
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 29 April 2021
Sec. Experimental Pharmacology and Drug Discovery
Volume 12 - 2021 | https://doi.org/10.3389/fphar.2021.593021
 Ryuichi Kambayashi1Hiroko Izumi-Nakaseko1Ai Goto1
Ryuichi Kambayashi1Hiroko Izumi-Nakaseko1Ai Goto1 Kazuya Tsurudome2Hironori Ohshiro2Taku Izumi2Mihoko Hagiwara-Nagasawa1Koki Chiba1
Kazuya Tsurudome2Hironori Ohshiro2Taku Izumi2Mihoko Hagiwara-Nagasawa1Koki Chiba1 Ryota Nishiyama3Satomi Oyama3Yoshio Nunoi1Yoshinori Takei4Akio Matsumoto5
Ryota Nishiyama3Satomi Oyama3Yoshio Nunoi1Yoshinori Takei4Akio Matsumoto5 Atsushi Sugiyama1,4,5,6*
Atsushi Sugiyama1,4,5,6*Oseltamivir has been shown to prolong the atrial conduction time and effective refractory period, and to suppress the onset of burst pacing-induced atrial fibrillation in vitro. To better predict its potential clinical benefit as an anti-atrial fibrillatory drug, we performed translational studies by assessing in vivo anti-atrial fibrillatory effect along with in vivo and in vitro electropharmacological analyses. Oseltamivir in intravenous doses of 3 (n = 6) and 30 mg/kg (n = 7) was administered in conscious state to the persistent atrial fibrillation model dogs to confirm its anti-atrial fibrillatory action. The model was prepared by tachypacing to the atria of chronic atrioventricular block dogs for > 6 weeks. Next, oseltamivir in doses of 0.3, 3 and 30 mg/kg was intravenously administered to the halothane-anesthetized intact dogs to analyze its in vivo electrophysiological actions (n = 4). Finally, its in vitro effects of 10–1,000 μM on IK,ACh, IKur, IKr, INa and ICaL were analyzed by using cell lines stably expressing Kir3.1/3.4, KV1.5, hERG, NaV1.5 or CaV1.2, respectively (n = 3 for IK,ACh and IKr or n = 6 for IKr, INa and ICaL). Oseltamivir in doses of 3 and 30 mg/kg terminated the atrial fibrillation in 1 out of 6 and in 6 out of 7 atrial fibrillation model dogs, respectively without inducing any lethal ventricular arrhythmia. Its 3 and 30 mg/kg delayed inter-atrial conduction in a frequency-dependent manner, whereas they prolonged atrial effective refractory period in a reverse frequency-dependent manner in the intact dogs. The current assay indicated that IC50 values for IK,ACh and IKr were 160 and 231 μM, respectively, but 1,000 µM inhibited INa, ICaL and IKur by 22, 19 and 13%, respectively. The extent of INa blockade was enhanced at faster beating rate and more depolarized resting membrane potential. Oseltamivir effectively terminated the persistent atrial fibrillation, which may be largely due to the prolongation of the atrial effective refractory period and inter-atrial conduction time induced by IK,ACh and IKr inhibitions along with INa suppression. Thus, oseltamivir can exert a powerful anti-atrial fibrillatory action through its ideal multi-channel blocking property; and oseltamivir would become a promising seed compound for developing efficacious and safe anti-atrial fibrillatory drugs.
Since existing anti-atrial fibrillation (AF) drugs have been known to block both atrial and ventricular ionic channels, adverse effects were largely induced by their ionic channel blockade in the ventricle, which may often blunt their therapeutic benefits for the atrial arrhythmias (Geng et al., 2020). The ideal anti-AF drug should selectively improve the pathophysiology in the atria without affecting the electrophysiological profile of the ventricles.
Oseltamivir is a pro-drug of oseltamivir carboxylate, which has been used for treating patients with influenza virus infection (McClellan and Perry, 2001). We have demonstrated that 30 mg/kg, i.v. of oseltamivir delayed the atrial and ventricular conductions as well as ventricular repolarization in halothane-anesthetized dogs (Kitahara et al., 2013), that 3 and 30 mg/kg, i.v. of oseltamivir did not induce torsade de pointes in chronic atrioventricular block dogs (Nakamura et al., 2016), and that 100 μM of oseltamivir prolonged the atrial conduction time as well as the effective refractory period in the isolated guinea pig hearts (Takahara et al., 2013). Moreover, in a previous study using the Langendorff-perfused rabbit hearts, 100 μM of oseltamivir suppressed the onset of AF induced by burst pacing with ACh and isoproterenol pretreatment (Frommeyer et al., 2017). However, those known basic findings (Kitahara et al., 2013; Takahara et al., 2013; Nakamura et al., 2016; Frommeyer et al., 2017) would not be enough to make the clinical development of oseltamivir as an anti-AF drug valid.
In order to facilitate its clinical application, we performed the following translational studies. Initially, we assessed antiarrhythmic effects of oseltamivir on persistent AF model dogs, which was prepared by tachypacing to the atria of chronic atrioventricular block dogs for > 6 weeks, inducing structural and electrical remodeling that enables to maintain AF (Wijffels et al., 1995; Gaspo et al., 1997). Next, we examined electrophysiological effects of oseltamivir on the atrium of halothane-anesthetized intact dogs, which have been demonstrated to be able to mimic the drug-induced electropharmacological responses in human subjects (Sugiyama, 2008). These in vivo results were compared with those of clinically available class Ic drug pilsicainide, since pilsicainide as well as flecainide are recommended for treatment of AF in JCS/JHRS 2020 Guideline on Pharmacotherapy of Cardiac Arrhythmias in Japan (Plosker, 2010; Ono et al., 2020). Finally, using patch clamp assay, we studied the effects of oseltamivir on each of IK,ACh, IKur, IKr, INa and ICaL to clarify which combination of the inhibitory action may play a pivotal role for suppressing AF.
Experiments were performed by using 29 beagle dogs of either sex, aging 1–3 years and weighing approximately 10 kg, which were obtained through Kitayama Labes Co., Ltd. (Nagano, Japan). All experiments were reviewed and approved by Animal Care and User Committee of Yamanashi Research Center of Clinical Pharmacology (No.2005-16) and Toho University (No.17-52-324), and performed according to the Guideline for the Care and Use of Laboratory Animals of University of Yamanashi and Toho University, and ARRIVE guidelines (Kilkenny et al., 2010; McGrath et al., 2010). Randomization and allocation concealment of the animals were performed for each of the in vivo experiments. Data were analyzed under the open-label for all experiments, which were confirmed once more by another evaluator to secure their reliability. Animals showing the heart rate of > 200 bpm or < 50 bpm, mean blood pressure of > 150 mmHg or < 50 mmHg, and/or electrocardiogram of other than normal sinus rhythm after the initial induction of anesthesia were excluded from the studies.
After the animals (n = 21) were anesthetized with thiopental sodium (30 mg/kg, i.v.), catheter electrodes (Cordis-Webster Inc. CA, United States) was positioned at the compact atrioventricular node. The radiofrequency energy of 20 W was delivered for 10 s from the tip electrode to develop the complete atrioventricular block. More than 1 month later, the dogs were initially anesthetized pentobarbital sodium (30 mg/kg, i.v.) (Sugiyama, 2008). After intubation, anesthesia was maintained by isoflurane inhalation (1.5% v/v) vaporized in oxygen with a volume-limited ventilator. Tidal volume and respiratory rate were set at 20 ml/kg and 15 breaths/min, respectively. Initially, the skin of the gluteal region was incised to make a subcutaneous pocket for implanting the pacemaker. Next, the left fourth intercostal thoracotomy was performed and the pericardium was incised to expose the atrium. Two sets of the electrode of temporary pacing lead (Streamline atrial 6492; Medtronic Inc. MN, United States) were sutured onto the Bachmann bundle while lifting the left atrial appendage. The pericardial incision was fixed, and the thorax was closed. The pacing leads were connected to the rapid pulse-generating pacemaker (TNT-002AL; Taisho Biomed Instruments Co., Ltd. Osaka, Japan) placed at the gluteal region through the subcutaneous tunnel. The incision at the gluteal region was sutured carefully.
More than 4 weeks after implanting a pacemaker, the atria were electrically driven at 400 bpm for 2 weeks. Then, the pacing rate was increased to 600 bpm to promote the atrial electrical remodeling. When the atrial pacing could not achieve 1:1 atrial capture at 600 bpm, the atrial pacing was maintained at 400–500 bpm until the atria could be electrically paced at 600 bpm. After confirming the onset of AF, the pacing was maintained at 600 bpm for 2 weeks. Then, the atrial pacing was stopped under the monitoring of Holter electrocardiogram (QR2100; Fukuda M-E Kogyo Co., Ltd. Tokyo, Japan). The duration of AF was assessed using the Holter analysis system (HS1000; Fukuda M-E Kogyo Co., Ltd.). When the AF persisted > 24 h, the dog was used as the persistent AF model animal to assess the anti-AF effect of drugs; whereas if it was terminated before 24 h, the pacing was restarted at 600 bpm for additional 1 week until AF persisted for > 24 h. In our preliminary study, once AF persisted for > 24 h, it did for > 1 week.
