderplanck et al., 2011; Lim et al., 2015). Interestingly, siRNAs against the promoter likely inhibited DUX4 expression through epigenetic silencing at the DNA level, since 2′-MOE gapmers against the same region did not affect DUX4 transcript levels (Holoch and Moazed, 2015; Lim et al., 2015). Meanwhile, one group screened a large number of miRNAs (Figure 3) and found two targeting exon 1 (mi1155, mi405) to knock down DUX4 expression the best at >75% in DUX4-luciferase reporter cells (Wallace et al., 2012; Wallace et al., 2018). Treatment of DUX4-transduced mice (i.m., TA) with 3 × 1010 adeno-associated viruses (AAVs) carrying mi405 constructs reduced DUX4 mRNA expression by 64%, and DUX4 protein levels by 90% (Wallace et al., 2012). Histopathology was improved with this miRNA, but not with mi1155 that instead showed signs of overt toxicity (Wallace et al., 2018).
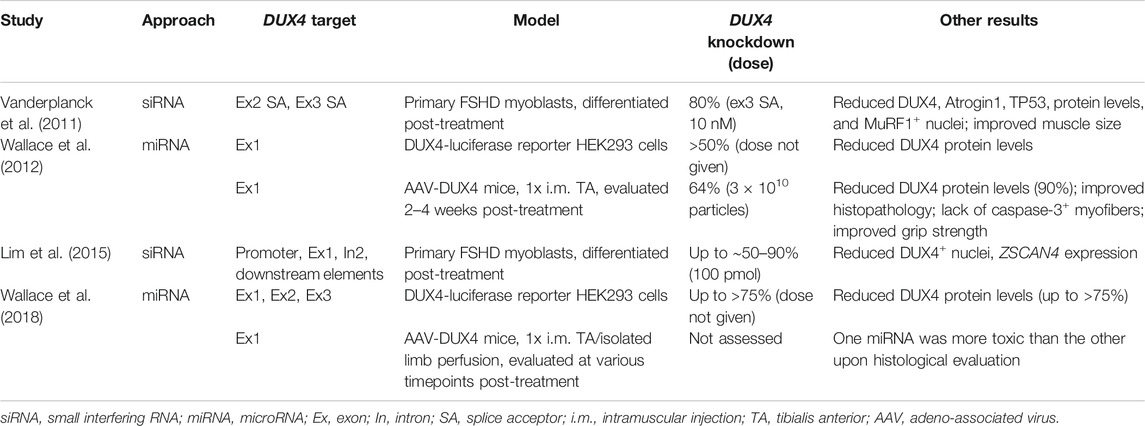
TABLE 2. Summary of results from pre-clinical studies on RNA interference for DUX4 knockdown.
Oligonucleotides can also be designed to target the DUX4 protein. Double-stranded DNA decoys containing the DUX4 binding motif have recently been developed to sequester and prevent DUX4 from activating its downstream targets (Mariot et al., 2020). Indeed, the expression levels of DUX4 downstream targets ZSCAN4 and TRIM43 were knocked down by 39–91% in primary patient myotubes upon treatment with these decoys. The DNA decoys were also tested in AAV-DUX4 mice, where administration either by intramuscular electroporation or AAV delivery led to decreased expression of Tm7sf4, another DUX4 downstream target. On a related note, single-stranded DNA aptamers have recently been developed with high, preferential affinity to the DUX4 DNA-binding domain (Klingler et al., 2020). However, these aptamers have yet to be tested for their therapeutic potential. Developing oligonucleotides for targets other than DUX4 may be useful as well. For instance, PITX1 is a direct transcriptional target of DUX4 whose overexpression induces an FSHD-like dystrophic phenotype in mice (Dixit et al., 2007; Pandey et al., 2012). Intravenous injection of AOs against Pitx1 in Pitx1-transgenic mice improved grip strength and decreased muscle pathology (Pandey et al., 2014). FRG1 is another direct transcriptional target of DUX4, whose knockdown by RNAi reversed dystrophic histopathology and improved treadmill performance in FRG1-overexpressing mice (Bortolanza et al., 2011; Ferri et al., 2015). It would be interesting to see if similar effects could be observed if these strategies were used to treat DUX4-overexpressing mouse models such as FLExDUX4 (Jones and Jones, 2018), the doxycycline-inducible iDUX4pA (Bosnakovski et al., 2017a; Bosnakovski et al., 2020), or the tamoxifen-inducible TIC-DUX4 (Giesige et al., 2018).
CRISPR
The bacterial defense system based on clustered regularly interspaced short palindromic repeats (CRISPR) has been adapted and developed to become perhaps one of the most revolutionary tools for targeted genome editing to date. In its most common configuration, CRISPR has two basic components: an endonuclease for cleaving DNA (the CRISPR-associated or Cas protein), and an RNA molecule that associates with this enzyme and tells it where in the genome to cut (the guide RNA or gRNA) (Jinek et al., 2012; Jiang and Doudna, 2017). The gRNA is designed complementary to the target DNA site, which additionally has to have a protospacer-adjacent motif sequence nearby to facilitate Cas binding (Mojica et al., 2009; O’Connell et al., 2014). Upon binding of the gRNA-Cas complex, a double-stranded break is introduced into the target DNA. This break is subsequently resolved by non-homologous end joining or homology-directed repair, which create random insertions/deletions or precise edits at the site, respectively, and form the basis of CRISPR-based genome editing.
CRISPR has been previously used to correct an FSHD2-associated SMCHD1 mutation, a missense variant in intron 34 that introduced an out-of-frame 53-bp pseudoexon in the final transcript (Goossens et al., 2019). CRISPR/Cas9 with gRNAs against the intronic sequences flanking this pseudoexon restored the SMCHD1 reading frame and increased wild-type SMCHD1 expression in primary and immortalized patient myotubes, resulting in reduced DUX4 mRNA expression. It has been suggested that CRISPR be used to edit the permissive 4qA to the restrictive 4qB haplotype (Cohen et al., 2020), but attempts on realizing this approach have not yet been reported in the literature. In addition to genome editing, CRISPR can also be used for the targeted modulation of gene expression. Using a catalytically-deficient version of Cas9 (dCas9) fused to a KRAB transcriptional repressor, together with gRNAs against the DUX4 promoter or exon 1, one group achieved ∼45% DUX4 knockdown in myotubes differentiated from treated primary patient myoblasts (Himeda et al., 2016). A trend toward increased chromatin repression of the DUX4 gene at the contracted locus was observed. When dCas9-KRAB was used with gRNAs solely targeting DUX4 exon 3 or various regions within/upstream of the D4Z4 repeat sequence, no significant DUX4 knockdown was observed. The same group used dCas9-KRAB to inhibit the expression of other genes—BRD2, BAZ1A, KDM4C, and SMARCA5—which led to about 40–60% DUX4 knockdown in primary patient myotubes (Himeda et al., 2018). These genes code for epigenetic regulators, and were previously identified from an RNAi screen as candidates whose knockdown lowered DUX4 transcript levels without negatively impacting the expression of genes involved in muscle development or homeostasis. On a related note, CRISPR/Cas9 has itself been employed for a genome-wide knockout screen to search for genes whose loss-of-function was protective against DUX4 cytotoxicity (Lek et al., 2020). Hypoxia signaling pathway members were identified as the most promising candidates, in accordance with the role of oxidative stress in DUX4-mediated pathogenesis (Dmitriev et al., 2016; Denny and Heather, 2017; Lim et al., 2020c).
