
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 31 May 2021
Sec. Inflammation Pharmacology
Volume 12 - 2021 | https://doi.org/10.3389/fphar.2021.599393
This article is part of the Research Topic Precision Medicine and Immuno-Mediated Inflammatory Diseases: Latest Progress and Next Challenges View all 15 articles
 Yan Chen1,2,3*
Yan Chen1,2,3* Yuan Ma1
Yuan Ma1 Jin Jin Feng1
Jin Jin Feng1 Yi He Wang1
Yi He Wang1 Tian Fang Li4
Tian Fang Li4 Katariina Nurmi2,3
Katariina Nurmi2,3 Kari K. Eklund2,3,5
Kari K. Eklund2,3,5 Jian Guo Wen1*
Jian Guo Wen1*NLRP3 inflammasome has been implicated in impaired post-injury muscle healing and in muscle atrophy. Histamine receptors play an important role in inflammation, but the role of histamine H3 receptor (H3R) in myocyte regeneration and in the regulation of NLRP3 inflammasome is not known. We studied the effects of H3R signaling on C2C12 myocyte viability, apoptosis, and tumor necrosis factor alpha (TNFα)-induced NLRP3 inflammasome activation during striated myogenic differentiation at three time points (days 0, 3, and 6). Expression of Nlrp3, interleukin-1β (IL-1β), and myogenesis markers were determined. TNFα reduced overall viability of C2C12 cells, and exposure to TNFα induced apoptosis of cells at D6. Activation of H3R had no effect on viability or apoptosis, whereas inhibition of H3R increased TNFα-induced apoptosis. Stimulation of C2C12 cells with TNFα increased Nlrp3 mRNA expression at D3 and D6. Moreover, TNFα reduced the expression of myogenesis markers MyoD1, Myogenin, and Myosin-2 at D3 and D6. H3R attenuated TNFα-induced expression of Nlrp3 and further inhibited the myogenesis marker expression; while H3R -blockage enhanced the proinflammatory effects of TNFα and increased the myogenesis marker expression. TNFα-induced secretion of mature IL-1β was dependent on the activation of the NLRP3 inflammasome, as shown by the reduced secretion of mature IL-1β upon treatment of the cells with the small molecule inhibitor of the NLRP3 inflammasome (MCC950). The activation of H3R reduced TNFα-induced IL-1β secretion, while the H3R blockage had an opposite effect. In conclusion, the modulation of H3R activity regulates the effects of TNFα on C2C12 myocyte differentiation and TNFα-induced activation of NLRP3 inflammasome. Thus, H3R signaling may represent a novel target for limiting postinjury muscle inflammation and muscle atrophy.
Inflammation can complicate post-injury muscle healing and slow down the regenerative repair process (Hofer et al., 2014). However, the factors underlying the post injury inflammation are poorly known. It is believed that activation of the inflammasomes, with ensuing production of highly proinflammatory cytokines, IL-1β and IL-18, may play a key role by initiating the inflammatory reaction (Mangan et al., 2018). Nucleotide-binding domain and leucine-rich repeat containing family (NLR), pyrin domain containing 3 (NLRP3) inflammasome is the most widely studied inflammasome. It is activated by a myriad of factors, including exogenous pathogens and endogenous danger signals, and has been implicated in a variety of immunological (Mulay et al., 2018) and non-immunological diseases (Hughes et al., 2016). The NLRP3 inflammasome is a multi-protein complex, consisting of the sensor protein NLRP3, apoptosis-associated speck-like protein containing a CARD (ASC), and the precursor of caspase 1 (pro-caspase-1). Upon activation, NLRP3 is associated with pro-caspase-1 via the adaptor protein ASC resulting in autoactivation of pro-caspase-1 into its active form. The active caspase-1 then catalyzes the cleavage of the inactive precursors of interleukin 1β (pro-IL-1β) and interleukin 18 (pro-IL-18) into their active secreted forms, thus triggering the inflammatory storm (Man and Kanneganti, 2015). NLRP3 inflammasome is mainly expressed in innate immune cells such as neutrophils, macrophages, monocytes, and dendritic cells, but low expression levels are seen in other cell types and tissues (Latz et al., 2013; Elliott and Sutterwala, 2015; Guo et al., 2015; Jo et al., 2016). NLRP3 has been shown to be expressed also in myocytes (Wei et al., 2020) and it has been implicated in the pathogenesis of inflammatory myopathies (Boursereau et al., 2018).
After exercise, skeletal muscle can secrete many bioactive molecules, known as myokines, and tumor necrosis factor alpha (TNFα) is one of the most common exercise-regulated myokines (also named adipo-myokines) (Görgens et al., 2015), which can induce autophagy and apoptosis in mouse C2C12 myoblasts and myotubes (Gallo et al., 2015). The inflammation-associated insulin resistance of C2C12 myoblasts and myotubes has been suggested to be mediated by the activation of NLRP3 inflammasome (Cho and Kang, 2015).
There are four known histamine receptors, of which histamine H3 receptor (H3R) is mainly expressed in immune and nerve cells, and is involved in nerve conduction, muscle contraction, gastrointestinal neuromodulation, and inflammation (Branco et al., 2018). We found previously that H3R is expressed in C2C12 myoblasts and in primary adult mid-urethral striated muscles (Chen et al., 2015; Chen et al., 2015). In C2C12 myoblasts the expression of H3R was increased during their myogenic differentiation into striated myocytes, and in myocytes the activation of the H3R facilitated the relaxation of the cells by limiting the cytoplasmic calcium peak (Chen et al., 2015). Histamine and histamine receptors possess immunomodulatory effects (Branco et al., 2018). Histamine has been reported to dampen the inflammatory effects of lipopolysaccharides, and in particular, histamine has been shown to inhibit the secretion of IL-1β in microglial cells (Barata-Antunes et al., 2017). In present study we explore further the role of H3R in regulation of myoblast differentiation and myocyte function. We hypothesized that H3R could modulate the secretion of IL-1β by regulating the activation of the NLRP3 inflammasome. By utilizing highly selective H3R agonists (methimepip, Met) (Chen et al., 2015; Panula et al., 2015), and H3R blocker (ciproxifan, CPX) (Chen et al., 2015; Panula et al., 2015) we show that activation of H3R signaling mitigates the TNFα-induced NLRP3 inflammasome activation, and thus H3R signaling could represent a potential target for treatment in conditions involving muscle inflammation and inflammation induced muscle atrophy.
The cell culture medium, Dulbecco’s modified Eagle’s medium (DMEM), was purchased from Biowhittaker/Lonza, penicillin-streptomycin from Cambrex/Lonza, Glutamine [Gibco® GlutaMAX™ Supplement (35050061)], and heat inactivated fetal bovine serum (FBS, 10100147) was from Thermo Fisher Scientific. The primers were designed and purchased from Sangon Biotech, China. Mouse IL-1β enzyme-linked immunosorbent assay (ELISA) Kit (D720335-0096) and recombinant murine TNFα (C600052-0005) were purchased from Sangon Biotech, China. The Cell Counting Kit (CCK-8) (C0038) and Annexin V-FITC/propidium iodide (PI) Apoptosis Detection Kit (C1063) were purchased from Beyotime Corporation, China. H3R agonist methimepip (Met) (sc-204080) was from Santa Cruz Corporation, and ciproxifan (CPX) (C6492), H3R blocker, was from SIGMA Inc. MCC950 (CP-456773, 210826-40-7, AbMole, China) was purchased from AbMole Inc.