The atria were electrically driven at 600 bpm for another one week. Under the monitoring of Holter electrocardiogram, the atrial pacing was stopped without anesthesia. Approximately 2 h after the termination of the atrial pacing, 3 (n = 6) and 30 mg/kg (n = 7) of oseltamivir as well as 3 mg/kg (n = 8) of pilsicainide hydrochloride hydrate were intravenously administered over 10 min.
The dogs were initially anesthetized with thiopental sodium (30 mg/kg, i.v.). After intubation, anesthesia was maintained by halothane inhalation (1% v/v) vaporized in oxygen with a volume-limited ventilator. Tidal volume and respiratory rate were set at 20 ml/kg and 15 breaths/min, respectively. The surface lead II electrocardiogram was continuously monitored, whereas the arterial blood pressure was measured through an indwelling needle placed at the right femoral artery.
Three sets of standard 6-French quad-polar electrodes catheter (Cordis-Webster Inc.) were used. The first one was positioned at the high right atrium through the right femoral vein to electrically pace the sinus nodal area and to simultaneously obtain the right atrial electrogram. The second one was positioned in the esophagus via the oral cavity to record the left atrial electrogram. The third one was positioned at the endocardium of the right ventricle through the left femoral vein to electrically pace the right ventricle.
The lead II electrocardiogram was obtained from the limb electrodes. The PR interval, QRS width, QT interval and P-wave duration were measured, and the QT interval was corrected with Van de Water’s formula: QTc = QT−0.087 × (RR−1,000) with RR given in ms, which was developed for anesthetized dogs (Van de Water et al., 1989). In addition, the J-Tpeak and Tpeak-Tend were separately measured. The J-Tpeak was corrected for heart rate with a coefficient as previously described (J-Tpeakc = J-Tpeak/RR0.58 with RR given in seconds) (Johannesen et al., 2014). Correction was not made on the Tpeak-Tend, since previous QT studies have shown that the Tpeak-Tend exhibited minimal heart rate dependency at resting heart rate (Johannesen et al., 2014). The heart was electrically driven with the cardiac stimulator (SEC-3102; Nihon Kohden Corporation, Tokyo, Japan) through pacing electrodes of the catheters placed at the sinus nodal area or at the right ventricle. The stimulation pulses were set rectangular in shape, consisting of 1–2 V amplitude (about twice the threshold voltage) and 1 ms duration. The inter-atrial conduction time (IACT) was defined as an interval between the right and left atrial electrograms, which was measured during spontaneous sinus rhythm (IACT(sinus)) and at pacing cycle lengths of 400 ms (IACT(CL400)), 300 ms (IACT(CL300)) and 200 ms (IACT(CL200)). The atrial (AERP) and ventricular effective refractory period (VERP) were assessed with programmed electrical stimulation on the sinus nodal area and the right ventricle, respectively. The pacing protocol consisted of five beats of basal stimuli in cycle lengths of 400 ms (AERP(CL400)), 300 ms (AERP(CL300)) and 200 ms (AERP(CL200)) for AERP, and 400 ms (VERP(CL400)) for VERP followed by an extra stimulus of various coupling intervals. The coupling interval was shortened in 5 ms decrements until the additional stimulus could no longer elicit a response. AERP and VERP were defined as the shortest coupling interval that can evoke stimulus response.
The right and left atrial electrograms, electrocardiogram and arterial blood pressure were monitored with a polygraph system (RM-6000; Nihon Kohden Corporation) and analyzed with real-time, fully automatic data analysis system (WinVAS3 ver. 1.1R24; Physio-Tech Co., Ltd. Tokyo, Japan). Each measurement of arterial pressure, electrocardiographic variables and IACT adopted mean of three recordings of consecutive complexes.
The electropharmacological variables were assessed in the following order. First, the right and left atrial electrograms, electrocardiogram and arterial blood pressure were recorded under the spontaneous sinus rhythm. Second, the sinus nodal area was paced at cycle lengths of 400, 300 and 200 ms to measure IACT. Third, AERP was assessed at basic pacing cycle lengths of 400, 300 and 200 ms. Fourth, VERP was measured at a basic pacing cycle length of 400 ms.
After the basal assessment, 0.3 mg/kg of oseltamivir was intravenously infused over 10 min, and each variable was assessed at 5, 10, 15, 20 and 30 min after the start of its administration (n = 4). Then, 3 mg/kg of oseltamivir was intravenously infused over 10 min, and each variable was assessed in the same manner. Finally, 30 mg/kg of oseltamivir was intravenously infused over 10 min, and each variable was assessed at 5, 10, 15, 20, 30, 45 and 60 min after the start of its administration. On the other hand, as another series of experiments, cardiovascular effects of 1 and 3 mg/kg of pilsicainide hydrochloride hydrate were assessed in the same manner (n = 4) as those for the middle and high doses of oseltamivir, respectively. We previously confirmed that the cardiohemodynamic and electrophysiological parameters in the in vivo experiments were stable > 2 h in the absence of pharmacological intervention (Sugiyama and Hashimoto, 1998; Usui et al., 1998; Satoh et al., 1999; Iwasaki et al., 2009).
Human embryonic kidney cell lines (HEK293 cells) stably co-expressing recombinant hKir3.1, hKir3.4 and hM2 (TOA EIYO LTD. Tokyo, Japan) were cultured in α-MEM supplemented with 10% fetal bovine serum (FBS), 12.5 μg/ml of G418 and 25 μg/ml of Zeocin. Chinese hamster ovary K1 (CHO-K1) cells stably expressing recombinant hKV1.5 (TOA EIYO LTD.) were cultured in Ham’s F12 medium supplemented with 10% FBS and 200 μg/ml of G418. CHO-K1 cells stably expressing recombinant hERG (B’SYS GmbH, Witterswill, Switzerland) were cultured in Ham’s F12 medium supplemented with 10% FBS, 100 μg/ml of G418, 100 μg/ml of hygromycin B and 2 mM of L-glutamine. CHO cells stably expressing recombinant hNaV1.5 (B’SYS) were cultured in Ham’s F12 medium supplemented with 10% FBS, 250 μg/ml of G418, 1 μg/ml of puromycin and 2 mM of L-glutamine. HEK293 cells stably co-expressing hCaV1.2 α, β2 and α2δ1 subunits (B’SYS) were cultured in D-MEM/Ham’s F12 medium supplemented with 10% FBS, 100 unit/mL of penicillin/streptomycin, 100 μg/ml of hygromycin B, 15 μg/ml of blasticidine S and 1 μg/ml of puromycin.
All ionic currents were recorded with automated whole-cell, patch-clamp technique. The inhibitory effects of 30–1000 μM of oseltamivir phosphate on Kir3.1/3.4 and KV1.5‐derived currents were assessed using port-a-patch automated patch-clamp technology (Nanion Technologies, Munich, Germany) at TOA EIYO LTD., whereas those of 10–1,000 μM on hERG, NaV1.5 and CaV1.2-derived currents were evaluated using Qube 384 automated patch-clamp system (Sophion Bioscience A/S, Ballerup, Denmark) at Sophion Bioscience K.K. Each current was recorded at room temperature (20–25°C) except for Kir3.1/3.4-derived current which was done at 30°C. The recording data was analyzed by PatchMaster (ver. 2 × 69, HEKA Electronik GmbH, Lambrecht, Germany) for Kir3.1/3.4 and KV1.5-derived currents, or by Sophion Analyzer (ver. 6.5, Sophion Bioscience A/S) for hERG, NaV1.5 and CaV1.2-derived ones. The cells for recording Kir3.1/3.4 or KV1.5-derived currents were exposed to oseltamivir phosphate solution in the order of rising concentration using a perfusion system. The cells for recording hERG, NaV1.5 or CaV1.2-derived currents were exposed to the three test solutions; namely, vehicle, oseltamivir phosphate at either concentration of 10, 30, 100, 300 or 1,000 μM, and selective ionic channel blockers or sodium-free bath solution with an interval of the replacement time of > 14 min. The IC50 values were calculated by fitting the data to Hill equation using GraphPad prism 8 (ver. 8.31; GraphPad Software, Inc. CA, United States).