Other Approaches
Preliminary findings from basic research studies are providing solid foundations for the development of more strategies for FSHD therapy. One interesting approach is to use other proteins to compete with DUX4 activity. DUX4-s is a short isoform of DUX4 that contains only the first 159 N-terminal amino acids of the protein, spanning both homeodomains (Mitsuhashi et al., 2018). It is non-pathogenic, and its expression has been detected in both healthy and FSHD skeletal muscle (Snider et al., 2010; Geng et al., 2011; Mitsuhashi et al., 2018). Since DUX4-s shares the exact same homeodomains as full-length DUX4, it is thought that overexpression of the former will prevent the latter from binding its usual genomic targets. Indeed, co-injection of DUX4-s and full-length DUX4 mRNA at a 20:1 ratio into fertilized zebrafish eggs decreased embryo mortality rates to ∼10%, improved musculature, and led to 70% of embryos having an overall normal phenotype (Mitsuhashi et al., 2013). In contrast, eggs injected with only full-length DUX4 mRNA had an embryo mortality rate of ∼40%, and less than 20% of resulting embryos were phenotypically wild-type. As the physiological functions of DUX4-s are unknown, more research into this area may help further develop this approach as an FSHD therapy. The DUX4 homeodomains are also highly similar and functionally interchangeable with those of PAX7 (Bosnakovski et al., 2017b). Overexpression of Pax7 or its homolog Pax3 considerably improved viability in DUX4-inducible C2C12 cells (Bosnakovski et al., 2008). This rescue was diminished in a dose-dependent manner when DUX4 expression was induced at higher levels, indicating that Pax7 or Pax3 may be exerting their effects via competition with the DUX4 protein. Although promising, pre-clinical testing of DUX4-s and PAX7/3 in FSHD mouse models have yet to be performed.
Research into understanding FSHD biology or DUX4-mediated cytotoxicity has also uncovered more potential targets for therapy. These include genes involved in apoptosis (CDKN1A, MYC), immune response activation (RNASEL, EIF2AK2), and epigenetic regulation (H3.X, H3.Y), to name a few (Lim et al., 2020c). As previously mentioned, RNAi and CRISPR screens have been instrumental in adding to this list by identifying genetic modifiers of DUX4 expression (Himeda et al., 2018; Lek et al., 2020). Modulating the expression of these genes, either by oligonucleotide- or CRISPR-based approaches, may be therapeutic avenues worth investigating. Developing treatments to alleviate FSHD symptoms may be beneficial as well. For instance, AAV delivery of a follistatin gene construct into TIC-DUX4 FSHD model mice (i.m.) increased mass and improved strength in injected muscles (Giesige et al., 2018). Follistatin is an inhibitor of myostatin, which in turn is a known inhibitor of muscle growth (Rodino-Klapac et al., 2009). It is important to note though that follistatin did not reverse DUX4-induced histopathology in treated mice, suggesting that treatments directed at secondary pathological features of FSHD are probably not curative and may be more useful when administered in conjunction with DUX4-targeting genetic therapies.
Challenges for FSHD Genetic Therapies
The development of genetic therapies for FSHD is at a relatively young phase, with most pre-clinical work limited to in vitro models. Only a handful of these experimental therapies have moved on to in vivo testing (Table 1), of which only one was evaluated for its efficacy in ameliorating FSHD symptoms (Wallace et al., 2012; Wallace et al., 2018). This may be explained by the lack of appropriate FSHD animal models at the time, an effort that was largely hindered by the lethal effects of DUX4 (DeSimone et al., 2020). Various groups have since capitalized on using conditional methods to overexpress DUX4 in mice, allowing for more refined control of DUX4 toxicity and ushering in the production of FSHD animal models amenable to pre-clinical study (Bosnakovski et al., 2017a; Bosnakovski et al., 2020; Giesige et al., 2018; Jones and Jones, 2018). With the impending progression of FSHD genetic therapies into in vivo testing, certain challenges will have to be considered and overcome to ensure treatment success not only in animal models but also and ultimately in patients. We consider challenges relevant to oligonucleotide- and CRISPR-based therapies, as these have advanced the most in pre-clinical development.
Delivery is perhaps one of the largest hurdles these therapies have to face in vivo. In order for genetic therapies to work, they will have to reach their target DNA/RNA sequences in the nuclei of their target cells. Preliminary work using AOs for instance have shown that levels of DUX4 knockdown in vivo are not usually as high as those observed in vitro (Wallace et al., 2012; Ansseau et al., 2017; Lim et al., 2020a; Lim et al., 2020b). The same divide in vitro and in vivo efficacy has been observed for similar genetic approaches in other muscular disorders, such as Duchenne muscular dystrophy (DMD) (Echigoya et al., 2017; Lim et al., 2019; Nguyen and Yokota, 2019). For oligonucleotide therapies, much of the challenge will be to get them into muscle cells and, once inside, to have them successfully escape from endosomes and reach the nucleus (Juliano, 2016). This is especially problematic for charge-neutral chemistries such as PMOs (Summerton and Weller, 1997), which are not readily recognized by cell surface receptors nor are they particularly disruptive toward cell membranes. Conjugation of cell-penetrating entities to oligonucleotides in order to enhance cellular uptake and endosomal escape are actively being investigated, as are the use of non-viral delivery vehicles like lipid nanoparticles, polymeric nanocarriers, and exosomes (Dominska and Dykxhoorn, 2010; Juliano, 2016; Tong et al., 2019). Chemically modifying the oligonucleotides themselves to appear more recognizable to muscle cell surface receptors is currently being looked into as well (Dominska and Dykxhoorn, 2010; Juliano, 2016; Tong et al., 2019). Viral vectors may be applicable for RNAi strategies using miRNAs or shRNAs, but their immunogenicity will have to be carefully considered when delivered in vivo.
CRISPR components, on the other hand, have mostly been delivered using viral vectors. This has mostly been effective, except for two major concerns: the packaging limit into viruses and immunogenicity. The SpCas9 (the most used Cas variant) gene construct is ∼4.2 kb long, which is quite near the packaging capacity of AAVs at ∼4.4–4.7 kb (Wu et al., 2010; Tong et al., 2019; Wilbie et al., 2019). Use of a two-vector delivery system has been necessary at times, one for Cas9 and another for the gRNA/s. How this division exactly affects CRISPR efficacy is still poorly understood, but could be unfavorable when considering that we have to maximize the likelihood of all CRISPR components being present in the same nuclei—a challenge that is perhaps made more difficult by the syncytial nature of mature muscle cells and the fact that only a very few nuclei actually express DUX4 (Tassin et al., 2013). Lentiviral and retroviral vectors have larger capacities at 7–10 kb, but are prone to inducing unwanted integration of their cargo into the host genome (Tong et al., 2019). Adenoviral vectors have even larger capacities at about 36 kb, and are emerging as effective vehicles for CRISPR delivery (Ehrke-Schulz et al., 2017; Tong et al., 2019; Ricobaraza et al., 2020). The discovery of smaller Cas variants such as CjCas9 (2.95 kb gene size) is also helping overcome this packaging limit (Kim et al., 2017). However, due to their immunogenicity, there is always the risk of complications and so the use of immunosuppressive agents remain important, especially since responses may still be activated at high doses of virus, and individual reactions to viral agents are difficult to predict (Tong et al., 2019; Xu et al., 2019; Shirley et al., 2020). Non-viral delivery approaches, similar to those described for oligonucleotide therapies, are also being developed for CRISPR to overcome both issues of packaging and immunogenicity (Lino et al., 2018; Wilbie et al., 2019).
Safety is another concern. Oligonucleotide and CRISPR therapies can be toxic through hybridization-dependent or -independent means (Frazier, 2015). One aspect of hybridization-dependent toxicity is off-target gene knockdown/knockout/editing, resulting from the recognition of non-target sequences that share imperfect complementarity with a given oligonucleotide or gRNA. Only a few studies on DUX4-targeting therapies have examined off-target effects—dose-dependent knockdown was found in some non-target genes, whereas no effect was observed in others (Lim et al., 2020a; Lim et al., 2020b). A more thorough analysis of off-target effects is recommended for future studies, perhaps using genome- or transcriptome-wide approaches (Kim et al., 2015; Tsai et al., 2015; Yoshida et al., 2019). Fortunately, the specificity of these genetic therapies is constantly being improved through chemical modification or by performing in silico screens to predict the off-targeting potential of certain oligonucleotide sequences prior and in addition to in vitro testing (Kamola et al., 2015; Tycko et al., 2016; Hendling and Barišić, 2019; Wang et al., 2019). In the case of CRISPR, splitting the Cas enzyme into two interdependent halves and limiting the duration of Cas activity via self-restricting mechanisms have emerged as possible approaches to reduce the chance of off-target effects (Ran et al., 2013; Shen et al., 2014; Moore et al., 2015; Chen et al., 2016b).
