 Fusheng Guo
Fusheng Guo Shuangshuang Xu
Shuangshuang Xu Yanlin Zhu1
Yanlin Zhu1 Xing Zheng
Xing Zheng Ying He
Ying He Lihua Jin
Lihua Jin- 1State Key Laboratory of Cellular Stress Biology, Innovation Center for Cell Signaling Network, School of Life Sciences, Xiamen University, Xiamen, China
- 2Department of Diabetes Complications and Metabolism, Diabetes and Metabolism Research Institute, Beckman Research Institute, City of Hope National Medical Center, Duarte, CA, United States
- 3Laboratory Animal Center, Xiamen University, Xiamen, China
Background: The transcriptional factor peroxisome proliferator–activated receptor γ (PPARγ) is an important therapeutic target for the treatment of type 2 diabetes. However, the role of the PPARγ transcriptional activity remains ambiguous in its metabolic regulation.
Methods: Based on the crystal structure of PPARγ bound with the DNA target of PPARγ response element (PPRE), Arg134, Arg135, and Arg138, three crucial DNA binding sites for PPARγ, were mutated to alanine (3RA), respectively. In vitro AlphaScreen assay and cell-based reporter assay validated that PPARγ 3RA mutant cannot bind with PPRE and lost transcriptional activity, while can still bind ligand (rosiglitazone) and cofactors (SRC1, SRC2, and NCoR). By using CRISPR/Cas9, we created mice that were heterozygous for PPARγ-3RA (PPARγ3RA/+). The phenotypes of chow diet and high-fat diet fed PPARγ3RA/+ mice were investigated, and the molecular mechanism were analyzed by assessing the PPARγ transcriptional activity.
Results: Homozygous PPARγ-3RA mutant mice are embryonically lethal. The mRNA levels of PPARγ target genes were significantly decreased in PPARγ3RA/+ mice. PPARγ3RA/+ mice showed more severe adipocyte hypertrophy, insulin resistance, and hepatic steatosis than wild type mice when fed with high-fat diet. These phenotypes were ameliorated after the transcription activity of PPARγ was restored by rosiglitazone, a PPARγ agonist.
Conclusion: The current report presents a novel mouse model for investigating the role of PPARγ transcription in physiological functions. The data demonstrate that the transcriptional activity plays an indispensable role for PPARγ in metabolic regulation.
Introduction
Obesity is a growing worldwide risk factor for many complications in health, such as type 2 diabetes (T2D) and non-alcoholic fatty liver disease (NAFLD) (Wild et al., 2004; Golay and Ybarra, 2005; Esser et al., 2014; Byrne and Targher, 2015). Peroxisome proliferator-activated receptors (PPARs) are a family of ligand-activated transcription factors that belong to the nuclear hormone receptor superfamily. There are two isoforms of PPARγ, γ1, and γ2. Both of the isoforms are transcribed from the same gene under the control of different promoters leading to a longer N-terminus in PPARγ2 (Fajas et al., 1997) (Supplementary Figure 1). PPARγ1 is expressed in various tissues and highly enriched in adipose tissues, while the expression of PPARγ2 is restricted to adipose tissues (Tontonoz et al., 1994). PPARγ is enriched in both white adipose tissue (WAT) and brown adipose tissue (BAT) (Tyagi et al., 2011). Like many nuclear receptors, PPARγ contains five functional regions: an N-terminal activation function-1 (AF-1) domain (A/B domain), a DNA-binding domain (DBD, domain C), a ligand-binding domain (LBD, domain E), a hinge region that links the DBD and the LBD (domain D), and a C-terminal AF-2 domain in LBD (Jin and Li, 2010). Upon the binding with ligands by its LBD, PPARγ locates to the specific PPAR response element (PPRE) via its DBD as a heterodimer with retinoid X receptor (RXR), and recruits cofactors to regulate the transcription of many direct downstream target genes (Larsen et al., 2003; Jin and Li, 2010; Seale, 2010; Ahmadian et al., 2013). These target genes include glucose transporter type 4 (Glut4), phosphoenolpyruvate carboxykinase (PEPCK) (Tontonoz et al., 1995), fatty acid translocase (FAT/CD36) (Teboul et al., 2001), aquaporin 7 (AQP7, also named AQPap) (Kishida et al., 2001), adipocyte fatty acid binding protein (aP2) gene (Rival et al., 2004), stearoyl-CoA desaturase 1 (SCD1) (Miller and Ntambi, 1996), and uncoupling protein 1 (UCP1) (Petrovic et al., 2010), etc., that are involved in a variety of processes including adipocyte differentiation, glucose metabolism, and insulin sensitivity (Larsen et al., 2003; Jin and Li, 2010; Seale, 2010; Ahmadian et al., 2013). Therefore, PPARγ has been a primary pharmacological target for drug discovery for the treatment of obesity and T2D.
Rosiglitazone (Avandia) and pioglitazone (Actos) belong to an anti-diabetic drug class that targets PPARγ, called thiazolidiniones (TZDs) (Larsen et al., 2003; Karak et al., 2013). As full agonists of PPARγ, TZDs induce the transcription and expression of hundreds of genes by activating PPARγ. Some of these activated genes enhance insulin sensitivity, leading to the therapeutic effects; while activation of some other genes are thought to be the causes of adverse effects of TZDs including weight gain, fluid retention, congestive heart failure, and bone fractures (Ahmadian et al., 2013; Wright et al., 2014). These adverse effects caused by PPARγ full agonists might override the glycemic benefits in T2D patients. In fact, rosiglitazone has ever been suspended by the European Medicines Agency and restricted by the U.S. FDA.
The concept of a disconnect between the agonism potency of PPARγ agonists and their therapeutic property has been proposed years ago (Jones, 2010). Much evidence show that partial agonists of PPARγ, also called selective PPARγ modulators (SPPARM) with poor agonist activities, such as MRL24, INT-131, and MBX-102, exert good anti-diabetic property with fewer adverse effects (Acton et al., 2005; Gregoire et al., 2009; Taygerly et al., 2013). Actually, ligands that do not possess transcriptional agonism can potentially exhibit anti-diabetic property with little adverse effect by blocking Cdk5-mediated phosphorylation of PPARγ (Choi et al., 2011). Therefore, the characterization of PPARγ transcriptional activity in drug discovery remains unclear till now.
The controversy over TZD drugs as diabetic treatment has weakened confidence in developing drugs that target the PPAR family of nuclear receptors. Reports have demonstrated that heterozygous PPARγ-deficient mice exhibit improved insulin sensitivity (Kubota et al., 1999; Miles et al., 2000), supporting the negative role of PPARγ. However, there are also other reports suggesting that PPARγ bearing mutations in DBD or LBD are associated with lipodystrophy (Barroso et al., 1999; Freedman et al., 2005; Agostini et al., 2006; Jeninga et al., 2007). Therefore, there is a dire need to create an applicable model to clarify the role of the transcriptional activity of PPARγ in metabolism. Based on the crystal structure of the PPARγ–RXRα complex bound to PPRE (Chandra et al., 2008), we identified three crucial residues (Arg134, Arg135, and Arg138) on PPARγ that control the binding ability of PPARγ with PPRE and the subsequent transcriptional activity of PPARγ. Therefore, we created a transgenic mouse model containing the three point mutations (R134/135/138A, 3RA) to study the role of PPARγ transcription in metabolism.
Materials and Methods
Protein Purification
Human PPARγ containing domains from DBD to the C-terminus (CDE domains, residues 103–477) was expressed as an N-terminal 6×His fusion protein (H6-PPARγ CDE) from the expression vector pET24a (Novagen, Germany). 3RA mutant plasmid was constructed by site-directed mutagenesis with forward primer: AGGATGCAAGGGTTTCTTCGCGGCAACAA
TCGCATTGAAGCTTATCTATGACAG, and reverse primer: CTGTCATAGATAAGCTTCA
ATGCGATTGTTGCCGCGAAGAAACCCTTGCATCCT, using Pfu DNA polymerase (Thermo Fisher Scientific, USA). BL21(DE3) cells transformed with the expression plasmids were grown in LB broth at 25°C to an OD600 of approximately 1.0 and induced with 0.1 mmol/L isopropyl 1-thio-β-D-galactopyranoside (IPTG) at 16°C. Cells were harvested and sonicated in 100 ml of extract buffer (20 mmol/L Tris pH8.0, 150 mmol/L NaCl, 10% glycerol, and 25 mmol/L imidazole) per 2 liters of cells. After sonication, the lysate was centrifuged at 20,000 rpm for 30 min, and the supernatant was loaded on a 5-ml NiSO4-loaded HiTrap HP column (GE Healthcare, PA, USA). The column was washed with extract buffer, and the protein was eluted with a gradient of 25 to 500 mmol/L imidazole. The PPARγ CDE was further purified with a SP-Sepharose column (GE Healthcare, PA, USA).
AlphaScreen Assay
The binding of H6-PPARγ CDE wild-type (WT) or H6-PPARγ CDE 3RA mutant protein with biotin-labeled PPRE was determined by AlphaScreen assay using a hexahistidine detection kit from Perkin-Elmer. PPRE was prepared by annealing biotin-PPRE-F: AGGGGACCAGGACAAAGGTCACGTTCGGGA and biotin-PPRE-R: TCCCGAACGTGACCTTTGTCCTGGTCCCCT, both of which with 5’ end biotin-labeled. The assay was performed in a buffer containing 50 mmol/L MOPS, 50 mmol/L NaF, 0.05 mmol/L CHAPS, and 0.1 mg/ml bovine serum albumin, all adjusted to a pH of 7.4. The binding assay was performed with 100 nM of protein with gradient doses of biotin-PPRE, or 1 nM of biotin-PPRE with gradient doses of H6-PPARγ CDE with or without 1 µM of rosiglitazone. For the binding of PPARγ CDE with cofactors peptide motifs in response to rosiglitazone, AlphaScreen assay was performed with 100 nM of PPARγ CDE, 100 nM biotin-labeled peptides with gradient doses of rosiglitazone. The sequences of the peptides: SRC1-2, SPSSHSSLTERHKILHRLLQEGSP; SRC2–3, QEPVSPKKKENALLRYLLDKDDTKD; and NCoR-2, GHSFADPASNLGLEDIIRKALMGSF.
