 Congrong Niu
Congrong Niu Bill Smith
Bill Smith Yurong Lai
Yurong Lai
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 21 January 2021
Sec. Drug Metabolism and Transport
Volume 11 - 2020 | https://doi.org/10.3389/fphar.2020.620197
The induction potentials of ligand-activated nuclear receptors on metabolizing enzyme genes are routinely tested for new chemical entities. However, regulations of drug transporter genes by the nuclear receptor ligands are underappreciated, especially in differentiated human hepatocyte cultures. In this study, gene induction by the ligands of constitutive androstane receptor (CAR) and aryl hydrocarbon receptor (AhR) was characterized in sandwich-cultured human hepatocytes (SCHH) from multiple donors. The cells were treated with 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), omeprazole (OP), 6-(4-chlorophenyl)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde O-(3,4-dichlorobenzyl)oxime (CITCO) and phenobarbital (PB) for three days. RNA samples were analyzed by qRT-PCR method. As expected, CITCO, the direct activator, and PB, the indirect activator of CAR, induced CYP3A4 (31 and 40-fold), CYP2B6 (24 and 28-fold) and UGT1A1 (2.9 and 4.2-fold), respectively. Conversely, TCDD and OP, the activators of AhR, induced CYP1A1 (38 and 37-fold), and UGT1A1 (4.3 and 5.0-fold), respectively. In addition, OP but not TCDD induced CY3A4 by about 61-fold. Twenty-four hepatic drug transporter genes were characterized, and of those, SLC51B was induced the most by PB and OP by about 3.3 and 6.5 fold, respectively. Marginal inductions (about 2-fold) of SLC47A1 and SLCO4C1 genes by PB, and ABCG2 gene by TCDD were observed. In contrast, SLC10A1 gene was suppressed about 2-fold by TCDD and CITCO. While clinical relevance of SLC51B gene induction or SLC10A1 gene suppression warrants further investigation, the results verified that the assessment of transporter gene inductions are not required for new drug entities, when a drug does not remarkably induce metabolizing enzyme genes by CAR and AhR activation.
There are about 450 membrane transporters divided into two superfamilies, the solute carrier (SLC) and the ATP Binding Cassette (ABC) transporters. Membrane transporters transport ions, essential nutrients, hormones etc. across a biological membrane. Among these membrane transporters, nearly thirty of them are involved in drug transport and currently are designated as clinically relevant drug transporters (International Transporter et al., 2010; Hillgren et al., 2013). The liver is the most important organ in the body for drug metabolism, accounting for about 70% of drugs and their metabolite elimination in humans (Patel et al., 2016). Many transporters are expressed on the sinusoidal and canalicular membrane of hepatocytes to transport poorly permeable endogenous substances into and out of the liver. About 20 of these membrane proteins are also involved in hepatic uptake and biliary excretion of a wide variety of prescription drugs and their metabolites (Vildhede et al., 2015), and play a key role in determining drug pharmacokinetics (PK) and hepatic exposure. Therefore, fluctuations of expressions and functional activities of drug transporters can result in changes of plasma level and/or liver exposure of substrate drugs. For example, active hepatic uptake mediated by sinusoidal transporters e.g., organic anion transporting polypeptides (OATP) can be the rate-determining step of drug elimination for the substrates that are either metabolically stable or unstable (Hagenbuch and Meier, 2004). The functional inhibition or induction of transporter gene expression can cause drug-drug interactions (DDIs) with co-administration drugs that are substrates of the transporters (International Transporter et al., 2010; Hillgren et al., 2013). Given the critical roles played by hepatic transporters in systemic clearance and liver exposure, it is important to understand the factors affecting transporter expression. Therefore, characterizing transporter gene regulation is a crucial step to improve the understanding of transporter-mediated drug absorption, disposition and elimination, and subsequently their association to drug efficacy and toxicity.
It is well-documented that metabolizing enzymes can be induced by activation of various cytoplasmic and orphan nuclear receptors, such as the pregnane X receptor (PXR) and constitutive androstane receptor (CAR). For example, phase I cytochrome P450 3A (CYP3A), CYP2C and phase II uridine diphosphate glucuronosyltransferase 1A1 (UGT1A1) are induced by PXR ligands (Niu et al., 2019). Similarly, CAR ligands induce CYP2B, 3A, 2C and UGT enzymes (Yang and Wang, 2014). Unfortunately, details in transporter gene regulations by these nuclear receptors remain sparse in the literature (Mottino and Catania, 2008; Tolson and Wang, 2010), and systemic evaluations have not been conducted to the same extent as metabolic enzyme inductions, especially in differentiated hepatocyte cultures with right transporter localizations. Recently we investigated transporter gene induction by rifampin, a known prototypical activator of PXR for enzymes and efflux transporter genes, in sandwich-cultured hepatocytes (Niu et al., 2019). Since sandwich-cultured human hepatocytes (SCHH) consist of collagen Magrigel™ on both sides of the hepatocytes, which allow the differentiation of hepatocytes to form the bile ducts, the hepatocytes can restore the expression of drug metabolizing enzyme and transporter proteins, and most importantly, with correct localization of hepatic drug transporters (Kimoto et al., 2012a; Kimoto et al., 2012b). We also observed similar induction patterns of metabolic enzyme genes by rifampin between sandwich-cultured hepatocytes and plated hepatocytes (Niu et al., 2019). Interestingly, hepatic transporter gene induction by the PXR ligand rifampin was generally not significant in both human and monkey hepatocytes, except for SLC51B, a greater than 10-fold induction was observed (Niu et al., 2019).
In order to further elucidate the regulation of hepatic transporter genes by CAR and AhR activation, dose-response induction studies were conducted in sandwich-cultured human hepatocytes from multiple donors treated with AhR or CAR activators. Omeprazole (OP) and 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) were selected as AhR activators, and 6-(4-chlorophenyl)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde O-(3,4-dichlorobenzyl)oxime (CITCO) and phenobarbital (PB) were used as the direct and indirect activators of CAR, respectively. The gene expressions were quantitated by real-time quantitative reverse transcription polymerase chain reaction (qRT-PCR).
