 Lisa Künzi
Lisa Künzi Molly Easter
Molly Easter Meghan June Hirsch1
Meghan June Hirsch1 Stefanie Krick
Stefanie Krick- 1Division of Pulmonary, Allergy and Critical Care Medicine, University of Alabama at Birmingham, Birmingham, AL, United States
- 2Epidemiology, Biostatistics and Prevention Institute, Department of Public and Global Health, University of Zürich, Zürich, Switzerland
- 3Gregory Fleming Cystic Fibrosis Research Center, University of Alabama at Birmingham, Birmingham, AL, United States
- 4Comprehensive Center for Healthy Aging, University of Alabama at Birmingham, Birmingham, AL, United States
The demographics of the population with cystic fibrosis (CF) is continuously changing, with nowadays adults outnumbering children and a median predicted survival of over 40 years. This leads to the challenge of treating an aging CF population, while previous research has largely focused on pediatric and adolescent patients. Chronic inflammation is not only a hallmark of CF lung disease, but also of the aging process. However, very little is known about the effects of an accelerated aging pathology in CF lungs. Several chronic lung disease pathologies show signs of chronic inflammation with accelerated aging, also termed “inflammaging”; the most notable being chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis (IPF). In these disease entities, accelerated aging has been implicated in the pathogenesis via interference with tissue repair mechanisms, alterations of the immune system leading to impaired defense against pulmonary infections and induction of a chronic pro-inflammatory state. In addition, CF lungs have been shown to exhibit increased expression of senescence markers. Sustained airway inflammation also leads to the degradation and increased turnover of cystic fibrosis transmembrane regulator (CFTR). This further reduces CFTR function and may prevent the novel CFTR modulator therapies from developing their full efficacy. Therefore, novel therapies targeting aging processes in CF lungs could be promising. This review summarizes the current research on CF in an aging population focusing on accelerated aging in the context of chronic airway inflammation and therapy implications.
Introduction
30 years ago, mutations in the cystic fibrosis transmembrane regulator (CFTR) gene were characterized as single gene defect leading to the pathology of cystic fibrosis (CF) (Kerem et al., 1989; Tsui, 1995). Since then, our understanding of the pathological processes has largely improved with CFTR targeted modulator therapies in clinical use (Wainwright et al., 2015; Rowe et al., 2017; Taylor-Cousar et al., 2017; Davies et al., 2018; Keating et al., 2018). Nevertheless, progressive lung disease is still the major cause for morbidity and mortality in individuals born with CF (Elborn, 2016). Functional failure of CFTR results in impaired ciliary function, mucus obstruction, bacterial colonization and chronic inflammation (Matsui et al., 1998; Mall et al., 2004; Boucher, 2007). This sets the stage for smoldering chronic infections interrupted by episodic exacerbations due to newly acquired pathogens leading to progressive loss of lung function (Nichols et al., 2008; Elborn, 2016). Elevated levels of pro-inflammatory mediators, reactive oxygen species (ROS), and proteases, which further propagate airway inflammation, can accelerate cellular senescence (Bezzerri et al., 2019).
Major improvements in supportive therapies and the development of CFTR targeted therapies increased life expectancy in CF patients to a median survival of 40 years or more in industrialized nations (Dodge et al., 2007; MacKenzie et al., 2014). Hence, CF is no longer considered primarily a pediatric disease. This entails new challenges, including treatment of CF in an aging population with CF dependent and independent comorbidities. Diabetes, osteopenia and osteoporosis, renal, and vestibulocholear complications from aminoglycoside toxicity, cardiovascular disease, and worsening lung disease are medical issues often emerging in the aging CF population (Hodson et al., 2008; Simmonds et al., 2009). Chronic local and systemic inflammation is a key pathogenic mechanisms suspected of promoting these comorbidities (Nichols et al., 2008; Bezzerri et al., 2019). Aging impairs lung function through several mechanisms, a central being chronic, low-grade inflammation termed “inflammaging” (Franceschi and Campisi, 2014). Accelerated aging has been implicated in several chronic inflammatory lung disease pathologies. As chronic inflammation is a hallmark of CF airway disease, the question arises whether this chronic inflammation may also cause premature aging in CF lung tissue; and if so whether novel therapies targeting “inflammaging” and aging pathomechanisms in CF lungs might prove to be beneficial.
Hallmarks of Aging in CF Lung Disease
Physiological aging is associated with considerable changes in lung structure and cell function, giving rise to reduced lung function, pulmonary remodeling, diminished regenerative capacity, and enhanced susceptibility to lung injury, and disease (Cho and Stout-Delgado, 2020). Considering the increased life expectancy of CF patients, the question whether chronic inflammation contributes to accelerated aging becomes more and more relevant. Interestingly, healthy aging lungs show a proinflammatory state with increased neutrophils and IL-8 (Meyer et al., 1998), bearing some similarity with chronic CF lung inflammation. In the following paragraphs we aim to provide an overview on the hallmarks of aging and their potential implications for CF lung disease. Central findings on accelerated aging in CF are also summarized in Figure 1.
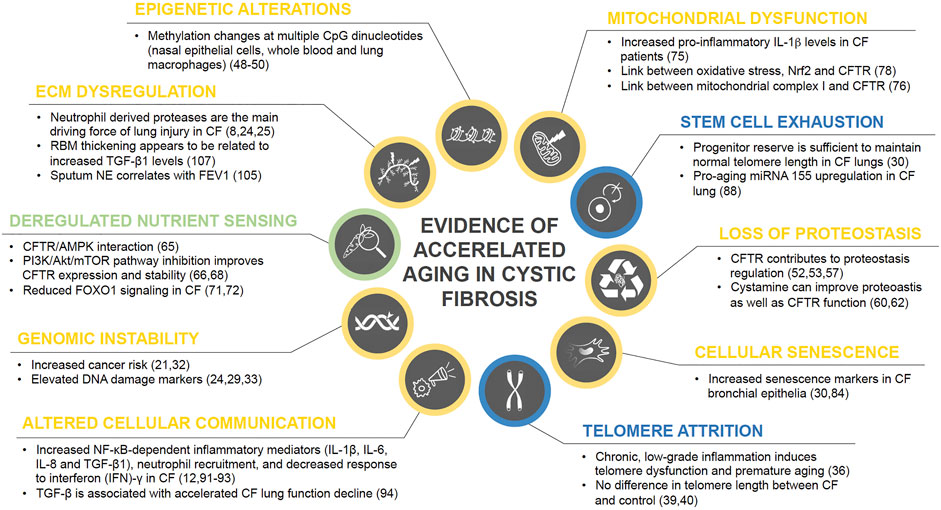
FIGURE 1. Current state of evidence on accelerated aging processes in CF. Research findings on the role of each of the ten hallmarks of aging in CF disease are summarized and color coded based on their level of evidence with blue = no evidence for involvement in CF pathology, green = weak evidence, yellow = strong evidence.
Genomic Instability
Non-transplanted CF patients have a higher risk for lymphoid leukemia, testicular cancer and digestive tract cancers than the general population (Maisonneuve et al., 2013). Similarly, cancer risk in CF lung transplant recipients is more increased than in non-CF recipients, particularly for colorectal cancer, esophageal cancer, and non-Hodgkin lymphoma (Fink et al., 2017). Defects in CFTR themselves have been linked to cancers (Scott et al., 2020). The striking overlap of organ systems with elevated cancer risk and those that are primarily affected by CF-associated mucus hypersecretion, i.e., the respiratory, digestive, and reproductive systems, strongly implicates chronic inflammation as a potential causative factor. In CF airway inflammation, large numbers of activated neutrophils release reactive oxygen species (ROS) and proteases, such as neutrophil elastase, thereby inducing additional ROS production in lung cells (Brown and Kelly, 1994; Aoshiba et al., 2001). In addition, reduced antioxidant defense with systemic glutathione deficiency, reduced vitamin E plasma levels, and increased mitochondrial oxidative stress has been reported in CF patients (Roum et al., 1985; Brown et al., 1995; Starosta et al., 2006; Velsor et al., 2006). Not surprisingly, CF lungs are exposed to significantly higher oxidative stress levels (Starosta et al., 2006). As marker of oxidant-induced DNA damage, urinary 8-hydroxydeoxyguanosine (8-OHdG) was found to be significantly elevated in children with CF (Brown and Kelly, 1994). Further, airway epithelia from CF patients show increased expression of two DNA damage markers (phospho-Histone H2A.X (γH2A.X) and phospho-checkpoint 2 kinase (phospho-Chk2)) (Fischer et al., 2013). However, it would be too simplistic to attribute the increased cancer risk solely to oxidative stress. CFTR has been proposed a tumor suppressor protein and CFTR deficiency is implicated in airway and intestinal cancer progression and poor prognosis (Than et al., 2016; Li et al., 2015; Tu et al., 2016). In summary, aging-associated DNA repair deficiency in combination with increased oxidative damage can lead to accumulating genome damage and thus to genomic instability (Moskalev et al., 2013).
Telomere Attrition
The proliferative capacity of cells is limited by the gradual loss of their protective telomeric DNA with each division ultimately leading to cellular senescence (Blackburn et al., 2006). Chronic inflammation can not only increase cell turnover, it has also been shown to lead to telomere dysfunction and accelerated aging (Jurk et al., 2014; Gorenjak et al., 2020). Telomeres are highly sensitive to oxidative damage as it is ineffectively repaired causing accelerated telomere loss (Gorenjak et al., 2020; Hewitt et al., 2012). This raises the question whether oxidative stress associated with chronic airway inflammation can cause accelerated telomere shortening in CF patients.
In CF, decreased telomere length in peripheral blood leukocytes has been reported to be associated with disease severity (Lammertyn et al., 2017). However, telomere length was not reduced in explant lungs from CF patients, and no correlation between telomere length and disease severity at the sampling location was found (Everaerts et al., 2018). Similarly, telomere length in CF airway epithelial cells was not significantly different from control donor lungs; only a small subgroup of CF subjects had shorter telomeres (Fischer et al., 2013). Based on the limited scientific data available, we cannot conclude whether telomere attrition is a common feature in CF lungs. Further research is needed to determine its contribution to CF airway disease pathogenesis.
Epigenetic Alterations
Epigenetic alterations are stable, heritable changes to the chemical DNA structure by alterations in its methylation pattern, post-translational histone modifications, and chromatin remodeling. They regulate gene expression without modifying the DNA sequence (Gonzalo, 1985; Berger et al., 2009). Specific changes in the histone modification pattern such as increased H4K20 and H3K4 trimethylation, and decreased H3K9 methylation as well as H3K27 trimethylation constitute markers of age-associated epigenetic alterations (Gonzalo, 1985; López-Otín et al., 2013). DNA methylation has been shown to play an important role in aging and disease. Global DNA methylation changes over the lifespan and methylation of specific CpG (cytosine guanine) sites correlate with age (Jones et al., 2015; Ashapkin et al., 2017; Levine et al., 2018). Alterations in DNA methylation patterns are associated with cancer, several genetic diseases and inflammatory lung conditions including CF (Robertson, 2005; Scott and De Sario, 2020). In CF, methylation changes were found at multiple CpG dinucleotides in nasal epithelial cells, whole blood and lung macrophages. Interestingly, these epigenetic alterations were preferentially located at genes important for the respective tissue, such as for cell adhesion, immune response or inflammation, and correlated with lung function (Scott and De Sario, 2020; Magalhães et al., 2018; Chen et al., 2018). Though the specific research is yet in its infancy, CFTR has been shown to be epigenetically regulated too which may open up novel therapeutic strategies to restore CFTR dysfunction in CF pathology (Sirinupong and Yang, 2015).