Mouse C2C12 myoblasts were obtained from American Type Culture Collection, and maintained in DMEM supplemented with 10% fetal bovine serum (FBS), 1% antibiotics (100 U/ml penicillin and 100 μg/ml streptomycin (GIBCO #15140-122), and glutamine at 37°C in humidified 5% CO2-in-air. In the differentiation medium, FBS was reduced from 10 to 1%, but otherwise the differentiation medium had the same composition as the growth medium. The cells were passaged at 80% confluence by trypsinization (0.5% trypsin in 0.5 mM EDTA, Gibco BRL, Life Technologies, Gaithersburg, MD).
C2C12 cells were seeded in 96-well plates (1.6 × 104 cells/well), 12-well plates (5 × 104 cells/well), or six-well plates (4 × 105/well) in the growth media and cultured for three days before changing to differentiation media. The differentiation time points are calculated as follows: D0 represent the cells that were collected 3 days after seeding and have not received differentiation media; D3 cells received differentiation media 3 days after seeding, and were cultured for 3 days in differentiation media; D6 received differentiation media 6 days after seeding, and were cultured for 6 days in differentiation media. Collected samples were subjected on analysis of proliferation and viability (CKK-8 assay), apoptosis and necrosis (AnnexinV/PI assay) and gene expression (qPCR). The experimental protocol is described as a flowchart in Supplementary Figure S1A.
Experimental groups 1–4 represent the non-differentiated myocytes, that were collected on differentiation day 0 (D0) 12 h after stimulations with TNFα, TNFα+ Met, TNFα+ CPX, or left unstimulated.
Groups 5–8 were cultured for 3 days (D3) in differentiation media, after which they were stimulated for 12 h with TNFα, TNFα+ Met, TNFα+ CPX, or left unstimulated (Differentiation media was changed on D1 and D2).
Groups 9–12 represent the fully differentiated myocytes that were cultured for 6 days in differentiation media. D6 cells were collected after 12 h stimulations with TNFα, TNFα+ Met, TNFα+ CPX, or left unstimulated (Differentiation media was changed on D3, D4, and D5).
All the above-mentioned treatments were applied on cells with identical treatments with or without 1 h preincubation of the cells in the presence of MCC950 (1 µM), which was left on the cells during stimulations. Experiment media were collected for ELISA, and cell lysates were subjected to western blot analysis (Supplementary Figure S1B). Simultaneously cultured cells with Met or CPX alone were analyzed for apoptosis by flow cytometry (FCM).
The cell count kit-8 (CCK-8), a rapid and highly sensitive detection kit for cell proliferation and cytotoxicity, was used to analyze the cell viability according to the manufacturer’s protocol (Beyotime Corporation, Shanghai, China).
Cell apoptosis was detected with FCM. AnnexinV/PI apoptosis detection kit was used according to the manufacturer’s protocols (Beyotime Corporation, Shanghai, China). The method is based on the cytofluorimetric assay established by Vermes et al. The kit discriminates intact cells (Annexin-/PI-) from apoptotic (Annexin +/PI−) and necrotic cells (Annexin +/PI+) (Vermes et al., 1995).
C2C12 cells were harvested and their total cellular RNA was purified using RNeasy Mini Kit (QIAGEN, Düsseldorf, Germany) according to the manufacturer’s instructions. Briefly, 1 μg of total RNA was reverse transcribed using iScript cDNA Synthesis Kit (BioRad Laboratories, Hercules, CA). Quantitative RT-PCR (qPCR) was performed with 100 ng first-strand cDNA using iQ SYBR Green Supermix (Bio-Rad Laboratories, Hercules, CA) in iCycler iQ5 Multicolor Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, CA). The copy numbers of mRNA in the samples analyzed were determined in triplicate and normalized against PBGD gene as an endogenous control and the relative units were calculated using the comparative Ct method. The following primers were designed with the NCBI Primer Blast program:
Il1b sense primer (F, forward): 5′-GCAACTGTTCCTGAACTCAACT-3′, anti-sense primer (R, reverse): 5′-ATCTTTTGGGGTCCGTCAACT-3′; Nlrp3 sense primer (F, forward): 5′-ATTACCCGCCCGAGAAAGG-3′, anti-sense primer (R, reverse): 5′-TCGCAGCAAAGATCCACACAG-3′; MyoD1 sense primer (F, forward): 5′-CCACTCCGGGACATAGACTTG-3′, anti-sense primer (R, reverse): 5′-AAAAGCGCAGGTCTGGTGAG-3′; Myogenin sense primer (F, forward): 5′-GAGACATCCCCCTATTTCTACCA-3′, anti-sense primer (R, reverse): 5′-GCTCAGTCCGCTCATAGCC-3′; Myosin-2 sense primer (F, forward): 5′-GTCAGCACCATGTCTTATGGG-3′, anti-sense primer (R, reverse): 5′-TTTGCCAAATCGGGAAGAGTT-3′; PBGD sense primer (F, forward): 5′-AGGTCGGTGTGAACGGATTTG-3′, anti-sense primer (R, reverse): 5′-GGGGTCGTTGATGGCAACA-3′.
Cells were lyzed in 150 µl of Mammal Cell Protein Extraction Reagent (CWBiotech, Beijing, China). The total protein concentration was determined with a Pierce BCA protein assay kit (Trans, Beijing, China). For each sample 40 µg of protein was loaded and separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis consisting of 5% stacking gel and 12% resolving gel, and the proteins were electrophoretically transferred to a polyvinylidene fluoride (PVDF) membrane. The membranes were blocked with 5% nonfat milk in Tris-buffered saline (pH 7.4) containing 0.1% Tween-20 (TBST). The membranes were incubated overnight at 4 °C with mouse monoclonal antibodies against IL-1β (3A6, mouse mAb #12242S, Cell Signaling Technology, Shanghai, China) in TBST with 5% nonfat milk. The secondary antibody goat anti-mouse IgG, HRP conjugated (CW0102S, CWBiotech, Beijing, China) was used to incubate the membrane for 2 h at room temperature (RT). Labeling was performed using an enhanced chemiluminescence (ECL) system (Thermo Fisher Scientific, Thermo, United States). The density and area of the bands was quantitated.
C2C12 cells were incubated in the presence of TNFα, with or without Met or TNFα or MCC950 at day D0, D3, D6 for 24 h. Mouse IL-1β concentrations in the cell culture media were analyzed using a commercial enzyme-linked immunosorbent assay (ELISA) kit according to the manufacturer’s protocols (Sangon Biotech, Shanghai, China).
Calculations were performed using GraphPad InStat3 for Macintosh software. Overall significance level between the stimulated and control groups or between stimulated and inhibited groups was analyzed using one-way ANOVA, with the Dunnett and Tukey-Kramer multiple comparisons tests when appropriate. Statistical significance was set at p < 0.05. The data are calculated as mean ± SEM of 12 samples (three repeated experiments, each treatment performed in four replicates).