Cryopreserved primary human hepatocytes isolated from deceased donor livers were purchased from In Vitro ADMET Laboratories (Columbia, MD 21045) and BioIVT (Westbury, NY). Pre-experiments were conducted to confirm the induction of CYP genes in selected hepatocyte lots. The donor demographics is listed in Table 1. Consent was obtained from the human donor or the donor's legal next of kin for use of these samples and their derivatives for research purposes using IRB-approved authorizations.
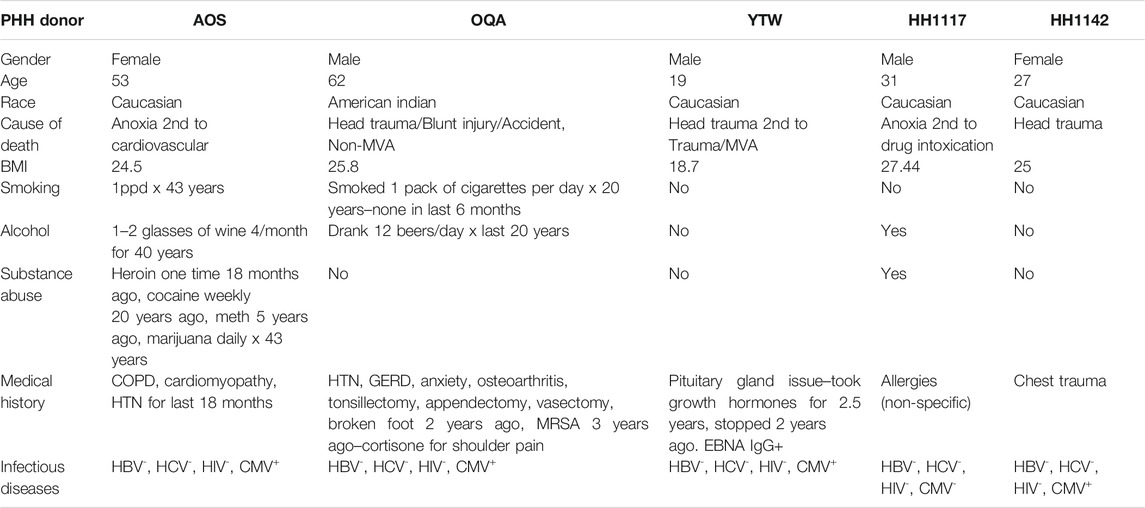
TABLE 1. Primary human hepatocyte donor demographics.
Omeprazole (OP), 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), 6-(4-chlorophenyl)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde O-(3,4-dichlorobenzyl)oxime (CITCO) and phenobarbital (PB) were purchased from Sigma-Aldrich (St. Louis, MO). Matrigel™ (phenol red free) was obtained from Corning (Bedford, MA). Collagen I coated 96-well plates were obtained from Life Technologies (Carlsbad, CA). The hepatocyte thawing medium (UCRM™) was purchased from In Vitro ADMET (Columbia, MD). The hepatocyte plating medium (InVitroGRO™ CP medium), incubation medium (InVitroGRO™ HI medium) and TorpedoTM Antibiotic Mix were from BioIVT (Westbury, NY). All oligonucleotide primer and probe sets were manufactured by Life Technologies (Table 2).

TABLE 2. Information of primer and probe sets manufactured by Life Technologies.
Cryopreserved human hepatocytes were quickly thawed at 37°C and then transferred into hepatocyte thawing medium. Cells were pelleted at 100 × g for 10 min and then resuspended in hepatocyte complete plating medium (CP medium). The cells were then seeded at 65,000 cells per well in 96-well collagen I coated plates. After cell attachment (4–6 h post-seeding), CP medium was exchanged for ice-cold complete incubation medium (HI medium) containing BD Matrigel™ at the concentration of 0.25 mg/ml. The cells were then incubated overnight at 37°C in a humidified 5% CO2 incubator. The HI medium was replaced by the fresh HI medium containing TCDD (0.08–50 nM), OP (0.08–50 μM), CITCO (0.008–5 μM), PB (1.6–1,000 μM) or dimethyl sulfoxide (DMSO) only. The media containing TCDD, Omeprazole, CITCO, PB or DMSO were subsequently changed daily for a total of three days. Cell death or loss of adhesion of hepatocytes during or after treatment were examined by microscopy. In addition, the β-actin mRNA levels were assessed to confirm that the treatments of transcript activators didn’t show significant cytotoxicity.
A RNeasy 96 Kit (Qiagen) was used to extract the total cellular RNA from sandwich cultured hepatocytes in 96-well plates following the manufacturer's instructions. A total volume of 200 μL RNA was obtained via the column elution provided in the RNeasy kit. Five μL RNA extraction was mixed with TaqMan® Fast Virus 1-Step Master Mix (Life Technologies) in a final reaction volume of 20 μL and qRT-PCR was achieved using a QuantStudio 6 Flex Real-Time PCR System (Life Technologies), following the manufacturer's instructions. β-actin mRNA expression in the SCHH was used as a house-keeping gene for the normalization of target gene expressions. The mRNA levels in SCHH treated with AhR or CAR activators were expressed by a fold change, compared to the gene expressions without the treatment. The 2−ΔΔCt method (Livak and Schmittgen, 2001) was used to analyze the relative changes in gene expression, where the Ct is the cycle number at which the fluorescence in the reaction crosses the preset arbitrary threshold; the ΔCt represents the difference between the Ct target and reference and the ΔΔCt is the difference between the ΔCt of the test and the ΔCt of the preassigned control. Gene expression for each condition is expressed as a fold-change of mean ± standard deviation (SD) from three independent donors (each performed in triplicate). All primer and probe sets were purchased from Life Technologies (Table 2). When concentration-dependent induction observed, the in vitro induction mRNA data were fitted using the sigmoid 3-parameter equation in GraphPad Prism (La Jolla, CA) to estimate the in vitro concentration of inducer that produced half the maximum induction values (EC50).