Loss of Proteostasis
Molecular chaperones and the proteolytic machinery assure continuous proteome renewal protecting cells from accumulation of misfolded or damaged proteins (López-Otín et al., 2013). Loss of adequate proteostasis occurs during the aging process and has been shown to be altered in CF epithelia. There is increasing evidence that CFTR function can regulate the proteostasis network (Villella et al., 2013; Esposito et al., 2016). CFTR carrying the ∆F508 mutation as well as other less frequent mutations does not mature into the fully glycosylated protein. Instead, the misfolded protein is mostly retained at the endoplasmic reticulum, where it is sequestered for degradation (Cheng et al., 1990; Van Goor et al., 2011). ∆F508 overexpression causes ER stress and activates the unfolded protein response in vitro leading to decreased wild type CFTR mRNA levels (Bartoszewski et al., 2008). This implicates defective CFTR leads to suppressed CFTR expression. The severity of the clinical disease spectrum varies considerably even within patients carrying the same mutation. Variations in the proteostasis network are suspected to be modifying factors contributing to those differences (Balch et al., 2011). A promising new therapeutic strategy aims at improving protein trafficking by the proteostasis network (Strub and McCray Jr, 2020). The proteostasis regulator cystamine promotes ∆F508-CFTR trafficking to the apical cell membrane and thereby allows it to respond to CFTR potentiators (Luciani et al., 2012). Cystamine combined with epigallocatechin gallate restored CFTR function, reduced lung inflammation and restored autophagy in a phase-2 trial, particularly in patients carrying the ∆F508 mutation (Tosco et al., 2016).
CFTR is not only subjected to degradation by the proteostasis network, it is a key player in its own regulation. Inhibition of CFTR function leads to CFTR protein ubiquitination and drastically reduced stability in the plasma membrane of bronchial epithelial cells (Esposito et al., 2016), with cystamine correcting this deranged proteostasis (Villella et al., 2013). In addition, siRNA depleting CFTR interferes with endosomal trafficking of cell surface proteins demonstrated for transferrin receptor, epidermal growth factor receptor, and CFTR itself (Villella et al., 2013). Hence, insufficient CFTR function derails the proteostasis network in a feed-forward loop fostering its own degradation. Dysfunctional autophagy also appears to contribute to the exaggerated CF lung inflammation. Improving autophagosome clearance attenuates the hyperinflammatory response (Mayer et al., 2013). Oxidative stress from defective CFTR function inhibits ubiquitination and proteasome degradation of the pro-fibrotic tissue trans-glutaminase 2, thereby driving chronic inflammation (Esposito et al., 2016; Luciani et al., 2009). Autophagy restoration by cystamine has been shown to suppress trans-glutaminase 2 and diminish inflammation in CF mice and in patient derived nasal epithelial cells in vitro. In summary, loss of proteostasis as an aging hallmark in CF disease has been established by current evidence and might be a valuable target for future therapies.
Deregulated Nutrient Sensing
Aging is regulated by nutrient-sensing pathways including insulin/insulin-like growth factor (IGF-1), mTOR, AMP kinase, sirtuins, and FOXO transcription factors (Vellai, 2009; Kenyon, 2010). Pro-inflammatory signals are closely integrated in stress and nutrient signaling (Jurk et al., 2014). Hence, both inflammaging and chronic inflammatory conditions such as CF lung disease have the potential to interfere with nutrient sensing signaling pathways. An in vitro study identified the α1 (catalytic) subunit of AMP-activated protein kinase (AMPK) as a dominant and novel protein interacting with CFTR demonstrating a potential link between transepithelial transport and cell metabolic state (Hallows et al., 2000).
Oxidative stress as seen in CF airway inflammation leads to increased pro-growth signaling by the mTOR pathway (Chen et al., 2011). So far, only little is known about deregulated nutrient sensing in CF lung disease. CFTR has been found to interact with mTOR signaling pathway components, which can be affected by CFTR mutations such that ∆F508 CFTR exhibits a specific interactome different from wild type CFTR (Pankow et al., 2015; Reilly et al., 2017). mTOR activity in CF bronchial epithelial cells is upregulated, and PI3K/Akt/mTOR pathway inhibition improves CFTR expression and stability (Reilly et al., 2017). FOXO transcription factors down stream of insulin/IGF-1 signaling (IIS) regulate cellular processes involved in stress resistance, metabolism as well as cell cycle arrest, and are central to IIS attenuation-mediated lifespan expansion (Martins et al., 2016). FOXO1 and 3 have been shown to regulate innate immune mechanisms in respiratory epithelia in response to bacterial infections (Seiler et al., 2013). In a human CF bronchial epithelial cell line, reduced FOXO1 was found related to loss of CFTR function (Smerieri et al., 2014). Interestingly, four miRNAs that are predicted FOXO1 regulators were differently expressed in CF patient sera (Montanini et al., 2016). However, the implications of altered FOXO transcription factor signaling in terms of accelerated aging in CF patients remains unclear and calls for further investigation.
Mitochondrial Dysfunction
Age-associated mitochondrial dysfunction is most commonly caused by increased ROS ultimately leading to cell senescence (Chapman et al., 2019). Excessive oxidant levels from dysfunctional mitochondria can trigger inflammatory cytokine release inducing chronic inflammation and progression of airway diseases such as CF (Prakash et al., 2017). In addition, as cells age, DNA damage occurs and activates DNA damage response pathways. These chronically stimulated pathways cause mitochondrial stress leading to increased release of mitochondrial damage-associated molecular patterns (DAMPs). DAMPs along with ROS production, and/or ATP and K+ efflux contribute to NLRP3 inflammasome activation (Prakash et al., 2017; dos Santos et al., 2012). This leads to caspase-1 activation and release of the proinflammatory cytokines IL-1β and IL-18, initiating a cycle of chronic inflammation, disease progression, and further accumulative damage to mitochondria resulting in accelerated aging (dos Santos et al., 2012). Recent research is showing that mitochondrial dysfunction could be involved in disease progression in CF. Cells with impaired CFTR function exhibit reduced mitochondrial complex I activity (Valdivieso et al., 2012). And increased IL-1β levels are very common among CF patients, which has been linked to inflammation triggered by underlying chronic Pseudomonas aeruginosa infections (Rimessi et al., 2015). As discussed, elevated IL-1β is not only a sign of inflammation, but also a hallmark of mitochondrial dysfunction. These two intrinsically connected hallmarks of aging both contribute to disease progression in CF. Furthermore, it will be of benefit to assess whether the triple CFTR modulator therapy affects these pathogenetic mechanisms favorably. A recent study has shown that CFTR modulation can restore Nrf2 phosphorylation, a major regulator of oxidative balance and inflammatory signaling (Borcherding et al., 2019).
Cellular Senescence
Accumulation of senescent cells is seen in normal aging and chronic pulmonary disease (Jeyapalan et al., 2007; Meiners et al., 2015). Senescence is triggered by a range of insults such as oxidative stress, DNA damage, telomere shortening, and inflammation, and is associated with a characteristic secretory profile termed senescence-associated secretory phenotype (SASP) (Naylor et al., 2013; Jurk et al., 2014). This includes release of proinflammatory mediators, growth factors, and matrix-remodeling proteases that may perpetuate inflammatory processes resulting in further accumulation of senescent cells (Naylor et al., 2013; Parikh et al., 2019). In CF, the liquid lung lining layer contains high amounts of neutrophil elastase that has been shown to trigger cell cycle arrest through elevated p27Kip1 expression resulting in G1 arrest in normal human bronchial epithelial cells in vitro (Nakamura et al., 1992; Fischer et al., 2007). Cell cycle arrest may lead to cellular senescence (Fischer et al., 2013; Naylor et al., 2013). Indeed, a study by Fischer and colleagues confirmed airway epithelia from CF lungs have increased expression of the senescence markers p16INK4a, γH2A.X, and phospho-Chk2 (Fischer et al., 2013). Their study showed that neutrophil elastase increases p16 expression resulting in inhibition of cyclin-dependent kinase 4 activity in vitro (Fischer et al., 2007). This suggests cellular senescence due to excessive neutrophil elastase release in CF lungs may contribute to accelerated aging. A recent review concluded there was consistent data supporting that cellular senescence may also be involved in CF lung disease, but the exact mechanisms leading from loss of CFTR function to cellular senescence and the precise role in CF lung disease remain unknown (Bezzerri et al., 2019).
Stem Cell Exhaustion
Lung tissue has a low steady-state cell turnover with the ability to increase proliferation of stem/progenitor cells in response to injury (Hogan et al., 2014). Aging is associated with a progressive decline of stem/progenitor cells that maintain homeostatic and regenerative capacity (Oh et al., 2014). Chronic inflammation is considered a main factor accelerating the deterioration of stem cell function with transforming growth factor (TGF)-β and ROS accumulation as important pathological factors (Oh et al., 2014). miRNA-155 is upregulated in aging and has been suggested to contribute to inflammation-associated stem cell dysfunction (Teramura and Onodera, 2018). High expression of this specific miRNA has been found in CF lung epithelial cells and circulating neutrophils (Bhattacharyya et al., 2011). Further, chronic airway inflammation in combination with recurrent exacerbations causes recurrent tissue damage thereby increasing the need for stem cell proliferation. This may exceed the supply the stem cell niche is capable of providing resulting in its depletion in CF lungs (Mora and Rojas, 2013). To this point, there are no reports on the exact role of stem cell exhaustion in CF lung disease. One study observed no telomere shortening in CF airway epithelial cells leading to the conclusion that the epithelial stem/progenitor reserve is sufficient to maintain normal telomere length despite enhanced cell turnover (Fischer et al., 2013). But this does not exclude an involvement of stem cell exhaustion in accelerated aging in CF lungs, and further studies are needed to draw definite conclusions.
Altered Cellular Communication
In addition to chronic lung inflammation, CFTR deficiency has been reported to causes abnormalities in diverse signaling pathways. Functional CFTR has been reported to downregulate NF-κB activity and CF associated hyper-inflammation may represent a consequence of insufficient inhibition of NF-κB signaling (Hunter et al., 2010). Chronic, progressive low-grade inflammation promoted by NF-κB can cause premature aging (Jurk et al., 2014). CF airway inflammation is associated with excessive production of NF-κB- dependent inflammatory mediators such as interleukin-1β (IL-1β), IL-6, IL-8, and TGF-β1, neutrophil recruitment, and decreased response to interferon (IFN)-γ as well as further abnormalities in various signaling pathways (Nichols et al., 2008; Harris et al., 2009; Peterson-Carmichael et al., 2009; Lara-Reyna et al., 2020). The profibrotic cytokine TGF-β is considered a pro-aging factor and is associated with accelerated lung function decline in CF patients (Arkwright et al., 2000). Several reports show that TGF-β causes CFTR dysfunction (Manzanares et al., 2015; Snodgrass et al., 2013; Sun et al., 2014). Chronic bacterial infections stimulate persistent IL-8 release from airway epithelia (McCuaig and Martin, 2013). Attracted neutrophils further potentiate airway inflammation by releasing high concentrations of inflammatory mediators such as TNFα and IL-8, oxidants, and proteases (Petit-Bertron et al., 2008). Neutrophil elastase promotes CFTR degradation and dysfunction by calpains (Le Gars et al., 2013). Recent drug developments have focused on restoring CFTR function; these therapies significantly improve lung function and reduce exacerbations in CF patients (Wainwright et al., 2015; Rowe et al., 2017; Davies et al., 2018; Keating et al., 2018). However, sustained airway inflammation leads to degradation and increased turnover of CFTR, hindering the targeted therapies from developing their full potential by inducing a state of accelerated aging at the tissue level (Rowe et al., 2014). Oxidative stress from ROS associated with neutrophilic airway inflammation can lead to accumulation of modified or damaged biomolecules impairing their function. Oxidatively damaged cellular structures can act as DAMPs that are recognized by receptors of the innate immune system, i.e., Toll-like-receptors (TLRs) and the NLRP3 inflammasome (Meiners et al., 2015). Hence, oxidative stress can further enhance cytokine production in the ongoing inflammation and maintain the proinflammatory state in inflammaging (Franceschi and Campisi, 2014; Meiners et al., 2015).
