
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol., 18 August 2020
Sec. Pharmacology of Ion Channels and Channelopathies
Volume 11 - 2020 | https://doi.org/10.3389/fphar.2020.01276
This article is part of the Research TopicVenoms, Animal and Microbial ToxinsView all 18 articles
 Luis Felipe Santos Menezes1
Luis Felipe Santos Menezes1 Elias Ferreira Sabiá Júnior1
Elias Ferreira Sabiá Júnior1 Diogo Vieira Tibery1
Diogo Vieira Tibery1 Lilian dos Anjos Carneiro2,3
Lilian dos Anjos Carneiro2,3 Elisabeth Ferroni Schwartz1*
Elisabeth Ferroni Schwartz1*Epilepsy is a disease characterized by abnormal brain activity and a predisposition to generate epileptic seizures, leading to neurobiological, cognitive, psychological, social, and economic impacts for the patient. There are several known causes for epilepsy; one of them is the malfunction of ion channels, resulting from mutations. Voltage-gated sodium channels (NaV) play an essential role in the generation and propagation of action potential, and malfunction caused by mutations can induce irregular neuronal activity. That said, several genetic variations in NaV channels have been described and associated with epilepsy. These mutations can affect channel kinetics, modifying channel activation, inactivation, recovery from inactivation, and/or the current window. Among the NaV subtypes related to epilepsy, NaV1.1 is doubtless the most relevant, with more than 1500 mutations described. Truncation and missense mutations are the most observed alterations. In addition, several studies have already related mutated NaV channels with the electrophysiological functioning of the channel, aiming to correlate with the epilepsy phenotype. The present review provides an overview of studies on epilepsy-associated mutated human NaV1.1, NaV1.2, NaV1.3, NaV1.6, and NaV1.7.
Epilepsy is a disease known worldwide, affecting around 70 million people in the world (Thijs et al., 2019). It has been considered a disease and no longer a disorder or a family of disorders since 2014 by International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE) (Falco-Walter et al., 2018). Epilepsy is conceptually defined as a disease in which an individual has at least two unprovoked or reflex seizures in a period greater than 24 h apart, one unprovoked or reflex seizure and a probability of having another seizure similar to the general recurrence risk after two unprovoked seizures (greater than or equal to 60%) over the next ten years or an epilepsy syndrome (Fisher et al., 2014).
When abnormal brain activity begins in one or more identified regions, epilepsy is called focal, whereas, when it occurs in both hemispheres with a wide distribution, it is called generalized. Finally, when it cannot be classified as either focal or generalized, it is called unknown (Devinsky et al., 2018).
Epilepsy can affect anyone, regardless of gender, age, and income levels (Saxena and Li, 2017). Understanding the etiology of epilepsy is crucial for clinical management of patients and for conducting neurobiological research that will direct future therapies (Thomas and Berkovic, 2014). The ILAE Task Force has defined six etiologic categories; they are not hierarchical and more than one might often apply (structural, genetic, infectious, metabolic, immune, and unknown) (Falco-Walter et al., 2018).
Among those genetically caused, it is possible to identify several epilepsy-related genes (Lindy et al., 2018). For example, voltage-gated potassium channel, voltage-gated calcium channel and voltage-gated chloride channel genes, GABA receptors, nicotinic acetylcholine receptors, polymerase (DNA) Gamma genes and voltage-gated sodium channel genes (Deng et al., 2014).
Voltage-gated sodium channels (NaV) can be found mainly in the central nervous system (CNS), peripheral nervous systems (PNS), skeletal, and cardiac muscles (Huang et al., 2017). NaVs are distributed throughout the body and play an important role in the generation and propagation of action potential (Wang et al., 2017b). Structurally, NaVs are composed by an α subunit organized in four homologous ligated domains (DI-DIV), each domain composed by six transmembrane segments (S1-S6), and one or more β subunits associated by non-covalent interactions or disulfide bond (Abdelsayed and Sokolov, 2013; Gilchrist et al., 2013; Catterall, 2017; Bouza and Isom, 2018; Jiang et al., 2020). The domains of an α subunit present a high degree of conservation with each other, presenting the region known as the voltage sensor domains (VSD) located in transmembranes S1-S4, especially S4 helix, which contains positively charged residues, and the pore-forming (PM) domain located in S5-S6 segments, structuring a four VSD around a central pore (Ahern et al., 2016).
The S4 helix of DI, DII, and DIII domains moves faster than the S4 helix of DIV during membrane depolarization, and this asynchronous movement is an essential feature in the steady activation voltage-dependent process, which provokes movement of S4-S5 intracellular links followed by the displacement of the S6 segments to initiate Na+ influx (Goldschen-Ohm et al., 2013; Oelstrom et al., 2014). The movement of the S4 helix of DIV initiates the process of fast inactivation, since the movement of the voltage sensor in domain DIV is associated with the displacement of an intracellular loop between DIII and DIV within an IFM (isoleucine, phenylalanine, and methionine) motif that binds intracellular to PM and terminate Na+ influx (Capes et al., 2013; Clairfeuille et al., 2019). A second type of reversible inactivation occurs after repetitive or prolonged stimulation and results in steady-state inactivation whose asymmetric movement of S6 segments collapses the pore (Payandeh et al., 2012; Zhang et al., 2012; Gamal El-Din et al., 2013; Silva and Goldstein, 2013; Ghovanloo et al., 2016). Consequently, electrophysiological changes such as increased current density, shifting steady-state activation, and inactivation to negative and positive values, respectively, enhanced persistent current, accelerated recovery from inactivation, and delayed fast inactivation can cause gain-of-function (GoF) in the channel. Also, decreased current density, positive shift in steady-state activation, negative shift in steady-state inactivation, and slower recovery from inactivation can cause loss-of-function (LoF) (Mantegazza et al., 2005; Liao et al., 2010; Lossin et al., 2012; Catterall, 2014b; Vanoye et al., 2014; Wagnon et al., 2017; Yang et al., 2018; Zaman et al., 2018; Wengert et al., 2019; Zhang S. et al., 2020).
Currently, there are nine different alpha subtypes of NaVs (NaV1.1-NaV1.9), and mutations in these channels can cause diseases known as channelopathies (Catterall et al., 2010). NaV1.1 (SCN1A), NaV1.2 (SCN2A), NaV1.3 (SCN3A), NaV1.6 (SCN8A) and NaV1.7 (SCN9A) are genes whose mutations are related to epilepsy. So far, there is no correlation of mutations in NaV1.4 (SCN4A), NaV1.5 (SCN5A), NaV1.8 (SCN10A), and NaV1.9 (SCN11A) with epilepsy, which is to be expected, since these channels are mainly expressed in skeletal muscles, cardiac tissues, dorsal root ganglia, trigeminal sensory neurons, nociceptive neurons of the dorsal root and trigeminal ganglia, respectively (Brunklaus et al., 2014). Both α and β subunits (SCN1B) have been reported as the cause of epilepsy phenotype (Meisler et al., 2010; Kaplan et al., 2016).
NaV channels rank amongst the 2% most conserved proteins in the human genome, with an extremely low rate of coding variation, accounting for nearly 5% of known epileptic encephalopathies (Petrovski et al., 2013; Mercimek-Mahmutoglu et al., 2015; Lek et al., 2016; Heyne et al., 2019). Pathogenic mutated residues are situated in the highly evolutionarily conserved portions of the channel: transmembrane segments, intracellular inactivation gate loop, and the proximal 2/3 of the C-terminal domain (Blanchard et al., 2015; Wagnon and Meisler, 2015). The final 1/3 portion of the C-terminal and cytoplasmic interdomain loops 1 and 2 are less conserved (Denis et al., 2019). The proximal 2/3 of the C-terminal are involved in the interaction of several binding sites for proteins and accessory molecules, like beta subunits β1 and β3, fibroblast growth factors (molecules implicated in neural development), calmodulin (regulatory protein in neuronal function and hyperexcitability) and G protein (Bähler and Rhoads, 2002; Spampanato, 2004; Wittmack et al., 2004; Laezza et al., 2009; Yang et al., 2010). Moreover, the C-terminal has been shown to interact with the inactivated channel via ionic interaction between its positively charged residues and negatively charged residues at the inactivation gate. A shift in any of the charges can brake electrostatic interaction and affect normal channel inactivation (Nguyen and Goldin, 2010; Shen et al., 2017; Johnson et al., 2018).
The N-terminal region seems to play a more important role on protein trafficking than on channel activity. This domain interacts with the light chain of microtubule-associated protein MAP1B, facilitating the traffic of the NaV channel to the neuronal cell surface (O’brien et al., 2012; Blanchard et al., 2015). In addition, mutation in the N-terminal leads to protein retention in the endoplasmic reticulum (Sharkey et al., 2009).
Newer genomic approaches, especially next generation sequencing (NGS), improve the rate and reduce the costs associated with genetic epilepsy diagnosis, since traditional cytogenetic and microarray-based tests are lengthy, expensive, and diagnostic yield is incredibly low (Veeramah et al., 2013; Allen et al., 2016; Sands and Choi, 2017; Orsini et al., 2018). The use of gene panels and whole-exome sequencing (WES) provides a powerful tool to change the paradigm of genetic epilepsy diagnosis (Ng et al., 2010; Clark et al., 2018). These techniques have been widely used to elucidate suspected inherited neurological diseases in the last years, contributing to dramatically increase the number of patients diagnosed with genetic epilepsy. Both mendelian and de novo genetic epilepsy can be detected with these methods, but doubtless, de novo mutations are the most prevalent mutations related to epilepsy-related voltage-gated sodium channel mutations.
Gene therapy is promising as an effective approach to treat genetic diseases. Personalized epilepsy therapies are in development and have shown promising results, ranging from antisense oligonucleotides and small peptides to modulation of gene expression through epigenetics (Riban et al., 2009; Tan et al., 2017; Stoke Therapeutics, 2018; Perucca and Perucca, 2019). Even eating habits may be related to an improvement in the patient's clinical condition. Ketogenic diet has been described as an effective treatment in epilepsy (Gardella et al., 2018). Moreover, the combination of traditional antiepileptic drugs with new compounds displayed a synergic and improved efficacy, since these molecules do not compete for the same interaction site (Bialer et al., 2018). Each specific epilepsy-related NaV isoform will be presented and discussed in detail in the following sections.
The SCN1A gene encodes for the α subunit NaV1.1, and is allocated at the 2q24.3 chromosome between 165,984,641 and 166,149,161 base pairs, same gene cluster of SCN2A-SCN3A genes, being the most frequent target of mutation in genetic epilepsy syndromes (OMIM#182389) (Malo et al., 1991; Malo et al., 1994; Catterall et al., 2010). NaV1.1 is widely expressed in the CNS, predominant in inhibitory GABAergic interneurons, regulating neuronal excitability, and the reduction of its activity is one of the factors that cause epileptic diseases due to imbalance between inhibition and excitation (Yu et al., 2006; Verret et al., 2012; Tai et al., 2014; Rubinstein et al., 2015).
Epilepsy syndromes, such as generalized epilepsy with febrile seizures plus (GEFS+; Online Mendelian Inheritance in Man [OMIM] #604233), severe myoclonic epilepsy (SME) and SMEI, also known as Dravet syndrome (OMIM #607208), are associated with mutations in the SCN1A gene (Escayg and Goldin, 2010; Meng et al., 2015; Huang et al., 2017).
In the SCN1A mutation database (http://www.caae.org.cn/gzneurosci/scn1adatabase/data), among 1727 mutations described for the SCN1A gene, 1528 are related to epileptic diseases (Table 1 and for the full description of mutations in the SCN1A gene, see Supplementary Table S1). Among the epilepsy-related mutations, 945 are related to severe myoclonic epilepsy of infancy (SMEI), 263 are related to severe myoclonic epilepsy (SME), 151 are related to severe myoclonic epilepsy borderline (SMEB), 18 are related to partial epilepsy (PE), 31 are related to partial epilepsy and febrile seizures plus (PEFS +), 8 are related to generalized epilepsy (GE), and 55 are related to generalized epilepsy with febrile seizures plus (GEFS +).
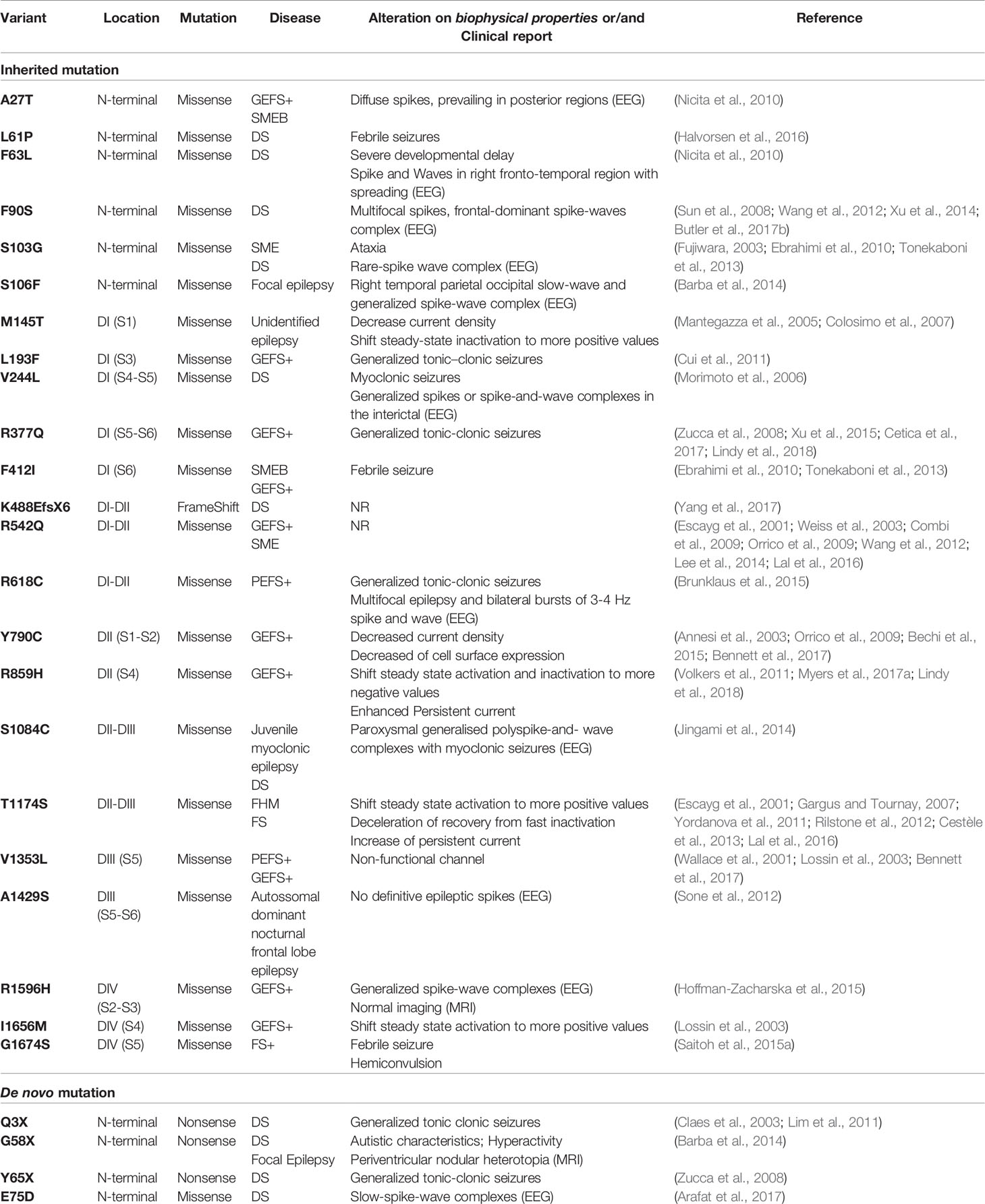
Table 1 SCN1A-related epilepsies identified in clinical patients through WES and/or NGS.
Mutations in the NaV1.1 channel are described in almost all regions of the protein and may cause GoF or LoF (Goldin and Escayg, 2010; Meng et al., 2015). Among the 52 mutations in SCN1A related to epilepsy with functional studies, 35 mutations (67.30%) exclusively display characteristics of LoF, 6 mutations (11.53%) display characteristics unique to GoF, and 11 mutations (21,15%) display characteristics of GoF+LoF, whereas, in GoF+LoF mutations, the main characteristic that gives GoF features is enhanced persistent current, present in 10 out of the 11 GoF+LoF mutations listed (Tables 1 and S1).
Due to the role of the NaV1.1 channels in the regulation of electrical excitability by the inhibitory interneurons, prescription of AEDs non-selective sodium channel blockers (SCB) for SMEI or GEFS + syndromes is contraindicated, for it may aggravate crises due to the enhanced suppress status of the NaV1.1 channels (Catterall, 2014a; Shi et al., 2016; Knupp and Wirrell, 2018; Ziobro et al., 2018). The first-line drug-based therapy for SCN1A epilepsy diseases is the enhancement of postsynaptic GABAergic transmission with allosteric activation of GABAA receptors as target by Clobazam and/or an increase in GABA concentration in synaptic cleft resulting from increased GABA production and decreased GABA degradation as target by Valproic acid (Catterall, 2014a; Hammer et al., 2016; Knupp and Wirrell, 2018; Musto et al., 2020). Antisense nucleotides (ASO) therapy to increase mRNA of SCN1A for NaV1.1 channel expression in normal levels is a promising strategy for genetic disorders involving haploinsufficiency (Hsiao et al., 2016; Stoke Therapeutics, 2018). Drug-resistant Dravet syndrome cases may thrive on alternative therapeutic strategies based on ketogenic diets (Nabbout et al., 2011; Wu et al., 2018). A recent study with 20 patients with medically intractable Dravet syndrome caused by missense, non-sense, insertion, deletions and splicing mutations presents efficacy during three months of treatment in 17 patients, decreasing seizure frequency in more than 50% (Yan et al., 2018). Besisdes that, Epidiolex is an FDA approved CBD-based drug approved in June 2018 for the treatment of severe forms of epilepsy, as Dravet and Lennox-Gastaut syndromes (U.S. Food and Drug Administration [website]., 2018). Clinical trials using CBD in DS and LGS shown reduced frequency of seizures in monthly average (Lattanzi et al., 2020; Morano et al., 2020). Voltage-gated sodium channel are inhibit by CBD in low micromolar concentrations, IC50 between 1.9 and 3.8 μM, NaV1.4 and NaV1.1 being the most sensitive channels to CBD, 1.9 and 2.0 μM respectively, probably the mechanism of action is reducing channel availability due shift to more hyperpolarized potential in steady-state inactivation (Ghovanloo et al., 2019).
NaV1.2 is encoded by the SCN2A gene (Wolff et al., 2017). It is located on chromosome 2q24.3 (Shi et al., 2009) and expressed in the CNS (Catterall, 2014a), especially in excitatory neurons (Syrbe et al., 2016) and glutamatergic neurons (Sanders et al., 2018), unlike the NaV1.1 channel, which is highly expressed in the GABAergic interneurons (Catterall, 2014a).