The potential induction of metabolizing enzyme genes was investigated in three donors of cryopreserved human hepatocytes. The inductions of metabolizing enzymes including CYP1A1, CYP1A2, CYP3A4, CYP2C9, CYP2B6 and UGT1A1 were used as positive or negative controls for CAR and AhR activators. SCHH were incubated with TCDD (0.08–50 nM), OP (0.08–50 µM), CITCO (0.008–5 µM) and PB (1.6–1,000 µM) for 72 h. TCDD induced CYP1A1 and UGT1A1, to a maximal increases of 38- and 4.3-fold, respectively (Table 3). The TCDD induction was concentration-dependent (Figure 1A), and the EC50 was estimated to be 0.5 and 1.56 nM for CYP1A1 and UGT1A1, respectively (Table 4). Likewise, CYP1A1, CYP3A4 and UGT1A1 mRNA inductions by OP were observed in a concentration-dependent manner to a maximum of 37, 61 and 5.0-fold increase at 50 μM, respectively (Table 3). The EC50s of OP induction were 3.34, 7.37, and 28.7 µM for CYP1A1, CYP3A4 and UGT1A1, respectively (Table 4). The mRNA of CYP2B6, CYP3A4 and UGT1A1, but not CYP1A2 was induced by CAR activators, CITCO and PB. The average of 24, 31 and 2.9-fold increase of CYP2B6, CYP3A4 and UGT1A1 mRNA expression were detected in the hepatocytes treated with CITCO at 5 µM (Table 3), with the EC50 of 0.16, 1.14 and >5 μM, respectively (Table 4). Similarly, PB induced CYP2B6, CYP3A4 and UGT1A1 mRNA expressions by about 40, 28 and 4.2-fold at the highest concentration tested (1,000 µM) (Table 3), and the EC50s were 726, 343 and 639 μM, respectively (Table 4). Although robust enzyme inductions were detected in SCHH treated with activators of AhR or CAR, it is worth noting that the donor-to-donor variabilities were observed among three lots of human hepatocytes. The CYP3A4 fold induction by CITCO (5 µM) in hepatocytes from the donor OQA was nearly 8-times greater than that of the donor AOS (Supplementary Figure 1). In addition, a greater than 50% reduction of CYP3A4 mRNA (<0.5-fold) by TCDD was detected in only two donors among three lots of hepatocytes, resulting in an average reduction of about 0.4-fold (Table 3) (Supplementary Figure 1).
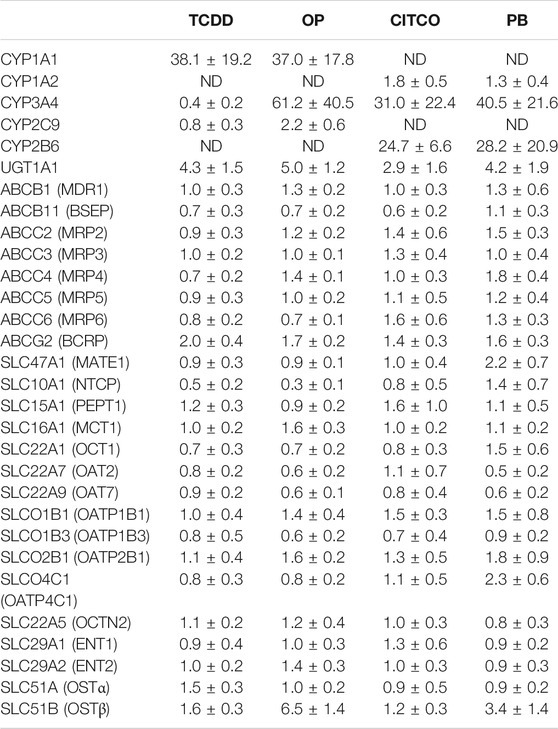
TABLE 3. Changes in gene expression of CYP enzymes and drug transporters in sandwich- cultured human hepatocytes treated with CAR or AhR activators.
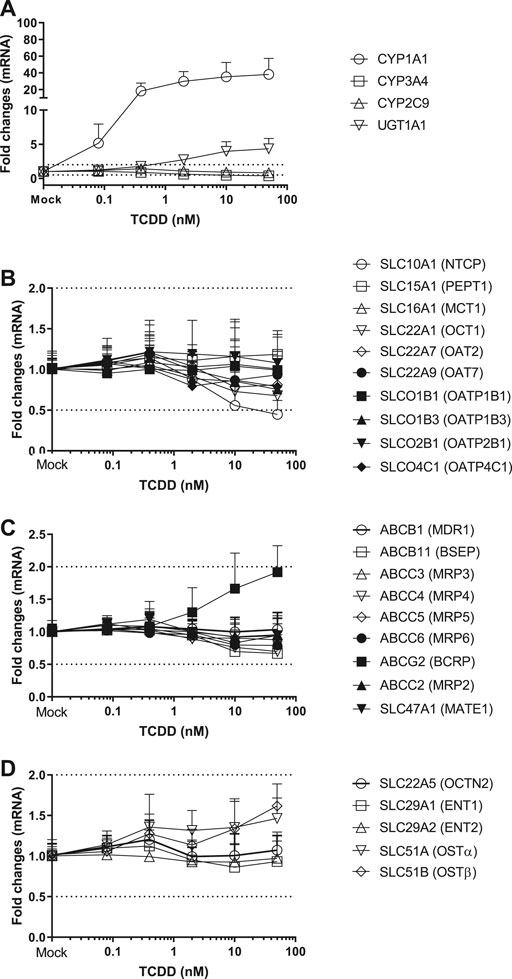
FIGURE 1. Induction of metabolizing enzyme and transporter mRNA by TCDD. The concentration-dependent induction studies were conducted in SCHH (lot HH1117, HH1142 and YTW) treated with TCDD at the concentrations ranging from 0.08 to 50 nM. The gene expressions were quantitated by qRT-PCR. Gene expression for each condition is expressed as a fold-change of mean ± standard deviation (SD) from three independent donors (each performed in triplicate). Dot lines indicate the change of 2-fold.