Extracellular Matrix Dysregulation
ECM remodeling in lung diseases is not only a consequence of tissue injury, it also contributes to disease progression by impaired repair mechanisms (Parker et al., 2014). The neutrophilic inflammation in CF is accompanied by protease/antiprotease imbalance with increased neutrophil elastase and matrix metalloprotease-9 and reduced tissue inhibitor of metalloprotease-1 (Gaggar et al., 2007). While proteolytic enzymes are beneficial for tissue repair, excessive release due to chronic inflammation may overwhelm anti-protease activity causing airway remodeling and obstruction (Gaggar et al., 2011). Neutrophil derived proteases are the main driving force of lung injury in CF (Elborn, 2016) and sputum neutrophil elastase correlates with FEV1 in children with CF (Sagel et al., 2002). Furthermore, neutrophil elastase and proteolytic collagen and elastin breakdown products play a pathogenic role in fibrotic lung remodeling (Chua and Laurent, 2006). Ultrastructural evidence for ECM degradation includes lysis of elastic and collagen fibers, loss of arborescent elastic network distribution and alterations in the reticular basement membrane structure (RBM) (Durieu et al., 1998; Hilliard et al., 2007; Regamey et al., 2011). In CF, RBM thickening appears to be related to increased TGF-β1 levels (Hilliard et al., 2007). The elastic network reduction combined with increased collagen deposition in bronchial walls of CF patients resembles fibrotic ECM changes in aging lungs (Hilliard et al., 2007). Together, these data suggest that ECM dysregulation plays an important role in accelerated lung aging in CF.
Therapeutic Implications
Current Effective CF Therapies Targeting Chronic Inflammation
Various drugs targeting inflammation have been studied in CF since 1990, but only few are recommended for clinical use. Due to the length of this review, only significant ones will be discussed here.
Corticosteroids were one of the first anti-inflammatory drugs studied as a chronic therapy attenuating CF airway inflammation (Auerbach et al., 1985; Eigen et al., 1995). Although beneficial effects on lung function were shown, overall adverse effects outweighed the benefits and long-term use of systemic corticosteroids to slow lung function decline is currently not recommended (Flume et al., 2007). Several studies investigated the effects of inhaled corticosteroids (ICS) on lung function decline, but their anti-inflammatory effect was never confirmed (Ren et al., 2008; De Boeck et al., 2011). The CF Foundation therefore is advising against the use of long-term ICS in patients with CF older than 6 years without coexistent asthma or allergic bronchopulmonary aspergillosis (Flume et al., 2007).
Nonsteroidal anti-inflammatory drugs (NSAIDs) have been studied as therapeutic options for CF due to similar properties as corticosteroids, but fewer adverse effects. Ibuprofen has specific activity against neutrophils and twice-daily high-dose ibuprofen use was linked to a slower lung function decline, a decreased exacerbation frequency and less weight loss in the pediatric CF population (Konstan et al., 1995; Konstan et al., 2003; Konstan et al., 2007; Lands et al., 2007). Nevertheless, ibuprofen is not widely used, but the CF Foundation expert panel recommends its use for CF patients with mild lung disease (Flume et al., 2007).
CFTR modulator therapy has demonstrated its efficacy and beneficial effect on restoration of CFTR function for approximately 90% CF genotypes (Wainwright et al., 2015; Rowe et al., 2017; Davies et al., 2018; Keating et al., 2018; Middleton et al., 2019). Interestingly, CFTR dysfunction has been linked to contribute to chronic inflammation as well as other hallmarks of aging, and two recent studies have demonstrated an anti-inflammatory effect of CFTR modulators (Jarosz-Griffiths et al., 2020; Hisert et al., 2017). A recent observational study though did not show any significant alterations in sputum inflammatory markers, but changes in ECM using whole proteome analysis (Kopp et al., 2020). Therefore, the validation of potential anti-inflammatory effects of CFTR correctors needs further investigation. CFTR correctors also have been shown to act as proteostasis regulators (Lopes-Pacheco et al., 2015; Lopes-Pacheco et al., 2016) and there is evidence that CFTR modulators can regulate mitochondrial dysfunction. Figure 2 summarizes current evidence of CFTR modulators and their link to aging hallmarks.
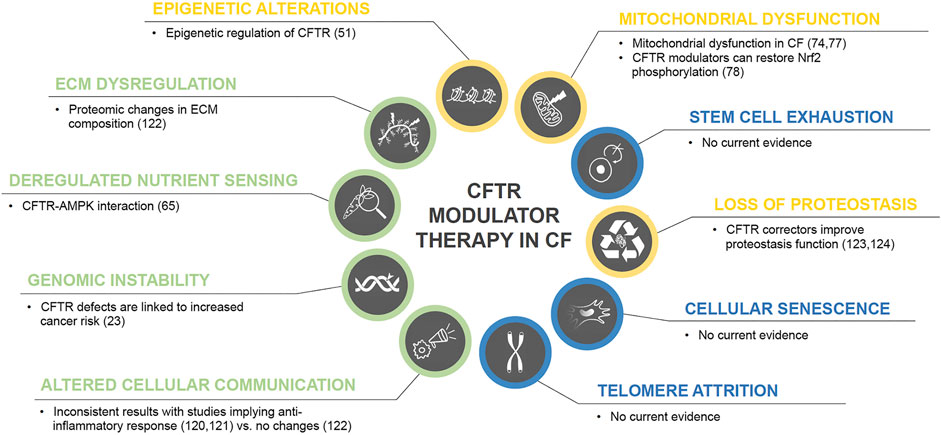
FIGURE 2. CFTR modulator therapy in CF and potential effects on the hallmarks of aging. Color code for current level of evidence with blue = no evidence for involvement in CF pathology, green = weak evidence, yellow = strong evidence.
Potential Novel CF Therapies
Modulation of ion channels other than CFTR has been evaluated as potential therapies. Most recently, potentiation of TMEM16A using ETX001 significantly increased the Ca2+-activated Cl− channel activity and anion secretion both in vitro and in vivo ovine models leading to improved mucociliary clearance without impacting calcium signaling (Danahay et al., 2020). Whether ETX001 also affects airway inflammation will be of great interest.
Anti-inflammatory cytokines and antibodies targeting cytokines may be valuable novel CF therapies. IL-10 has been demonstrated to terminate the inflammatory response and is deficient in CF patients (Bonfield et al., 1999). And its administration is anti-inflammatory in Pseudomonas infected mice (Chmiel et al., 1999; Soltys et al., 2002). Treatment with the anti-inflammatory cytokine IFN-γ was tested in a multicenter clinical trial but did not show any beneficial effect on pulmonary function or inflammatory sputum markers (Moss et al., 2005). This is in line with the above-mentioned reduced response of CF airways to IFN-γ. As an alternative to the treatment with anti-inflammatory cytokines, various blocking antibodies targeting pro-inflammatory cytokines have been assessed. Anti-IL-17 antibodies have been shown to decrease neutrophil recruitment in murine airways when exposed to lipopolysaccharide (Ferretti et al., 2003). Anti-IL-17 has been tested clinically in rheumatoid arthritis and psoriasis (Ly et al., 2019; Taams, 2020) and might be a potential novel therapeutic strategy for CF-associated airway inflammation. Anti-ICAM-1 and -IL-8 have been assessed preclinically, but never reached the clinical stage. Because of high concentrations of LTB4 in CF airways (Konstan et al., 1993) BIIL 284 BS (amelubant), a specific LTB4 receptor antagonist, has been tested in CF. Due to serious pulmonary adverse events, the clinical trial was terminated early (Konstan MW et al., 2005).
Fatty acid modulation may proof beneficial as alterations in fatty acid concentrations contributing to airway inflammation have been demonstrated in CF such as increases in arachidonic acid and decreases in docosahexaenoic acid (DHA) (Freedman et al., 1999; Horati et al., 2020). Cumulative data from in vitro studies and some small, non-placebo controlled trials using oral DHA supplementation imply this may be beneficial for pulmonary function preservation (Beharry et al., 2007; Van Biervliet et al., 2008; Aldámiz-Echevarría et al., 2009). To validate these results, larger placebo-controlled trials will be needed. Furthermore, Myriocin, an inhibitor of sphingolipid synthesis, induced changes in a transcriptional program of cell metabolism in vitro in CF airway epithelial cells. Therefore, the authors speculated that sphingolipid de novo synthesis could attenuate chronic inflammation, optimize energy supply, and anti-oxidant responses, implying a novel future therapeutic strategy for CF (Mingione et al., 2020)
Inhibiton of NF-κB activation has been demonstrated by both ibuprofen at high doses and IL-10. Furthermore, NF-κB signaling can be attenuated by glitazones through peroxisome proliferator activating receptor (PPAR) elevation (Zingarelli et al., 2003; Ruan et al., 2003) and there seems to be a deficiency of PPAR in CF (Perez et al., 2008). Both tiglitazone and troglitazone can activate PPAR in the CF epithelium and attenuate the inflammatory response to P. aeruginosa (Perez et al., 2008). One clinical trial using glitazones has not shown a beneficial effect towards inflammation, but this trial was very small and did not assess lung function outcomes (Konstan MW et al., 2009). Long term use of Azithromycin has not only been shown to improve functional outcomes, but some of the underlying mechanisms can be attributed to STAT and NF-κB inhibition (Haydar et al., 2019; Nichols et al., 2020). In summary, targeting NF-κB signaling in CF disease seems to be an attractive future therapeutic direction.
Inhibition of the NLRP3 Inflammasome has also been discussed as a potential anti-inflammatory strategy (Scambler et al., 2019). A recent report utilized MCC950 as a specific NLRP3 inhibitor in vivo in CF animal models, which resulted in significantly reduced airway inflammation and improved Pseudomonas clearance (McElvaney et al., 2019).
Statins exhibit anti-inflammatory effects through various mechanisms including inhibition of neutrophil migration, RhoGTPase/increase in nitric oxide (NO)/IL-8 production and other proinflammatory cytokines as well as increase of PPAR transcription (Dunzendorfer et al., 1997; Kraynack et al., 2002; Zelvyte et al., 2002). Agents that can increase NO, such as arginine, have been studied, since it has been shown that there is increased arginase activity in blood and sputum of CF patients. This leads to degradation of L-arginine, which is a substrate for NO production (Grasemann et al., 2005a). A small pilot study assessed oral supplementation with L-arginine in CF patients, which led to increased exhaled NO (Grasemann et al., 2005b). In order to show efficacy, large prospective clinical trials are needed.
The antioxidant N-Acetyl cysteine (NAC) is frequently used as a mucolytic, but has spiked recent interest as antioxidant due to its ability to increase glutathione levels and inhibit H2O2-induced damage. Supplementation of oral NAC was linked to significantly increased blood glutathione levels and decreased neutrophils, IL-8, and elastase activity in sputum of CF patients (Tirouvanziam et al., 2006) A randomized double-blind placebo-controlled trial including 70 CF patients for 24 weeks showed stability or a slight increase in spirometric lung function in NAC treated participants compared to the control group (Conrad et al., 2015). A second open-label randomized controlled trial demonstrated a non-significant improvement in lung function in adults with CF and chronic Pseudomonas infection (Skov et al., 2015). P3001, a mucolytic agent was also assessed and directly compared with NAC in a more recent study using both in vitro and in vivo CF models. Results indicated that P3001 acted faster and was more effective than NAC and DNAse, but investigators did not study its anti-oxidant or anti-inflammatory properties (Ehre et al., 2019). Since glutathione is transported by CFTR (Linsdell and Hanrahan, 1998), CFTR defects could potentially decrease levels of this antioxidant in the airway epithelium increasing susceptibility to oxidative stress. Indeed, decreased glutathione levels have been observed in CF mouse models and patients (Roum et al., 1985; Velsor et al., 2001). A small pilot study using twice daily treatment of aerosolized glutathione in CF patients led to a reduction in superoxide production, but no changes in oxidative stress markers in bronchoalveolar lavage fluid (Bishop et al., 2005; Hartl et al., 2005). A more recent Cochrane meta-analysis summarized antioxidant therapy approaches in CF and concluded that there does not seem to be a beneficial effect of antioxidant micronutrients on clinical outcomes; but oral supplementation with glutathione showed some benefit to lung function, nutritional status, and decrease in oxidative stress (Ciofu et al., 2019). Nevertheless, due to concurrent treatments including intensive antibiotic therapies, there is not sufficient evidence yet justifying their use in the CF population without larger trials and longer follow up.