More than 100 mutations have already been described for this gene, with approximately 300 patients studied yet (Reynolds et al., 2020) (Table 2). The most common diseases related with SCN2A mutation are West syndrome (WS; OMIM #308350), epilepsy of infancy with migrating focal seizures (EIMFS; OMIM #616645), and benign familial neonatal-infantile seizures (BFNIS; OMIM #607745) (Perucca and Perucca, 2019). Although epilepsy-related mutations are present throughout the channel, several hotspots such as the ion selectivity filter, the voltage-sensing domain, the intracellular N-terminal, and the C-terminal domain can be highlighted (Sanders et al., 2018).
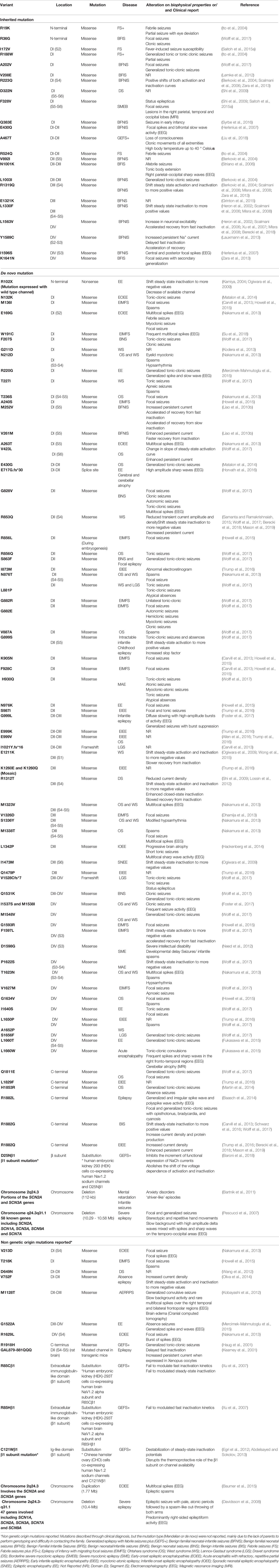
Table 2 SCN2A-related epilepsies identified in clinical patients through WES and/or NGS.
NaV1.2 channels are expressed in the excitatory neurons; therefore, GoF mutations are related to epilepsy because it causes neuronal hyperexcitability. On the other hand, LoF mutations are related to autism and intellectual disability phenotype (Ben-Shalom et al., 2017). Nevertheless, some studies have already related loss of function to epilepsy, as described by Lossin and co-workers (2012) with R1312T mutation (Lossin et al., 2012). Normally, LoF SCN2A gene mutations for epilepsy are related to late-onset epilepsy; however, the mechanism of action is unclear (Mason et al., 2019).
In some cases, NaV1.2 seizures are not controlled not even by various antiepileptic drugs, as with the patient described by Syrbe and colleagues (2016). The proband, even after being treated with oxcarbazepine (OXC), valproic acid, topiramate, sulthiame, phenytoin, among other drugs, kept on having seizures (Syrbe et al., 2016). Furthermore, the SCB drugs can assist the patient during the treatment as described by Gorman and King (2017). The patient had seizures controlled after administration of phenytoin (Gorman and King, 2017). In addition, Musto et al. (2020) cite benefits treatments using SCB such as carbamazepine, mexiletine, oxcarbazepine, phenytoin, lidocaine, and lamotrigine for patients with early onset epilepsies (Musto et al., 2020). Besides, Peters and colleagues studied a substance commercially used as an antianginal drug (human heart) called ranolazine that has been shown to affect NaV1.2 channels, reducing macroscopic currents and delaying the recovery of fast and slow inactivation of the NaV1.2 channel, consequently with more future studies ranolazine could be a efficacious therapy for epilepsy (Peters et al., 2013).
Drugs can be important to modulate channel kinetics for both GoF and LoF, but some precautions must be observed. For example, the degree of conservation between subtypes, such as NaV1.2 and other sodium channels as NaV1.5 and the excessive decrease in channel function or the excessive increase in function obtained by the drug (Sanders et al., 2018).
Organizations like the FamilieSCN2A Foundation (www.scn2a.org) might be essential in the search for new treatments. Understanding the genotype-phenotype of gain and loss of function is essential because science-patient relationship may be helpful in the search for new therapies (Sanders et al., 2018).
SCN3A is a gene that encodes for type 3 voltage-gated Na+ channel α subunit, the NaV1.3, located on human chromosome 2q24, in a cluster with SCN1A and SCN2A (Holland et al., 2008). NaV1.3 is expressed predominantly in the CNS during embryonic and neonatal development, being extremely low or sometimes undetectable in postnatal individuals. Subsequently, during infancy, it is gradually replaced by increased expression of the NaV1.1 isoform (Felts et al., 1997; Whitaker et al., 2000; Cheah et al., 2013; Zaman et al., 2018). On the other hand, studies regarding nervous system injury and neuropathic pain showed an increasing presence of NaV1.3 channels in affected tissues, suggesting a pivotal hole of these transmembrane proteins in these processes and diseases (Hains et al., 2003; Waxman and Hains, 2006; Black et al., 2008). For the reasons mentioned above, in the last decades, NaV1.3-associated pathogenesis has been restricted to pain. Recently, a genetic linkage between NaV1.3 mutated variants and epilepsy has been suggested, especially in cryptogenic epilepsy cases (OMIM#182391).
K354Q was the first described NaV1.3 epilepsy-related mutation that revealed harmful electrophysiological alterations (Holland et al., 2008; Estacion et al., 2010). In fact, mutations can change many functional characteristics of NaV1.3 affecting biophysical properties differently; however, these changes result predominantly in neuronal hyper-responsiveness (Table 3) (Cummins and Waxman, 1997; Chen et al., 2000; Cummins et al., 2001; Sun et al., 2007). Previous reports correlate heterozygous variants in SCN3A in association with moderate forms of epilepsy, while homozygosis is related with severe cognitive damage and premature mortality, resulting in a broad range of epileptic phenotypes (Estacion and Waxman, 2013; Vanoye et al., 2014; Lamar et al., 2017).
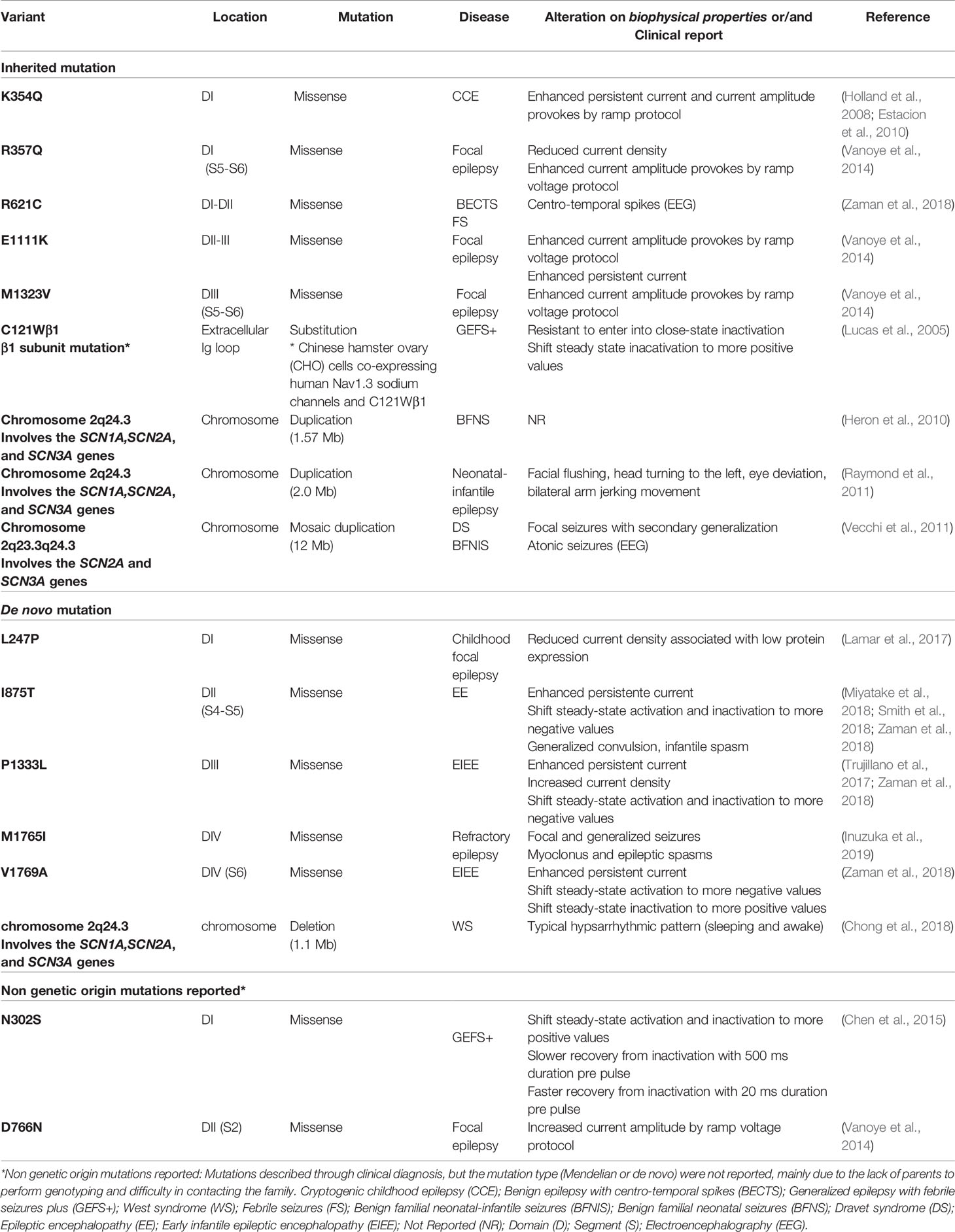
Table 3 SCN3A-related epilepsies identified in clinical patients through WES and/or NGS.
Different hereditary mutations on NaV1.3 have been reported to date in patients with epilepsy. In general, the biophysical characterization of these mutations reveals GoF, only one mutation (N302S) is related with LoF (Chen et al., 2015), but both GoF and LoF may lead to an increased seizure susceptibility (Lamar et al., 2017).
Moreover, several de novo mutations in SCN3A have been described in the last three years, related with severe infantile neurological dysfunctions and cognitive impairments. These mutations may alter the functionality of NaV1.3 channels, neurons organization, migration, and proliferation during the embryonic development (Smith et al., 2018). Epileptic encephalopathy and polymicrogyria are the main features related with these pathogenic variants, and, so far, polymicrogyria was not reported in other channelopathies, being an exclusive characteristic of SCN3A mutants (Inuzuka et al., 2019).
There is a lack of clinical data on SCN3A-related epilepsies, especially regarding treatment and the use of specific medication. However, in vitro studies reported that mutations related with GoF effect respond favorably to treatment using SCB, like phenytoin, carbamazepine, lacosamide, and topiramate (Sun et al., 2007; Sheets et al., 2008; Colombo et al., 2013; Zaman et al., 2018). The anticonvulsant valproic acid represents a novel and promising epigenetic therapeutic approach (Tan et al., 2017). The compound modulates the SCN3A gene through methylation, downregulating the expression of NaV1.3 and, consequently, decreasing biophysical alterations in the channel.
The SCN8A gene encodes for type 8 voltage-gated Na+ channel α subunit, the NaV1.6, located in chromosome 12q13.13. The first case of SCN8A pathogenic variant associated with epilepsy was reported eight years ago (Veeramah et al., 2012). Thereafter, due to advances in genome sequencing technology, especially the WES, the number of epilepsy diagnosis associated with NaV1.6 mutations has increased significantly (OMIM #600702), with more than 300 patients diagnosed with SCN8A epilepsy mutations and nearly 200 different putative spots of mutations described, totaling over 100 published reports (Table 4). A website developed especially to present SCN8A epilepsy and related diseases (www.scn8a.net) was created to provide information to families, clinicians, and researchers, gathering news and recent publications on the subject in a private forum for family interaction, to answer questions, strengthening the ties between the community and the researchers.
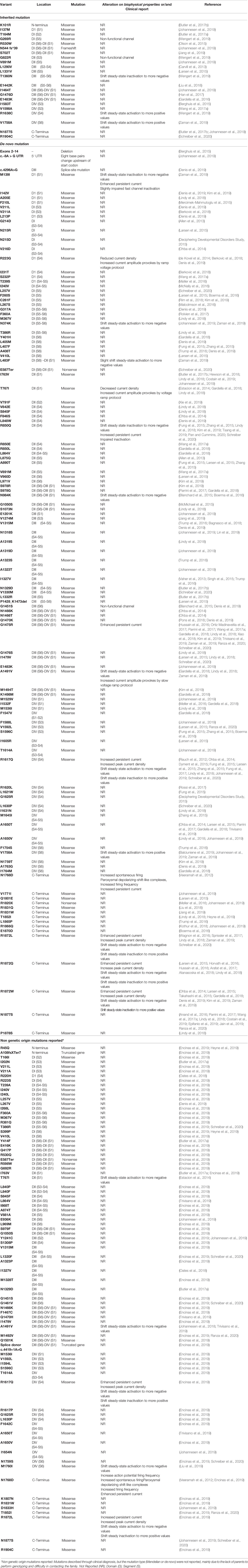
Table 4 SCN8A-related epilepsies identified in clinical patients through WES and/or NGS.
NaV1.6 is expressed since prenatal, during fetal development (Plummer et al., 1997). Shortly after birth, expression begins to increase, reaching maximum levels during the first years of life. This channel is widely expressed in the nodes of Ranvier of myelinated axons and in the distal part of the axon initial segments (AIS), although they are also ubiquitously present throughout the central and peripheral nervous systems, in both excitatory and inhibitory neurons (Caldwell et al., 2000; Oliva et al., 2012). For these reasons, NaV1.6 is one of the most common subtype of voltage-gated sodium channels found in the central nervous system (Caldwell et al., 2000). In humans, the distal AIS is the specialized membrane region in neurons where action potentials are triggered. Overexpression of Nav1.6 in the AIS has been shown to cause an increase in spontaneous and repetitive firing (Hu et al., 2009; Sun et al., 2013), a possible explanation for why SCN8A mutations in epilepsy patients are predominantly GoF and affect the action potential threshold. On the other hand, the functional importance of Nav1.6 in inhibitory interneurons is not clear yet, but evidence indicates a role for Nav1.6 in establishing synaptic inhibition in the thalamic network (Makinson et al., 2017), supporting the LoF results caused by missense mutations in the mature protein. These attributes lead to different network effects in distinct nervous system circuits. Mutations in SCN8A are associated with early-infantile epileptic encephalopathy type 13 (EIEE13; OMIM #614558), a phenotypically heterogeneous early onset epilepsy, with seizure onset happening before 18 months of age (Hammer et al., 2016). Patients typically develop intellectual disability, developmental delay, and movement disorders (Ohba et al., 2014; Gardella et al., 2016; Johannesen et al., 2018). Co-occurrence of autism spectrum disorders, severe juvenile osteoporosis, bradyarrhythmia, cortical visual impairment, and gastrointestinal disorders have been reported in rare cases (Larsen et al., 2015; Hammer et al., 2016; Rolvien et al., 2017; Gardella et al., 2018). Sudden unexpected death in epilepsy (SUDEP) has also been linked to SCN8A mutations, described as the most common cause of death in epilepsy patients. Reports have suggested that patients with SCN8A-related epilepsy have increased risk of SUDEP, ranging from 1% to 10% (Hammer et al., 2016; Wang et al., 2017a; Gardella et al., 2018; Johannesen et al., 2018). One possible correlation of SUDEP with SCN8A-related epilepsy is the presence of NaV1.6 in heart muscles and tissues, being broadly expressed within ventricular myocytes (Maier et al., 2002). Single mutations may affect heart function, causing failure of the cardiorespiratory system and, consequently, death (Haufe et al., 2005; Noujaim et al., 2012). Most recently, few cases of SCN8A-related epilepsies with “milder” phenotype were associated with benign familial infantile seizures-5 (BFIS5; OMIM #617080) (Anand et al., 2016; Gardella et al., 2016; Han et al., 2017).
An increase in new described variants made some mutation patterns visible. Wagnon and co-workers observed numerous cases of the same epiletogenic mutation, and suggested that CpG dinucleotides are mutation hotspots that, through enzymatic processing and epigenetic methylation, can convert cytosine to thymine, such as arginine residues 1617 and 1872 (Wagnon and Meisler, 2015). The prominent number of new variant cases in Arg850 indicates this residue as a new hotspot, since the arginine codon holds a CpG dinucleotide. In addition to these mutation hotspots, residues I763, I1327, G1475, A1650, and N1877 do not present CpG dinucleotides in their codon; however, they can be considered recurrent mutations in view of its high repetition cases in literature (Table 4).
The mutation at position c.- 8A>G produces a pathogenic variant, despite not being inside the gene, or promoter regions, transcriptional and translational sites. This mutation was detected in an untranslated region outside of the Kozak consensus sequence (Johannesen et al., 2019). Its role in SCN8A-related epilepsy is still unclear; however, it may change RNA stability, modulate transcriptional factors and promoters, modify the initiation of translation, or work as an enhancer or silencer in the splicing pattern. For all the reasons mentioned above, Nav1.6 variants are predominantly harmful, and the same mutation can lead to different phenotypes, hampering the correlation of genotypes with phenotypes (Blanchard et al., 2015).
SCN8A mutations can be both GoF and LoF, which will likely require different approaches and targets. Even in patients with the same SCN8A mutation, the response to the same drug treatment can differ. Surprisingly, most SCN8A-related epilepsies respond favorably to channel blockers. Phenytoin and lacosamide are SBCs widely used in SCN8A mutations with GoF effect, while carbamazepine exhibited positive seizure control in a patient with NaV 1.6 mutation and LoF effect. (Blanchard et al., 2015; Wagnon and Meisler, 2015; Hammer et al., 2016; Perucca and Perucca, 2019). Phenytoin demonstrated effectiveness in decreasing seizure episodes in several patients with SCN8A-related epilepsies, however, side effects during prolonged use are very common (Boerma et al., 2016; Braakman et al., 2017). A recent study of a DS model using zebrafish demonstrated the use of the channel blocking compound MV1312, which is 5–6 fold selectivity of NaV1.6 over NaV1.1–1.7, reduced burst movement phenotype and the number of epileptiform events, activity similar to that described with the use of a selective NaV1.1 activator AA43279 (Weuring et al., 2020). Selective Nav1.6 blockers may represent a new therapeutic strategy for DS patients. In addition, two precise and promising drugs have been described recently: XEN901 and GS967. XEN901 is an arylsulfonamide highly selective and potent NaV1.6 inhibitor that binds specifically in voltage sensor domain IV, avoiding recovery from inactivation. GS967 is a NaV1.6 modulator that inhibits the persistent sodium current and exhibits a protective effect (Baker et al., 2018; Bialer et al., 2018).
The SCN9A gene encodes for the NaV1.7 channel, located in chromosome 2q24 (Yang et al., 2018). NaV1.7 is expressed preferably in the PNS, but it is also expressed in the CNS (Cen et al., 2017). Consequently, mutations in this channel are generally related to pain disorders (Young, 2007; Han et al., 2009; Doty, 2010; Rush et al., 2018); however, current studies have described a correlation between epilepsy and this channel (OMIM #603415).
