
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
TECHNOLOGY AND CODE article
Front. Pharmacol., 18 December 2020
Sec. Translational Pharmacology
Volume 11 - 2020 | https://doi.org/10.3389/fphar.2020.565644
This article is part of the Research TopicArtificial intelligence for Drug Discovery and DevelopmentView all 18 articles
 Daniil Polykovskiy1*
Daniil Polykovskiy1* Alexander Zhebrak1
Alexander Zhebrak1 Benjamin Sanchez-Lengeling2
Benjamin Sanchez-Lengeling2 Sergey Golovanov3
Sergey Golovanov3 Oktai Tatanov3
Oktai Tatanov3 Stanislav Belyaev3
Stanislav Belyaev3 Rauf Kurbanov3
Rauf Kurbanov3 Aleksey Artamonov3
Aleksey Artamonov3 Vladimir Aladinskiy1
Vladimir Aladinskiy1 Mark Veselov1
Mark Veselov1 Artur Kadurin1
Artur Kadurin1 Simon Johansson4
Simon Johansson4 Hongming Chen4
Hongming Chen4 Sergey Nikolenko1,3,5*
Sergey Nikolenko1,3,5* Alán Aspuru-Guzik6,7,8,9*
Alán Aspuru-Guzik6,7,8,9* Alex Zhavoronkov1*
Alex Zhavoronkov1*Generative models are becoming a tool of choice for exploring the molecular space. These models learn on a large training dataset and produce novel molecular structures with similar properties. Generated structures can be utilized for virtual screening or training semi-supervized predictive models in the downstream tasks. While there are plenty of generative models, it is unclear how to compare and rank them. In this work, we introduce a benchmarking platform called Molecular Sets (MOSES) to standardize training and comparison of molecular generative models. MOSES provides training and testing datasets, and a set of metrics to evaluate the quality and diversity of generated structures. We have implemented and compared several molecular generation models and suggest to use our results as reference points for further advancements in generative chemistry research. The platform and source code are available at https://github.com/molecularsets/moses.
The discovery of new molecules for drugs and materials can bring enormous societal and technological progress, potentially curing rare diseases and providing a pathway for personalized precision medicine (Lee et al., 2018). However, complete exploration of the huge space of potential chemicals is computationally intractable; it has been estimated that the number of pharmacologically-sensible molecules is in the order of
Over the last few years, advances in machine learning, and especially in deep learning, have driven the design of new computational systems for modeling increasingly complex phenomena. One approach that has been proven fruitful for modeling molecular data is deep generative models. Deep generative models have found applications in a wide range of settings, from generating synthetic images (Karras et al., 2018) and natural language texts (Yu et al., 2017), to the applications in biomedicine, including the design of DNA sequences (Killoran et al., 2017), and aging research (Zhavoronkov et al., 2019b). One important field of application for deep generative models lies in the inverse design of drug compounds (Sanchez-Lengeling and Aspuru-Guzik, 2018) for a given functionality (solubility, ease of synthesis, toxicity). Deep learning also found other applications in biomedicine (Mamoshina et al., 2016; Ching et al., 2018), including target identification (Mamoshina et al., 2018), antibacterial drug discovery (Ivanenkov et al., 2019), and drug repurposing (Aliper et al., 2016; Vanhaelen et al., 2017).
Part of the success of deep learning in different fields has been driven by ever-growing availability of large datasets and standard benchmark sets. These sets serve as a common measuring stick for newly developed models and optimization strategies (LeCun et al., 1998; Deng et al., 2009). In the context of organic molecules, MoleculeNet (Wu et al., 2018) was introduced as a standardized benchmark suite for regression and classification tasks. Brown et al. (2019) proposed to evaluate generative models on goal-oriented and distribution learning tasks with a focus on the former. We focus on standardizing metrics and data for the distribution learning problem that we introduce below.
In this work, we provide a benchmark suite—Molecular Sets (MOSES)—for molecular generation: a standardized dataset, data preprocessing utilities, evaluation metrics, and molecular generation models. We hope that our platform will serve as a clear and unified testbed for current and future generative models. We illustrate the main components of MOSES in Figure 1.
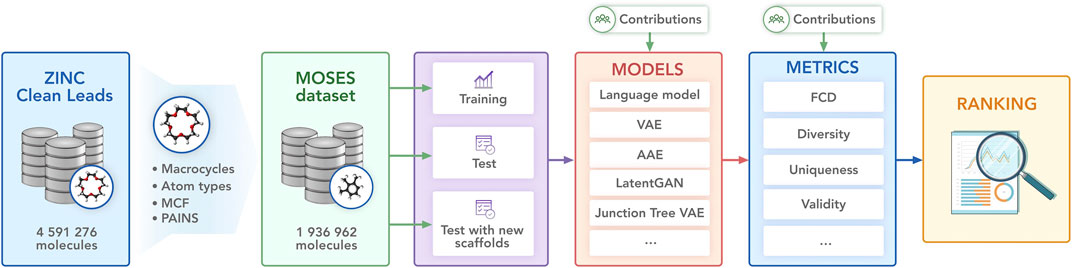
FIGURE 1. Molecular Sets (MOSES) pipeline. The open-source library provides a dataset, baseline models, and evaluation metrics.
In MOSES, we study distribution learning models. Formally, given a set of training samples
Distribution learning models are mainly used for building virtual libraries (van Hilten et al., 2019) for computer-assisted drug discovery. While imposing simple rule-based restrictions on a virtual library (such as maximum or minimum weight) is straightforward, it is unclear how to apply implicit or soft restrictions on the library. For example, a medicinal chemist might expect certain substructures to be more prevalent in generated structures. Relying on a set of manually or automatically selected compounds, distribution learning models produce a larger dataset, preserving implicit rules from the dataset. Another application of distribution learning models is extending the training set for downstream semi-supervized predictive tasks: one can add new unlabeled data by sampling compounds from a generative model.
The quality of a distribution learning model is a deviation measure between
In this section, we discuss different approaches to representing a molecule in a machine learning-friendly way (Figure 2): string and graph representations.