Hybridization-dependent toxicity can also come from on-target effects. This is not much of an issue for the FSHD-associated DUX4 isoform, since it is not supposed to be expressed in the first place, and its expression appears to be muscle-specific (Himeda et al., 2014). Non-pathogenic DUX4 isoforms are, however, expressed in healthy tissues (Snider et al., 2010; Himeda and Jones, 2019). As the physiological functions of these isoforms are currently unknown, it would be hard to predict the consequences of their reduced expression. Regardless, as a form of caution, genetic therapies should be designed to preferentially target skeletal muscle or regions specific to the pathogenic DUX4 isoform. Use of viruses with muscle-specific tropism or ligand-directed oligonucleotides/non-viral delivery vehicles can help ensure tissue-specific treatment (Juliano, 2016). As for target sequence design, exon 3 is the region most specific to the pathogenic, full-length DUX4 isoform. Most oligonucleotide therapies target this exon and so are not particularly concerning; however, therapies targeting other exons, the DUX4 promoter region, or D4Z4 sequences on other tissues will have to be closely monitored for potential adverse effects. Another on-target effect is the potential integration of genetic material from viral delivery vectors—however, this threat can be minimized by using AAVs or non-viral delivery methods.
On the other hand, hybridization-independent toxicity refers to effects caused by the therapies themselves that are not attributed to their intended sequence-dependent genetic activities. Certain secondary structures on oligonucleotides and gRNAs can be recognized by pattern recognition receptors and lead to an innate immune response (Agrawal and Kandimalla, 2004; Kim et al., 2004; Lee and Yokota, 2013; Kim et al., 2018; Wienert et al., 2018; Wilbie et al., 2019). The formation of such structures therefore has to be considered during the sequence design process for these modalities. Pre-existing adaptive immunity against Cas proteins is also common in the population (Chew, 2018; Crudele and Chamberlain, 2018; Wagner et al., 2019). Whereas Cas proteins are now being engineered to become less immunogenic (Ferdosi et al., 2019; Mehta and Merkel, 2020), efforts to reduce the persistence of Cas activity as described previously are also potential solutions. Aside from immune response activation, hepatotoxicity and nephrotoxicity have been previously linked to phosphorothioated AOs (Frazier, 2015). This toxicity has been partially attributed to the propensity of AOs to bind cellular proteins (Brown et al., 1994; Liang et al., 2015; Kakiuchi-Kiyota et al., 2016; Shen et al., 2018). Accordingly in one study, chemical modifications that reduced the overall protein-binding affinity of gapmers prevented hepatotoxicity in mice, without sacrificing therapeutic efficacy (Shen et al., 2019). Certain sequence patterns have also been found to correlate with the hepatotoxic potential of oligonucleotides (Hagedorn et al., 2013; Burdick et al., 2014). The development of predictive in silico tools may help prevent the integration of such sequences during oligonucleotide design (Hagedorn et al., 2013); use of in vitro screening for potential AO toxicity prior to therapeutic evaluation would be recommended (Dieckmann et al., 2018; Shen et al., 2019).
Another challenge to consider is that DUX4 expression is very rare in skeletal muscle, only detected in 1/1000 or 1/200 nuclei in FSHD myoblasts and myotubes, respectively (Snider et al., 2010; Tassin et al., 2013). This poses an issue when evaluating the efficacy of DUX4-targeting therapies, by preventing a robust read-out of DUX4 knockdown. This explains why some studies have instead opted to use DUX4-transfected models, which may create an overly toxic, non-representative environment compared to what is seen in patients. Fortunately, we now have robust protocols that enable reliable detection of endogenous DUX4 mRNA levels, based on real-time quantitative reverse transcription PCR or RNA in situ hybridization (Lim et al., 2020b, Lim et al., 2015; Amini Chermahini et al., 2019). Induction of endogenous DUX4 expression using media supplements such as KOSR has been likewise helpful (Pandey et al., 2015). DUX4 protein detection has shown some success, owing to the development of good antibodies (Dixit et al., 2007; Snider et al., 2010; Geng et al., 2011), but may remain a challenge in certain conditions when considering the potentially short half-life of the DUX4 protein (Geng et al., 2011; Rickard et al., 2015) or the proposed stochastic model of DUX4 expression in muscle cells (Snider et al., 2010; Rickard et al., 2015). Of course, evaluating effects on DUX4 downstream transcriptional targets or FSHD biomarkers as proxies for DUX4 knockdown remain options for therapeutic evaluation.
Finally, one has to recognize the unique clinical presentation of FSHD when translating these genetic therapies into patients. One of the most distinctive features of FSHD is its asymmetric phenotype (Wang and Tawil, 2016). We are far from understanding how this occurs—what determines which parts of the muscle get affected first, why certain muscle groups are spared more than others, why the pattern of muscle weakness differs between patients, and so on. The best strategy at the moment would probably be to develop an approach that equally benefits all muscle groups. This requires that therapeutic efficacy be evaluated in representative muscles across the body during in vivo testing, and not only on commonly assessed muscles such as the TA. If anything, therapeutic effects on muscles in the upper parts of the body have to be prioritized, given their early involvement in FSHD (Wang and Tawil, 2016). Another approach would be to administer genetic therapies locally, injecting only the affected muscles. While this takes advantage of the patchy nature of FSHD pathology, this method requires a way to reliably locate affected muscle areas and may not be practical if too many muscles have become involved; it will also not improve the state of any extramuscular symptoms. Needless to say, developing therapies that would address the unique characteristics of FSHD would require the use of animal models that resemble the disease fairly well. As previously mentioned, recent advancements in the conditional control of DUX4 expression have helped us get closer to generating such models, resulting in mice with dystrophic histopathology, impaired muscle strength, and asymmetric muscle degeneration similar to those seen in patients (DeSimone et al., 2020). Due to the inducible nature of DUX4 expression in these models, the severity of resulting phenotypes is also tunable, allowing for therapeutic testing in a variety of disease states. This is a rather helpful aspect, considering the wide variability in disease presentation seen in patients with FSHD.
Conclusion
The discovery of DUX4 as the genetic cause of FSHD has greatly accelerated efforts not only to understand the disorder but also to treat it. As we have seen, DUX4 has proven itself to be a useful target for genetic therapy. Oligonucleotide- and CRISPR-based approaches have demonstrated the feasibility of DUX4 knockdown in reversing muscle-specific FSHD pathology, with promising preliminary results in vitro and in vivo. However, there is much to be done before these therapies can reach the patients they are meant for. Challenges in delivery, efficacy, safety, and in dealing with the unique pathology of FSHD all have to be taken into consideration in vivo. The field can use the lessons learned from the application of these therapies in other muscular disorders to help overcome such hurdles. Development of alternative genetic approaches for FSHD therapy should also be encouraged. For instance, strategies to interfere with DUX4 transcription factor activity or to modulate the expression of genes that impart protection against the cytotoxicity of DUX4 appear to have therapeutic potential. The ongoing identification of genes involved in FSHD pathogenesis by basic research and transcriptomic studies are providing new targets for genetic therapies as well. Finally, although FSHD is predominantly a skeletal muscle disease, it will be necessary to create therapies that address its extramuscular symptoms, especially since these tend to manifest in the more severe cases of the disease. Further investigations into both the pathological and physiological roles of DUX4 in other tissues are steps toward achieving this goal. Such studies should also help us better assess the possible effects of DUX4-targeting genetic therapies in non-muscle tissues. Appreciating all the work that has been accomplished thus far, we have certainly come a long way in the development of genetic therapies for FSHD. With continued efforts from both basic and translational research teams in the FSHD community, it may only be a matter of time until we see these therapies making their way into clinical trials.
Author Contributions
KL performed a literature review and wrote the manuscript draft. KL and TY edited the manuscript. TY supervised the work and acquired funding.
Funding
This work was funded by the Friends of Garrett Cumming Research Chair Fund, HM Toupin Neurological Science Research Chair Fund, Muscular Dystrophy Canada, Canadian Institutes of Health Research (CIHR) FDN 143251, Alberta Innovates, the University of Alberta Faculty of Medicine and Dentistry, University Hospital Foundation, and the Women and Children’s Health Research Institute (WCHRI) IG 3279. Muscular Dystrophy Association 629268, FSHD Canada Foundation 30004574-01, FSHD Society 30003892-17-01.