Therapeutic use of protease inhibitors has been investigated in CF since 1990. Several groups demonstrated that α1-antitrypsin delivered as an aerosol formulary decreases inflammatory markers and neutrophils (McElvaney et al., 1991; Griese et al., 2007). rSLPI and EPI-hNE4 are two neutrophil elastase inhibitors, which have shown beneficial effects in CF, but large cohort trials are still lacking (McElvaney et al., 1993; Grimbert et al., 2003).
Other potential anti-inflammatories are also being investigated. SB-656933, a CXCR2 antagonist, has shown a trend toward attenuating airway inflammation (Moss et al., 2013). Low-dose cyclosporin A was characterized as a potential steroid sparing agent in a small case series (Bhal et al., 2001). Furthermore, methotrexate was studied as a potential anti-inflammatory agent in cystic fibrosis, but studies were inconclusive with one showing beneficial effects on lung function and total serum immunoglobulin levels, whereas another study showed less tolerance and increased exacerbations (Ballmann et al., 2003; Oermann and Wheeler C, 2007).
Micro RNAs as a potential anti-inflammatory approach have recently become a focus of interest. mi-RNAs have been implicated in various diseases including cystic fibrosis (Bardin et al., 2019; Bartoszewska et al., 2017). Vencken et al. suggested the use of nebulized lipid-polymer hybrid nanoparticles for the delivery of a therapeutic anti-inflammatory microRNA, miR-17, to reduce IL-8 secretion, which was successfully tested in vitro (Vencken et al., 2019) In addition, targeting miRNAs to restore CFTR activity has also been suggested as a novel therapeutic strategy (De Santi et al., 2020). Please see Table 1 for a summary of the evidence for accelerated aging in CF shown in in vitro and in vivo studies.
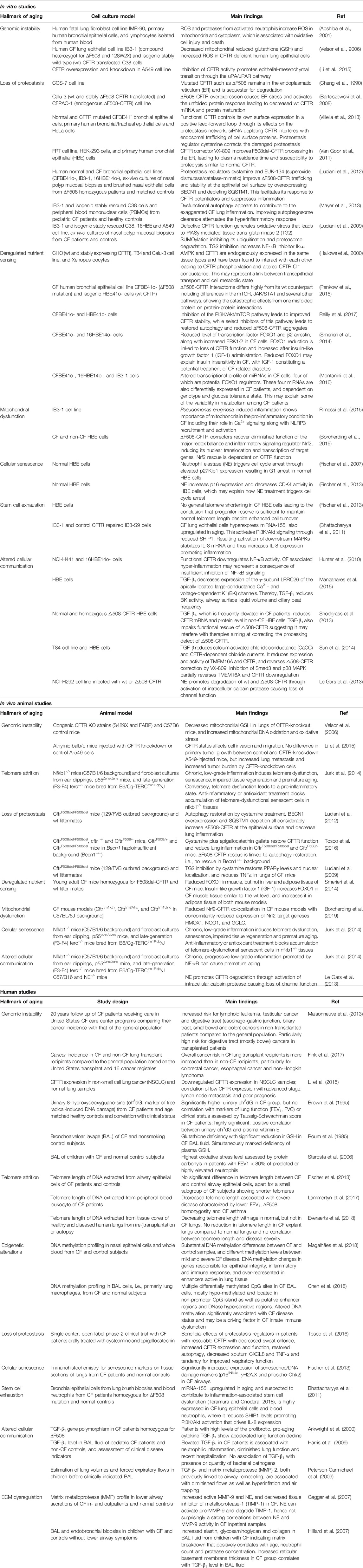
TABLE 1. Summary of in vitro, animal and human studies pointing towards an involvement of accelerated aging in CF disease.
Conclusion and Further Directions
Increasing evidence suggests that accelerated aging processes are involved in CF lung pathology. Based on the current state of research summarized in the paragraphs above, it appears sufficiently established that accelerated aging in CF patients is not only a consequence of chronic lung inflammation, but aging-associated processes are also driving disease progression. Of the ten hallmarks of aging, according to recent literature all but stem cell depletion and telomere attrition appear to be involved to varying degrees in CF lung disease (cp. Figure 1). For some hallmarks there is already considerable data on the specific mechanisms of their pathologic involvement, as in the case of altered proteostasis and intercellular communication. Others, such as mitochondrial dysfunction, deregulated nutrient sensing and cellular senescence, have been indicated to play a role in CF lung pathology and accelerated aging, but further research is needed to elucidate underlying pathomechanisms. And although each of the ten hallmarks was discussed individually, they should be considered as interdependent processes that influence each other.
Growing evidence on the role of accelerated aging processes in CF lung disease and progress in deciphering involved mechanisms also provide new therapeutic targets for future treatment strategy complementing established therapies as summarized in Therapeutic Implications. Some therapies targeting the ten hallmarks of aging are already in clinical use, and further can be expected to follow which may aid further reducing mortality and improving quality of life in CF patients.
Author Contributions
LK, ME, MH, and SK contributed to the writing of the manuscript. LK designed the figures and helped to draft the manuscript. LK, ME, and MH contributed to the literature search. SK critically revised the manuscript. All authors read and approved the manuscript for publication.
Funding
This work was supported by the Flight Attendant Medical Research Institute (YFAC152003 to SK), the Cystic Fibrosis Foundation (P30 DK072482 and Rowe19RO to SK), and the National Institutes of Health (R03AG059994 to SK).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Eva Maria Künzi for her assistance in creating the illustrations.
Abbreviations
BAL, bronchoalveolar lavage; BK, large-conductance Ca2+- and voltage-dependent K+; ECM, extracellular matrix; FEV1, forced expiratory volume in 1s; FVC, forced ventilator capacity; GSH, reduced glutathione; HBE, human bronchial epithelial; IGF-1, Insulin-like growth factor 1; MAPK, mitogen-activated protein kinase; NE, neutrophil elastase; oh8dG, urinary 8-hydroxydeoxyguanosine; PBMCs, peripheral blood mononuclear cells; PMN, polymorphnuclear leukocyte; ROS, reactive oxygen species; TG2, tissue trans-glutaminase 2; TIMP-1 tissue inhibitor of metalloprotease-1; wt, wild-type.
References
Aldámiz-Echevarría, L., Prieto, J. A., Andrade, F., Elorz, J., Sojo, A., Lage, S., et al. (2009). Persistence of essential fatty acid deficiency in cystic fibrosis despite nutritional therapy. Pediatr. Res. 66 (5), 585–589. doi:10.1203/pdr.0b013e3181b4e8d3
Aoshiba, K., Yasuda, K., Yasui, S., Tamaoki, J., and Nagai, A. (2001). Serine proteases increase oxidative stress in lung cells. Am. J. Physiology-Lung Cell Mol. Physiol. 281 (3), L556–L564. doi:10.1152/ajplung.2001.281.3.l556
Arkwright, P. D., Laurie, S., Super, M., Pravica, V., Schwarz, M. J., Webb, A. K., et al. (2000). TGF-beta 1 genotype and accelerated decline in lung function of patients with cystic fibrosis. Thorax 55 (6), 459–462. doi:10.1136/thorax.55.6.459
Ashapkin, V. V., Kutueva, L. I., and Vanyushin, B. F. (2017). Aging as an epigenetic phenomenon. Curr. Genomics 18 (5), 385–407. doi:10.2174/1389202918666170412112130
Auerbach, H., Kirkpatrick, J., Williams, M., and Colten, H. (1985). Alternate-day prednisone reduces morbidity and improves pulmonary function in cystic fibrosis. The Lancet 326 (8457), 686–688. doi:10.1016/s0140-6736(85)92929-0
Balch, W. E., Roth, D. M., and Hutt, D. M. (2011). Emergent properties of proteostasis in managing cystic fibrosis. Cold Spring Harb Perspect. Biol. 3 (2). doi:10.1101/cshperspect.a004499
Ballmann, M., Junge, S., and von der Hardt, H. (2003). Low-dose methotrexate for advanced pulmonary disease in patients with cystic fibrosis. Respir. Med. 97 (5), 498–500. doi:10.1053/rmed.2002.1471
Bardin, P., Foussignière, T., Rousselet, N., Rebeyrol, C., Porter, J. C., Corvol, H., et al. (2019). miR-636: a newly-identified actor for the regulation of pulmonary inflammation in cystic fibrosis. Front. Immunol. 10, 2643. doi:10.3389/fimmu.2019.02643
Bartoszewska, S., Kamysz, W., Jakiela, B., Sanak, M., Króliczewski, J., Bebok, Z., et al. (2017). miR-200b downregulates CFTR during hypoxia in human lung epithelial cells. Cell Mol Biol Lett 22, 23. doi:10.1186/s11658-017-0054-0
Bartoszewski, R., Rab, A., Jurkuvenaite, A., Mazur, M., Wakefield, J., Collawn, J. F., et al. (2008). Activation of the unfolded protein response by ΔF508 CFTR. Am. J. Respir. Cel Mol Biol 39 (4), 448–457. doi:10.1165/rcmb.2008-0065oc
Beharry, S., Ackerley, C., Corey, M., Kent, G., Heng, Y.-M., Christensen, H., et al. (2007). Long-term docosahexaenoic acid therapy in a congenic murine model of cystic fibrosis. Am. J. Physiology-Gastrointestinal Liver Physiol. 292 (3), G839–G848. doi:10.1152/ajpgi.00582.2005
Berger, S. L., Kouzarides, T., Shiekhattar, R., and Shilatifard, A. (2009). An operational definition of epigenetics. Genes Development 23 (7), 781–783. doi:10.1101/gad.1787609
Bezzerri, V., Piacenza, F., Caporelli, N., Malavolta, M., Provinciali, M., and Cipolli, M. (2019). Is cellular senescence involved in cystic fibrosis? Respir. Res. 20 (1), 32. doi:10.1186/s12931-019-0993-2
Bhal, G. K., Maguire, S. A., and Bowler, I. M. (2001). Use of cyclosporin A as a steroid sparing agent in cystic fibrosis. Arch. Dis. Child. 84 (1), 89. doi:10.1136/adc.84.1.89
Bhattacharyya, S., Balakathiresan, N. S., Dalgard, C., Gutti, U., Armistead, D., Jozwik, C., et al. (2011). Elevated miR-155 promotes inflammation in cystic fibrosis by driving hyperexpression of interleukin-8. J. Biol. Chem. 286 (13), 11604–11615. doi:10.1074/jbc.m110.198390
Bishop, C., Hudson, V. M., Hilton, S. C., and Wilde, C. (2005). A pilot study of the effect of inhaled buffered reduced glutathione on the clinical status of patients with cystic fibrosis. Chest 127 (1), 308–317. doi:10.1378/chest.127.1.308
Blackburn, E. H., Greider, C. W., and Szostak, J. W. (2006). Telomeres and telomerase: the path from maize, Tetrahymena and yeast to human cancer and aging. Nat. Med. 12 (10), 1133–1138. doi:10.1038/nm1006-1133
Bonfield, T. L., Konstan, M. W., and Berger, M. (1999). Altered respiratory epithelial cell cytokine production in cystic fibrosis☆☆☆★. J. Allergy Clin. Immunol. 104 (1), 72–78. doi:10.1016/s0091-6749(99)70116-8
Borcherding, D. C., Siefert, M. E., Lin, S., Brewington, J., Sadek, H., Clancy, J. P., et al. (2019). Clinically approved CFTR modulators rescue Nrf2 dysfunction in cystic fibrosis airway epithelia. J. Clin. Invest. 129 (8), 3448–3463. doi:10.1172/jci96273
Boucher, R. C. (2007). Cystic fibrosis: a disease of vulnerability to airway surface dehydration. Trends Mol. Med. 13 (6), 231–240. doi:10.1016/j.molmed.2007.05.001
