 Francesco Fiorentino
Francesco Fiorentino Antonello Mai
Antonello Mai Dante Rotili
Dante Rotili
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 12 August 2020
Sec. Translational Pharmacology
Volume 11 - 2020 | https://doi.org/10.3389/fphar.2020.01243
This article is part of the Research Topic Natural Product Epigenetic Modulators and Inhibitors View all 7 articles
Acetylation of histone and non-histone protein lysine residues has been widely described as a critical modulator of several cell functions in humans. Lysine acetyltransferases (KATs) catalyse the transfer of acetyl groups on substrate proteins and are involved in multiple physiological processes such as cell signalling, metabolism, gene regulation, and apoptosis. Given the pivotal role of acetylation, the alteration of KATs enzymatic activity has been clearly linked to various cellular dysfunctions leading to several inflammatory, metabolic, neurological, and cancer diseases. Hence, the use KAT inhibitors (KATi) has been suggested as a potentially successful strategy to reverse or prevent these conditions. To date, only a few KATi have proven to be potential drug candidates, and there is still a keen interest in designing molecules showing drug-like properties from both pharmacodynamics and pharmacokinetics point of view. Increasing literature evidence has been highlighting natural compounds as a wide source of molecular scaffolds for developing therapeutic agents, including KATi. In fact, several polyphenols, catechins, quinones, and peptides obtained from natural sources (including nuts, oils, root extracts, and fungi metabolites) have been described as promising KATi. Here we summarize the features of this class of compounds, describing their modes of action, structure-activity relationships and (semi)-synthetic derivatives, with the aim of assisting the development of novel more potent, isoform selective and drug-like KATi.
The acetylation of the ϵ-amino groups of lysine residues is one of the most common post translation modifications (PTMs) occurring at cellular level to influence the protein function. The equilibrium between transfer and removal of acetyl groups is regulated by two classes of enzymes: lysine acetyltransferases (KATs) and lysine deacetylases (KDACs) (Falkenberg and Johnstone, 2014; Carafa et al., 2016; Fiorentino et al., 2018). KATs catalyse the transfer of an acetyl group from the co-substrate acetyl-coenzyme A (Ac-CoA) to selected lysine residues of the substrate proteins, while KDACs catalyse the hydrolysis of the acetyl group (Figure 1).
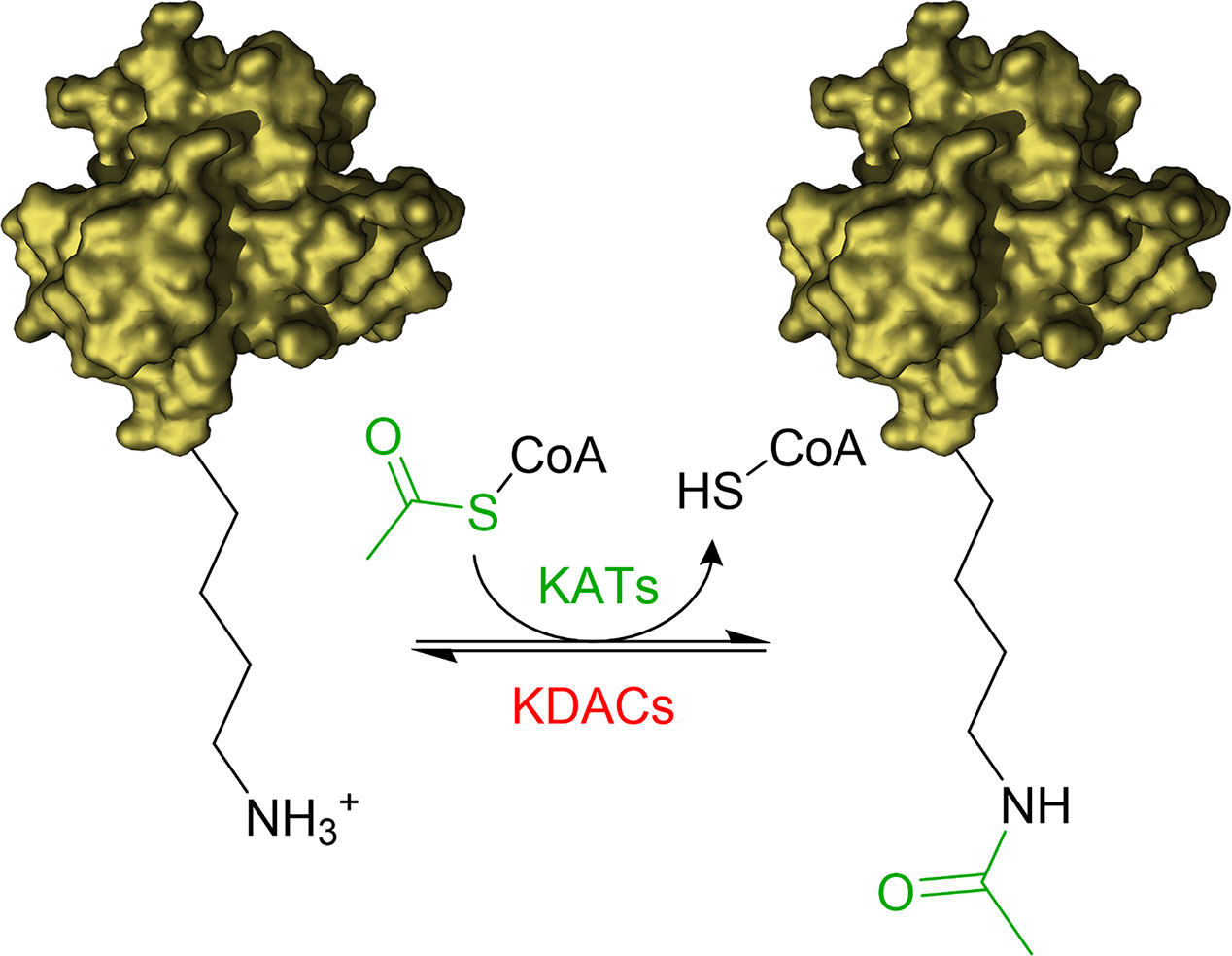
Figure 1 Illustration of acetylation/deacetylation equilibrium. KATs transfer an acetyl group from Acetyl-CoA to the ϵ-amino groups of lysine residues of the substrate proteins. KDACs catalyse the removal of the acetyl group.