TABLE 4. The EC50 of gene induction.
Twenty-four hepatic drug transporters were tested for potential gene induction in SCHH treated with prototypical activators of AhR (TCDD and OP) or CAR (CITCO and PB) for 3 days. All 24 transporter genes were found to be expressed in hepatocytes from five donors. In the US FDA guidance for industry on in vitro DDI studies, CYP induction is presumed when ≥ 2-fold concentration-dependent increase in CYP mRNA is observed (USFDA-Guidance-2020, 2020). Accordingly, the average fold changes of transporter genes by CAR or AhR activators in SCHH were generally less than 2-fold, with the exception of SLC51B, SLC47A1, SLCO4C1, ABCG2 and SLC10A1 genes (Table 3 and Figures 1–4). A modest (∼2-fold) and concentration-dependent induction of ABCG2 mRNA by TCDD (50 nM) was detected (Figure 1C). In contrast, marginal suppression of SLC10A1 mRNA by TCDD and OP was observed at the highest concentration treated and the gene suppression appeared to be concentration-dependent (Figure 1B and Figure 2B). A dose-dependent induction of SLC51B gene by PB and OP was detected (Figure 2D and Figure 3D), and at the highest concentration tested, the induction of SLC51B gene was about 3.4 and 6.5-fold, respectively. Additionally, about 2-fold increases of SLC47A1 and SLCO4C1 mRNA expression were also found in SCHH treated with PB at 1,000 μM. The fold induction by CITCO for all transporter genes in SCHH was generally less than 2-fold (Table 3).
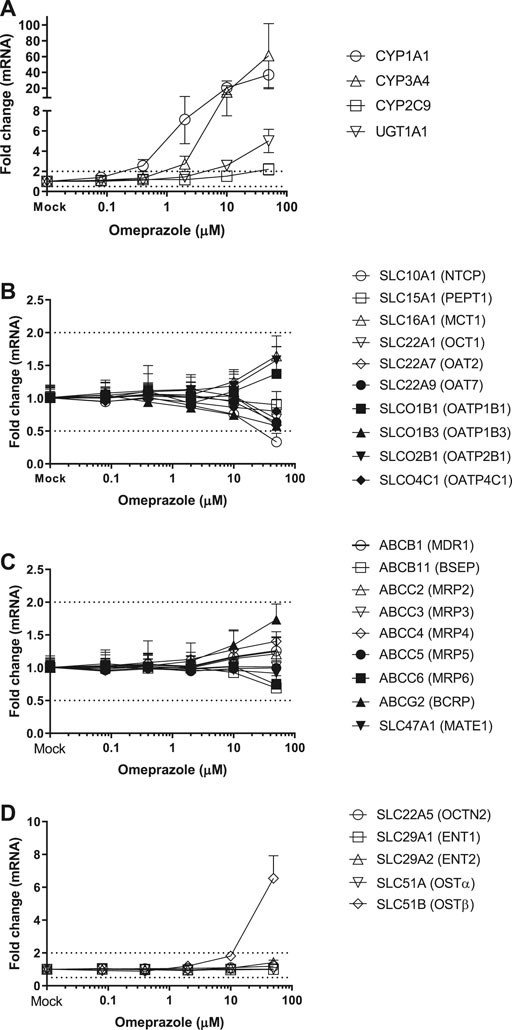
FIGURE 2. Induction of metabolizing enzyme and transporter mRNA by OP. The concentration-dependent induction studies were conducted in SCHH (Lot HH1117, HH1142 and YTW) treated with OP at the concentrations ranging from 0.08 to 50 μM. The gene expressions were quantitated by qRT-PCR. Gene expression for each condition is expressed as a fold-change of mean ± standard deviation (SD) from three independent donors (each performed in triplicate). Dot lines indicate the change of 2-fold.
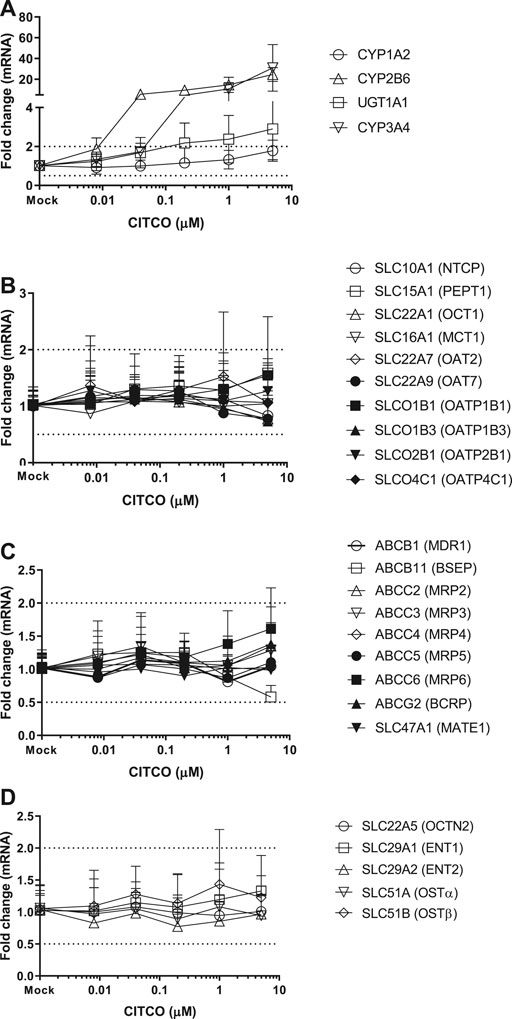
FIGURE 3. Induction of metabolizing enzyme and transporter mRNA by CITCO. The concentration-dependent induction studies were conducted in SCHH (Lot AOS, OQA and YTW) treated with CITCO at the concentrations ranging from 0.008 to 5 μM. The gene expressions were quantitated by qRT-PCR. Gene expression for each condition is expressed as a fold-change of mean ± standard deviation (SD) from three independent donors (each performed in triplicate). Dot lines indicate the change of 2-fold.