The protocol for recording Kir3.1/3.4-derived K+ current consisted of a 500 ms prepulse at –120 mV, followed by a 1,000 ms ramp pulse from –120 mV to + 60 mV with an interval of 10 s at a holding potential of −40 mV. The current amplitude was measured at + 60 mV for analyzing the concentration-response relationship (n = 3). The bath solution was composed of (in mM) 100 NaCl, 40 KCl, 1 MgCl2, 1.8 CaCl2, 10 D (+)-glucose and 10 HEPES at pH 7.4 with NaOH. The internal recording solution was composed of (in mM) 50 KCl, 10 NaCl, 60 KF, 2 MgCl2, 20 EGTA and 10 HEPES at pH 7.2 titrated with KOH. The KV1.5-derived K+ current was evoked by a 500 ms step pulse at 0 mV with an interval of 20 s at a holding potential of –70 mV. The peak amplitude was measured for analyzing the concentration-response relationship (n = 3). The external recording solution was composed of (in mM) 140 NaCl, 4 KCl, 1 MgCl2, 2 CaCl2, 5 D (+)-glucose and 10 HEPES at pH 7.4 titrated with NaOH, whereas the internal recording solution was the same as that used for recording Kir3.1/3.4 current. Tail current of hERG-derived K+ current was elicited by a 2,000 ms step pulse at –50 mV after a 1,000 ms prepulse at +40 mV with an interval of 30 s at a holding potential of −70 mV (n = 6). The peak amplitude of the tail currents was normalized with the following equation (Ipeak–Ib)/(If–Ib); Ipeak is the peak amplitude in the presence of oseltamivir; Ib is that with vehicle (1% PBS containing bath solution, basal); and If is that in the presence of 100 μM quinidine (fully blocked). The bath solution was composed of (in mM) 145 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 10 D (+)-glucose and 10 HEPES at pH 7.4 titrated with NaOH. The internal recording solution was composed of (in mM) 120 KF, 20 KCl, 10 EGTA and 10 HEPES at pH 7.2 titrated with KOH. The NaV1.5‐derived Na+ current was evoked by a 20 ms step pulse at −10 mV every 10 s at a holding potential of −90 mV (n = 6). The peak amplitude in the presence of oseltamivir was measured and normalized using that with vehicle and that obtained in a sodium-free bath solution. To assess the frequency-dependent block of NaV1.5 by oseltamivir, a train of 30 step pulses at −10 mV for 20 ms at 1 Hz, 5 Hz or 10 Hz was applied, and the peak amplitude at the 30th test pulse (P30) was normalized by that at the first pulse (P1). The steady-state inactivation curves of NaV1.5 channels before and after application of vehicle and oseltamivir phosphate were obtained by the following protocol; a 500 ms prepulse ranging from –120 mV to –10 mV in 10 mV increments followed by a 20 ms test pulse at –10 mV with an interval of 20 s at a holding potential of –120 mV. The peak amplitude was normalized with the maximum peak amplitude for each cell in the same condition and was fitted with Boltzmann equation to calculate the half-maximum voltage of inactivation (V1/2, inact). The bath solution was composed of (in mM) 145 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 10 D (+)-glucose and 10 HEPES at pH 7.4 titrated with NaOH. The sodium-free bath solution was composed of (in mM) 145 NMDG-Cl, 4 KCl, 2 CaCl2, 1 MgCl2, 10 D (+)-glucose and 10 HEPES at pH 7.4 titrated with NaOH. The internal recording solution was composed of (in mM) 10 NaCl, 140 CsF, 1 EGTA and 10 HEPES at pH 7.3 titrated with CsOH. The CaV1.2-derived inward Ca2+ current was evoked by a 200 ms step pulse at 0 mV every 30 s at a holding potential of –80 mV (n = 6). The peak amplitude in the presence of oseltamivir was normalized using that with vehicle and that in the presence of 30 μM nifedipine. The bath solution was composed of (in mM) 145 NaCl, 4 KCl, 10 CaCl2 and 10 HEPES at pH 7.4 with NaOH. The internal recording solution was composed of (in mM) 112 CsCl, 2 NaCl, 28 CsF, 8.2 EGTA, 4 MgATP and 10 HEPES at pH 7.2 titrated with CsOH.
Oseltamivir phosphate (Tamiflu®, 75 mg capsule as oseltamivir, Chugai Pharmaceutical Co., Ltd. Tokyo, Japan) of six capsules were decapsulated, mixed with 15 ml of saline and sonicated for 30 min. The mixture was centrifuged at 1,200 × g for 30 min and its supernatant was treated with syringe filters of 0.45 μm pore size (EB-DISK 25, Kanto Chemical Co., Ltd. Tokyo, Japan). The solution containing 30 mg/ml of oseltamivir (MW: 312.41) was diluted to 3 and 0.3 mg/ml with saline for the in vivo experiment. Pilsicainide hydrochloride hydrate (MW: 317.85) (Sunrythm®, Daiichi-Sankyo Co., Ltd. Tokyo, Japan) was dissolved with saline in a concentration of 3 mg/ml, which was diluted to 1 mg/ml for the experiment. The dose of pilsicainide was calculated as its hydrochloride hydrate form. Oseltamivir phosphate (AK Scientific Inc. CA, United States) was dissolved in distilled water or PBS and diluted with each bath solution for patch clamp assay. The other drugs used were thiopental sodium (Ravonal®, Mitsubishi-Tanabe Pharma Co., Osaka, Japan), pentobarbital sodium (Tokyo Chemical Industry Co., Ltd. Tokyo, Japan), isoflurane (Pfizer Japan Inc. Tokyo, Japan) and halothane (Fluothane®, Takeda Pharmaceutical Co., Ltd. Osaka, Japan).
Data are presented as mean ± S.E.M. Differences within a parameter were evaluated with one-way, repeated-measures analysis of variance (ANOVA) followed by Contrasts as a post hoc-test for mean values comparison, whereas those between the groups were assessed by unpaired t-test. A p value < 0.05 was considered to be significant, and those among the groups were done by one-way, factorial ANOVA followed by Fisher’s LSD test.
There was no animal excluded from cardioversion experiments (Exp. 1) or electrophysiological studies (Exp. 2) after its randomization. No episode of lethal ventricular arrhythmias or hemodynamic collapse was observed during cardioversion experiments (Exp. 1) or electrophysiological studies (Exp. 2).
Typical tracings of Holter electrocardiogram before and during the intravenous infusion of oseltamivir are depicted in Figure 1A, whereas time courses of anti-AF effects of oseltamivir and pilsicainide are summarized in Figure 1B. Three mg/kg of oseltamivir terminated the AF in 1 out of 6 animals at 30 min after the start of administration. Its 30 mg/kg did it in 6 out of 7 animals at 8 (#7), 12 (#8), 7 (#9), 6 (#10), 9 (#11) and 19 min (#12) after the start of administration. The AF recurred in 2 animals at 8.6 (#10) and 16 h (#11) after the termination of AF. Meanwhile, 3 mg/kg of pilsicainide hydrochloride hydrate terminated the AF in 2 out of 8 animals at 15 (#17) and 11 min (#18) after the start of administration.
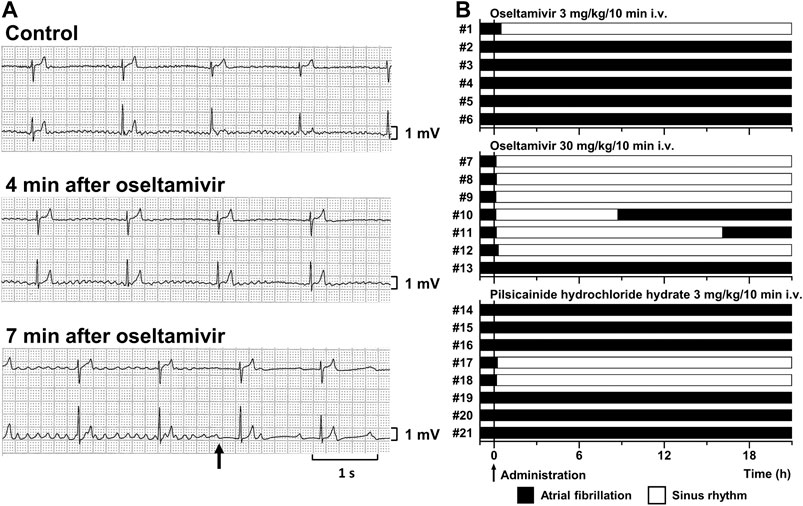
FIGURE 1. Antiarrhythmic effects of oseltamivir and pilsicainide hydrochloride hydrate on the conscious dogs with persistent AF. (A) Typical tracings of Holter electrocardiogram (NASA lead: upper; and CM5 lead: lower) before (Control; top), 4 min (4 min after oseltamivir; middle) and 7 min (7 min after oseltamivir; bottom) after the start of intravenous administration of 30 mg/kg/10 min of oseltamivir (animal number #9). The AF was terminated at 7 min (arrow). (B) Time courses of the antiarrhythmic effects of oseltamivir in doses of 3 (top, n = 6) and 30 mg/kg/10 min i.v. (middle, n = 7) and pilsicainide hydrochloride hydrate in a dose of 3 mg/kg/10 min i.v. (bottom, n = 8).
Time courses of changes in the variables during sinus rhythm after the administration of oseltamivir and pilsicainide are summarized in Figures 2, 3. The pre-drug basal control values (C) of the heart rate, mean blood pressure, PR interval, QRS width, QT interval, QTc, J-Tpeakc, Tpeak-Tend, P-duration and IACT(sinus) were 93 ± 1 beats/min, 103 ± 5 mmHg, 112 ± 5 ms, 61 ± 3 ms, 274 ± 7 ms, 305 ± 7, 192 ± 9, 63 ± 5 ms, 62 ± 3 ms and 38 ± 2 ms in the oseltamivir-treated animals (n = 4), whereas those were 93 ± 6 beats/min, 101 ± 3 mmHg, 126 ± 3 ms, 64 ± 2 ms, 304 ± 6 ms, 334 ± 5, 192 ± 10, 91 ± 6 ms, 70 ± 5 ms and 34 ± 4 ms in the pilsicainide-treated animals (n = 4), respectively. There was no significant difference in these values between the groups except for PR interval, QT interval, QTc or Tpeak-Tend.