Conflict of Interest
TY is a co-founder and shareholder of OligomicsTx Inc., which aims to commercialize antisense technology.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Agrawal, S., and Kandimalla, E. R. (2004). Antisense and siRNA as agonists of Toll-like receptors. Nat. Biotechnol. 22, 1533–1537. doi:10.1038/nbt1042 |
Amini Chermahini, G., Rashnonejad, A., and Harper, S. Q. (2019). RNAscope in situ hybridization-based method for detecting DUX4 RNA expression in vitro. RNA 25, 1211–1217. doi:10.1261/rna.070177.118 |
Ansseau, E., Vanderplanck, C., Wauters, A., Harper, S. Q., Coppée, F., and Belayew, A. (2017). Antisense oligonucleotides used to target the DUX4 mRNA as therapeutic approaches in FaciosScapuloHumeral muscular dystrophy (FSHD). Genes 8, 93. doi:10.3390/genes8030093
Bakker, E., Wijmenga, C., Vossen, R. H., Padberg, G. W., Hewitt, J., van der Wielen, M., et al. (1995). The FSHD-linked locus D4F104S1 (p13E-11) on 4q35 has a homologue on 10qter. Muscle Nerve Suppl. 2, S39–S44. doi:10.1002/mus.880181309 |
Bao, B., Maruyama, R., and Yokota, T. (2016). Targeting mRNA for the treatment of facioscapulohumeral muscular dystrophy. Intractable Rare Dis. Res. 5, 168–176. doi:10.5582/irdr.2016.01056 |
Bittel, A. J., Sreetama, S. C., Bittel, D. C., Horn, A., Novak, J. S., Yokota, T., et al. (2020). Membrane repair deficit in facioscapulohumeral muscular dystrophy. Int. J. Mol. Sci. 21, 5575. doi:10.3390/ijms21155575
Bortolanza, S., Nonis, A., Sanvito, F., Maciotta, S., Sitia, G., Wei, J., et al. (2011). AAV6-mediated systemic shRNA delivery reverses disease in a mouse model of facioscapulohumeral muscular dystrophy. Mol. Ther. 19, 2055–2064. doi:10.1038/mt.2011.153 |
Bosnakovski, D., Chan, S. S. K., Recht, O. O., Hartweck, L. M., Gustafson, C. J., Athman, L. L., et al. (2017a). Muscle pathology from stochastic low level DUX4 expression in an FSHD mouse model. Nat. Commun. 8, 550. doi:10.1038/s41467-017-00730-1
Bosnakovski, D., Toso, E. A., Hartweck, L. M., Magli, A., Lee, H. A., Thompson, E. R., et al. (2017b). The DUX4 homeodomains mediate inhibition of myogenesis and are functionally exchangeable with the Pax7 homeodomain. J. Cel Sci. 130, 3685–3697. doi:10.1242/jcs.205427
Bosnakovski, D., Shams, A. S., Yuan, C., da Silva, M. T., Ener, E. T., Baumann, C. W., et al. (2020). Transcriptional and cytopathological hallmarks of FSHD in chronic DUX4-expressing mice. J. Clin. Invest. 130, 2465–2477. doi:10.1172/JCI133303 |
Bosnakovski, D., Xu, Z., Gang, E. J., Galindo, C. L., Liu, M., Simsek, T., et al. (2008). An isogenetic myoblast expression screen identifies DUX4-mediated FSHD-associated molecular pathologies. EMBO J. 27, 2766–2779. doi:10.1038/emboj.2008.201 |
Bouwman, L. F., van der Maarel, S. M., and de Greef, J. C. (2020). The prospects of targeting DUX4 in facioscapulohumeral muscular dystrophy. Curr. Opin. Neurol. 33, 635–640. doi:10.1097/WCO.0000000000000849 |
Brown, D. A., Kang, S. H., Gryaznov, S. M., DeDionisio, L., Heidenreich, O., Sullivan, S., et al. (1994). Effect of phosphorothioate modification of oligodeoxynucleotides on specific protein binding. J. Biol. Chem. 269, 26801–26805. doi:10.1016/S0021-9258(18)47090-1 |
Burdick, A. D., Sciabola, S., Mantena, S. R., Hollingshead, B. D., Stanton, R., Warneke, J. A., et al. (2014). Sequence motifs associated with hepatotoxicity of locked nucleic acid--modified antisense oligonucleotides. Nucleic Acids Res. 42, 4882–4891. doi:10.1093/nar/gku142 |
Chen, J. C., King, O. D., Zhang, Y., Clayton, N. P., Spencer, C., Wentworth, B. M., et al. (2016a). Morpholino-mediated knockdown of DUX4 toward facioscapulohumeral muscular dystrophy therapeutics. Mol. Ther. 24, 1405–1411. doi:10.1038/mt.2016.111 |
Chen, Y., Liu, X., Zhang, Y., Wang, H., Ying, H., Liu, M., et al. (2016b). A self-restricted CRISPR system to reduce off-target effects. Mol. Ther. 24, 1508–1510. doi:10.1038/mt.2016.172 |
Chew, W. L. (2018). Immunity to CRISPR Cas9 and Cas12a therapeutics. Wiley Interdiscip. Rev. Syst. Biol. Med. 10 (1). doi:10.1002/wsbm.1408
Cohen, J., DeSimone, A., Lek, M., and Lek, A. (2020). Therapeutic approaches in facioscapulohumeral muscular dystrophy. Trends Mol. Med. 27, 123–137. doi:10.1016/j.molmed.2020.09.008 |
Crudele, J. M., and Chamberlain, J. S. (2018). Cas9 immunity creates challenges for CRISPR gene editing therapies. Nat. Commun. 9, 3497. doi:10.1038/s41467-018-05843-9 |
Das, S., and Chadwick, B. P. (2016). Influence of repressive histone and DNA methylation upon D4Z4 transcription in non-myogenic cells. PLoS One 11, e0160022, doi:10.1371/journal.pone.0160022 |
de Greef, J. C., Krom, Y. D., den Hamer, B., Snider, L., Hiramuki, Y., van den Akker, R. F. P., et al. (2018). Smchd1 haploinsufficiency exacerbates the phenotype of a transgenic FSHD1 mouse model. Hum. Mol. Genet. 27, 716–731. doi:10.1093/hmg/ddx437 |
de Greef, J. C., Lemmers, R. J., Camaño, P., Day, J. W., Sacconi, S., Dunand, M., et al. (2010). Clinical features of facioscapulohumeral muscular dystrophy 2. Neurology 75, 1548–1554. doi:10.1212/WNL.0b013e3181f96175 |
De Iaco, A., Planet, E., Coluccio, A., Verp, S., Duc, J., and Trono, D. (2017). DUX-family transcription factors regulate zygotic genome activation in placental mammals. Nat. Genet. 49, 941–945. doi:10.1038/ng.3858 |
Deidda, G., Cacurri, S., Grisanti, P., Vigneti, E., Piazzo, N., and Felicetti, L. (1995). Physical mapping evidence for a duplicated region on chromosome 10qter showing high homology with the facioscapulohumeral muscular dystrophy locus on chromosome 4qter. Eur. J. Hum. Genet. 3, 155–167. doi:10.1159/000472291 |
Denny, A. P., and Heather, A. K. (2017). Are antioxidants a potential therapy for FSHD? A review of the literature. Oxid. Med. Cel. Longev. 2017, 7020295. doi:10.1155/2017/7020295
Derenne, A., Tassin, A., Nguyen, T. H., De Roeck, E., Jenart, V., Ansseau, E., et al. (2020). Induction of a local muscular dystrophy using electroporation in vivo: an easy tool for screening therapeutics. Sci. Rep. 10, 11301. doi:10.1038/s41598-020-68135-7 |
DeSimone, A. M., Cohen, J., Lek, M., and Lek, A. (2020). Cellular and animal models for facioscapulohumeral muscular dystrophy. Dis. Model. Mech. 13, dmm046904. doi:10.1242/dmm.046904 |