Dual Luciferase Report Assay
HEK-293T cells (ATCC, USA) were maintained in DMEM containing 10% fetal bovine serum (FBS) and were transiently transfected using Lipofectamine 2000 reagent (Thermo Fisher Scientific, USA). 24-well plates were plated 24 h prior to transfection (5 x 104 cells per well). 200 ng of pcDNA3.1-Flag-PPARγ WT or 3RA mutant plasmid was co-transfected with 200 ng of PPRE-luc reporter plasmid into cells (Zheng et al., 2013). Renilla was co-transfected as an internal control. 1 µM of rosiglitazone or DMSO was added 5 h after transfection. Cells were harvested 24 h later for the luciferase assays. Luciferase activities were analyzed as the instruction of CheckMate™ Mammalian Two-Hybrid System (Promega, USA).
Generation of PPARγ3RA/+ Mice
A PPARγ BAC clone was screened and isolated from BAC library, mapped by restriction digests and sequenced. The arginine 134, 135, and 138 residues in exon 5 were all paralleled mutated to alanine (3RA) using overlap PCR, and the fragment was cloned into a targeting vector that contains exon 5 homology arm. Meanwhile, the vector contains cassette with a floxed pGK-neor. The vector was then delivered to embryonic stem (ES) cells (C57BL/6) via electroporation, followed by G418 selection, PCR screening, and Southern blot confirmation. Targeted lines were expanded and electroporated with a Cre recombinants expression vector to delete the neor-cassette. Some correct targeted ES clones were selected for blastocyst microinjection, followed by chimera production in C57BL6 background. These mice were then interbred to obtain different genotypes littermate mice for experiments. Mice were maintained under environmentally controlled conditions with free access to diet and water. Animal experiments were conducted in the barrier facility of the Laboratory Animal Center, Xiamen University, approved by the Institutional Animal Use and Care Committee of Xiamen University, China. The methods were carried out in accordance with the approved guidelines.
Mice Treatment
8 week-old male PPARγ3RA/+ and WT littermates were fed with a high-fat diet (HFD, 60% kcal fat, D12492, Research Diets Inc, USA). The body weight of mice were weighed weekly and the food intake was assessed every 4 weeks. Blood samples were obtained by the tail-cut method for small samples every 4 weeks for detecting blood glucose and insulin levels. After 15 weeks of HFD, mice were euthanized after 6 h of fasting. For rosiglitazone treatment study, mice were divided into two groups after a 15-week HFD, and intraperitoneally (i.p.) injected once daily with vehicle (40% of 2-hydroxypropyl-β-cyclodextrin, HBC, Sigma, USA) or 3 mg/kg of rosiglitazone for 6 days. Mice were euthanized after 6 h of fasting. For all mice research, part of liver and fat tissues was fixed in 4% paraformaldehyde for hematoxylin and eosin (H&E) staining by standard procedures. Other tissues were collected and frozen in liquid nitrogen for use. Serum was collected for the measurement of metabolic parameters. Animal experiments were conducted in the barrier facility of the Laboratory Animal Center, Xiamen University, approved by the Institutional Animal Use and Care Committee of Xiamen University, China.
Metabolic Parameters
Serum glucose level was analyzed using glucose oxidase method (Applygen, Beijing, China) (Wang C. et al., 2016). Serum insulin level was determined by ELISA using an ultra-sensitive mouse insulin kit (Crystal Chem, USA) (Ding et al., 2016). Serum levels of total cholesterol, triglycerides, LDL-C, HDL-C, and free fatty acids (FFA) levels were assayed using the calorimetric kits from Nanjing Jiancheng Bioengineering Institute (Nanjing, China) (Jiang et al., 2016; Wang et al., 2019; Liu et al., 2020). Liver TG was analyzed using Tissue triglyceride assay kit (Applygen, Beijing, China) (Wang C. et al., 2016).
GTT and ITT
Glucose tolerance test (GTT) and insulin tolerance test (ITT) were performed in mice before and after a 15-week HFD feeding. For the GTT, mice were fasted for 16 h with free access to water, and then orally gavaged with 1 g/kg body weight of glucose. Blood glucose level was assessed with the Accu-Check Performa (Roche Applied Science, Mannheim, Germany) at 0, 15, 30, 60, 90, and 120 min. For the ITT, mice were fasted for 6 h with free access to water, and then i.p. injected with 1 U/kg of recombinant human insulin (Novolin 30R; Novo Nordisk, Bagsvaerd, Denmark). Blood glucose level was measured at 0, 15, 30, 60, and 120 min after insulin injection.
Gene Expression
The protein level of PPARγ in inguinal WAT (iWAT) was assessed by western blot using mouse monoclonal anti- PPARγ (Santa Cruz, Cat. No. sc-7273, 1:1000) (Chakraborty et al., 2019; Jung et al., 2019) and mouse monoclonal anti-β-actin (Protein Tech, Cat. No. 60008-1-Ig, 1:2000). Total RNA was isolated from liver and fat tissues using Tissue RNA kit (Omega Bio-Tek, GA). The first strand cDNA was reverse-transcribed using TAKARA reverse transcription kit. Real-time quantitative PCR reactions were performed with SYBR Premix Ex TaqTM (TAKARA) on a CFX96™ Real-Time PCR Detection System (Bio-Rad). Relative mRNA expression levels were normalized to β-actin levels. The sequences of the primers used were listed in Supplementary Table 1.
Statistical Analysis
Values were expressed as mean ± standard error of mean (SEM). Statistical differences were calculated by one-way ANOVA followed by the Dunn’s test or Student’s t test. Statistical significance was shown as *p<0.05, **p<0.01 or ***p<0.001.
Result
R134/135/138A Mutations Abolish the PPRE-Binding Ability and the Transcriptional Activity of PPARγ
To evaluate the role of the transcriptional function of PPARγ on metabolism, we attempted to create a mouse model that is deficient in the transcriptional activity of PPARγ while the DBD-independent actions of PPARγ are intact. Because PPARγ needs to bind to PPRE to activate the downstream transcription, we searched for crucial sites in PPARγ to destroy its binding on PPRE. Based on the crystal structure of the PPARγ–RXRα complex bound to PPRE (Chandra et al., 2008), we found that the Arg134, Arg135, and Arg138 residues in PPARγ form six hydrogen bonds in the major groove of the PPRE double helix (Figures 1A–C). However, if the three arginine residues were mutated into alanine, the six hydrogen bonds will not form (Figure 1D). The absence of the hydrogen bonds is predicted to abolish the binding between PPARγ and PPRE while sparing other functions crucial for the transcriptional activity of PPARγ including the zinc finger structure of PPARγ DBD and the ligand-binding LBD (Chandra et al., 2008).
