 Marloes Sol1
Marloes Sol1 Jan A. A. M. Kamps1
Jan A. A. M. Kamps1 Jacob van den Born2
Jacob van den Born2 Marius C. van den Heuvel3
Marius C. van den Heuvel3 Johan van der Vlag4
Johan van der Vlag4 Guido Krenning1
Guido Krenning1 Jan-Luuk Hillebrands3*
Jan-Luuk Hillebrands3*- 1Department of Pathology and Medical Biology, Division of Medical Biology, University of Groningen, University Medical Center Groningen, Groningen, Netherlands
- 2Department of Internal Medicine, Division of Nephrology, University of Groningen, University Medical Center Groningen, Groningen, Netherlands
- 3Department of Pathology and Medical Biology, Division of Pathology, University of Groningen, University Medical Center Groningen, Groningen, Netherlands
- 4Department of Nephrology, Radboud Institute for Molecular Life Sciences, Radboud University Medical Center, Nijmegen, Netherlands
Glomerular endothelial cell (GEnC) dysfunction is important in the pathogenesis of glomerular sclerotic diseases, including Focal Segmental Glomerulosclerosis (FSGS) and overt diabetic nephropathy (DN). GEnCs form the first cellular barrier in direct contact with cells and factors circulating in the blood. Disturbances in these circulating factors can induce GEnC dysfunction. GEnC dysfunction occurs in early stages of FSGS and DN, and is characterized by a compromised endothelial glycocalyx, an inflammatory phenotype, mitochondrial damage and oxidative stress, aberrant cell signaling, and endothelial-to-mesenchymal transition (EndMT). GEnCs are in an interdependent relationship with podocytes and mesangial cells, which involves bidirectional cross-talk via intercellular signaling. Given that GEnC behavior directly influences podocyte function, it is conceivable that GEnC dysfunction may culminate in podocyte damage, proteinuria, subsequent mesangial activation, and ultimately glomerulosclerosis. Indeed, GEnC dysfunction is sufficient to cause podocyte injury, proteinuria and activation of mesangial cells. Aberrant gene expression patterns largely contribute to GEnC dysfunction and epigenetic changes seem to be involved in causing aberrant transcription. This review summarizes literature that uncovers the importance of cross-talk between GEnCs and podocytes, and GEnCs and mesangial cells in the context of the development of FSGS and DN, and the potential use of GEnCs as efficacious cellular target to pharmacologically halt development and progression of DN and FSGS.
The Kidney and the Glomerulus
The kidneys have a vital role in fluid homeostasis and osmoregulation. Additionally, the kidneys are important for control of blood pressure and mineral metabolism. By filtering blood in the glomeruli, the kidneys produce about 150 liter glomerular filtrate per day of which 99% is reabsorbed in the tubules, to eventually generate approximately 1 liter of urine per day. By blood filtration and tubular excretion, waste products such as urea, minerals and toxic substances, are excreted from the body.
The glomerulus is a network of capillary loops, known as the glomerular tuft, and is enclosed by the Bowman’s capsule. Blood flows into the glomerulus via the afferent arteriole and leaves the glomerulus via the efferent arteriole (Scott and Quaggin, 2015). The glomerulus is assembled by four different cell types: parietal epithelial cells, glomerular endothelial cells (GEnCs), podocytes (visceral epithelial cells), and mesangial cells (Figures 1A, B). Parietal epithelial cells line the Bowman’s capsule, where the pre-urine is collected and forwarded to the proximal tubule. GEnCs cover the luminal surface of glomerular capillaries and are the cells of the glomerulus in direct contact with the blood. GEnCs are characterized by transcellular pores (i.e., fenestrae), essential for blood filtration. At the adluminal side, GEnCs are covered with the endothelial glycocalyx, filling the fenestrae (Satchell, 2013; Scott and Quaggin, 2015; Hegermann et al., 2016) (Figure 1C). The endothelial glycocalyx is a gel-like layer consisting of glycoproteins, proteoglycans with bound glycosaminoglycans (GAGs) (Reitsma et al., 2007; Slater et al., 2012; Garsen et al., 2014; Dane et al., 2015) and plasma proteins loosely adherent within the meshwork of the glycocalyx. The endothelial glycocalyx prevents leakage of circulating plasma proteins by size and steric hindrance and electrostatic repulsion (Ryan and Karnovsky, 1976; Singh et al., 2007; Patrakka and Tryggvason, 2010; Friden et al., 2011; Dane et al., 2013), and inhibits adhesion and extravasation of inflammatory cells.
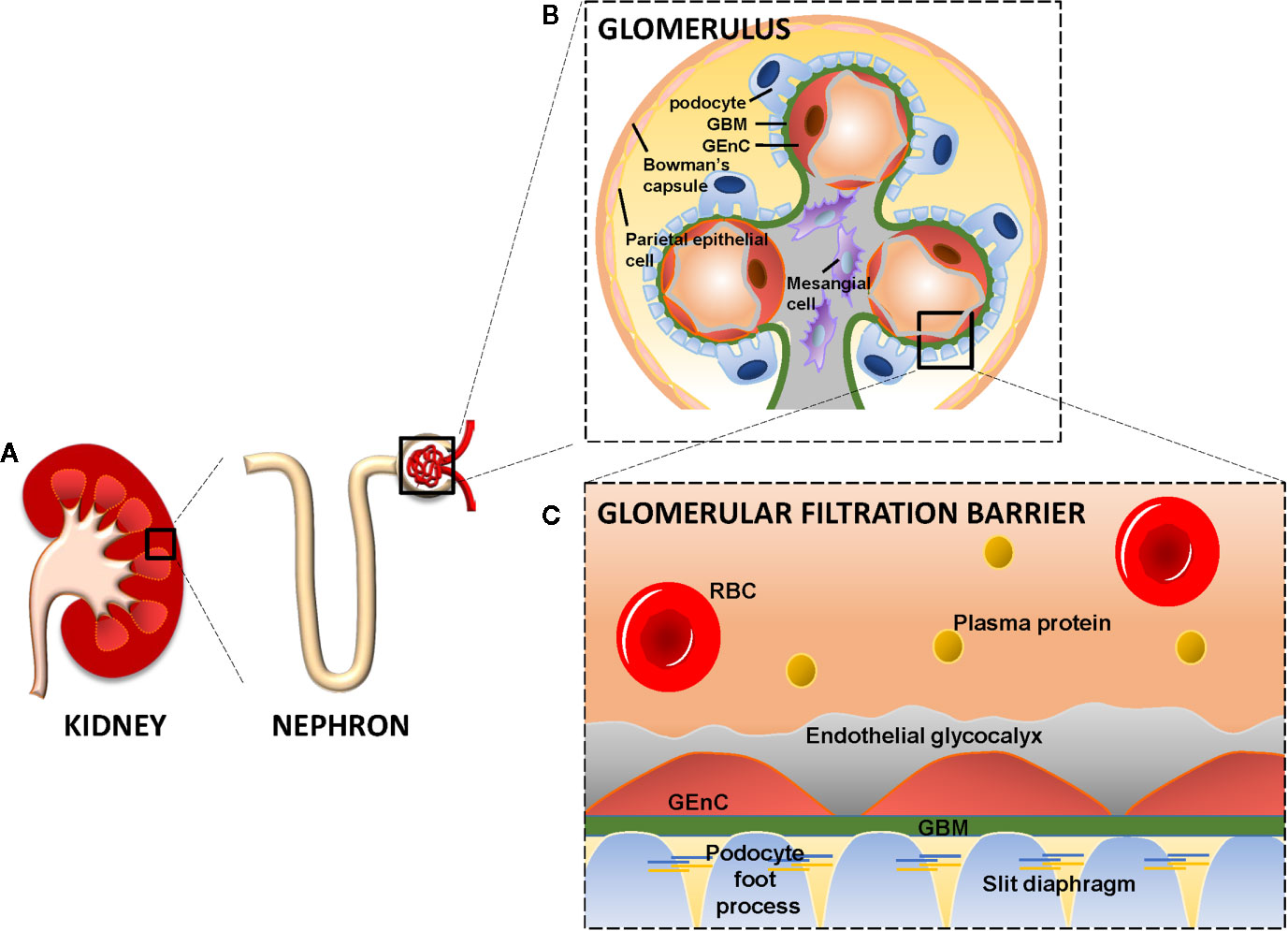
Figure 1 The kidney, glomerulus, and the glomerular filtration barrier. Each kidney consists of about 1 million nephrons. Each nephron consists of a glomerulus and a tubular compartment (A). The glomerulus is assembled by four different cell types, namely parietal epithelial cells, glomerular endothelial cells (GEnC), podocytes (visceral epithelial cells), and mesangial cells (B). GEnC and podocytes share a common extracellular matrix, the glomerular basement membrane (GBM). GEnC and their fenestrae are covered by the endothelial glycocalyx. Podocytes contain foot processes with slit diaphragms that are wrapped around the exterior of glomerular capillaries. Together, the GEnC and the endothelial glycocalyx, GBM and podocytes comprise the glomerular filtration barrier to filter the blood and remaining essential plasma proteins in the circulation (C). RBC, Red Blood Cell; GBM, Glomerular Basement Membrane; GEnC, Glomerular Endothelial Cell.
The endothelial glycocalyx serves as the primary sensor of wall shear stress through the initiation of signal transduction in GEnCs (Tarbell and Ebong, 2008). Wall shear stress, the hydrodynamic frictional force created from blood flow, transmits through the endothelial glycocalyx into the GEnC, leading to signal transduction that subsequently regulates the expression of Krüppel Like Factor 2 (KLF2), KLF4 and the transcription of eNOS and the production of nitric oxide (NO) which are crucial to maintain GEnC function (Dekker et al., 2006; Weinbaum et al., 2007; Ohnesorge et al., 2010; Slater et al., 2012; Dogne et al., 2018). In addition, GEnCs also function as a sink for factors essential for the regulation of the vascular tone and cross-talk with other glomerular cell types, such as vasoactive factors (endothelin-1 (ET-1), and NO) (Feliers et al., 2005; Dhaun et al., 2012).
Podocytes are specialized perivascular epithelial cells with elaborate projections called foot processes that are intimately wrapped around the exterior of glomerular capillaries (Figures 1B, C). The foot processes leave slits between them, called slit diaphragms, which are instrumental for proper blood filtration. GEnCs and podocytes share a common extracellular matrix, referred to as the glomerular basement membrane (GBM), which separates the GEnCs from the podocytes. Together, the GEnCs and the endothelial glycocalyx, the GBM, and the podocytes constitute the glomerular filtration barrier (GFB). The GFB is responsible for size-selective and charge-dependent filtration of the blood. Small and positively charged molecules such as urea, glucose, amino acids, and minerals can pass the GFB freely, whereas circulating cells and large and negatively charged proteins, including albumin, cannot pass the GFB. Mesangial cells are located in between the capillaries and form the mesangium together with their extracellular matrix (ECM). The mesangium provides structural stability to the glomerular vasculature and modulates capillary blood flow (Scott and Quaggin, 2015). The functionality and integrity of the GFB depends on proper function of GEnCs, podocytes and mesangial cells. Dysfunction of any of the cellular or extracellular components of the GFB culminates in a decreased filtration and eventually glomerulosclerosis (Haraldsson and Nystrom, 2012; Fu et al., 2015).
Cross-Talk Between Glomerular Cells Is Essential for Glomerular Integrity
There is a growing understanding of the interdependent relationship between GEnCs, podocytes and mesangial cells, which involves bidirectional cross-talk at a molecular level. To exemplify the importance of cross-talk between glomerular cells, the signaling of Vascular Endothelial Growth Factor A (VEGFA), Endothelin-1 (ET-1), and endothelial Nitric Oxide Synthase (eNOS) between GEnCs and podocytes are described. These molecules together form the VEGFA-eNOS/NO-ET-1 axis between GEnCs and podocytes.