Dieckmann, A., Hagedorn, P. H., Burki, Y., Brügmann, C., Berrera, M., Ebeling, M., et al. (2018). A sensitive in vitro approach to assess the hybridization-dependent toxic potential of high affinity gapmer oligonucleotides. Mol. Ther. Nucleic Acids 10, 45–54. doi:10.1016/j.omtn.2017.11.004 |
Dixit, M., Ansseau, E., Tassin, A., Winokur, S., Shi, R., Qian, H., et al. (2007). DUX4, a candidate gene of facioscapulohumeral muscular dystrophy, encodes a transcriptional activator of PITX1. Proc. Natl. Acad. Sci. USA 104, 18157–18162. doi:10.1073/pnas.0708659104 |
Dmitriev, P., Bou Saada, Y., Dib, C., Ansseau, E., Barat, A., Hamade, A., et al. (2016). DUX4-induced constitutive DNA damage and oxidative stress contribute to aberrant differentiation of myoblasts from FSHD patients. Free Radic. Biol. Med. 99, 244–258. doi:10.1016/j.freeradbiomed.2016.08.007 |
Dominska, M., and Dykxhoorn, D. M. (2010). Breaking down the barriers: siRNA delivery and endosome escape. J. Cel Sci. 123, 1183–1189. doi:10.1242/jcs.066399
Echigoya, Y., Lim, K. R. Q., Trieu, N., Bao, B., Miskew Nichols, B., Vila, M. C., et al. (2017). Quantitative antisense screening and optimization for exon 51 skipping in Duchenne muscular dystrophy. Mol. Ther. 25, 2561. doi:10.1016/j.ymthe.2017.07.014 |
Ehrke-Schulz, E., Schiwon, M., Leitner, T., Dávid, S., Bergmann, T., Liu, J., et al. (2017). CRISPR/Cas9 delivery with one single adenoviral vector devoid of all viral genes. Sci. Rep. 7, 17113. doi:10.1038/s41598-017-17180-w |
Ferdosi, S. R., Ewaisha, R., Moghadam, F., Krishna, S., Park, J. G., Ebrahimkhani, M. R., et al. (2019). Multifunctional CRISPR-Cas9 with engineered immunosilenced human T cell epitopes. Nat. Commun. 10, 1842. doi:10.1038/s41467-019-09693-x |
Ferri, G., Huichalaf, C. H., Caccia, R., and Gabellini, D. (2015). Direct interplay between two candidate genes in FSHD muscular dystrophy. Hum. Mol. Genet. 24, 1256–1266. doi:10.1093/hmg/ddu536 |
Fitzsimons, R. B., Gurwin, E. B., and Bird, A. C. (1987). Retinal vascular abnormalities in facioscapulohumeral muscular dystrophy. A general association with genetic and therapeutic implications. Brain 110 (Pt 3), 631–648. doi:10.1093/brain/110.3.631 |
Frazier, K. S. (2015). Antisense oligonucleotide therapies: the promise and the challenges from a toxicologic pathologist’s perspective. Toxicol. Pathol. 43, 78–89. doi:10.1177/0192623314551840 |
Gabriëls, J., Beckers, M. C., Ding, H., De Vriese, A., Plaisance, S., van der Maarel, S. M., et al. (1999). Nucleotide sequence of the partially deleted D4Z4 locus in a patient with FSHD identifies a putative gene within each 3.3 kb element. Gene 236, 25–32. doi:10.1016/s0378-1119(99)00267-x |
Geng, L. N., Tyler, A. E., and Tapscott, S. J. (2011). Immunodetection of human double homeobox 4. Hybridoma 30, 125–130. doi:10.1089/hyb.2010.0094 |
Giesige, C. R., Wallace, L. M., Heller, K. N., Eidahl, J. O., Saad, N. Y., Fowler, A. M., et al. (2018). AAV-mediated follistatin gene therapy improves functional outcomes in the TIC-DUX4 mouse model of FSHD. JCI Insight 3, e123538. doi:10.1172/jci.insight.123538
Goossens, R., van den Boogaard, M. L., Lemmers, R. J. L. F., Balog, J., van der Vliet, P. J., Willemsen, I. M., et al. (2019). Intronic SMCHD1 variants in FSHD: testing the potential for CRISPR-Cas9 genome editing. J. Med. Genet. 56, 828–837. doi:10.1136/jmedgenet-2019-106402 |
Goselink, R. J. M., Voermans, N. C., Okkersen, K., Brouwer, O. F., Padberg, G. W., Nikolic, A., et al. (2017). Early onset facioscapulohumeral dystrophy—a systematic review using individual patient data. Neuromuscul. Disord. 27, 1077–1083. doi:10.1016/j.nmd.2017.09.007 |
Hagedorn, P. H., Yakimov, V., Ottosen, S., Kammler, S., Nielsen, N. F., Høg, A. M., et al. (2013). Hepatotoxic potential of therapeutic oligonucleotides can Be predicted from their sequence and modification pattern. Nucleic Acid Ther. 23, 302–310. doi:10.1089/nat.2013.0436 |
Hamanaka, K., Šikrová, D., Mitsuhashi, S., Masuda, H., Sekiguchi, Y., Sugiyama, A., et al. (2020). Homozygous nonsense variant in LRIF1 associated with facioscapulohumeral muscular dystrophy. Neurology 94, e2441–e2447. doi:10.1212/WNL.0000000000009617 |
Hamel, J., and Tawil, R. (2018). Facioscapulohumeral muscular dystrophy: update on pathogenesis and future treatments. Neurotherapeutics 15, 863–871. doi:10.1007/s13311-018-00675-3 |
Hendling, M., and Barišić, I. (2019). In-silico design of DNA oligonucleotides: challenges and approaches. Comput. Struct. Biotechnol. J. 17, 1056–1065. doi:10.1016/j.csbj.2019.07.008 |
Hendrickson, P. G., Doráis, J. A., Grow, E. J., Whiddon, J. L., Lim, J. W., Wike, C. L., et al. (2017). Conserved roles of mouse DUX and human DUX4 in activating cleavage-stage genes and MERVL/HERVL retrotransposons. Nat. Genet. 49, 925–934. doi:10.1038/ng.3844 |
Hewitt, J. E. (2015). Loss of epigenetic silencing of the DUX4 transcription factor gene in facioscapulohumeral muscular dystrophy. Hum. Mol. Genet. 24, R17–R23. doi:10.1093/hmg/ddv237 |
Hewitt, J. E., Lyle, R., Clark, L. N., Valleley, E. M., Wright, T. J., Wijmenga, C., et al. (1994). Analysis of the tandem repeat locus D4Z4 associated with facioscapulohumeral muscular dystrophy. Hum. Mol. Genet. 3, 1287–1295. doi:10.1093/hmg/3.8.1287 |
Himeda, C. L., Debarnot, C., Homma, S., Beermann, M. L., Miller, J. B., Jones, P. L., et al. (2014). Myogenic enhancers regulate expression of the facioscapulohumeral muscular dystrophy-associated DUX4 gene. Mol. Cel. Biol. 34, 1942–1955. doi:10.1128/MCB.00149-14
Himeda, C. L., and Jones, P. L. (2019). The genetics and epigenetics of facioscapulohumeral muscular dystrophy. Annu. Rev. Genomics Hum. Genet. 20, 265–291. doi:10.1146/annurev-genom-083118-014933 |
Himeda, C. L., Jones, T. I., and Jones, P. L. (2016). CRISPR/dCas9-mediated transcriptional inhibition ameliorates the epigenetic dysregulation at D4Z4 and represses DUX4-fl in FSH muscular dystrophy. Mol. Ther. 24, 527–535. doi:10.1038/mt.2015.200 |
Himeda, C. L., Jones, T. I., Virbasius, C. M., Zhu, L. J., Green, M. R., and Jones, P. L. (2018). Identification of epigenetic regulators of DUX4-fl for targeted therapy of facioscapulohumeral muscular dystrophy. Mol. Ther. 26, 1797–1807. doi:10.1016/j.ymthe.2018.04.019 |