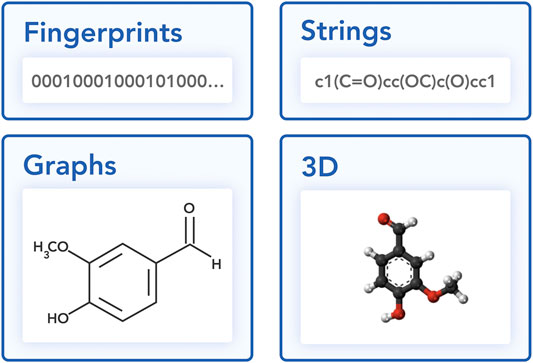
FIGURE 2. Different views on a vanillin molecule.
String representations. Representing a molecular structure as a string have been quickly adopted (Jaques et al., 2016; Guimaraes et al., 2017; Kadurin et al., 2017; Olivecrona et al., 2017; Yang et al., 2017; Kang and Cho, 2018; Popova et al., 2018; Putin et al., 2018; Segler et al., 2018) for generative models due to the abundance of sequence modeling tools such as recurrent neural networks, attention mechanisms, and dilated convolutions. Simplified molecular input line entry system (SMILES) (Weininger, 1988) is the most widely used string representation for generative machine learning models. SMILES algorithm traverses a spanning tree of a molecular graph in depth-first order and stores atom and edge tokens. SMILES also uses special tokens for branching and edges not covered with a spanning tree. Note that since a molecule can have multiple spanning trees, different SMILES strings can represent a single molecule. While there is a canonicalization procedure to uniquely construct a SMILES string from a molecule (Weininger et al., 1989), ambiguity of SMILES can also serve as augmentation and improve generative models (Arús-Pous et al., 2019).
DeepSMILES (O’Boyle and Dalke, 2018) was introduced as an extension of SMILES that seeks to reduce invalid sequences by altering syntax for branches and ring closures. Some methods try to incorporate SMILES syntax into a network architecture to increase the fraction of valid molecules (Kusner et al., 2017; Dai et al., 2018). SELFIES (Krenn et al., 2019) defines a new syntax based on a Chomsky type-2 grammar augmented with self-referencing functions. International Chemical Identifier (InChI) (Stein et al., 2003) is a more verbose string representation which explicitly specifies a chemical formula, atoms’ charges, hydrogens, and isotopes. However, Gómez-Bombarelli et al. (2018) reported that InChI-based models perform substantially worse than SMILES-based models in generative modeling—presumably due to a more complex syntax.
Molecular graphs. Graph representations have long been used in chemoinformatics for storing and processing molecular data. In a molecular graph, each node corresponds to an atom and each edge corresponds to a bond. Such graph can specify hydrogens either explicitly or implicitly. In the latter case, the number of hydrogens can be deduced from atoms’ valencies.
Classical machine learning methods mostly utilize molecular descriptors extracted from such graphs. Deep learning models, however, can learn from graphs directly with models such as Graph Convolutional Networks (Duvenaud et al., 2015), Weave Networks (Wu et al., 2018), and Message Passing Networks (Gilmer et al., 2017). Molecular graph can also be represented as adjacency matrix and node feature matrix; this approach has been successfully employed in the MolGAN model (De Cao and Kipf, 2018) for the QM9 dataset (Ramakrishnan et al., 2014). Other approaches such as Junction Tree VAE (Jin et al., 2018) process molecules in terms of their subgraphs.
In this section, we propose a set of metrics to assess the quality of generative models. The proposed metrics detect common issues in generative models such as overfitting, imbalance of frequent structures or mode collapse. Each metric depends on a generated set G and a test (reference) set R. We compute all metrics (except for validity) only for valid molecules from the generated set. We suggest generating 30, 000 molecules and obtaining G as valid molecules from this set.
Fraction of valid (Valid) and unique (Unique@k) molecules report validity and uniqueness of the generated SMILES strings. We define validity using RDKit’s molecular structure parser that checks atoms’ valency and consistency of bonds in aromatic rings. In the experiments, we compute Unique@K and for the first
Novelty is the fraction of the generated molecules that are not present in the training set. Low novelty indicates overfitting.
Filters is the fraction of generated molecules that pass filters applied during dataset construction (see Section 5). While the generated molecules are often chemically valid, they may contain unwanted fragments: when constructing the training dataset, we removed molecules with such fragments and expect the models to avoid producing them.
Fragment similarity (Frag) compares distributions of BRICS fragments (Degen et al., 2008) in generated and reference sets. Denoting
If molecules in both sets have similar fragments, Frag metric is large. If some fragments are over- or underrepresented (or never appear) in the generated set, the metric will be lower. Limits of this metric are [0,1].
Scaffold similarity (Scaff) is similar to fragment similarity metric, but instead of fragments we compare frequencies of Bemis–Murcko scaffolds (Bemis and Murcko, 1996). Bemis–Murcko scaffold contains all molecule’s ring structures and linker fragments connecting rings. We use RDKit implementation of this algorithm which additionally considers carbonyl groups attached to rings as part of a scaffold. Denoting
The purpose of this metric is to show how similar are the scaffolds present in generated and reference datasets. For example, if the model rarely produces a certain chemotype from a reference set, the metric will be low. Limits of this metric are [0,1].
Note that both fragment and scaffold similarities compare molecules at a substructure level. Hence, it is possible to have a similarity one even when G and R contain different molecules.
Similarity to a nearest neighbor (SNN) is an average Tanimoto similarity T(mG,mR) (also known as the Jaccard index) between fingerprints of a molecule
In this work, we used standard Morgan (extended connectivity) fingerprints (Rogers and Hahn, 2010) with radius 2 and 1024 bits computed using RDKit library (Landrum, 2006). The resulting similarity metric can be interpreted as precision: if generated molecules are far from the manifold of the reference set, similarity to the nearest neighbor will be low. Limits of this metric are [0,1].
Internal diversity (IntDivp) (Benhenda, 2017) assesses the chemical diversity within the generated set of molecules G.
This metric detects a common failure case of generative models—mode collapse. With mode collapse, the model produces a limited variety of samples, ignoring some areas of the chemical space. A higher value of this metric corresponds to higher diversity in the generated set. In the experiments, we report IntDiv1 (G) and IntDiv2 (G). Limits of this metric are [0,1].