VEGFA-eNOS/NO-ET-1 Axis
VEGFA is synthesized by podocytes and binds to its receptors VEGFR1 and VEGFR2 expressed on GEnCs (Eremina et al., 2008). Under physiological conditions, VEGFA induces eNOS activation in GEnCs and a subsequent increase in NO production. The increase of NO may negatively regulates the amount of VEGFA produced by podocytes (Mooyaart et al., 2011). Via this crosstalk, the glomerular cells ensure that sufficient VEGFA is produced to maintain viability of GEnCs, without VEGFA levels rising to a level that induces sprouting angiogenesis by GEnCs. In addition to NO, VEGFA also regulates ET-1 production by GEnCs, since VEGFA blockage in podocytes induces ET-1 release from GEnCs (Collino et al., 2008). GEnCs are considered the principal source of ET-1 within the glomeruli (Herman et al., 1998). ET-1 exerts its effect via ET-1 receptors (ETR) A and ETRB. Low levels of ET-1 induce an increase in NO, whereas high levels of ET-1 inhibit NO production (Watschinger et al., 1995; Dong et al., 2005; Sud and Black, 2009). ET-1 release from GEnCs associates with cytoskeleton redistribution with a decrease of nephrin in podocytes (Lenoir et al., 2014; Yuan et al., 2019). NO, in its turn, inhibits ET-1 expression (Khimji and Rockey, 2010) and exerts protective effects in podocytes (Sun et al., 2013). An illustration of cross-talk between GEnCs and podocytes in the VEGFA-eNOS/NO-ET-1 axis is provided in Figure 2. Next to the effect of ET-1 on podocytes, ET-1 also exerts effects on mesangial cells. ETRA signaling is associated with inflammation, contraction and proliferation of mesangial cells (Barton and Sorokin, 2015), and fibrosis. ETRB signaling has a reciprocal effect and is associated with vasorelaxation via eNOS-derived NO release (Barton and Yanagisawa, 2008).
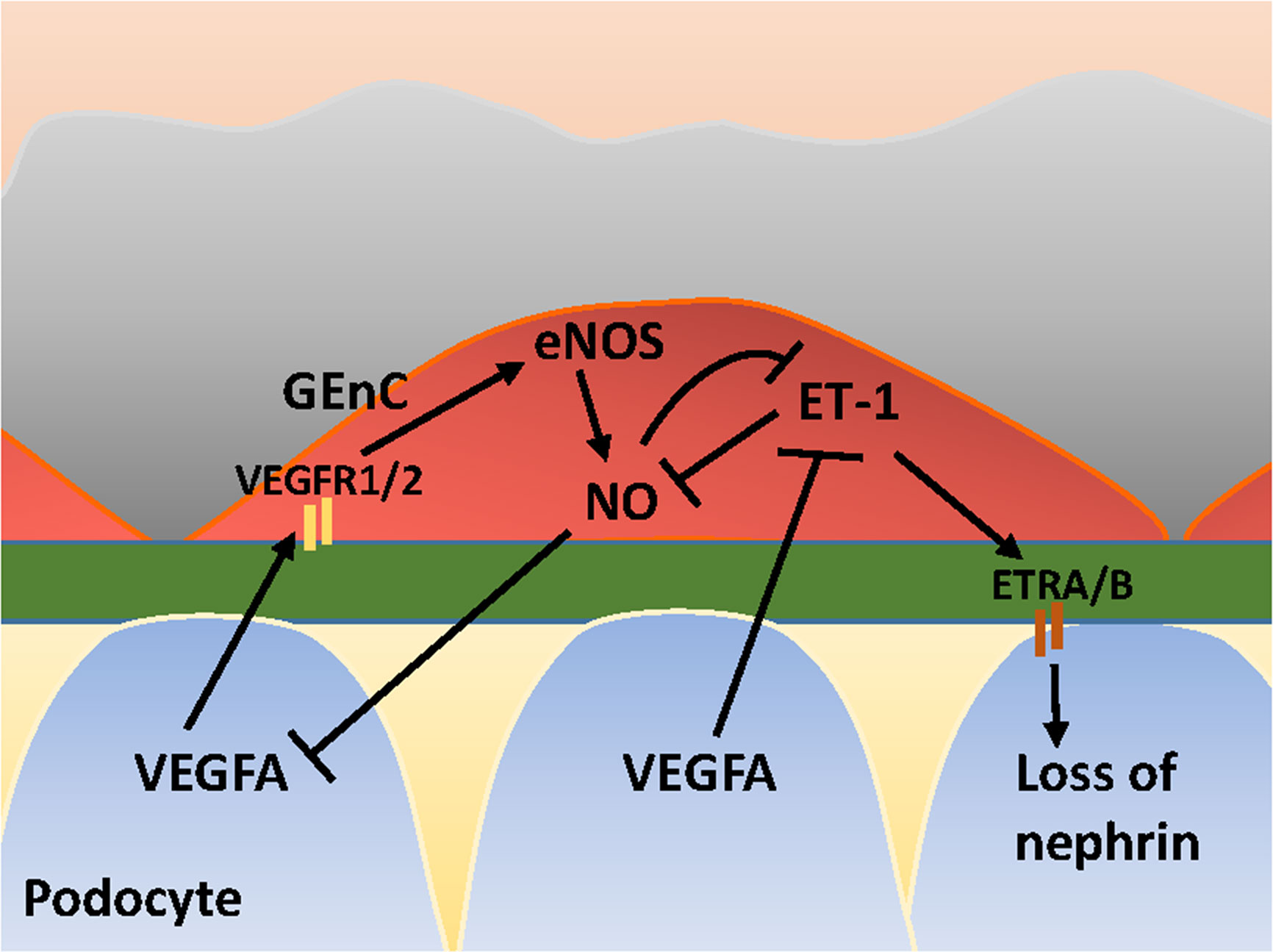
Figure 2 Glomerular cross-talk between GEnC and podocytes via the VEGFA-eNOS/NO-ET-1 axis. VEGFA, Vascular Endothelial Growth Factor A; VEGFR1/2, VEGF Receptor 1 and 2; eNOS, endothelial Nitric Oxide Synthase; NO, Nitric Oxide; ET-1, Endothelin-1; ETRA/B, ET-1 Receptor A and B. Stimulating and inhibitory effects are indicated with arrows and blunt lines, respectively.
Glomerular Sclerotic Diseases: Histopathology of FSGS and DN
DN is a long-term complication of both type 1 and type 2 diabetes mellitus and develops in 20%–40% of all diabetes mellitus patients (Rossing et al., 2018). DN, together with Focal Segmental Glomerulosclerosis (FSGS), is the most important cause of chronic kidney disease (CKD). Two types of FSGS exist: primary (or idiopathic) FSGS and secondary FSGS. In primary FSGS, which comprises 80% of all FSGS cases, the etiology is unknown. Secondary FSGS is induced by a preexisting pathologic condition, e.g., hypertension (Jefferson and Shankland, 2014), a viral infection, such as human immunodeficiency virus, drug-induced, or induced by genetic mutations (Lim et al., 2016). In case of primary or mutation-induced FSGS, mutations in genes encoding proteins expressed in podocytes, which are mostly related to slit diaphragm structure, the actin cytoskeleton, or foot processes, such as nephrin (NPHS1), podocin (NPHS2), actinin α4 (ACTN4), and TRPC6 are commonly observed (Lim et al., 2016). No mutations are known in GEnC-specific genes that would cause FSGS.
FSGS and overt diabetic nephropathy (DN) both are characterized by scarring (sclerosis) of the glomerular tuft, i.e., glomerulosclerosis (Figure 3). Glomerulosclerosis causes obliteration of the glomerular capillaries eventually (Fioretto and Mauer, 2007; De Vriese et al., 2018). In FSGS, only a fraction of the glomeruli (i.e., focal) is affected in a segmental manner, i.e., part of a glomerulus is affected. Sclerosis in FSGS is characterized by deposition of extracellular matrix (ECM) at the capillary loops. DN is the specific histopathology associated with reduced renal function in patients suffering from diabetic kidney disease (Yamanouchi et al., 2020). Overt DN comprises diffuse and sometimes nodular glomerulosclerosis in many glomeruli, caused by mesangial cell proliferation and mesangial sclerosis, and develops primarily in patients with proteinuria. Of note, nonproteinuric diabetic kidney disease also exists and which is characterized by minor histopathological changes without DN and with better prognosis compared with proteinuric diabetic kidney disease (Yamanouchi et al., 2020). So, particularly FSGS but also DN are accompanied by proteinuria (macroalbuminuria: >300 mg/gr creatinine), as well as by glomerular hypertension and hyperfiltration, and activation of glomerular inflammatory pathways (Fioretto and Mauer, 2007; Reidy and Kaskel, 2007). At the ultrastructural level, damage to podocytes and GEnCs is observed. Podocyte injury is observed as extensive effacement of the foot processes, ultimately leading to detachment of podocytes from the GBM (podocyte loss). GEnC dysfunction is characterized morphologically as a reduction of the endothelial glycocalyx, loss of fenestrae, widening of the subendothelial space, and swelling of the cytoplasm (Weil et al., 2012; Eleftheriadis et al., 2013; Morita et al., 2015; Taneda et al., 2015; Boels et al., 2016). In many patients, DN and FSGS progresses into end-stage renal disease (ESRD). Therapy resistance and the failure to adequately treat proteinuria, a glomerular inflammatory phenotype and hypertension are the main reasons for progression towards ESRD (Kiffel et al., 2011; Collins et al., 2013). Renal replacement therapy (dialysis or kidney transplantation), is the only effective treatment to postpone premature death in ESRD patients (Collins et al., 2014).
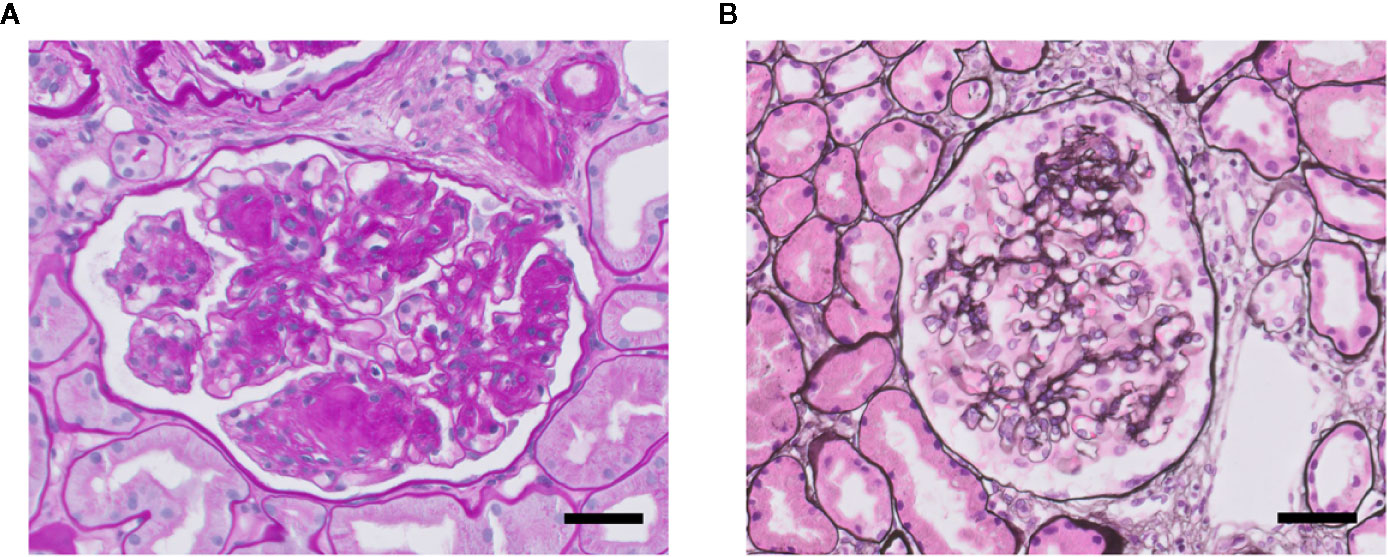
Figure 3 Glomerulosclerosis in DN (A) and FSGS (B). Light microscopy photomicrographs of a glomerulus showing DN with characteristic nodular mesangial expansion (Kimmelstiel-Wilson lesions) (Periodic acid–Schiff staining) (A), and of a glomerulus with mild FSGS (methenamine-silver staining) (B). Nuclei are stained in blue. Both glomeruli presenting glomerulosclerosis with increased glomerular extracellular matrix deposition and obliteration of capillaries. Scale bars represent 50 µm.