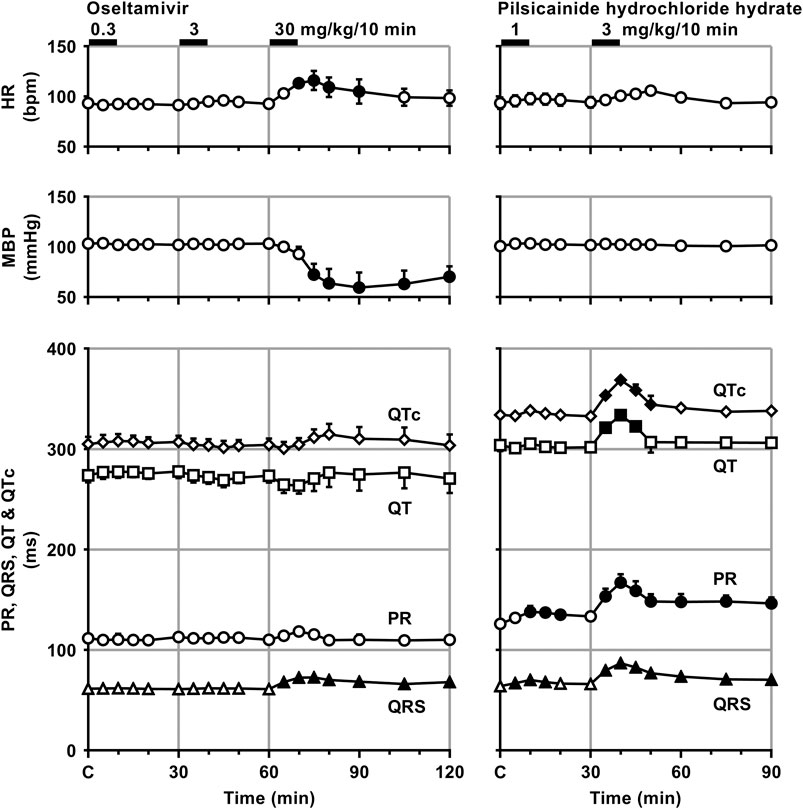
FIGURE 2. Time courses of changes in the heart rate (HR), mean blood pressure (MBP), PR interval (PR), QRS width (QRS), QT interval (QT) and QTc after the administrations of oseltamivir (left) and pilsicainide hydrochloride hydrate (right). QT interval was corrected with Van de Water’s formula: QTc = QT−0.087 × (RR−1,000) with RR given in ms (Van de Water et al., 1989). Data are presented as mean ± S.E.M. (n = 4 for each drug). Closed symbols represent significant differences from their corresponding pre-drug basal control value (C) by p < 0.05.
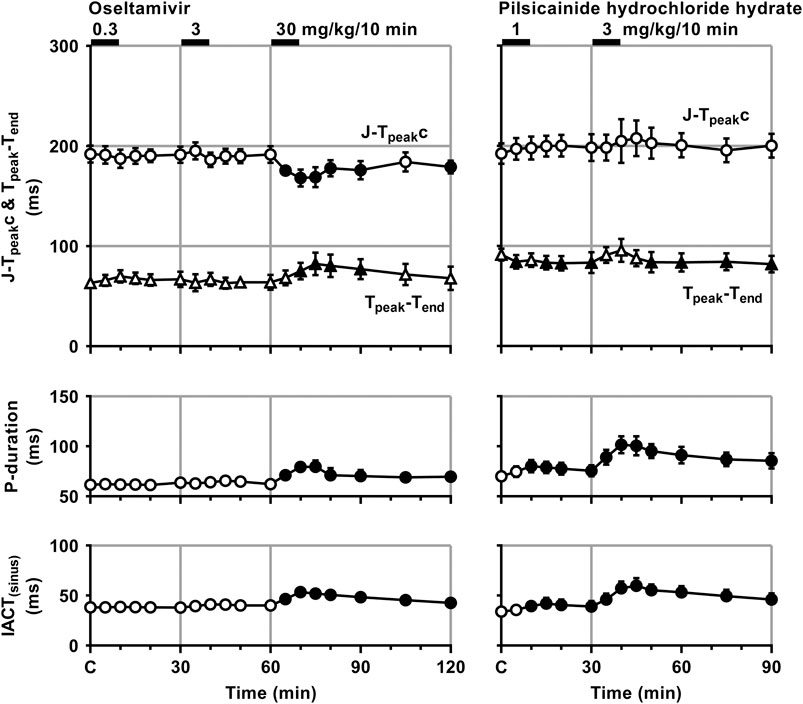
FIGURE 3. Time courses of changes in the J-Tpeakc, Tpeak-Tend, P-wave duration (P-duration) and inter-atrial conduction time (IACT) during sinus rhythm (IACT(sinus)) after the administrations of oseltamivir (left) and pilsicainide hydrochloride hydrate (right). J-Tpeak was corrected by the following formula: J-Tpeakc = J-Tpeak/RR0.58 with RR given in seconds (Johannesen et al., 2014). Data are presented as mean ± S.E.M. (n = 4 for each drug). Closed symbols represent significant differences from their corresponding pre-drug basal control value (C) by p < 0.05.
The low and middle doses of oseltamivir hardly altered any of the variables during sinus rhythm. The high dose increased the heart rate for 10–30 min, but decreased the mean blood pressure for 15–60 min. It also prolonged the QRS width, P-duration and IACT(sinus) for 5–60 min and Tpeak-Tend for 10–30 min, but shortened the J-Tpeakc for 5–30 min and at 60 min, whereas no significant change was detected in the PR interval, QT interval or QTc. Meanwhile, the low dose of pilsicainide prolonged the PR interval for 10–20 min, QRS width for 5–15 min and P-duration and IACT(sinus) for 10–30 min, but shortened the Tpeak-Tend at 5 min and for 15–30 min, whereas no significant change was detected in the other variables. The high dose prolonged the PR interval, QRS width, P-duration and IACT(sinus) for 5–60 min, QT interval for 5–15 min and QTc for 5–20 min, but shortened the Tpeak-Tend for 20–60 min, whereas no significant change was detected in the other variables.
Time courses of changes in the inter-atrial conduction time and atrial as well as ventricular effective refractory period after the administration of oseltamivir and pilsicainide are summarized in Figure 4. Typical tracings of the right as well as left atrial electrogram, electrocardiogram and aortic pressure during the atrial programmed electrical stimulation for the assessment of AERP(CL400) are depicted in Figure 5. The pre-drug basal control values (C) of the IACT(CL400), IACT(CL300), IACT(CL200), AERP(CL400), AERP(CL300), AERP(CL200) and VERP(CL400) were 60 ± 6, 58 ± 6, 61 ± 6, 138 ± 10, 138 ± 6, 129 ± 7 and 210 ± 5 ms in the oseltamivir-treated animals (n = 4), whereas those were 56 ± 5, 54 ± 4, 56 ± 4, 138 ± 5, 133 ± 3, 121 ± 6 and 201 ± 5 ms in the pilsicainide-treated animals (n = 4), respectively. There was no significant difference in these values between the groups.
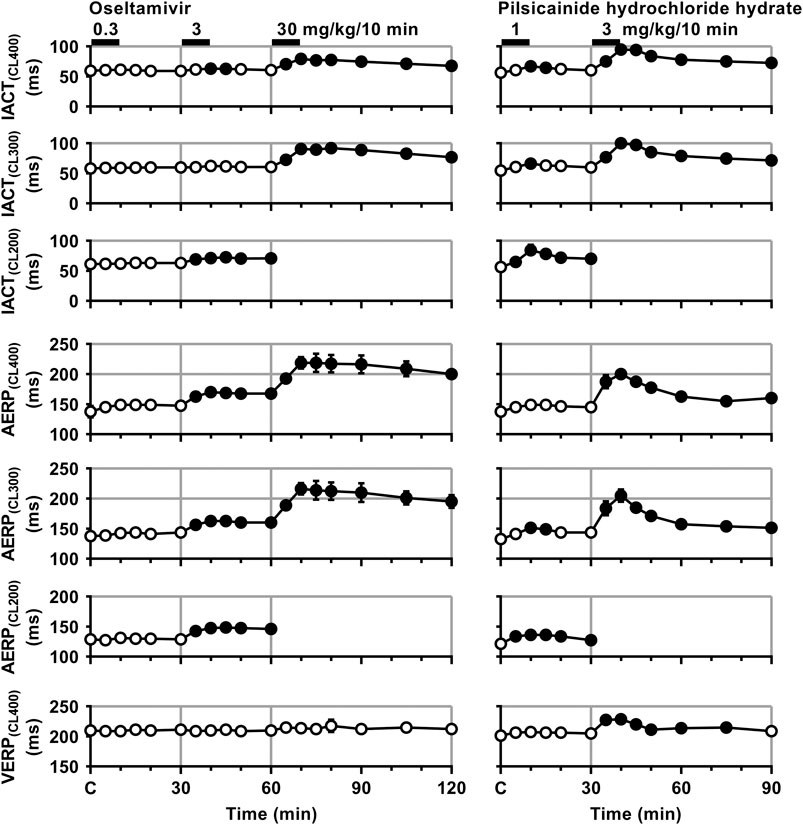
FIGURE 4. Time courses of changes in the inter-atrial conduction time (IACT) at pacing cycle lengths of 400 ms (IACT(CL400)), 300 ms (IACT(CL300)) and 200 ms (IACT(CL200)); atrial effective refractory period (AERP) at basic pacing cycle lengths of 400 ms (AERP(CL400)), 300 ms (AERP(CL300)) and 200 ms (AERP(CL200)); and ventricular effective refractory period (VERP) at a basic pacing cycle length of 400 ms (VERP(CL400)) after the administrations of oseltamivir (left) and pilsicainide hydrochloride hydrate (right). Data are presented as mean ± S.E.M. (n = 4 for each drug). Closed symbols represent significant differences from their corresponding pre-drug basal control value (C) by p < 0.05.