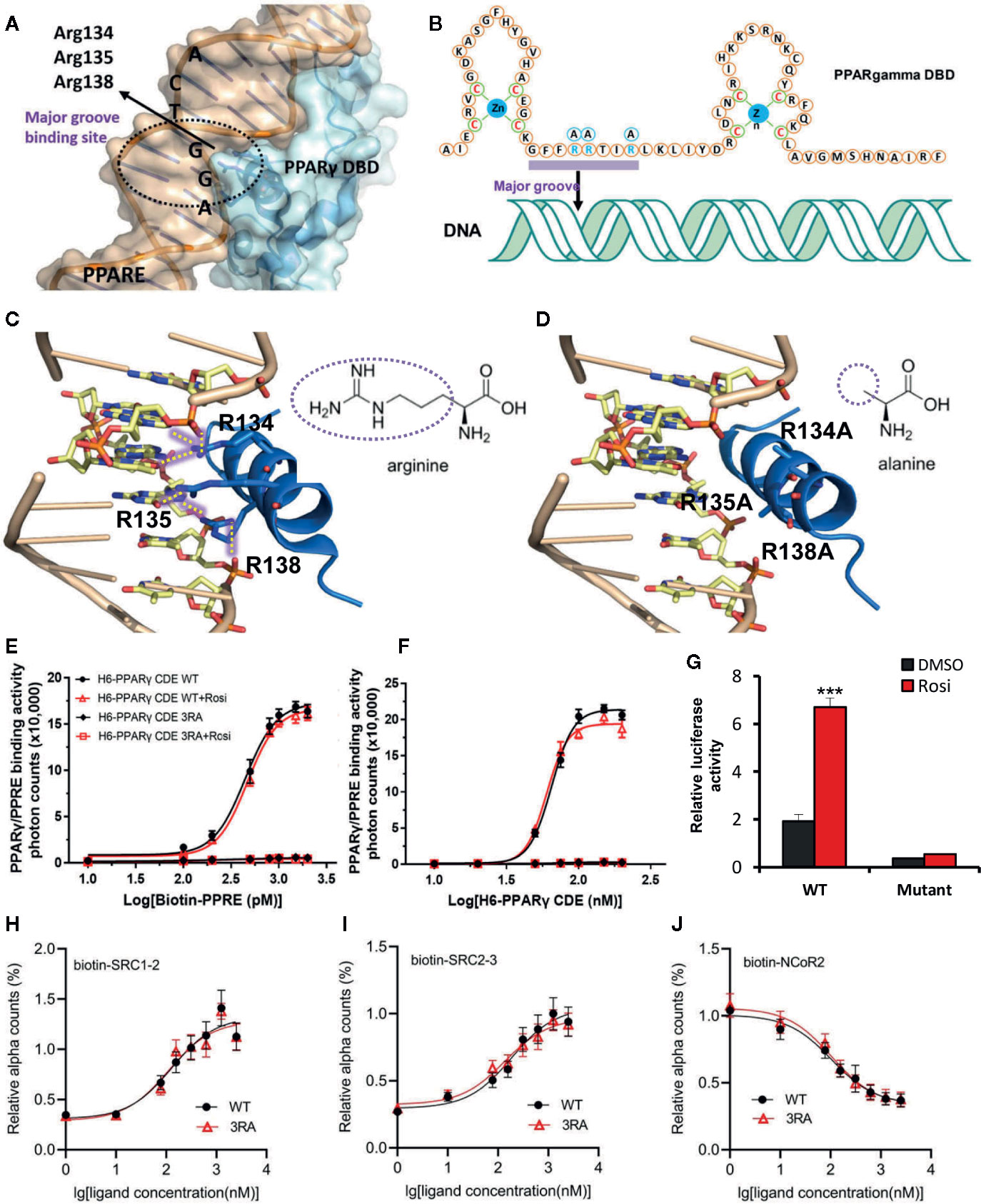
Figure 1 R134/135/138A mutations abolish the PPRE-binding ability and the transcriptional activity of PPARγ. (A) The structure of PPRE and PPARγ interaction shows that Arg134, Arg135 and Arg138 locate into the major groove of PPRE. PPRE is colored in light orange, and PPARγ DBD is in sky blue. (B) Binding model of PPARγ DBD on PPRE. Zinc finger domains of PPARγ DBD and Arg134, Arg135 and Arg138 are shown in the sequence of PPARγ DBD. (C, D) The binding of wild type (WT) PPARγ with PPRE (C) and the predicted binding model of R134/135/138A (3RA) mutant PPARγ with PPRE (D). Hydrogen bonds are indicated by dotted yellow lines. PPRE is colored in light orange, and the helix 1 of PPARγ DBD is in blue. The different groups of arginine and alanine are shown. The structure images in Figures 1A, C, D were generated by open source software PyMOL 099rc6 (www.pymol.org) and the chemical structures were drawn by chemdraw2014. (E) Dose response curve of biotin-PPRE with 100 nM of WT or 3RA mutant PPARγ CDE by AlphaScreen assay. (F) Dose response curve of WT or 3RA mutant PPARγ CDE with 1 nM of biotin-PPRE by AlphaScreen assay. Rosiglitazone (1 μM) does not affect the binding affinity of both WT and 3RA mutant PPARγ in (E, F). (G) Transcriptional activity of WT or 3RA mutant PPARγ by rosiglitazone. HEK-293T cells were co-transfected with WT or 3RA mutant pcDNA3.1-Flag-PPARγ plasmid together with PPRE-luc reporter plasmid. Renilla was co-transfected as an internal control. 1 µM of rosiglitazone or DMSO was added 5 h after transfection. Cells were harvested 24 h later for the luciferase assays. (H–J) 3RA mutation does not affect the interaction of PPARγ CDE with rosiglitazone and co-factors. Dose curves of the interaction between PPARγ CDE WT/3RA and biotin-labeled cofactors: coactivator peptides SRC1-2 (H) and SRC2-3 (I), and corepressor peptide NCoR2 (J), in response to rosiglitazone by AlphaScreen assay. For (E–J), experiments were performed in triplicate and repeated three times with similar results. Data show a representative experiment. Values are means ± SEM, ***p < 0.001 by one-way ANOVA followed by the Dunn’s test.
Therefore, we mutated the three arginine residues at Arg134, Arg135, and Arg138 of PPARγ to alanine (named PPARγ-3RA). The binding ability of PPAR-3RA with PPRE was studied by using AlphaScreen assay. 6 × His tag- PPARγ WT or PPARγ-3RA that contains the domains ranging from DBD, hinge, and the C-terminus LBD of PPARγ (H6-PPARγ CDE) was expressed in BL21 (DE3) and purified for the assay. As expected, the binding signal of the H6-PPARγ CDE WT increased in a PPRE concentration-dependent manner (Figure 1E). In contrast, H6-PPARγ CDE 3RA did not show any binding signal with increasing concentration of PPRE. We obtained similar results when a gradient concentration of H6-PPARγ CDE WT or H6-PPARγ CDE 3RA was used to bind PPRE (Figure 1F). Additionally, the same result was produced with the treatment of rosiglitazone, an agonist for PPARγ (Figures 1E, F), due to the ligand-independent nature of the binding ability of DBD domain to DNA, which is distinct from LBD (Rastinejad et al., 2013). These data confirmed that the PPARγ-3RA mutant lost the binding ability toward PPRE. Next, we tested the transcriptional activity of the PPARγ-3RA mutant. WT or 3RA mutant pcDNA3.1-Flag-PPARγ was co-transfected with PPRE-luc plasmid into HEK-293T cells for luciferase reporter assay. The result showed that the transcriptional activity of PPARγ dramatically decreased after 3RA mutation. Rosiglitazone treatment significantly induced the transcriptional activity of WT PPARγ, but failed to activate PPARγ-3RA (Figure 1G).
To test if the 3RA mutations affect the binding ability of PPARγ with ligands and cofactors, we performed an AlphaScreen assay using PPARγ CDE WT or 3RA protein and biotin-labeled cofactors peptides in response to rosiglitazone. The results showed that both WT and 3RA mutant of PPARγ CDE can recruit co-activators SRC1 and SRC2 and release co-repressor NCoR in response to rosiglitazone (Figures 1H–J). Our data demonstrate that although PPARγ 3RA mutant cannot bind with PPRE (Figures 1E, F), the mutant is still able to bind ligands and cofactors such as SRC1, SRC2, and NCoR.
Together, these results demonstrate that the PPARγ-3RA lost the binding ability toward PPRE and further the DBD-dependent ligand-regulated transcriptional activity of PPARγ, while maintains the ability to bind ligands such as rosiglitazone and cofactors, such as SRC1, SRC2, and NCoR.
Decreased Transcriptional Ability of PPARγ in Heterozygous PPARγ3RA/+ Mice
We mutated codons for amino acids 134, 135, and 138 of the mouse PPARγ gene from CGA (arginine), AGA (arginine) and CGA (arginine) to GCA (alanine), respectively, via gene targeting in mouse ES cells and created genetically modified mouse carrying the 3RA mutations (Figures 2A–C). In 74 progenies born from PPARγ3RA/+ intercrosses, no PPARγ3RA/3RA homozygous mice were obtained. WT and PPARγ3RA/+ littermates were born at the expected Mendelian ratio (26:48 ≈ 1:2) (Figure 2D), indicating that the PPARγ 3RA mutations cause embryonic lethality due to the loss of PPARγ transcriptional activity. These results suggest that the transcriptional function of PPARγ is essential for embryonic development.