Brown, R. K., and Kelly, F. J. (1994). Evidence for increased oxidative damage in patients with cystic fibrosis. Pediatr. Res. 36 (4), 487–492. doi:10.1203/00006450-199410000-00013
Brown, R. K., McBurney, A., Lunec, J., and Kelly, F. J. (1995). Oxidative damage to DNA in patients with cystic fibrosis. Free Radic. Biol. Med. 18 (4), 801–806. doi:10.1016/0891-5849(94)00172-g
Chapman, J., Fielder, E., and Passos, J. F. (2019). Mitochondrial dysfunction and cell senescence: deciphering a complex relationship. FEBS Lett. 593 (13), 1566–1579. doi:10.1002/1873-3468.13498
Chen, T., Shen, L., Yu, J., Wan, H., Guo, A., Chen, J., et al. (2011). Rapamycin and other longevity-promoting compounds enhance the generation of mouse induced pluripotent stem cells. Aging Cell 10 (5), 908–911. doi:10.1111/j.1474-9726.2011.00722.x
Chen, Y., Armstrong, D. A., Salas, L. A., Hazlett, H. F., Nymon, A. B., Dessaint, J. A., et al. (2018). Genome-wide DNA methylation profiling shows a distinct epigenetic signature associated with lung macrophages in cystic fibrosis. Clin. Epigenetics 10 (1), 152. doi:10.1186/s13148-018-0580-2
Cheng, S. H., Gregory, R. J., Marshall, J., Paul, S., Souza, D. W., White, G. A., et al. (1990). Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 63 (4), 827–834. doi:10.1016/0092-8674(90)90148-8
Chmiel, J. F., Konstan, M. W., Knesebeck, J. E., Hilliard, J. B., Bonfield, T. L., Dawson, D. V., et al. (1999). IL-10 attenuates excessive inflammation in ChronicPseudomonasInfection in mice. Am. J. Respir. Crit. Care Med. 160 (6), 2040–2047. doi:10.1164/ajrccm.160.6.9901043
Cho, S. J., and Stout-Delgado, H. W. (2020). Aging and lung disease. Annu. Rev. Physiol. 82, 433–459. doi:10.1146/annurev-physiol-021119-034610
Chua, F., and Laurent, G. J. (2006). Neutrophil elastase: mediator of extracellular matrix destruction and accumulation. Proc. Am. Thorac. Soc. 3 (5), 424–427. doi:10.1513/pats.200603-078aw
Ciofu, O., Lykkesfeldt, J., and Lykkesfeldt, J. (2019). Antioxidant supplementation for lung disease in cystic fibrosis. Cochrane Database Syst. Rev. 10, CD007020. doi:10.1002/14651858.CD007020.pub3
Conrad, C., Lymp, J., Thompson, V., Dunn, C., Davies, Z., Chatfield, B., et al. (2015). Long-term treatment with oral N-acetylcysteine: affects lung function but not sputum inflammation in cystic fibrosis subjects. A phase II randomized placebo-controlled trial. J. Cystic Fibrosis 14 (2), 219–227. doi:10.1016/j.jcf.2014.08.008
Danahay, H. L., Lilley, S., Fox, R., Charlton, H., Sabater, J., Button, B., et al. (2020). TMEM16A potentiation: a novel therapeutic approach for the treatment of cystic fibrosis. Am. J. Respir. Crit. Care Med. 201 (8), 946–954. doi:10.1164/rccm.201908-1641oc
Davies, J. C., Moskowitz, S. M., Brown, C., Horsley, A., Mall, M. A., McKone, E. F., et al. (2018). VX-659-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 379 (17), 1599–1611. doi:10.1056/nejmoa1807119
De Boeck, K., Vermeulen, F., Wanyama, S., and Thomas, M. (2011). Inhaled corticosteroids and lower lung function decline in young children with cystic fibrosis. Eur. Respir. J. 37 (5), 1091–1095. doi:10.1183/09031936.00077210
De Santi, C., Fernández Fernández, E., Gaul, R., Vencken, S., Glasgow, A., Oglesby, I. K., et al. (2020). Precise targeting of miRNA sites restores CFTR activity in CF bronchial epithelial cells. Mol. Ther. 28 (4), 1190–1199. doi:10.1016/j.ymthe.2020.02.001
Dodge, J. A., Lewis, P. A., Stanton, M., and Wilsher, J. (2007). Cystic fibrosis mortality and survival in the UK: 1947-2003. Eur. Respir. J. 29 (3), 522–526. doi:10.1183/09031936.00099506
dos Santos, G., Kutuzov, M. A., and Ridge, K. M. (2012). The inflammasome in lung diseases. Am. J. Physiology-Lung Cell Mol. Physiol. 303 (8), L627–L633. doi:10.1152/ajplung.00225.2012
Dunzendorfer, S., Rothbucher, D., Schratzberger, P., Reinisch, N., Kähler, C. M., and Wiedermann, C. J. (1997). Mevalonate-dependent inhibition of transendothelial migration and chemotaxis of human peripheral blood neutrophils by pravastatin. Circ. Res. 81 (6), 963–969. doi:10.1161/01.res.81.6.963
Durieu, I., Peyrol, S., Gindre, D., Bellon, G., Durand, D. V., and Pacheco, Y. (1998). Subepithelial fibrosis and degradation of the bronchial extracellular matrix in cystic fibrosis. Am. J. Respir. Crit. Care Med. 158 (2), 580–588. doi:10.1164/ajrccm.158.2.9707126
Ehre, C., Rushton, Z. L., Wang, B., Hothem, L. N., Morrison, C. B., Fontana, N. C., et al. (2019). An improved inhaled mucolytic to treat airway muco-obstructive diseases. Am. J. Respir. Crit. Care Med. 199 (2), 171–180. doi:10.1164/rccm.201802-0245oc
Eigen, H., Rosenstein, B. J., FitzSimmons, S., and Schidlow, D. V. (1995). A multicenter study of alternate-day prednisone therapy in patients with cystic fibrosis. J. Pediatr. 126 (4), 515–523. doi:10.1016/s0022-3476(95)70343-8
Elborn, J. S. (2016). Cystic fibrosis. The Lancet 388 (10059), 2519–2531. doi:10.1016/s0140-6736(16)00576-6
Esposito, S., Tosco, A., Villella, V. R., Raia, V., Kroemer, G., and Maiuri, L. (2016). Manipulating proteostasis to repair the F508del-CFTR defect in cystic fibrosis. Mol. Cel Pediatr 3 (1), 13. doi:10.1186/s40348-016-0040-z
Everaerts, S., Lammertyn, E., Martens, D. S., Sadeleer, L. D., Maes, K., Goldschmeding, R., et al. (2018). The aging lung: tissue telomere shortening in health and disease. Respir. Res. 19 (1), 95. doi:10.1186/s12931-018-0794-z
Ferretti, S., Bonneau, O., Dubois, G. R., Jones, C. E., and Trifilieff, A. (2003). IL-17, produced by lymphocytes and neutrophils, is necessary for lipopolysaccharide-induced airway neutrophilia: IL-15 as a possible trigger. J. Immunol. 170 (4), 2106–2112. doi:10.4049/jimmunol.170.4.2106
Fink, A. K., Yanik, E. L., Marshall, B. C., Wilschanski, M., Lynch, C. F., Austin, A. A., et al. (2017). Cancer risk among lung transplant recipients with cystic fibrosis. J. Cystic Fibrosis 16 (1), 91–97. doi:10.1016/j.jcf.2016.07.011
Fischer, B. M., Wong, J. K., Degan, S., Kummarapurugu, A. B., Zheng, S., Haridass, P., et al. (2013). Increased expression of senescence markers in cystic fibrosis airways. Am. J. Physiology-Lung Cell Mol. Physiol. 304 (6), L394–L400. doi:10.1152/ajplung.00091.2012
Fischer, B. M., Zheng, S., Fan, R., and Voynow, J. A. (2007). Neutrophil elastase inhibition of cell cycle progression in airway epithelial cells in vitro is mediated by p27kip1. Am. J. Physiology-Lung Cell Mol. Physiol. 293 (3), L762–L768. doi:10.1152/ajplung.00067.2007
Flume, P. A., O'Sullivan, B. P., Robinson, K. A., Goss, C. H., Mogayzel, P. J., Willey-Courand, D. B., et al. (2007). Cystic fibrosis pulmonary guidelines. Am. J. Respir. Crit. Care Med. 176 (10), 957–969. doi:10.1164/rccm.200705-664oc
Franceschi, C., and Campisi, J. (2014). Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. Journals Gerontol. Ser. A: Biol. Sci. Med. Sci. 69 (Suppl. 1), S4–S9. doi:10.1093/gerona/glu057
Freedman, S. D., Katz, M. H., Parker, E. M., Laposata, M., Urman, M. Y., and Alvarez, J. G. (1999). A membrane lipid imbalance plays a role in the phenotypic expression of cystic fibrosis in cftr-/- mice. Proc. Natl. Acad. Sci. 96 (24), 13995–14000. doi:10.1073/pnas.96.24.13995
Gaggar, A., Hector, A., Bratcher, P. E., Mall, M. A., Griese, M., and Hartl, D. (2011). The role of matrix metalloproteinases in cystic fibrosis lung disease. Eur. Respir. J. 38 (3), 721–727. doi:10.1183/09031936.00173210
Gaggar, A., Li, Y., Weathington, N., Winkler, M., Kong, M., Jackson, P., et al. (2007). Matrix metalloprotease-9 dysregulation in lower airway secretions of cystic fibrosis patients. Am. J. Physiology-Lung Cell Mol. Physiol. 293 (1), L96–L104. doi:10.1152/ajplung.00492.2006
Gonzalo, S. (1985). Epigenetic alterations in aging. J. Appl. Physiol. 109 (2), 586–597. doi:10.1152/japplphysiol.00238.2010
Gorenjak, V., Petrelis, A. M., Stathopoulou, M. G., and Visvikis-Siest, S. (2020). Telomere length determinants in childhood. Clin. Chem. Lab. Med. 58 (2), 162–177. doi:10.1515/cclm-2019-0235
Grasemann, H., Grasemann, C., Kurtz, F., Tietze-Schillings, G., Vester, U., and Ratjen, F. (2005a). Oral L-arginine supplementation in cystic fibrosis patients: a placebo-controlled study. Eur. Respir. J. 25 (1), 62–68. doi:10.1183/09031936.04.00086104
Grasemann, H., Schwiertz, R., Matthiesen, S., Racké, K., and Ratjen, F. (2005b). Increased arginase activity in cystic fibrosis airways. Am. J. Respir. Crit. Care Med. 172 (12), 1523–1528. doi:10.1164/rccm.200502-253oc
Griese, M., Latzin, P., Kappler, M., Weckerle, K., Heinzlmaier, T., Bernhardt, T., et al. (2007). alpha1-Antitrypsin inhalation reduces airway inflammation in cystic fibrosis patients. Eur. Respir. J. 29 (2), 240–250. doi:10.1183/09031936.00047306
Grimbert, D., Vecellio, L., Delépine, P., Attucci, S., Boissinot, E., Poncin, A., et al. (2003). Characteristics of EPI-hNE4 aerosol: a new elastase inhibitor for treatment of cystic fibrosis. J. Aerosol Med. 16 (2), 121–129. doi:10.1089/089426803321919889
Hallows, K. R., Raghuram, V., Kemp, B. E., Witters, L. A., and Foskett, J. K. (2000). Inhibition of cystic fibrosis transmembrane conductance regulator by novel interaction with the metabolic sensor AMP-activated protein kinase. J. Clin. Invest. 105 (12), 1711–1721. doi:10.1172/jci9622