Pain disorder mutations with GoF are related with diseases such as erythromelalgia (EMI), small-fiber neuropathy (SFN) and paroxysmal extreme pain disorder (PEPD), and mutations with LoF are related with congenital insensitivity to pain (CIP) (Cen et al., 2017). Epilepsy studies such as Zhang S. et al. (2020) showed mutations with GoF phenotype: W1150R, N641Y, and K655R mutations (Table 5). Being that, after treatment with OXC (120 µmol/L), N641Y and K655R reduced sodium current and decreased the opening time of the channel, while W1150R did not alter that (Zhang S. et al., 2020). However, in a study conducted by Yang et al. (2018), one of the patients presented generalized tonic-clonic seizure with fever, treated with sodium valproic acid, and a LoF mutation I1901fs was observed (Yang et al., 2018) (Table 5).
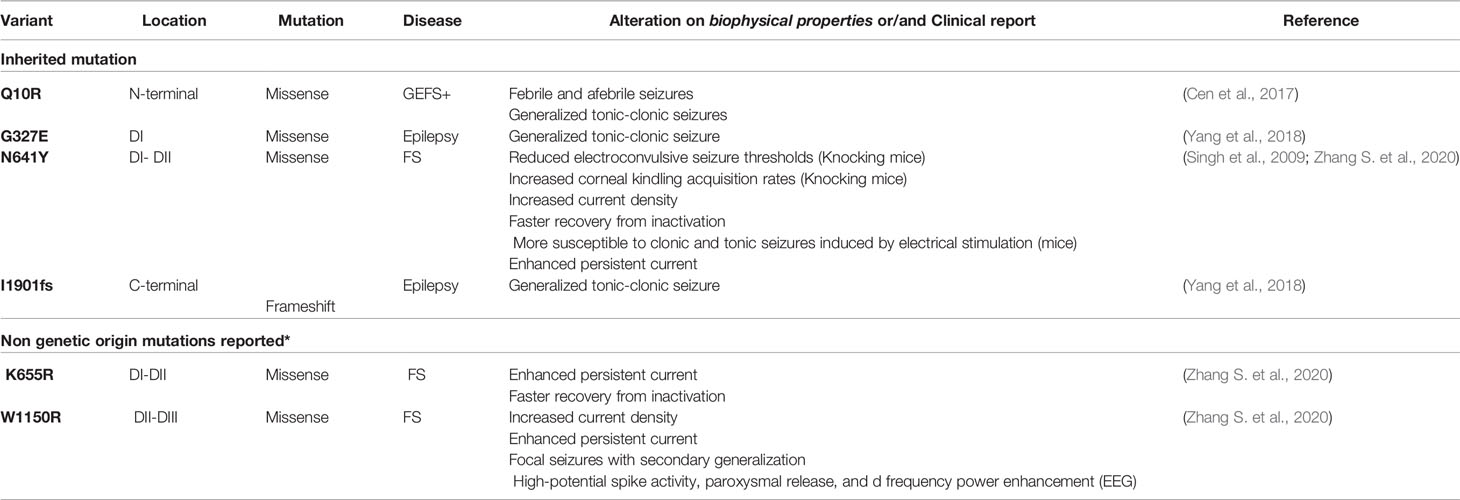
Table 5 SCN9A-related epilepsies identified in clinical patients through WES and/or NGS.
Variants of NaV1.7 have been related with febrile seizure or GEFS+ (Cen et al., 2017; Zhang S. et al., 2020) and even as asymptomatic (Singh et al., 2009). However, SCN9A can act as a putative modifier of NaV1.1 gene; consequently, it can elevate the severity of patients’ phenotype (Guerrini et al., 2010; Parihar and Ganesh, 2013). Some NaV1.7 mutations could probably contribute to generate a genetic susceptibility to a known epilepsy disease called Dravet syndrome, in a multifactorial way, as a modifier gene (Singh et al., 2009; Doty, 2010; Mulley et al., 2013; Cen et al., 2017; Zhang T. et al., 2020). That said, some rare cases of DS found in patients can be understood (Mulley et al., 2013). For example, even parents with mild phenotype had children with severe cases (Guerrini et al., 2010).
The past two decades have enabled remarkable progress in understanding monogenic epilepsies. NaV-related epilepsies are diseases of phenotypic heterogeneity, since sodium channels are found in both the CNS and the PNS, but with different expression ranges. The lack of a clear genotype-phenotype correlation to help guide patient counseling and management by healthcare professionals makes it very complex, and often expensive, to determine a correct diagnosis. Consequently, identify the monogenic mutation in individual patients with epilepsy is important not only for diagnosis and prognosis, but also for a correct treatment approach (Mei et al., 2017; Reif et al., 2017).
Susceptibility to specific treatments may be different depending on the disease’s features, diverging even in patients who share the same phenotype and/or mutation (Weber et al., 2014). The use of innovative tools that facilitate and prevent diagnostic delay in patients with epilepsy of unknown etiology onset is crucial. WES has proved to be a valuable tool to circumvent the lack of an accurate and fast diagnosis to epilepsies caused by monogenic mutation, and also cheapen and drastically anticipate diagnosis. This genetic diagnostic tool may reduce traditional investigation costs by 55 to 70%, besides avoiding further pre-surgical evaluation and epilepsy surgery (Kothur et al., 2018; Oates et al., 2018). In addition to the financial impact, it can anticipate diagnosis from nearly 3.5 years to 21 days, optimizing management and health care support (Oates et al., 2018).
Effective and safe drugs for the treatment of monogenic epilepsy are still an unmet clinical need. The drugs currently available in the pharmaceutical market are only palliative methods for a temporary control of the disease symptoms, and few patients will benefit from the existing pharmacotherapy, since a great number of patients treated with antiepileptic channel blockers showed no improvement in clinical conditions. Also, most treated patients exhibited manifold side effects, and the prolonged use of these medications proved to be harmful (Boerma et al., 2016; Braakman et al., 2017). Several examples of novel and promising candidate compounds to be used in personalized medicine, such as precision therapies, have been suggested. A previously study demonstrated that CBD at 1μM inhibit preferably resurgent currents than transient current in Nav1.6 WT and also inhibit peak resurgent current in Nav1.6 mutant N1768D, with less effect in current density and without alters voltage dependence of activation (Patel et al., 2016) Possibly the modulation of CBD over mutations in SCN8A that promotes a phenotype with increased resurgent currents would cause a reduction in the causative excitability of epileptic seizures. CBD also showed its ability to preferential inhibit resurgent currents in the NaV1.2 channel (Mason and Cummins, 2020). Due the role of Nav1.2 and Nav1.6 in excitatory neurons, preferentially inhibition in resurgent currents by CBD could possibly reduce the excitability in that subset of neurons and decrease the frequency of seizures by a change in threshold of activation and repetitive fire (Lewis and Raman, 2014). Peptides derived from scorpion and spider venom are well known modulator tools in neuroscience and showed specific capacity to regulate most NaV subtypes related with monogenic epilepsy, unlike the available promiscuous drugs that generally interact with any NaV channel isoform (Schiavon et al., 2006; Israel et al., 2018; Richards et al., 2018; Tibery et al., 2019; Zhang et al., 2019). Bioengineering tools, like antisense oligonucleotides capable to regulate NaV1.1 channels expression, and the peptide Hm1, that modulates the function of this subtype of sodium channel, are some innovative treatment examples (Richards et al., 2018; Stoke Therapeutics, 2018).
However, there is still a long path toward the development of efficacious treatments for NaV-related epilepsies. Recent studies offered a better understanding of the complexity of the phenotypic and genetic spectrum, which has only just begun to be elucidated. Biomolecular diagnostic tools will drastically reduce the developmental and cognitive effects caused by misdiagnosis and late diagnosis, and maybe, in the upcoming years, the treatment for inherited NaV-related epilepsies will be conducted ideally in utero, during the prenatal stage. Moreover, further functional studies, with greater cohorts of patients, represent an urgent medical need for a better understanding of the correlations between genotype and clinical symptoms, as well as the different NaV-related epilepsies mechanisms. These studies will improve clinical efficacy and promote safety diagnostic strategies, as well as develop prognosis prediction in the near future.
All authors made an intellectual and direct contribution for this article and approved it for publication.
This study was supported by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) [407625/2013-5] and the Fundação de Apoio à Pesquisa do Distrito Federal (FAPDF) [grants 193.001.202/2016 and 00193.0000109/2019-17].
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
CNPq, CAPES, and the Molecular Biology postgraduate program of the University of Brasilia. LM received scholarships from CNPq and DT from CAPES. EFS was supported by CNPq.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2020.01276/full#supplementary-material
Abdelsayed, M., Sokolov, S. (2013). Voltage-gated sodium channels. Channels 7, 146–152. doi: 10.4161/chan.24380
Ahern, C. A., Payandeh, J., Bosmans, F., Chanda, B. (2016). The hitchhiker’s guide to the voltage-gated sodium channel galaxy. J. Gen. Physiol. 147, 1–24. doi: 10.1085/jgp.201511492
Allen, A. S., Berkovic, S. F., Cossette, P., Delanty, N., Dlugos, D., Eichler, E. E., et al. (2013). De novo mutations in epileptic encephalopathies. Nature 501, 217–221. doi: 10.1038/nature12439
Allen, N. M., Conroy, J., Shahwan, A., Lynch, B., Correa, R. G., Pena, S. D. J., et al. (2016). Unexplained early onset epileptic encephalopathy: Exome screening and phenotype expansion. Epilepsia 57, e12–e17. doi: 10.1111/epi.13250
Anand, G., Collett-White, F., Orsini, A., Thomas, S., Jayapal, S., Trump, N., et al. (2016). Autosomal dominant SCN8A mutation with an unusually mild phenotype. Eur. J. Paediatr. Neurol. 20, 761–765. doi: 10.1016/j.ejpn.2016.04.015
Annesi, G., Gambardella, A., Carrideo, S., Incorpora, G., Labate, A., Pasqua, A. A., et al. (2003). Two Novel SCN1A Missense Mutations in Generalized Epilepsy with Febrile Seizures Plus. Epilepsia 44, 1257–1258. doi: 10.1046/j.1528-1157.2003.22503.x
Arafat, A., Jing, P., Ma, Y., Pu, M., Nan, G., Fang, H., et al. (2017). Unexplained Early Infantile Epileptic Encephalopathy in Han Chinese Children: Next-Generation Sequencing and Phenotype Enriching. Sci. Rep. 7:46227. doi: 10.1038/srep46227
Atanasoska, M., Vazharova, R., Ivanov, I., Balabanski, L., Andonova, S., Ivanov, S., et al. (2018). SCN8A p.Arg1872Gln mutation in early infantile epileptic encephalopathy type 13: Review and case report. Biotechnol. Biotechnol. Equip. 32, 1345–1351. doi: 10.1080/13102818.2018.1532815
Bähler, M., Rhoads, A. (2002). Calmodulin signaling via the IQ motif. FEBS Lett. 513, 107–113. doi: 10.1016/S0014-5793(01)03239-2
Baasch, A. L., Hüning, I., Gilissen, C., Klepper, J., Veltman, J. A., Gillessen-Kaesbach, G., et al. (2014). Exome sequencing identifies a de novo SCN2A mutation in a patient with intractable seizures, severe intellectual disability, optic atrophy, muscular hypotonia, and brain abnormalities. Epilepsia 55, e25–e29. doi: 10.1111/epi.12554
Bagnasco, I., Dassi, P., Blé, R., Vigliano, P. (2018). A relatively mild phenotype associated with mutation of SCN8A. Seizure 56, 47–49. doi: 10.1016/j.seizure.2018.01.021
Baker, E. M., Thompson, C. H., Hawkins, N. A., Wagnon, J. L., Wengert, E. R., Patel, M. K., et al. (2018). The novel sodium channel modulator GS-458967 (GS967) is an effective treatment in a mouse model of SCN8A encephalopathy. Epilepsia 59, 1166–1176. doi: 10.1111/epi.14196
Balciuniene, J., DeChene, E. T., Akgumus, G., Romasko, E. J., Cao, K., Dubbs, H. A., et al. (2019). Use of a Dynamic Genetic Testing Approach for Childhood-Onset Epilepsy. JAMA Netw. Open 2, e192129. doi: 10.1001/jamanetworkopen.2019.2129
Barba, C., Parrini, E., Coras, R., Galuppi, A., Craiu, D., Kluger, G., et al. (2014). Co-occurring malformations of cortical development and SCN1A gene mutations. Epilepsia 55, 1009–1019. doi: 10.1111/epi.12658
Baroni, D., Picco, C., Moran, O. (2018). A mutation of SCN1B associated with GEFS+ causes functional and maturation defects of the voltage-dependent sodium channel. Hum. Mutat. 39, 1402–1415. doi: 10.1002/humu.23589
Bartnik, M., Chun-Hui Tsai, A., Xia, Z., Cheung, S., Stankiewicz, P. (2011). Disruption of the SCN2A and SCN3A genes in a patient with mental retardation, neurobehavioral and psychiatric abnormalities, and a history of infantile seizures. Clin. Genet. 80, 191–195. doi: 10.1111/j.1399-0004.2010.01526.x
Baumer, F. M., Peters, J. M., El Achkar, C. M., Pearl, P. L. (2015). SCN2A-Related Early-Onset Epileptic Encephalopathy Responsive to Phenobarbital. J. Pediatr. Epilepsy 05, 042–046. doi: 10.1055/s-0035-1567853
Bechi, G., Rusconi, R., Cestèle, S., Striano, P., Franceschetti, S., Mantegazza, M. (2015). Rescuable folding defective NaV1.1 (SCN1A) mutants in epilepsy: Properties, occurrence, and novel rescuing strategy with peptides targeted to the endoplasmic reticulum. Neurobiol. Dis. 75, 100–114. doi: 10.1016/j.nbd.2014.12.028
Bennett, C. A., Petrovski, S., Oliver, K. L., Berkovic, S. F. (2017). ExACtly zero or once. Neurol. Genet. 3, e163. doi: 10.1212/NXG.0000000000000163
Ben-Shalom, R., Keeshen, C. M., Berrios, K. N., An, J. Y., Sanders, S. J., Bender, K. J. (2017). Opposing Effects on NaV1.2 Function Underlie Differences Between SCN2A Variants Observed in Individuals With Autism Spectrum Disorder or Infantile Seizures. Biol. Psychiatry 82, 224–232. doi: 10.1016/j.biopsych.2017.01.009
Berecki, G., Howell, K. B., Deerasooriya, Y. H., Cilio, M. R., Oliva, M. K., Kaplan, D., et al. (2018). Dynamic action potential clamp predicts functional separation in mild familial and severe de novo forms of SCN2A epilepsy. Proc. Natl. Acad. Sci. U. S. A. 115, E5516–E5525. doi: 10.1073/pnas.1800077115
Berghuis, B., de Kovel, C. G. F., van Iterson, L., Lamberts, R. J., Sander, J. W., Lindhout, D., et al. (2015). Complex SCN8A DNA-abnormalities in an individual with therapy resistant absence epilepsy. Epilepsy Res. 115, 141–144. doi: 10.1016/j.eplepsyres.2015.06.007
Berkovic, S. F., Heron, S. E., Giordano, L., Marini, C., Guerrini, R., Kaplan, R. E., et al. (2004). Benign Familial Neonatal-Infantile Seizures: Characterization of a New Sodium Channelopathy. Ann. Neurol. 55, 550–557. doi: 10.1002/ana.20029
Berkovic, S. F., Grinton, B., Dixon-Salazar, T., Laughlin, B. L., Lubbers, L., Milder, J., et al. (2018). De novo variants in the alternative exon 5 of SCN8A cause epileptic encephalopathy. Genet. Med. 20, 275–281. doi: 10.1038/gim.2017.100
Bialer, M., Johannessen, S. I. I., Koepp, M. J., Levy, R. H., Perucca, E., Tomson, T., et al. (2018). Progress report on new antiepileptic drugs: A summary of the Fourteenth Eilat Conference on New Antiepileptic Drugs and Devices (EILAT XIV). I. Drugs preclinical early clinical development. Epilepsia 59, 1811–1841. doi: 10.1111/epi.14557
Black, J. A., Nikolajsen, L., Kroner, K., Jensen, T. S., Waxman, S. G. (2008). Multiple sodium channel isoforms and mitogen-activated protein kinases are present in painful human neuromas. Ann. Neurol. 64, 644–653. doi: 10.1002/ana.21527
Blanchard, M. G., Willemsen, M. H., Walker, J. B., Dib-Hajj, S. D., Waxman, S. G., Jongmans, M. C. J., et al. (2015). De novo gain-of-function and loss-of-function mutations of SCN8A in patients with intellectual disabilities and epilepsy. J. Med. Genet. 52, 330–337. doi: 10.1136/jmedgenet-2014-102813
Boerma, R. S., Braun, K. P., van de Broek, M. P. H., van Berkestijn, F. M. C., Swinkels, M. E., Hagebeuk, E. O., et al. (2016). Remarkable Phenytoin Sensitivity in 4 Children with SCN8A-related Epilepsy: A Molecular Neuropharmacological Approach. Neurotherapeutics 13, 192–197. doi: 10.1007/s13311-015-0372-8
Bouza, A. A., Isom, L. L. (2018). “Voltage-Gated Sodium Channel b Subunits and Their Related Diseases,” in Handbook of experimental pharmacology (Springer International Publishing), 423–450. doi: 10.1007/164_2017_48
Braakman, H. M., Verhoeven, J. S., Erasmus, C. E., Haaxma, C. A., Willemsen, M. H., Schelhaas, H. J. (2017). Phenytoin as a last-resort treatment in SCN8A encephalopathy. Epilepsia Open 2, 343–344. doi: 10.1002/epi4.12059
Brunklaus, A., Ellis, R., Reavey, E., Semsarian, C., Zuberi, S. M. (2014). Genotype phenotype associations across the voltage-gated sodium channel family. J. Med. Genet. 51, 650–658. doi: 10.1136/jmedgenet-2014-102608
Brunklaus, A., Ellis, R., Stewart, H., Aylett, S., Reavey, E., Jefferson, R., et al. (2015). Homozygous mutations in the SCN1A gene associated with genetic epilepsy with febrile seizures plus and Dravet syndrome in 2 families. Eur. J. Paediatr. Neurol. 19, 484–488. doi: 10.1016/j.ejpn.2015.02.001
Buoni, S., Orrico, A., Galli, L., Zannolli, R., Burroni, L., Hayek, J., et al. (2006). SCN1delG) novel truncating mutation with benign outcome of severe myoclonic epilepsy of infancy. Neurology 66, 606–607. doi: 10.1212/01.WNL.0000198504.41315.B1
Butler, K. M., da Silva, C., Alexander, J. J., Hegde, M., Escayg, A. (2017a). Diagnostic Yield From 339 Epilepsy Patients Screened on a Clinical Gene Panel. Pediatr. Neurol. 77, 61–66. doi: 10.1016/j.pediatrneurol.2017.09.003
Butler, K. M., da Silva, C., Shafir, Y., Weisfeld-Adams, J. D., Alexander, J. J., Hegde, M., et al. (2017b). De novo and inherited SCN8A epilepsy mutations detected by gene panel analysis. Epilepsy Res. 129, 17–25. doi: 10.1016/j.eplepsyres.2016.11.002
Caldwell, J. H., Schaller, K. L., Lasher, R. S., Peles, E., Levinson, S. R. (2000). Sodium channel Nav1.6 is localized at nodes of Ranvier, dendrites, and synapses. Proc. Natl. Acad. Sci. 97, 5616–5620. doi: 10.1073/pnas.090034797
Capes, D. L., Goldschen-Ohm, M. P., Arcisio-Miranda, M., Bezanilla, F., Chanda, B. (2013). Domain IV voltage-sensor movement is both sufficient and rate limiting for fast inactivation in sodium channels. J. Gen. Physiol. 142, 101–112. doi: 10.1085/jgp.201310998
Carranza Rojo, D., Hamiwka, L., McMahon, J. M., Dibbens, L. M., Arsov, T., Suls, A., et al. (2011). De novo SCN1A mutations in migrating partial seizures of infancy. Neurology 77, 380–383. doi: 10.1212/WNL.0b013e318227046d
Carvill, G. L., Heavin, S. B., Yendle, S. C., McMahon, J. M., O’Roak, B. J., Cook, J., et al. (2013). Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat. Genet. 45, 825–830. doi: 10.1038/ng.2646
Catterall, W. A., Kalume, F., Oakley, J. C. (2010). NaV1.1 channels and epilepsy. J. Physiol. 588, 1849–1859. doi: 10.1113/jphysiol.2010.187484