The C2C12 cells were fully differentiated at D6 in each stimulation (Figure 1A). During C2C12 myogenesis, TNFα decreased cell viability at all the measured time points, days 0, 3, 6 (D0, D3, D6), by 20.5% (p < 0.05), 29.8% (p < 0.05), and 29.7% (p < 0.05), respectively, as compared to the controls. Addition of H3R antagonist ciproxifan (CPX) together with TNFα had no additional effect on cell viability at the time points D0 and D3, but further reduced the viability at time point D6 (40.3%, p < 0.05). Meanwhile, addition of H3R agonist methimepip (Met) together with TNFα had no additional effect on cell viability at any of the time points of D0, D3, and D6 (Figure 1).
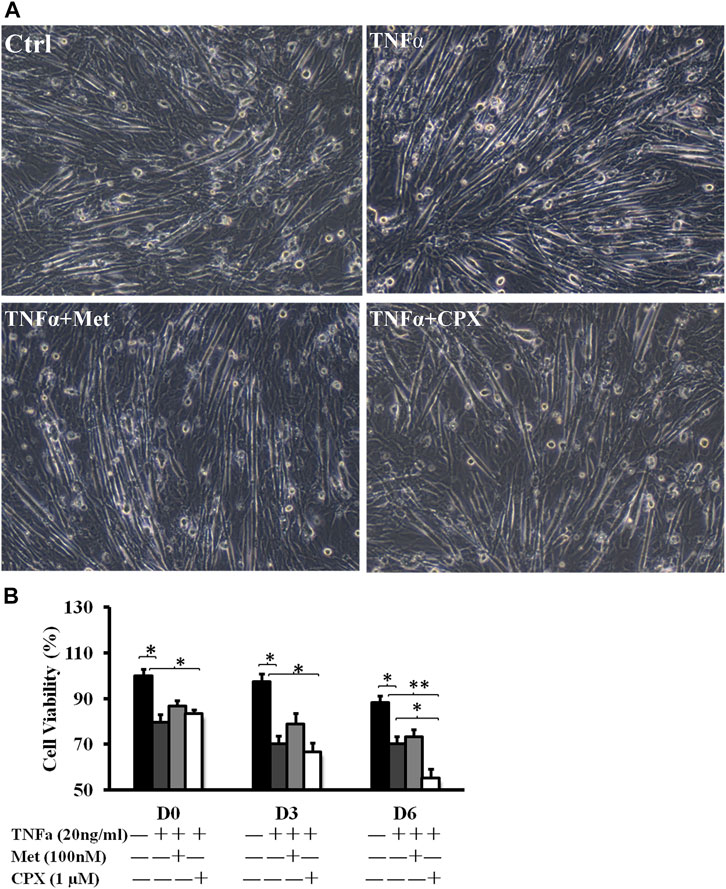
FIGURE 1. The effect of TNFα, Met and CXP on cell viability. (A): D6 C2C12 cells incubated in the presence of TNFa (20 ng/ml), TNFa + Met (100 nM) or TNFa + CPX (1 µM) for 12 h and observed by phase contrast microscopy (100×). (B): Viability at D0, D3, and D6. C2C12 cells were incubated for 12 h in the presence of TNFa, TNFa + Met or TNFa + CPX. The cell death was detected by cell count kit-8 assay; *p < 0.05, **p < 0.01 vs. control of the same differentiation day (black columns); normalizing D0 ctrl = 100%. There was no statistical difference when TNFa + Met or TNFa + CPX was compared with TNFa-treatment of 0- and 3-differentiation day (dark gray columns). Ctrl, control; TNFa, murine Tumor Necrosis Factor-alpha; Met, methimepip; CPX, ciproxifan. D0, undifferentiated cells; D3, cells differentiated for 3 days; D6, cells differentiated for 6 days.
Met alone did not induce apoptosis, however, 15.3% of the cells stimulated with CPX were apoptotic (p < 0.05) at D6 (Figures 2A,B). Next, we studied the effect of H3R inhibition on TNFα-induced apoptosis. During C2C12 myogenesis, apoptosis and necrosis were detected in 4–6.9% of the control cells. Compared to the control cells, TNFα or TNFα +Met did not cause an obvious increase in apoptosis or necrosis during myogenic differentiation (Figure 3). However, treatment with combination of TNFα +CXP induced overall 61.3% (p < 0.01) apoptotic cells on D6, among which early apoptosis was observed in 29.9% (T = 4.2, p < 0.05) of the cells and late apoptosis in 31.4% of the cells (T = 5.0, p < 0.01) (Figure 3).
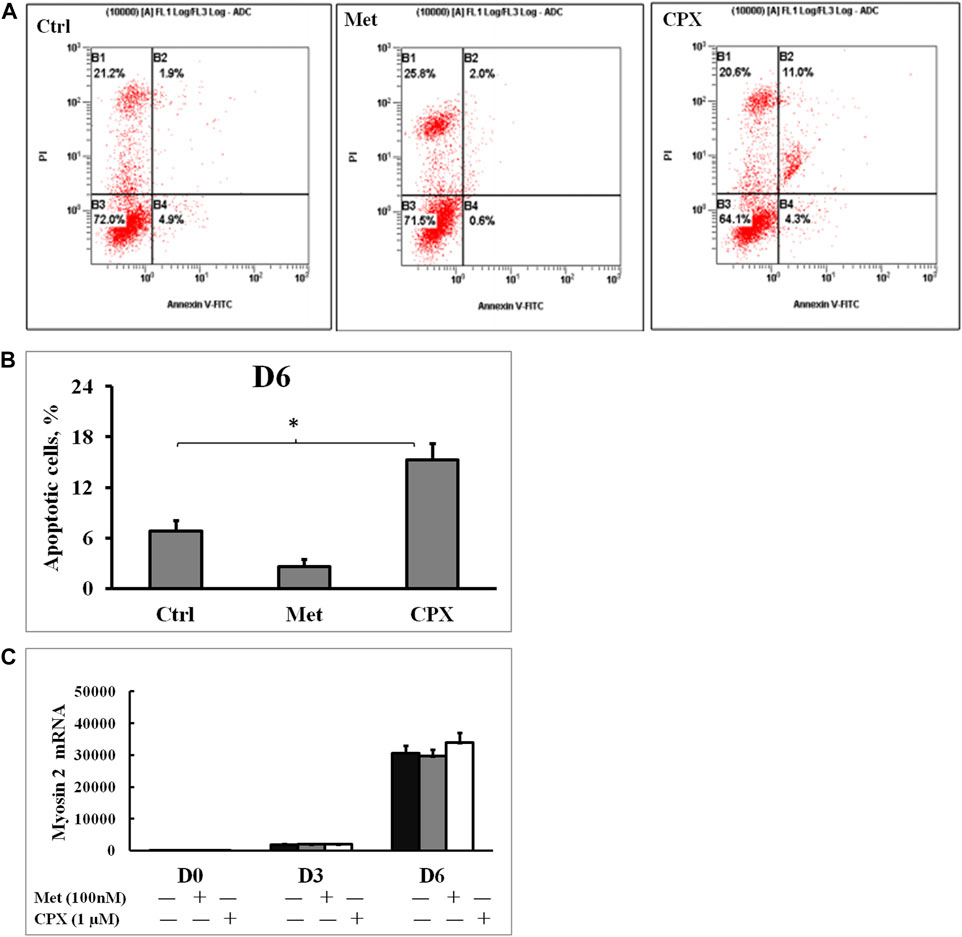
FIGURE 2. The effect of histamine H3 receptor modulators on cell apoptosis and myogenesis. (A): Apoptotic cells were detected by flow cytometry using Annexin V and propidium iodide staining; percentage of mechanically damaged cells (top left quadrant), live cells (bottom left), early apoptotic cells (bottom right), late apoptotic cells (top right). (B): Percentage of apoptotic cells was calculated by adding the early and the late apoptotic cells for each experimental condition.*p < 0.05 vs. control. (C): The C2C12 myocytes were incubated in the presence of Met (100 nM) or CPX (1 μM) for 12 h at D6, and the effect of H3 receptor inhibition or activation on myogenesis was assessed by analyzing the expression of the late myogenesis marker, myosin 2, mRNA by real time RT‐PCR.*p < 0.05 vs. control of the same differentiation day (black columns); normalizing D0 ctrl=1. There was no statistical difference when Met or CPX was compared with control group (dark gray columns). Ctrl, control; Met, methimepip; CPX, ciproxifan. D0, undifferentiated cells; D3, cells differentiated for 3 days; D6, cells differentiated for 6 days.