Holoch, D., and Moazed, D. (2015). RNA-mediated epigenetic regulation of gene expression. Nat. Rev. Genet. 16, 71–84. doi:10.1038/nrg3863 |
Jiang, F., and Doudna, J. A. (2017). CRISPR-Cas9 structures and mechanisms. Annu. Rev. Biophys. 46, 505–529. doi:10.1146/annurev-biophys-062215-010822 |
Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., and Charpentier, E. (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. doi:10.1126/science.1225829 |
Jones, T., and Jones, P. L. (2018). A cre-inducible DUX4 transgenic mouse model for investigating facioscapulohumeral muscular dystrophy. PLoS One 13, e0192657. doi:10.1371/journal.pone.0192657 |
Juliano, R. L. (2016). The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 44, 6518–6548. doi:10.1093/nar/gkw236 |
Kakiuchi-Kiyota, S., Whiteley, L. O., Ryan, A. M., and Mathialagan, N. (2016). Development of a method for profiling protein interactions with LNA-modified antisense oligonucleotides using protein microarrays. Nucleic Acid Ther. 26, 93–101. doi:10.1089/nat.2015.0576 |
Kamola, P. J., Kitson, J. D., Turner, G., Maratou, K., Eriksson, S., Panjwani, A., et al. (2015). In silico and in vitro evaluation of exonic and intronic off-target effects form a critical element of therapeutic ASO gapmer optimization. Nucleic Acids Res. 43, 8638–8650. doi:10.1093/nar/gkv857 |
Kim, D., Bae, S., Park, J., Kim, E., Kim, S., Yu, H. R., et al. (2015). Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat. Methods 12, 237–243. doi:10.1038/nmeth.3284 |
Kim, D. H., Longo, M., Han, Y., Lundberg, P., Cantin, E., and Rossi, J. J. (2004). Interferon induction by siRNAs and ssRNAs synthesized by phage polymerase. Nat. Biotechnol. 22, 321–325. doi:10.1038/nbt940 |
Kim, E., Koo, T., Park, S. W., Kim, D., Kim, K., Cho, H. Y., et al. (2017). In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nat. Commun. 8, 14500. doi:10.1038/ncomms14500 |
Kim, S., Koo, T., Jee, H.-G., Cho, H.-Y., Lee, G., Lim, D.-G., et al. (2018). CRISPR RNAs trigger innate immune responses in human cells. Genome Res. 28, 367–373. doi:10.1101/gr.231936.117
Klinge, L., Eagle, M., Haggerty, I. D., Roberts, C. E., Straub, V., and Bushby, K. M. (2006). Severe phenotype in infantile facioscapulohumeral muscular dystrophy. Neuromuscul. Disord. 16, 553–558. doi:10.1016/j.nmd.2006.06.008 |
Klingler, C., Ashley, J., Shi, K., Stiefvater, A., Kyba, M., Sinnreich, M., et al. (2020). DNA aptamers against the DUX4 protein reveal novel therapeutic implications for FSHD. FASEB J. 34, 4573–4590. doi:10.1096/fj.201902696 |
Laforêt, P., de Toma, C., Eymard, B., Becane, H. M., Jeanpierre, M., Fardeau, M., et al. (1998). Cardiac involvement in genetically confirmed facioscapulohumeral muscular dystrophy. Neurology 51, 1454–1456. doi:10.1212/wnl.51.5.1454 |
Le Gall, L., Sidlauskaite, E., Mariot, V., and Dumonceaux, J. (2020). Therapeutic strategies targeting DUX4 in FSHD. J. Clin. Med. 9, 2886. doi:10.3390/jcm9092886
Lee, J. J., and Yokota, T. (2013). Antisense therapy in neurology. J. Pers. Med. 3, 144–176. doi: doi:10.3390/jpm3030144 |
Lek, A., Zhang, Y., Woodman, K. G., Huang, S., DeSimone, A. M., Cohen, J., et al. (2020). Applying genome-wide CRISPR-Cas9 screens for therapeutic discovery in facioscapulohumeral muscular dystrophy. Sci. Transl. Med. 12, eaay0271. doi:10.1126/scitranslmed.aay0271 |
Lemmers, R. J., de Kievit, P., Sandkuijl, L., Padberg, G. W., van Ommen, G. J., Frants, R. R., et al. (2002). Facioscapulohumeral muscular dystrophy is uniquely associated with one of the two variants of the 4q subtelomere. Nat. Genet. 32, 235–236. doi:10.1038/ng999 |
Lemmers, R. J., Goeman, J. J., van der Vliet, P. J., van Nieuwenhuizen, M. P., Balog, J., Vos-Versteeg, M., et al. (2015). Inter-individual differences in CpG methylation at D4Z4 correlate with clinical variability in FSHD1 and FSHD2. Hum. Mol. Genet. 24, 659–669. doi:10.1093/hmg/ddu486 |
Lemmers, R. J., Tawil, R., Petek, L. M., Balog, J., Block, G. J., Santen, G. W., et al. (2012). Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat. Genet. 44, 1370–1374. doi:10.1038/ng.2454 |
Lemmers, R. J., van der Vliet, P. J., Klooster, R., Sacconi, S., Camaño, P., Dauwerse, J. G., et al. (2010). A unifying genetic model for facioscapulohumeral muscular dystrophy. Science 329, 1650–1653. doi:10.1126/science.1189044 |
Lemmers, R. J., Wohlgemuth, M., Frants, R. R., Padberg, G. W., Morava, E., and van der Maarel, S. M. (2004). Contractions of D4Z4 on 4qB subtelomeres do not cause facioscapulohumeral muscular dystrophy. Am. J. Hum. Genet. 75, 1124–1130. doi:10.1086/426035 |
Liang, X., Sun, H., Shen, W., and Crooke, S. T. (2015). Identification and characterization of intracellular proteins that bind oligonucleotides with phosphorothioate linkages. Nucleic Acids Res. 43, 2927–2945. doi:10.1093/nar/gkv143 |
Lim, J. W., Snider, L., Yao, Z., Tawil, R., Van Der Maarel, S. M., Rigo, F., et al. (2015). DICER/AGO-dependent epigenetic silencing of D4Z4 repeats enhanced by exogenous siRNA suggests mechanisms and therapies for FSHD. Hum. Mol. Genet. 24, 4817–4828. doi:10.1093/hmg/ddv206 |
Lim, K. R. Q., Bittel, A., Maruyama, R., Echigoya, Y., Nguyen, Q., Huang, Y., et al. (2020a). DUX4 transcript knockdown with antisense 2′-O-methoxyethyl gapmers for the treatment of facioscapulohumeral muscular dystrophy. Mol. Ther. 29, 848–858. doi:10.1016/j.ymthe.2020.10.010 |
Lim, K. R. Q., Maruyama, R., Echigoya, Y., Nguyen, Q., Zhang, A., Khawaja, H., et al. (2020b). Inhibition of DUX4 expression with antisense LNA gapmers as a therapy for facioscapulohumeral muscular dystrophy. Proc. Natl. Acad. Sci. U S A. 117, 16509–16515. doi:10.1073/pnas.1909649117 |
Lim, K. R. Q., Nguyen, Q., and Yokota, T. (2020c). DUX4 signalling in the pathogenesis of facioscapulohumeral muscular dystrophy. Int. J. Mol. Sci. 21, 729. doi:10.3390/ijms21030729
Lim, K. R. Q., Echigoya, Y., Nagata, T., Kuraoka, M., Kobayashi, M., Aoki, Y., et al. (2019). Efficacy of multi-exon skipping treatment in Duchenne muscular dystrophy dog model neonates. Mol. Ther. 27. doi:10.1016/j.ymthe.2018.10.011
Lim, K. R. Q., and Yokota, T. (2018). “Invention and early history of exon skipping and splice modulation,” in Exon skipping and inclusion therapies: methods and protocols. Editors T. Yokota, and R. Maruyama (New York, NY: Springer), 3–30.