The targets of the acetylation/deacetylation reaction include different classes of proteins such as enzymes (for instance kinases), transcription factors, and histones (Falkenberg and Johnstone, 2014; Carafa et al., 2016; Fiorentino et al., 2018). Recent mass spectrometric studies have shown that proteins undergoing acetylation are present in approximately all subcellular compartments (Choudhary et al., 2014). Therefore, it is not surprising that acetylation is virtually related to every cellular function, ranging from signal transduction to metabolism and cell cycle regulation (Ali et al., 2018).
Acetylation prevents positive charges from forming on the amino group, thus having a significant impact on the electrostatic properties of the protein and influencing protein-protein interaction networks, as well as the cellular localisation and the sensitivity to degradation. In the case of histones, the neutralization of the positive charge of lysine residues weakens their interaction with the negatively charged DNA, thus causing chromatin decompaction and facilitating transcription (Smith and Denu, 2009). Acetylated lysine residues are also recognized by domains present in “reader” proteins called bromodomains, thus translating the acetylation reaction in specific downstream signals (Filippakopoulos and Knapp, 2014).
KAT enzymes have been arranged into three major families based on their homology to yeast orthologues, as well as mechanism of catalysis. The families comprise: the p300/CREB-binding proteins (p300/CBP); the GCN5-related N-acetyltransferases (GNAT), and the MOZ, Ybf2, Sas2, and Tip60 (MYST) family. Acetyltransferase activity has also been found in further proteins not classified in any of the major families, such as the transcription factor complexes TAF1/TBP and TFIIIC90, circadian locomotor output cycles protein kaput CLOCK (KAT13D), and nuclear receptor coactivator-1 (NCoA-1, also referred as SRC-1).
All KATs are characterised by a similar tertiary structure in the catalytic domain, consisting of an α/β fold, essential for co-substrate binding and catalysis, while the adjacent regions contribute to the determination of substrate specificity. Furthermore, KATs are often part of heteromultimeric complexes, and the interacting protein partners also play a key role in determining the target specificity and functions (Lee and Workman, 2007).
The p300/CBP family comprises p300 (KAT3B, Figure 2A) and its paralog CBP (KAT3A). The two proteins have interchangeable functions and present high sequence and structural similarity which reflects the same mechanism of catalysis (Chan and La Thangue, 2001). The acetylation reaction follows a “hit and run” (Theorell−Chance) mechanism consisting of initial binding of acetyl-CoA to the enzyme, followed by a weak and transient interaction with the histone substrate essential for acetyl transfer. Interestingly, in this proposed mechanism the ternary complex is kinetically irrelevant for catalysis (Liu et al., 2008). Not only p300/CBP possess KAT activity, but they also have additional domains, specifically three cysteine-histidine rich domains (TAZ, PHD, and ZZ) and a bromodomain (BRD), which are critical for protein-protein interactions.