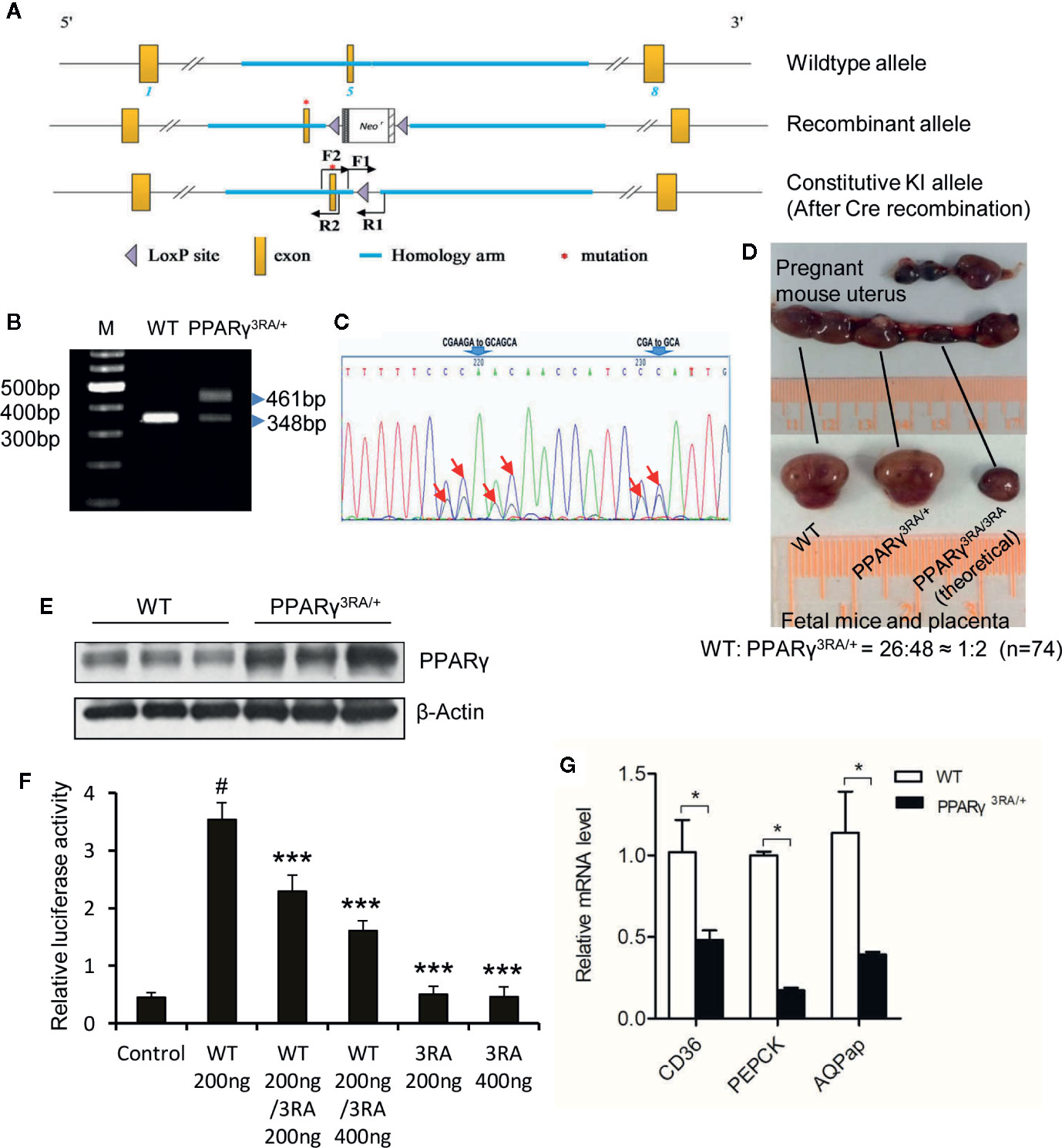
Figure 2 Generation of PPARγ 3RA mutant mouse model. (A) Schematics of the targeting strategy. Top: WT allele. Middle: recombinant allele. Bottom: constitutive knock-in allele after Cre recombination. (B) Genotyping. Genomic DNA extracted from mouse tail was used for PCR with primer pair F1/R1. The PCR product with one band of 348 bp represents the WT PPARγ, and PCR product with two bands (348 bp and 461 bp) represents the heterozygotes. (C) Sequencing analyses of PCR product using primer pair F2/R2 verified the mutations in PPARγ gene. Heterozygotes show two peaks in the codes of Arg134, Arg135 and Arg138, where one peak indicates the allele of wild-type (CGAAGA and CGA), the other is 3RA mutant (GCAGCA and GCA). The mutated nucleotides were indicated by arrows. (D) Homozygous PPARγ 3RA mutant (PPARγ3RA/3RA) mice are embryonic death. Heterozygous pregnant (mate with a heterozygous male) was dissected, about a quarter of the fetal mice in the womb were absorbed by the mother, leaving only the placenta. The ratio of newborn cubs (homozygote/heterozygote) is about 1/2 when we calculated total 74 cubs (n=74). (E) The protein level of PPARγ in the fat tissues of WT and PPARγ3RA/+ mice. (F) In vitro reporter assay. HEK-293T cells were co-transfected with indicated quantity of WT and 3RA mutant pcDNA3.1-Flag-PPARγ plasmid together with PPRE-luc reporter plasmid. Empty pcDNA3.1-Flag vector was the control. Renilla was co-transfected as an internal control. 1 µM of rosiglitazone was added 5 h after transfection. Cells were harvested 24 h later for the luciferase assays. Experiments were performed in triplicate and repeated three times with similar results. Data show a representative experiment. Values are means ± SEM, #p<0.001 versus vector control, ***p < 0.001 versus WT 200 ng, one-way ANOVA followed by the Dunn’s test. (G) Relative mRNA level of PPARγ direct target genes in fat tissues of WT and PPARγ3RA/+ mice by quantitative RT-PCR. Experiments were repeated three times with similar results. Data show a representative experiment. Values are means ± SEM, n=6 per group. *p < 0.05 by Student’s t test.
To confirm the decreased transcriptional ability of PPARγ in heterozygous PPARγ3RA/+ mice, we analyzed gene expression in iWAT from WT and PPARγ3RA/+ mice. The expression level of total PPARγ was higher in iWAT from PPARγ3RA/+ than WT mice (Figure 2E) which might be the compensatory expression due to the loss of transcriptional activity for PPARγ 3RA mutation in vivo. Our in vitro reporter assay indicated that co-existence of PPARγ 3RA mutant significantly reduced the transcriptional activity of WT PPARγ (Figure 2F), further supporting the decreased transcriptional activity in heterozygous PPARγ3RA/+ mice. As expected, the mRNA levels of genes that are directly downstream of PPARγ, such as FAT/CD36, PEPCK, and AQPap, were significantly lower in iWAT of PPARγ3RA/+ mice compared to those of WT littermates (Figure 2G). The differential mRNA expression level of PPARγ downstream genes in WT and PPARγ3RA/+ mice confirmed the impaired transcriptional activity of PPARγ in the PPARγ3RA/+ mice.
PPARγ 3RA Mutations in Mice Exacerbate HFD-Induced Obesity and Adipocyte Hypertrophy
Under chow diet, the WT and PPARγ3RA/+ littermates showed similar phenotypes in body weight, liver/body weight ratio, fat/body weight ratio, as well as the histological analysis of the brown adipose tissue (BAT), WATs and the liver tissue (Figure 3A and Supplementary Figure 2). The similarity in these parameters suggests that the transcriptional activity of one PPARγ allele is enough for maintaining basic metabolism in the absence of external stimuli. Interestingly, when fed HFD, PPARγ3RA/+ mice gained significantly more body weight than WT mice did (Figure 3A) even though food intake was similar (Supplementary Figure 3). After 15 weeks of HFD feeding, the WATs and livers of PPARγ3RA/+ mice weighed significantly more than those of WT mice, while the BAT of PPARγ3RA/+ mice weighed significantly less than that of WT mice (Figures 3B, C). Histological analysis by H&E staining showed larger adipocytes size in the sections of iWAT, gonadal WAT (gWAT), and BAT from PPARγ3RA/+ mice than those of WT mice (Figures 3D, E). These results indicate that heterozygous PPARγ deficiency leads to more severe hypertrophy in white adipocytes and more whitening in brown adipocytes in mice under HFD. Notably, there was also visible inflammatory infiltration in iWAT of PPARγ3RA/+ mice (Figure 3D). Additionally, histological examination showed that heterozygous PPARγ deficiency leads to more lipid accumulation in the liver of mice under HFD (Figure 3F), which was further confirmed by the biochemical analysis of the hepatic triglycerides level (Figure 3G). As the results shown, the fasting plasma levels of total cholesterol (TCHO) (Figure 3H), triglyceride (TG) (Figure 3I), LDL-C, and FFA (Figures 3J, K) of PPARγ3RA/+ were all significantly higher than those of WT littermates, whereas the level of HDL-C (Figure 3L) was significantly lower. These results demonstrate that PPARγ 3RA mutations exacerbated HFD–induced obesity and adipocyte hypertrophy in mice.
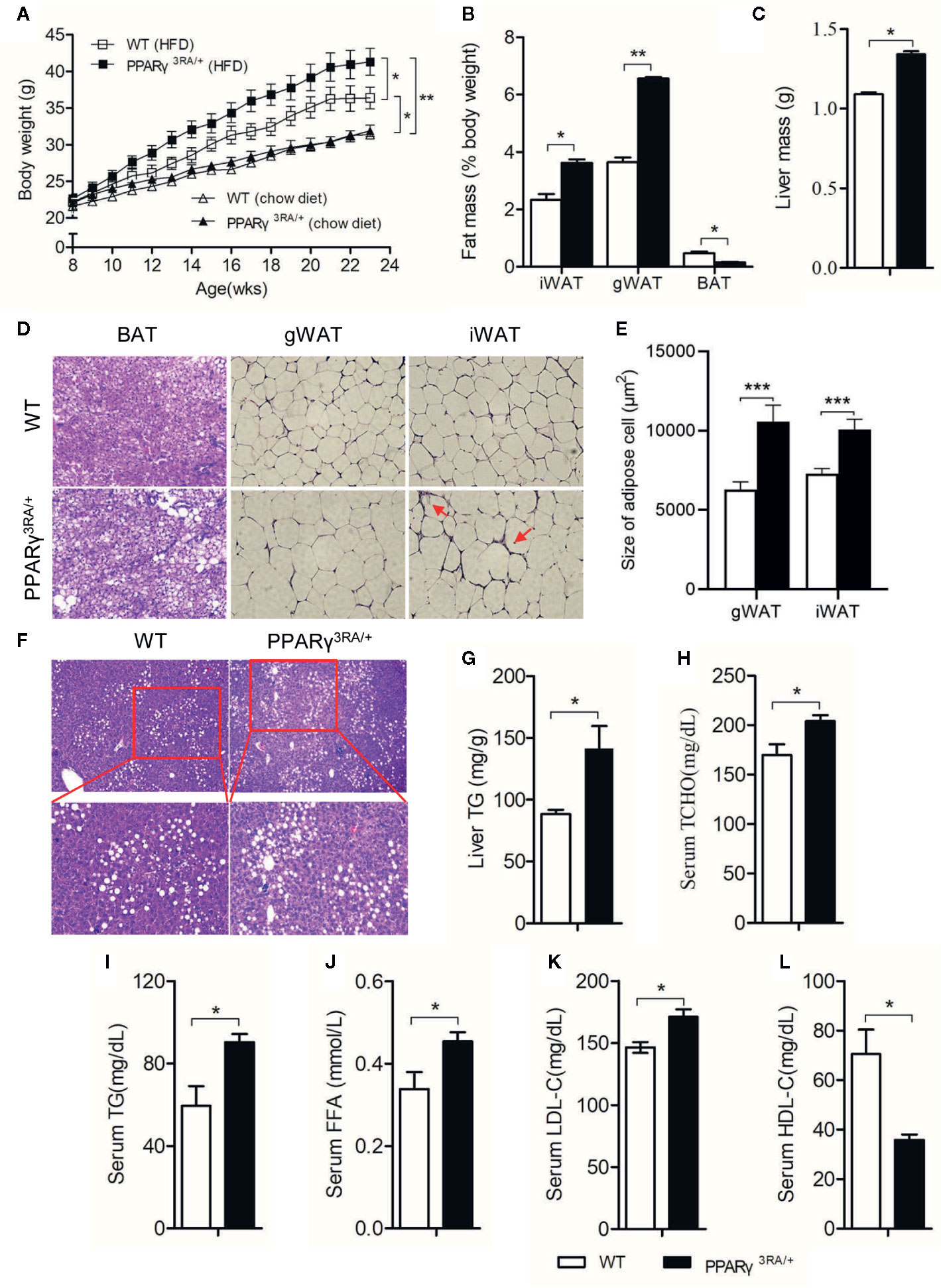
Figure 3 PPARγ 3RA mutations in mice exacerbate HFD-induced obesity and adipocyte hypertrophy. (A) Body weight of PPARγ3RA/+ and WT littermates during a 15 weeks HFD feeding or normal chow diet. Data from the final time point were compared. Samples were collected from 15-week HFD-fed mice for assays. (B) Weights of white adipose tissue (inguinal white fat, iWAT; gonadal white fat, gWAT), brown fat (BAT) and (C) liver mass from PPARγ3RA/+ and WT littermates. (D) Representative histological analysis of iWAT, gWAT and BAT from PPARγ3RA/+ and WT littermates under a HFD by H&E staining. Inflammatory infiltration was labeled by red arrows. Original magnification, 200×. (E) Quantitative statistics of adipocyte size in (D). (F) Representative images of H&E stained liver sections of PPARγ3RA/+ and WT littermates. Original magnification: top, 100×; bottom, 200×. (G) Hepatic triglyceride level of PPARγ3RA/+ and WT littermates. (H, I) Fasting serum levels of total cholesterol (TCHO) (H), triglycerides (TG) (I), free fatty acids (FFA) (J), LDL-C (K) and HDL-C (L) of PPARγ3RA/+ and WT littermates. Values are means ± SEM, n=12 per group, *p < 0.05, **p < 0.01 and ***p < 0.001 by Student’s t test. For (G–L), measurement were repeated three times with similar results. Data show a representative experiment.