Drugs or xenobiotics can induce the expression of metabolizing enzyme and/or transporter proteins through activating gene transcription receptors. As a result, fluctuation of systemic exposure can occur for drugs that are transported or metabolized by the induced enzymes or transporters. Thus, assessing the potentials for CYP and transporter inductions early become important de-risk steps for planning appropriate clinical DDI studies in drug research and development cycle. Primary cultures of hepatocytes are commonly used as tools to investigate induction potential of metabolizing enzyme genes, and the model is adapted to investigate transporter gene regulation (Nishimura et al., 2002; Williamson et al., 2013). However, when hepatocytes are grown in a conventional plated monolayer format, many drug metabolism and transport genes are substantially downregulated (Sun et al., 2019), which can result in false positive or negative outcomes, particularly for drug transporters that require the unique polarized architecture present in the liver. In fact, Courtois et al. (Courtois et al., 2002) observed remarkable in vitro induction of multidrug resistance-associated protein 2 (MRP2) mRNA and protein levels in conventional plated rat and human hepatocytes exposed to phenobarbital. Surprisingly, MRP2 induction observed in vitro was not reproduced in rats in vivo treated by the bartiturates (Courtois et al., 2002), which suggests that the positive in vitro induction of MRP2 could be a false positive event. Additionally, inconsistent results are reported in the literature when using the conventional plated hepatocytes for characterizing transporter gene inductions by PXR or CAR ligands, as compared with the results obtained from sandwich cultured hepatocytes (Jigorel et al., 2006). With that in mind, in the current investigation, transporter induction potential by AhR or CAR activators was accessed in SCHH to overcome the disadvantage of the conventional hepatocyte cultures suffering from loss of polarized nature of hepatocytes.
OP is a proton pump inhibitor used for the treatment of acid-related disorders. TCDD, also known as dioxin, was used as an herbicide infamously known as Agent Orange and is found in animals and humans through environmental polluted food chain. OP and TCDD are the AhR ligands (Quattrochi and Tukey, 1993). Ligands bind to the AhR receptor and form a heterodimeric complex with the AhR nuclear translocator protein, known as a transcription factor, to stimulate the transcription of CYP1A family genes. As a result, induction of CYP1A1 and UGT1A1 genes in SCHH by TCDD and OP was observed following a concentration-dependent manner (Figure 1, 2). In addition, OP, but not TCDD, also induced CYP3A4 gene to a level that was comparable to the induction of CYP1A1 (61-fold at 50 μM) (Table 3). The results agreed with the literature reports (Yoshinari et al., 2008; Donato et al., 2010), and confirmed the reliability and robustness of SCHH for investigating gene induction. Regarding the drug transporter regulation, it is known that efflux transporters ABCG2 (BCRP) and ABCB1 (P-gp) are induced by TCDD mediated AhR activation in Caco-2 and other human carcinoma cells including HepG2, LS180, LS174T and MCF7 cells (Ebert et al., 2005; Tan et al., 2010). The induction of ABCG2 mRNA in primary hepatocytes is also observed; however, the magnitude of induction is relatively small in human hepatocytes compared with those of the human carcinoma cell lines, which suggests a cell-dependent induction (Tan et al., 2010). Accordingly, a modest concentration-dependent induction of ABCG2, but not ABCB1 mRNA, by TCDD and OP was observed in SCHH (Figure 1, 2). Additionally, the SLC10A1 gene that codes the sodium-dependent taurocholate transporter protein (NTCP) for hepatic uptake of bile acid, was reduced about 2-fold by TCDD and OP (Figure 1, 2). Although we can not rule out if the maginal effects observed is due to slight cell stress at the high concentrations, this observation was supported by previous studies that TCDD can alter genes involved in cholesterol metabolism and bile acids homeostasis (Fletcher et al., 2005). Interestingly, SLC51B gene was found to be the most highly induced transporter gene by OP, but not TCDD (Figure 1, 2). SLC51A and SLC51B genes code OSTα and β protein respectively, to form the heteromeric transport protein OSTα/β. This is an important transporter in bile acid homeostasis, and can undergo adaptive regulation in the disease progression of obstructive cholestasis or primary biliary cholangitis (Ballatori et al., 2009). The OSTα/β expression can be regulated by other transcription factors including small heterodimer partner (Shp), liver receptor homolog-1 and LXR/the retinoid X receptor (RXR) heterodimer (Ballatori et al., 2009). In addition, the farnesoid X receptor (FXR), also called the bile acid receptor, and LXR may exert a coordinated role in maintaining bile acid homeostasis in the hepatocytes, which result in a novel negative feedback loop of bile acids to induce or suppress bile acid transporter genes including OSTα/β. In fact, we previously reported that SLC51B gene was also induced in hepatocytes treated with rifampin (Niu et al., 2019). Rifampicin induces CYP3A4 and CYP2C9 through activation of PXR (Maglich et al., 2002). The observation of CYP3A4 and SLC51B induction was consistent with the current literature (Novotna and Dvorak, 2014; Benson et al., 2016; Niu et al., 2019), suggesting that the induction is involved in PXR. Since AhR and PXR can share common regulation pathways (Maglich et al., 2002; Lim and Huang, 2008), our results implicate the crosstalk of AhR-PXR in human hepatocytes (Gerbal-Chaloin et al., 2006).