Fréchet ChemNet Distance (FCD) (Preuer et al., 2018) is calculated using activations of the penultimate layer of a deep neural network ChemNet trained to predict biological activities of drugs. We compute activations for canonical SMILES representations of molecules. These activations capture both chemical and biological properties of the compounds. For two sets of molecules G and R, FCD is defined as
where
Properties distribution is a useful tool for visually assessing the generated structures. To quantitatively compare the distributions in the generated and test sets, we compute a 1D Wasserstein-1 distance between property distributions of generated and test sets. We also visualize a kernel density estimation of these distributions in the Experiments section. We use the following four properties:
Molecular weight (MW): the sum of atomic weights in a molecule. By plotting histograms of molecular weight for the generated and test sets, one can judge if a generated set is biased toward lighter or heavier molecules.
LogP: the octanol-water partition coefficient, a ratio of a chemical’s concentration in the octanol phase to its concentration in the aqueous phase of a two-phase octanol/water system; computed with RDKit’s Crippen (Wildman and Crippen, 1999) estimation.
Synthetic Accessibility Score (SA): a heuristic estimate of how hard (10) or how easy (1) it is to synthesize a given molecule. SA score is based on a combination of the molecule’s fragments contributions (Ertl and Schuffenhauer, 2009). Note that SA score does not adequately assess up-to-date chemical structures, but it is useful for assessing distribution learning models.
Quantitative Estimation of Drug-likeness (QED): a [0,1] value estimating how likely a molecule is a viable candidate for a drug. QED is meant to capture the abstract notion of esthetics in medicinal chemistry (Bickerton et al., 2012). Similar to SA, descriptor limits in QED have been changing during the last decade and current limits may not cover latest drugs (Shultz, 2018).
The proposed dataset used for training and testing is based on the ZINC Clean Leads (Sterling and Irwin, 2015) collection which contains 4, 591, 276 molecules with molecular weight in the range from 250 to 350 Da, a number of rotatable bonds not greater than 7, and XlogP (Wang et al., 1997) not greater then 3.5. Clean-leads dataset consists of structures suitable for identifying hit compounds and they are small enough to allow for further ADMET optimization of generated molecules (Teague et al., 1999). We removed molecules containing charged atoms, atoms besides C, N, S, O, F, Cl, Br, H, or cycles larger than eight atoms. The molecules were filtered via custom medicinal chemistry filters (MCFs) and PAINS filters (Baell and Holloway, 2010). We describe MCFs and discuss PAINS in Supplementary Information 1. We removed charged molecules to avoid ambiguity with tautomers and pH conditions. Note that in the initial set of molecules, functional groups were present in both ionized and unionized forms.
The final dataset contains molecules, with internal diversity IntDiv1 = 0.857; it contains
FIGURE 3. Examples of molecules from MOSES dataset.
We implemented several models that cover different approaches to molecular generation, such as character-level recurrent neural networks (CharRNN) (Preuer et al., 2018; Segler et al., 2018), Variational Autoencoders (VAE) (Kadurin et al., 2016; Blaschke et al., 2018; Gómez-Bombarelli et al., 2018), Adversarial Autoencoders (AAE) (Kadurin et al., 2016; Polykovskiy et al., 2018b), Junction Tree Variational Autoencoders (JTN-VAE) (Jin et al., 2018), LatentGAN (Prykhodko et al., 2019), and non-neural baselines.
Model comparison can be challenging since different training parameters (number of epochs, batch size, learning rate, initial state, optimizer) and architecture hyperparameters (hidden layer dimension, number of layers, etc.) can significantly alter their performance. For each model, we attempted to preserve its original architecture as published and tuned the hyperparameters to improve the performance. We used random search over multiple architectures for every model and selected the architecture that produced the best value of FCD. Models are implemented in Python 3 utilizing PyTorch (Paszke et al., 2017) framework. Please refer to the Supplementary Information three for the training details and hyperparameters.
Character-level recurrent neural network (CharRNN) (Segler et al., 2018) models a distribution over the next token given previously generated ones. We train this model by maximizing log-likelihood of the training data represented as SMILES strings.
Variational autoencoder (VAE) (Kingma and Welling, 2013) consists of two neural networks—an encoder and a decoder—that infer a mapping from high-dimensional data representation onto a lower-dimensional space and back. The lower-dimensional space is called the latent space, which is often a continuous vector space with normal prior distribution. VAE parameters are optimized to encode and decode data by minimizing reconstruction loss and regularization term in a form of Kullback-Leibler divergence. VAE-based architecture for the molecular generation was studied in multiple previous works (Kadurin et al. 2016; Blaschke et al. 2018; Gómez-Bombarelli et al. 2018). We combine aspects from these implementations and use SMILES as input and output representations.
Adversarial Autoencoder (AAE) (Makhzani et al., 2016) replaces the Kullback-Leibler divergence from VAE with an adversarial objective. An auxiliary discriminator network is trained to distinguish samples from a prior distribution and model’s latent codes. The encoder then adapts its latent codes to minimize discriminator’s predictive accuracy. The training process oscillates between training the encoder-decoder pair and the discriminator. Unlike Kullback-Leibler divergence that has a closed-form analytical solution only for a handful of distributions, a discriminator can be used for any prior distribution. AAE-based models for molecular design were studied in (Kadurin et al., 2016; Kadurin et al., 2017; Polykovskiy et al., 2018b). Similar to VAE, we use SMILES as input and output representations.
Junction Tree VAE (JTN-VAE) (Jin et al., 2018) generates molecules in two phases by exploiting valid subgraphs as components. In the first phase, it generates a tree-structured object (a junction tree) whose role is to represent the scaffold of subgraph components and their coarse relative arrangements. The components are valid chemical substructures automatically extracted from the training set. In the second phase, the subgraphs (nodes of the tree) are assembled together into a coherent molecular graph.
Latent Vector Based Generative Adversarial Network (LatentGAN) (Prykhodko et al., 2019) combines an autoencoder and a generative adversarial network. LatentGAN pretrains an autoencoder to map SMILES structures onto latent vectors. A generative adversarial network is then trained to produce latent vectors for the pre-trained decoder.