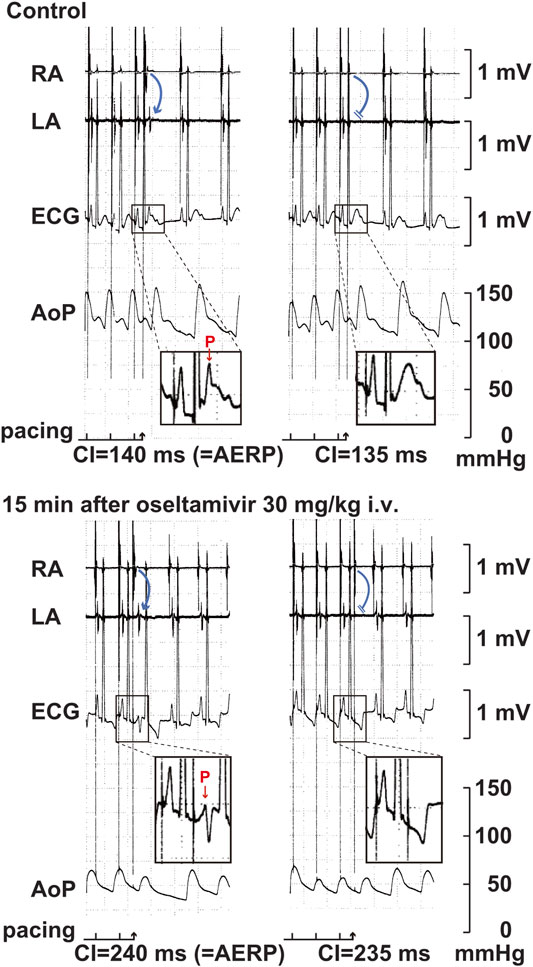
FIGURE 5. Typical tracings of the right (RA) as well as left (LA) atrial electrograms, electrocardiogram (ECG) and aortic pressure (AoP) during the atrial programed electrical stimulation for the assessment of atrial effective refractory period (AERP) at a basic pacing cycle length of 400 ms at pre-drug control (Control; upper panels) and 15 min after the start of intravenous administration of 30 mg/kg of oseltamivir (15 min after oseltamivir 30 mg/kg i.v.; lower panels). A premature right atrial stimulation with a coupling interval (CI) of 140 ms induced electrical activity in LA electrogram (blue arrow) and p wave in ECG (red arrow) (upper left), whereas that of 135 ms failed to develop either of them (upper right), indicating that the AERP was 140 ms. After the administration oseltamivir, a premature right atrial stimulation with a CI of 240 ms induced the electrical activity and p wave (lower left), whereas that of 235 ms did not develop either of them (lower right), indicating that the AERP was 240 ms. Administration of 30 mg/kg of oseltamivir decreased the aortic pressure. Moreover, the programmed atrial stimulation-induced atrial tachycardia followed by atrial premature contraction could have enhanced the hypotensive action.
The low dose of oseltamivir hardly altered any of the variables. The middle dose prolonged the IACT(CL400) for 10–15 min and IACT(CL200) for 5–30 min; and AERP(CL400), AERP(CL300) and AERP(CL200) for 5–30 min; whereas no significant change was detected in the IACT(CL300) or VERP(CL400). The high dose prolonged the IACT(CL400) and IACT(CL300) for 5–60 min; and AERP(CL400) and AERP(CL300) for 5–60 min; whereas no significant change was detected in VERP(CL400). Meanwhile, the low dose of pilsicainide prolonged the IACT(CL400) for 10–15 min, IACT(CL300) at 10 min, IACT(CL200) for 5–30 min; AERP(CL300) for 10–15 min and AERP(CL200) for 5–30 min, whereas no significant change was detected in the AERP(CL400) or VERP(CL400). The high dose prolonged the IACT(CL400) and IACT(CL300) for 5–60 min; AERP(CL400) and AERP(CL300) for 5–60 min; and VERP(CL400) for 5–45 min. Since the atrium could not be paced at a cycle length of 200 ms after the administration of the high dose of both drugs due to the prolongation of AERP to > 200 ms, we could not obtain the IACT(CL200) or AERP(CL200) for 5–60 min.
The representative current waveforms showing the inhibitory actions of oseltamivir on Kir3.1/3.4 (IK,ACh), KV1.5 (IKur), hERG (IKr), NaV1.5 (INa) and CaV1.2-derived currents (ICaL) along with voltage pulse protocols are shown in Figure 6, whereas its concentration-response curves showing its inhibitory action (%) on these currents are summarized in Figure 7. Oseltamivir suppressed each of the currents in a concentration-related manner. The IC50 values for Kir3.1/3.4 and hERG-derived currents were 160 and 231 μM, respectively. Meanwhile, oseltamivir in a concentration of 1,000 µM inhibited the KV1.5, NaV1.5 and CaV1.2‐derived currents by 13 ± 8, 22 ± 9 and 19 ± 6%, respectively. Frequency- and voltage-dependent properties of INa block by oseltamivir were further analyzed, which are summarized in Figure 8 and Table 1. Oseltamivir enhanced its INa block in a frequency-dependent manner, and achieved significant differences at 1, 5 and 10 Hz when the concentrations were 300 (Δnormalized current: −0.052, −0.103 and −0.127) and 1,000 µM (Δnormalized current: −0.206, −0.340 and −0.394, respectively) (Figure 8A). Meanwhile, oseltamivir negatively shifted the steady-state inactivation curve of NaV1.5 channels in a concentration-related manner (Figure 8B), and achieved significant differences in V1/2, inact when the concentrations were 300 and 1,000 μM (Table 1). Oseltamivir tended to suppress the INa at prepulse voltage of −90, −80 and −70 mV when the concentrations were 100 (Δnormalized current: −0.021, −0.045 and −0.093), 300 (Δnormalized current: −0.075, −0.128 and −0.147) and 1,000 (Δnormalized current: −0.109, −0.184 and −0.227, respectively).
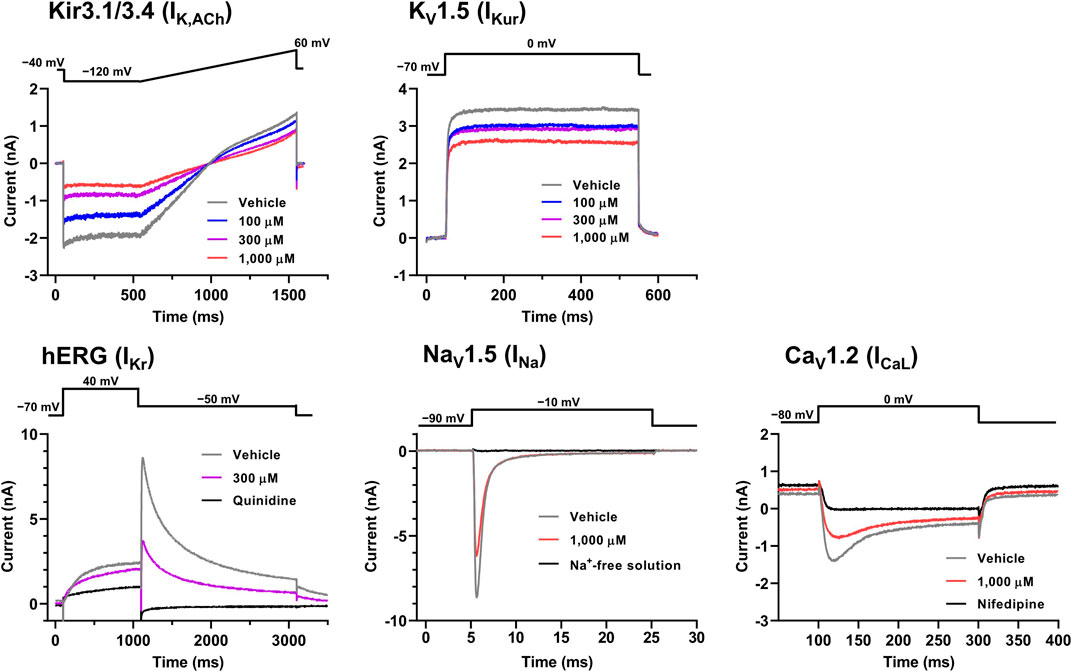
FIGURE 6. Voltage protocols (upper) and representative current waveforms (lower) showing the inhibitory actions of oseltamivir phosphate on Kir3.1/3.4, KV1.5, hERG, NaV1.5 and CaV1.2-derived currents. The current waveforms of Kir3.1/3.4 and KV1.5-derived currents were recorded with vehicle and 100–1,000 μM of oseltamivir, whereas those of hERG, NaV1.5, CaV1.2-derived currents were done with vehicle, 300 or 1,000 μM oseltamivir, and relevant ion channel blockers (100 μM quinidine or 30 μM nifedipine) or Na+-free solution.