Lim, K. R. Q., and Yokota, T. (2020). Invention and early history of gapmers. Methods Mol. Biol. 2176, 3–19. doi:10.1007/978-1-0716-0771-8_1 |
Lino, C. A., Harper, J. C., Carney, J. P., and Timlin, J. A. (2018). Delivering CRISPR: a review of the challenges and approaches. Drug Deliv. 25, 1234–1257. doi:10.1080/10717544.2018.1474964 |
Lunt, P. W., Jardine, P. E., Koch, M. C., Maynard, J., Osborn, M., Williams, M., et al. (1995). Correlation between fragment size at D4F104S1 and age at onset or at wheelchair use, with a possible generational effect, accounts for much phenotypic variation in 4q35-facioscapulohumeral muscular dystrophy (FSHD). Hum. Mol. Genet. 4, 951–958. doi:10.1093/hmg/4.5.951 |
Lutz, K. L., Holte, L., Kliethermes, S. A., Stephan, C., and Mathews, K. D. (2013). Clinical and genetic features of hearing loss in facioscapulohumeral muscular dystrophy. Neurology 81, 1374–1377. doi:10.1212/WNL.0b013e3182a84140 |
Mariot, V., Joubert, R., Marsollier, A.-C., Hourdé, C., Voit, T., and Dumonceaux, J. (2020). A deoxyribonucleic acid decoy trapping DUX4 for the treatment of facioscapulohumeral muscular dystrophy. Mol. Ther. Nucleic Acids. doi:10.1016/j.omtn.2020.10.028
Marsollier, A. C., Ciszewski, L., Mariot, V., Popplewell, L., Voit, T., Dickson, G., et al. (2016). Antisense targeting of 3' end elements involved in DUX4 mRNA processing is an efficient therapeutic strategy for facioscapulohumeral dystrophy: a new gene-silencing approach. Hum. Mol. Genet. 25, 1468–1478. doi:10.1093/hmg/ddw015 |
Mehta, A., and Merkel, O. M. (2020). Immunogenicity of Cas9 protein. J. Pharm. Sci. 109, 62–67. doi:10.1016/j.xphs.2019.10.003 |
Mitsuhashi, H., Ishimaru, S., Homma, S., Yu, B., Honma, Y., Beermann, M. L., et al. (2018). Functional domains of the FSHD-associated DUX4 protein. Biol. Open 7, bio033977. doi:10.1242/bio.033977 |
Mitsuhashi, H., Mitsuhashi, S., Lynn-Jones, T., Kawahara, G., and Kunkel, L. M. (2013). Expression of DUX4 in zebrafish development recapitulates facioscapulohumeral muscular dystrophy. Hum. Mol. Genet. 22, 568–577. doi:10.1093/hmg/dds467 |
Mojica, F. J., Díez-Villaseñor, C., García-Martínez, J., and Almendros, C. (2009). Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 155, 733–740. doi:10.1099/mic.0.023960-0 |
Moore, R., Spinhirne, A., Lai, M. J., Preisser, S., Li, Y., Kang, T., et al. (2015). CRISPR-based self-cleaving mechanism for controllable gene delivery in human cells. Nucleic Acids Res. 43, 1297–1303. doi:10.1093/nar/gku1326 |
Morcos, P. A., Li, Y., and Jiang, S. (2008). Vivo-Morpholinos: a non-peptide transporter delivers Morpholinos into a wide array of mouse tissues. Biotechniques 45, 613–passim. doi:10.2144/000113005 |
Morosetti, R., Gidaro, T., Broccolini, A., Gliubizzi, C., Sancricca, C., Tonali, P. A., et al. (2011). Mesoangioblasts from facioscapulohumeral muscular dystrophy display in Vivo a variable myogenic ability predictable by their in Vitro behavior. Cell Transplant. 20, 1299–1313. doi:10.3727/096368910X546571 |
Nguyen, Q., and Yokota, T. (2019). Antisense oligonucleotides for the treatment of cardiomyopathy in Duchenne muscular dystrophy. Am. J. Transl. Res. 11, 1202–1218.
Nikolic, A., Ricci, G., Sera, F., Bucci, E., Govi, M., Mele, F., et al. (2016). Clinical expression of facioscapulohumeral muscular dystrophy in carriers of 1-3 D4Z4 reduced alleles: experience of the FSHD Italian national registry. BMJ Open 6, e007798. doi:10.1136/bmjopen-2015-007798 |
O’Connell, M. R., Oakes, B. L., Sternberg, S. H., East-Seletsky, A., Kaplan, M., and Doudna, J. A. (2014). Programmable RNA recognition and cleavage by CRISPR/Cas9. Nature 516, 263–266. doi:10.1038/nature13769 |
Padberg, G. W., Brouwer, O. F., de Keizer, R. J., Dijkman, G., Wijmenga, C., Grote, J. J., et al. (1995). On the significance of retinal vascular disease and hearing loss in facioscapulohumeral muscular dystrophy. Muscle Nerve Suppl. 2, S73–S80. doi:10.1002/mus.880181314 |
Pandey, S. N., Cabotage, J., Shi, R., Dixit, M., Sutherland, M., Liu, J., et al. (2012). Conditional over-expression of PITX1 causes skeletal muscle dystrophy in mice. Biol. Open 1, 629–639. doi:10.1242/bio.20121305 |
Pandey, S. N., Khawaja, H., and Chen, Y. W. (2015). Culture conditions affect expression of DUX4 in FSHD myoblasts. Molecules 20, 8304–8315. doi:10.3390/molecules20058304 |
Pandey, S. N., Lee, Y. C., Yokota, T., and Chen, Y. W. (2014). Morpholino treatment improves muscle function and pathology of Pitx1 transgenic mice. Mol. Ther. 22, 390–396. doi:10.1038/mt.2013.263 |
Ran, F. A., Hsu, P. D., Lin, C. Y., Gootenberg, J. S., Konermann, S., Trevino, A. E., et al. (2013). Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154, 1380–1389. doi:10.1016/j.cell.2013.08.021 |
Ricci, G., Scionti, I., Sera, F., Govi, M., D'Amico, R., Frambolli, I., et al. (2013). Large scale genotype-phenotype analyses indicate that novel prognostic tools are required for families with facioscapulohumeral muscular dystrophy. Brain 136, 3408–3417. doi:10.1093/brain/awt226 |
Richards, M., Coppée, F., Thomas, N., Belayew, A., and Upadhyaya, M. (2012). Facioscapulohumeral muscular dystrophy (FSHD): an enigma unravelled?. Hum. Genet. 131, 325–340. doi:10.1007/s00439-011-1100-z |
Rickard, A. M., Petek, L. M., and Miller, D. G. (2015). Endogenous DUX4 expression in FSHD myotubes is sufficient to cause cell death and disrupts RNA splicing and cell migration pathways. Hum. Mol. Genet. 24, 5901–5914. doi:10.1093/hmg/ddv315 |
Ricobaraza, A., Gonzalez-Aparicio, M., Mora-Jimenez, L., Lumbreras, S., and Hernandez-Alcoceba, R. (2020). High-capacity adenoviral vectors: expanding the scope of gene therapy. Int. J. Mol. Sci. 21, 3643. doi:10.3390/ijms21103643
Rodino-Klapac, L. R., Haidet, A. M., Kota, J., Handy, C., Kaspar, B. K., and Mendell, J. R. (2009). Inhibition of myostatin with emphasis on follistatin as a therapy for muscle disease. Muscle Nerve 39, 283–296. doi:10.1002/mus.21244 |
Sacconi, S., Briand-Suleau, A., Gros, M., Baudoin, C., Lemmers, R. J. L. F., Rondeau, S., et al. (2019). FSHD1 and FSHD2 form a disease continuum. Neurology 92, e2273–e2285. doi:10.1212/WNL.0000000000007456 |
Sacconi, S., Lemmers, R. J., Balog, J., van der Vliet, P. J., Lahaut, P., van Nieuwenhuizen, M. P., et al. (2013). The FSHD2 gene SMCHD1 is a modifier of disease severity in families affected by FSHD1. Am. J. Hum. Genet. 93, 744–751. doi:10.1016/j.ajhg.2013.08.004 |