GEnC Dysfunction in DN and FSGS
GEnC dysfunction is important in the pathogenesis of glomerular sclerotic diseases, including FSGS and overt DN. GEnCs, covered by a thick glycocalyx, form the first cellular barrier in direct contact with all circulating factors. Changes in these circulating factors, such as high glucose levels and advanced glycation end-products, can induce GEnC dysfunction (Singh et al., 2011; Singh et al., 2013; Peng et al., 2016). In FSGS, the development of mesangial matrix expansion and sclerosis by parietal epithelial cells appears to be secondary to podocyte injury, whereas in DN mesangial matrix expansion is the key morphologic finding (Najafian et al., 2015). It is likely that GEnC dysfunction precedes and possibly also contributes to podocyte damage and mesangial expansion. In the past decade, evidence has been provided that also GEnC dysfunction is present and plays an important role in FSGS and DN development. GEnC dysfunction occurs in the early stages of FSGS and DN, and is sufficient to cause podocyte injury, proteinuria and activation of mesangial cells, as will be discussed in detail below. An interdependent relationship between GEnCs, podocytes and mesangial cells exists, which involves bidirectional cross-talk with intercellular signaling. Disturbed molecular cross-talk involving for example endothelial nitric oxide synthase (eNOS) may result in reduced GEnC-derived NO exposure to podocytes and can induce podocyte damage, and eventually compromise glomerular integrity (Yuen et al., 2012). Therapies aiming to prevent endothelial injury have shown to reduce DN in animal models. For example ETRA blockers have shown to restore the endothelial glycocalyx and to reduce albuminuria in diabetic mice (Boels et al., 2016). In diabetic patients, the ETRA blocker atrasentan reduced urinary albumin to creatinine ratios (Lin et al., 2018). Furthermore, renal elevation of cGMP, a key messenger for NO signaling, resulted in a reduction of glomerulosclerosis in rats with DN (Boustany-Kari et al., 2016). Given that GEnCs are the first cells exposed to changes in circulating factors and that GEnC behavior directly influences podocyte function, it is conceivable that GEnC dysfunction may culminate in podocyte damage and mesangial activation. It is, however, elusive which molecular mechanisms underlie GEnC dysfunction and the subsequent altered cross-talk with podocytes and mesangial cells. To develop new treatment options in order to halt the progression of glomerular sclerotic disease, a deeper understanding of the pathogenetic mechanisms underlying GEnC dysfunction and the disturbed cross-talk is required. Hereunder, it is described that GEnC dysfunction comprises multiple facets and is a pivotal and early factor in the development of glomerulosclerosis and is at the basis of developing proteinuria, podocyte dysfunction and mesangial expansion in FSGS and DN.
Compromised Endothelial Glycocalyx in DN and FSGS
Healthy GEnCs are covered with an endothelial glycocalyx. The endothelial glycocalyx consists of glycoproteins, glycolipids and proteoglycans with bound GAGs. Proteoglycans with bound GAGs, of which heparan sulphate and hyaluronan constitute up to 90%, are the main contributors to the function and structure of the endothelial glycocalyx (Reitsma et al., 2007; Slater et al., 2012; Garsen et al., 2014; Dane et al., 2015). In DN and FSGS, the endothelial glycocalyx is reduced, characterized by a loss of essential GAGs, including heparan sulphate and hyaluronan, and reduced thickness (Nieuwdorp et al., 2006a; Kuwabara et al., 2010; Satoh et al., 2010; van den Berg et al., 2019). Environmental factors, such as elevated levels of glucose, oxidative stress, or inflammatory stimuli, can modulate the endothelial glycocalyx (Singh et al., 2011; Singh et al., 2013; Kolarova et al., 2014). Inflammatory mediators like cytokines and chemokines cause degradation of the endothelial glycocalyx. Under physiological conditions, adhesion molecules on endothelial are covered by the endothelial glycocalyx, and only become accessible to leukocytes upon degradation of the glycocalyx (Kolarova et al., 2014). In vivo, intravenous administration of the bacterial heparan sulphate-degrading enzyme heparinase enhances leukocyte adherence to endothelial cells (Constantinescu et al., 2003). High glucose and oxidative stress cause a reduction of heparan sulphate in the endothelial glycocalyx on GEnCs in vitro (Singh et al., 2011; Singh et al., 2013). Furthermore, high glucose reduces GAG biosynthesis in GEnCs (Singh et al., 2011). Reduction of heparan sulphate culminates in increased passage of albumin across a GEnC monolayer (Singh et al., 2011; Singh et al., 2013). In line with these in vitro data, a reduced endothelial glycocalyx instantly causes proteinuria in vivo (Gil et al., 2012). Preservation of the endothelial glycocalyx by the genetic deletion of the heparan sulphate-degrading enzyme heparanase prevents proteinuria and kidney failure in experimental DN and glomerulonephritis (Gil et al., 2012; Garsen et al., 2016a). Loss of endothelial hyaluronan and thereby the endothelial glycocalyx induced by an endothelial-specific deletion of the hyaluronan synthesis enzyme hyaluronan synthase 2 (HAS2) (van den Berg et al., 2019) or by treatment with the hyaluronan-degrading enzyme hyaluronidase (Meuwese et al., 2010) also induces proteinuria (Meuwese et al., 2010; van den Berg et al., 2019) and progressive glomerulopathy (van den Berg et al., 2019), phenocopying the events in DN. In addition to the induction of leukocyte adherence and proteinuria, degradation of the endothelial glycocalyx also compromises GEnC signaling via the loss of mechanosensing. Fluid shear stress induces the production of NO in endothelial cells via activation of eNOS (Boo et al., 2002). Fluid shear stress-induced NO production is almost completely inhibited upon enzymatic removal of heparan sulphate in the endothelial glycocalyx (Florian et al., 2003), due to loss of eNOS activation. Impaired eNOS activation has negative effects on both GEnCs and podocytes in vivo as this results in GEnC dysfunction and disturbed cross-talk with podocytes (Yuen et al., 2012). A reduced endothelial glycocalyx on GEnCs, in response to noxious stimuli, clearly induces glomerular inflammation, proteinuria, and disturbs GEnC signaling. Loss of the endothelial glycocalyx coincides with coagulation activation (Nieuwdorp et al., 2006b) and could possibly also be linked with complement activation (Boels et al., 2013), which is described elsewhere (Nieuwdorp et al., 2006b; Boels et al., 2013) and will not further be addressed here.
Compromised Barrier Function by Endothelial Cell-Selective Adhesion Molecule (ESAM)
The barrier function of GEnCs in FSGS or DN is mainly compromised by a reduction of the endothelial glycocalyx but additional factors that contribute to an increased permeability have been described as well. The altered expression of endothelial cell-selective adhesion molecule (ESAM) has been implied in the loss of the endothelial cell barrier in DN. ESAM is a surface protein laterally expressed on GEnCs that is part of the endothelial tight junctions, and mediates the interaction between endothelial cells. ESAM expression is reduced in the early course of DN (4 weeks) and is associated with increased vascular permeability in vitro. In vivo, genetic ablation of ESAM causes proteinuria, a decrease in GEnC fenestrations and an increased space between GEnCs through expanded tight junctions, while no structural changes are observed in podocytes, the GBM and mesangium (Hara et al., 2009). Therefore, these observations provide evidence that solely GEnC dysfunction (induced by ESAM deficiency) already leads to glomerular paracellular albumin leakage with preserved podocyte structure (Hara et al., 2009).
Pro-Inflammatory Phenotype
GEnC dysfunction also contributes to glomerulosclerosis via obtaining a pro-inflammatory phenotype without having direct effects on podocytes and mesangial cells. Inflammatory pathways are involved in the pathogenesis of DN and FSGS (Navarro-Gonzalez et al., 2011; Wada and Makino, 2013; Moreno et al., 2018; Wilkening et al., 2020). Inflammation-related molecules and pathways (but without pronounced inflammation) may promote fibrotic and proliferative responses of mesangial cells, culminating in glomerulosclerosis (Furuta et al., 1993; Fogo, 2007). GEnC activation plays an important role in glomerular leukocyte infiltration as GEnC activation enables leukocyte rolling, adhesion, arrest and transmigration across the endothelial cell lining (Ley et al., 2007). Upon GEnC activation, the expression of chemokines and adhesion molecules on the cell surface of GEnCs, such as E-selectin (Hirata et al., 1998), intercellular adhesion molecule 1 (ICAM-1) and monocyte chemoattractant protein 1 (MCP-1), are increased (Rao et al., 2017). The high glucose-induced toxic metabolites advanced glycation end-products (AGEs), induce the expression of ICAM-1 and MCP-1 in a Rho-kinase dependent manner. AGE-induced activation of Rho-kinase could be a result of activation of the receptor for AGEs (RAGE) (Hirose et al., 2010). Also ET-1 can activate Rho-kinase in endothelial cells (Gien et al., 2013). Blockage of Rho-kinase in DN mice reduces the expression of ICAM-1 and MCP-1, and ablates concomitant glomerular infiltration of macrophages and glomerulosclerosis. Since macrophages also display Rho-kinase, an endothelial-specific inducible Rho-kinase gene targeting approach would be needed to confirm the role of endothelial Rho-kinase in the increased expression of ICAM-1 and MCP-1 in DN. This implies that AGEs-induced expression of adhesion molecules on GEnCs plays a key role in the development of diabetic glomerulosclerosis (Rao et al., 2017). Indeed, inhibition of AGEs reduces glomerulosclerosis in diabetic mice (Wilkinson-Berka et al., 2002; Forbes et al., 2003). In addition to the increased expression of adhesion molecules, GEnCs show a reduced expression of endothelial-specific molecule-1 (ESM-1), already in very early stages of DN. Under physiological conditions, GEnCs constitutively express ESM-1 that functions as an anti-inflammatory molecule and inhibits migration and rolling of leukocytes. Four weeks after the induction of diabetes, before the development of histological glomerular changes indicative of DN, ESM-1 expression was decreased in glomeruli of DN-susceptible mice compared to glomeruli of DN-resistant mice. These observations demonstrate that in early stages of DN, GEnCs display a pro-inflammatory phenotype which precedes glomerular damage (Zheng et al., 2017).
Mitochondrial Damage
In DN and FSGS, GEnCs display oxidative mitochondrial DNA lesions and mitochondrial oxidative stress, which is associated with loss of GEnC fenestrations (Daehn et al., 2014; Qi et al., 2017) and a loss of the endothelial glycocalyx (Ebefors et al., 2019). Mitochondrial oxidative stress in GEnCs was mediated by release of ET-1 by podocytes and the subsequent paracrine ETRA activation in GEnCs (Daehn et al., 2014; Ebefors et al., 2019). ET-1 induced an increase in heparanase mRNA expression in GEnCs in vitro, which could explain the loss of the endothelial glycocalyx upon release of ET-1 by podocytes in vivo (Ebefors et al., 2019). Mitochondrial oxidative stress was only observed in GEnCs and not in podocytes in streptozotocin (STZ)-induced DN (Qi et al., 2017). Interestingly, mitochondrial damage in GEnCs preceded podocyte loss, proteinuria, and glomerulosclerosis in adriamycin-induced FSGS and STZ-induced DN (Daehn et al., 2014; Qi et al., 2017). Scavenging of mitochondrial superoxide by systemic administration of the mitochondria-targeted potent antioxidant mitoTEMPO prevented GEnC mitochondrial oxidative stress (Daehn et al., 2014; Qi et al., 2017), the loss of fenestrations (Qi et al., 2017) and the loss of the endothelial glycocalyx (Ebefors et al., 2019). Attenuation of GEnC mitochondrial stress results in ameliorated podocyte loss, demonstrating that mitochondrial damage in GEnCs and the resulting production of mitochondrial superoxide are important triggers for podocyte loss (Daehn et al., 2014; Nagasu et al., 2016; Qi et al., 2017).
eNOS Inactivation
eNOS inactivation, due to impaired dimerization and phosphorylation, has been suggested to play an important role in experimental DN (Cheng et al., 2012). In mice, resistant for adriamycin-induced glomerulopathy, administration of adriamycin induced massive proteinuria and severe glomerulosclerosis upon eNOS deficiency. This observation shows that loss of eNOS increases the susceptibility for the development of adriamycin-induced nephropathy. GEnC dysfunction, observed as loss of CD31 and apoptosis, appeared 3 days after adriamycin administration. Notably, podocyte damage (i.e., loss of synaptopodin expression and apoptosis), occurred only after 7 days, demonstrating that GEnC dysfunction preceded podocyte damage in this model (Sun et al., 2013). Part of these in vivo results could be explained by adriamycin’s ability to induce inflammatory effects (Abou El Hassan et al., 2003). In line with these findings it has been shown that eNOS prevents heparanase expression and the development of proteinuria in adriamycin-induced experimental FSGS (Garsen et al., 2016b). In vitro, conditioned medium from eNOS-overexpressing microvascular endothelial cells protected podocytes from TNF-α-induced synaptopodin loss, suggesting that “healthy” GEnCs protect podocytes from an inflammatory insult in a paracrine manner by secreting protective mediators. Which mediators are secreted by GEnCs and how these mediators affect podocytes is not known (Sun et al., 2013).