CITCO and PB are known as CAR direct and indirect activators, respectively. As one of the xenosensors, CAR regulates hepatic drug metabolizing enzyme genes and plays a role in mediating various hepatic functions including fatty acid oxidation, insulin signaling and biotransformation of bile acids (Gao et al., 2009; Lynch et al., 2013). For example, induction of CYP2B6 gene expression is mediated by CAR via responsive elements located in the promoter regions of CYP2B6 gene. PB does not directly bind to CAR and is considered as an indirect activator to translocate CAR into the nucleus, where it leads to transcriptional activation of target genes (Kawamoto et al., 1999; Wang et al., 2004). PB is also a human PXR ligands to induce gene expression of numerous hepatic metabolizing enzymes and transporters (Li et al., 2019). In contrast, CITCO binds to the CAR and is a direct activator of CAR. It also activates human PXR (Li et al., 2019). Here we observed that both CYP2B6 and CYP3A4 were greatly induced by PB and CITCO, and to a less extent, UGT1A1 was also induced in a concentration-dependent manner (Figure 3, 4), which is consistent with the literature reports (Li et al., 2019). Burk et al. reported that CAR can also regulate MDR1 gene in cells stably expressing CAR (Burk et al., 2005). Our findings suggested that CITCO and PB may also activate the PXR receptor to induce CYP3A gene (Novotna and Dvorak, 2014; Li et al., 2019). Controversially, neither CITCO nor PB induced the ABCB1 (MDR1) gene in SCHH (Figure 3 and 4). This suggests that transporter induction can be cell line (or organ) specific. Interestingly, SLC51B gene was also induced by PB, but not CITCO (Table 3). Together with the SLC51B induction by OP, PB and rifampin that are PXR ligands (Benson et al., 2016; Niu et al., 2019), the SLC51B induction appears to likely not be mediated by CAR/AhR receptors. In addition, OSTβ heterodimerizes with OSTα to form the functional transporter complex, OSTα/β. Therefore, the outcome of SLC51B (OSTβ) induction, but not SLC51A (OSTα), remains unclear and further investigation on OSTβ induction is warranted.
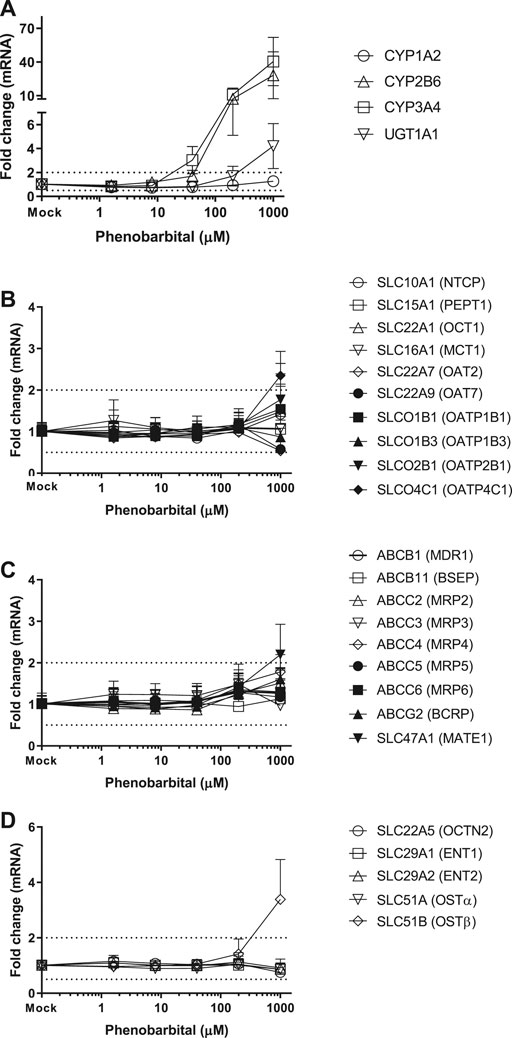
FIGURE 4. Induction of metabolizing enzyme and transporter mRNA by PB. The concentration-dependent induction studies were conducted in SCHH (Lot AOS, OQA and YTW) treated with PB at the concentrations ranging from 1.6 to 1,000 μM. The gene expressions were quantitated by qRT-PCR. Gene expression for each condition is expressed as a fold-change of mean ± standard deviation (SD) from three independent donors (each performed in triplicate). Dot lines indicate the change of 2-fold.
Collectively, according to the regulatory guidance that a concentration-dependent 2-fold increase in mRNA is considered the threshold for a positive in vitro induction signal (USFDA-Guidance-2020, 2020), our results revealed that except SLC51B gene, hepatic transporter gene regulations by CAR and AhR ligands were generally marginal, and significantly lower than the inductions of metabolizing enzyme genes. The current findings suggest that the assessment of transporter gene inductions is not required when a drug does not remarkably induce metabolizing enzyme genes by CAR and AhR activation.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
YL, CN, and BS participated in research design. CN conducted experiments. YL and CN performed data analysis. YL, CN, and BS wrote or contributed to the writing of the manuscript.
All authors were employed by Gilead Sciences Inc.
We thank Dr Xiaomin Liang, Eisuke Murakami for helpfully scientific discussions and Dr Monica Yang for proofreading.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2020.620197/full#supplementary-material.
CAR, the constitutive androstane receptor; AhR, the aryl hydrocarbon receptor. qRT-PCR, real-time quantitative reverse transcription polymerase chain reaction. SLC transporters, solute carrier transporters; ABC transporters, ATP-binding cassette transporters. TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin; OP, omeprazole; CITCO, 6-(4-chlorophenyl)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde O-(3,4-dichlorobenzyl)oxime; PB, phenobarbital.