Scully, M. A., Eichinger, K. J., Donlin-Smith, C. M., Tawil, R., and Statland, J. M. (2014). Restrictive lung involvement in facioscapulohumeral muscular dystrophy. Muscle Nerve 50, 739–743. doi:10.1002/mus.24218 |
Shen, B., Zhang, W., Zhang, J., Zhou, J., Wang, J., Chen, L., et al. (2014). Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat. Methods 11, 399–402. doi:10.1038/nmeth.2857 |
Shen, W., De Hoyos, C. L., Sun, H., Vickers, T. A., Liang, X., and Crooke, S. T. (2018). Acute hepatotoxicity of 2' fluoro-modified 5-10-5 gapmer phosphorothioate oligonucleotides in mice correlates with intracellular protein binding and the loss of DBHS proteins. Nucleic Acids Res. 46, 2204–2217. doi:10.1093/nar/gky060 |
Shen, W., De Hoyos, C. L., Migawa, M. T., Vickers, T. A., Sun, H., Low, A., et al. (2019). Chemical modification of PS-ASO therapeutics reduces cellular protein-binding and improves the therapeutic index. Nat. Biotechnol. 37, 640–650. doi:10.1038/s41587-019-0106-2 |
Shirley, J. L., de Jong, Y. P., Terhorst, C., and Herzog, R. W. (2020). Immune responses to viral gene therapy vectors. Mol. Ther. 28, 709–722. doi:10.1016/j.ymthe.2020.01.001 |
Snider, L., Geng, L. N., Lemmers, R. J., Kyba, M., Ware, C. B., Nelson, A. M., et al. (2010). Facioscapulohumeral dystrophy: incomplete suppression of a retrotransposed gene. Plos Genet. 6, e1001181. doi:10.1371/journal.pgen.1001181 |
Summerton, J., and Weller, D. (1997). Morpholino antisense oligomers: design, preparation, and properties. Antisense Nucleic Acid Drug Dev. 7, 187–195. doi:10.1089/oli.1.1997.7.187 |
Tassin, A., Laoudj-Chenivesse, D., Vanderplanck, C., Barro, M., Charron, S., Ansseau, E., et al. (2013). DUX4 expression in FSHD muscle cells: how could such a rare protein cause a myopathy?. J. Cel. Mol. Med. 17, 76–89. doi:10.1111/j.1582-4934.2012.01647.x
Tawil, R., van der Maarel, S. M., and Tapscott, S. J. (2014). Facioscapulohumeral dystrophy: the path to consensus on pathophysiology. Skelet. Muscle 4, 12. doi:10.1186/2044-5040-4-12 |
Tong, S., Moyo, B., Lee, C. M., Leong, K., and Bao, G. (2019). Engineered materials for in vivo delivery of genome-editing machinery. Nat. Rev. Mater. 4, 726–737. doi:10.1038/s41578-019-0145-9
Tsai, S. Q., Zheng, Z., Nguyen, N. T., Liebers, M., Topkar, V. V., Thapar, V., et al. (2015). GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 33, 187–197. doi:10.1038/nbt.3117 |
Tycko, J., Myer, V. E., and Hsu, P. D. (2016). Methods for optimizing CRISPR-cas9 genome editing specificity. Mol. Cell 63, 355–370. doi:10.1016/j.molcel.2016.07.004 |
van den Boogaard, M. L., Lemmers, R. J. L. F., Balog, J., Wohlgemuth, M., Auranen, M., Mitsuhashi, S., et al. (2016). Mutations in DNMT3B modify epigenetic repression of the D4Z4 repeat and the penetrance of facioscapulohumeral dystrophy. Am. J. Hum. Genet. 98, 1020–1029. doi:10.1016/j.ajhg.2016.03.013 |
Van Overveld, P. G., Enthoven, L., Ricci, E., Rossi, M., Felicetti, L., Jeanpierre, M., et al. (2005). Variable hypomethylation of D4Z4 in facioscapulohumeral muscular dystrophy. Ann. Neurol. 58, 569–576. doi:10.1002/ana.20625 |
van Overveld, P. G., Lemmers, R. J., Sandkuijl, L. A., Enthoven, L., Winokur, S. T., Bakels, F., et al. (2003). Hypomethylation of D4Z4 in 4q-linked and non-4q-linked facioscapulohumeral muscular dystrophy. Nat. Genet. 35, 315–317. doi:10.1038/ng1262 |
Vanderplanck, C., Ansseau, E., Charron, S., Stricwant, N., Tassin, A., Laoudj-Chenivesse, D., et al. (2011). The FSHD atrophic myotube phenotype is caused by DUX4 expression. PLoS One 6, e26820. doi:10.1371/journal.pone.0026820 |
Vilquin, J. T., Marolleau, J. P., Sacconi, S., Garcin, I., Lacassagne, M. N., Robert, I., et al. (2005). Normal growth and regenerating ability of myoblasts from unaffected muscles of facioscapulohumeral muscular dystrophy patients. Gene Ther. 12, 1651–1662. doi:10.1038/sj.gt.3302565 |
Wagner, D. L., Amini, L., Wendering, D. J., Burkhardt, L. M., Akyüz, L., Reinke, P., et al. (2019). High prevalence of Streptococcus pyogenes Cas9-reactive T cells within the adult human population. Nat. Med. 25, 242–248. doi:10.1038/s41591-018-0204-6 |
Wallace, L. M., Liu, J., Domire, J. S., Garwick-Coppens, S. E., Guckes, S. M., Mendell, J. R., et al. (2012). RNA interference inhibits DUX4-induced muscle toxicity in vivo: implications for a targeted FSHD therapy. Mol. Ther. 20, 1417–1423. doi:10.1038/mt.2012.68 |
Wallace, L. M., Saad, N. Y., Pyne, N. K., Fowler, A. M., Eidahl, J. O., Domire, J. S., et al. (2018). Pre-clinical safety and off-target studies to support translation of AAV-mediated RNAi therapy for FSHD. Mol. Ther. Methods Clin. Dev. 8, 121–130. doi:10.1016/j.omtm.2017.12.005 |
Wang, D., Zhang, C., Wang, B., Li, B., Wang, Q., Liu, D., et al. (2019). Optimized CRISPR guide RNA design for two high-fidelity Cas9 variants by deep learning. Nat. Commun. 10, 4284. doi:10.1038/s41467-019-12281-8 |
Wang, L. H., and Tawil, R. (2016). Facioscapulohumeral dystrophy. Curr. Neurol. Neurosci. Rep. 16, 66. doi:10.1007/s11910-016-0667-0 |
Wienert, B., Shin, J., Zelin, E., Pestal, K., and Corn, J. E. (2018). In vitro-transcribed guide RNAs trigger an innate immune response via the RIG-I pathway. PLoS Biol. 16, e2005840. doi:10.1371/journal.pbio.2005840 |
Wilbie, D., Walther, J., and Mastrobattista, E. (2019). Delivery aspects of CRISPR/Cas for in Vivo genome editing. Acc. Chem. Res. 52, 1555–1564. doi:10.1021/acs.accounts.9b00106 |
Wu, Z., Yang, H., and Colosi, P. (2010). Effect of genome size on AAV vector packaging. Mol. Ther. 18, 80–86. doi:10.1038/mt.2009.255 |
Xu, C. L., Ruan, M. Z. C., Mahajan, V. B., and Tsang, S. H. (2019). Viral delivery systems for CRISPR. Viruses 11, 28. doi:10.3390/v11010028
Keywords: facioscapulohumeral muscular dystrophy, DUX4, antisense oligonucleotides, RNAi, DNA decoys, DNA aptamers, CRISPR, gene modulation
Citation: Lim KRQ and Yokota T (2021) Genetic Approaches for the Treatment of Facioscapulohumeral Muscular Dystrophy. Front. Pharmacol. 12:642858. doi: 10.3389/fphar.2021.642858
Received: 16 December 2020; Accepted: 01 February 2021;
Published: 12 March 2021.
Edited by:
Carlo Rinaldi, University of Oxford, United KingdomReviewed by:
Qing Lyu, Chongqing Institute of Green and Intelligent Technology, ChinaJudit Balog, Leiden University Medical Center, Netherlands
Copyright © 2021 Lim and Yokota. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Toshifumi Yokota, dG9zaGlmdW1pLnlva290YUB1YWxiZXJ0YS5jYQ==