Non-neural baselines implemented in MOSES are n-gram generative model, Hidden Markov Model (HMM), and a combinatorial generator. N-gram model collects statistics of n-grams frequencies in the training set and uses such distribution to sequentially sample new strings. Hidden Markov models utilize Baum-Welch algorithm to learn a probabilistic distribution over the SMILES strings. The model consists of several states (s1,...,sK), transition probabilities between states p(si+1 | si), and token emission probabilities p(xi | si). Beginning from a “start” state, at each iteration the model samples a next token and state from emission and transition probabilities correspondingly. A combinatorial generator splits molecular graphs of the training data into BRICS fragments and generates new molecules by randomly connecting random substructures. We sample fragments according to their frequencies in the training set to model the distribution better.
The dataset, metrics and baseline models are provided in a GitHub repository https://github.com/molecularsets/moses and as a PyPI package molsets. To contribute a new model, one should train a model on MOSES train set, generate 30, 000 samples and compute metrics using the provided utilities. We recommend running the experiment at least three times with different random seeds to estimate sensitivity of the model to random parameter initialization. We store molecular structures in SMILES format; molecular graphs can be reconstructed using RDKit (Landrum, 2006).
We trained the baseline models on MOSES train set and provide results in this section. In Table 1 we compare models with respect to the validity and uniqueness metrics. Hidden Markov Model and NGram models fail to produce valid molecules since they have a limited context. Combinatorial generator and JTN-VAE have built-in validity constraints, so their validity is 100%.
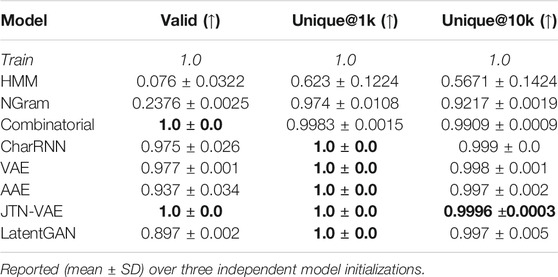
TABLE 1. Performance metrics for baseline models: fraction of valid molecules, fraction of unique molecules from and molecules.
Table 2 reports additional properties of the generated set: fraction of molecules passing filters, fraction of molecules not present in the training set, and internal diversity. All modules successfully avoid forbidden structures (MCF and PAINS) even though such restrictions were only defined implicitly—using a training dataset. Combinatorial generator has higher diversity than the training dataset, which might be favorable for discovering new chemical structures. Autoencoder-based models show low novelty, indicating that these models overfit to the training set.
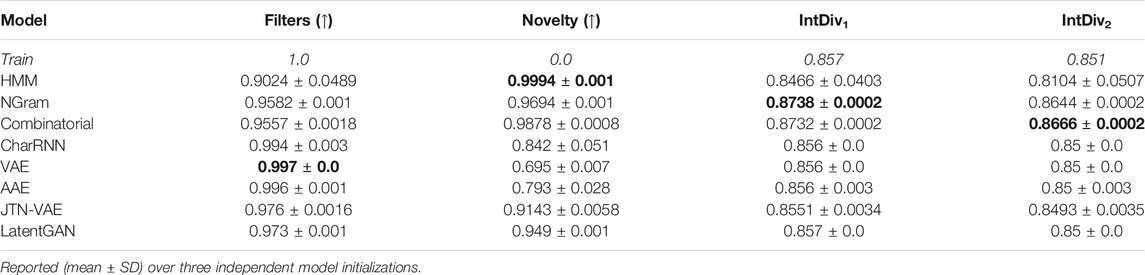
TABLE 2. Performance metrics for baseline models: fraction of molecules passing filters (MCF, PAINS, ring sizes, charge, atom types), novelty, and internal diversity.
Table 3 reports Fréchet ChemNet Distance (FCD) and similarity to a nearest neighbor (SNN). All neural network-based models show low FCD, indicating that the models successfully captured the statistics of the dataset. Surprisingly, a simple language model, character level RNN, shows the best results in terms of the FCD measure. Variational autoencoder (VAE) showed the best results in terms of SNN, but combined with low novelty we suppose that the model overfitted on the training set.
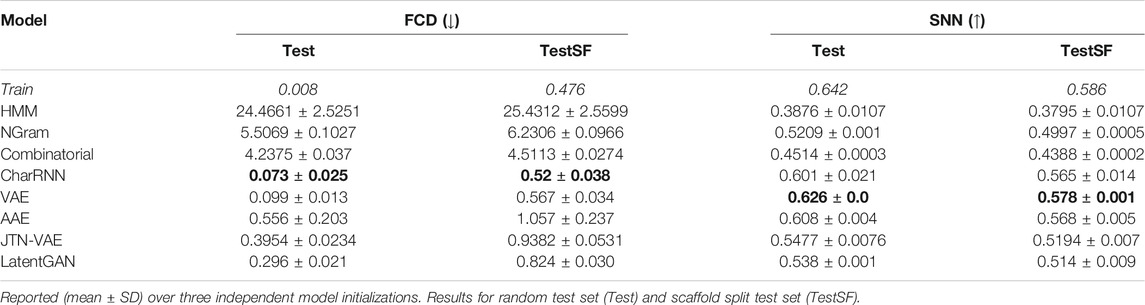
TABLE 3. Performance metrics for baseline models: Fréchet ChemNet Distance (FCD) and Similarity to a nearest neighbor (SNN).
In Table 4 we report similarities of substructure distributions—fragments and scaffolds. Scaffold similarity from the training set to the scaffold test set (TestSF) is zero by design. Note that CharRNN successfully discovered many novel scaffolds (11%), suggesting that the model generalizes well.