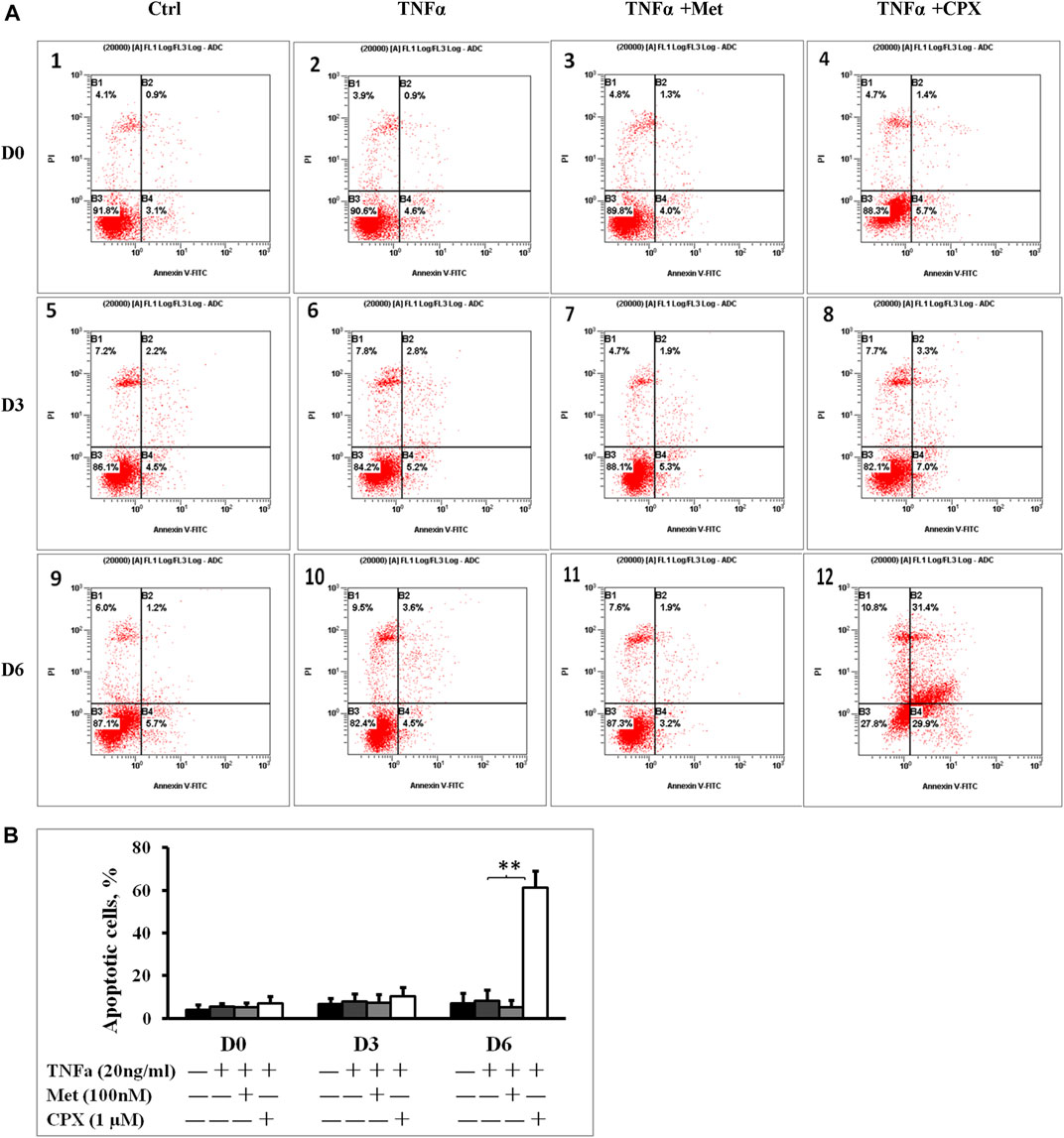
FIGURE 3. Inhibition of histamine H3 receptor enhances apoptosis. (A): C2C12 cells were incubated in the presence of TNFa (20 ng/ml), TNFa + Met (100 nM) or TNFa + CPX (1 µM) at D0, D3, or D6. Apoptotic cells were detected by Annexin V and propidium iodide staining and analyzed by flow cytometry: percentage of cells mechanically damaged (top left quadrant), live cells (bottom left), early apoptotic cells (bottom right), late apoptotic cells (top right). (B): Percentage of apoptotic cells was calculated by adding the early and the late apoptotic cells for each experimental condition. **p < 0.01 vs. TNFα of the same differentiation day (dark gray columns). Ctrl, control; TNFa, murine Tumor Necrosis Factor-alpha; Met, methimepip; CPX, ciproxifan. D0, undifferentiated cells; D3, cells differentiated for 3 days; D6, cells differentiated for 6 days.
As previous study has shown that another proinflammatory cytokine, IL-1β, induces the expression of Nlrp3 in C2C12 cells (Huang et al., 2017), we studied the involvement of the NLRP3 inflammasome in the TNFα-induced inflammation. We analyzed the Nlrp3 expression during C2C12 myogenesis on D0, D3 and D6. TNFα induced the expression of Nlrp3 mRNA up to 201% at D3 (p < 0.01) and 190% at D6 (p < 0.01) as compared to the controls (Figure 4A). Met reduced the TNFα-induced expression of Nlrp3 by 33% at D3 (p < 0.05) and 30% at D6 (p < 0.05), while CPX enhanced the effects of TNFα on Nlrp3 expression by 24% (p < 0.05) at D6 (Figure 4A).