PPARγ 3RA Mutations in Mice Exacerbate HFD-Induced Insulin Resistance
Chow diet-fed PPARγ3RA/+ and WT littermates showed similar fasting blood glucose level, glucose tolerance, and insulin tolerance (Figures 4A, B). When fed HFD, PPARγ3RA/+ and WT mice maintained similar fasting blood glucose levels (Figure 4C). However PPARγ3RA/+ mice showed significantly higher level of fasting blood insulin from 8 weeks after HFD-feeding (Figure 4D). Furthermore, PPARγ3RA/+ mice showed impaired glucose tolerance and insulin tolerance compared to their WT counterparts, suggesting that PPARγ 3RA mutations in mice exacerbate HFD-induced insulin resistance (Figures 4E, F). Taken together, our data demonstrate that the decreased transcriptional activity of PPARγ in PPARγ3RA/+ mice led to impairment of lipid and glucose metabolism under HFD.
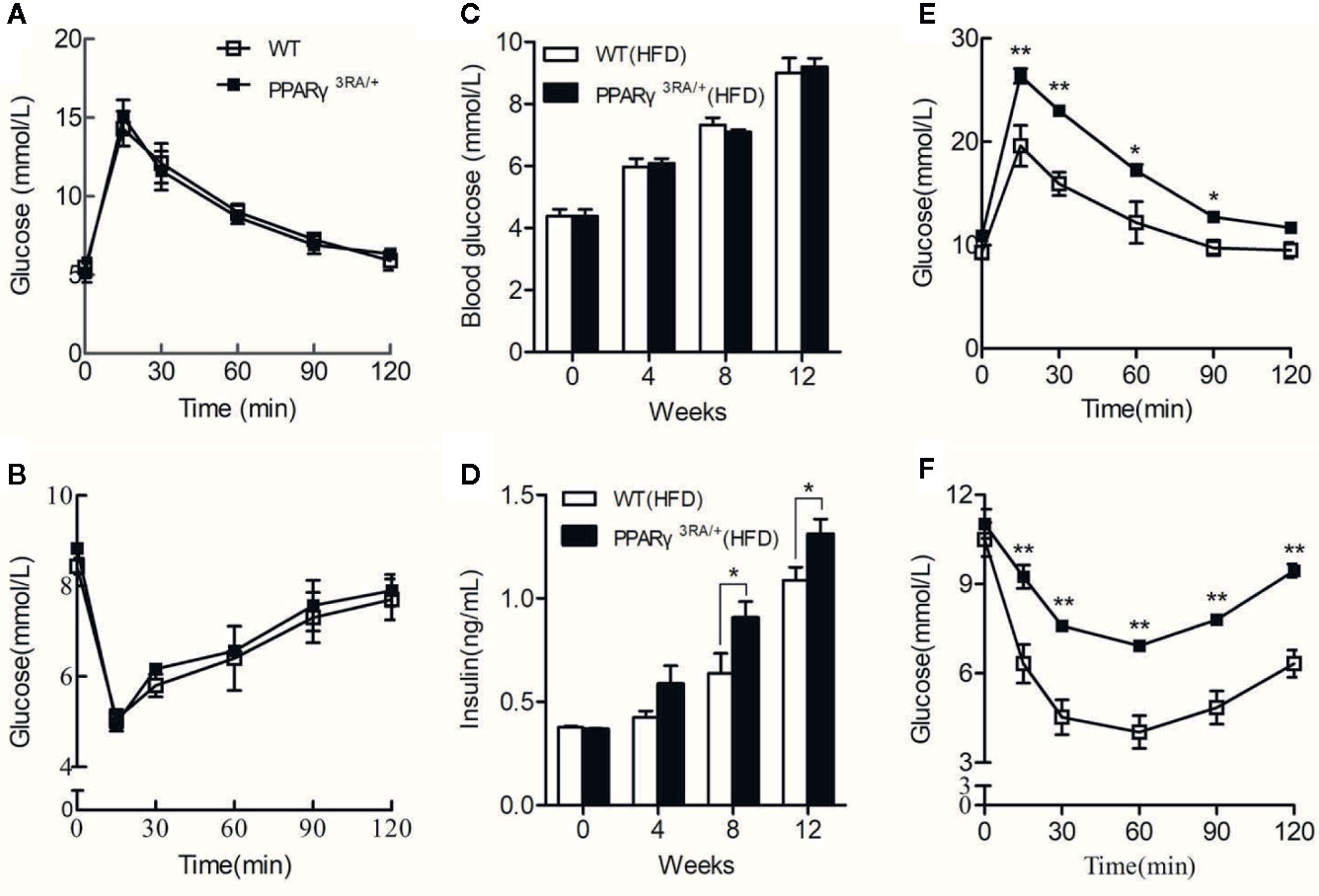
Figure 4 PPARγ 3RA mutations exacerbate HFD-induced insulin resistance in mice. Oral glucose tolerance test (OGTT) (A) and intraperitoneal insulin tolerance test (IPITT) (B) of 8-week old PPARγ3RA/+ and WT littermates with chow diet. From 8 weeks age, mice were fed with HFD for 15 weeks. Fasting blood glucose (C) and insulin (D) levels of mice fed with HFD. OGTT (E) and IPITT (F) of mice fed with HFD for 15 weeks. For (C, D), values are means ± SEM, n=6 per group, *p < 0.05, **p < 0.01 by Student’s t test.
Metabolic Disorders in HFD-Fed PPARγ3RA/+ Mice Were Improved by Rosiglitazone Treatment
Next, we aimed to investigate whether or not the metabolic disorders induced by HFD in PPARγ3RA/+ mice could be reversed by increasing the transcriptional activity of PPARγ. Rosiglitazone was administered at 3 mg/kg once daily for 6 days to PPARγ3RA/+ and WT littermates that had been fed HFD for 15 weeks. Metabolic parameters were studied after the treatment. As shown in Figure 5, rosiglitazone treatment significantly reduced or showed the tendency to reduce the levels of cholesterol, triglyceride, FFA, LDL-C, and glucose in the serum, while increased the level of HDL-C in the serum of both PPARγ3RA/+ and WT littermates (Figures 5A–F). Histological examination further showed that after HFD feeding, both WT and PPARγ3RA/+ mice administrated with rosiglitazone showed less fat vacuoles in BAT and smaller adipocyte size in WAT (Figures 5G, H). Notably, rosiglitazone administration significantly improved the inflammation in WAT of HFD-fed PPARγ3RA/+ mice (Figure 5G). Additionally, rosiglitazone treatment not only efficaciously improved the hepatic steatosis in WT mice, but also in PPARγ3RA/+ mice (Figure 5I).