Disturbed Crosstalk in the VEGFA-eNOS/NO-ET-1
Disturbances in paracrine signaling of VEGFA, eNOS/NO, and ET-1 between podocytes to GEnCs are critical and may compromise glomerular integrity. Either increased or decreased VEGFA expression, decreased eNOS signaling and increased ET-1 signaling are all implicated in glomerular pathology. In mice, gain of VEGFA in podocytes and lack of eNOS causes the development of proteinuria and nodular glomerulosclerosis (Veron et al., 2014). Podocyte-specific deletion of VEGFA causes GEnC damage, observed as swelling of GEnCs, necrosis and culminating in capillary obliteration (Eremina et al., 2008) and loss of fenestrae (Eremina et al., 2003). Additionally, podocyte-specific deletion of VEGFA also causes a loss of GEnCs in diabetic mice (Sivaskandarajah et al., 2012). Whole-body deletion of VEGFR2 results in a loss of viable GEnCs (Sison et al., 2010). Also podocyte-specific VEGFA overexpression results in loss of GEnCs and collapse of capillary loops (Eremina et al., 2003) and causes advanced DN with endothelial swelling (Veron et al., 2011), suggesting the existence of a delicate balance between the protective and deleterious effects of VEGFA, depending on the strength of signaling. Deletion of eNOS causes GEnC dysfunction and subsequently podocyte damage (Yuen et al., 2012). Administration of NO to cultured podocytes increases the production of cyclic guanosine monophosphate (cGMP), which controls the cytoskeletal structure of podocytes and limits podocyte retraction (Sharma et al., 1992). Deletion of eNOS and decreased availability of NO probably causes decreased cGMP production and subsequent podocyte retraction and foot process effacement. Maintenance of endothelial eNOS levels by the essential eNOS cofactor tetrahydrobiopterin ameliorates DN (Kidokoro et al., 2013). Furthermore, treatment with sepiapterin, a stable precursor of the eNOS cofactor tetrahydrobiopterin or L-arginine, the nitric oxide precursor induces a correction of eNOS dimerization and phosphorylation and decreases albuminuria (Cheng et al., 2012). In a recent paper, it was shown that ET-1 induces heparanase expression in podocytes, which was associated with a reduced glomerular endothelial glycocalyx in experimental diabetes and which could be prevented in a podocyte-specific ETR deficient mouse model nephropathy (Garsen et al., 2016c). The mechanisms underlying the trafficking of podocyte-derived VEGFA and heparanase against the filtration direction remain to be identified, but may involve heparan sulfate present in the GBM.
These studies demonstrate that the VEGFA-eNOS/NO-ET-1 signaling pathway is important for intraglomerular cross-talk between podocytes and GEnCs, and the strength and direction of signaling is critical for glomerular health. Disturbed cross-talk causes glomerular damage. GEnCs are the first cells in contact with all circulating factors in the blood. It is therefore likely that GEnC dysfunction, culminating in altered secretion of signaling molecules, occurs prior to, and is in fact (partly) responsible for podocyte damage and activation of mesangial cells. GEnC dysfunction might therefore be a leading initiating factor in the development of both FSGS and DN.
Other Aberrant Molecular Signaling and Expression Patterns
LRG1 and Enhancement of TGF-β/ALK1 Signaling
Recently, transcriptome profiling of GEnCs obtained from diabetic mice showed increased gene expression of leucine-rich α-2-glycoprotein (LRG1) in early stages of DN (Fu et al., 2018). LRG1 is a protein present in the glomeruli and is predominantly expressed by GEnCs. LRG1 is involved in angiogenesis and the pathogenesis of DN by enhancement of endothelial Tumor Growth Factor β (TGF-β)/activin receptor-like kinase 1 (ALK1) signaling. TGF-β signaling has previously been found to be involved in the pathogenesis of DN by promoting cell hypertrophy, ECM accumulation in the mesangium, and increasing glomerular permeability (Chang et al., 2016). Global genetic ablation of LRG1 led to a reduction of oxidative damage and glomerular angiogenesis in diabetic mice. Concomitantly, podocyte foot process effacement, podocyte loss, proteinuria, and glomerulosclerosis were attenuated. These results exemplify that alterations in GEnC gene expression and molecular pathways in early disease mediate podocyte damage and glomerulopathy (Hong et al., 2019). How increased LRG1 expression and TGF-β signaling in GEnCs specifically relate to podocyte damage was not addressed in these studies.
GEnC-Derived Exosomes
As a consequence of high glucose concentration, GEnCs show an increased secretion of exosomes containing TGF-β1 mRNA. In vitro, these exosomes induced mesangial cells to proliferate and produce ECM (Wu et al., 2016) and caused the induction of epithelial-mesenchymal-transition in podocytes (Wu et al., 2017). Injection of exosomes, derived from high glucose-treated GEnCs in vitro, caused glomerulosclerosis in mice (Wu et al., 2016). These studies together suggest that high glucose-induced GEnC dysfunction increases the production of GEnC exosomes, which induce phenotypic changes in mesangial cells and podocytes in vitro, and culminate in glomerulosclerosis in vivo (Wu et al., 2016).
Hypoxia-Induced Dysregulation of GEnCs
DN is associated with renal cortical hypoxia (O’Neill et al., 2015). Hypoxia and concomitant dysregulation of hypoxia-regulated transcriptional mechanisms in GEnCs are associated with the pathogenetic mechanisms involved in both FSGS and DN development. Endothelial PAS domain-containing protein 1 (EPAS1) is an isoform of hypoxia inducible factor (HIF), also known as HIF-2α. Endothelial-specific deletion of EPAS1 induced the loss of GEnC fenestrations and enhanced endothelial swelling in experimental hypertension-induced secondary FSGS. Additionally, GEnC dysfunction was associated with podocyte foot process effacement and worsening of proteinuria and glomerulosclerosis. In the presence of hypertension and EPAS1, podocyte lesions were not observed, demonstrating that aberrant EPAS1-mediated endothelial signaling associates with podocyte damage and exacerbates FSGS (Luque et al., 2017). Potential mechanisms for aforementioned results include a direct effect of EPAS1 on endothelial-dependent vasoreactivity and modulation of glomerular pressure resulting in hyperfiltration, as mechanical stress is thought to contribute to FSGS. Hyperfiltration results in glomerular hypertrophy, culminating in loss of podocytes and aggravation of mechanical stress and glomerular damage. Furthermore, EPAS1 was previously shown to associate with the assembly of intercellular adherens junctions and enhanced endothelial barrier integrity (Gong et al., 2015). The involvement of dysregulation of hypoxia-associated mechanisms in GEnCs in the pathogenetic pathways leading to glomerular disease is further substantiated by a study showing that endothelial-specific knockout of hypoxia inducible factor 1α (HIF1α) prevents the development of proteinuria and collagen deposition in hypertensive FSGS (Luo et al., 2015). These and the previous mentioned results show that HIF1α is detrimental, whereas EPAS1/HIF2α confers protection in glomerular disease. An explanation could be that the target genes of HIF1α and HIF2α differ in a context-dependent manner (Dengler et al., 2014). Collectively, the aforementioned studies show that disturbed hypoxia-driven signaling in GEnCs contributes to the pathogenesis of glomerular damage in FSGS and DN.
GEnC Plasticity: Endothelial-to-Mesenchymal Transition
GEnC dysfunction can induce the process of endothelial-to-mesenchymal transition (EndMT). Whether EndMT is an initiating event in glomerulosclerosis, and to which extent EndMT contributes to glomerulosclerosis is not known. EndMT is a process in which endothelial cells show an abrogated endothelial phenotype (such as loss of the expression of endothelial cell markers CD31 and VE-cadherin) and loss of endothelial characteristics such as an increased vascular permeability. Loss of endothelial marker expression coincides with an increase of mesenchymal marker expression such as α-smooth muscle actin (αSMA) and fibroblast specific protein 1 (FSP-1), and the production of ECM proteins (Dejana et al., 2017). In general, endothelial cells are suggested to contribute to the number of activated fibroblasts via EndMT. EndMT most probably contributes to fibrosis and is observed in cardiac and cancer-related fibrosis (Zeisberg et al., 2007), fibro-proliferative vascular disease (Moonen et al., 2015), but also in experimental kidney disease as shown in streptozotocin (STZ)-induced DN, unilateral ureteral obstruction, and a mouse model for Alport’s syndrome (Zeisberg et al., 2008). In these models, ~30%–50% of the activated fibroblasts co-express the endothelial cell marker CD31 and mesenchymal markers, such as αSMA and FSP-1 (Zeisberg et al., 2008). In lineage tracing experiments in STZ-induced diabetic mice, interstitial endothelial cells acquired a more mesenchymal-like phenotype by expressing αSMA, already early in development of renal interstitial fibrosis (Li et al., 2009). Also in glomeruli of DN patients, EndMT is observed as demonstrated by co-expression of endothelial and mesenchymal markers (Peng et al., 2016; Liu et al., 2018). High glucose conditions and advanced oxidation protein products will stimulate GEnCs to undergo EndMT (Liang et al., 2016; Peng et al., 2016; Shang et al., 2017). Together, aforementioned observations provide evidence that GEnCs can acquire a mesenchymal-like phenotype and may contribute to glomerular fibrosis in DN. The process of EndMT is shown to be controlled by autophagy in endothelial cells (Patschan et al., 2016; Wang et al., 2017). In diabetic mice, deletion of autophagy in endothelial cells induced by the endothelial-specific genetic deletion of Autophagy-Related Gene 5 (ATG5) caused endothelial cell lesions, podocyte foot process broadening and effacement, and an increase of microalbuminuria. These results exemplify the tight intercellular cross-talk between GEnC and podocytes, in which GEnC dysfunction (induced by ATG5 deficiency) leads to podocyte injury (Lenoir et al., 2015).
Epigenetic Modifications: A Potential Mechanism Involved in GEnC Dysfunction
The above mentioned facets of GEnC dysfunction in FSGS and DN associate with altered gene and protein expression. A quiescent endothelial phenotype is harbored by tight regulation of the endothelial transcriptome, i.e., the full array of mRNA transcripts produced (Brooks et al., 2002; Passerini et al., 2004; Gimbrone and Garcia-Cardena, 2013). Epigenetic mechanisms are involved in this regulation of the transcriptome of cells (Eccleston et al., 2013). Epigenetic modifications can cause changes in gene expression, without changing the DNA sequence (Gibney and Nolan, 2010) and are self-perpetuating, dynamic, and reversible in response to the environment (Beckerman et al., 2014). Many factors can influence epigenetic profiles, including hyperglycemia, hypoxia, and inflammation (Lu et al., 2017). Epigenetic modifications can either be beneficial, or hamper GEnC function by changing the transcriptome, resulting in GEnC dysfunction and potentially disturbed cross-talk and pathogenesis of FSGS and DN.
Epigenetic modifications include DNA methylation and histone modifications. In general, DNA methylation is associated with gene repression by changing the biophysical characteristics of the DNA to bind transcription factors. DNA methylation can also inhibit gene expression via methyl binding proteins, which in turn recruit transcriptional co-repressors. DNA methylation at genes can modulate transcriptional elongation and alternative splicing (Gibney and Nolan, 2010; Lu et al., 2017).
In addition to DNA methylation, epigenetic mechanisms also include modifications of histones. The best-characterized histone modifications involve methylation, acetylation, and phosphorylation. Histone modifications stably alter the conformation of chromatin, and thereby either enhance or inhibit gene transcriptional activity depending on the type of modification and the position of the modified residue within the histone (Kouzarides, 2007; Berger et al., 2009). DN is associated with aberrant DNA methylation in proximal tubules and peripheral blood cells (Maghbooli et al., 2014), and DNA methylation is recently shown to be present in GEnCs (Fu et al., 2018). Histone modifications have previously been shown to be involved in the pathogenesis of DN and FSGS (Sun et al., 2017; Majumder et al., 2018), but not much is known about altered histone modification patterns in GEnCs in DN or FSGS. Recently, transcriptome profiling of GEnCs obtained from diabetic mice with early DN, showed that many of the genes with decreased expression were involved in epigenetic regulation, suggesting altered epigenetic regulation in GEnCs in early stages of DN (Fu et al., 2018). Lysine-specific demethylase 6A (KDM6a), also known as Ubiquitously Transcribed Tetratricopeptide Repeat X Chromosome (UTX) was one of the genes found to be downregulated. KDM6a is a histone demethylase that specifically demethylates lysine 27 of histone 3. Methylation of lysine 27 of histone 3 (H3K27me3), mediated by the methyltransferase Enhancer of Zeste Homolog 2 (EZH2), is associated with gene repression (Tan et al., 2014). The role of EZH2 and H3K27me3 in GEnCs in DN and FSGS is yet unknown. In podocytes, H3K27me3 was previously shown to be decreased in DN, which associated with the extent of podocyte damage due to activation of Notch signaling and loss of quiescence (Majumder et al., 2018). Previous studies showed that EZH2 plays a role in endothelial homeostasis and is a modulator of a number of endothelial cell functions, such as endothelial-leukocytes interactions and angiogenesis (Dreger et al., 2012; Maleszewska et al., 2016). This is indicative for a role of altered epigenetic modifications in GEnCs resulting in aberrant and pathologic gene expression contributing to the pathogenesis of DN. Alteration of epigenetic modifications is shown to be beneficial. For example, inhibition of the demethylases Jumonji C domain–containing demethylases (JMJD3) and UTX attenuated podocyte injury in diabetic mice (Majumder et al., 2018). Also in an unilateral ureteric obstruction mouse model, inhibition of EZH2 and H3K27me3 attenuated renal fibrosis (Zhou et al., 2016). Our current knowledge about the contribution of an altered epigenetic landscape to GEnC dysfunction and disturbed cross-talk in DN and FSGS is limited. Therefore, expanding our knowledge on the potential causative role of epigenetic modifications in GEnCs is highly needed. Herewith, specific mediators involved in epigenetic pathways involved in GEnC dysfunction and disturbed cross-talk can be considered potential targets for future therapies in the pathogenesis of DN and FSGS.