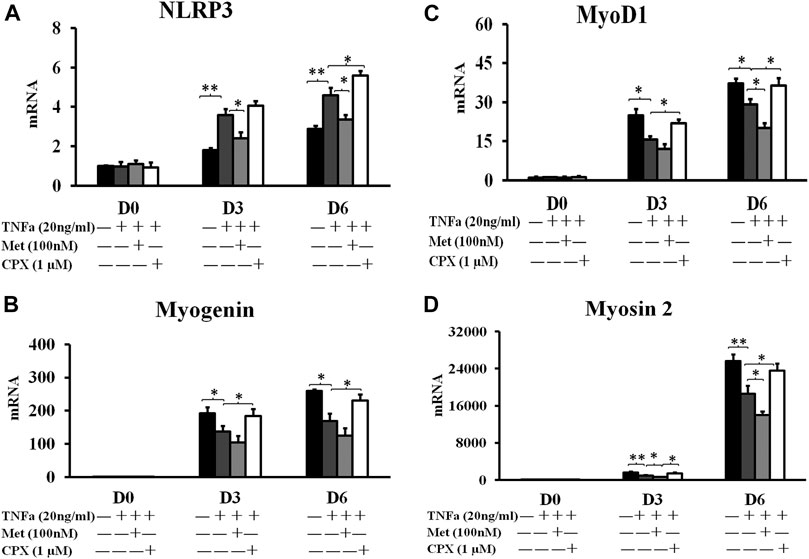
FIGURE 4. The mRNA expression of inflammasome NLRP3 protein and myogenesis markers. C2C12 cells were incubated in the presence of TNFa (20 ng/ml), TNFa + Met (100 nM) or TNFa + CPX (1 µM) for 12 h at D0, D3, or D6. The mRNA expressions were assessed by real time RT-PCR. *p < 0.05, and **p < 0.01 between two compared groups. Normalizing D0 ctrl = 1 after compared with housekeeping gene PBGD. (A): NLRP3 mRNA expression. (B) The mRNA expression of early myogenesis marker, MyoD1. (C) The mRNA expression of intermediate myogenesis marker, Myogenin. (D) The mRNA expression of late myogenesis marker, myosin 2. Ctrl, control; TNFa, murine Tumor Necrosis Factor-alpha; Met, methimepip; CPX, ciproxifan. D0, undifferentiated cells; D3, cells differentiated for 3 days; D6, cells differentiated for 6 days. NLRP3, Nucleotide-binding domain and leucine-rich repeat containing family, pyrin domain containing three.
To study the effects of TNFα and H3R modulation in myogenic differentiation, we analyzed the expression of the conventional myogenic markers. Myogenic Differentiation 1 (MyoD1) is considered as one of the earliest myogenic markers. MyoD1 is a nuclear protein that belongs to the subfamily of myogenic factors, and it regulates muscle cell differentiation by inducing cell cycle arrest, which is a prerequisite for myogenic initiation (Blais et al., 2005). Myogenin is usually regarded as an early myogenic marker. It is a muscle-specific transcription factor that is involved in coordination of skeletal muscle development or myogenesis and repair, and it is essential for the development of functional skeletal muscle (Blais et al., 2005). Myosin heavy chain 2 (Myosin-2) is a late myogenic marker. It is a member of the class II or conventional myosin heavy chains, and functions in skeletal muscle contraction (Stuart et al., 2016). TNFα reduced the expression of myogenesis markers: MyoD1 at D3 and D6 by 38% (p < 0.05) and 22% (p < 0.05) (Figure 4C), Myogenin at D3 and D6 by 29% (p < 0.05) vs. 35% (p < 0.05) (Figure 4B), and Myosin-2 at D3 and D6 by 42% (p < 0.01) and 28% (p < 0.01) (Figure 4D), respectively. Met enhanced the inhibitory effect of TNFα of the above three markers by 23 and 31% (p < 0.05), 22 and 26%, 30% (p < 0.05) and 25% (p < 0.05) at D3 and D6, respectively, while CPX reduced the inhibitory effect of TNFα on the myogenic marker expression by 40% (p < 0.05) and 25%, 35% (p < 0.05) and 37% (p < 0.05), 50% (p < 0.05) and 27% (p < 0.05) (Figures 4B–D) respectively. Neither Met nor CPX alone had a clear effect on the mRNA expression of the late myocyte differentiation marker, myosin 2 (p > 0.05) (Figure 2C).
To study the effects of TNFα and H3R modulation on the protein expression IL-1β, we detected the levels of pro-IL-1β (31 kDa) and IL-1β (17 kDa) proteins from cell lysates by western blot. TNFα increased the levels of both the pro-IL-1β (31 kDa) and IL-1β (17 kDa) proteins (p < 0.05), and neither Met nor CPX had an effect on TNFα-induced pro-IL-1β expression in D0, D3 and D6 cells (p > 0.05). However, Met and CPX modulated the TNFα-induced expression of mature IL-1β protein; Met reduced the level of the mature IL-1β at all the time points (p < 0.05) in TNFα-induced D0 and D6 cells, while CPX increased it (p < 0.05) at D0, and D6 time points. MCC950, a new specific small molecule NLRP3 inhibitor, reduced the pro-IL-1β (31 kDa) and mature IL-1β (17 kDa) protein levels in each treatment group, except for control group (p < 0.05) (Figure 5).
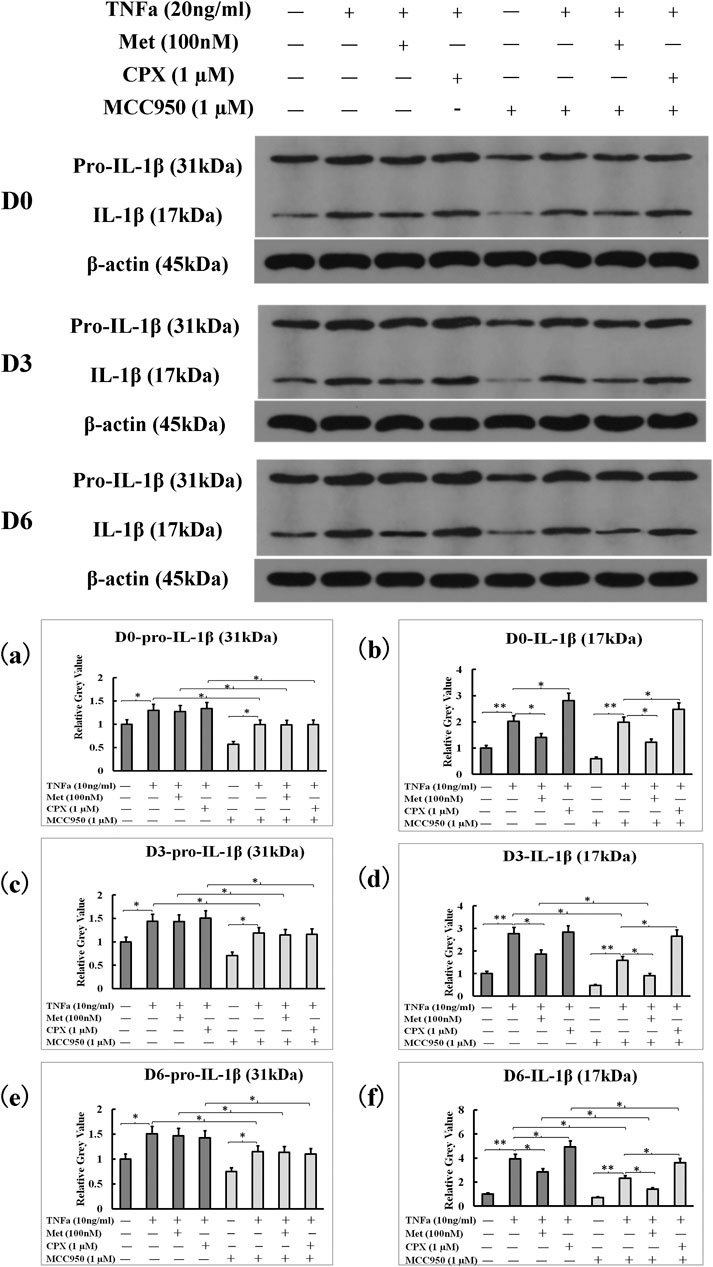
FIGURE 5. NLRP3 inhibitor, MCC950, inhibited overall pro-IL-1β and IL-1β protein levels induced by TNFα. C2C12 cells were incubated in the presence of TNFa (20 ng/ml), TNFa + Met (100 nM) or TNFa + CPX (1 µM) or overall MCC950 (1 µM) for 12 h at D0, D3, or D6. The pro-IL-1β (31 kDa) and mature IL-1β protein (17 kDa) levels were assessed by westernblot. *p < 0.05, and **p < 0.01 between two compared groups. (A,B): The grey-values of westernblot on pro-IL-1β and IL-1β proteins at D0 were calculated by normalized protein expressions to β-actin. (C,D): The grey-values of westernblot on pro-IL-1β and IL-1β proteins at D3 were calculated by normalized protein expressions to β-actin. (E,F): The grey-values of westernblot on pro-IL-1β and IL-1β proteins at D6 were calculated by normalized protein expressions to β-actin. Ctrl, control; TNFa, murine Tumor Necrosis Factor-alpha; Met, methimepip; pro-IL-1β, the precursor of interleukin 1beta; IL-1β, interleukin 1beta; CPX, ciproxifan. D0, undifferentiated cells; D3, cells differentiated for 3 days; D6, cells differentiated for 6 days.