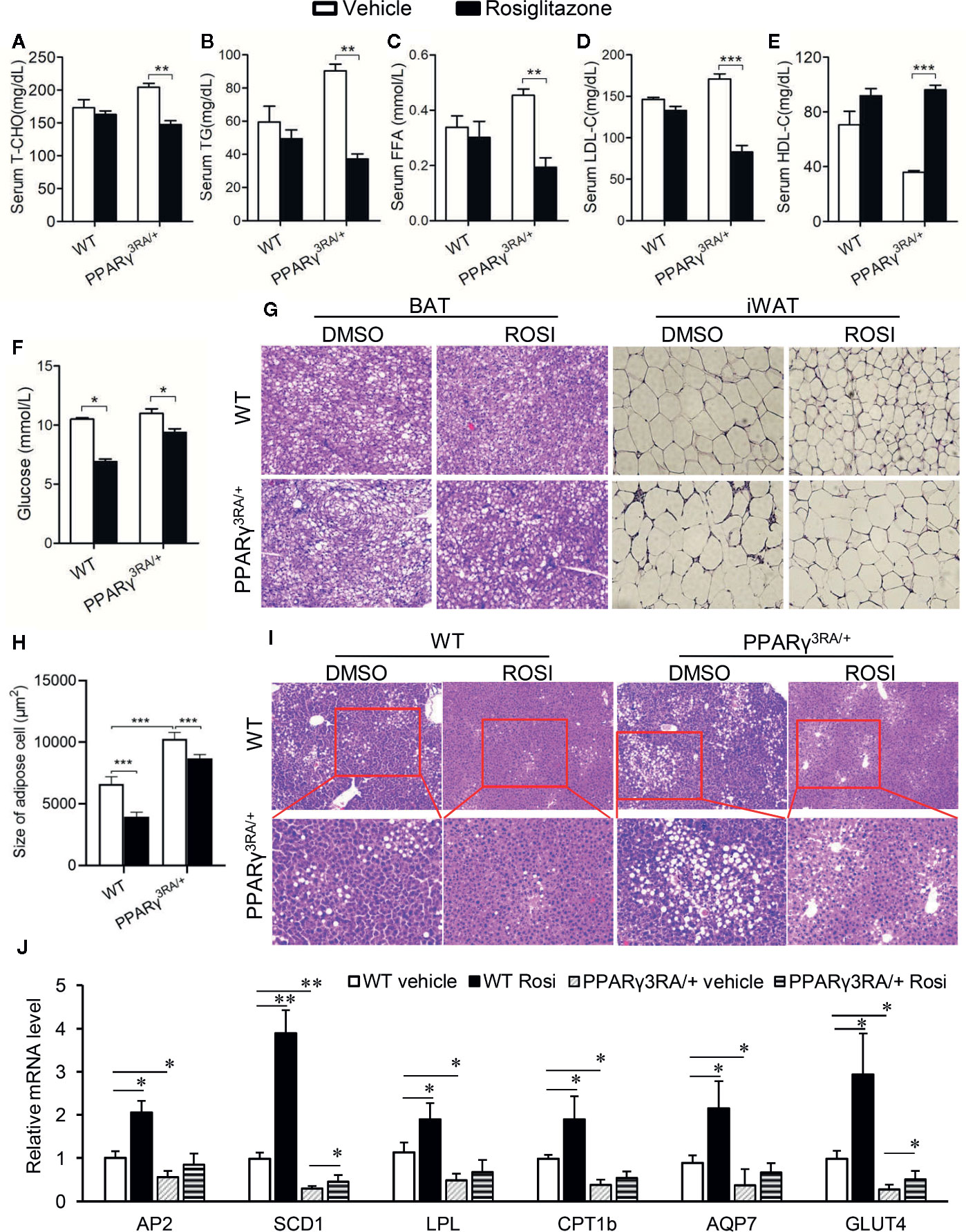
Figure 5 Metabolic disorders in HFD-fed PPARγ3RA/+ mice were improved by rosiglitazone treatment. 8-week old male PPARγ3RA/+ and WT littermates were fed with HFD for 15 weeks, and then i.p. injected with 3 mg/kg of rosiglitazone once daily for 6 days. (A–F) Fasting serum levels of TCHO (A), TG (B) FFA (C), LDL-C (D) HDL-C (E) and glucose (F). (G) Representative images of H&E stained BAT and iWAT (original magnification, 200×). (H) Quantitative statistics of adipose cell size in (G). (I) Representative images of H&E stained liver sections (original magnification: top, 100×; bottom, 200×). (J) mRNA levels of PPARγ direct target genes involved in glucose and lipid metabolism in liver tissues of mice. For (A–F, J), measurement were repeated three times with similar results. Data show a representative experiment. Values are means ± SEM, n=6 per group, *p < 0.05, **p < 0.01 and ***p < 0.001 by one-way ANOVA followed by the Dunn’s test.
We further investigated the mRNA levels of PPARγ target genes in the adipose tissue. As the results showed, the mRNA levels of the target genes of PPARγ we tested were significantly decreased in PPARγ3RA/+ mice compared to those in WT mice (Figure 5J). Rosiglitazone treatment significantly induced the expression PPARγ target genes in WT mice; in PPARγ3RA/+ mice, rosiglitazone treatment restored the expression level of PPARγ target genes in PPARγ3RA/+ mice to levels similar to those of vehicle-treated WT mice (Figure 5J). Taken the rosiglitazone-induced metabolic improvement and gene expression restoration together, our results suggest that increasing PPARγ transcriptional activity could overcome the HFD-induced obesity and adipocyte hypertrophy in PPARγ3RA/+ mice.
Discussion
In this study, based on the structural analysis of PPARγ/PPRE complex (Chandra et al., 2008), the roles of arginine at 134, 135, and 138 residues of PPARγ in binding PPRE, ligand (rosiglitazone) and cofactors (SRC1, SRC2, and NCoR) were verified by in vitro biochemical AlphaScreen and cell-based reporter assays. PPARγ may also possess regulatory mechanisms independent of DBD, such as PPARγ Ser273 phosphorylation mediated by CDK5 (Choi et al., 2011). Because previous reports have demonstrated that the regulation of PPARγ Ser273 phosphorylation can be detected by in vitro kinase assay in a reaction system including PPARγ LBD, CDK5, and ATP (Choi et al., 2011; Zheng et al., 2013), and the results from in vitro assay are consistent with that from in vivo assay, these results suggest that the post-translational phosphorylation of PPARγ at Ser273 by CDK5 is not rely on the existence of PPARγ DBD. Thus, the phosphorylation mediated by CDK5 may also be preserved in the 3RA mutant, although this has not been confirmed experimentally. Together, these data suggest that DBD-independent PPARγ regulations are intact in the PPARγ 3RA mutant. Therefore, we created a knock-in mouse model containing the PPARγ 3RA mutations. Homozygous PPARγ3RA/3RA leads to embryonic death, suggesting the necessary role of the transcriptional activity of PPARγ for development. Chow diet fed PPARγ3RA/+ mice showed decreased transcriptional activity of PPARγ, while maintained comparable phenotypes with the WT littermate mice, suggesting that the one PPARγ alleles is sufficient to maintain the organismal metabolic network without stimuli. However, due to impaired transcriptional activity, PPARγ3RA/+ mice cannot sustain the burden of HFD stimuli, and appeared more severe insulin resistance and obesity. Accordingly, PPARγ agonist treatment rescued the activity of PPARγ, and restored the metabolic disorders in HFD-fed PPARγ3RA/+ mice. These results would indicate the important role of the transcriptional activity of PPARγ in protecting mice from HFD-stimulated metabolic disorders.
PPARγ plays crucial roles in maintaining the homeostasis of glucose and lipid metabolism. Over activating or debilitating its downstream signaling may cause the imbalance of the homeostasis (Rubenstrunk et al., 2007). For example, a water/glycerol transporting protein AQP7 regulates adipocyte glycerol efflux and influences lipid and glucose homeostasis. The deletion of AQP7 gene in mice leads to obesity and T2D (Rodriguez et al., 2006). However, it has also been reported that either increased or decreased AQP7 expression may lead to impaired glycerol dynamics and adipocyte hypertrophy (Oikonomou et al., 2018). Another example is that GLUT-4 is necessary for the insulin-regulated glucose uptake into muscle and fat cells which keeps the glucose homeostasis (Wu et al., 1998). However, if GLUT4 is over-expressed, it will send excess glucose into adipose tissue, leading to increased adipose cell hypertrophy and obesity (Shepherd et al., 1993). Also, overexpression of SCD1 in humans may be involved in the development of hypertriglyceridemia, atherosclerosis, and diabetes (Mar-Heyming et al., 2008). While inhibiting SCD1 function may also result in the accumulation of fatty acid metabolites that are deleterious to insulin signaling, and accordingly, the development of fatty acid-induced insulin resistance (Pinnamaneni et al., 2006). Thus, disorders of these genes will result in an imbalance of nutrients distribution and lead to obesity and diabetes. Rosiglitazone induces the expression of PPARγ target genes, which may provide a potential lighthouse to explain the adverse effects of long-term administration of TZD drugs (Rubenstrunk et al., 2007), as well as the metabolic improvement in HFD-fed PPARγ3RA/+ mice.
A number of laboratories have reported metabolic changes observed in heterozygous PPARγ-deficient mice (Barak et al., 1999; Kubota et al., 1999; Miles et al., 2000). Contrary to our finding, Kubota and colleagues reported that heterozygous PPARγ-deficient mice were protected from the development of insulin resistance caused by adipocyte hypertrophy after HFD-feeding. After administration of pioglitazone, the mice showed worsened phenotypes. Similarly, another group has reported improved insulin-sensitivity in an independently generated heterozygous PPARγ-deficient mouse model (Miles et al., 2000). These reports seem to approbate the negative roles of PPARγ transcriptional activity in metabolic regulation. However, PPARγ is required for adipose tissue development. Barak et al. found that the absence of PPARγ in mice leads to complete lipodystrophy (Barak et al., 1999), indicating the necessary role of PPARγ in lipid metabolism. It should be noted that besides transcriptional regulation, all of the five domains of PPARγ are involved in modulating the PPARγ signaling cascades (Wang S. B. et al., 2016). In this process, except transcriptional regulation by binding to PPREs, cofactors binding, and post-translational modifications including phosphorylation, acetylation, sumoylation, and ubiquitination throughout the full length of PPARγ also contribute to the functions regulated by PPARγ (Jin and Li, 2010; Ahmadian et al., 2013). However, both of the heterozygeous PPARγ-deficient mouse models by groups of Kubota and Barak eliminate most of the domains of PPARγ from DBD to the C-terminus (Barak et al., 1999), and thus the PPARγ in these models lost not only the transcription activity, but also other functional regulations. On the contrary, our PPARγ 3RA model is only mutated at three residues in DBD which contribute to PPARγ transcription deficiency, therefore may represent a suitable tool for the research of the role of transcriptional function of PPARγ in metabolism. The different phenotypes between PPARγ-deficient mice and PPARγ 3RA mutant mice further suggest that PPARγ needs to coordinate its transcriptional activity and its non-transcriptional regulatory actions for metabolic regulation.