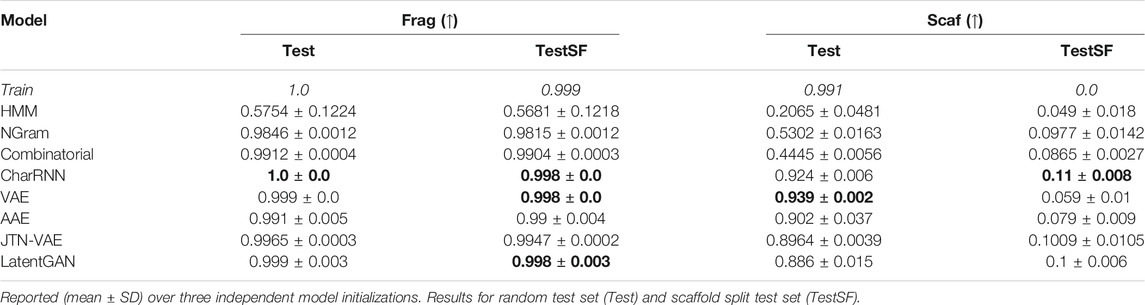
TABLE 4. Fragment similarity (Frag), Scaffold similarity (Scaff).
Finally, we compared distributions of four molecular properties in generated and test sets (Figure 4): molecular weight (MW), octanol-water partition coefficient (logP), quantitative estimation of drug-likeness (QED), and synthetic accessibility score (SA). Deep generative models closely match the data distribution; hidden Markov Model is biased toward lighter molecules, which is consistent with low validity: larger molecules impose more validity constraints. A combinatorial generator has higher variance in molecular weight, producing larger and smaller molecules than those present in the training set.
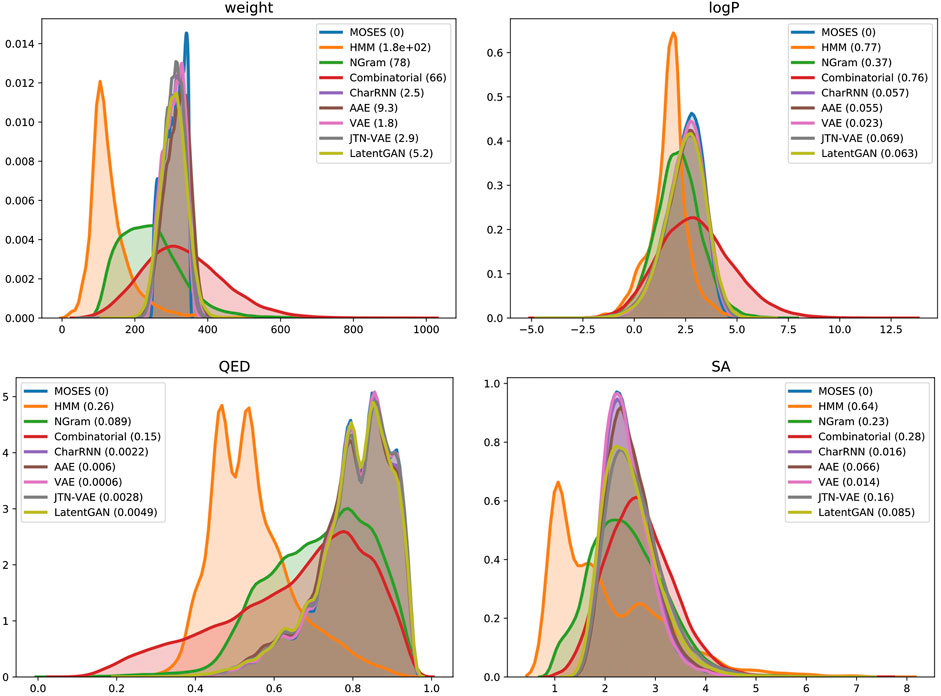
FIGURE 4. Distribution of chemical properties for MOSES dataset and sets of generated molecules. In brackets—Wasserstein-1 distance to MOSES test set. Parameters: molecular weight, octanol-water partition coefficient (logP), quantitative estimation of drug-likeness (QED) and synthetic accessibility score (SA).
From a wide range of presented models, CharRNN currently performs the best in terms of the key metrics. Specifically, it produces the best FCD, Fragment, and Scaffold scores, indicating that the model not only captured the training distribution well, but also did not overfit on the training set.
The presented set of metrics assesses models’ performance from different perspectives; therefore, for each specific downstream task, one could consider the most relevant metric. For example, evaluation based on Scaf/TestSF score could be relevant when model’s objective is to discover novel scaffolds. For a general evaluation, we suggest using FCD/Test metric that captures multiple aspects of other metrics in a single number. However, it does not give insights into specific issues that cause high FCD/Test values, hence more interpretable metrics presented in this paper are necessary to investigate the model’s performance thoroughly.
With MOSES, we have designed a molecular generation benchmark platform that provides a dataset with molecular structures, an implementation of baseline models, and metrics for their evaluation. While standardized comparative studies and test sets are essential for the progress of machine learning applications, the current field of de novo drug design lacks evaluation protocols for generative machine learning models. Being on the intersection of mathematics, computer science, and chemistry, these applications are often too challenging to explore for research scientists starting in the field. Hence, it is necessary to develop a transparent approach to implementing new models and assessing their performance. We presented a benchmark suite with unified and extendable programming interfaces for generative models and evaluation metrics.
This platform should allow for a fair and comprehensive comparison of new generative models. For future work on this project, we will keep extending the MOSES repository with new baseline models and new evaluation metrics. We hope this work will attract researchers interested in tackling drug discovery challenges.
The data and code of the MOSES platform is available at https://github.com/molecularsets/moses.
DP, AZhe, SG, OT, SB, RK, AA, AK, SJ, and HC designed and conducted the experiments; DP and AZhe, BS-L, VA, MV, SJ, HC, SN, AA-G, AZha wrote the manuscript.
DP, AZhe, VA, MV, and AZha work for Insilico Medicine, a commercial artificial intelligence company. SG, OT, SB, RK, AA, and SN work for Neuromation OU, a company engaged in AI development through synthetic data and generative models. SJ and HC work for a pharmaceutical company AstraZeneca. AA-G is a cofounder and board member of, and consultant for, Kebotix, an artificial intelligence-driven molecular discovery company and a member of the science advisory board of Insilico Medicine.
This manuscript has been released as a pre-print at https://arxiv.org/abs/1811.12823 (Polykovskiy et al., 2018a).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2020.565644/full#supplementary-material.