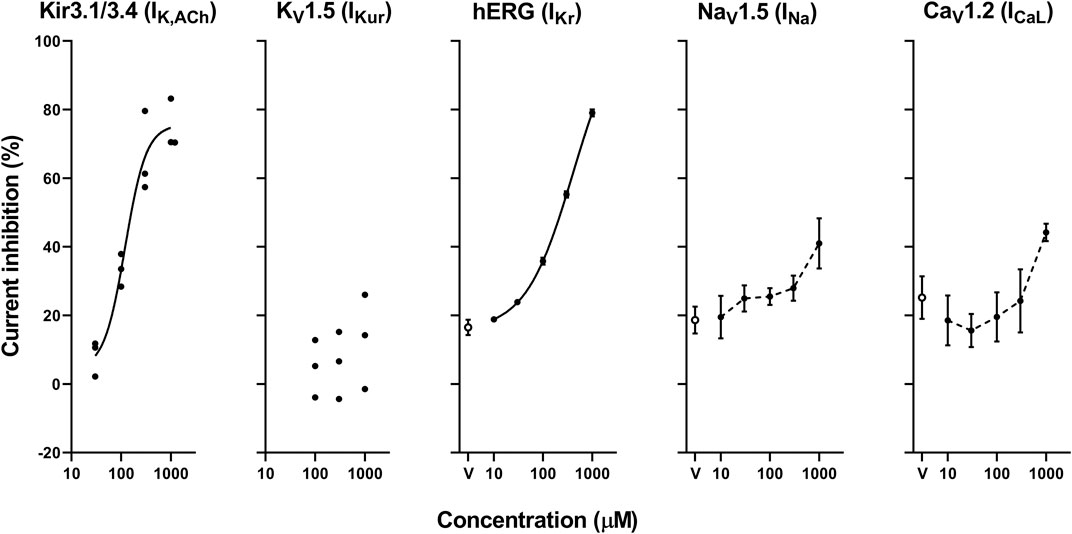
FIGURE 7. Concentration-response curves showing the inhibitory action (%) of oseltamivir phosphate or vehicle (V) on Kir3.1/3.4, KV1.5, hERG, NaV1.5 and CaV1.2-derived currents. Data are presented as individual data (n = 3 for Kir3.1/3.4 and KV1.5-derived currents) or mean ± S.E.M. (n = 6 for hERG, NaV1.5 and CaV1.2-derived currents). The concentration-response curves of Kir3.1/3.4 and hERG-derived currents were fitted with Hill equation with solid line and their IC50 values were 160 and 231 μM, respectively, whereas those of NaV1.5 and CaV1.2-derived currents were made by interpolating those values of current inhibition at each concentration with dashed line.
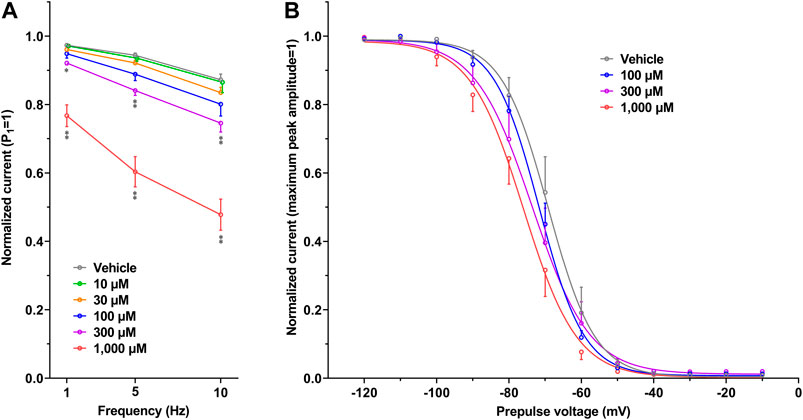
FIGURE 8. Frequency- and Voltage-dependent block of NaV1.5 by oseltamivir. (A) The normalized peak amplitudes of NaV1.5-derived current at 30th test pulse in the presence of vehicle (n = 5) and oseltamivir (n = 6 for each concentration) were compared at the pulse frequencies of 1, 5 and 10 Hz. The peak amplitude was normalized by that at 1st step pulse (P1 = 1). The reduction of NaV1.5 current by oseltamivir was augmented in a frequency-dependent manner. *p < 0.05 and **p < 0.01 vs. vehicle at the same frequency. (B) The steady-state inactivation curves of NaV1.5 channels in the presence of vehicle (n = 6) and oseltamivir (n = 6 for each concentration). The peak amplitude was normalized by the maximum peak amplitude (maximum peak amplitude = 1). Oseltamivir negatively shifted the curves in a concentration-related manner. Data are presented as mean ± S.E.M.

TABLE 1. Effect of oseltamivir on the steady-state inactivation curve of NaV1.5.
In our previous study using the halothane-anesthetized dogs, 3 and 30 mg/kg, i.v. of oseltamivir prolonged the P-wave duration in a dose-related manner, reflecting its inhibitory action on INa (Kitahara et al., 2013), which is known to play an important role in the anti-AF effect (Harvey and Grant, 2018). Thus, we selected those doses for cardioversion experiments (Exp. 1). In order to better characterize effects of oseltamivir on the atrial electrophysiological variables in electrophysiological studies (Exp. 2), we adopted the same protocol as that used in the standard electropharmacological study (Kitahara et al., 2013). Since 0.3, 3 and 30 mg/kg, i.v. over 10 min of oseltamivir provided peak plasma concentrations of 1.2 (3.8), 10.6 (33.9) and 117.5 (376.1) μg/ml (μM), respectively in our previous study and its protein binding ratio was < 50% (Kitahara et al., 2013), their free plasma concentrations could be estimated to be > 2, > 17 and > 188 μM, respectively. Accordingly, we chose the concentrations of 10–1,000 μM of oseltamivir for patch-clamp experiments (Exp. 3). On the other hand, pilsicainide hydrochloride hydrate has been clinically used in an intravenous dose of 1 mg/kg over 10 min, providing peak plasma concentration of 1.74 μg/ml in healthy human subjects according to the interview form from manufacturer. Thus, we assessed 3 mg/kg, i.v. over 10 min of pilsicainide hydrochloride hydrate in cardioversion experiments (Exp. 1) to better detect its anti-AF action, and its 1 and 3 mg/kg, i.v. over 10 min in electrophysiological studies (Exp. 2).
In order to better characterize the canine model of persistent AF, in this study we calibrated it using anti-AF drug pilsicainide (Plosker, 2010). Three mg/kg of pilsicainide hydrochloride hydrate terminated AF in 2 out of 8 animals, resulting in sinus rhythm (25%). Meanwhile, 3 and 30 mg/kg of oseltamivir did it in 1 out of 6 (17%) and 6 out of 7 animals (86%), respectively, indicating its potent anti-AF efficacy. It should be noted that AF recurred in 2 animals after the high dose of oseltamivir, reflecting a causal relationship between the plasma concentration of oseltamivir and its anti-AF action. Since we previously reported that the time course of the plasma concentration of oseltamivir followed a pattern that was predicted by the two-compartment theory of pharmacokinetics in the halothane-anesthetized dog (Kitahara et al., 2013), the recurrence of AF in 2 animals may be associated with the decrease of the plasma concentration of oseltamivir. In addition, lethal ventricular arrhythmia or hemodynamic collapse was not induced during the experimental period, indicating favorable safety pharmacological profile of oseltamivir, which may confirm previously described cardiovascular safety profile (Nakamura et al., 2016). Thus, oseltamivir may become a promising seed compound to develop efficacious and safe anti-AF drugs.
The mechanisms of anti-AF effects of oseltamivir were analyzed using the anesthetized dogs with the intact hearts. In our previous study, 30 mg/kg of oseltamivir decreased the mean blood pressure by 7 mmHg possibly through INa and/or ICaL inhibitions along with the heart rate reduction (−9 bpm) (Kitahara et al., 2013). Meanwhile in this study, the same dose of oseltamivir decreased the mean blood pressure by 44 mmHg, but increased the heart rate by 23 bpm, which may indicate occurrence of the reflex-mediated increase of sympathetic tone. The larger depressor action in the currently used animals than the previous ones might be partly associated with differences in their breeding companies (TOYO Beagle vs. Iar:Beagle) and/or ages (2 years vs. 12 months). This difference needs further analysis; however, such depressor action can be expected to be smaller in conscious state.