Among the domains in nuclear receptors, the sequence of DBD shows the highest evolutionary conservation (Jin and Li, 2010; Helsen et al., 2012). Importantly, the three arginine residues we selected for mutation are conserved from birds to mammals including rodents and humans (Supplementary Figure 4), suggesting the conserved function or the PPARγ 3RA mutant in evolution, including humans. Additionally, the PPRE for PPARγ binding is also conserved with a direct repeats of hexameric sequence AGGTCA in different target genes, although each gene has distinct flanking sequence for its selective regulation (Khorasanizadeh and Rastinejad, 2001). Therefore, our PPARγ 3RA mutant model is a suitable tool for the research of PPARγ transcription in evolution.
In conclusion, we provide an alternative mouse model for further research on the transcriptional activity of PPARγ, and also for the drug discovery by targeting PPARγ. It should be noted that more detailed investigation about the DBD-independent PPARγ actions will further improve the significance of this mouse model. Considering the embryonic death of the PPARγ-3RA mice, future research will focus on creating homozygous conditional knockout mouse model with tissue specific PPARγ 3RA mutations to completely investigate the role of PPARγ transcriptional activity in specific tissues, particularly the adipose tissues and liver.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
Ethics Statement
The animal study was reviewed and approved by the Laboratory Animal Center, Xiamen University.
Author Contributions
YLi, LJ and YH designed the experiment, LJ and FG wrote and revised the manuscript. FG, SX, and YZ performed experiments. YLi and JT discussed and revised the manuscript. XZ assisted with the mice experiments. YLu and FG contributed to the structural analysis. All authors contributed to the article and approved the submitted version.
Funding
work was supported by grants from the National Natural Science Foundation of China (81773793 and 31770814), the Fundamental Research Funds for the Central Universities (20720150052), the Programme of Introducing Talents of Discipline to Universities (B12001), and the National Science Foundation of China for Fostering Talents in Basic Research (J1310027).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2020.01285/full#supplementary-material
References
Acton, J. J. 3., Black, R. M., Jones, A. B., Moller, D. E., Colwell, L., Doebber, T. W., et al. (2005). Benzoyl 2-methyl indoles as selective PPARgamma modulators. Bioorg. Med. Chem. Lett. 15, 357–362. doi: 10.1016/j.bmcl.2004.10.068
Agostini, M., Schoenmakers, E., Mitchell, C., Szatmari, I., Savage, D., Smith, A., et al. (2006). Non-DNA binding, dominant-negative, human PPARgamma mutations cause lipodystrophic insulin resistance. Cell Metab. 4, 303–311. doi: 10.1016/j.cmet.2006.09.003
Ahmadian, M., Suh, J. M., Hah, N., Liddle, C., Atkins, A. R., Downes, M., et al. (2013). PPAR gamma signaling and metabolism: the good, the bad and the future. Nat. Med. 19, 557–566. doi: 10.1038/nm.3159
Barak, Y., Nelson, M. C., Ong, E. S., Jones, Y. Z., Ruiz-Lozano, P., Chien, K. R., et al. (1999). PPAR gamma is required for placental, cardiac, and adipose tissue development. Mol. Cell 4, 585–595. doi: 10.1016/S1097-2765(00)80209-9
Barroso, I., Gurnell, M., Crowley, V. E., Agostini, M., Schwabe, J. W., Soos, M. A., et al. (1999). Dominant negative mutations in human PPARgamma associated with severe insulin resistance, diabetes mellitus and hypertension. Nature 402, 880–883. doi: 10.1038/47254
Byrne, C. D., Targher, G. (2015). NAFLD: a multisystem disease. J. Hepatol. 62, S47–S64. doi: 10.1016/j.jhep.2014.12.012
Chakraborty, P., Vaena, S. G., Thyagarajan, K., Chatterjee, S., Al-Khami, A., Selvam, S. P., et al. (2019). Pro-Survival Lipid Sphingosine-1-Phosphate Metabolically Programs T Cells to Limit Anti-tumor Activity. Cell Rep. 28, 1879–187+. doi: 10.1016/j.celrep.2019.07.044
Chandra, V., Huang, P., Hamuro, Y., Raghuram, S., Wang, Y., Burris, T. P., et al. (2008). Structure of the intact PPAR-gamma-RXR- nuclear receptor complex on DNA. Nature 456, 350–356. doi: 10.1038/nature07413
Choi, J. H., Banks, A. S., Kamenecka, T. M., Busby, S. A., Chalmers, M. J., Kumar, N., et al. (2011). Antidiabetic actions of a non-agonist PPAR gamma ligand blocking Cdk5-mediated phosphorylation. Nature 477, 477–U131. doi: 10.1038/nature10383
Ding, L., Sousa, K. M., Jin, L., Dong, B., Kim, B. W., Ramirez, R., et al. (2016). Vertical sleeve gastrectomy activates GPBAR-1/TGR5 to sustain weight loss, improve fatty liver, and remit insulin resistance in mice. Hepatology 64, 760–773. doi: 10.1002/hep.28689
Esser, N., Legrand-Poels, S., Piette, J., Scheen, A. J., Paquot, N. (2014). Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res. Clin. Pract. 105, 141–150. doi: 10.1016/j.diabres.2014.04.006
Fajas, L., Auboeuf, D., Raspe, E., Schoonjans, K., Lefebvre, A. M., Saladin, R., et al. (1997). The organization, promoter analysis, and expression of the human PPARgamma gene. J. Biol. Chem. 272, 18779–18789. doi: 10.1074/jbc.272.30.18779
Freedman, B. D., Lee, E. J., Park, Y., Jameson, J. L. (2005). A dominant negative peroxisome proliferator-activated receptor-gamma knock-in mouse exhibits features of the metabolic syndrome. J. Biol. Chem. 280, 17118–17125. doi: 10.1074/jbc.M407539200
Golay, A., Ybarra, J. (2005). Link between obesity and type 2 diabetes. Best Pract. Res. Clin. Endocrinol. Metab. 19, 649–663. doi: 10.1016/j.beem.2005.07.010
Gregoire, F. M., Zhang, F., Clarke, H. J., Gustafson, T. A., Sears, D. D., Favelyukis, S., et al. (2009). MBX-102/JNJ39659100, a novel peroxisome proliferator-activated receptor-ligand with weak transactivation activity retains antidiabetic properties in the absence of weight gain and edema. Mol. Endocrinol. 23, 975–988. doi: 10.1210/me.2008-0473
Helsen, C., Kerkhofs, S., Clinckemalie, L., Spans, L., Laurent, M., Boonen, S., et al. (2012). Structural basis for nuclear hormone receptor DNA binding. Mol. Cell Endocrinol. 348, 411–417. doi: 10.1016/j.mce.2011.07.025
Jeninga, E. H., Van Beekum, O., Van Dijk, A. D., Hamers, N., Hendriks-Stegeman, B. I., Bonvin, A. M., et al. (2007). Impaired peroxisome proliferator-activated receptor gamma function through mutation of a conserved salt bridge (R425C) in familial partial lipodystrophy. Mol. Endocrinol. 21, 1049–1065. doi: 10.1210/me.2006-0485
Jiang, J., Wu, Y., Wang, X., Lu, L., Wang, L., Zhang, B., et al. (2016). Blood Free Fatty Acids Were Not Increased in High-Fat Diet Induced Obese Insulin-Resistant Animals. Obes. Res. Clin. Pract. 10, 207–210. doi: 10.1016/j.orcp.2015.06.005
Jin, L., Li, Y. (2010). Structural and functional insights into nuclear receptor signaling. Adv. Drug Delivery Rev. 62, 1218–1226. doi: 10.1016/j.addr.2010.08.007
Jones, D. (2010). Potential remains for PPAR-targeted drugs. Nat. Rev. Drug Discovery 9, 668–669. doi: 10.1038/nrd3271
Jung, S. M., Hung, C. M., Hildebrand, S. R., Sanchez-Gurmaches, J., Martinez-Pastor, B., Gengatharan, J. M., et al. (2019). Non-canonical mTORC2 Signaling Regulates Brown Adipocyte Lipid Catabolism through SIRT6-FoxO1. Mol. Cell 75, 807–80+. doi: 10.1016/j.molcel.2019.07.023
Karak, M., Bal, N. C., Bal, C., Sharon, A. (2013). Targeting peroxisome proliferator-activated receptor gamma for generation of antidiabetic drug. Curr. Diabetes Rev. 9, 275–285. doi: 10.2174/15733998113099990065
Khorasanizadeh, S., Rastinejad, F. (2001). Nuclear-receptor interactions on DNA-response elements. Trends Biochem. Sci. 26, 384–390. doi: 10.1016/S0968-0004(01)01800-X