Summary and Future Perspectives
As outlined above, podocytes and mesangial cells have previously received a lot of attention in research on the pathogenesis of FSGS and DN. However, the studies summarized in this review show that GEnC dysfunction occurs in the early stages of FSGS and DN, and contributes to podocyte damage and mesangial activation, eventually culminating in glomerulosclerosis. Several of the studies described here show that GEnC dysfunction precedes podocyte damage, and is sufficient to develop proteinuria. This provides a new insight on the role of GEnCs in the early phase in development of FSGS and DN. GEnC dysfunction is characterized by a compromised endothelial glycocalyx, an inflammatory phenotype, mitochondrial damage and oxidative stress, aberrant signaling and EndMT, resulting in proteinuria, podocyte damage or loss, mesangial activation, and ultimately glomerulosclerosis (Figure 4). The glomerular endothelium poses a potential efficacious cellular target to pharmacologically halt disease development and progression in DN and FSGS. Aberrant gene expression patterns largely contribute to GEnC dysfunction and altered epigenetic mechanisms seem involved in this aberrant transcriptome. To expand our understanding of the cross-talk between GEnCs and other glomerular cells in health and disease, isolated systems could be useful, such as co-cultured cells and organoids. Co-culture systems of differentiated GEnCs and podocytes (Li et al., 2016) and organoids (Hale et al., 2018) with subsequent endothelial genetic and epigenetic characterization and manipulation could be instrumental for understanding the pathways involved in GEnC-podocyte cross-talk. Until now, the knowledge of the epigenetic mechanisms involved in GEnC dysfunction in DN and FSGS is scarce and needs to be expanded.
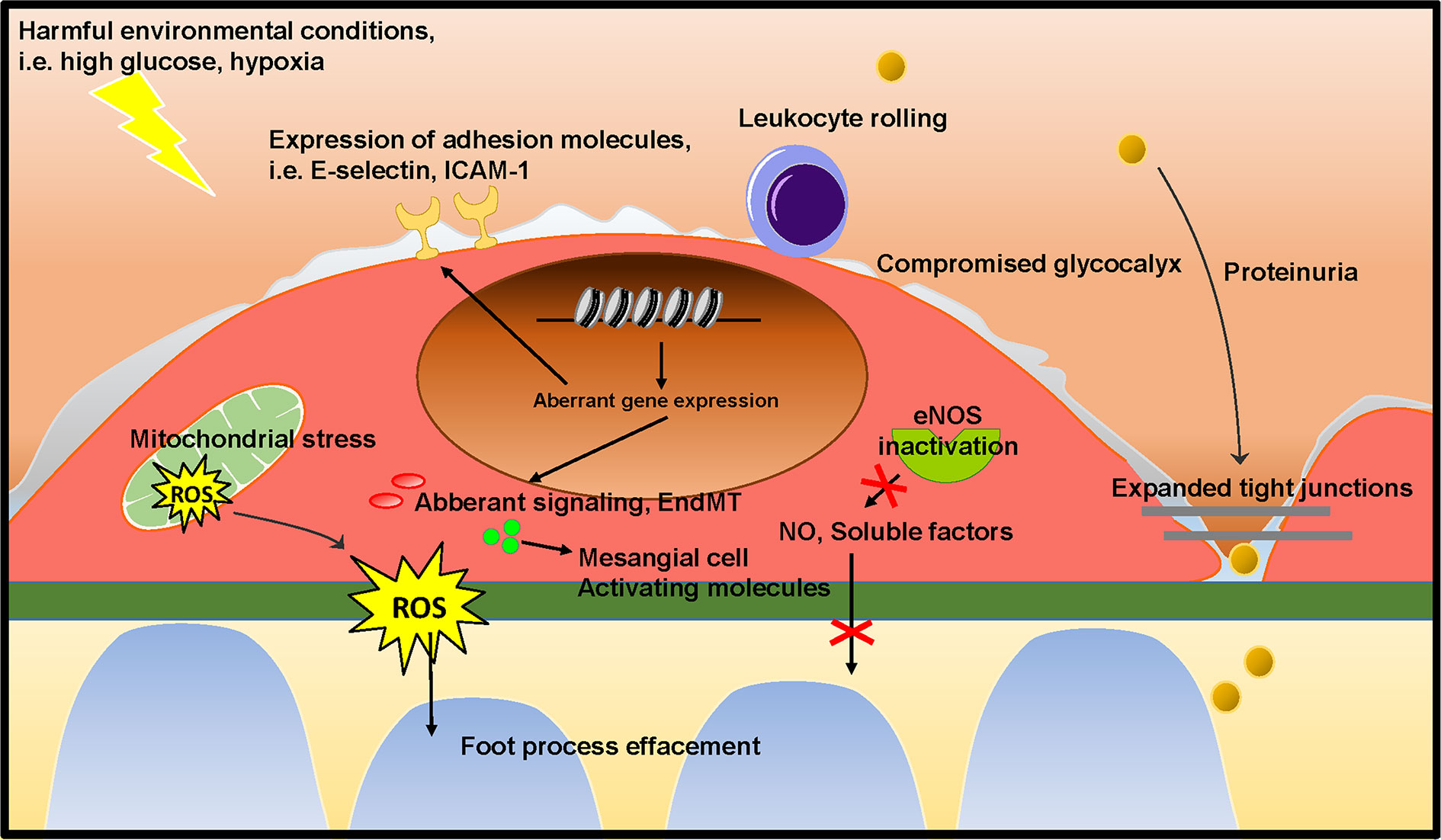
Figure 4 Proposed mechanism on the role of GEnC in the development of glomerular sclerotic diseases. Harmful environmental conditions, such as hyperglycemia and hypoxia cause GEnC dysfunction. GEnC dysfunction is characterized by a compromised endothelial glycocalyx, an inflammatory phenotype, mitochondrial damage and oxidative stress, aberrant signaling and EndMT, resulting in proteinuria, podocyte damage or loss, mesangial activation, and ultimately glomerulosclerosis.
Transcriptome profiling of GEnCs in DN and FSGS is of utmost importance to identify aberrantly expressed genes and associated regulatory pathways. Epigenomic databases, such as encyclopedia of DNA elements (ENCODE), in which chromatin modifications on both DNA and histone proteins are mapped in various cell lines (Consortium, 2012), could reveal potential epigenetic modifications responsible for aberrant expression patterns. Cell-specific delivery is needed to therapeutically intervene in the epigenetic mechanisms involved in GEnC dysfunction to avoid off-target cell effects. The identification of epigenetic mechanisms involved in GEnC dysfunction can effectively be studied with CRISPR-Cas9 technology in vitro (Adli, 2018). However, cell-specific delivery of CRISPR-Cas is still a huge challenge (Adli, 2018). The delivery of nucleotides, such as siRNAs therefore is an approach with great potential for intervention in GEnCs. As epigenetic modifications are regulated by epigenetic enzymes, intervening in the expression of epigenetic enzymes can influence the amount of epigenetic modifications. Endothelial cell-specific delivery of siRNA is feasible and this strategy has previously been used to successfully deliver siRNA to inflamed endothelial cells, including specifically GEnCs, and to decrease the expression of the target gene of interest (Kowalski et al., 2014; Choi et al., 2017).
Author Contributions
MS searched articles, drafted and wrote the manuscript. MS, JK, GK, and J-LH created the outline of the manuscript. JK, JB, MH, JV, GK, and J-LH supervised the manuscript writing and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was financially supported by the Dutch Kidney Foundation (grant 15OP13).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Illustrations were assembled using the Motifolio Biology Illustration Toolkit (motifolio.com).
References
Abou El Hassan, M. A., Verheul, H. M., Jorna, A. S., Schalkwijk, C., van Bezu, J., van der Vijgh, W. J., et al. (2003). The new cardioprotector Monohydroxyethylrutoside protects against doxorubicin-induced inflammatory effects in vitro. Br. J. Cancer 89 (2), 357–362. doi: 10.1038/sj.bjc.6601022
Adli, M. (2018). The CRISPR tool kit for genome editing and beyond. Nat. Commun. 9 (1), 1911. doi: 10.1038/s41467-018-04252-2
Barton, M., Sorokin, A. (2015). Endothelin and the glomerulus in chronic kidney disease. Semin. Nephrol. 35 (2), 156–167. doi: 10.1016/j.semnephrol.2015.02.005
Barton, M., Yanagisawa, M. (2008). Endothelin: 20 years from discovery to therapy. Can. J. Physiol. Pharmacol. 86 (8), 485–498. doi: 10.1139/Y08-059
Beckerman, P., Ko, Y. A., Susztak, K. (2014). Epigenetics: a new way to look at kidney diseases. Nephrol. Dial Transplant. 29 (10), 1821–1827. doi: 10.1093/ndt/gfu026
Berger, S. L., Kouzarides, T., Shiekhattar, R., Shilatifard, A. (2009). An operational definition of epigenetics. Genes Dev. 23 (7), 781–783. doi: 10.1101/gad.1787609
Boels, M. G., Lee, D. H., van den Berg, B. M., Dane, M. J., van der Vlag, J., Rabelink, T. J. (2013). The endothelial glycocalyx as a potential modifier of the hemolytic uremic syndrome. Eur. J. Intern Med. 24 (6), 503–509. doi: 10.1016/j.ejim.2012.12.016
Boels, M. G., Avramut, M. C., Koudijs, A., Dane, M. J., Lee, D. H., van der Vlag, J., et al. (2016). Atrasentan Reduces Albuminuria by Restoring the Glomerular Endothelial Glycocalyx Barrier in Diabetic Nephropathy. Diabetes. 65 (8), 2429–2439. doi: 10.2337/db15-1413
Boo, Y. C., Hwang, J., Sykes, M., Michell, B. J., Kemp, B. E., Lum, H., et al. (2002). Shear stress stimulates phosphorylation of eNOS at Ser(635) by a protein kinase A-dependent mechanism. Am. J. Physiol. Heart Circ. Physiol. 283 (5), H1819–H1828. doi: 10.1152/ajpheart.00214.2002
Boustany-Kari, C. M., Harrison, P. C., Chen, H., Lincoln, K. A., Qian, H. S., Clifford, H., et al. (2016). A Soluble Guanylate Cyclase Activator Inhibits the Progression of Diabetic Nephropathy in the ZSF1 Rat. J. Pharmacol. Exp. Ther. 356 (3), 712–719. doi: 10.1124/jpet.115.230706
Brooks, A. R., Lelkes, P. I., Rubanyi, G. M. (2002). Gene expression profiling of human aortic endothelial cells exposed to disturbed flow and steady laminar flow. Physiol. Genomics 9 (1), 27–41. doi: 10.1152/physiolgenomics.00075.2001
Chang, A. S., Hathaway, C. K., Smithies, O., Kakoki, M. (2016). Transforming growth factor-beta1 and diabetic nephropathy. Am. J. Physiol. Renal Physiol. 310 (8), F689–FF96. doi: 10.1152/ajprenal.00502.2015
Cheng, H., Wang, H., Fan, X., Paueksakon, P., Harris, R. C. (2012). Improvement of endothelial nitric oxide synthase activity retards the progression of diabetic nephropathy in db/db mice. Kidney Int. 82 (11), 1176–1183. doi: 10.1038/ki.2012.248
Choi, M., Schreiber, A., Eulenberg-Gustavus, C., Scheidereit, C., Kamps, J., Kettritz, R. (2017). Endothelial NF-kappaB Blockade Abrogates ANCA-Induced GN. J. Am. Soc. Nephrol. 28 (11), 3191–3204. doi: 10.1681/ASN.2016060690
Collino, F., Bussolati, B., Gerbaudo, E., Marozio, L., Pelissetto, S., Benedetto, C., et al. (2008). Preeclamptic sera induce nephrin shedding from podocytes through endothelin-1 release by endothelial glomerular cells. Am. J. Physiol. Renal Physiol. 294 (5), F1185–F1194. doi: 10.1152/ajprenal.00442.2007
Collins, A. J., Foley, R. N., Herzog, C., Chavers, B., Gilbertson, D., Herzog, C., et al. (2013). US Renal Data System 2012 Annual Data Report. Am. J. Kidney Dis. 61 (1 Suppl 1), A7, e1–A7,476. doi: 10.1053/j.ajkd.2012.11.031
Collins, A. J., Foley, R. N., Chavers, B., Gilbertson, D., Herzog, C., Ishani, A., et al. (2014). US Renal Data System 2013 Annual Data Report. Am. J. Kidney Dis. 63 (1 Suppl), A7. doi: 10.1053/j.ajkd.2013.11.001
Consortium, E. P. (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489 (7414), 57–74. doi: 10.1038/nature11247
Constantinescu, A. A., Vink, H., Spaan, J. A. (2003). Endothelial cell glycocalyx modulates immobilization of leukocytes at the endothelial surface. Arterioscler. Thromb. Vasc. Biol. 23 (9), 1541–1547. doi: 10.1161/01.ATV.0000085630.24353.3D
Daehn, I., Casalena, G., Zhang, T., Shi, S., Fenninger, F., Barasch, N., et al. (2014). Endothelial mitochondrial oxidative stress determines podocyte depletion in segmental glomerulosclerosis. J. Clin. Invest. 124 (4), 1608–1621. doi: 10.1172/JCI71195
Dane, M. J., van den Berg, B. M., Avramut, M. C., Faas, F. G., van der Vlag, J., Rops, A. L., et al. (2013). Glomerular endothelial surface layer acts as a barrier against albumin filtration. Am. J. Pathol. 182 (5), 1532–1540. doi: 10.1016/j.ajpath.2013.01.049
Dane, M. J., van den Berg, B. M., Lee, D. H., Boels, M. G., Tiemeier, G. L., Avramut, M. C., et al. (2015). A microscopic view on the renal endothelial glycocalyx. Am. J. Physiol. Renal Physiol. 308 (9), F956–F966. doi: 10.1152/ajprenal.00532.2014
De Vriese, A. S., Sethi, S., Nath, K. A., Glassock, R. J., Fervenza, F. C. (2018). Differentiating Primary, Genetic, and Secondary FSGS in Adults: A Clinicopathologic Approach. J. Am. Soc. Nephrol. 29 (3), 759–774. doi: 10.1681/ASN.2017090958
Dejana, E., Hirschi, K. K., Simons, M. (2017). The molecular basis of endothelial cell plasticity. Nat. Commun. 8, 14361. doi: 10.1038/ncomms14361
Dekker, R. J., Boon, R. A., Rondaij, M. G., Kragt, A., Volger, O. L., Elderkamp, Y. W., et al. (2006). KLF2 provokes a gene expression pattern that establishes functional quiescent differentiation of the endothelium. Blood 107 (11), 4354–4363. doi: 10.1182/blood-2005-08-3465