Harris, W. T., Muhlebach, M. S., Oster, R. A., Knowles, M. R., and Noah, T. L. (2009). Transforming growth factor-β1 in bronchoalveolar lavage fluid from children with cystic fibrosis. Pediatr. Pulmonol. 44 (11), 1057–1064. doi:10.1002/ppul.21079
Hartl, D., Starosta, V., Maier, K., Beck-Speier, I., Rebhan, C., Becker, B. F., et al. (2005). Inhaled glutathione decreases PGE2 and increases lymphocytes in cystic fibrosis lungs. Free Radic. Biol. Med. 39 (4), 463–472. doi:10.1016/j.freeradbiomed.2005.03.032
Haydar, D., Cory, T. J., Birket, S. E., Murphy, B. S., Pennypacker, K. R., Sinai, A. P., et al. (2019). Azithromycin polarizes macrophages to an M2 phenotype via inhibition of the STAT1 and NF-κB signaling pathways. J.I. 203 (4), 1021–1030. doi:10.4049/jimmunol.1801228
Hewitt, G., Jurk, D., Marques, F. D. M., Correia-Melo, C., Hardy, T., Gackowska, A., et al. (2012). Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 3, 708. doi:10.1038/ncomms1708
Hilliard, T. N., Regamey, N., Shute, J. K., Nicholson, A. G., Alton, E. W. F. W., Bush, A., et al. (2007). Airway remodelling in children with cystic fibrosis. Thorax 62 (12), 1074–1080. doi:10.1136/thx.2006.074641
Hisert, K. B., Heltshe, S. L., Pope, C., Jorth, P., Wu, X., Edwards, R. M., et al. (2017). Restoring cystic fibrosis transmembrane conductance regulator function reduces airway bacteria and inflammation in people with cystic fibrosis and chronic lung infections. Am. J. Respir. Crit. Care Med. 195 (12), 1617–1628. doi:10.1164/rccm.201609-1954oc
Hodson, M. E., Simmonds, N. J., Warwick, W. J., Tullis, E., Castellani, C., Assael, B., et al. (2008). An international/multicentre report on patients with cystic fibrosis (CF) over the age of 40 years. J. Cystic Fibrosis 7 (6), 537–542. doi:10.1016/j.jcf.2008.06.003
Hogan, B. L. M., Barkauskas, C. E., Chapman, H. A., Epstein, J. A., Jain, R., Hsia, C. C. W., et al. (2014). Repair and regeneration of the respiratory system: complexity, plasticity, and mechanisms of lung stem cell function. Cell Stem Cell 15 (2), 123–138. doi:10.1016/j.stem.2014.07.012
Horati, H., Janssens, H. M., Margaroli, C., Veltman, M., Stolarczyk, M., Kilgore, M. B., et al. (2020). Airway profile of bioactive lipids predicts early progression of lung disease in cystic fibrosis. J. Cystic Fibrosis 19 (6), 902–909. doi:10.1016/j.jcf.2020.01.010
Hunter, M. J., Treharne, K. J., Winter, AK, Cassidy, D. M., and Land, S. (2010). Expression of wild-type CFTR suppresses NF-kappaB-driven inflammatory signalling. PLoS One 5 (7), e11598. doi:10.1371/journal.pone.0011598
Jarosz-Griffiths, H. H., Scambler, T., Wong, C. H., Lara-Reyna, S., Holbrook, J., Martinon, F., et al. (2020). Different CFTR modulator combinations downregulate inflammation differently in cystic fibrosis. Elife 9. doi:10.7554/elife.54556
Jeyapalan, J. C., Ferreira, M., Sedivy, J. M., and Herbig, U. (2007). Accumulation of senescent cells in mitotic tissue of aging primates. Mech. Ageing Development 128 (1), 36–44. doi:10.1016/j.mad.2006.11.008
Jones, M. J., Goodman, S. J., and Kobor, M. S. (2015). DNA methylation and healthy human aging. Aging Cell 14 (6), 924–932. doi:10.1111/acel.12349
Jurk, D., Wilson, C., Passos, J. F., Oakley, F., Correia-Melo, C., Greaves, L., et al. (2014). Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nat. Commun. 2, 4172. doi:10.1038/ncomms5172
Keating, D., Marigowda, G., Burr, L., Daines, C., Mall, M. A., McKone, E. F., et al. (2018). VX-445-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 379 (17), 1612–1620. doi:10.1056/nejmoa1807120
Kerem, B., Rommens, J., Buchanan, J., Markiewicz, D., Cox, T., Chakravarti, A., et al. (1989). Identification of the cystic fibrosis gene: genetic analysis. Science 245 (4922), 1073–1080. doi:10.1126/science.2570460
Konstan Mw, D. G., Lands, L. C., Hilliard, K. A., Koker, P., Bhattacharya, S., Staab, A., et al. (2005). Results of a phase II clinical trial of BIIL 284 BS (a LTB4 receptor antagonist) for the treatment of CF lung disease. Pediatr. Pulmonol 40, 125–127.
Konstan, M. W., Krenicky, J. E., Hilliard, K. A., and Hilliard, J. B. (2009). A pilot study evaluating the effect of pioglitazone, simvastatin and ibuprofen on neutrophil migration in vivo in healthy subjects. Pediatr. Pulmonol 44, 289–290.
Konstan, M. W., Byard, P. J., Hoppel, C. L., and Davis, P. B. (1995). Effect of high-dose ibuprofen in patients with cystic fibrosis. N. Engl. J. Med. 332 (13), 848–854. doi:10.1056/nejm199503303321303
Konstan, M. W., Krenicky, J. E., Finney, M. R., Kirchner, H. L., Hilliard, K. A., Hilliard, J. B., et al. (2003). Effect of ibuprofen on neutrophil migration in vivo in cystic fibrosis and healthy subjects. J. Pharmacol. Exp. Ther. 306 (3), 1086–1091. doi:10.1124/jpet.103.052449
Konstan, M. W., Schluchter, M. D., Xue, W., and Davis, P. B. (2007). Clinical use of ibuprofen is associated with slower FEV1Decline in children with cystic fibrosis. Am. J. Respir. Crit. Care Med. 176 (11), 1084–1089. doi:10.1164/rccm.200702-181oc
Konstan, M. W., Walenga, R. W., Hilliard, K. A., and Hilliard, J. B. (1993). Leukotriene B4Markedly elevated in the epithelial lining fluid of patients with cystic fibrosis. Am. Rev. Respir. Dis. 148 (4 Pt 1), 896–901. doi:10.1164/ajrccm/148.4_pt_1.896
Kopp, B. T., Fitch, J., Jaramillo, L., Shrestha, C. L., Robledo-Avila, F., Zhang, S., et al. (2020). Whole-blood transcriptomic responses to lumacaftor/ivacaftor therapy in cystic fibrosis. J. Cystic Fibrosis 19 (2), 245–254. doi:10.1016/j.jcf.2019.08.021
Kraynack, N. C., Corey, D. A., Elmer, H. L., and Kelley, T. J. (2002). Mechanisms of NOS2 regulation by Rho GTPase signaling in airway epithelial cells. Am. J. Physiology-Lung Cell Mol. Physiol. 283 (3), L604–L611. doi:10.1152/ajplung.00459.2001
Lammertyn, E. M. D., Colpaert, K., Goeminne, P., Proesmans, M., Vanaudenaerde, B., Nawrot, T., et al. (2017). The association between leukocyte telomere length, telomere attrition and disease severity in cystic fibrosis patients. Eur. Respir. J., OA4402.
Lands, L. C., Milner, R., Cantin, A. M., Manson, D., and Corey, M. (2007). High-dose ibuprofen in cystic fibrosis: Canadian safety and effectiveness trial. J. Pediatr. 151 (3), 249–254. doi:10.1016/j.jpeds.2007.04.009
Lara-Reyna, S., Holbrook, J., Jarosz-Griffiths, H. H., Peckham, D., and McDermott, M. F. (2020). Dysregulated signalling pathways in innate immune cells with cystic fibrosis mutations. Cell. Mol. Life Sci. 77 (22), 4485–4503. doi:10.1007/s00018-020-03540-9
Le Gars, M., Descamps, D., Roussel, D., Saussereau, E., Guillot, L., Ruffin, M., et al. (2013). Neutrophil elastase degrades cystic fibrosis transmembrane conductance regulator via calpains and disables channel FunctionIn VitroandIn vivo. Am. J. Respir. Crit. Care Med. 187 (2), 170–179. doi:10.1164/rccm.201205-0875oc
Levine, M. E., Lu, A. T., Quach, A., Chen, B. H., Assimes, T. L., Bandinelli, S., et al. (2018). An epigenetic biomarker of aging for lifespan and healthspan. Aging 10 (4), 573–591. doi:10.18632/aging.101414
Li, J., Zhang, J. T., Jiang, X., Shi, X., Shen, J., Feng, F., et al. (2015). The cystic fibrosis transmembrane conductance regulator as a biomarker in non-small cell lung cancer. Int. J. Oncol. 46 (5), 2107–2115. doi:10.3892/ijo.2015.2921
Linsdell, P., and Hanrahan, J. W. (1998). Glutathione permeability of CFTR. Am. J. Physiology-Cell Physiol. 275 (1), C323–C326. doi:10.1152/ajpcell.1998.275.1.c323
Lopes-Pacheco, M., Boinot, C., Sabirzhanova, I., Morales, M. M., Guggino, W. B., and Cebotaru, L. (2015). Combination of correctors rescue df508-CFTR by reducing its association with Hsp40 and Hsp27. J. Biol. Chem. 290 (42), 25636–25645. doi:10.1074/jbc.m115.671925
Lopes-Pacheco, M., Sabirzhanova, I., Rapino, D., Morales, M. M., Guggino, W. B., and Cebotaru, L. (2016). Correctors rescue CFTR mutations in nucleotide-binding domain 1 (NBD1) by modulating proteostasis. Chembiochem 17 (6), 493–505. doi:10.1002/cbic.201500620
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M., and Kroemer, G. (2013). The hallmarks of aging. Cell 153 (6), 1194–1217. doi:10.1016/j.cell.2013.05.039
Luciani, A., Villella, V. R., Esposito, S., Gavina, M., Russo, I., Silano, M., et al. (2012). Targeting autophagy as a novel strategy for facilitating the therapeutic action of potentiators on ΔF508 cystic fibrosis transmembrane conductance regulator. Autophagy 8 (11), 1657–1672. doi:10.4161/auto.21483
Luciani, A., Villella, V. R., Vasaturo, A., Giardino, I., Raia, V., Pettoello-Mantovani, M., et al. (2009). SUMOylation of tissue transglutaminase as link between oxidative stress and inflammation. J. Immunol. 183 (4), 2775–2784. doi:10.4049/jimmunol.0900993
Ly, K., Smith, M. P., Thibodeaux, Q., Reddy, V., Liao, W., and Bhutani, T. (2019). Anti IL-17 in psoriasis. Expert Rev. Clin. Immunol. 15 (11), 1185–1194. doi:10.1080/1744666x.2020.1679625
MacKenzie, T., Gifford, A. H., Sabadosa, K. A., Quinton, H. B., Knapp, E. A., Goss, C. H., et al. (2014). Longevity of patients with cystic fibrosis in 2000 to 2010 and beyond: survival analysis of the Cystic Fibrosis Foundation patient registry. Ann. Intern. Med. 161 (4), 233–241. doi:10.7326/m13-0636
Magalhães, M., Tost, J., Pineau, F., Rivals, I., Busato, F., Alary, N., et al. (2018). Dynamic changes of DNA methylation and lung disease in cystic fibrosis: lessons from a monogenic disease. Epigenomics 10 (8), 1131–1145. doi:10.2217/epi-2018-0005