In the previous study (Kitahara et al., 2013), the same doses of oseltamivir as used in this study delayed the atrioventricular conduction and repolarization in a dose-related manner possibly through direct ICaL and IKr inhibition, respectively (Takahara et al., 2013). In this study, however, they tended to prolong these variables, which did not achieve a statistical significance, since the increase of sympathetic tone could have counteracted the actions of oseltamivir by indirectly enhancing ICaL and IKs (Harvey and Grant, 2018). The high dose of oseltamivir delayed the intra-atrial and intra-ventricular conductions by 18 and 11 ms, whereas that of pilsicainide did the variables by 32 and 23 ms, respectively, showing the higher atrial selectivity of oseltamivir than that of pilsicainide. Importantly, their increments were 16 and 8 ms for oseltamivir, respectively in the previous study (Kitahara et al., 2013), indicating that sympathetic tone would have hardly modified these effects.
There is a discrepancy between the significant IKr-blocking effect of oseltamivir in vitro as discussed below and no change in ventricular refractory period or QTc interval in vivo. In order to better understand these findings, we analyzed the early (J-Tpeakc) and late (Tpeak-Tend) ventricular repolarization periods of electrocardiogram; the former could reflect the net balance between inward ICaL and outward IKs plus IKr, and the latter would indicate the extent of IKr modification along with transmural dispersion of the ventricular repolarization period. As shown in Figure 3, oseltamivir significantly prolonged the Tpeak-Tend, indicating its IKr blocking action in vivo, which was in accordance with that demonstrated in patch-clamp experiments (Exp. 3). More importantly, oseltamivir significantly shortened the J-Tpeakc possibly through direct ICaL suppression and/or indirect IKs enhancement induced by the hypotension-induced, reflex-mediated, increase of sympathetic tone, which may have counteracted the IKr blockade-dependent J-Tpeakc and QT/QTc prolongation.
In order to better understand how oseltamivir terminated the AF, we assessed its effects on the IACT and AERP; surrogate markers for anti-AF action (Kambayashi et al., 2020), which was compared with those of pilsicainide as summarized in Table 2. The middle and high doses of oseltamivir increased the IACT in a frequency-dependent manner but prolonged the AERP in a reverse frequency-dependent manner, suggesting that the former may be related to INa inhibition and that the latter would be more associated with IKr inhibition than INa suppression. Meanwhile, pilsicainide prolonged the IACT and AERP in a frequency-dependent manner except for AERP(CL200), indicating that INa inhibition may play an important role for both of them. It should be noted that the high dose of oseltamivir prolonged the AERP greater than that of pilsicainide, whereas its reverse was true for the IACT as shown in Table 2. The anti-AF action of the high dose of oseltamivir may more depend on the prolongation of AERP than IACT. Taking the result of cardioversion experiments (Exp. 1) into account, the AERP may be more sensitive marker than the IACT for predicting antiarrhythmic effects against the persistent AF. In addition, the high dose of oseltamivir prolonged the AERP(CL400) and VERP(CL400) by 81 and 4 ms (ΔAERP/ΔVERP = 20.3), whereas that of pilsicainide did the variables by 63 and 28 ms (ΔAERP/ΔVERP = 2.3), respectively, indicating that oseltamivir may have much higher atrial selectivity than pilsicainide, which was shown in Table 2.
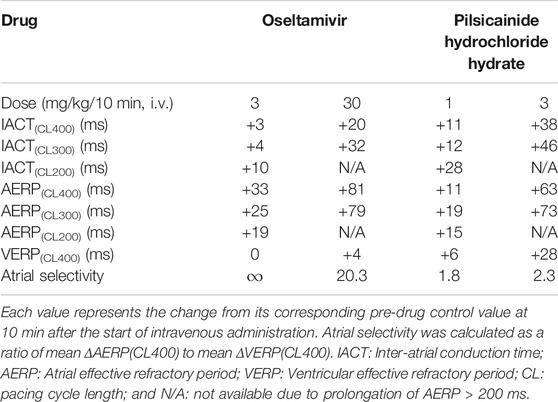
TABLE 2. Electrophysiological effects on the inter-atrial conduction and effective refractory period.
The patch clamp assay indicated that oseltamivir has multi-channel blocking action and suppressed the currents in the order of IK,ACh > IKr >> INa > ICaL > IKur, of which IC50 values of IK,ACh and IKr were 160 and 231 μM, respectively. The estimated free peak plasma concentration of oseltamivir would be > 188 μM after its high dose administration in cardioversion experiments (Exp. 1) and electrophysiological studies (Exp. 2), indicating that oseltamivir may have potently inhibited IK,ACh and moderately suppressed IKr in those experiments. Although IK,ACh as well as IKur has been shown to be predominantly expressed in the atrium (Gaborit et al., 2007), IK,ACh and IKur inhibitors have been clinically shown to have a limited efficacy against AF (Heijman et al., 2017; Camm et al., 2019; Peyronnet and Ravens, 2019), implying the limitation of selective atrial ionic channel inhibitors. Importantly, the IK,ACh suppression would depolarize the resting potential of atrial myocytes, which may result in a delay of Na+ channel recovery and an increase of inactivated Na+ channels (Yamada et al., 1998; Harvey and Grant, 2018). Similar synergic effect of INa and IKr inhibition has been shown in dogs (Aguilar et al., 2015). As shown in patch-clamp experiments (Exp. 3), oseltamivir blocked INa in a frequency-dependent manner, whereas it negatively shifted the steady-state inactivation curve of NaV1.5 in a concentration-related manner. These results can partly explain why oseltamivir inhibited the conduction of the atria in vivo at atrial beating rate of 100–150 bpm in electrophysiological studies (Exp. 2), in which resting membrane potential could be around −80 mV (Pandit, 2018). Moreover, constitutively active IK,ACh in the atrium under chronic AF has been reported to increase, which may contribute to the shortening of the AERP that will promote the onset of reentry leading to persistence of AF (Dobrev et al., 2005; Heijman et al., 2017). Thus, IK,ACh inhibitory action of oseltamivir may play a pivotal role in terminating the persistent AF, which was confirmed in cardioversion experiments (Exp. 1). In addition, IK,ACh inhibition in addition to IKr suppression by oseltamivir may have prolonged AERP, also contributing to its anti-AF.
The estimated peak plasma concentrations of oseltamivir in cardioversion experiments (Exp. 1) and electrophysiological studies (Exp. 2) would be 100–1,000 and 10–1,000 times higher than that attained after its clinically recommended oral dose against influenza virus infection which was 115 ng/ml in healthy human subjects (Kitahara et al., 2013). Although oseltamivir could be well tolerated in the dogs having persistent AF beside the chronic atrioventricular conduction block, a frequent use of this drug for AF in clinical may increase the possibility to induce oseltamivir–resistant influenza virus. Thus, daily use of oseltamivir for patient having persistent AF should not be recommended for protecting the public health. Oseltamivir is metabolized extensively and predominantly by hepatic carboxylesterase to its sole active metabolite showing anti-influenza virus action (McClellan and Perry, 2001) which lacks electrophysiological activity on the heart (Fukushima et al., 2015). Developing a novel formulation that can avoid the hepatic first-pass effect and/or modulating its chemical structure that will be resistant to carboxylesterase would be able to reduce the effective dose against AF, and may help facilitate the development of ideal anti-AF drugs.
Oseltamivir can exert a powerful anti-AF action through its ideal multi-channel blocking effects on IK,ACh, IKr and INa. Although developing a novel formulation that can reduce the effective dose against AF may be mandatory, oseltamivir would be a promising seed compound for developing efficacious and safe anti-AF drugs.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
The animal study was reviewed and approved by the Animal Care and User Committee of Yamanashi Research Center of Clinical Pharmacology and the Animal Care and User Committee of Toho University.
RK and AS supervised the project and designed the experiments. RK, AG, KT, HO, TI, MH-N, KC, RN, and SO performed and analyzed the experiments. HI-N, YN, YT, and AM provided technical support and discussions. RK, HI-N, and AS wrote the manuscript. All the authors reviewed the manuscript.
This study was supported in part by research grants from Japan Society for the Promotion of Science (JSPS KAKENHI grant number 19K16505 to MH-N and 20K16136 to RK).
KT, HO, and TI were employed by Sophion Bioscience K.K. RN and SO were employed by TOA EIYO LTD.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank Yuji Nakamura (deceased on March 26, 2018) and Yuri Ichikawa for their technical assistances during preparation of the manuscript and Yuji Tsurubuchi and Yasuki Akie for their great contribution to the current study.