Dengler, V. L., Galbraith, M., Espinosa, J. M. (2014). Transcriptional regulation by hypoxia inducible factors. Crit. Rev. Biochem. Mol. Biol. 49 (1), 1–15. doi: 10.3109/10409238.2013.838205
Dhaun, N., Webb, D. J., Kluth, D. C. (2012). Endothelin-1 and the kidney–beyond BP. Br. J. Pharmacol. 167 (4), 720–731. doi: 10.1111/j.1476-5381.2012.02070.x
Dogne, S., Flamion, B., Caron, N. (2018). Endothelial Glycocalyx as a Shield Against Diabetic Vascular Complications: Involvement of Hyaluronan and Hyaluronidases. Arterioscler. Thromb. Vasc. Biol. 38 (7), 1427–1439. doi: 10.1161/ATVBAHA.118.310839
Dong, F., Zhang, X., Wold, L. E., Ren, Q., Zhang, Z., Ren, J. (2005). Endothelin-1 enhances oxidative stress, cell proliferation and reduces apoptosis in human umbilical vein endothelial cells: role of ETB receptor, NADPH oxidase and caveolin-1. Br. J. Pharmacol. 145 (3), 323–333. doi: 10.1038/sj.bjp.0706193
Dreger, H., Ludwig, A., Weller, A., Stangl, V., Baumann, G., Meiners, S., et al. (2012). Epigenetic regulation of cell adhesion and communication by enhancer of zeste homolog 2 in human endothelial cells. Hypertension 60 (5), 1176–1183. doi: 10.1161/HYPERTENSIONAHA.112.191098
Ebefors, K., Wiener, R. J., Yu, L., Azeloglu, E. U., Yi, Z., Jia, F., et al. (2019). Endothelin receptor-A mediates degradation of the glomerular endothelial surface layer via pathologic crosstalk between activated podocytes and glomerular endothelial cells. Kidney Int. 96 (4), 957–970. doi: 10.1016/j.kint.2019.05.007
Eccleston, A., Cesari, F., Skipper, M. (2013). Transcription and epigenetics. Nature. 502 (7472), 461. doi: 10.1038/502461a
Eleftheriadis, T., Antoniadi, G., Pissas, G., Liakopoulos, V., Stefanidis, I. (2013). The renal endothelium in diabetic nephropathy. Ren Fail. 35 (4), 592–599. doi: 10.3109/0886022X.2013.773836
Eremina, V., Sood, M., Haigh, J., Nagy, A., Lajoie, G., Ferrara, N., et al. (2003). Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J. Clin. Invest. 111 (5), 707–716. doi: 10.1172/JCI17423
Eremina, V., Jefferson, J. A., Kowalewska, J., Hochster, H., Haas, M., Weisstuch, J., et al. (2008). VEGF inhibition and renal thrombotic microangiopathy. N Engl. J. Med. 358 (11), 1129–1136. doi: 10.1056/NEJMoa0707330
Feliers, D., Chen, X., Akis, N., Choudhury, G. G., Madaio, M., Kasinath, B. S. (2005). VEGF regulation of endothelial nitric oxide synthase in glomerular endothelial cells. Kidney Int. 68 (4), 1648–1659. doi: 10.1111/j.1523-1755.2005.00575.x
Fioretto, P., Mauer, M. (2007). Histopathology of diabetic nephropathy. Semin. Nephrol. 27 (2), 195–207. doi: 10.1016/j.semnephrol.2007.01.012
Florian, J. A., Kosky, J. R., Ainslie, K., Pang, Z., Dull, R. O., Tarbell, J. M. (2003). Heparan sulfate proteoglycan is a mechanosensor on endothelial cells. Circ. Res. 93 (10), e136–e142. doi: 10.1161/01.RES.0000101744.47866.D5
Fogo, A. B. (2007). Mechanisms of progression of chronic kidney disease. Pediatr. Nephrol. 22 (12), 2011–2022. doi: 10.1007/s00467-007-0524-0
Forbes, J. M., Thallas, V., Thomas, M. C., Founds, H. W., Burns, W. C., Jerums, G., et al. (2003). The breakdown of preexisting advanced glycation end products is associated with reduced renal fibrosis in experimental diabetes. FASEB J. 17 (12), 1762–1764. doi: 10.1096/fj.02-1102fje
Friden, V., Oveland, E., Tenstad, O., Ebefors, K., Nystrom, J., Nilsson, U. A., et al. (2011). The glomerular endothelial cell coat is essential for glomerular filtration. Kidney Int. 79 (12), 1322–1330. doi: 10.1038/ki.2011.58
Fu, J., Lee, K., Chuang, P. Y., Liu, Z., He, J. C. (2015). Glomerular endothelial cell injury and cross talk in diabetic kidney disease. Am. J. Physiol. Renal Physiol. 308 (4), F287–F297. doi: 10.1152/ajprenal.00533.2014
Fu, J., Wei, C., Zhang, W., Schlondorff, D., Wu, J., Cai, M., et al. (2018). Gene expression profiles of glomerular endothelial cells support their role in the glomerulopathy of diabetic mice. Kidney Int. 94 (2), 326–345. doi: 10.1016/j.kint.2018.02.028
Furuta, T., Saito, T., Ootaka, T., Soma, J., Obara, K., Abe, K., et al. (1993). The role of macrophages in diabetic glomerulosclerosis. Am. J. Kidney Dis. 21 (5), 480–485. doi: 10.1016/S0272-6386(12)80393-3
Garsen, M., Rops, A. L., Rabelink, T. J., Berden, J. H., van der Vlag, J. (2014). The role of heparanase and the endothelial glycocalyx in the development of proteinuria. Nephrol. Dial Transplant. 29 (1), 49–55. doi: 10.1093/ndt/gft410
Garsen, M., Benner, M., Dijkman, H. B., van Kuppevelt, T. H., Li, J. P., Rabelink, T. J., et al. (2016a). Heparanase Is Essential for the Development of Acute Experimental Glomerulonephritis. Am. J. Pathol. 186 (4), 805–815. doi: 10.1016/j.ajpath.2015.12.008
Garsen, M., Rops, A. L., Li, J., van Beneden, K., van den Branden, C., Berden, J. H., et al. (2016b). Endothelial Nitric Oxide Synthase Prevents Heparanase Induction and the Development of Proteinuria. PloS One 11 (8), e0160894. doi: 10.1371/journal.pone.0160894
Garsen, M., Lenoir, O., Rops, A. L., Dijkman, H. B., Willemsen, B., van Kuppevelt, T. H., et al. (2016c). Endothelin-1 Induces Proteinuria by Heparanase-Mediated Disruption of the Glomerular Glycocalyx. J. Am. Soc. Nephrol. 27 (12), 3545–3551. doi: 10.1681/ASN.2015091070
Gibney, E. R., Nolan, C. M. (2010). Epigenetics and gene expression. Heredity (Edinb). 105 (1), 4–13. doi: 10.1038/hdy.2010.54
Gien, J., Tseng, N., Seedorf, G., Roe, G., Abman, S. H. (2013). Endothelin-1 impairs angiogenesis in vitro through Rho-kinase activation after chronic intrauterine pulmonary hypertension in fetal sheep. Pediatr. Res. 73 (3), 252–262. doi: 10.1038/pr.2012.177
Gil, N., Goldberg, R., Neuman, T., Garsen, M., Zcharia, E., Rubinstein, A. M., et al. (2012). Heparanase is essential for the development of diabetic nephropathy in mice. Diabetes 61 (1), 208–216. doi: 10.2337/db11-1024
Gimbrone, M. A., Jr., Garcia-Cardena, G. (2013). Vascular endothelium, hemodynamics, and the pathobiology of atherosclerosis. Cardiovasc. Pathol. 22 (1), 9–15. doi: 10.1016/j.carpath.2012.06.006
Gong, H., Rehman, J., Tang, H., Wary, K., Mittal, M., Chaturvedi, P., et al. (2015). HIF2alpha signaling inhibits adherens junctional disruption in acute lung injury. J. Clin. Invest. 125 (2), 652–664. doi: 10.1172/JCI77701
Hale, L. J., Howden, S. E., Phipson, B., Lonsdale, A., Er, P. X., Ghobrial, I., et al. (2018). 3D organoid-derived human glomeruli for personalised podocyte disease modelling and drug screening. Nat. Commun. 9 (1), 5167. doi: 10.1038/s41467-018-07594-z
Hara, T., Ishida, T., Cangara, H. M., Hirata, K. (2009). Endothelial cell-selective adhesion molecule regulates albuminuria in diabetic nephropathy. Microvasc Res. 77 (3), 348–355. doi: 10.1016/j.mvr.2009.01.002
Haraldsson, B., Nystrom, J. (2012). The glomerular endothelium: new insights on function and structure. Curr. Opin. Nephrol. Hypertens. 21 (3), 258–263. doi: 10.1097/MNH.0b013e3283522e7a
Hegermann, J., Lunsdorf, H., Ochs, M., Haller, H. (2016). Visualization of the glomerular endothelial glycocalyx by electron microscopy using cationic colloidal thorium dioxide. Histochem Cell Biol. 145 (1), 41–51. doi: 10.1007/s00418-015-1378-3
Herman, W. H., Emancipator, S. N., Rhoten, R. L., Simonson, M. S. (1998). Vascular and glomerular expression of endothelin-1 in normal human kidney. Am. J. Physiol. 275 (1), F8–17. doi: 10.1152/ajprenal.1998.275.1.F8
Hirata, K., Shikata, K., Matsuda, M., Akiyama, K., Sugimoto, H., Kushiro, M., et al. (1998). Increased expression of selectins in kidneys of patients with diabetic nephropathy. Diabetologia 41 (2), 185–192. doi: 10.1007/s001250050888
Hirose, A., Tanikawa, T., Mori, H., Okada, Y., Tanaka, Y. (2010). Advanced glycation end products increase endothelial permeability through the RAGE/Rho signaling pathway. FEBS Lett. 584 (1), 61–66. doi: 10.1016/j.febslet.2009.11.082
Hong, Q., Zhang, L., Fu, J., Verghese, D. A., Chauhan, K., Nadkarni, G. N., et al. (2019). LRG1 Promotes Diabetic Kidney Disease Progression by Enhancing TGF-beta-Induced Angiogenesis. J. Am. Soc. Nephrol. 30 (4), 546–562. doi: 10.1681/ASN.2018060599
Jefferson, J. A., Shankland, S. J. (2014). The pathogenesis of focal segmental glomerulosclerosis. Adv. Chronic Kidney Dis. 21 (5), 408–416. doi: 10.1053/j.ackd.2014.05.009
Khimji, A. K., Rockey, D. C. (2010). Endothelin–biology and disease. Cell Signal. 22 (11), 1615–1625. doi: 10.1016/j.cellsig.2010.05.002
Kidokoro, K., Satoh, M., Channon, K. M., Yada, T., Sasaki, T., Kashihara, N. (2013). Maintenance of endothelial guanosine triphosphate cyclohydrolase I ameliorates diabetic nephropathy. J. Am. Soc. Nephrol. 24 (7), 1139–1150. doi: 10.1681/ASN.2012080783
Kiffel, J., Rahimzada, Y., Trachtman, H. (2011). Focal segmental glomerulosclerosis and chronic kidney disease in pediatric patients. Adv. Chronic Kidney Dis. 18 (5), 332–338. doi: 10.1053/j.ackd.2011.03.005
Kolarova, H., Ambruzova, B., Svihalkova Sindlerova, L., Klinke, A., Kubala, L. (2014). Modulation of endothelial glycocalyx structure under inflammatory conditions. Mediators Inflamm. 2014, 694312. doi: 10.1155/2014/694312
Kouzarides, T. (2007). Chromatin modifications and their function. Cell 128 (4), 693–705. doi: 10.1016/j.cell.2007.02.005
Kowalski, P. S., Zwiers, P. J., Morselt, H. W., Kuldo, J. M., Leus, N. G., Ruiters, M. H., et al. (2014). Anti-VCAM-1 SAINT-O-Somes enable endothelial-specific delivery of siRNA and downregulation of inflammatory genes in activated endothelium in vivo. J. Control Release 176, 64–75. doi: 10.1016/j.jconrel.2013.12.029
Kuwabara, A., Satoh, M., Tomita, N., Sasaki, T., Kashihara, N. (2010). Deterioration of glomerular endothelial surface layer induced by oxidative stress is implicated in altered permeability of macromolecules in Zucker fatty rats. Diabetologia. 53 (9), 2056–2065. doi: 10.1007/s00125-010-1810-0
Lenoir, O., Milon, M., Virsolvy, A., Henique, C., Schmitt, A., Masse, J. M., et al. (2014). Direct action of endothelin-1 on podocytes promotes diabetic glomerulosclerosis. J. Am. Soc. Nephrol. 25 (5), 1050–1062. doi: 10.1681/ASN.2013020195