Maisonneuve, P., Marshall, B. C., Knapp, E. A., and Lowenfels, A. B. (2013). Cancer risk in cystic fibrosis: a 20-year nationwide study from the United States. J. Natl. Cancer Inst. 105 (2), 122–129. doi:10.1093/jnci/djs481
Mall, M., Grubb, B. R., Harkema, J. R., O'Neal, W. K., and Boucher, R. C. (2004). Increased airway epithelial Na+ absorption produces cystic fibrosis-like lung disease in mice. Nat. Med. 10 (5), 487–493. doi:10.1038/nm1028
Manzanares, D., Krick, S., Baumlin, N., Dennis, J. S., Tyrrell, J., Tarran, R., et al. (2015). Airway surface dehydration by transforming growth factor β (TGF-β) in cystic fibrosis is due to decreased function of a voltage-dependent potassium channel and can Be rescued by the drug pirfenidone. J. Biol. Chem. 290 (42), 25710–25716. doi:10.1074/jbc.m115.670885
Martins, R., Lithgow, G. J., and Link, W. (2016). Long live FOXO : unraveling the role of FOXO proteins in aging and longevity. Aging Cell 15 (2), 196–207. doi:10.1111/acel.12427
Matsui, H., Grubb, B. R., Tarran, R., Randell, S. H., Gatzy, J. T., Davis, C. W., et al. (1998). Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell 95 (7), 1005–1015. doi:10.1016/s0092-8674(00)81724-9
Mayer, M. L., Blohmke, C. J., Falsafi, R., Fjell, C. D., Madera, L., Turvey, S. E., et al. (2013). Rescue of dysfunctional autophagy attenuates hyperinflammatory responses from cystic fibrosis cells. J.I. 190 (3), 1227–1238. doi:10.4049/jimmunol.1201404
McCuaig, S., and Martin, J. G. (2013). How the airway smooth muscle in cystic fibrosis reacts in proinflammatory conditions: implications for airway hyper-responsiveness and asthma in cystic fibrosis. Lancet Respir. Med. 1 (2), 137–147. doi:10.1016/s2213-2600(12)70058-9
McElvaney, N. G., Doujaiji, B., Moan, M. J., Burnham, M. R., Wu, M. C., and Crystal, R. G. (1993). Pharmacokinetics of recombinant secretory leukoprotease inhibitor aerosolized to normals and individuals with cystic fibrosis. Am. Rev. Respir. Dis. 148 (4 Pt 1), 1056–1060. doi:10.1164/ajrccm/148.4_pt_1.1056
McElvaney, N. G., Hubbard, R. C., Birrer, P., Crystal, R. G., Chernick, M. S., Frank, M. M., et al. (1991). Aerosol α1 -antitrypsin treatment for cystic fibrosis. The Lancet 337 (8738), 392–394. doi:10.1016/0140-6736(91)91167-s
McElvaney, O. J., Zaslona, Z., Becker-Flegler, K., Palsson-McDermott, E. M., Boland, F., Gunaratnam, C., et al. (2019). Specific inhibition of the NLRP3 inflammasome as an antiinflammatory strategy in cystic fibrosis. Am. J. Respir. Crit. Care Med. 200 (11), 1381–1391. doi:10.1164/rccm.201905-1013oc
Meiners, S., Eickelberg, O., and Königshoff, M. (2015). Hallmarks of the ageing lung. Eur. Respir. J. 45 (3), 807–827. doi:10.1183/09031936.00186914
Meyer, K. C., Rosenthal, N. S., Soergel, P., and Peterson, K. (1998). Neutrophils and low-grade inflammation in the seemingly normal aging human lung. Mech. Ageing Development 104 (2), 169–181. doi:10.1016/s0047-6374(98)00065-7
Middleton, P. G., Mall, M. A., Dřevínek, P., Lands, L. C., McKone, E. F., Polineni, D., et al. (2019). Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 381 (19), 1809–1819. doi:10.1056/nejmoa1908639
Mingione, A., Dei Cas, M., Bonezzi, F., Caretti, A., Piccoli, M., Anastasia, L., et al. (2020). Inhibition of sphingolipid synthesis as a phenotype-modifying therapy in cystic fibrosis. Cell Physiol Biochem 54 (1), 110–125. doi:10.33594/000000208
Montanini, L., Smerieri, A., Gullì, M., Cirillo, F., Pisi, G., Sartori, C., et al. (2016). miR-146a, miR-155, miR-370, and miR-708 are CFTR-dependent, predicted FOXO1 regulators and change at onset of CFRDs. J. Clin. Endocrinol. Metab. 101 (12), 4955–4963. doi:10.1210/jc.2016-2431
Mora, A. L., and Rojas, M. (2013). Adult stem cells for chronic lung diseases. Respirology 18 (7), 1041–1046. doi:10.1111/resp.12112
Moskalev, A. A., Shaposhnikov, M. V., Plyusnina, E. N., Zhavoronkov, A., Budovsky, A., Yanai, H., et al. (2013). The role of DNA damage and repair in aging through the prism of Koch-like criteria. Ageing Res. Rev. 12 (2), 661–684. doi:10.1016/j.arr.2012.02.001
Moss, R. B., Mayer-Hamblett, N., Wagener, J., Daines, C., Hale, K., Ahrens, R., et al. (2005). Randomized, double-blind, placebo-controlled, dose-escalating study of aerosolized interferon gamma-1b in patients with mild to moderate cystic fibrosis lung disease. Pediatr. Pulmonol. 39 (3), 209–218. doi:10.1002/ppul.20152
Moss, R. B., Mistry, S. J., Konstan, M. W., Pilewski, J. M., Kerem, E., Tal-Singer, R., et al. (2013). Safety and early treatment effects of the CXCR2 antagonist SB-656933 in patients with cystic fibrosis. J. Cystic Fibrosis 12 (3), 241–248. doi:10.1016/j.jcf.2012.08.016
Nakamura, H., Yoshimura, K., McElvaney, N. G., and Crystal, R. G. (1992). Neutrophil elastase in respiratory epithelial lining fluid of individuals with cystic fibrosis induces interleukin-8 gene expression in a human bronchial epithelial cell line. J. Clin. Invest. 89 (5), 1478–1484. doi:10.1172/jci115738
Naylor, R. M., Baker, D. J., and van Deursen, J. M. (2013). Senescent cells: a novel therapeutic target for aging and age-related diseases. Clin. Pharmacol. Ther. 93 (1), 105–116. doi:10.1038/clpt.2012.193
Nichols, D., Chmiel, J., and Berger, M. (2008). Chronic inflammation in the cystic fibrosis lung: alterations in inter- and intracellular signaling. Clinic Rev. Allerg Immunol. 34 (2), 146–162. doi:10.1007/s12016-007-8039-9
Nichols, D. P., Odem-Davis, K., Cogen, J. D., Goss, C. H., Ren, C. L., Skalland, M., et al. (2020). Pulmonary outcomes associated with long-term Azithromycin therapy in cystic fibrosis. Am. J. Respir. Crit. Care Med. 201 (4), 430–437. doi:10.1164/rccm.201906-1206oc
Oermann, C. M. K. M., and Wheeler C, Cumming. S. (2007). A pilot study evaluating the potential use of low-dose methotrexate as an anti-inflammatory therapy for cystic fibrosis lung disease. Pediatr. Pulmonol 42, 292–293.
Oh, J., Lee, Y. D., and Wagers, A. J. (2014). Stem cell aging: mechanisms, regulators and therapeutic opportunities. Nat. Med. 20 (8), 870–880. doi:10.1038/nm.3651
Pankow, S., Bamberger, C., Calzolari, D., Martínez-Bartolomé, S., Lavallée-Adam, M., Balch, W. E., et al. (2015). ∆F508 CFTR interactome remodelling promotes rescue of cystic fibrosis. Nature 528 (7583), 510–516. doi:10.1038/nature15729
Parikh, P., Wicher, S., Khandalavala, K., Pabelick, C. M., Britt, R. D., and Prakash, Y. S. (2019). Cellular senescence in the lung across the age spectrum. Am. J. Physiology-Lung Cell Mol. Physiol. 316 (5), L826–L842. doi:10.1152/ajplung.00424.2018
Parker, M. W., Rossi, D., Peterson, M., Smith, K., Sikström, K., White, E. S., et al. (2014). Fibrotic extracellular matrix activates a profibrotic positive feedback loop. J. Clin. Invest. 124 (4), 1622–1635. doi:10.1172/jci71386
Perez, A., van Heeckeren, A. M., Nichols, D., Gupta, S., Eastman, J. F., and Davis, P. B. (2008). Peroxisome proliferator-activated receptor-γ in cystic fibrosis lung epithelium. Am. J. Physiology-Lung Cell Mol. Physiol. 295 (2), L303–L313. doi:10.1152/ajplung.90276.2008
Peterson-Carmichael, S. L., Harris, W. T., Goel, R., Noah, T. L., Johnson, R., Leigh, M. W., et al. (2009). Association of lower airway inflammation with physiologic findings in young children with cystic fibrosis. Pediatr. Pulmonol. 44 (5), 503–511. doi:10.1002/ppul.21044
Petit-Bertron, A.-F., Tabary, O., Corvol, H., Jacquot, J., Clément, A., Cavaillon, J.-M., et al. (2008). Circulating and airway neutrophils in cystic fibrosis display different TLR expression and responsiveness to interleukin-10. Cytokine 41 (1), 54–60. doi:10.1016/j.cyto.2007.10.012
Prakash, Y. S., Pabelick, C. M., and Sieck, G. C. (2017). Mitochondrial dysfunction in airway disease. Chest 152 (3), 618–626. doi:10.1016/j.chest.2017.03.020
Regamey, N., Jeffery, P. K., Alton, E. W. F. W., Bush, A., and Davies, J. C. (2011). Airway remodelling and its relationship to inflammation in cystic fibrosis. Thorax 66 (7), 624–629. doi:10.1136/thx.2009.134106
Reilly, R., Mroz, M. S., Dempsey, E., Wynne, K., Keely, S. J., McKone, E. F., et al. (2017). Targeting the PI3K/Akt/mTOR signalling pathway in Cystic Fibrosis. Sci. Rep. 7 (1), 7642. doi:10.1038/s41598-017-06588z
Ren, C. L., Pasta, D. J., Rasouliyan, L., Wagener, J. S., Konstan, M. W., and Morgan, W. J. (2008). Relationship between inhaled corticosteroid therapy and rate of lung function decline in children with cystic fibrosis. J. Pediatr. 153 (6), 746–751. doi:10.1016/j.jpeds.2008.07.010
Rimessi, A., Bezzerri, V., Patergnani, S., Marchi, S., Cabrini, G., and Pinton, P. (2015). Mitochondrial Ca2+-dependent NLRP3 activation exacerbates the Pseudomonas aeruginosa-driven inflammatory response in cystic fibrosis. Nat. Commun. 6, 6201. doi:10.1038/ncomms7201
Robertson, K. D. (2005). DNA methylation and human disease. Nat. Rev. Genet. 6 (8), 597–610. doi:10.1038/nrg1655
Roum, J. H., Buhl, R., McElvaney, N. G., Borok, Z., and Crystal, R. G. (1985). Systemic deficiency of glutathione in cystic fibrosis. J. Appl. Physiol. 75 (6), 2419–2424. doi:10.1152/jappl.1993.75.6.24191993