Ballatori, N., Li, N., Fang, F., Boyer, J. L., Christian, W. V., and Hammond, C. L. (2009). OST alpha-OST beta: a key membrane transporter of bile acids and conjugated steroids. Front. Biosci. 14, 2829–2844. doi:10.2741/3416
Benson, E. A., Eadon, M. T., Desta, Z., Liu, Y., Lin, H., Burgess, K. S., et al. (2016). Rifampin regulation of drug transporters gene expression and the association of MicroRNAs in human hepatocytes. Front. Pharmacol. 7, 111. doi:10.3389/fphar.2016.00111
Burk, O., Arnold, K. A., Geick, A., Tegude, H., and Eichelbaum, M. (2005). A role for constitutive androstane receptor in the regulation of human intestinal MDR1 expression. Biol. Chem. 386 (6), 503–513. doi:10.1515/BC.2005.060
Courtois, A., Payen, L., Le Ferrec, E., Scheffer, G. L., Trinquart, Y., Guillouzo, A., et al. (2002). Differential regulation of multidrug resistance-associated protein 2 (MRP2) and cytochromes P450 2B1/2 and 3A1/2 in phenobarbital-treated hepatocytes. Biochem. Pharmacol. 63 (2), 333–341. doi:10.1016/s0006-2952(01)00829-2
Donato, M. T., Montero, S., Castell, J. V., Gomez-Lechon, M. J., and Lahoz, A. (2010). Validated assay for studying activity profiles of human liver UGTs after drug exposure: inhibition and induction studies. Anal. Bioanal. Chem. 396 (6), 2251–2263. doi:10.1007/s00216-009-3441-1
Ebert, B., Seidel, A., and Lampen, A. (2005). Identification of BCRP as transporter of benzo[a]pyrene conjugates metabolically formed in Caco-2 cells and its induction by Ah-receptor agonists. Carcinogenesis 26 (10), 1754–1763. doi:10.1093/carcin/bgi139
Fletcher, N., Wahlstrom, D., Lundberg, R., Nilsson, C. B., Nilsson, K. C., Stockling, K., et al. (2005). 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) alters the mRNA expression of critical genes associated with cholesterol metabolism, bile acid biosynthesis, and bile transport in rat liver: a microarray study. Toxicol. Appl. Pharmacol. 207 (1), 1–24. doi:10.1016/j.taap.2004.12.003
Gao, J., He, J., Zhai, Y., Wada, T., and Xie, W. (2009). The constitutive androstane receptor is an anti-obesity nuclear receptor that improves insulin sensitivity. J. Biol. Chem. 284 (38), 25984–25992. doi:10.1074/jbc.M109.016808
Gerbal-Chaloin, S., Pichard-Garcia, L., Fabre, J. M., Sa-Cunha, A., Poellinger, L., Maurel, P., et al. (2006). Role of CYP3A4 in the regulation of the aryl hydrocarbon receptor by omeprazole sulphide. Cell. Signal. 18 (5), 740–750. doi:10.1016/j.cellsig.2005.07.007
Hagenbuch, B., and Meier, P. J. (2004). Organic anion transporting polypeptides of the OATP/SLC21 family: phylogenetic classification as OATP/SLCO superfamily, new nomenclature and molecular/functional properties. Pflügers Archiv. 447 (5), 653–665. doi:10.1007/s00424-003-1168-y
Hillgren, K. M., Keppler, D., Zur, A. A., Giacomini, K. M., Stieger, B., Cass, C. E., et al. (2013). Emerging transporters of clinical importance: an update from the International Transporter Consortium. Clin. Pharmacol. Ther. 94 (1), 52–63. doi:10.1038/clpt.2013.74
International Transporter, C., Giacomini, K. M., Huang, S. M., Tweedie, D. J., Benet, L. Z., Brouwer, K. L., et al. (2010). Membrane transporters in drug development. Nat. Rev. Drug Discov. 9 (3), 215–236. doi:10.1038/nrd3028
Jigorel, E., Le Vee, M., Boursier-Neyret, C., Parmentier, Y., and Fardel, O. (2006). Differential regulation of sinusoidal and canalicular hepatic drug transporter expression by xenobiotics activating drug-sensing receptors in primary human hepatocytes. Drug Metab. Dispos. 34 (10), 1756–1763. doi:10.1124/dmd.106.010033
Kawamoto, T., Sueyoshi, T., Zelko, I., Moore, R., Washburn, K., and Negishi, M. (1999). Phenobarbital-responsive nuclear translocation of the receptor CAR in induction of the CYP2B gene. Mol. Cell Biol. 19 (9), 6318–6322. doi:10.1128/mcb.19.9.6318
Kimoto, E., Walsky, R., Zhang, H., Bi, Y. A., Whalen, K. M., Yang, Y. S., et al. (2012a). Differential modulation of cytochrome P450 activity and the effect of 1-aminobenzotriazole on hepatic transport in sandwich-cultured human hepatocytes. Drug Metab. Dispos. 40 (2), 407–411. doi:10.1124/dmd.111.039297
Kimoto, E., Yoshida, K., Balogh, L. M., Bi, Y. A., Maeda, K., El-Kattan, A., et al. (2012b). Characterization of organic anion transporting polypeptide (OATP) expression and its functional contribution to the uptake of substrates in human hepatocytes. Mol. Pharm. 9 (12), 3535–3542. doi:10.1021/mp300379q
Li, L., Welch, M. A., Li, Z., Mackowiak, B., Heyward, S., Swaan, P. W., et al. (2019). Mechanistic insights of phenobarbital-mediated activation of human but not mouse pregnane X receptor. Mol. Pharmacol. 96 (3), 345–354. doi:10.1124/mol.119.116616
Lim, Y. P., and Huang, J. D. (2008). Interplay of pregnane X receptor with other nuclear receptors on gene regulation. Drug Metabol. Pharmacokinet. 23 (1), 14–21. doi:10.2133/dmpk.23.14
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25 (4), 402–408. doi:10.1006/meth.2001.1262