Lenoir, O., Jasiek, M., Henique, C., Guyonnet, L., Hartleben, B., Bork, T., et al. (2015). Endothelial cell and podocyte autophagy synergistically protect from diabetes-induced glomerulosclerosis. Autophagy 11 (7), 1130–1145. doi: 10.1080/15548627.2015.1049799
Ley, K., Laudanna, C., Cybulsky, M. I., Nourshargh, S. (2007). Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat. Rev. Immunol. 7 (9), 678–689. doi: 10.1038/nri2156
Li, J., Qu, X., Bertram, J. F. (2009). Endothelial-myofibroblast transition contributes to the early development of diabetic renal interstitial fibrosis in streptozotocin-induced diabetic mice. Am. J. Pathol. 175 (4), 1380–1388. doi: 10.2353/ajpath.2009.090096
Li, M., Corbelli, A., Watanabe, S., Armelloni, S., Ikehata, M., Parazzi, V., et al. (2016). Three-dimensional podocyte-endothelial cell co-cultures: Assembly, validation, and application to drug testing and intercellular signaling studies. Eur. J. Pharm. Sci. 86, 1–12. doi: 10.1016/j.ejps.2016.02.013
Liang, X., Duan, N., Wang, Y., Shu, S., Xiang, X., Guo, T., et al. (2016). Advanced oxidation protein products induce endothelial-to-mesenchymal transition in human renal glomerular endothelial cells through induction of endoplasmic reticulum stress. J. Diabetes Complications 30 (4), 573–579. doi: 10.1016/j.jdiacomp.2016.01.009
Lim, B. J., Yang, J. W., Do, W. S., Fogo, A. B. (2016). Pathogenesis of Focal Segmental Glomerulosclerosis. J. Pathol. Transl. Med. 50 (6), 405–410. doi: 10.4132/jptm.2016.09.21
Lin, C. W., Mostafa, N. M., Andress, D. L., Brennan, J. J., CE, K., Awni, W. M. (2018). Relationship Between Atrasentan Concentrations and Urinary Albumin to Creatinine Ratio in Western and Japanese Patients With Diabetic Nephropathy. Clin. Ther. 40 (2), 242–251. doi: 10.1016/j.clinthera.2017.07.011
Liu, F., Zhang, S., Xu, R., Gao, S., Yin, J. (2018). Melatonin Attenuates Endothelial-to-Mesenchymal Transition of Glomerular Endothelial Cells via Regulating miR-497/ROCK in Diabetic Nephropathy. Kidney Blood Press Res. 43 (5), 1425–1436. doi: 10.1159/000493380
Lu, Z., Liu, N., Wang, F. (2017). Epigenetic Regulations in Diabetic Nephropathy. J. Diabetes Res. 2017, 7805058. doi: 10.1155/2017/7805058
Luo, R., Zhang, W., Zhao, C., Zhang, Y., Wu, H., Jin, J., et al. (2015). Elevated Endothelial Hypoxia-Inducible Factor-1alpha Contributes to Glomerular Injury and Promotes Hypertensive Chronic Kidney Disease. Hypertension 66 (1), 75–84. doi: 10.1161/HYPERTENSIONAHA.115.05578
Luque, Y., Lenoir, O., Bonnin, P., Hardy, L., Chipont, A., Placier, S., et al. (2017). Endothelial Epas1 Deficiency Is Sufficient To Promote Parietal Epithelial Cell Activation and FSGS in Experimental Hypertension. J. Am. Soc. Nephrol. 28 (12), 3563–3578. doi: 10.1681/ASN.2016090960
Maghbooli, Z., Larijani, B., Emamgholipour, S., Amini, M., Keshtkar, A., Pasalar, P. (2014). Aberrant DNA methylation patterns in diabetic nephropathy. J. Diabetes Metab. Disord. 13, 69. doi: 10.1186/2251-6581-13-69
Majumder, S., Thieme, K., Batchu, S. N., Alghamdi, T. A., Bowskill, B. B., Kabir, M. G., et al. (2018). Shifts in podocyte histone H3K27me3 regulate mouse and human glomerular disease. J. Clin. Invest. 128 (1), 483–499. doi: 10.1172/JCI95946
Maleszewska, M., Vanchin, B., Harmsen, M. C., Krenning, G. (2016). The decrease in histone methyltransferase EZH2 in response to fluid shear stress alters endothelial gene expression and promotes quiescence. Angiogenesis. 19 (1), 9–24. doi: 10.1007/s10456-015-9485-2
Meuwese, M. C., Broekhuizen, L. N., Kuikhoven, M., Heeneman, S., Lutgens, E., Gijbels, M. J., et al. (2010). Endothelial surface layer degradation by chronic hyaluronidase infusion induces proteinuria in apolipoprotein E-deficient mice. PloS One 5 (12), e14262. doi: 10.1371/journal.pone.0014262
Moonen, J. R., Lee, E. S., Schmidt, M., Maleszewska, M., Koerts, J. A., Brouwer, L. A., et al. (2015). Endothelial-to-mesenchymal transition contributes to fibro-proliferative vascular disease and is modulated by fluid shear stress. Cardiovasc. Res. 108 (3), 377–386. doi: 10.1093/cvr/cvv175
Mooyaart, A. L., Valk, E. J., van Es, L. A., Bruijn, J. A., de Heer, E., Freedman, B. I., et al. (2011). Genetic associations in diabetic nephropathy: a meta-analysis. Diabetologia 54 (3), 544–553. doi: 10.1007/s00125-010-1996-1
Moreno, J. A., Gomez-Guerrero, C., Mas, S., Sanz, A. B., Lorenzo, O., Ruiz-Ortega, M., et al. (2018). Targeting inflammation in diabetic nephropathy: a tale of hope. Expert Opin. Invest. Drugs 27 (11), 917–930. doi: 10.1080/13543784.2018.1538352
Morita, M., Mii, A., Shimizu, A., Yasuda, F., Shoji, J., Masuda, Y., et al. (2015). Glomerular endothelial cell injury and focal segmental glomerulosclerosis lesion in idiopathic membranous nephropathy. PloS One 10 (4), e0116700. doi: 10.1371/journal.pone.0116700
Nagasu, H., Satoh, M., Kiyokage, E., Kidokoro, K., Toida, K., Channon, K. M., et al. (2016). Activation of endothelial NAD(P)H oxidase accelerates early glomerular injury in diabetic mice. Lab. Invest. 96 (1), 25–36. doi: 10.1038/labinvest.2015.128
Najafian, B., Fogo, A. B., Lusco, M. A., Alpers, C. E. (2015). AJKD Atlas of Renal Pathology: diabetic nephropathy. Am. J. Kidney Dis. 66 (5), e37–e38. doi: 10.1053/j.ajkd.2015.08.010
Navarro-Gonzalez, J. F., Mora-Fernandez, C., Muros de Fuentes, M., Garcia-Perez, J. (2011). Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nat. Rev. Nephrol. 7 (6), 327–340. doi: 10.1038/nrneph.2011.51
Nieuwdorp, M., Mooij, H. L., Kroon, J., Atasever, B., Spaan, J. A., Ince, C., et al. (2006a). Endothelial glycocalyx damage coincides with microalbuminuria in type 1 diabetes. Diabetes 55 (4), 1127–1132. doi: 10.2337/diabetes.55.04.06.db05-1619
Nieuwdorp, M., van Haeften, T. W., Gouverneur, M. C., Mooij, H. L., van Lieshout, M. H., Levi, M., et al. (2006b). Loss of endothelial glycocalyx during acute hyperglycemia coincides with endothelial dysfunction and coagulation activation in vivo. Diabetes 55 (2), 480–486. doi: 10.2337/diabetes.55.02.06.db05-1103
Ohnesorge, N., Viemann, D., Schmidt, N., Czymai, T., Spiering, D., Schmolke, M., et al. (2010). Erk5 activation elicits a vasoprotective endothelial phenotype via induction of Kruppel-like factor 4 (KLF4). J. Biol. Chem. 285 (34), 26199–26210. doi: 10.1074/jbc.M110.103127
O’Neill, J., Fasching, A., Pihl, L., Patinha, D., Franzen, S., Palm, F. (2015). Acute SGLT inhibition normalizes O2 tension in the renal cortex but causes hypoxia in the renal medulla in anaesthetized control and diabetic rats. Am. J. Physiol. Renal Physiol. 309 (3), F227–F234. doi: 10.1152/ajprenal.00689.2014
Passerini, A. G., Polacek, D. C., Shi, C., Francesco, N. M., Manduchi, E., Grant, G. R., et al. (2004). Coexisting proinflammatory and antioxidative endothelial transcription profiles in a disturbed flow region of the adult porcine aorta. Proc. Natl. Acad. Sci. U.S.A. 101 (8), 2482–2487. doi: 10.1073/pnas.0305938101
Patrakka, J., Tryggvason, K. (2010). Molecular make-up of the glomerular filtration barrier. Biochem. Biophys. Res. Commun. 396 (1), 164–169. doi: 10.1016/j.bbrc.2010.04.069
Patschan, D., Schwarze, K., Henze, E., Patschan, S., Muller, G. A. (2016). Endothelial autophagy and Endothelial-to-Mesenchymal Transition (EndoMT) in eEPC treatment of ischemic AKI. J. Nephrol. 29 (5), 637–644. doi: 10.1007/s40620-015-0222-0
Peng, H., Li, Y., Wang, C., Zhang, J., Chen, Y., Chen, W., et al. (2016). ROCK1 Induces Endothelial-to-Mesenchymal Transition in Glomeruli to Aggravate Albuminuria in Diabetic Nephropathy. Sci. Rep. 6, 20304. doi: 10.1038/srep20304
Qi, H., Casalena, G., Shi, S., Yu, L., Ebefors, K., Sun, Y., et al. (2017). Glomerular Endothelial Mitochondrial Dysfunction Is Essential and Characteristic of Diabetic Kidney Disease Susceptibility. Diabetes 66 (3), 763–778. doi: 10.2337/db16-0695
Rao, J., Ye, Z., Tang, H., Wang, C., Peng, H., Lai, W., et al. (2017). The RhoA/ROCK Pathway Ameliorates Adhesion and Inflammatory Infiltration Induced by AGEs in Glomerular Endothelial Cells. Sci. Rep. 7, 39727. doi: 10.1038/srep39727
Reidy, K., Kaskel, F. J. (2007). Pathophysiology of focal segmental glomerulosclerosis. Pediatr. Nephrol. 22 (3), 350–354. doi: 10.1007/s00467-006-0357-2
Reitsma, S., Slaaf, D. W., Vink, H., van Zandvoort, M. A., oude Egbrink, M. G. (2007). The endothelial glycocalyx: composition, functions, and visualization. Pflugers Arch. 454 (3), 345–359. doi: 10.1007/s00424-007-0212-8
Rossing, P., Persson, F., Frimodt-Moller, M. (2018). Prognosis and treatment of diabetic nephropathy: Recent advances and perspectives. Nephrol. Ther. 14 Suppl 1, S31–SS7. doi: 10.1016/j.nephro.2018.02.007
Ryan, G. B., Karnovsky, M. J. (1976). Distribution of endogenous albumin in the rat glomerulus: role of hemodynamic factors in glomerular barrier function. Kidney Int. 9 (1), 36–45. doi: 10.1038/ki.1976.5
Satchell, S. (2013). The role of the glomerular endothelium in albumin handling. Nat. Rev. Nephrol. 9 (12), 717–725. doi: 10.1038/nrneph.2013.197
Satoh, M., Kobayashi, S., Kuwabara, A., Tomita, N., Sasaki, T., Kashihara, N. (2010). In vivo visualization of glomerular microcirculation and hyperfiltration in streptozotocin-induced diabetic rats. Microcirculation. 17 (2), 103–112. doi: 10.1111/j.1549-8719.2009.00010.x
Scott, R. P., Quaggin, S. E. (2015). Review series: The cell biology of renal filtration. J. Cell Biol. 209 (2), 199–210. doi: 10.1083/jcb.201410017
Shang, J., Zhang, Y., Jiang, Y., Li, Z., Duan, Y., Wang, L., et al. (2017). NOD2 promotes endothelial-to-mesenchymal transition of glomerular endothelial cells via MEK/ERK signaling pathway in diabetic nephropathy. Biochem. Biophys. Res. Commun. 484 (2), 435–441. doi: 10.1016/j.bbrc.2017.01.155
Sharma, R., Lovell, H. B., Wiegmann, T. B., Savin, V. J. (1992). Vasoactive substances induce cytoskeletal changes in cultured rat glomerular epithelial cells. J. Am. Soc. Nephrol. 3 (5), 1131–1138.