Rowe, S. M., Daines, C., Ringshausen, F. C., Kerem, E., Wilson, J., Tullis, E., et al. (2017). Tezacaftor-Ivacaftor in residual-function heterozygotes with cystic fibrosis. N. Engl. J. Med. 377 (21), 2024–2035. doi:10.1056/nejmoa1709847
Rowe, S. M., Heltshe, S. L., Gonska, T., Donaldson, S. H., Borowitz, D., Gelfond, D., et al. (2014). Clinical mechanism of the cystic fibrosis transmembrane conductance regulator potentiator ivacaftor in G551D-mediated cystic fibrosis. Am. J. Respir. Crit. Care Med. 190 (2), 175–184. doi:10.1164/rccm.201404-0703oc
Ruan, X. Z., Moorhead, J. F., Fernando, R., Wheeler, D. C., Powis, S. H., and Varghese, Z. (2003). PPAR agonists protect mesangial cells from interleukin 1 -induced intracellular lipid accumulation by activating the ABCA1 cholesterol efflux pathway. J. Am. Soc. Nephrol. 14 (3), 593–600. doi:10.1097/01.asn.0000050414.52908.da
Sagel, S. D., Sontag, M. K., Wagener, J. S., Kapsner, R. K., Osberg, I., and Accurso, F. J. (2002). Induced sputum inflammatory measures correlate with lung function in children with cystic fibrosis. J. Pediatr. 141 (6), 811–817. doi:10.1067/mpd.2002.129847
Scambler, T., Jarosz-Griffiths, H. H., Lara-Reyna, S., Pathak, S., Wong, C., Holbrook, J., et al. (2019). ENaC-mediated sodium influx exacerbates NLRP3-dependent inflammation in cystic fibrosis. Elife 8. doi:10.7554/elife.49248
Scott, M., and De Sario, A. (2020). DNA methylation changes in cystic fibrosis: cause or consequence? Clin. Genet. 98 (1), 3–9. doi:10.1111/cge.13731
Scott, P., Anderson, K., Singhania, M., and Cormier, R. (2020). Cystic fibrosis, CFTR, and colorectal cancer. Int. J. Mol. Sci. 21 (8). doi:10.3390/ijms21082891
Seiler, F., Hellberg, J., Lepper, P. M., Kamyschnikow, A., Herr, C., Bischoff, M., et al. (2013). FOXO transcription factors regulate innate immune mechanisms in respiratory epithelial cells. J.I. 190 (4), 1603–1613. doi:10.4049/jimmunol.1200596
Simmonds, N. J., Cullinan, P., and Hodson, M. E. (2009). Growing old with cystic fibrosis - the characteristics of long-term survivors of cystic fibrosis. Respir. Med. 103 (4), 629–635. doi:10.1016/j.rmed.2008.10.011
Sirinupong, N., and Yang, Z. (2015). Epigenetics in cystic fibrosis: epigenetic targeting of a genetic disease. Cdt 16 (9), 976–987. doi:10.2174/1389450116666150416114514
Skov, M., Pressler, T., Lykkesfeldt, J., Poulsen, H. E., Jensen, P. Ø., Johansen, H. K., et al. (2015). The effect of short-term, high-dose oral N-acetylcysteine treatment on oxidative stress markers in cystic fibrosis patients with chronic P. aeruginosa infection - a pilot study. J. Cystic Fibrosis 14 (2), 211–218. doi:10.1016/j.jcf.2014.09.015
Smerieri, A., Montanini, L., Maiuri, L., Bernasconi, S., and Street, M. (2014). FOXO1 content is reduced in cystic fibrosis and increases with IGF-I treatment. Ijms 15 (10), 18000–18022. doi:10.3390/ijms151018000
Snodgrass, S. M., Cihil, K. M., Cornuet, P. K., Myerburg, M. M., and Swiatecka-Urban, A. (2013). Tgf-beta1 inhibits Cftr biogenesis and prevents functional rescue of DeltaF508-Cftr in primary differentiated human bronchial epithelial cells. PLoS One 8 (5), e63167. doi:10.1371/journal.pone.0063167
Soltys, J., Bonfield, T., Chmiel, J., and Berger, M. (2002). Functional IL-10 deficiency in the lung of cystic fibrosis (cftr−/−) and IL-10 knockout mice causes increased expression and function of B7 costimulatory molecules on alveolar macrophages. J. Immunol. 168 (4), 1903–1910. doi:10.4049/jimmunol.168.4.1903
Starosta, V., Rietschel, E., Paul, K., Baumann, U., and Griese, M. (2006). Oxidative changes of bronchoalveolar proteins in cystic fibrosis. Chest 129 (2), 431–437. doi:10.1378/chest.129.2.431
Strub, M. D., and McCray, P. B. (2020). Transcriptomic and proteostasis networks of CFTR and the development of small molecule modulators for the treatment of cystic fibrosis lung disease. Genes (Basel) 11 (5). doi:10.3390/genes11050546
Sun, H., Harris, W. T., Kortyka, S., Kotha, K., Ostmann, A. J., Rezayat, A., et al. (2014). Tgf-beta downregulation of distinct chloride channels in cystic fibrosis-affected epithelia. PLoS One 9 (9), e106842. doi:10.1371/journal.pone.0106842
Taams, L. S. (2020). Interleukin-17 in rheumatoid arthritis: trials and tribulations. J. Exp. Med. 217 (3). doi:10.1084/jem.20192048
Taylor-Cousar, J. L., Munck, A., McKone, E. F., van der Ent, C. K., Moeller, A., Simard, C., et al. (2017). Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N. Engl. J. Med. 377 (21), 2013–2023. doi:10.1056/nejmoa1709846
Teramura, T., and Onodera, Y. (2018). Stem cell depletion by inflammation-associated miR-155. Aging 10 (1), 17–18. doi:10.18632/aging.101374
Than, B. L., Linnekamp, J. F., Starr, T. K., Largaespada, D. A., Rod, A., Zhang, Y., et al. (2016). CFTR is a tumor suppressor gene in murine and human intestinal cancer. Oncogene 35 (32), 4179–4187. doi:10.1038/onc.2015.483
Tirouvanziam, R., Conrad, C. K., Bottiglieri, T., Herzenberg, L. A., Moss, R. B., and Herzenberg, L. A. (2006). High-dose oral N-acetylcysteine, a glutathione prodrug, modulates inflammation in cystic fibrosis. Proc. Natl. Acad. Sci. 103 (12), 4628–4633. doi:10.1073/pnas.0511304103
Tosco, A., De Gregorio, F., Esposito, S., De Stefano, D., Sana, I., Ferrari, E., et al. (2016). A novel treatment of cystic fibrosis acting on-target: cysteamine plus epigallocatechin gallate for the autophagy-dependent rescue of class II-mutated CFTR. Cell Death Differ 23 (8), 1380–1393. doi:10.1038/cdd.2016.22
Tsui, L.-C. (1995). The cystic fibrosis transmembrane conductance regulator gene. Am. J. Respir. Crit. Care Med. 151 (3 Pt 2), S47–S53. doi:10.1164/ajrccm/151.3_pt_2.s47
Tu, Z., Chen, Q., Zhang, J. T., Jiang, X., Xia, Y., and Chan, H. C. (2016). CFTR is a potential marker for nasopharyngeal carcinoma prognosis and metastasis. Oncotarget 7 (47), 76955–76965. doi:10.18632/oncotarget.12762
Valdivieso, A. G., Clauzure, M., Marín, M. C., Taminelli, G. L., Massip Copiz, M. M., Sánchez, F., et al. (2012). The mitochondrial complex I activity is reduced in cells with impaired cystic fibrosis transmembrane conductance regulator (CFTR) function. PLoS One 7 (11), e48059. doi:10.1371/journal.pone.0048059
Van Biervliet, S., Devos, M., Delhaye, T., Van Biervliet, J. P., Robberecht, E., and Christophe, A. (2008). Oral DHA supplementation in ΔF508 homozygous cystic fibrosis patients. Prostaglandins, Leukot. Essent. Fatty Acids 78 (2), 109–115. doi:10.1016/j.plefa.2007.12.005
Van Goor, F., Hadida, S., Grootenhuis, P. D. J., Burton, B., Stack, J. H., Straley, K. S., et al. (2011). Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. 108 (46), 18843–18848. doi:10.1073/pnas.1105787108
Vellai, T. (2009). Autophagy genes and ageing. Cel Death Differ 16 (1), 94–102. doi:10.1038/cdd.2008.126
Velsor, L. W., Kariya, C., Kachadourian, R., and Day, B. J. (2006). Mitochondrial oxidative stress in the lungs of cystic fibrosis transmembrane conductance regulator protein mutant mice. Am. J. Respir. Cel Mol Biol 35 (5), 579–586. doi:10.1165/rcmb.2005-0473oc
Velsor, L. W., van Heeckeren, A., and Day, B. J. (2001). Antioxidant imbalance in the lungs of cystic fibrosis transmembrane conductance regulator protein mutant mice. Am. J. Physiology-Lung Cell Mol. Physiol. 281 (1), L31–L38. doi:10.1152/ajplung.2001.281.1.l31
Vencken, S., Foged, C., Ramsey, J. M., Sweeney, L., Cryan, S. A., MacLoughlin, R. J., et al. (2019). Nebulised lipid-polymer hybrid nanoparticles for the delivery of a therapeutic anti-inflammatory microRNA to bronchial epithelial cells. ERJ Open Res. 5 (2). doi:10.1183/23120541.00161-2018
Villella, V. R., Esposito, S., Bruscia, E. M., Vicinanza, M., Cenci, S., Guido, S., et al. (2013). Disease-relevant proteostasis regulation of cystic fibrosis transmembrane conductance regulator. Cel Death Differ 20 (8), 1101–1115. doi:10.1038/cdd.2013.46
Wainwright, C. E., Elborn, J. S., Ramsey, B. W., Marigowda, G., Huang, X., Cipolli, M., et al. (2015). Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 373 (3), 220–231. doi:10.1056/nejmoa1409547
Zelvyte, I., Dominaitiene, R., Crisby, M., and Janciauskiene, S. (2002). Modulation of inflammatory mediators and pparγand nfκb expression by pravastatin in response to lipoproteins in human monocytes in vitro. Pharmacol. Res. 45 (2), 147–154. doi:10.1006/phrs.2001.0922
Zingarelli, B., Sheehan, M., Hake, P. W., O’Connor, M., Denenberg, A., and Cook, J. A. (2003). Peroxisome proliferator activator receptor-γ ligands, 15-deoxy-d12,14-prostaglandin J2 and ciglitazone, reduce systemic inflammation in polymicrobial sepsis by modulation of signal transduction pathways. J. Immunol. 171 (12), 6827–6837. doi:10.4049/jimmunol.171.12.6827
Keywords: cystic fibrosis, aging, inflammaging, oxidative stress, mitochondrial dysfunction, senescence
Citation: Künzi L, Easter M, Hirsch MJ and Krick S (2021) Cystic Fibrosis Lung Disease in the Aging Population. Front. Pharmacol. 12:601438. doi: 10.3389/fphar.2021.601438
Received: 27 November 2020; Accepted: 15 March 2021;
Published: 15 April 2021.
Edited by:
Noel Gerard McElvaney, Royal College of Surgeons in Ireland, IrelandReviewed by:
Isaac Kirubakaran Sundar, University of Kansas Medical Center, United StatesAntonio Molino, University of Naples Federico II, Italy
Copyright © 2021 Künzi, Easter, Hirsch and Krick. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefanie Krick, c2tyaWNrQHVhYm1jLmVkdQ==