Catterall, W. A. (2014a). Sodium Channels, Inherited Epilepsy, and Antiepileptic Drugs. Annu. Rev. Pharmacol. Toxicol. 54, 317–338. doi: 10.1146/annurevpharmtox-011112-140232
Catterall, W. A. (2014b). Structure and function of voltage-gated sodium channels at atomic resolution. Exp. Physiol. 99, 35–51. doi: 10.1113/expphysiol.2013.071969
Catterall, W. A. (2017). Forty Years of Sodium Channels: Structure, Function, Pharmacology, and Epilepsy. Neurochem. Res. 42, 2495–2504. doi: 10.1007/s11064-017-2314-9
Cen, Z., Lou, Y., Guo, Y., Wang, J., Feng, J. (2017). Q10R mutation in SCN9A gene is associated with generalized epilepsy with febrile seizures plus. Seizure 50, 186–188. doi: 10.1016/j.seizure.2017.06.023
Cheah, C. S., Westenbroek, R. E., Roden, W. H., Kalume, F., Oakley, J. C., Jansen, L. A., et al. (2013). Correlations in timing of sodium channel expression, epilepsy, and sudden death in Dravet syndrome. Channels 7, 468–472. doi: 10.4161/chan.26023
Chen, Y. H., Dale, T. J., Romanos, M. A., Whitaker, W. R. J., Xie, X. M., Clare, J. J. (2000). Cloning, distribution and functional analysis of the type III sodium channel from human brain. Eur. J. Neurosci. 12, 4281–4289. doi: 10.1046/j.1460-9568.2000.01336.x
Cestèle, S., Labate, A., Rusconi, R., Tarantino, P., Mumoli, L., Franceschetti, S., et al. (2013). Divergent effects of the T1174S SCN1A mutation associated with seizures and hemiplegic migraine. Epilepsia 54, 927–935. doi: 10.1111/epi.12123
Cetica, V., Chiari, S., Mei, D., Parrini, E., Grisotto, L., Marini, C., et al. (2017). Clinical and genetic factors predicting Dravet syndrome in infants with SCN1A mutations. Neurology 88, 1037–1044. doi: 10.1212/WNL.0000000000003716
Chen, Y. J., Shi, Y. W., Xu, H. Q., Chen, M. L., Gao, M. M., Sun, W. W., et al. (2015). Electrophysiological Differences between the Same Pore Region Mutation in SCN1A and SCN3A. Mol. Neurobiol. 51, 1263–1270. doi: 10.1007/s12035-014-8802-x
Chong, P. F., Saitsu, H., Sakai, Y., Imagi, T., Nakamura, R., Matsukura, M., et al. (2018). Deletions of SCN2A and SCN3A genes in a patient with West syndrome and autistic spectrum disorder. Seizure 60, 91–93. doi: 10.1016/j.seizure.2018.06.012
Claes, L., Del-Favero, J., Ceulemans, B., Lagae, L., Van Broeckhoven, C., De Jonghe, P. (2001). De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am. J. Hum. Genet. 68, 1327–1332. doi: 10.1086/320609
Claes, L., Ceulemans, B., Audenaert, D., Smets, K., Löfgren, A., Del-Favero, J., et al. (2003). De novo SCN1A mutations are a major cause of severe myoclonic epilepsy of infancy. Hum. Mutat. 21, 615–621. doi: 10.1002/humu.10217
Clairfeuille, T., Cloake, A., Infield, D. T., Llongueras, J. P., Arthur, C. P., Li, Z. R., et al. (2019). Structural basis of a-scorpion toxin action on Nav channels. Science 363, 1–25. doi: 10.1126/science.aav8573
Clark, M. M., Stark, Z., Farnaes, L., Tan, T. Y., White, S. M., Dimmock, D., et al. (2018). Meta-analysis of the diagnostic and clinical utility of genome and exome sequencing and chromosomal microarray in children with suspected genetic diseases. NPJ Genomic Med. 3, 16. doi: 10.1038/s41525-018-0053-8
Colombo, E., Franceschetti, S., Avanzini, G., Mantegazza, M. (2013). Phenytoin Inhibits the Persistent Sodium Current in Neocortical Neurons by Modifying Its Inactivation Properties. PloS One 8, e55329. doi: 10.1371/journal.pone.0055329
Colosimo, E., Gambardella, A., Mantegazza, M., Labate, A., Rusconi, R., Schiavon, E., et al. (2007). Electroclinical Features of a Family with Simple Febrile Seizures and Temporal Lobe Epilepsy Associated with SCN1A Loss-of-Function Mutation. Epilepsia 48, 1691–1696. doi: 10.1111/j.1528-1167.2007.01153.x
Combi, R., Grioni, D., Contri, M., Redaelli, S., Redaelli, F., Bassi, M. T., et al. (2009). Clinical and genetic familial study of a large cohort of Italian children with idiopathic epilepsy. Brain Res. Bull. 79, 89–96. doi: 10.1016/j.brainresbull.2009.01.008
Costain, G., Cordeiro, D., Matviychuk, D., Mercimek-Andrews, S. (2019). Clinical Application of Targeted Next-Generation Sequencing Panels and Whole Exome Sequencing in Childhood Epilepsy. Neuroscience 418, 291–310. doi: 10.1016/j.neuroscience.2019.08.016
Cui, X., Zeng, F., Liu, Y., Zhang, J., Archacki, S., Zhan, T., et al. (2011). A novel SCN1A missense mutation causes generalized epilepsy with febrile seizures plus in a Chinese family. Neurosci. Lett. 503, 27–30. doi: 10.1016/j.neulet.2011.08.001
Cummins, T. R., Waxman, S. G. (1997). Downregulation of tetrodotoxinresistant sodium currents and upregulation of a rapidly repriming tetrodotoxin-sensitive sodium current in small spinal sensory neurons after nerve injury. J. Neurosci. 17, 3503–3514. doi: 10.1523/jneurosci.17-10-03503.1997
Cummins, T. R., Aglieco, F., Renganathan, M., Herzog, R. I. I., Dib-Hajj, S. D., Waxman, S. G. (2001). Nav1.3 sodium channels: Rapid repriming and slow closed-state inactivation display quantitative differences after expression in a mammalian cell line and in spinal sensory neurons. J. Neurosci. 21, 5952–5961. doi: 10.1523/jneurosci.21-16-05952.2001
Daoud, H., Luco, S. M., Li, R., Bareke, E., Beaulieu, C., Jarinova, O., et al. (2016). Next-generation sequencing for diagnosis of Rare diseases in the neonatal intensive care unit. Cmaj 188, E254–E260. doi: 10.1503/cmaj.150823
Davidsson, J., Collin, A., Olsson, M. E., Lundgren, J., Soller, M. (2008). Deletion of the SCN gene cluster on 2q24.4 is associated with severe epilepsy: An array-based genotype–phenotype correlation and a comprehensive review of previously published cases. Epilepsy Res. 81, 69–79. doi: 10.1016/j.eplepsyres.2008.04.018
de Kovel, C. G. F., Meisler, M. H., Brilstra, E. H., van Berkestijn, F. M. C., van Lieshout, S., et al. (2014). Characterization of a de novo SCN8A mutation in a patient with epileptic encephalopathy. Epilepsy Res. 108, 1511–1518. doi: 10.1016/j.eplepsyres.2014.08.020
Deciphering Developmental Disorders Study (2015). Large-scale discovery of novel genetic causes of developmental disorders. Nature 519, 223–228. doi: 10.1038/nature14135
Deng, H., Xiu, X., Song, Z. (2014). The molecular biology of genetic-based epilepsies. Mol. Neurobiol. 49, 352–367. doi: 10.1007/s12035-013-8523-6
Denis, J., Villeneuve, N., Cacciagli, P., Mignon-Ravix, C., Lacoste, C., Lefranc, J., et al. (2019). Clinical study of 19 patients with SCN8A-related epilepsy: Two modes of onset regarding EEG and seizures. Epilepsia 60, 845–856. doi: 10.1111/epi.14727
Depienne, C., Trouillard, O., Saint-Martin, C., Gourfinkel-An, I., Bouteiller, D., Carpentier, W., et al. (2008). Spectrum of SCN1A gene mutations associated with Dravet syndrome: analysis of 333 patients. J. Med. Genet. 46, 183–191. doi: 10.1136/jmg.2008.062323
Devinsky, O., Vezzani, A., Jette, N., De Curtis, M., Perucca, P. (2018). Epilepsy. Nat. Rev. 3, 1–24. doi: 10.1038/nrdp.2018.24
Dhamija, R., Wirrell, E., Falcao, G., Kirmani, S., Wong-Kisiel, L. C. (2013). Novel de novo SCN2A Mutation in a Child With Migrating Focal Seizures of Infancy. Pediatr. Neurol. 49, 486–488. doi: 10.1016/j.pediatrneurol.2013.07.004
Dhamija, R., Erickson, M. K., St Louis, E. K., Wirrell, E., Kotagal, S. (2014). Sleep Abnormalities in Children With Dravet Syndrome. Pediatr. Neurol. 50, 474–478. doi: 10.1016/j.pediatrneurol.2014.01.017
Djémié, T., Weckhuysen, S., von Spiczak, S., Carvill, G. L., Jaehn, J., Anttonen, A.-K., et al. (2016). Pitfalls in genetic testing: the story of missed SCN1A mutations. Mol. Genet. Genomic Med. 4, 457–464. doi: 10.1002/mgg3.217
Doty, C. N. (2010). SCN9A: Another sodium channel excited to play a role in human epilepsies. Clin. Genet. 77, 326–328. doi: 10.1111/j.1399-0004.2009.01366_1.x
Dyment, D. A., Tétreault, M., Beaulieu, C. L., Hartley, T., Ferreira, P., Chardon, J. W., et al. (2015). Whole-exome sequencing broadens the phenotypic spectrum of rare pediatric epilepsy: A retrospective study. Clin. Genet. 88, 34–40. doi: 10.1111/cge.12464
Ebach, K., Joos, H., Doose, H., Stephani, U., Kurlemann, G., Fiedler, B., et al. (2005). SCN1A mutation analysis in myoclonic astatic epilepsy and severe idiopathic generalized epilepsy of infancy with generalized tonic-clonic seizures. Neuropediatrics 36, 210–213. doi: 10.1055/s-2005-865607
Ebrahimi, A., Houshmand, M., Tonekaboni, S. H., Fallah Mahboob Passand, M. S., Zainali, S., Moghadasi, M. (2010). Two Novel Mutations in SCN1A Gene in Iranian Patients with Epilepsy. Arch. Med. Res. 41, 207–214. doi: 10.1016/j.arcmed.2010.04.007
Egri, C., Vilin, Y. Y., Ruben, P. C. (2012). A thermoprotective role of the sodium channel β 1 subunit is lost with the β 1(C121W) mutation. Epilepsia 53, 494–505. doi: 10.1111/j.1528-1167.2011.03389.x
Encinas, A. C., Moore, I., (Ki), M., Watkins, J. C., Hammer, M. F. (2019). Influence of age at seizure onset on the acquisition of neurodevelopmental skills in an SCN8A cohort. Epilepsia 60, 1711–1720. doi: 10.1111/epi.16288
Epifanio, R., Zanotta, N., Giorda, R., Bardoni, A., Zucca, C. (2019). Novel epilepsy phenotype associated to a known SCN8A mutation. Seizure 67, 15–17. doi: 10.1016/j.seizure.2019.01.017
Escayg, A., Heils, A., MacDonald, B. T., Haug, K., Sander, T., Meisler, M. H. (2001). A Novel SCN1A Mutation Associated with Generalized Epilepsy with Febrile Seizures Plus—and Prevalence of Variants in Patients with Epilepsy. Am. J. Hum. Genet. 68, 866–873. doi: 10.1086/319524
Escayg, A., Goldin, A. L. (2010). Sodium channel SCN1A and epilepsy : Mutations and mechanisms. Epilepsia 51, 1650–1658. doi: 10.1111/j.1528-1167.2010.02640.x
Estacion, M., Gasser, A., Dib-Hajj, S. D., Waxman, S. G. (2010). A sodium channel mutation linked to epilepsy increases ramp and persistent current of Nav1.3 and induces hyperexcitability in hippocampal neurons. Exp. Neurol. 224, 362–368. doi: 10.1016/j.expneurol.2010.04.012
Estacion, M., Waxman, S. G. (2013). The response of NaV1.3 sodium channels to ramp stimuli: Multiple components and mechanisms. J. Neurophysiol. 109, 306–314. doi: 10.1152/jn.00438.2012
Estacion, M., O’Brien, J. E., Conravey, A., Hammer, M. F., Waxman, S. G., Dib-Hajj, S. D., et al. (2014). A novel de novo mutation of SCN8A (Nav1.6) with enhanced channel activation in a child with epileptic encephalopathy. Neurobiol. Dis. 69, 117–123. doi: 10.1016/j.nbd.2014.05.017
Esterhuizen, A., II, Mefford, H. C., Ramesar, R. S., Wang, S., Carvill, G. L., Wilmshurst, J. M. (2018). Dravet syndrome in South African infants: Tools for an early diagnosis. Seizure 62, 99–105. doi: 10.1016/j.seizure.2018.09.010
Falco-Walter, J. J., Scheffer, I. E., Fisher, R. S. (2018). The new definition and classification of seizures and epilepsy. Epilepsy Res. 139, 73–79. doi: 10.1016/j.eplepsyres.2017.11.015
Felts, P. A., Yokoyama, S., Dib-Hajj, S., Black, J. A., Waxman, S. G. (1997). Sodium channel a-subunit mRNAs I, II, III, NaG, Na6 and hNE (PN1): different expression patterns in developing rat nervous system. Mol. Brain Res. 45, 71–82. doi: 10.1016/S0169-328X(96)00241-0
Fisher, R. S., Acevedo, C., Arzimanoglou, A., Bogacz, A., Cross, J. H., Elger, C. E., et al. (2014). ILAE Official Report: A practical clinical definition of epilepsy. Epilepsia 55, 475–482. doi: 10.1111/epi.12550
Foster, L. A., Johnson, M. R., MacDonald, J. T., Karachunski, P., II, Henry, T. R., Nascene, D. R., et al. (2017). Infantile Epileptic Encephalopathy Associated With SCN2A Mutation Responsive to Oral Mexiletine. Pediatr. Neurol. 66, 108–111. doi: 10.1016/j.pediatrneurol.2016.10.008
Fry, A. E., Rees, E., Thompson, R., Mantripragada, K., Blake, P., Jones, G., et al. (2016). Pathogenic copy number variants and SCN1A mutations in patients with intellectual disability and childhood-onset epilepsy. BMC Med. Genet. 17, 34. doi: 10.1186/s12881-016-0294-2
Fujiwara, T. (2003). Mutations of sodium channel alpha subunit type 1 (SCN1A) in intractable childhood epilepsies with frequent generalized tonic-clonic seizures. Brain 126, 531–546. doi: 10.1093/brain/awg053
Fukasawa, T., Kubota, T., Negoro, T., Saitoh, M., Mizuguchi, M., Ihara, Y., et al. (2015). A case of recurrent encephalopathy with SCN2A missense mutation. Brain Dev. 37, 631–634. doi: 10.1016/j.braindev.2014.10.001
Fukuma, G., Oguni, H., Shirasaka, Y., Watanabe, K., Miyajima, T., Yasumoto, S., et al. (2004). Mutations of Neuronal Voltage-gated Na+ Channel alpha1 Subunit Gene SCN1A in Core Severe Myoclonic Epilepsy in Infancy (SMEI) and in Borderline SMEI (SMEB). Epilepsia 45, 140–148. doi: 10.1111/j.0013-9580.2004.15103.x
Fung, L.-W. E., Kwok, S.-L. J., Tsui, K.-W. S. (2015). SCN8A mutations in Chinese children with early onset epilepsy and intellectual disability. Epilepsia 56, 1319–1320. doi: 10.1111/epi.12925
Fung, C. W., Kwong, A. K. Y., Wong, V. C. N. (2017). Gene panel analysis for nonsyndromic cryptogenic neonatal/infantile epileptic encephalopathy. Epilepsia Open 2, 236–243. doi: 10.1002/epi4.12055
Gamal El-Din, T. M., Martinez, G. Q., Payandeh, J., Scheuer, T., Catterall, W. A. (2013). A gating charge interaction required for late slow inactivation of the bacterial sodium channel NavAb. J. Gen. Physiol. 142, 181–190. doi: 10.1085/jgp.201311012
Gardella, E., Becker, F., Møller, R. S., Schubert, J., Lemke, J. R., Larsen, L. H. G., et al. (2016). Benign infantile seizures and paroxysmal dyskinesia caused by an SCN8A mutation. Ann. Neurol. 79, 428–436. doi: 10.1002/ana.24580
Gardella, E., Marini, C., Trivisano, M., Fitzgerald, M. P., Alber, M., Howell, K. B., et al. (2018). The phenotype of SCN8A developmental and epileptic encephalopathy. Neurology 91, E1112–E1124. doi: 10.1212/WNL.0000000000006199
Gargus, J. J., Tournay, A. (2007). Novel Mutation Confirms Seizure Locus SCN1A is Also Familial Hemiplegic Migraine Locus FHM3. Pediatr. Neurol. 37, 407–410. doi: 10.1016/j.pediatrneurol.2007.06.016
Ghovanloo, M. R., Aimar, K., Ghadiry-Tavi, R., Yu, A., Ruben, P. C. (2016). Physiology and Pathophysiology of Sodium Channel Inactivation. Curr. Top. Membr. 78, 479–509. doi: 10.1016/bs.ctm.2016.04.001
Gokben, S., Onay, H., Yilmaz, S., Atik, T., Serdaroglu, G., Tekin, H., et al. (2017). Targeted next generation sequencing: the diagnostic value in early-onset epileptic encephalopathy. Acta Neurol. Belg. 117, 131–138. doi: 10.1007/s13760-016-0709-z
Gilchrist, J., Das, S., Van Petegem, F., Bosmans, F. (2013). Crystallographic insights into sodium-channel modulation by the b4 subunit. Proc. Natl. Acad. Sci. 110, E5016–E5024. doi: 10.1073/pnas.1314557110
Goldin, A. L., Escayg, A. (2010). Sodium channel SCN1A and epilepsy: mutations and mechanisms. Epilepsia 51:16. doi: 10.1111/j.1528-1167.2010.02640.x
Goldschen-Ohm, M. P., Capes, D. L., Oelstrom, K. M., Chanda, B. (2013). Multiple pore conformations driven by asynchronous movements of voltage sensors in a eukaryotic sodium channel. Nat. Commun. 4, 1350. doi: 10.1038/ncomms2356
Gorman, K. M., King, M. D. (2017). SCN2A p.Ala263Val Variant a Phenotype of Neonatal Seizures Followed by Paroxysmal Ataxia in Toddlers. Pediatr. Neurol. 67, 111–112. doi: 10.1016/j.pediatrneurol.2016.11.008
Grinton, B. E., Heron, S. E., Pelekanos, J. T., Zuberi, S. M., Kivity, S., Afawi, Z., et al. (2015). Familial neonatal seizures in 36 families: Clinical and genetic features correlate with outcome. Epilepsia 56, 1071–1080. doi: 10.1111/epi.13020
Guerrini, R., Cellini, E., Mei, D., Metitieri, T., Petrelli, C., Pucatti, D., et al. (2010). Variable epilepsy phenotypes associated with a familial intragenic deletion of the SCN1A gene. Epilepsia 51, 2474–2477. doi: 10.1111/j.1528-1167.2010.02790.x
Hackenberg, A., Baumer, A., Sticht, H., Schmitt, B., Kroell-Seger, J., Wille, D., et al. (2014). Infantile Epileptic Encephalopathy, Transient Choreoathetotic Movements, and Hypersomnia due to a De Novo Missense Mutation in the SCN2A Gene. Neuropediatrics 45, 261–264. doi: 10.1055/s-0034-1372302
Haginoya, K., Togashi, N., Kaneta, T., Hino-Fukuyo, N., Ishitobi, M., Kakisaka, Y., et al. (2018). [18F]fluorodeoxyglucose-positron emission tomography study of genetically confirmed patients with Dravet syndrome. Epilepsy Res. 147, 9–14. doi: 10.1016/j.eplepsyres.2018.08.008
Hains, B. C., Klein, J. P., Saab, C. Y., Craner, M. J., Black, J. A., Waxman, S. G. (2003). Upregulation of sodium channel Nav1.3 and functional involvement in neuronal hyperexcitability associated with central neuropathic pain after spinal cord injury. J. Neurosci. 23, 8881–8892. doi: 10.1523/jneurosci.23-26-08881.2003
Halvorsen, M., Petrovski, S., Shellhaas, R., Tang, Y., Crandall, L., Goldstein, D., et al. (2016). Mosaic mutations in early-onset genetic diseases. Genet. Med. 18, 746–749. doi: 10.1038/gim.2015.155
Han, J. Y., Jang, J. H., Lee, I. G., Shin, S., Park, J. (2017). A novel inherited mutation of SCN8a in a korean family with benign familial infantile epilepsy using diagnostic exome sequencing. Ann. Clin. Lab. Sci. 47, 747–753.