Since the secretion of mature IL-1β is a marker of inflammasome activation, we next assessed the effect of TNFα and H3R modulation on the inflammasome activation by analyzing IL-1β secretion during C2C12 myogenesis. TNFα increased the secretion of IL-1β at D0, D3 and D6 cells to 56.8 (p < 0.01), 66.8 (p < 0.01), and 85.8 pg/ml (p < 0.01), respectively. Met reduced the TNFα-induced secretion of IL-1β to 38.4 (p < 0.05), 48.4 (p < 0.05), 65.8 (p < 0.05) pg/ml, while CPX enhanced the TNFα-induced secretion of IL-1β to 67.5 (p < 0.05), 77.5 (p < 0.05), 98.9 (p < 0.05) pg/ml on D0, D3 and D6 cells, respectively (Figure 6). MCC950 reduced TNFα-induced IL-1β secretion to 41.2 (p < 0.05), 56.8 (p < 0.05), and 66.8 pg/ml (p < 0.05); and MCC950 reduced Met-TNFα-induced IL-1β secretion to 31.9 (p < 0.05), 38.4 (p < 0.05), 48.4 (p < 0.05) pg/ml. MCC950 reduced also CPX-TNFα-induced IL-1β secretion to 49.8 (p < 0.05), 67.5 (p < 0.05), 77.5 (p < 0.05) pg/ml (Figure 6). Thus, our results suggest that H3R plays a role in regulation of NLRP3 inflammasome. H3R inhibition reduces the TNFα-induced secretion of IL-1β whereas H3R activation increases IL-1β secretion in C2C12 myocytes. The secretion of IL-1β was dependent on the NLRP3 inflammasome, as the NLRP3 inhibitor, MCC950, reduced TNFα induced IL-1β secretion in each groups of D0, D3 and D6 cells.
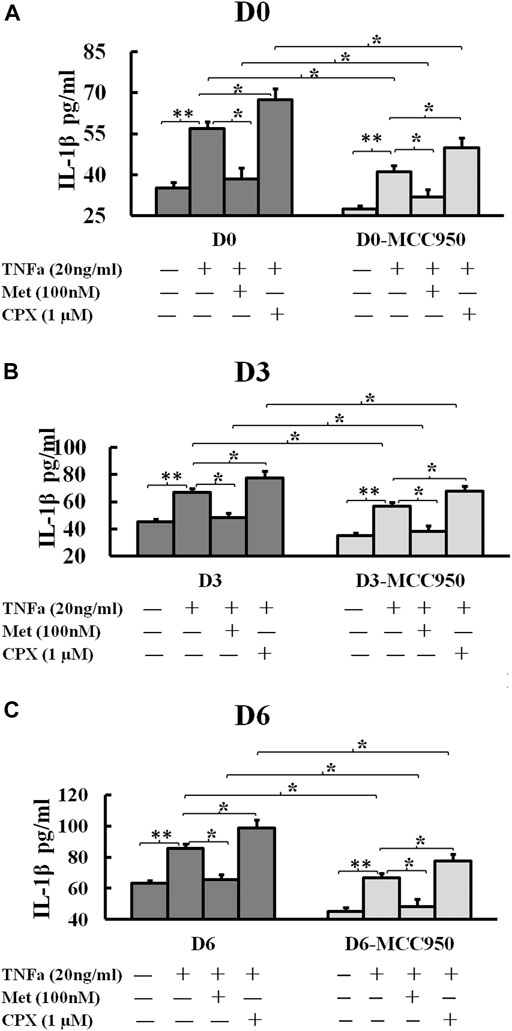
FIGURE 6. Histamine H3 receptor modulates the secretion of IL-1β. C2C12 cells were incubated in the presence of TNFa (20 ng/ml), TNFa + Met (100 nM) or TNFa + CPX (1 µM) or MCC950 (1 µM) for 24 h at D0, D3, or D6. The mature IL-1β protein secretions were assessed by ELISA. *p < 0.05, and **p < 0.01 between two compared groups. (A): IL-1β secretion in D0 cells. (B) IL-1β secretion in D3 cells. (C) IL-1β secretion in D6 cells. Ctrl, control; TNFα, murine Tumor Necrosis Factor-alpha; Met, methimepip; CPX, ciproxifan; IL-1β, interleukin 1beta.
Inflammation is an immunological response to injury and the concomitant release of damage associated molecular patterns (Hofer et al., 2014). The underlying molecular mechanisms are complex but recent studies implicate activation of inflammasomes in sterile inflammatory reactions (Mangan et al., 2018). The present study verified the presence of functional NLRP3-inflammasome in mouse striated C2C12 myocytes (Cho and Kang, 2015; Gallo et al., 2015). TNFα induced both the expressions of pro-IL-1β and also the activation of NLRP3 inflammasome in C2C12 myocytes resulting in secretion of the mature IL-1β protein. TNFα also reduced cell viability and the expression of myogenesis markers, suggesting that it inhibited myocyte differentiation. These findings are in line with previous studies which have shown that IL-1β induces markers of atrophy in C2C12 cells (Huang et al., 2017). Together these findings suggest that TNFα mediated inflammasome activation with ensuing IL-1β secretion could play an important role in postinjury muscle inflammation and inflammation associated muscle atrophy.