Singh, A., Satchell, S. C., Neal, C. R., McKenzie, E. A., Tooke, J. E., Mathieson, P. W. (2007). Glomerular endothelial glycocalyx constitutes a barrier to protein permeability. J. Am. Soc. Nephrol. 18 (11), 2885–2893. doi: 10.1681/ASN.2007010119
Singh, A., Friden, V., Dasgupta, I., Foster, R. R., Welsh, G. I., Tooke, J. E., et al. (2011). High glucose causes dysfunction of the human glomerular endothelial glycocalyx. Am. J. Physiol. Renal Physiol. 300 (1), F40–F48. doi: 10.1152/ajprenal.00103.2010
Singh, A., Ramnath, R. D., Foster, R. R., Wylie, E. C., Friden, V., Dasgupta, I., et al. (2013). Reactive oxygen species modulate the barrier function of the human glomerular endothelial glycocalyx. PloS One 8 (2), e55852. doi: 10.1371/journal.pone.0055852
Sison, K., Eremina, V., Baelde, H., Min, W., Hirashima, M., Fantus, I. G., et al. (2010). Glomerular structure and function require paracrine, not autocrine, VEGF-VEGFR-2 signaling. J. Am. Soc. Nephrol. 21 (10), 1691–1701. doi: 10.1681/ASN.2010030295
Sivaskandarajah, G. A., Jeansson, M., Maezawa, Y., Eremina, V., Baelde, H. J., Quaggin, S. E. (2012). Vegfa protects the glomerular microvasculature in diabetes. Diabetes. 61 (11), 2958–2966. doi: 10.2337/DB11-1655
Slater, S. C., Ramnath, R. D., Uttridge, K., Saleem, M. A., Cahill, P. A., Mathieson, P. W., et al. (2012). Chronic exposure to laminar shear stress induces Kruppel-like factor 2 in glomerular endothelial cells and modulates interactions with co-cultured podocytes. Int. J. Biochem. Cell Biol. 44 (9), 1482–1490. doi: 10.1016/j.biocel.2012.05.020
Sud, N., Black, S. M. (2009). Endothelin-1 impairs nitric oxide signaling in endothelial cells through a protein kinase Cdelta-dependent activation of STAT3 and decreased endothelial nitric oxide synthase expression. DNA Cell Biol. 28 (11), 543–553. doi: 10.1089/dna.2009.0865
Sun, Y. B., Qu, X., Zhang, X., Caruana, G., Bertram, J. F., Li, J. (2013). Glomerular endothelial cell injury and damage precedes that of podocytes in adriamycin-induced nephropathy. PloS One 8 (1), e55027. doi: 10.1371/journal.pone.0055027
Sun, J., Wang, Y., Cui, W., Lou, Y., Sun, G., Zhang, D., et al. (2017). Role of Epigenetic Histone Modifications in Diabetic Kidney Disease Involving Renal Fibrosis. J. Diabetes Res. 2017, 7242384. doi: 10.1155/2017/7242384
Tan, J. Z., Yan, Y., Wang, X. X., Jiang, Y., Xu, H. E. (2014). EZH2: biology, disease, and structure-based drug discovery. Acta Pharmacol. Sin. 35 (2), 161–174. doi: 10.1038/aps.2013.161
Taneda, S., Honda, K., Ohno, M., Uchida, K., Nitta, K., Oda, H. (2015). Podocyte and endothelial injury in focal segmental glomerulosclerosis: an ultrastructural analysis. Virchows Arch. 467 (4), 449–458. doi: 10.1007/s00428-015-1821-9
Tarbell, J. M., Ebong, E. E. (2008). The endothelial glycocalyx: a mechano-sensor and -transducer. Sci. Signal. 1 (40), pt8. doi: 10.1126/scisignal.140pt8
van den Berg, B. M., Wang, G., Boels, M. G. S., Avramut, M. C., Jansen, E., Sol, W., et al. (2019). Glomerular Function and Structural Integrity Depend on Hyaluronan Synthesis by Glomerular Endothelium. J. Am. Soc. Nephrol. 30 (10), 1886–1897. doi: 10.1681/ASN.2019020192
Veron, D., Bertuccio, C. A., Marlier, A., Reidy, K., Garcia, A. M., Jimenez, J., et al. (2011). Podocyte vascular endothelial growth factor (Vegf(1)(6)(4)) overexpression causes severe nodular glomerulosclerosis in a mouse model of type 1 diabetes. Diabetologia 54 (5), 1227–1241. doi: 10.1007/s00125-010-2034-z
Veron, D., Aggarwal, P. K., Velazquez, H., Kashgarian, M., Moeckel, G., Tufro, A. (2014). Podocyte-specific VEGF-a gain of function induces nodular glomerulosclerosis in eNOS null mice. J. Am. Soc. Nephrol. 25 (8), 1814–1824. doi: 10.1681/ASN.2013070752
Wada, J., Makino, H. (2013). Inflammation and the pathogenesis of diabetic nephropathy. Clin. Sci. (Lond). 124 (3), 139–152. doi: 10.1042/CS20120198
Wang, J., Feng, Y., Wang, Y., Xiang, D., Zhang, X., Yuan, F. (2017). Autophagy regulates Endothelial-Mesenchymal transition by decreasing the phosphorylation level of Smad3. Biochem. Biophys. Res. Commun. 487 (3), 740–747. doi: 10.1016/j.bbrc.2017.04.130
Watschinger, B., Sayegh, M. H., Hancock, W. W., Russell, M. E. (1995). Up-regulation of endothelin-1 mRNA and peptide expression in rat cardiac allografts with rejection and arteriosclerosis. Am. J. Pathol. 146 (5), 1065–1072.
Weil, E. J., Lemley, K. V., Mason, C. C., Yee, B., Jones, L. I., Blouch, K., et al. (2012). Podocyte detachment and reduced glomerular capillary endothelial fenestration promote kidney disease in type 2 diabetic nephropathy. Kidney Int. 82 (9), 1010–1017. doi: 10.1038/ki.2012.234
Weinbaum, S., Tarbell, J. M., Damiano, E. R. (2007). The structure and function of the endothelial glycocalyx layer. Annu. Rev. BioMed. Eng. 9, 121–167. doi: 10.1146/annurev.bioeng.9.060906.151959
Wilkening, A., Krappe, J., Muhe, A. M., Lindenmeyer, M. T., Eltrich, N., Luckow, B., et al. (2020). C-C chemokine receptor type 2 mediates glomerular injury and interstitial fibrosis in focal segmental glomerulosclerosis. Nephrol. Dial Transplant. 35 (2), 227–239. doi: 10.1093/ndt/gfy380
Wilkinson-Berka, J. L., Kelly, D. J., Koerner, S. M., Jaworski, K., Davis, B., Thallas, V., et al. (2002). ALT-946 and aminoguanidine, inhibitors of advanced glycation, improve severe nephropathy in the diabetic transgenic (mREN-2)27 rat. Diabetes 51 (11), 3283–3289. doi: 10.2337/diabetes.51.11.3283
Wu, X. M., Gao, Y. B., Cui, F. Q., Zhang, N. (2016). Exosomes from high glucose-treated glomerular endothelial cells activate mesangial cells to promote renal fibrosis. Biol. Open 5 (4), 484–491. doi: 10.1242/bio.015990
Wu, X., Gao, Y., Xu, L., Dang, W., Yan, H., Zou, D., et al. (2017). Exosomes from high glucose-treated glomerular endothelial cells trigger the epithelial-mesenchymal transition and dysfunction of podocytes. Sci. Rep. 7 (1), 9371. doi: 10.1038/s41598-017-09907-6
Yamanouchi, M., Furuichi, K., Hoshino, J., Ubara, Y., Wada, T. (2020). Nonproteinuric diabetic kidney disease. Clin. Exp. Nephrol. 24 (7), 573–581. doi: 10.1007/s10157-020-01881-0
Yuan, M., Tan, Y., Wang, Y., Wang, S. X., Yu, F., Zhao, M. H. (2019). The associations of endothelial and podocyte injury in proliferative lupus nephritis: from observational analysis to in vitro study. Lupus. 28 (3), 347–358. doi: 10.1177/0961203319828509
Yuen, D. A., Stead, B. E., Zhang, Y., White, K. E., Kabir, M. G., Thai, K., et al. (2012). eNOS deficiency predisposes podocytes to injury in diabetes. J. Am. Soc. Nephrol. 23 (11), 1810–1823. doi: 10.1681/ASN.2011121170
Zeisberg, E. M., Tarnavski, O., Zeisberg, M., Dorfman, A. L., McMullen, J. R., Gustafsson, E., et al. (2007). Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 13 (8), 952–961. doi: 10.1038/nm1613
Zeisberg, E. M., Potenta, S. E., Sugimoto, H., Zeisberg, M., Kalluri, R. (2008). Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J. Am. Soc. Nephrol. 19 (12), 2282–2287. doi: 10.1681/ASN.2008050513
Zheng, X., Soroush, F., Long, J., Hall, E. T., Adishesha, P. K., Bhattacharya, S., et al. (2017). Murine glomerular transcriptome links endothelial cell-specific molecule-1 deficiency with susceptibility to diabetic nephropathy. PloS One 12 (9), e0185250. doi: 10.1371/journal.pone.0185250
Keywords: Kidney glomerulus (MeSH: D007678), Glycocalyx (MeSH: D019276), Endothelial cells (MeSH: D042783), Podocytes (MeSH: D050199), Proteinuria (MeSH: D011507), Diabetic Nephropathy (MeSH: D003928), Focal Segmental Glomerulosclerosis (MeSH: D005923)
Citation: Sol M, Kamps JAAM, van den Born J, van den Heuvel MC, van der Vlag J, Krenning G and Hillebrands J-L (2020) Glomerular Endothelial Cells as Instigators of Glomerular Sclerotic Diseases. Front. Pharmacol. 11:573557. doi: 10.3389/fphar.2020.573557
Received: 17 June 2020; Accepted: 14 September 2020;
Published: 06 October 2020.
Edited by:
Rohan Samarakoon, Albany Medical College, United StatesReviewed by:
Ilse Sofia Daehn, Icahn School of Medicine at Mount Sinai, United StatesPierre-Louis Tharaux, Institut National de la Santé et de la Recherche Médicale (INSERM), France
Copyright © 2020 Sol, Kamps, van den Born, van den Heuvel, van der Vlag, Krenning and Hillebrands. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jan-Luuk Hillebrands, ai5sLmhpbGxlYnJhbmRzQHVtY2cubmw=