Aliper, A., Plis, S., Artemov, A., Ulloa, A., Mamoshina, P., and Zhavoronkov, A. (2016). Deep learning applications for predicting pharmacological properties of drugs and drug repurposing using transcriptomic data. Mol. Pharm. 13, 2524–2530. doi:10.1021/acs.molpharmaceut.6b00248
Arús-Pous, J., Johansson, S. V., Prykhodko, O., Bjerrum, E. J., Tyrchan, C., Reymond, J.-L., et al. (2019). Randomized smiles strings improve the quality of molecular generative models. J. Cheminf. 11, 1–13. doi:10.1186/s13321-019-0393-0
Baell, J. B., and Holloway, G. A. (2010). New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 53, 2719–2740. doi:10.1021/jm901137j
Bemis, G. W., and Murcko, M. A. (1996). The properties of known drugs. 1. molecular frameworks. J. Med. Chem. 39, 2887–2893. doi:10.1021/jm9602928
Benhenda, M. (2017). ChemGAN challenge for drug discovery: can AI reproduce natural chemical diversity? Available from: https://arxiv.org/abs/1708.08227.
Bickerton, G. R., Paolini, G. V., Besnard, J., Muresan, S., and Hopkins, A. L. (2012). Quantifying the chemical beauty of drugs. Nat. Chem. 4, 90–98. doi:10.1038/nchem.1243
Blaschke, T., Olivecrona, M., Engkvist, O., Bajorath, J., and Chen, H. (2018). Application of generative autoencoder in de novo molecular design. Mol. Inform. 37, 1700123. doi:10.1002/minf.201700123
Brown, N., Fiscato, M., Segler, M. H. S., and Vaucher, A. C. (2019). Guacamol: benchmarking models for de novo molecular design. J. Chem. Inf. Model. 59, 1096–1108. doi:10.1021/acs.jcim.8b00839
Ching, T., Himmelstein, D. S., Beaulieu-Jones, B. K., Kalinin, A. A., Do, B. T., Way, G. P., et al. (2018). Opportunities and obstacles for deep learning in biology and medicine. J. R. Soc. Interface 15, 20170387. doi:10.1098/rsif.2017.0387
Dai, H., Tian, Y., Dai, B., Skiena, S., and Song, L. (2018). “Syntax-directed variational autoencoder for structured data,” in International conference on learning representations.
De Cao, N., and Kipf, T. (2018). “MolGAN: an implicit generative model for small molecular graphs,” in ICML 2018 workshop on Theoretical Foundations and Applications of Deep Generative Models.
Degen, J., Wegscheid-Gerlach, C., Zaliani, A., and Rarey, M. (2008). On the art of compiling and using 'drug-like' chemical fragment spaces. ChemMedChem 3, 1503–1507. doi:10.1002/cmdc.200800178
Deng, J., Dong, W., Socher, R., Li, L.-J., Li, K., and Fei-Fei, L. (2009). CVPR09.ImageNet: a large-scale hierarchical image database,” in 2009 IEEE Conference on Computer Vision and Pattern Recognition, Miami, FL, June 20–25, 2009IEEE.
Dinh, L., Sohl-Dickstein, J., and Bengio, S. (2017). Density estimation using real NVP. Available at: https://library.seg.org/doi/10.1190/segam2017-17559486.1
Duvenaud, D. K., Maclaurin, D., Iparraguirre, J., Bombarell, R., Hirzel, T., Aspuru-Guzik, A., et al. (2015). “Convolutional networks on graphs for learning molecular fingerprints,” in Advances in neural information processing systems 28. Editors C. Cortes, N. D. Lawrence, D. D. Lee, M. Sugiyama, and R. Garnett (New York, NY: Curran Associates, Inc.), 2224–2232.
Ertl, P., and Schuffenhauer, A. (2009). Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminf. 1, 8. doi:10.1186/1758-2946-1-8
Ferrero, S., Hart, G. L. W., Nardelli, M. B., Mingo, N., Sanvito, S., and Levy, O. (2013). The high-throughput highway to computational materials design. Nat. Mater. 12, 191–201. doi:10.1038/nmat3568
Gilmer, J., Schoenholz, S. S., Riley, P. F., Vinyals, O., and Dahl, G. E. (2017). Neural message passing for quantum chemistry,” in Proceedings of the 34th international conference on machine learning. JMLR, 1263–1272
Gómez-Bombarelli, R., Wei, J. N., Duvenaud, D., Hernández-Lobato, J. M., Sánchez-Lengeling, B., Sheberla, D., et al. (2018). Automatic chemical design using a Data-Driven continuous representation of molecules. ACS Cent. Sci. 4, 268–276. doi:10.1021/acscentsci.7b00572
Grisoni, F., Neuhaus, C. S., Gabernet, G., Müller, A. T., Hiss, J. A., and Schneider, G. (2018). Designing anticancer peptides by constructive machine learning. ChemMedChem 13, 1300–1302. doi:10.1002/cmdc.201800204
Guimaraes, G. L., Sanchez-Lengeling, B., Farias, P. L. C., and Aspuru-Guzik, A. (2017). Objective-Reinforced generative adversarial networks (ORGAN) for sequence generation models. Available at: https://arxiv.org/abs/1705.10843.
Hu, X., Beratan, D. N., and Yang, W. (2009). Emergent strategies for inverse molecular design. Sci. China Ser. B-Chem. 52, 1769–1776. doi:10.1007/s11426-009-0260-3
Ivanenkov, Y. A., Zhavoronkov, A., Yamidanov, R. S., Osterman, I. A., Sergiev, P. V., Aladinskiy, V. A., et al. (2019). Identification of novel antibacterials using machine learning techniques. Front. Pharmacol. 10, 913. doi:10.3389/fphar.2019.00913
Jaques, N., Gu, S., Bahdanau, D., Hernández-Lobato, J. M., Turner, R. E., and Eck, D. (2016). Sequence tutor: conservative fine-tuning of sequence generation models with KL-control. Available at: https://arxiv.org/abs/1611.02796.
Jin, W., Barzilay, R., and Jaakkola, T. (2018). “Junction tree variational autoencoder for molecular graph generation,” in Proceedings of the 35th international conference on machine learning. Editors J. Dy, and A. Krause (Stockholmsmässan, Stockholm Sweden: PMLR), 2323–2332.