Lynch, C., Pan, Y., Li, L., Ferguson, S. S., Xia, M., Swaan, P. W., et al. (2013). Identification of novel activators of constitutive androstane receptor from FDA-approved drugs by integrated computational and biological approaches. Pharm. Res. (N. Y.). 30 (2), 489–501. doi:10.1007/s11095-012-0895-1
Maglich, J. M., Stoltz, C. M., Goodwin, B., Hawkins-Brown, D., Moore, J. T., and Kliewer, S. A. (2002). Nuclear pregnane x receptor and constitutive androstane receptor regulate overlapping but distinct sets of genes involved in xenobiotic detoxification. Mol. Pharmacol. 62 (3), 638–646. doi:10.1124/mol.62.3.638
Mottino, A. D., and Catania, V. A. (2008). Hepatic drug transporters and nuclear receptors: regulation by therapeutic agents. World J. Gastroenterol. 14 (46), 7068–7074. doi:10.3748/wjg.14.7068
Nishimura, M., Yoshitsugu, H., Naito, S., and Hiraoka, I. (2002). Evaluation of gene induction of drug-metabolizing enzymes and transporters in primary culture of human hepatocytes using high-sensitivity real-time reverse transcription PCR. Yakugaku Zasshi 122 (5), 339–361. doi:10.1248/yakushi.122.339
Niu, C., Wang, Y., Zhao, X., Tep, S., Murakami, E., Subramanian, R., et al. (2019). Organic anion-transporting polypeptide genes are not induced by the pregnane X receptor activator rifampin: studies in hepatocytes in vitro and in monkeys in vivo. Drug Metab. Dispos. 47 (12), 1433–1442. doi:10.1124/dmd.119.088922
Novotna, A., and Dvorak, Z. (2014). Omeprazole and lansoprazole enantiomers induce CYP3A4 in human hepatocytes and cell lines via glucocorticoid receptor and pregnane X receptor axis. PloS One 9 (8), e105580. doi:10.1371/journal.pone.0105580
Patel, M., Taskar, K. S., and Zamek-Gliszczynski, M. J. (2016). Importance of hepatic transporters in clinical disposition of drugs and their metabolites. J. Clin. Pharmacol. 56 (Suppl. 7), S23–S39. doi:10.1002/jcph.671
Quattrochi, L. C., and Tukey, R. H. (1993). Nuclear uptake of the Ah (dioxin) receptor in response to omeprazole: transcriptional activation of the human CYP1A1 gene. Mol. Pharmacol. 43 (4), 504–508.
Sun, P., Zhang, G., Su, X., Jin, C., Yu, B., Yu, X., et al. (2019). Maintenance of primary hepatocyte functions in vitro by inhibiting mechanical tension-induced YAP activation. Cell Rep. 29 (10), 3212–3222. doi:10.1016/j.celrep.2019.10.128
Tan, K. P., Wang, B., Yang, M., Boutros, P. C., Macaulay, J., Xu, H., et al. (2010). Aryl hydrocarbon receptor is a transcriptional activator of the human breast cancer resistance protein (BCRP/ABCG2). Mol. Pharmacol. 78 (2), 175–185. doi:10.1124/mol.110.065078
Tolson, A. H., and Wang, H. (2010). Regulation of drug-metabolizing enzymes by xenobiotic receptors: PXR and CAR. Adv. Drug Deliv. Rev. 62 (13), 1238–1249. doi:10.1016/j.addr.2010.08.006
USFDA-Guidance-2020 (2020). Available at: https://www.fda.gov/media/134582/download (Accessed December 17, 2020).
Vildhede, A., Wisniewski, J. R., Noren, A., Karlgren, M., and Artursson, P. (2015). Comparative proteomic analysis of human liver tissue and isolated hepatocytes with a focus on proteins determining drug exposure. J. Proteome Res. 14 (8), 3305–3314. doi:10.1021/acs.jproteome.5b00334
Wang, H., Faucette, S., Moore, R., Sueyoshi, T., Negishi, M., and LeCluyse, E. (2004). Human constitutive androstane receptor mediates induction of CYP2B6 gene expression by phenytoin. J. Biol. Chem. 279 (28), 29295–29301. doi:10.1074/jbc.M400580200
Williamson, B., Dooley, K. E., Zhang, Y., Back, D. J., and Owen, A. (2013). Induction of influx and efflux transporters and cytochrome P450 3A4 in primary human hepatocytes by rifampin, rifabutin, and rifapentine. Antimicrob. Agents Chemother. 57 (12), 6366–6369. doi:10.1128/AAC.01124-13
Yang, H., and Wang, H. (2014). Signaling control of the constitutive androstane receptor (CAR). Protein Cell 5 (2), 113–123. doi:10.1007/s13238-013-0013-0
Yoshinari, K., Ueda, R., Kusano, K., Yoshimura, T., Nagata, K., and Yamazoe, Y. (2008). Omeprazole transactivates human CYP1A1 and CYP1A2 expression through the common regulatory region containing multiple xenobiotic-responsive elements. Biochem. Pharmacol. 76 (1), 139–145. doi:10.1016/j.bcp.2008.04.005
Keywords: gene induction, transporters, hepatocytes, constitutive androstane receptor, aryl hydrocarbon receptor
Citation: Niu C, Smith B and Lai Y (2021) Transporter Gene Regulation in Sandwich Cultured Human Hepatocytes Through the Activation of Constitutive Androstane Receptor (CAR) or Aryl Hydrocarbon Receptor (AhR). Front. Pharmacol. 11:620197. doi: 10.3389/fphar.2020.620197
Received: 22 October 2020; Accepted: 15 December 2020;
Published: 21 January 2021.
Edited by:
Petr Pavek, Charles University, CzechiaReviewed by:
Paavo Honkakoski, University of Eastern Finland, FinlandCopyright © 2021 Niu, Smith and Lai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yurong Lai, eXVyb25nLmxhaUBnaWxlYWQuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.