Kishida, K., Shimomura, I., Nishizawa, H., Maeda, N., Kuriyama, H., Kondo, H., et al. (2001). Enhancement of the aquaporin adipose gene expression by a peroxisome proliferator-activated receptor gamma. J. Biol. Chem. 276, 48572–48579. doi: 10.1074/jbc.M108213200
Kubota, N., Terauchi, Y., Miki, H., Tamemoto, H., Yamauchi, T., Komeda, K., et al. (1999). PPAR gamma mediates high-fat diet-induced adipocyte hypertrophy and insulin resistance. Mol. Cell 4, 597–609. doi: 10.1016/S1097-2765(00)80210-5
Larsen, T. M., Toubro, S., Astrup, A. (2003). PPARgamma agonists in the treatment of type II diabetes: is increased fatness commensurate with long-term efficacy? Int. J. Obes. 27, 147–161. doi: 10.1038/sj.ijo.802223
Liu, L., Tan, L., Yao, J., Yang, L. (2020). Long non-coding RNA MALAT1 regulates cholesterol accumulation in ox-LDL-induced macrophages via the microRNA-17-5p/ABCA1 axis. Mol. Med. Rep. 21, 1761–1770. doi: 10.3892/mmr.2020.10987
Mar-Heyming, R., Miyazaki, M., Weissglas-Volkov, D., Kolaitis, N. A., Sadaat, N., Plaisier, C., et al. (2008). Association of stearoyl-CoA desaturase 1 activity with familial combined hyperlipidemia. Arterioscler. Thromb. Vasc. Biol. 28, 1193–1199. doi: 10.1161/ATVBAHA.107.160150
Miles, P. D., Barak, Y., He, W., Evans, R. M., Olefsky, J. M. (2000). Improved insulin-sensitivity in mice heterozygous for PPAR-gamma deficiency. J. Clin. Invest. 105, 287–292. doi: 10.1172/JCI8538
Miller, C. W., Ntambi, J. M. (1996). Peroxisome proliferators induce mouse liver stearoyl-CoA desaturase 1 gene expression. Proc. Natl. Acad. Sci. U.S.A. 93, 9443–9448. doi: 10.1073/pnas.93.18.9443
Oikonomou, E., Kostopoulou, E., Rojas-Gil, A. P., Georgiou, G., Spiliotis, B. E. (2018). Adipocyte aquaporin 7 (AQP7) expression in lean children and children with obesity. Possible involvement in molecular mechanisms of childhood obesity. J. Pediatr. Endocrinol. Metab. 31, 1081–1089. doi: 10.1515/jpem-2018-0281
Petrovic, N., Walden, T. B., Shabalina, I. G., Timmons, J. A., Cannon, B., Nedergaard, J. (2010). Chronic peroxisome proliferator-activated receptor gamma (PPARgamma) activation of epididymally derived white adipocyte cultures reveals a population of thermogenically competent, UCP1-containing adipocytes molecularly distinct from classic brown adipocytes. J. Biol. Chem. 285, 7153–7164. doi: 10.1074/jbc.M109.053942
Pinnamaneni, S. K., Southgate, R. J., Febbraio, M. A., Watt, M. J. (2006). Stearoyl CoA desaturase 1 is elevated in obesity but protects against fatty acid-induced skeletal muscle insulin resistance in vitro. Diabetologia 49, 3027–3037. doi: 10.1007/s00125-006-0427-9
Rastinejad, F., Huang, P., Chandra, V., Khorasanizadeh, S. (2013). Understanding nuclear receptor form and function using structural biology. J. Mol. Endocrinol. 51, T1–T21. doi: 10.1530/JME-13-0173
Rival, Y., Stennevin, A., Puech, L., Rouquette, A., Cathala, C., Lestienne, F., et al. (2004). Human adipocyte fatty acid-binding protein (aP2) gene promoter-driven reporter assay discriminates nonlipogenic peroxisome proliferator-activated receptor gamma ligands. J. Pharmacol. Exp. Ther. 311, 467–475. doi: 10.1124/jpet.104.068254
Rodriguez, A., Catalan, V., Gomez-Ambrosi, J., Fruhbeck, G. (2006). Role of aquaporin-7 in the pathophysiological control of fat accumulation in mice. FEBS Lett. 580, 4771–4776. doi: 10.1016/j.febslet.2006.07.080
Rubenstrunk, A., Hanf, R., Hum, D. W., Fruchart, J. C., Staels, B. (2007). Safety issues and prospects for future generations of PPAR modulators. Biochim. Biophys. Acta 1771, 1065–1081. doi: 10.1016/j.bbalip.2007.02.003
Seale, P. (2010). Transcriptional control of brown adipocyte development and thermogenesis. Int. J. Obes. (Lond) 34 (Suppl 1), S17–S22. doi: 10.1038/ijo.2010.178
Shepherd, P. R., Gnudi, L., Tozzo, E., Yang, H., Leach, F., Kahn, B. B. (1993). Adipose cell hyperplasia and enhanced glucose disposal in transgenic mice overexpressing GLUT4 selectively in adipose tissue. J. Biol. Chem. 268, 22243–22246.
Taygerly, J. P., Mcgee, L. R., Rubenstein, S. M., Houze, J. B., Cushing, T. D., Li, Y., et al. (2013). Discovery of INT131: a selective PPARgamma modulator that enhances insulin sensitivity. Bioorg. Med. Chem. 21, 979–992. doi: 10.1016/j.bmc.2012.11.058
Teboul, L., Febbraio, M., Gaillard, D., Amri, E. Z., Silverstein, R., Grimaldi, P. A. (2001). Structural and functional characterization of the mouse fatty acid translocase promoter: activation during adipose differentiation. Biochem. J. 360, 305–312. doi: 10.1042/bj3600305
Tontonoz, P., Hu, E., Graves, R. A., Budavari, A. I., Spiegelman, B. M. (1994). mPPAR gamma 2: tissue-specific regulator of an adipocyte enhancer. Genes Dev. 8, 1224–1234. doi: 10.1101/gad.8.10.1224
Tontonoz, P., Hu, E., Devine, J., Beale, E. G., Spiegelman, B. M. (1995). PPAR gamma 2 regulates adipose expression of the phosphoenolpyruvate carboxykinase gene. Mol. Cell Biol. 15, 351–357. doi: 10.1128/MCB.15.1.351
Tyagi, S., Gupta, P., Saini, A. S., Kaushal, C., Sharma, S. (2011). The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J. Adv. Pharm. Technol. Res. 2, 236–240. doi: 10.4103/2231-4040.90879
Wang, C., Jiang, J.-D., Wu, W., Kong, W.-J. (2016). The Compound of Mangiferin-Berberine Salt Has Potent Activities in Modulating Lipid and Glucose Metabolisms in HepG2 Cells. BioMed. Res. Int. 5, 1–14. doi: 10.1155/2016/8753436
Wang, S. B., Dougherty, E. J., Danner, R. L. (2016). PPAR gamma signaling and emerging opportunities for improved therapeutics. Pharmacol. Res. 111, 76–85. doi: 10.1016/j.phrs.2016.02.028
Wang, L., Fan, W., Zhang, M., Zhang, Q., Li, L., Wang, J., et al. (2019). Antiobesity, Regulation of Lipid Metabolism, and Attenuation of Liver Oxidative Stress Effects of Hydroxy-alpha-sanshool Isolated from Zanthoxylum bungeanum on High-Fat Diet-Induced Hyperlipidemic Rats. Oxid. Med. Cell Longev. 2019, 5852494. doi: 10.1155/2019/5852494
Wild, S., Roglic, G., Green, A., Sicree, R., King, H. (2004). Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care 27, 1047–1053. doi: 10.2337/diacare.27.5.1047
Wright, M. B., Bortolini, M., Tadayyon, M., Bopst, M. (2014). Minireview: Challenges and opportunities in development of PPAR agonists. Mol. Endocrinol. 28, 1756–1768. doi: 10.1210/me.2013-1427
Wu, Z. D., Xie, Y. H., Morrison, R. F., Bucher, N. L. R., Farmer, S. R. (1998). PPAR gamma induces the insulin-dependent glucose transporter GLUT4 in the absence of C/EBP alpha during the conversion of 3T3 fibroblasts into adipocytes. J. Clin. Invest. 101, 22–32. doi: 10.1172/JCI1244
Zheng, W., Feng, X., Qiu, L., Pan, Z., Wang, R., Lin, S., et al. (2013). Identification of the antibiotic ionomycin as an unexpected peroxisome proliferator-activated receptor gamma (PPAR gamma) ligand with a unique binding mode and effective glucose-lowering activity in a mouse model of diabetes. Diabetologia 56, 401–411. doi: 10.1007/s00125-012-2777-9
Keywords: PPARγ, DNA binding, transcriptional activity, metabolic disorder, mutant, obesity, insulin resistance
Citation: Guo F, Xu S, Zhu Y, Zheng X, Lu Y, Tu J, He Y, Jin L and Li Y (2020) PPARγ Transcription Deficiency Exacerbates High-Fat Diet-Induced Adipocyte Hypertrophy and Insulin Resistance in Mice. Front. Pharmacol. 11:1285. doi: 10.3389/fphar.2020.01285
Received: 19 March 2020; Accepted: 03 August 2020;
Published: 19 August 2020.
Edited by:
Andres Trostchansky, Universidad de la República, UruguayReviewed by:
Ana María Ferreira, Universidad de la República, UruguayFederica Gilardi, Centre Universitaire de Médecine Légale, CHUV, Switzerland
Copyright © 2020 Guo, Xu, Zhu, Zheng, Lu, Tu, He, Jin and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ying He, aGV5QHhtdS5lZHUuY24=; Lihua Jin, amlubGhAeG11LmVkdS5jbg==; Yong Li, eW9uZ2xpQHhtdS5lZHUuY24=