Stimulation of H3R signaling by Met attenuated the TNFα-induced activation of NLRP3 inflammasome, whereas, inhibition of H3R signaling enhanced the inflammasome activation by TNFα. Recently Gao et al. showed that loratadine, a histamine H1 receptor blocker, inhibits the activation of NLRP3 inflammasome (Gao and Zhang, 2020), but there are no studies addressing the role of H3R in regulation of NLRP3 inflammasome. The effect of TNFα on myogenic differentiation is affected by the conditions used and also on concentration of TNFα. Low levels of TNFα are required for the differentiation, but on the other hand, especially at higher concentration, such as during inflammation, TNFα has been shown to increase myoblast proliferation, and to prevent the cells entering to cell cycle arrest that is a prerequisite for the initiation of myoblast differentiation (Grounds and Torrisi, 2004; Chen et al., 2007; Alter et al., 2008). TNFα reduced the expression of the earliest differentiation markers on the earlier day three time point slightly more than at later sixth day time point. The differences between the time points are probably explained by the number of the cells that have entered the cell cycle arrest before TNFα treatment, because TNFα does not inhibit the differentiation after the cells have entered the cell cycle arrest (Alter et al., 2008), and by day sixmore cells had entered the cell cycle arrest and started differentiation than by day three, and probably therefore the effect of TNFα on differentiation was slightly mitigated.
Activation of H3R further slowed myogenic differentiation in the presence of TNFα. The mechanism remains to be elucidated but one possibility is that H3R activation directs cell resources into processes of reducing inflammation, thus halting the differentiation. In carcinoma cells H3R -mediated activation of protein kinase C α inhibited the growth of cholangiocarcinoma and hepatocellular carcinoma cells (Francis et al., 2009; Yu et al., 2019). The finding that blocking the H3R by CPX in the presence of TNFα enhanced cell differentiation, is also in line with studies on cancer cells reporting that inhibition of histamine receptor H3 suppresses the growth and metastasis in human non-small cell lung cancer cells (Zhao et al., 2020) thus potentially promoting differentiation. Moreover, the enhanced inflammation induced by CPX + TNFα could also direct cells into apoptosis resulting in the release of ATP, which further activates the NLRP3 inflammasome (Jo et al., 2016).
TNFα induces NLRP3 inflammasome activation and inhibits myogenesis in striated C2C12 cells during myogenic differentiation. Histamine H3R signaling slowed the differentiation and inhibited TNFα induced NLRP3 inflammasome activation. Inhibition of H3R signaling enhanced the differentiation and TNFα-induced inflammation. These findings implicate a regulatory role for histamine H3R in NLRP3 inflammasome activation. This beneficial effect of H3R activation should be balanced with the potentially disadvantageous effect of further slowing down the myocyte differentiation in the presence of TNFα. H3R modulation could represent a lucrative target in the treatment of postinjury muscle inflammation and atrophy.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
All authors substantially contributed to the drafting and revising of the article, as well as the final approval of the version to be submitted. YC got the financial supports from the Foundations, conceived and designed the experiments, performed the experiments, analyzed the data, wrote the paper, prepared figures, reviewed drafts of the paper, redid the experiments and revised the paper. YM repeated the experiments, analyzed the data, wrote the paper, prepared figures, and reviewed drafts of the paper. JF performed the experiments, analyzed the data, wrote the paper and reviewed drafts of the paper. YW performed the experiments, and reviewed drafts of the paper. TL contributed reagents, materials and analysis tools, and reviewed drafts of the paper. KN contributed reagents, materials and analysis tools, and reviewed drafts of the paper. KE helped to design the experiments, and reviewed drafts of the paper. JW provided the experimental platform and all the equipments, conceived and designed the experiments, and reviewed drafts of the paper.
This work was supported by grants from the National Natural Sciences Foundation of China (No. 81500588) and an independent research grant from the Finnish Cultural Foundation-Central Grant (No. 00130167). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Xiaoping Shang at Medical Records Department for the statistical assistance. We also would like to thank Paul L. Chazot at School of Biological and Biomedical Sciences, Durham University, United Kingdom for the excellent technical instructions and advice.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2021.599393/full#supplementary-material
Alter, J., Rozentzweig, D., and Bengal, E. (2008). Inhibition of Myoblast Differentiation by Tumor Necrosis Factor α Is Mediated by C-Jun N-Terminal Kinase 1 and Leukemia Inhibitory Factor. J. Biol. Chem. 283, 23224–23234. doi:10.1074/jbc.m801379200
Barata-Antunes, S., Cristóvão, A. C., Pires, J., Rocha, S. M., and Bernardino, L. (2017). Dual Role of Histamine on Microglia-Induced Neurodegeneration. Biochim. Biophys. Acta (Bba) - Mol. Basis Dis. 1863, 764–769. doi:10.1016/j.bbadis.2016.12.016
Blais, A., Tsikitis, M., Acosta-Alvear, D., Sharan, R., Kluger, Y., and Dynlacht, B. D. (2005). An Initial Blueprint for Myogenic Differentiation. Genes Dev. 19, 553–569. doi:10.1101/gad.1281105
Boursereau, R., Abou-Samra, M., Lecompte, S., Noel, L., and Brichard, S. M. (2018). Downregulation of the NLRP3 Inflammasome by Adiponectin Rescues Duchenne Muscular Dystrophy. BMC Biol. 16, 33. doi:10.1186/s12915-018-0501-z
Branco, A. C. C. C., Yoshikawa, F. S. Y., Pietrobon, A. J., and Sato, M. N. (2018). Role of Histamine in Modulating the Immune Response and Inflammation. Mediators Inflamm. 2018, 9524075. doi:10.1155/2018/9524075
Chen, S.-E., Jin, B., and Li, Y.-P. (2007). TNF-α Regulates Myogenesis and Muscle Regeneration by Activating P38 MAPK. Am. J. Physiology-Cell Physiol. 292, C1660–C1671. doi:10.1152/ajpcell.00486.2006