Han, C., Dib-Hajj, S. D., Lin, Z., Li, Y., Eastman, E. M., Tyrrell, L., et al. (2009). Early- and late-onset inherited erythromelalgia: genotypephenotype correlation. Brain 132, 1711–1722. doi: 10.1093/brain/awp078
Hammer, M. F., Wagnon, J. L., Mefford, H. C., Meisler, M. H., et al. (2016). “SCN8A-Related Epilepsy with Encephalopathy,” in GeneReviews® [Internet]. Eds. Adam, M. P., Ardinger, H. H., Pagon, R. A. (Seattle (WA): University of Washington).
Harkin, L. A., McMahon, J. M., Iona, X., Dibbens, L., Pelekanos, J. T., Zuberi, S. M., et al. (2007). The spectrum of SCN1A-related infantile epileptic encephalopathies. Brain 130, 843–852. doi: 10.1093/brain/awm002
Haug, K., Hallmann, K., Rebstock, J., Dullinger, J., Muth, S., Haverkamp, F., et al. (2001). The voltage-gated sodium channel gene SCN2A and idiopathic generalized epilepsy. Epilepsy Res. 47, 243–246. doi: 10.1016/S0920-1211(01)00312-6
Haufe, V., Camacho, J. A., Dumaine, R., Günther, B., Bollensdorff, C., von Banchet, G. S., et al. (2005). Expression pattern of neuronal and skeletal muscle voltage-gated Na+ channels in the developing mouse heart. J. Physiol. 564, 683–696. doi: 10.1113/jphysiol.2004.079681
Herlenius, E., Heron, S. E., Grinton, B. E., Keay, D., Scheffer, I. E., Mulley, J. C., et al. (2007). SCN2A mutations and benign familial neonatal-infantile seizures: The phenotypic spectrum. Epilepsia 48, 1138–1142. doi: 10.1111/j.1528-1167.2007.01049.x
Hernández Chávez, M., Mesa Latorre, T., Pedraza Herrera, M., Troncoso Schifferli, M. (2014). ¿Crisis febriles complejas o síndrome de Dravet?: Descripción de 3 casos clínicos. Rev. Chil. pediatría 85, 588–593. doi: 10.4067/S0370-41062014000500010
Heron, S. E., Crossland, K. M., Andermann, E., Phillips, H. A., Hall, A. J., Bleasel, A., et al. (2002). Sodium-channel defects in benign familial neonatal-infantile seizures. Lancet 360, 851–852. doi: 10.1016/S0140-6736(02)09968-3
Heron, S. E., Scheffer, I. E., Grinton, B. E., Eyre, H., Oliver, K. L., Bain, S., et al. (2010). Familial neonatal seizures with intellectual disability caused by a microduplication of chromosome 2q24.3. Epilepsia 51, 1865–1869. doi: 10.1111/j.1528-1167.2010.02558.x
Hewson, S., Brunga, L., Ojeda, M. F., Imhof, E., Patel, J., Zak, M., et al. (2018). Prevalence of Genetic Disorders and GLUT1 Deficiency in a Ketogenic Diet Clinic. Can. J. Neurol. Sci. 45, 93–96. doi: 10.1017/cjn.2017.246
Heyne, H. O., Artomov, M., Battke, F., Bianchini, C., Smith, D. R., Liebmann, N., et al. (2019). Targeted gene sequencing in 6994 individuals with neurodevelopmental disorder with epilepsy. Genet. Med. 21, 2496–2503. doi: 10.1038/s41436-019-0531-0
Hoffman-Zacharska, D., Szczepanik, E., Terczynska, I., Goszczanska-Ciuchta, A., Zalewska-Miszkurka, Z., Tataj, R., et al. (2015). From focal epilepsy to dravet syndrome –heterogeneity of the phenotype due to SCN1A mutations of the p.Arg1596 amino acid residue in the nav1.1 subunit. Neurol. Neurochir. Pol. 49, 258–266. doi: 10.1016/j.pjnns.2015.06.006
Holland, K. D., Kearney, J. A., Glauser, T. A., Buck, G., Keddache, M., Blankston, J. R., et al. (2008). Mutation of sodium channel SCN3A in a patient with cryptogenic pediatric partial epilepsy. Neurosci. Lett. 433, 65–70. doi: 10.1016/j.neulet.2007.12.064
Horvath, G. A., Demos, M., Shyr, C., Matthews, A., Zhang, L., Race, S., et al. (2016). Secondary neurotransmitter deficiencies in epilepsy caused by voltage-gated sodium channelopathies: A potential treatment target? Mol. Genet. Metab. 117, 42–48. doi: 10.1016/j.ymgme.2015.11.008
Howell, K. B., McMahon, J. M., Carvill, G. L., Tambunan, D., Mackay, M. T., Rodriguez-Casero, V., et al. (2015). SCN2A encephalopathy. Neurology 85, 958–966. doi: 10.1212/WNL.0000000000001926
Hsiao, J., Yuan, T. Y., Tsai, M. S., Lu, C. Y., Lin, Y. C., Lee, M. L., et al. (2016). Upregulation of Haploinsufficient Gene Expression in the Brain by Targeting a Long Non-coding RNA Improves Seizure Phenotype in a Model of Dravet Syndrome. EBioMedicine 9, 257–277. doi: 10.1016/j.ebiom.2016.05.011
Hu, W., Tian, C., Li, T., Yang, M., Hou, H., Shu, Y. (2009). Distinct contributions of Nav1.6 and Nav1.2 in action potential initiation and backpropagation. Nat. Neurosci. 12, 996–1002. doi: 10.1038/nn.2359
Huang, W., Liu, M., Yan, S. F., Yan, N. (2017). Structure-based assessment of disease-related mutations in human voltage-gated sodium channels. Protein Cell 8, 401–438. doi: 10.1007/s13238-017-0372-z
Hussain, A., Seinfeld, S., Morton, L. (2016). Genetic association with ictal cardiorespiratory phenomena: SCN8A case series. J. Pediatr. Neurol. 14, 151–155. doi: 10.1055/s-0036-1593744
Iannetti, P., Parisi, P., Spalice, A., Ruggieri, M., Zara, F. (2009). Addition of verapamil in the treatment of severe myoclonic epilepsy in infancy. Epilepsy Res. 85, 89–95. doi: 10.1016/j.eplepsyres.2009.02.014
Inuzuka, L. M., Macedo-Souza, L., II, Della-Ripa, B., Cabral, K. S. S., Monteiro, F., Kitajima, J. P., et al. (2019). Neurodevelopmental disorder associated with de novo SCN3A pathogenic variants: two new cases and review of the literature. Brain Dev. 42, 211–216. doi: 10.1016/j.braindev.2019.09.004
Israel, M. R., Thongyoo, P., Deuis, J. R., Craik, D. J., Vetter, I., Durek, T. (2018). The E15R Point Mutation in Scorpion Toxin Cn2 Uncouples Its Depressant and Excitatory Activities on Human Na V 1.6. J. Med. Chem. 61, 1730–1736. doi: 10.1021/acs.jmedchem.7b01609
Ito, M., Shirasaka, Y., Hirose, S., Sugawara, T., Yamakawa, K. (2004). Seizure phenotypes of a family with missense mutations in SCN2A. Pediatr. Neurol. 31, 150–152. doi: 10.1016/j.pediatrneurol.2004.02.013
Jain, P., Gulati, P., Morrison-Levy, N., Yau, I., Alsowat, D., Otsubo, H., et al. (2019). “Breath holding spells” in a child with SCN8A-related epilepsy: Expanding the clinical spectrum. Seizure 65, 129–130. doi: 10.1016/j.seizure.2019.01.020
Jang, S. S., Kim, S. Y., Kim, H., Hwang, H., Chae, J. H., Kim, K. J., et al. (2019). Diagnostic Yield of Epilepsy Panel Testing in Patients With Seizure Onset Within the First Year of Life. Front. Neurol. 10, 988. doi: 10.3389/fneur.2019.00988
Jiang, D., Shi, H., Tonggu, L., Gamal El-Din, T. M., Lenaeus, M. J., Zhao, Y., et al. (2020). Structure of the Cardiac Sodium Channel. Cell 180, 122–134.e10. doi: 10.1016/j.cell.2019.11.041
Jingami, N., Matsumoto, R., Ito, H., Ishii, A., Ihara, Y., Hirose, S., et al. (2014). A novel SCN1A mutation in a cytoplasmic loop in intractable juvenile myoclonic epilepsy without febrile seizures. Epileptic Disord. 16, 227–231. doi: 10.1684/epd.2014.0657
Johannesen, K. M., Gardella, E., Scheffer, I., Howell, K., Smith, D. M., Helbig, I., et al. (2018). Early mortality in SCN8A -related epilepsies. Epilepsy Res. 143, 79–81. doi: 10.1016/j.eplepsyres.2018.04.008
Johannesen, K. M., Gardella, E., Encinas, A. C., Lehesjoki, A. E., Linnankivi, T., Petersen, M. B., et al. (2019). The spectrum of intermediate SCN8A-related epilepsy. Epilepsia 60, 830–844. doi: 10.1111/epi.14705
Johnson, C. N., Potet, F., Thompson, M. K., Kroncke, B. M., Glazer, A. M., Voehler, M. W., et al. (2018). A Mechanism of Calmodulin Modulation of the Human Cardiac Sodium Channel. Structure 26, 683–694.e3. doi: 10.1016/j.str.2018.03.005
Kamiya, K. (2004). A Nonsense Mutation of the Sodium Channel Gene SCN2A in a Patient with Intractable Epilepsy and Mental Decline. J. Neurosci. 24, 2690–2698. doi: 10.1523/JNEUROSCI.3089-03.2004
Kaplan, D. I. I., Isom, L. L., Petrou, S. (2016). Role of sodium channels in epilepsy. Cold Spring Harb. Perspect. Med. 6:a022814. doi: 10.1101/cshperspect.a022814
Kearney, J., Plummer, N., Smith, M., Kapur, J., Cummins, T., Waxman, S., et al. (2001). A gain-of-function mutation in the sodium channel gene Scn2a results in seizures and behavioral abnormalities. Neuroscience 102, 307–317. doi: 10.1016/S0306-4522(00)00479-6
Kim, Y. O., Bellows, S., Mcmahon, J. M., Iona, X., Damiano, J., Dibbens, L., et al. (2014). Atypical multifocal Dravet syndrome lacks generalized seizures and may show later cognitive decline. Dev. Med. Child Neurol. 56, 85–90. doi: 10.1111/dmcn.12322
Kim, H. J., Yang, D., Kim, S. H., Kim, B., Kim, H. D., Lee, J. S., et al. (2019). Genetic and clinical features of SCN8A developmental and epileptic encephalopathy. Epilepsy Res. 158, 106222. doi: 10.1016/j.eplepsyres.2019.106222
Knupp, K. G., Wirrell, E. C. (2018). Treatment Strategies for Dravet Syndrome. CNS Drugs 32, 335–350. doi: 10.1007/s40263-018-0511-y
Kobayashi, K., Ohzono, H., Shinohara, M., Saitoh, M., Ohmori, I., Ohtsuka, Y., et al. (2012). Acute encephalopathy with a novel point mutation in the SCN2A gene. Epilepsy Res. 102, 109–112. doi: 10.1016/j.eplepsyres.2012.04.016
Kodera, H., Kato, M., Nord, A. S., Walsh, T., Lee, M., Yamanaka, G., et al. (2013). Targeted capture and sequencing for detection of mutations causing early onset epileptic encephalopathy. Epilepsia 54, 1262–1269. doi: 10.1111/epi.12203
Kothur, K., Holman, K., Farnsworth, E., Ho, G., Lorentzos, M., Troedson, C., et al. (2018). Diagnostic yield of targeted massively parallel sequencing in children with epileptic encephalopathy. Seizure 59, 132–140. doi: 10.1016/j.seizure.2018.05.005
Kwong, A. K. Y., Fung, C. W., Chan, S. Y., Wong, V. C. N. (2012). Identification of SCN1A and PCDH19 mutations in Chinese children with Dravet syndrome. PloS One 7, e41802. doi: 10.1371/journal.pone.0041802
Laezza, F., Lampert, A., Kozel, M. A., Gerber, B. R., Rush, A. M., Nerbonne, J. M., et al. (2009). FGF14 N-terminal splice variants differentially modulate Nav1.2 and Nav1.6-encoded sodium channels. Mol. Cell. Neurosci. 42, 90–101. doi: 10.1016/j.mcn.2009.05.007
Lal, D., Reinthaler, E. M., Dejanovic, B., May, P., Thiele, H., Lehesjoki, A.-E., et al. (2016). Evaluation of Presumably Disease Causing SCN1A Variants in a Cohort of Common Epilepsy Syndromes. PloS One 11, e0150426. doi: 10.1371/journal.pone.0150426
Lamar, T., Vanoye, C. G., Calhoun, J., Wong, J. C., Dutton, S. B., Jorge, B. S., et al. (2017). SCN3A deficiency associated with increased seizure susceptibility. Neurobiol. Dis. 102, 38–48. doi: 10.1016/j.nbd.2017.02.006
Larsen, J., Carvill, G. L., Gardella, E., Kluger, G., Schmiedel, G., Barisic, N., et al. (2015). The phenotypic spectrum of SCN8A encephalopathy. Neurology 84, 480–489. doi: 10.1212/WNL.0000000000001211
Lattanzi, S., Brigo, F., Trinka, E., Zaccara, G., Striano, P., Del Giovane, C., et al. (2020). Adjunctive Cannabidiol in Patients with Dravet Syndrome: A Systematic Review and Meta-Analysis of Efficacy and Safety. CNS Drugs 34, 229–241. doi: 10.1007/s40263-020-00708-6
Lauxmann, S., Boutry-Kryza, N., Rivier, C., Mueller, S., Hedrich, U. B. S., Maljevic, S., et al. (2013). An SCN2A mutation in a family with infantile seizures from Madagascar reveals an increased subthreshold Na+ current. Epilepsia 54, e117–e121. doi: 10.1111/epi.12241
Lek, M., Karczewski, K. J., Minikel, E. V., Samocha, K. E., Banks, E., Fennell, T., et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. doi: 10.1038/nature19057
Le Gal, F., Lebon, S., Ramelli, G. P., Datta, A. N., Mercati, D., Maier, O., et al. (2014). When is a child with status epilepticus likely to have Dravet syndrome? Epilepsy Res. 108, 740–747. doi: 10.1016/j.eplepsyres.2014.02.019
Lee, H.-F., Chi, C.-S., Tsai, C.-R., Chen, C.-H., Wang, C.-C. (2014). Electroencephalographic features of patients with SCN1A-positive Dravet syndrome. Brain Dev. 37, 599–611. doi: 10.1016/j.braindev.2014.10.003
Lemke, J. R., Riesch, E., Scheurenbrand, T., Schubach, M., Wilhelm, C., Steiner, I., et al. (2012). Targeted next generation sequencing as a diagnostic tool in epileptic disorders. Epilepsia 53, 1387–1398. doi: 10.1111/j.1528-1167.2012.03516.x
Lewis, A. H., Raman, I. M. (2014). Resurgent current of voltage-gated Na+ channels. J. Physiol. 592, 4825–4838. doi: 10.1113/jphysiol.2014.277582
Liao, W.-P., Shi, Y.-W., Long, Y.-S., Zeng, Y., Li, T., Yu, M.-J., et al. (2010a). Partial epilepsy with antecedent febrile seizures and seizure aggravation by antiepileptic drugs: Associated with loss of function of Nav1.1. Epilepsia 51, 1669–1678. doi: 10.1111/j.1528-1167.2010.02645.x
Liao, Y., Deprez, L., Maljevic, S., Pitsch, J., Claes, L., Hristova, D., et al. (2010b). Molecular correlates of age-dependent seizures in an inherited neonatal-infantile epilepsy. Brain 133, 1403–1414. doi: 10.1093/brain/awq057
Lim, B. C., Hwang, H., Chae, J. H., Choi, J.-E., Hwang, Y. S., Kang, S.-H., et al. (2011). SCN1A mutational analysis in Korean patients with Dravet syndrome. Seizure 20, 789–794. doi: 10.1016/j.seizure.2011.08.002
Lin, K. M., Su, G., Wang, F., Zhang, X., Wang, Y., Ren, J., et al. (2019). A de novo SCN8A heterozygous mutation in a child with epileptic encephalopathy: A case report. BMC Pediatr. 19, 400. doi: 10.1186/s12887-019-1796-9
Lindy, A. S., Stosser, M. B., Butler, E., Downtain-Pickersgill, C., Shanmugham, A., Retterer, K., et al. (2018). Diagnostic outcomes for genetic testing of 70 genes in 8565 patients with epilepsy and neurodevelopmental disorders. Epilepsia 59, 1062–1071. doi: 10.1111/epi.14074
Liu, J., Tong, L., Song, S., Niu, Y., Li, J., Wu, X., et al. (2018). Novel and de novo mutations in pediatric refractory epilepsy. Mol. Brain 11, 48. doi: 10.1186/s13041-018-0392-5
Liu, Y., Schubert, J., Sonnenberg, L., Helbig, K. L., Hoei-Hansen, C. E., Koko, M., et al. (2019). Neuronal mechanisms of mutations in SCN8A causing epilepsy or intellectual disability. Brain 142, 376–390. doi: 10.1093/brain/awy326
Lossin, C., Rhodes, T. H., Desai, R. R., Vanoye, C. G., Wang, D., Carniciu, S., et al. (2003). Epilepsy-Associated Dysfunction in the Voltage-Gated Neuronal Sodium Channel SCN1A. J. Neurosci. 23, 11289–11295. doi: 10.1523/jneurosci.23-36-11289.2003
Lossin, C., Shi, X., Rogawski, M. A., Hirose, S. (2012). Compromised function in the Nav1.2 Dravet syndrome mutation R1312T. Neurobiol. Dis. 47, 378–384. doi: 10.1016/j.nbd.2012.05.017
Lucas, P. T., Meadows, L. S., Nicholls, J., Ragsdale, D. S. (2005). An epilepsy mutation in the β1 subunit of the voltage-gated sodium channel results in reduced channel sensitivity to phenytoin. Epilepsy Res. 64, 77–84. doi: 10.1016/j.eplepsyres.2005.03.003
Maier, S. K. G., Westenbroek, R. E., Schenkman, K. A., Feigl, E. O., Scheuer, T., Catterall, W. A. (2002). An unexpected role for brain-type sodium channels in coupling of cell surface depolarization to contraction in the heart. Proc. Natl. Acad. Sci. U. S. A. 99, 4073–4078. doi: 10.1073/pnas.261705699
Mak, C. M., Chan, K. Y. W., Yau, E. K. C., Chen, S. P. L., Siu, W. K., Law, C. Y., et al. (2011). Genetic diagnosis of severe myoclonic epilepsy of infancy (Dravet syndrome) with SCN1A mutations in the Hong Kong Chinese patients. Hong Kong Med. J. = Xianggang yi xue za zhi 17, 500–502.