Aguilar, M., Xiong, F., Qi, X. Y., Comtois, P., and Nattel, S. (2015). Potassium channel blockade enhances atrial fibrillation-selective antiarrhythmic effects of optimized state-dependent sodium channel blockade. Circulation 132, 2203–2211. doi:10.1161/CIRCULATIONAHA.115.018016
Camm, A. J., Dorian, P., Hohnloser, S. H., Kowey, P. R., Tyl, B., Ni, Y., et al. (2019). A randomized, double-blind, placebo-controlled trial assessing the efficacy of S66913 in patients with paroxysmal atrial fibrillation. Eur. Heart J. Cardiovasc. Pharmacother. 5, 21–28. doi:10.1093/ehjcvp/pvy022
Dobrev, D., Friedrich, A., Voigt, N., Jost, N., Wettwer, E., Christ, T., et al. (2005). The G protein-gated potassium current I(K,ACh) is constitutively active in patients with chronic atrial fibrillation. Circulation 112, 3697–3706. doi:10.1161/CIRCULATIONAHA.105.575332
Frommeyer, G., Mittelstedt, A., Wolfes, J., Ellermann, C., Kochhäuser, S., Leitz, P., et al. (2017). The anti-influenza drug oseltamivir reduces atrial fibrillation in an experimental whole-heart model. Naunyn Schmiedebergs Arch. Pharmacol. 390, 1155–1161. doi:10.1007/s00210-017-1415-y
Fukushima, A., Chazono, K., Hashimoto, Y., Iwajima, Y., Yamamoto, S., Maeda, Y., et al. (2015). Oseltamivir produces hypothermic and neuromuscular effects by inhibition of nicotinic acetylcholine receptor functions: comparison to procaine and bupropion. Eur. J. Pharmacol. 762, 275–282. doi:10.1016/j.ejphar.2015.06.004
Gaborit, N., Le Bouter, S., Szuts, V., Varro, A., Escande, D., Nattel, S., et al. (2007). Regional and tissue specific transcript signatures of ion channel genes in the non-diseased human heart. J. Physiol. 582, 675–693. doi:10.1113/jphysiol.2006.126714
Gaspo, R., Bosch, R. F., Talajic, M., and Nattel, S. (1997). Functional mechanisms underlying tachycardia-induced sustained atrial fibrillation in a chronic dog model. Circulation 96, 4027–4035. doi:10.1161/01.cir.96.11.4027
Geng, M., Lin, A., and Nguyen, T. P. (2020). Revisiting antiarrhythmic drug therapy for atrial fibrillation: reviewing lessons learned and redefining therapeutic paradigms. Front. Pharmacol. 11, 581837. doi:10.3389/fphar.2020.581837
Harvey, R. D., and Grant, A. O. (2018). “Agents used in cardiac arrhythmias,” in The basic & clinical Pharmacology. 14th Edn, Editor B. G. Katzung (New York, NY: McGraw Hill Education), 228–253.
Heijman, J., Ghezelbash, S., and Dobrev, D. (2017). Investigational antiarrhythmic agents: promising drugs in early clinical development. Expert Opin. Investig. Drugs 26, 897–907. doi:10.1080/13543784.2017.1353601
Iwasaki, H., Takahara, A., Nakamura, Y., Satoh, Y., Nagai, T., Shinkai, N., et al. (2009). Simultaneous assessment of pharmacokinetics of pilsicainide transdermal patch and its electropharmacological effects on atria of chronic atrioventricular block dogs. J. Pharmacol. Sci. 110, 410–414. doi:10.1254/jphs.09061sc
Johannesen, L., Vicente, J., Mason, J. W., Sanabria, C., Waite-Labott, K., Hong, M., et al. (2014). Differentiating drug-induced multichannel block on the electrocardiogram: randomized study of dofetilide, quinidine, ranolazine, and verapamil. Clin. Pharmacol. Ther. 96, 549–558. doi:10.1038/clpt.2014.155
Kambayashi, R., Hagiwara-Nagasawa, M., Ichikawa, T., Goto, A., Chiba, K., Nunoi, Y., et al. (2020). Analysis of electropharmacological effects of AVE0118 on the atria of chronic atrioventricular block dogs: characterization of anti-atrial fibrillatory action by atrial repolarization-delaying agent. Heart Vessels 35, 1316–1322. doi:10.1007/s00380-020-01612-1
Kilkenny, C., Browne, W., Cuthill, I. C., Emerson, M., and Altman, D. G. (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br. J. Pharmacol. 160, 1577–1579. doi:10.1111/j.1476-5381.2010.00872.x
Kitahara, K., Nakamura, Y., Tsuneoka, Y., Adachi-Akahane, S., Tanaka, H., Yamazaki, H., et al. (2013). Cardiohemodynamic and electrophysiological effects of anti-influenza drug oseltamivir in vivo and in vitro. Cardiovasc. Toxicol. 13, 234–243. doi:10.1007/s12012-013-9202-6
McClellan, K., and Perry, C. M. (2001). Oseltamivir: a review of its use in influenza. Drugs 61, 263–283. doi:10.2165/00003495-200161020-00011
McGrath, J., Drummond, G., McLachlan, E., Kilkenny, C., and Wainwright, C. (2010). Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br. J. Pharmacol. 160, 1573–1576. doi:10.1111/j.1476-5381.2010.00873.x
Nakamura, Y., Sasaki, R., Cao, X., Wada, T., Hamaguchi, S., Izumi-Nakaseko, H., et al. (2016). Intravenous anti-influenza drug oseltamivir will not induce torsade de pointes: evidences from proarrhythmia model and action-potential assay. J. Pharmacol. Sci. 131, 72–75. doi:10.1016/j.jphs.2016.04.018
Ono, K., Iwasaki, Y., Shimizu, W., Akao, M., Ikeda, T., Ishii, K., et al. (2020). JCS/JHRS 2020 Guideline on pharmacotherapy of cardiac arrhythmias. The japanese circulation society/japanese heart rhythm society Joint Guidelines, Available at https://www.j-circ.or.jp/old/guideline/pdf/JCS2020_Ono.pdf (Accessed January 14, 2021).
Pandit, S. V. (2018). in “Ionic mechanisms of atrial action potentials” in The cardiac electrophysiology from cell to bedside. 7th Edn, Editors D. P. Zipes, J. Jalife, and W. G. Stevenson (Philadelphia, PA: Elsevier Inc.), 293–303.
Peyronnet, R., and Ravens, U. (2019). Atria-selective antiarrhythmic drugs in need of alliance partners. Pharmacol. Res. 145, 104262. doi:10.1016/j.phrs.2019.104262
Satoh, Y., Sugiyama, A., Tamura, K., and Hashimoto, K. (1999). Effects of a class III antiarrhythmic drug, dofetilide, on the in situ canine heart assessed by the simultaneous monitoring of hemodynamic and electrophysiological parameters. Jpn. J. Pharmacol. 81, 79–85. doi:10.1254/jjp.81.79
Sugiyama, A., and Hashimoto, K. (1998). Effects of gastrointestinal prokinetic agents, TKS159 and cisapride, on the in situ canine heart assessed by cardiohemodynamic and electrophysiological monitoring. Toxicol. Appl. Pharmacol. 152, 261–269. doi:10.1006/taap.1998.8494
Sugiyama, A. (2008). Sensitive and reliable proarrhythmia in vivo animal models for predicting drug-induced torsades de pointes in patients with remodelled hearts. Br. J. Pharmacol. 154, 1528–1537. doi:10.1038/bjp.2008.240
Takahara, A., Suzuki, S., Hagiwara, M., Nozaki, S., and Sugiyama, A. (2013). Electrophysiological effects of an anti-influenza drug oseltamivir on the Guinea-pig atrium: comparison with those of pilsicainide. Biol. Pharm. Bull. 36, 1650–1652. doi:10.1248/bpb.b13-00347
Usui, T., Sugiyama, A., Ishida, Y., Satoh, Y., Sasaki, Y., and Hashimoto, K. (1998). Simultaneous assessment of the hemodynamic, cardiomechanical, and electrophysiological effects of terfenadine on the in vivo canine model. Heart Vessels 13, 49–57. doi:10.1007/BF01744586
Van de Water, A., Verheyen, J., Xhonneux, R., and Reneman, R. S. (1989). An improved method to correct the QT interval of the electrocardiogram for changes in heart rate. J. Pharmacol. Methods 22, 207–217. doi:10.1016/0160-5402(89)90015-6
Wijffels, M. C., Kirchhof, C. J., Dorland, R., and Allessie, M. A. (1995). Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation 92, 1954–1968. doi:10.1161/01.cir.92.7.1954
Keywords: oseltamivir, pilsicainide, persistent atrial fibrillation, patch clamp assay, IK,ACh, dogs, ECG, safety pharmacology
Citation: Kambayashi R, Izumi-Nakaseko H, Goto A, Tsurudome K, Ohshiro H, Izumi T, Hagiwara-Nagasawa M, Chiba K, Nishiyama R, Oyama S, Nunoi Y, Takei Y, Matsumoto A and Sugiyama A (2021) Translational Studies on Anti-Atrial Fibrillatory Action of Oseltamivir by its in vivo and in vitro Electropharmacological Analyses. Front. Pharmacol. 12:593021. doi: 10.3389/fphar.2021.593021
Received: 09 August 2020; Accepted: 04 February 2021;
Published: 29 April 2021.
Edited by:
Apostolos Zarros, University of Glasgow, United KingdomReviewed by:
Ilya Kolb, Janelia Research Campus, United StatesCopyright © 2021 Kambayashi, Izumi-Nakaseko, Goto, Tsurudome, Ohshiro, Izumi, Hagiwara-Nagasawa, Chiba, Nishiyama, Oyama, Nunoi, Takei, Matsumoto and Sugiyama. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Atsushi Sugiyama, YXRzdXNoaS5zdWdpeWFtYUBtZWQudG9oby11LmFjLmpw
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.