Chen, Y., Paavola, J., Stegajev, V., Stark, H., Chazot, P. L., Wen, J. G., et al. (2015). Activation of Histamine H3 Receptor Decreased Cytoplasmic Ca2+ Imaging during Electrical Stimulation in the Skeletal Myotubes. Eur. J. Pharmacol. 754, 173–178. doi:10.1016/j.ejphar.2015.02.035
Chen, Y., Stegaev, V., Kouri, V.-P., Sillat, T., Chazot, P. L., Stark, H., et al. (2015). Identification of Histamine Receptor Subtypes in Skeletal Myogenesis. Mol. Med. Rep. 11, 2624–2630. doi:10.3892/mmr.2014.3073
Cho, K.-A., and Kang, P. B. (2015). PLIN2 Inhibits Insulin-Induced Glucose Uptake in Myoblasts through the Activation of the NLRP3 Inflammasome. Int. J. Mol. Med. 36, 839–844. doi:10.3892/ijmm.2015.2276
Elliott, E. I., and Sutterwala, F. S. (2015). Initiation and Perpetuation of NLRP3 Inflammasome Activation and Assembly. Immunol. Rev. 265, 35–52. doi:10.1111/imr.12286
Francis, H., Onori, P., Gaudio, E., Franchitto, A., DeMorrow, S., Venter, J., et al. (2009). H3 Histamine Receptor-Mediated Activation of Protein Kinase Cα Inhibits the Growth of Cholangiocarcinoma In Vitro and In Vivo. Mol. Cancer Res. 7, 1704–1713. doi:10.1158/1541-7786.mcr-09-0261
Gallo, D., Gesmundo, I., Trovato, L., Pera, G., Gargantini, E., Minetto, M. A., et al. (2015). GH-releasing Hormone Promotes Survival and Prevents TNF-α-Induced Apoptosis and Atrophy in C2C12 Myotubes. Endocrinology 156, 3239–3252. doi:10.1210/en.2015-1098
Gao, F., and Zhang, S. (2020). Loratadine Alleviates Advanced Glycation End Product-Induced Activation of NLRP3 Inflammasome in Human Chondrocytes. Dddt Vol. 14, 2899–2908. doi:10.2147/dddt.s243512
Görgens, S. W., Eckardt, K., Jensen, J., Drevon, C. A., and Eckel, J. (2015). Exercise and Regulation of Adipokine and Myokine Production. Prog. Mol. Biol. Transl Sci. 135, 313–336. doi:10.1016/bs.pmbts.2015.07.002
Grounds, M. D., and Torrisi, J. (2004). Anti‐Tnfα (Remicade) Therapy Protects Dystrophic Skeletal Muscle from Necrosis. FASEB j. 18, 676–682. doi:10.1096/fj.03-1024com
Guo, H., Callaway, J. B., and Ting, J. P.-Y. (2015). Inflammasomes: Mechanism of Action, Role in Disease, and Therapeutics. Nat. Med. 21, 677–687. doi:10.1038/nm.3893
Hofer, M. D., Cheng, E. Y., Bury, M. I., Park, E., Xu, W., Hong, S. J., et al. (2014). Analysis of Primary Urethral Wound Healing in the Rat. Urology 84 (1), 246.e1–246.e7. doi:10.1016/j.urology.2014.04.012
Huang, N., Kny, M., Riediger, F., Busch, K., Schmidt, S., Luft, F. C., et al. (2017). Deletion of Nlrp3, Protects from Inflammation-Induced Skeletal Muscle Atrophy. Intensive Care Med. Exp. 5 (1), 3. doi:10.1186/s40635-016-0115-0
Hughes, F. M., Hill, H. M., Wood, C. M., Edmondson, A. T., Dumas, A., Foo, W.-C., et al. (2016). The NLRP3 Inflammasome Mediates Inflammation Produced by Bladder Outlet Obstruction. J. Urol. 195, 1598–1605. doi:10.1016/j.juro.2015.12.068
Jo, E.-K., Kim, J. K., Shin, D.-M., and Sasakawa, C. (2016). Molecular Mechanisms Regulating NLRP3 Inflammasome Activation. Cell Mol Immunol 13, 148–159. doi:10.1038/cmi.2015.95
Latz, E., Xiao, T. S., and Stutz, A. (2013). Activation and Regulation of the Inflammasomes. Nat. Rev. Immunol. 13, 397–411. doi:10.1038/nri3452
Man, S. M., and Kanneganti, T.-D. (2015). Regulation of Inflammasome Activation. Immunol. Rev. 265, 6–21. doi:10.1111/imr.12296
Mangan, M. S. J., Olhava, E. J., Roush, W. R., Seidel, H. M., Glick, G. D., and Latz, E. (2018). Targeting the NLRP3 Inflammasome in Inflammatory Diseases. Nat. Rev. Drug Discov. 17 (9), 588–606. doi:10.1038/nrd.2018.97
Mulay, S. R., Shi, C., Ma, X., and Anders, H. J. (2018). Novel Insights into Crystal-Induced Kidney Injury. Kidney Dis. 4, 49–57. doi:10.1159/000487671
Panula, P., Chazot, P. L., Cowart, M., Gutzmer, R., Leurs, R., Liu, W. L. S., et al. (2015). International Union of Basic and Clinical Pharmacology. XCVIII. Histamine Receptors. Pharmacol. Rev. 67, 601–655. doi:10.1124/pr.114.010249
Stuart, C. A., Stone, W. L., Howell, M. E., Brannon, M. F., Hall, H. K., Gibson, A. L., et al. (2016). Myosin Content of Individual Human Muscle Fibers Isolated by Laser Capture Microdissection. Am. J. Physiol. Cel Physiol 310, 381–389. doi:10.1152/ajpcell.00317.2015
Vermes, I., Haanen, C., Steffens-Nakken, H., and Reutellingsperger, C. (1995). A Novel Assay for Apoptosis Flow Cytometric Detection of Phosphatidylserine Expression on Early Apoptotic Cells Using Fluorescein Labelled Annexin V. J. Immunological Methods 184, 39–51. doi:10.1016/0022-1759(95)00072-i
Wei, S., Ma, W., Li, X., Jiang, C., Sun, T., Li, Y., et al. (2020). Involvement of ROS/NLRP3 Inflammasome Signaling Pathway in Doxorubicin-Induced Cardiotoxicity. Cardiovasc. Toxicol. 20, 507–519. doi:10.1007/s12012-020-09576-4
Yu, D., Zhao, J., Wang, Y., Hu, J., Zhao, Q., Li, J., et al. (2019). Upregulated Histamine Receptor H3 Promotes Tumor Growth and Metastasis in Hepatocellular Carcinoma. Oncol. Rep. 41, 3347–3354. doi:10.3892/or.2019.7119
Zhao, Y.-y., Jia, J., Zhang, J.-j., Xun, Y.-p., Xie, S.-j., Liang, J.-f., et al. (2020). Inhibition of Histamine Receptor H3 Suppresses the Growth and Metastasis of Human Non-small Cell Lung Cancer Cells via Inhibiting PI3K/Akt/mTOR and MEK/ERK Signaling Pathways and Blocking EMT. Acta Pharmacol. Sin, 1–10. [Epub ahead of print]. doi:10.1038/s41401-020-00548-6
Keywords: histamine H3 receptor, myogenesis, inflammation, NLRP3 inflammasome, IL-1β, TNFα, C2C12 myocyte
Citation: Chen Y, Ma Y, Feng JJ, Wang YH, Li TF, Nurmi K, Eklund KK and Wen JG (2021) Histamine H3 Receptor Signaling Regulates the NLRP3 Inflammasome Activation in C2C12 Myocyte During Myogenic Differentiation. Front. Pharmacol. 12:599393. doi: 10.3389/fphar.2021.599393
Received: 27 August 2020; Accepted: 05 May 2021;
Published: 31 May 2021.
Edited by:
Luca Antonioli, University of Pisa, ItalyReviewed by:
Simone Guarnieri, University of Studies G. d’Annunzio Chieti and Pescara, ItalyCopyright © 2021 Chen, Ma, Feng, Wang, Li, Nurmi, Eklund and Wen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan Chen, Y2hlbnlhbi5hYmJ5QDE2My5jb20=; Jian Guo Wen, d2VuamcxOTYzQDE2My5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.