Makinson, C. D., Tanaka, B. S., Sorokin, J. M., Wong, J. C., Christian, C. A., Goldin, A. L., et al. (2017). Regulation of Thalamic and Cortical Network Synchrony by Scn8a. Neuron 93, 1165–1179.e6. doi: 10.1016/j.neuron.2017.01.031
Malcolmson, J., Kleyner, R., Tegay, D., Adams, W., Ward, K., Coppinger, J., et al. (2016). SCN8A mutation in a child presenting with seizures and developmental delays. Cold Spring Harb. Mol. Case Stud. 2, a001073. doi: 10.1101/mcs.a001073
Malo, D., Schurr, E., Dorfman, J., Canfield, V., Levenson, R., Gros, P. (1991). Three brain sodium channel a-subunit genes are clustered on the proximal segment of mouse chromosome 2. Genomics 10, 666–672. doi: 10.1016/0888-7543(91)90450-S
Malo, M. S., Blanchard, B. J., Andresen, J. M., Srivastava, K., Chen, X.-N., Li, X., et al. (1994). Localization of a putative human brain sodium channel gene (SCN1A) to chromosome band 2q24. Cytogenet. Genome Res. 67, 178–186. doi: 10.1159/000133818
Mantegazza, M., Gambardella, A., Rusconi, R., Schiavon, E., Annesi, F., Cassulini, R. R., et al. (2005). Identification of an Nav1.1 sodium channel (SCN1A) loss-of-function mutation associated with familial simple febrile seizures. Proc. Natl. Acad. Sci. U. S. A. 102, 18177–18182. doi: 10.1073/pnas.0506818102
Marini, C., Mei, D., Temudo, T., Ferrari, A. R., Buti, D., Dravet, C., et al. (2007). Idiopathic Epilepsies with Seizures Precipitated by Fever and SCN1A Abnormalities. Epilepsia 48, 1678–1685. doi: 10.1111/j.1528-1167.2007.01122.x
Martin, H. C., Kim, G. E., Pagnamenta, A. T., Murakami, Y., Carvill, G. L., Meyer, E., et al. (2014). Clinical whole-genome sequencing in severe early-onset epilepsy reveals new genes and improves molecular diagnosis. Hum. Mol. Genet. 23, 3200–3211. doi: 10.1093/hmg/ddu030
Mason, E. R., Wu, F., Patel, R. R., Xiao, Y., Cannon, S. C., Cummins, T. R. (2019). Resurgent and gating pore currents induced by De Novo SCN2A epilepsy mutations. eNeuro 6, 1–17, ENEURO.0141–19.2019. doi: 10.1523/ENEURO.0141-19.2019
Mason, E. R., Cummins, T. R. (2020). Differential inhibition of human Nav1.2 resurgent and persistent sodium currents by cannabidiol and GS967. Int. J. Mol. Sci. 21, 1–21. doi: 10.3390/ijms21072454
Matalon, D., Goldberg, E., Medne, L., Marsh, E. D. (2014). Confirming an expanded spectrum of SCN2A mutations: a case series. Epileptic Disord. 16, 13–18. doi: 10.1684/epd.2014.0641
McMichael, G., Bainbridge, M. N., Haan, E., Corbett, M., Gardner, A., Thompson, S., et al. (2015). Whole-exome sequencing points to considerable genetic heterogeneity of cerebral palsy. Mol. Psychiatry 20, 176–182. doi: 10.1038/mp.2014.189
McNally, M. A., Johnson, J., Huisman, T. A., Poretti, A., Baranano, K. W., Baschat, A. A., et al. (2016). SCN8A Epileptic Encephalopathy: Detection of Fetal Seizures Guides Multidisciplinary Approach to Diagnosis and Treatment. Pediatr. Neurol. 64, 87–91. doi: 10.1016/j.pediatrneurol.2016.08.003
Mei, D., Parrini, E., Marini, C., Guerrini, R. (2017). The Impact of Next- Generation Sequencing on the Diagnosis and Treatment of Epilepsy in Paediatric Patients. Mol. Diagnosis Ther. 21, 357–373. doi: 10.1007/s40291-017-0257-0
Meisler, M. H., O’Brien, J. E., Sharkey, L. M. (2010). Sodium channel gene family: Epilepsy mutations, gene interactions and modifier effects. J. Physiol. 588, 1841–1848. doi: 10.1113/jphysiol.2010.188482
Meng, H., Xu, H. Q., Yu, L., Lin, G. W., He, N., Su, T., et al. (2015). The SCN1A Mutation Database: Updating Information and Analysis of the Relationships among Genotype, Functional Alteration, and Phenotype. Hum. Mutat. 36, 573–580. doi: 10.1002/humu.22782
Mercimek-Mahmutoglu, S., Patel, J., Cordeiro, D., Hewson, S., Callen, D., Donner, E. J., et al. (2015). Diagnostic yield of genetic testing in epileptic encephalopathy in childhood. Epilepsia 56, 707–716. doi: 10.1111/epi.12954
Misra, S. N., Kahlig, K. M., George, A. L. (2008). Impaired Na V 1.2 function and reduced cell surface expression in benign familial neonatal-infantile seizures. Epilepsia 49, 1535–1545. doi: 10.1111/j.1528-1167.2008.01619.x
Miyatake, S., Kato, M., Sawaishi, Y., Saito, T., Nakashima, M., Mizuguchi, T., et al. (2018). Recurrent SCN3A p.Ile875Thr variant in patients with polymicrogyria. Ann. Neurol. 84, 159–161. doi: 10.1002/ana.25256
Møller, R. S., Larsen, L. H. G., Johannesen, K. M., Talvik, I., Talvik, T., Vaher, U., et al. (2016). Gene panel testing in epileptic encephalopathies and familial epilepsies. Mol. Syndromol. 7, 210–219. doi: 10.1159/000448369
Morano, A., Fanella, M., Albini, M., Cifelli, P., Palma, E., Giallonardo, A. T., et al. (2020). Cannabinoids in the treatment of epilepsy: Current status and future prospects. Neuropsychiatr. Dis. Treat. 16, 381–396. doi: 10.2147/NDT.S203782
Morimoto, M., Mazaki, E., Nishimura, A., Chiyonobu, T., Sawai, Y., Murakami, A., et al. (2006). SCN1A Mutation Mosaicism in a Family with Severe Myoclonic Epilepsy in Infancy. Epilepsia 47, 1732–1736. doi: 10.1111/j.1528-1167.2006.00645.x
Mulley, J. C., Hodgson, B., McMahon, J. M., Iona, X., Bellows, S., Mullen, S. A., et al. (2013). Role of the sodium channel SCN9A in genetic epilepsy with febrile seizures plus and Dravet syndrome. Epilepsia 54, e122–e126. doi: 10.1111/epi.12323
Musto, E., Gardella, E., Møller, R. S. (2020). Recent advances in treatment of epilepsy-related sodium channelopathies. Eur. J. Paediatr. Neurol. 24, 123–128. doi: 10.1016/j.ejpn.2019.12.009
Myers, K. A., Burgess, R., Afawi, Z., Damiano, J. A., Berkovic, S. F., Hildebrand, M. S., et al. (2017a). De novo SCN1A pathogenic variants in the GEFS+ spectrum: Not always a familial syndrome. Epilepsia 58, e26–e30. doi: 10.1111/epi.13649
Myers, K. A., McMahon, J. M., Mandelstam, S. A., Mackay, M. T., Kalnins, R. M., Leventer, R. J., et al. (2017b). Fatal Cerebral Edema With Status Epilepticus in Children With Dravet Syndrome: Report of 5 Cases. Pediatrics 139, e20161933. doi: 10.1542/peds.2016-1933
Nabbout, R., Gennaro, E., Dalla Bernardina, B., Dulac, O., Madia, F., Bertini, E., et al. (2003). Spectrum of SCN1A mutations in severe myoclonic epilepsy of infancy. Neurology 60, 1961–1967. doi: 10.1212/01.WNL.0000069463.41870.2F
Nabbout, R., Copioli, C., Chipaux, M., Chemaly, N., Desguerre, I., Dulac, O., et al. (2011). Ketogenic diet also benefits Dravet syndrome patients receiving stiripentol: A prospective pilot study. Epilepsia 52, 54–57. doi: 10.1111/j.1528-1167.2011.03107.x
Nakamura, K., Kato, M., Osaka, H., Yamashita, S., Nakagawa, E., Haginoya, K., et al. (2013). Clinical spectrum of SCN2A mutations expanding to Ohtahara syndrome. Neurology 81, 992–998. doi: 10.1212/WNL.0b013e3182a43e57
Need, A. C., Shashi, V., Hitomi, Y., Schoch, K., Shianna, K. V., McDonald, M. T., et al. (2012). Clinical application of exome sequencing in undiagnosed genetic conditions. J. Med. Genet. 49, 353–361. doi: 10.1136/jmedgenet-2012-100819
Ng, S. B., Buckingham, K. J., Lee, C., Bigham, A. W., Tabor, H. K., Dent, K. M., et al. (2010). Exome sequencing identifies the cause of a mendelian disorder. Nat. Genet. 42, 30–35. doi: 10.1038/ng.499
Nguyen, H. M., Goldin, A. L. (2010). Sodium channel carboxyl-terminal residue regulates fast inactivation. J. Biol. Chem. 285, 9077–9089. doi: 10.1074/jbc.M109.054940
Nicita, F., Spalice, A., Papetti, L., Ursitti, F., Parisi, P., Gennaro, E., et al. (2010). Genotype-phenotype correlations in a group of 15 SCN1A-mutated italian patients with GEFS+ spectrum (seizures plus, classical and borderline severe myoclonic epilepsy of infancy). J. Child Neurol. 25, 1369–1376. doi: 10.1177/0883073810365737
Nishri, D., Blumkin, L., Lev, D., Leshinsky-Silver, E., Abu-Rashid, M., Birch, R., et al. (2010). Hepatic coma culminating in severe brain damage in a child with a SCN1A mutation. Eur. J. Paediatr. Neurol. 14, 456–459. doi: 10.1016/j.ejpn.2010.03.002
Noujaim, S. F., Kaur, K., Milstein, M., Jones, J. M., Furspan, P., Jiang, D., et al. (2012). A null mutation of the neuronal sodium channel Na V 1.6 disrupts action potential propagation and excitation-contraction coupling in the mouse heart. FASEB J. 26, 63–72. doi: 10.1096/fj.10-179770
Oates, S., Tang, S., Rosch, R., Lear, R., Hughes, E. F., Williams, R. E., et al. (2018). Incorporating epilepsy genetics into clinical practice: A 360°evaluation. NPJ Genomic Med. 3, 13. doi: 10.1038/s41525-018-0052-9
O’Brien, J. E., Sharkey, L. M., Vallianatos, C. N., Han, C., Blossom, J. C., Yu, T., et al. (2012). Interaction of Voltage-gated Sodium Channel Na v 1.6 ( SCN8A ) with Microtubule-associated Protein Map1b. J. Biol. Chem. 287, 18459–18466. doi: 10.1074/jbc.M111.336024
Oelstrom, K., Goldschen-ohm, M. P., Holmgren, M., Chanda, B. (2014). Evolutionarily conserved intracellular gate of voltage-dependent sodium channels. Nat. Commun. 5, 1–9. doi: 10.1038/ncomms4420
Ogiwara, I., Ito, K., Sawaishi, Y., Osaka, H., Mazaki, E., Inoue, I., et al. (2009). De novo mutations of voltage-gated sodium channel αiI gene SCN2A in intractable epilepsies. Neurology 73, 1046–1053. doi: 10.1212/WNL.0b013e3181b9cebc
Ohashi, T., Akasaka, N., Kobayashi, Y., Magara, S., Kawashima, H., Matsumoto, N., et al. (2014). Infantile epileptic encephalopathy with a hyperkinetic movement disorder and hand stereotypies associated with a novel SCN1A mutation. Epileptic Disord. 16, 208–212. doi: 10.1684/epd.2014.0649
Ohba, C., Kato, M., Takahashi, S., Lerman-Sagie, T., Lev, D., Terashima, H., et al. (2014). Early onset epileptic encephalopathy caused by de novo SCN8A mutations. Epilepsia 55, 994–1000. doi: 10.1111/epi.12668
Ohmori, I., Kahlig, K. M., Rhodes, T. H., Wang, D. W., George, A. L. (2006). Nonfunctional SCN1A Is Common in Severe Myoclonic Epilepsy of Infancy. Epilepsia 47, 1636–1642. doi: 10.1111/j.1528-1167.2006.00643.x
Oliva, M., Berkovic, S. F., Petrou, S. (2012). Sodium channels and the neurobiology of epilepsy. Epilepsia 53, 1849–1859. doi: 10.1111/j.1528-1167.2012.03631.x
Oliva, M. K., Mcgarr, T. C., Beyer, B. J., Gazina, E., Kaplan, D., II, Cordeiro, L., et al. (2014). Physiological and genetic analysis of multiple sodium channel variants in a model of genetic absence epilepsy. Neurobiol. Dis. 67, 180–190. doi: 10.1016/j.nbd.2014.03.007
Olson, H. E., Tambunan, D., LaCoursiere, C., Goldenberg, M., Pinsky, R., Martin, E., et al. (2015). Mutations in epilepsy and intellectual disability genes in patients with features of Rett syndrome. Am. J. Med. Genet. Part A 167, 2017–2025. doi: 10.1002/ajmg.a.37132
Orrico, A., Galli, L., Grosso, S., Buoni, S., Pianigiani, R., Balestri, P., et al. (2009). Mutational analysis of the SCN1A, SCN1B and GABRG2 genes in 150 Italian patients with idiopathic childhood epilepsies. Clin. Genet. 75, 579–581. doi: 10.1111/j.1399-0004.2009.01155.x
Orsini, A., Zara, F., Striano, P. (2018). Recent advances in epilepsy genetics. Neurosci. Lett. 667, 4–9. doi: 10.1016/j.neulet.2017.05.014
Ortiz Madinaveitia, S., Serrano Madrid, M. L., Conejo Moreno, D., Sagarra Mur, D., Jiménez Corral, C., Gutiérrez Álvarez, Á.M. (2017). Encefalopatía epiléptica de inicio precoz en un paciente con mutación en SCN8A. Rev. Neurol. 65, 572. doi: 10.33588/rn.6512.2017426
Pan, Y., Cummins, T. R. (2020). Distinct functional alterations in SCN8A epilepsy mutant channels. J. Physiol. 598, 381–401. doi: 10.1113/JP278952
Parrini, E., Marini, C., Mei, D., Galuppi, A., Cellini, E., Pucatti, D., et al. (2017). Diagnostic Targeted Resequencing in 349 Patients with Drug-Resistant Pediatric Epilepsies Identifies Causative Mutations in 30 Different Genes. Hum. Mutat. 38, 216–225. doi: 10.1002/humu.23149
Patel, R. R., Barbosa, C., Brustovetsky, T., Brustovetsky, N., Cummins, T. R. (2016). Aberrant epilepsy-associated mutant Nav1.6 sodium channel activity can be targeted with cannabidiol. Brain 139, 2164–2181. doi: 10.1093/brain/aww129
Payandeh, J., Gamal El-Din, T. M., Scheuer, T., Zheng, N., Catterall, W. A. (2012). Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature 486, 135–139. doi: 10.1038/nature11077
Perucca, P., Perucca, E. (2019). Identifying mutations in epilepsy genes:Impact on treatment selection. Epilepsy Res. 152, 18–30. doi: 10.1016/j.eplepsyres.2019.03.001
Pescucci, C., Caselli, R., Grosso, S., Mencarelli, M. A., Mari, F., Farnetani, M. A., et al. (2007). 2q24–q31 Deletion: Report of a case and review of the literature. Eur. J. Med. Genet. 50, 21–32. doi: 10.1016/j.ejmg.2006.09.001
Peters, C. H., Sokolov, S., Rajamani, S., Ruben, P. C. (2013). Effects of the antianginal drug, ranolazine, on the brain sodium channel NaV1.2 and its modulation by extracellular protons. Br. J. Pharmacol. 169, 704–716. doi: 10.1111/bph.12150
Petrelli, C., Passamonti, C., Cesaroni, E., Mei, D., Guerrini, R., Zamponi, N., et al. (2012). Early clinical features in Dravet syndrome patients with and without SCN1A mutations. Epilepsy Res. 99, 21–27. doi: 10.1016/j.eplepsyres.2011.10.010
Pons, L., Lesca, G., Sanlaville, D., Chatron, N., Labalme, A., Manel, V., et al. (2018). Neonatal tremor episodes and hyperekplexia-like presentation at onset in a child with SCN8A developmental and epileptic encephalopathy. Epileptic Disord. 20, 289–294. doi: 10.1684/epd.2018.0988
Petrovski, S., Wang, Q., Heinzen, E. L., Allen, A. S., Goldstein, D. B. (2013). Genic Intolerance to Functional Variation and the Interpretation of Personal Genomes. PloS Genet. 9, 1–13. doi: 10.1371/journal.pgen.1003709
Plummer, N. W., McBurney, M. W., Meisler, M. H. (1997). Alternative Splicing of the Sodium Channel SCN8A Predicts a Truncated Two-domain Protein in Fetal Brain and Non-neuronal Cells. J. Biol. Chem. 272, 24008–24015. doi: 10.1074/jbc.272.38.24008
Poryo, M., Clasen, O., Oehl-Jaschkowitz, B., Christmann, A., Gortner, L., Meyer, S. (2017). Dravet syndrome: a new causative SCN1A mutation? Clin. Case Rep. 5, 613–615. doi: 10.1002/ccr3.787
Ranza, E., Z’Graggen, W., Lidgren, M., Beghetti, M., Guipponi, M., Antonarakis, S. E., et al. (2020). SCN8A heterozygous variants are associated with anoxic-epileptic seizures. Am. J. Med. Genet. Part A.. doi: 10.1002/ajmg.a.61513
Rauch, A., Wieczorek, D., Graf, E., Wieland, T., Endele, S., Schwarzmayr, T., et al. (2012). Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet 380, 1674–1682. doi: 10.1016/S0140-6736(12)61480-9
Raymond, G., Wohler, E., Dinsmore, C., Cox, J., Johnston, M., Batista, D., et al. (2011). An interstitial duplication at 2q24.3 involving the SCN1A, SCN2A, SCN3A genes associated with infantile epilepsy. Am. J. Med. Genet. Part A 155, 920–923. doi: 10.1002/ajmg.a.33929