Kadurin, A., Aliper, A., Kazennov, A., Mamoshina, P., Vanhaelen, Q., Khrabrov, K., et al. (2016). The cornucopia of meaningful leads: applying deep adversarial autoencoders for new molecule development in oncology. Oncotarget 8, 10883–10890. doi:10.18632/oncotarget.14073
Kadurin, A., Nikolenko, S., Khrabrov, K., Aliper, A., and Zhavoronkov, A. (2017). druGAN: an advanced generative adversarial autoencoder model for de novo generation of new molecules with desired molecular properties in silico. Mol. Pharm. 14, 3098–3104. doi:10.1021/acs.molpharmaceut.7b00346
Kang, S., and Cho, K. (2018). Conditional molecular design with deep generative models. J. Chem. Inf. Model. 59, 43–52. doi:10.1021/acs.jcim.8b00263
Karras, T., Aila, T., Laine, S., and Lehtinen, J. (2018). Progressive growing of gans for improved quality, stability, and variation,” in International conference on learning representations. ICLR. 1–26.
Killoran, N., Lee, L. J., Delong, A., Duvenaud, D., and Frey, B. J. (2017). Generating and designing DNA with deep generative models. Available from: https://arxiv.org/abs/1712.06148.
Kingma, D. P., and Welling, M. (2013). Auto-Encoding variational bayes,” in International conference on learning representations.
Krenn, M., Häse, F., Nigam, A., Friederich, P., and Aspuru-Guzik, A. (2019). Selfies: a robust representation of semantically constrained graphs with an example application in chemistry. Available at: https://grlearning.github.io/papers/59.pdf.
Kusner, M. J., Paige, B., and Hernández-Lobato, J. M. (2017). “Grammar variational autoencoder,”in Proceedings of the 34th international conference on machine learning. Editors D. Precup, and Y. W. Teh (Sydney, Australia: Proceedings of Machine Learning Research), Vol. 70. 1945–1954.
Labat, R., Fu, Y., and Lai, L. (1997). A new atom-additive method for calculating partition coefficients. J. Chem. Inf. Comput. Sci. 37, 615–621. doi:10.1021/ci960169p
Landrum, G. (2006). RDKit: open-source cheminformatics. Available at: http://www.rdkit.org/.
Le, T. C., and Winkler, D. A. (2016). Discovery and optimization of materials using evolutionary approaches. Chem. Rev. 116, 6107–6132. doi:10.1021/acs.chemrev.5b00691
LeCun, Y., Bottou, L., Bengio, Y., and Haffner, P. (1998). Gradient-based learning applied to document recognition. Proc. IEEE 86, 2278–2324. doi:10.1109/5.726791
Lee, S.-I., Celik, S., Logsdon, B. A., Lundberg, S. M., Martins, T. J., Oehler, V. G., et al. (2018). A machine learning approach to integrate big data for precision medicine in acute myeloid leukemia. Nat. Commun. 9, 42. doi:10.1038/s41467-017-02465-5
Makhzani, A., Shlens, J., Jaitly, N., and Goodfellow, I. (2016). “Adversarial autoencoders,” in International conference on learning representations.
Mamoshina, P., Vieira, A., Putin, E., and Zhavoronkov, A. (2016). Applications of deep learning in biomedicine. Mol. Pharm. 13, 1445–1454. doi:10.1021/acs.molpharmaceut.5b00982
Mamoshina, P., Volosnikova, M., Ozerov, I. V., Putin, E., Skibina, E., Cortese, F., et al. (2018). Machine learning on human muscle transcriptomic data for biomarker discovery and tissue-specific drug target identification. Front. Genet. 9, 242. doi:10.3389/fgene.2018.00242
Merk, D., Friedrich, L., Grisoni, F., and Schneider, G. (2018a). De novo design of bioactive small molecules by artificial intelligence. Mol. Inf. 37, 1700153. doi:10.1002/minf.201700153
Merk, D., Grisoni, F., Friedrich, L., and Schneider, G. (2018b). Tuning artificial intelligence on the de novo design of natural-product-inspired retinoid x receptor modulators. Commun. Chem. 1, 68. doi:10.1038/s42004-018-0068-1
Olivecrona, M., Blaschke, T., Engkvist, O., and Chen, H. (2017). Molecular de-novo design through deep reinforcement learning. J. Cheminf. 9, 48. doi:10.1186/s13321-017-0235-x
O’Boyle, N., and Dalke, A. (2018). DeepSMILES: an adaptation of SMILES for use in machine-learning of chemical structures. ChemRxiv. doi:10.26434/chemrxiv.7097960
Paszke, A., Gross, S., Chintala, S., Chanan, G., Yang, E., DeVito, Z., et al. (2017). “Automatic differentiation in pytorch,” in NIPS workshop.
Polykovskiy, D., Zhebrak, A., Sanchez-Lengeling, B., Golovanov, S., Tatanov, O., Belyaev, S., et al. (2018a). Molecular sets (moses): a benchmarking platform for molecular generation models. Available from: https://arxiv.org/abs/1811.12823.