Reif, P. S., Tsai, M. H., Helbig, I., Rosenow, F., Klein, K. M. (2017). Precision medicine in genetic epilepsies: break of dawn? Expert Rev. Neurother. 17, 381–392. doi: 10.1080/14737175.2017.1253476
Reyes, I. S., Hsieh, D. T., Laux, L. C., Wilfong, A. A. (2011). Alleged Cases of Vaccine Encephalopathy Rediagnosed Years Later as Dravet Syndrome. Pediatrics 128. doi: 10.1542/peds.2010-0887
Reynolds, C., King, M. D., Gorman, K. M. (2020). The phenotypic spectrum of SCN2A-related epilepsy. Eur. J. Paediatr. Neurol. 24, 117–122. doi: 10.1016/j.ejpn.2019.12.016
Rhodes, T. H., Vanoye, C. G., Ohmori, I., Ogiwara, I., Yamakawa, K., George, A. L. (2005). Sodium channel dysfunction in intractable childhood epilepsy with generalized tonic-clonic seizures. J. Physiol. 569, 433–445. doi: 10.1113/jphysiol.2005.094326
Riban, V., Fitzsimons, H. L., During, M. J. (2009). Gene therapy in epilepsy. Epilepsia 50, 24–32. doi: 10.1111/j.1528-1167.2008.01743.x
Richards, K. L., Milligan, C. J., Richardson, R. J., Jancovski, N., Grunnet, M., Jacobson, L. H., et al. (2018). Selective NaV1.1 activation rescues Dravet syndrome mice from seizures and premature death. Proc. Natl. Acad. Sci. U.S. A. 115, E8077–E8085. doi: 10.1073/pnas.1804764115
Rilstone, J. J., Coelho, F. M., Minassian, B. A., Andrade, D. M. (2012). Dravet syndrome: Seizure control and gait in adults with different SCN1A mutations. Epilepsia 53, 1421–1428. doi: 10.1111/j.1528-1167.2012.03583.x
Rim, J. H., Kim, S. H., Hwang, I. S., Kwon, S. S., Kim, J., Kim, H. W., et al. (2018). Efficient strategy for the molecular diagnosis of intractable early-onset epilepsy using targeted gene sequencing. BMC Med. Genomics 11, 6. doi: 10.1186/s12920-018-0320-7
Riva, D., Vago, C., Pantaleoni, C., Bulgheroni, S., Mantegazza, M., Franceschetti, S. (2009). Progressive neurocognitive decline in two children with Dravet syndrome, de novo SCN1A truncations and different epileptic phenotypes. Am. J. Med. Genet. Part A 149A, 2339–2345. doi: 10.1002/ajmg.a.33029
Rolvien, T., Butscheidt, S., Jeschke, A., Neu, A., Denecke, J., Kubisch, C., et al. (2017). Severe bone loss and multiple fractures in SCN8A-related epileptic encephalopathy. Bone 103, 136–143. doi: 10.1016/j.bone.2017.06.025
Rossi, M., El-Khechen, D., Black, M. H., Farwell Hagman, K. D., Tang, S., Powis, Z. (2017). Outcomes of Diagnostic Exome Sequencing in Patients With Diagnosed or Suspected Autism Spectrum Disorders. Pediatr. Neurol. 70, 34–43.e2. doi: 10.1016/j.pediatrneurol.2017.01.033
Rubinstein, M., Westenbroek, R. E., Yu, F. H., Jones, C. J., Scheuer, T., Catterall, W. A. (2015). Genetic background modulates impaired excitability of inhibitory neurons in a mouse model of Dravet syndrome. Neurobiol. Dis. 73, 106–117. doi: 10.1016/j.nbd.2014.09.017
Rush, A. M., Dib-Hajj, S. D., Liu, S., Cummins, T. R., Black, J. A., Waxman, S. G. (2018). “A Single Sodium Channel Mutation Produces Hyperor Hypoexcitability In Different Types Of Neurons,” in Chasing Men on Fire (PNAS: The MIT Press), 89–101. doi: 10.7551/mitpress/10310.003.0014
Saitoh, M., Shinohara, M., Hoshino, H., Kubota, M., Amemiya, K., Takanashi, J., et al. (2012). Mutations of the SCN1A gene in acute encephalopathy. Epilepsia 53, 558–564. doi: 10.1111/j.1528-1167.2011.03402.x
Saitoh, M., Ishii, A., Ihara, Y., Hoshino, A., Terashima, H., Kubota, M., et al. (2015a). Missense mutations in sodium channel SCN1A and SCN2A predispose children to encephalopathy with severe febrile seizures. Epilepsy Res. 117, 1–6. doi: 10.1016/j.eplepsyres.2015.08.001
Saitoh, M., Shinohara, M., Ishii, A., Ihara, Y., Hirose, S., Shiomi, M., et al. (2015b). Clinical and genetic features of acute encephalopathy in children taking theophylline. Brain Dev. 37, 463–470. doi: 10.1016/j.braindev.2014.07.010
Samanta, D., Ramakrishnaiah, R. (2015). De novo R853Q mutation of SCN2A gene and West syndrome. Acta Neurol. Belg. 115, 773–776. doi: 10.1007/s13760-015-0454-8
Sanders, S. J., Campbell, A. J., Cottrell, J. R., Moller, R. S., Wagner, F. F., Auldridge, A. L., et al. (2018). Progress in Understanding and Treating SCN2A –Mediated Disorders. Trends Neurosci. 41, 442–456. doi: 10.1016/j.tins.2018.03.011
Sands, T. T., Choi, H. (2017). Genetic Testing in Pediatric Epilepsy. Curr. Neurol. Neurosci. Rep. 17, 1–11. doi: 10.1007/s11910-017-0753-y
Saxena, S., Li, S. (2017). Defeating epilepsy: A global public health commitment. Epilepsia Open 2, 153–155. doi: 10.1002/epi4.12010
Scalmani, P., Rusconi, R., Armatura, E., Zara, F., Avanzini, G., Franceschetti, S., et al. (2006). Effects in neocortical neurons of mutations of the Nav1.2 Na+ channel causing benign familial neonatal-infantile seizures. J. Neurosci. 26, 10100–10109. doi: 10.1523/JNEUROSCI.2476-06.2006
Schreiber, J. M., Tochen, L., Brown, M., Evans, S., Ball, L. J., Bumbut, A., et al. (2020). A multi-disciplinary clinic for SCN8A-related epilepsy. Epilepsy Res. 159, 106261. doi: 10.1016/j.eplepsyres.2019.106261
Schwarz, N., Hahn, A., Bast, T., Müller, S., Löffler, H., Maljevic, S., et al. (2016). Mutations in the sodium channel gene SCN2A cause neonatal epilepsy with late-onset episodic ataxia. J. Neurol. 263, 334–343. doi: 10.1007/s00415-015-7984-0
Schiavon, E., Sacco, T., Cassulini, R. R., Gurrola, G., Tempia, F., Possani, L. D., et al. (2006). Resurgent Current and Voltage Sensor Trapping Enhanced Activation by a b-Scorpion Toxin Solely in Na v 1.6 Channel. J. Biol. Chem. 281, 20326–20337. doi: 10.1074/jbc.M600565200
Sharkey, L. M., Jones, J. M., Hedera, P., Meisler, M. H. (2009). Evaluation of SCN8A as a candidate gene for autosomal dominant essential tremor. Park. Relat. Disord. 15, 321–323. doi: 10.1016/j.parkreldis.2008.06.010
Sheets, P. L., Heers, C., Stoehr, T., Cummins, T. R. (2008). Differential block of sensory neuronal voltage-gated sodium channels by lacosamide [(2R)-2- (acetylamino)-N-benzyl-3-methoxypropanamide], lidocaine, and carbamazepine. J. Pharmacol. Exp. Ther. 326, 89–99. doi: 10.1124/jpet.107.133413
Shen, H., Zhou, Q., Pan, X., Li, Z., Wu, J., Yan, N. (2017). Structure of a eukaryotic voltage-gated sodium channel at near-atomic resolution. Science 355, 1–12. doi: 10.1126/science.aal4326
Shi, X., Yasumoto, S., Nakagawa, E., Fukasawa, T., Uchiya, S., Hirose, S. (2009). Missense mutation of the sodium channel gene SCN2A causes Dravet syndrome. Brain Dev. 31, 758–762. doi: 10.1016/j.braindev.2009.08.009
Shi, X. Y., Tomonoh, Y., Wang, W. Z., Ishii, A., Higurashi, N., Kurahashi, H., et al. (2016). Efficacy of antiepileptic drugs for the treatment of Dravet syndrome with different genotypes. Brain Dev. 38, 40–46. doi: 10.1016/j.braindev.2015.06.008
Silva, J. R., Goldstein, S. A. N. (2013). Voltage-sensor movements describe slow inactivation of voltage-gated sodium channels I: Wild-type skeletal muscle NAv1.4. J. Gen. Physiol. 141, 309–321. doi: 10.1085/jgp.201210909
Singh, N. A., Pappas, C., Dahle, E. J., Claes, L. R. F., Pruess, T. H., De Jonghe, P., et al. (2009). A Role of SCN9A in Human Epilepsies, As a Cause of Febrile Seizures and As a Potential Modifier of Dravet Syndrome. PloS Genet. 5, e1000649. doi: 10.1371/journal.pgen.1000649
Singh, R., Jayapal, S., Goyal, S., Jungbluth, H., Lascelles, K. (2015). Early-onset movement disorder and epileptic encephalopathy due to de novo dominant SCN8A mutation. Seizure 26, 69–71. doi: 10.1016/j.seizure.2015.01.017
Skjei, K. L., Church, E. W., Harding, B. N., Santi, M., Holland-Bouley, K. D., Clancy, R. R., et al. (2015). Clinical and histopathological outcomes in patients with SCN1A mutations undergoing surgery for epilepsy. J. Neurosurg. Pediatr. 16, 668–674. doi: 10.3171/2015.5.PEDS14551
Smith, R. S., Kenny, C. J., Ganesh, V., Jang, A., Borges-Monroy, R., Partlow, J. N., et al. (2018). Sodium Channel SCN3A (NaV1.3) Regulation of Human Cerebral Cortical Folding and Oral Motor Development. Neuron 99, 905–913.e7. doi: 10.1016/j.neuron.2018.07.052
Spampanato, J. (2004). A Novel Epilepsy Mutation in the Sodium Channel SCN1A Identifies a Cytoplasmic Domain for Subunit Interaction. J. Neurosci. 24, 10022–10034. doi: 10.1523/JNEUROSCI.2034-04.2004
Sone, D., Sugawara, T., Sakakibara, E., Tomioka, Y., Taniguchi, G., Murata, Y., et al. (2012). A case of autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) coexisting with pervasive developmental disorder harboring SCN1A mutation in addition to CHRNB2 mutation. Epilepsy Behav. 25, 192–195. doi: 10.1016/j.yebeh.2012.07.027
Stoke Therapeutics (2018). Stoke Therapeutics Presents Data Showing Single Dose of ASO Therapy Restores Normal Protein Levels in Animal Model of Genetic Epilepsy. Available at: https://www.stoketherapeutics.com/press-releases/stoketherapeutics-presents-data-showing-single-dose-of-aso-therapy-restoresnormal-protein-levels-in-animal-model-of-genetic-epilepsy/ (Accessed December 2, 2020).
Sprissler, R. S., Wagnon, J. L., Bunton-Stasyshyn, R. K., Meisler, M. H., Hammer, M. F. (2017). Altered gene expression profile in a mouse model of SCN8A encephalopathy. Exp. Neurol. 288, 134–141. doi: 10.1016/j.expneurol.2016.11.002
Striano, P., Bordo, L., Lispi, M. L., Specchio, N., Minetti, C., Vigevano, F., et al. (2006). A novel SCN2A mutation in family with benign familial infantile seizures. Epilepsia 47, 218–220. doi: 10.1111/j.1528-1167.2006.00392.x
Su, D. J., Lu, J. F., Lin, L. J., Liang, J. S., Hung, K. L. (2018). SCN2A mutation in an infant presenting with migrating focal seizures and infantile spasm responsive to a ketogenic diet. Brain Dev. 40, 724–727. doi: 10.1016/j.braindev.2018.03.005
Sugawara, T., Mazaki-Miyazaki, E., Fukushima, K., Shimomura, J., Fujiwara, T., Hamano, S., et al. (2002). Frequent mutations of SCN1A in severe myoclonic epilepsy in infancy. Neurology 58, 1122–1124. doi: 10.1212/WNL.58.7.1122
Sugiura, Y., Ogiwara, I., Hoshi, A., Yamakawa, K., Ugawa, Y. (2012). Different degrees of loss of function between GEFS+ and SMEI Na v1.1 missense mutants at the same residue induced by rescuable folding defects. Epilepsia 53, 111–114. doi: 10.1111/j.1528-1167.2012.03467.x
Sun, G., Werkman, T. R., Battefeld, A., Clare, J. J., Wadman, W. J. (2007). Carbamazepine and Topiramate Modulation of Transient and Persistent Sodium Currents Studied in HEK293 Cells Expressing the Na v 1.3?? Subunit. Epilepsia 48, 774–782. doi: 10.1111/j.1528-1167.2007.01001.x
Sun, H., Zhang, Y., Liang, J., Liu, X., Ma, X., Qin, J., et al. (2008). Seven novel SCN1A mutations in Chinese patients with severe myoclonic epilepsy of infancy. Epilepsia 49, 1104–1107. doi: 10.1111/j.1528-1167.2008.01549_2.x
Sun, H., Zhang, Y., Liu, X., Ma, X., Yang, Z., Qin, J., et al. (2010). Analysis of SCN1A mutation and parental origin in patients with Dravet syndrome. J. Hum. Genet. 55, 421–427. doi: 10.1038/jhg.2010.39
Sun, W., Wagnon, J. L., Mahaffey, C. L., Briese, M., Ule, J., Frankel, W. N. (2013). Aberrant sodium channel activity in the complex seizure disorder of Celf4 mutant mice. J. Physiol. 591, 241–255. doi: 10.1113/jphysiol.2012.240168
Syrbe, S., Zhorov, B. S., Bertsche, A., Bernhard, M. K., Hornemann, F., Mütze, U., et al. (2016). Phenotypic Variability from Benign Infantile Epilepsy to Ohtahara Syndrome Associated with a Novel Mutation in SCN2A. Mol. Syndromol. 7, 182–188. doi: 10.1159/000447526
Tai, C., Abe, Y., Westenbroek, R. E., Scheuer, T., Catterall, W. A. (2014). Impaired excitability of somatostatin- and parvalbumin-expressing cortical interneurons in a mouse model of Dravet syndrome. Proc. Natl. Acad. Sci. U. S. A. 111, 3139–3148. doi: 10.1073/pnas.1411131111
Takahashi, S., Yamamoto, S., Okayama, A., Araki, A., Saitsu, H., Matsumoto, N., et al. (2015). Electroclinical features of epileptic encephalopathy caused by SCN8A mutation. Pediatr. Int. 57, 758–762. doi: 10.1111/ped.12622
Tan, N.-N., Tang, H.-L., Lin, G.-W., Chen, Y.-H., Lu, P., Li, H.-J., et al. (2017). Epigenetic Downregulation of Scn3a Expression by Valproate: a Possible Role in Its Anticonvulsant Activity. Mol. Neurobiol. 54, 2831–2842. doi: 10.1007/s12035-016-9871-9
Thijs, R. D., Surges, R., O’Brien, T. J., Sander, J. W. (2019). Epilepsy in adults. Lancet 393, 689–701. doi: 10.1016/S0140-6736(18)32596-0
Thomas, R. H., Berkovic, S. F. (2014). The hidden genetics of epilepsy – A clinically important new paradigm. Nat. Rev. Neurol. 10, 283–292. doi: 10.1038/nrneurol.2014.62
Tibery, D. V., Campos, L. A., Mourão, C. B. F., Peigneur, S., Tytgat, J., Schwartz, E. F., et al. (2019). Electrophysiological characterization of Tityus obscurus b toxin 1 (To1) on Na+-channel isoforms. Biochim. Biophys. Acta - Biomembr. 1861, 142–150. doi: 10.1016/j.bbamem.2018.08.005
Tiefes, A. M., Hartlieb, T., Tacke, M., von Stülpnagel-Steinbeis, C., Larsen, L. H. G., Hao, Q., et al. (2019). Mesial Temporal Sclerosis in SCN1A -Related Epilepsy: Two Long-Term EEG Case Studies. Clin. EEG Neurosci. 50, 267–272. doi: 10.1177/1550059418794347
Tonekaboni, S. H., Ebrahimi, A., Bakhshandeh Bali, M. K., Taheri Otaghsara, S. M., Houshmand, M., Nasehi, M. M., et al. (2013). Sodium channel gene mutations in Children with GEFS+ and Dravet syndrome: A cross sectional study. Iran. J. Child Neurol. 7, 31–36. doi: 10.22037/ijcn.v7i2.4074
Trivisano, M., Pavia, G. C., Ferretti, A., Fusco, L., Vigevano, F., Specchio, N. (2019). Generalized tonic seizures with autonomic signs are the hallmark of SCN8A developmental and epileptic encephalopathy. Epilepsy Behav. 96, 219–223. doi: 10.1016/j.yebeh.2019.03.043
Trujillano, D., Bertoli-Avella, A. M., Kumar Kandaswamy, K., Weiss, M. E., Köster, J., Marais, A., et al. (2017). Clinical exome sequencing: Results from 2819 samples reflecting 1000 families. Eur. J. Hum. Genet. 25, 176–182. doi: 10.1038/ejhg.2016.146
Trump, N., McTague, A., Brittain, H., Papandreou, A., Meyer, E., Ngoh, A., et al. (2016). Improving diagnosis and broadening the phenotypes in early-onset seizure and severe developmental delay disorders through gene panel analysis. J. Med. Genet. 53, 310–317. doi: 10.1136/jmedgenet-2015-103263
Tsang, M. H.-Y., Leung, G. K.-C., Ho, A. C.-C., Yeung, K.-S., Mak, C. C.-Y., Pei, S. L.-C., et al. (2019). Exome sequencing identifies molecular diagnosis in children with drug-resistant epilepsy. Epilepsia Open 4, 63–72. doi: 10.1002/epi4.12282
U.S. Food and Drug Administration [website] (2018). FDA Approves First Drug Comprised of an Active Ingredient Derived from Marijuana to Treat Rare, Severe Forms of Epilepsy. Available at: https://www.fda.gov/news-events/pressannouncements/fda-approves-first-drug-comprised-active-ingredientderived-marijuana-treat-rare-severe-forms (Accessed March 7, 2020).