Polykovskiy, D., Zhebrak, A., Vetrov, D., Ivanenkov, Y., Aladinskiy, V., Mamoshina, P., et al. (2018b). Entangled conditional adversarial autoencoder for de novo drug discovery. Mol. Pharm. 15, 4398–4405. doi:10.1021/acs.molpharmaceut.8b00839
Popova, M., Isayev, O., and Tropsha, A. (2018). Deep reinforcement learning for de novo drug design. Sci. Adv. 4, eaap7885. doi:10.1126/sciadv.aap7885
Preuer, K., Renz, P., Unterthiner, T., Hochreiter, S., and Klambauer, G. (2018). Fréchet ChemNet distance: a metric for generative models for molecules in drug discovery. J. Chem. Inf. Model. 58, 1736–1741. doi:10.1021/acs.jcim.8b00234
Prykhodko, O., Johansson, S. V., Kotsias, P.-C., Arús-Pous, J., Bjerrum, E. J., Engkvist, O., et al. (2019). A de novo molecular generation method using latent vector based generative adversarial network. J. Cheminf. 11, 74. doi:10.1186/s13321-019-0397-9
Putin, E., Asadulaev, A., Vanhaelen, Q., Ivanenkov, Y., Aladinskaya, A. V., Aliper, A., et al. (2018). Adversarial threshold neural computer for molecular de novo design. Mol. Pharm. 15, 4386–4397. doi:10.1021/acs.molpharmaceut.7b01137
Pyzer-Knapp, E. O., Suh, C., Gómez-Bombarelli, R., Aguilera-Iparraguirre, J., and Aspuru-Guzik, A. (2015). What is High-Throughput virtual screening? a perspective from organic materials discovery. Annu. Rev. Mater. Res. 45, 195–216. doi:10.1146/annurev-matsci-070214-020823
Ramakrishnan, R., Dral, P. O., Rupp, M., and von Lilienfeld, O. A. (2014). Quantum chemistry structures and properties of 134 kilo molecules. Scientific Data 1, 140022. doi:10.1038/sdata.2014.22
Reymond, J.-L. (2015). The chemical space project. Acc. Chem. Res. 48, 722–730. doi:10.1021/ar500432k
Rogers, D., and Hahn, M. (2010). Extended-connectivity fingerprints. J. Chem. Inf. Model. 50, 742–754. doi:10.1021/ci100050t
Sanchez-Lengeling, B., and Aspuru-Guzik, A. (2018). Inverse molecular design using machine learning: generative models for matter engineering. Science 361, 360–365. doi:10.1126/science.aat2663
Segler, M. H. S., Kogej, T., Tyrchan, C., and Waller, M. P. (2018). Generating focused molecule libraries for drug discovery with recurrent neural networks. ACS Cent. Sci. 4, 120–131. doi:10.1021/acscentsci.7b00512
Shi, C., Xu, M., Zhu, Z., Zhang, W., Zhang, M., and Tang, J. (2019). “Graphaf: a flow-based autoregressive model for molecular graph generation,” in International conference on learning representations.
Shultz, M. D. (2018). Two decades under the influence of the rule of five and the changing properties of approved oral drugs. J. Med. Chem. 62, 1701–1714. doi:10.1021/acs.jmedchem.8b00686
Stein, S. E., Heller, S. R., and Tchekhovskoi, D. V. (2003). “An open standard for chemical structure representation: the iupac chemical identifier.” in International chemical information conference.
Sterling, T., and Irwin, J. J. (2015). Zinc 15 - ligand discovery for everyone. J. Chem. Inf. Model. 55, 2324–2337. doi:10.1021/acs.jcim.5b00559
Teague, S. J., Davis, A. M., Leeson, P. D., and Oprea, T. (1999). The design of leadlike combinatorial libraries. Angew. Chem. Int. Ed. 38, 3743–3748. doi:10.1002/(SICI)1521-3773(19991216)38:24%3C3743::AID-ANIE3743%3E3.0.CO;2-U
van Hilten, N., Chevillard, F., and Kolb, P. (2019). Virtual compound libraries in computer-assisted drug discovery. J. Chem. Inf. Model. 59, 644–651. doi:10.1021/acs.jcim.8b00737
Vanhaelen, Q., Mamoshina, P., Aliper, A. M., Artemov, A., Lezhnina, K., Ozerov, I., et al. (2017). Design of efficient computational workflows for in silico drug repurposing. Drug Discov. Today 22, 210–222. doi:10.1016/j.drudis.2016.09.019
Weininger, D. (1988). Smiles, a chemical language and information system. 1. introduction to methodology and encoding rules. J. Chem. Inf. Model. 28, 31–36. doi:10.1021/ci00057a005
Weininger, D., Weininger, A., and Weininger, J. L. (1989). Smiles. 2. algorithm for generation of unique smiles notation. J. Chem. Inf. Model. 29, 97–101. doi:10.1021/ci00062a008
Wildman, S. A., and Crippen, G. M. (1999). Prediction of physicochemical parameters by atomic contributions. J. Chem. Inf. Comput. Sci. 39, 868–873. doi:10.1021/ci990307l
Wu, Z., Ramsundar, B., Feinberg, E. N., Gomes, J., Geniesse, C., Pappu, A. S., et al. (2018). MoleculeNet: a benchmark for molecular machine learning. Chem. Sci. 9, 513–530. doi:10.1039/c7sc02664a
Yang, X., Zhang, J., Yoshizoe, K., Terayama, K., and Tsuda, K. (2017). ChemTS: an efficient python library for de novo molecular generation. Sci. Technol. Adv. Mater. 18, 972–976. doi:10.1080/14686996.2017.1401424
Yu, L., Zhang, W., Wang, J., and Yu, Y. (2017). “Seqgan: sequence generative adversarial nets with policy gradient,” in Thirty-first AAAI conference on artificial intelligence.
Keywords: generative models, drug discovery, deep learning, benchmark, distribution learning
Citation: Polykovskiy D, Zhebrak A, Sanchez-Lengeling B, Golovanov S, Tatanov O, Belyaev S, Kurbanov R, Artamonov A, Aladinskiy V, Veselov M, Kadurin A, Johansson S, Chen H, Nikolenko S, Aspuru-Guzik A and Zhavoronkov A (2020) Molecular Sets (MOSES): A Benchmarking Platform for Molecular Generation Models. Front. Pharmacol. 11:565644. doi: 10.3389/fphar.2020.565644
Received: 25 May 2020; Accepted: 26 October 2020;
Published: 18 December 2020.
Edited by:
Jianxun Ding, Chinese Academy of Sciences, ChinaReviewed by:
Nazareno Paolocci, Johns Hopkins University, United StatesCopyright © 2020 Polykovskiy, Zhebrak, Sanchez-Lengeling, Golovanov, Tatanov, Belyaev, Kurbanov, Artamonov, Aladinskiy, Veselov, Kadurin, Johansson, Chen, Nikolenko, Aspuru-Guzik and Zhavoronkov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniil Polykovskiy, ZGFuaWlsQGluc2lsaWNvLmNvbQ==; Alex Zhavoronkov, YWxleEBpbnNpbGljby5jb20=; Alán Aspuru-Guzik, YWxhbkBhc3B1cnUuY29t; Sergey Nikolenko, c25pa29sZW5rb0BnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.