Usluer, S., Salar, S., Arslan, M., Yiş, U., Kara, B., Tektürk, P., et al. (2016). SCN1A gene sequencing in 46 Turkish epilepsy patients disclosed 12 novel mutations. Seizure 39, 34–43. doi: 10.1016/j.seizure.2016.05.008
Vaher, U., Nõukas, M., Nikopensius, T., Kals, M., Annilo, T., Nelis, M., et al. (2013). De Novo SCN8A Mutation Identified by Whole-Exome Sequencing in a Boy With Neonatal Epileptic Encephalopathy, Multiple Congenital Anomalies, and Movement Disorders. J. Child Neurol. 29, NP202–NP206. doi: 10.1177/0883073813511300
Vanoye, C. G., Gurnett, C. A., Holland, K. D., George, A. L., Kearney, J. A. (2014). Novel SCN3A variants associated with focal epilepsy in children. Neurobiol. Dis. 62, 313–322. doi: 10.1016/j.nbd.2013.10.015
Vecchi, M., Cassina, M., Casarin, A., Rigon, C., Drigo, P., De Palma, L., et al. (2011). Infantile epilepsy associated with mosaic 2q24 duplication including SCN2A and SCN3A. Seizure 20, 813–816. doi: 10.1016/j.seizure.2011.07.008
Veeramah, K. R., O’Brien, J. E., Meisler, M. H., Cheng, X., Dib-Hajj, S. D., Waxman, S. G., et al. (2012). De novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. Am. J. Hum. Genet. 90, 502–510. doi: 10.1016/j.ajhg.2012.01.006
Veeramah, K. R., Johnstone, L., Karafet, T. M., Wolf, D., Sprissler, R., Salogiannis, J., et al. (2013). Exome sequencing reveals new causal mutations in children with epileptic encephalopathies. Epilepsia 54, 1270–1281. doi: 10.1111/epi.12201
Verbeek, N. E., van Kempen, M., Gunning, W. B., Renier, W. O., Westland, B., Lindhout, D., et al. (2011). Adults with a history of possible Dravet syndrome: An illustration of the importance of analysis of the SCN1A gene. Epilepsia 52, e23–e25. doi: 10.1111/j.1528-1167.2011.02982.x
Verbeek, N. E., van der Maas, N. A. T., Jansen, F. E., van Kempen, M. J. A., Lindhout, D., Brilstra, E. H. (2013). Prevalence of SCN1A-Related Dravet Syndrome among Children Reported with Seizures following Vaccination: A Population-Based Ten-Year Cohort Study. PloS One 8, e65758. doi: 10.1371/journal.pone.0065758
Verret, L., Mann, E. O., Hang, G. B., Barth, A. M. I. I., Cobos, I., Ho, K., et al. (2012). Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in alzheimer model. Cell 149, 708–721. doi: 10.1016/j.cell.2012.02.046
Villeneuve, N., Laguitton, V., Viellard, M., Lépine, A., Chabrol, B., Dravet, C., et al. (2014). Cognitive and adaptive evaluation of 21 consecutive patients with Dravet syndrome. Epilepsy Behav. 31, 143–148. doi: 10.1016/j.yebeh.2013.11.021
Volkers, L., Kahlig, K. M., Verbeek, N. E., Das, J. H. G., van Kempen, M. J. A., Stroink, H., et al. (2011). Na v1.1 dysfunction in genetic epilepsy with febrile seizures-plus or Dravet syndrome. Eur. J. Neurosci. 34, 1268–1275. doi: 10.1111/j.1460-9568.2011.07826.x
Wagnon, J. L., Meisler, M. H. (2015). Recurrent and non-recurrent mutations of SCN8A in epileptic encephalopathy. Front. Neurol. 6, 104. doi: 10.3389/fneur.2015.00104
Wagnon, J. L., Barker, B. S., Hounshell, J. A., Haaxma, C. A., Shealy, A., Moss, T., et al. (2016). Pathogenic mechanism of recurrent mutations of SCN8A in epileptic encephalopathy. Ann. Clin. Transl. Neurol. 3, 114–123. doi: 10.1002/acn3.276
Wagnon, J. L., Barker, B. S., Ottolini, M., Park, Y., Volkheimer, A., Valdez, P., et al. (2017). Loss-of-function variants of SCN8A in intellectual disability without seizures. Neurol. Genet. 3, e170. doi: 10.1212/NXG.0000000000000170
Wallace, R. H., Scheffer, I. E., Barnett, S., Richards, M., Dibbens, L., Desai, R. R., et al. (2001). Neuronal sodium-channel β1-subunit mutations in generalized epilepsy with febrile seizures plus. Am. J. Hum. Genet. 68, 859–865. doi: 10.1086/319516
Wang, J. W., Shi, X. Y., Kurahashi, H., Hwang, S. K., Ishii, A., Higurashi, N., et al. (2012). Prevalence of SCN1A mutations in children with suspected Dravet syndrome and intractable childhood epilepsy. Epilepsy Res. 102, 195–200. doi: 10.1016/j.eplepsyres.2012.06.006
Wang, J., Gao, H., Bao, X., Zhang, Q., Li, J., Wei, L., et al. (2017a). SCN8A mutations in Chinese patients with early onset epileptic encephalopathy and benign infantile seizures. BMC Med. Genet. 18, 104. doi: 10.1186/s12881-017-0460-1
Wang, Y., Du, X., Bin, R., Yu, S., Xia, Z., Zheng, G., et al. (2017b). Genetic Variants Identified from Epilepsy of Unknown Etiology in Chinese Children by Targeted Exome Sequencing. Sci. Rep. 7, 40319. doi: 10.1038/srep40319
Waxman, S. G., Hains, B. C. (2006). Fire and phantoms after spinal cord injury: Na+ channels and central pain. Trends Neurosci. 29, 207–215. doi: 10.1016/j.tins.2006.02.003
Weber, Y. G., Nies, A. T., Schwab, M., Lerche, H. (2014). Genetic Biomarkers in Epilepsy. Neurotherapeutics 11, 324–333. doi: 10.1007/s13311-014-0262-5
Weiss, L. A., Escayg, A., Kearney, J. A., Trudeau, M., MacDonald, B. T., Mori, M., et al. (2003). Sodium channels SCN1A, SCN2A and SCN3A in familial autism. Mol. Psychiatry 8, 186–194. doi: 10.1038/sj.mp.4001241
Wengert, E. R., Tronhjem, C. E., Wagnon, J. L., Johannesen, K. M., Petit, H., Krey, I., et al. (2019). Biallelic inherited SCN8A variants, a rare cause of SCN8A -related developmental and epileptic encephalopathy. Epilepsia 60, 2277–2285. doi: 10.1111/epi.16371
Weuring, W. J., Singh, S., Volkers, L., Rook, M. B., Van’t Slot, R. H., Bosma, M., et al. (2020). NaV1.1 and NaV1.6 selective compounds reduce the behavior phenotype and epileptiform activity in a novel zebrafish model for Dravet syndrome. PloS One 15, 1–17. doi: 10.1371/journal.pone.0219106
Whitaker, W. R. J., Clare, J. J., Powell, A. J., Chen, Y. H., Faull, R. L. M., Emson, P. C. (2000). Distribution of voltage-gated sodium channel?-subunit and?-subunit mRNAs in human hippocampal formation, cortex, and cerebellum. J. Comp. Neurol. 422, 123–139. doi: 10.1002/(SICI)1096-9861(20000619)422:1<123::AID-CNE8>3.0.CO;2-X
Willemsen, M. H., Rensen, J. H. M., van Schrojenstein-Lantman de Valk, H. M. J., Hamel, B. C. J., Kleefstra, T. (2012). Adult Phenotypes in Angelman- and Rett-Like Syndromes. Mol. Syndromol. 2, 217–234. doi: 10.1159/000335661
Wittmack, E. K., Rush, A. M., Craner, M. J., Goldfarb, M., Waxman, S. G., Dib-Hajj, S. D. (2004). Fibroblast growth factor homologous factor 2B: Association with Na v1.6 and selective colocalization at nodes of Ranvier of dorsal root axons. J. Neurosci. 24, 6765–6775. doi: 10.1523/JNEUROSCI.1628-04.2004
Wolff, M., Johannesen, K. M., Hedrich, U. B. S., Masnada, S., Rubboli, G., Gardella, E., et al. (2017). Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain 140, 1316–1336. doi: 10.1093/brain/awx054
Wong, V. C. N., Fung, C. W., Kwong, A. K. Y. (2015). SCN2A mutation in a Chinese boy with infantile spasm - response to Modified Atkins Diet. Brain Dev. 37, 729–732. doi: 10.1016/j.braindev.2014.10.008
Wu, Y. W., Sullivan, J., McDaniel, S. S., Meisler, M. H., Walsh, E. M., Li, S. X., et al. (2015). Incidence of dravet syndrome in a US population. Pediatrics 136, e1310–e1315. doi: 10.1542/peds.2015-1807
Wu, Q., Wang, H., Fan, Y. Y., Zhang, J. M., Liu, X. Y., Fang, X. Y., et al. (2018). Ketogenic diet effects on 52 children with pharmacoresistant epileptic encephalopathy: A clinical prospective study. Brain Behav. 8, 1–8. doi: 10.1002/brb3.973
Xiao, Y., Xiong, J., Mao, D., Liu, L., Li, J., Li, X., et al. (2018). Early-onset epileptic encephalopathy with de novo SCN8A mutation. Epilepsy Res. 139, 9–13. doi: 10.1016/j.eplepsyres.2017.10.017
Xie, H., Su, W., Pei, J., Zhang, Y., Gao, K., Li, J., et al. (2019). De novo SCN1A, SCN8A, and CLCN2 mutations in childhood absence epilepsy. Epilepsy Res. 154, 55–61. doi: 10.1016/j.eplepsyres.2019.04.005
Xu, R., Thomas, E. A., Gazina, E. V., Richards, K. L., Quick, M., Wallace, R. H., et al. (2007). Generalized epilepsy with febrile seizures plus-associated sodium channel β1 subunit mutations severely reduce beta subunit-mediated modulation of sodium channel function. Neuroscience 148, 164–174. doi: 10.1016/j.neuroscience.2007.05.038
Xu, X., Zhang, Y., Sun, H., Liu, X., Yang, X., Xiong, H., et al. (2014). Early clinical features and diagnosis of Dravet syndrome in 138 Chinese patients with SCN1A mutations. Brain Dev. 36, 676–681. doi: 10.1016/j.braindev.2013.10.004
Xu, X., Yang, X., Wu, Q., Liu, A., Yang, X., Ye, A. Y., et al. (2015). Amplicon Resequencing Identified Parental Mosaicism for Approximately 10% of “ de novo “ SCN1A Mutations in Children with Dravet Syndrome. Hum. Mutat. 36, 861–872. doi: 10.1002/humu.22819
Yan, N., Xin-Hua, W., Lin-Mei, Z., Yi-Ming, C., Wen-Hui, L., Yuan-Feng, Z., et al. (2018). Prospective study of the efficacy of a ketogenic diet in 20 patients with Dravet syndrome. Seizure 60, 144–148. doi: 10.1016/j.seizure.2018.06.023
Yang, Y.-C., Huang, C.-S., Kuo, C.-C. (2010). Lidocaine, Carbamazepine, and Imipramine Have Partially Overlapping Binding Sites and Additive Inhibitory Effect on Neuronal Na+ Channels. Anesthesiology 113, 160–174. doi: 10.1097/ALN.0b013e3181dc1dd6
Yang, X., Liu, A., Xu, X., Yang, X., Zeng, Q., Ye, A. Y., et al. (2017). Genomic mosaicism in paternal sperm and multiple parental tissues in a Dravet syndrome cohort. Sci. Rep. 7, 15677. doi: 10.1038/s41598-017-15814-7
Yang, C., Hua, Y., Zhang, W., Xu, J., Xu, L., Gao, F., et al. (2018). Variable epilepsy phenotypes associated with heterozygous mutation in the SCN9A gene: report of two cases. Neurol. Sci. 39, 1113–1115. doi: 10.1007/s10072-018-3300-y
Yordanova, I., Todorov, T., Dimova, P., Hristova, D., Tincheva, R., Litvinenko, I., et al. (2011). One novel Dravet syndrome causing mutation and one recurrent MAE causing mutation in SCN1A gene. Neurosci. Lett. 494, 180–183. doi: 10.1016/j.neulet.2011.03.008
Young, F. (2007). When adaptive processes go awry: gain-of-function in SCN9A. Clin. Genet. 73, 34–36. doi: 10.1111/j.1399-0004.2007.00922.x
Yu, F. H., Mantegazza, M., Westenbroek, R. E., Robbins, C. A., Kalume, F., Burton, K. A., et al. (2006). Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci. 9, 1142–1149. doi: 10.1038/nn1754
Yu, M.-J., Shi, Y.-W., Gao, M.-M., Deng, W.-Y., Liu, X.-R., Chen, L., et al. (2010). Milder phenotype with SCN1A truncation mutation other than SMEI. Seizure 19, 443–445. doi: 10.1016/j.seizure.2010.06.010
Zaman, T., Helbig, I., Božović, I. B., DeBrosse, S. D., Bergqvist, A. C., Wallis, K., et al. (2018). Mutations in SCN3A cause early infantile epileptic encephalopathy. Ann. Neurol. 83, 703–717. doi: 10.1002/ana.25188
Zaman, T., Abou Tayoun, A., Goldberg, E. M. (2019). A single-center SCN8A- related epilepsy cohort: clinical, genetic, and physiologic characterization. Ann. Clin. Transl. Neurol. 6. doi: 10.1002/acn3.50839. acn3.50839.
Zara, F., Specchio, N., Striano, P., Robbiano, A., Gennaro, E., Paravidino, R., et al. (2013). Genetic testing in benign familial epilepsies of the first year of life: Clinical and diagnostic significance. Epilepsia 54, 425–436. doi: 10.1111/epi.12089
Zhang, X., Ren, W., Decaen, P., Yan, C., Tao, X., Tang, L., et al. (2012). Crystal structure of an orthologue of the NaChBac voltage-gated sodium channel. Nature 486, 130–135. doi: 10.1038/nature11054
Zhang, Y., Kong, W., Gao, Y., Liu, X., Gao, K., Xie, H., et al. (2015). Gene Mutation Analysis in 253 Chinese Children with Unexplained Epilepsy and Intellectual/Developmental Disabilities. PloS One 10, e0141782. doi: 10.1371/journal.pone.0141782
Zhang, S., Zhang, Z., Shen, Y., Zhu, Y., Du, K., Guo, J., et al. (2020). SCN9A Epileptic Encephalopathy Mutations Display a Gain-of-function Phenotype and Distinct Sensitivity to Oxcarbazepine. Neurosci. Bull. 36, 11–24. doi: 10.1007/s12264-019-00413-5
Zhang, F., Wu, Y., Zou, X., Tang, Q., Zhao, F., Cao, Z. (2019). BmK AEP, an Anti-Epileptic Peptide Distinctly Affects the Gating of Brain Subtypes of Voltage-Gated Sodium Channels. Int. J. Mol. Sci. 20, 729. doi: 10.3390/ijms20030729
Zhang, T., Chen, M., Zhu, A., Zhang, X., Fang, T. (2020). Novel mutation of SCN9A gene causing generalized epilepsy with febrile seizures plus in a Chinese family. Neurol. Sci. 41, 1913–1917. doi: 10.1007/s10072-020-04284-x
Zhou, P., He, N., Zhang, J. W., Lin, Z. J., Wang, J., Yan, L. M., et al. (2018). Novel mutations and phenotypes of epilepsy-associated genes in epileptic encephalopathies. Genes Brain Behav. 17, e12456. doi: 10.1111/gbb.12456
Ziobro, J., Eschbach, K., Sullivan, J. E., Knupp, K. G. (2018). Current Treatment Strategies and Future Treatment Options for Dravet Syndrome. Curr. Treat. Options Neurol. 20, 1–15. doi: 10.1007/s11940-018-0537-y
Zuberi, S. M., Brunklaus, A., Birch, R., Reavey, E., Duncan, J., Forbes, G. H. (2011). Genotype-phenotype associations in SCN1A-related epilepsies. Neurology 76, 594–600. doi: 10.1212/WNL.0b013e31820c309b
Keywords: channelopathies, epilepsy, ion channel, mutation, sodium channel
Citation: Menezes LFS, Sabiá Júnior EF, Tibery DV, Carneiro LdA and Schwartz EF (2020) Epilepsy-Related Voltage-Gated Sodium Channelopathies: A Review. Front. Pharmacol. 11:1276. doi: 10.3389/fphar.2020.01276
Received: 21 April 2020; Accepted: 31 July 2020;
Published: 18 August 2020.
Edited by:
Jean-Marc Sabatier, Aix-Marseille Université, FranceReviewed by:
Rikke Steensbjerre Møller, Filadelfia, DenmarkCopyright © 2020 Menezes, Sabiá Júnior, Tibery, Carneiro and Schwartz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elisabeth Ferroni Schwartz, ZWZzY2h3YUB1bmIuYnI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.