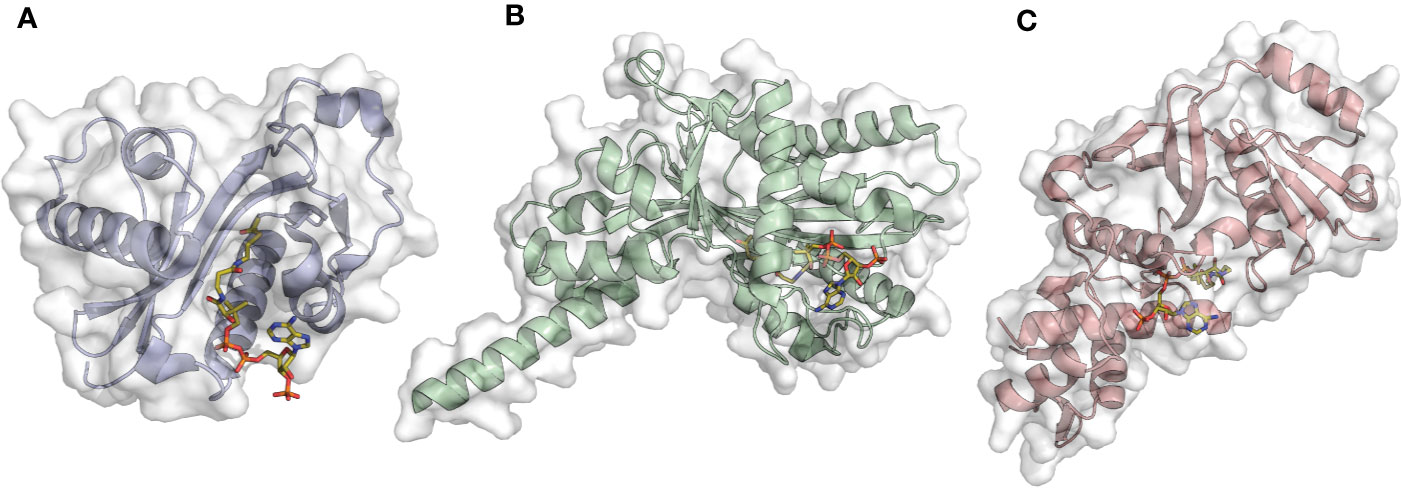
Figure 2 Examples of acetyltransferase domain structures from the three main KAT families in complex with the co-substrate Ac-CoA (orange sticks). (A) Gcn5 (PDB ID: 1Z4R); (B) p300 (PDB ID: 4PZS); (C) MOF (PDB ID: 6BA4).
The main substrates of p300/CBP are the histone proteins H2A, H2B, H3, and H4, however, given their intricate structures, they have more than 400 interacting partners. For instance, in the context of transcriptional activation, the two KATs may act as a bridge to connect transcription factors to the transcription machinery, and acetylates histones and/or transcription factors in order to enable a transcriptional response (Dancy and Cole, 2015). Consequently, p300/CBP regulates the cellular localization and activities of a large number of proteins, particularly transcription factors (Sano and Ishii, 2001; Wang et al., 2005), thus affecting cell growth and senescence, DNA damage repair and apoptosis (Kalkhoven, 2004).
The GNAT (Gcn5 related N-acetyltransferase) family includes the Gcn5 (General Control Nonderepressible 5, KAT2A, Figure 2B) (Brownell and Allis, 1995), PCAF (p300/CBP associated factor, KAT2B) (Yang et al., 1996), the Elp3 (Elongator complex protein 3, KAT9) (Wittschieben et al., 1999), Hpa2 (KAT10), Hpa3 (Sampath et al., 2013), the mediator-complex subunit Nut1 (Lorch et al., 2000), and α-tubulin acetyltransferase 1 (α-TAT1) (Friedmann et al., 2012).
GNAT family members share four conserved 15–33 amino acids motifs indicated as A, B, C, and D, as well as various chromo- and bromodomains. The A motif contains the conserved R/Q-X-X-G-X-G/A sequence, indispensable for acetyl-CoA recognition and binding (Dyda et al., 2000). The catalytic mechanism of GNAT proteins, differently from p300/CBP, involves the formation of a ternary complex composed of the enzyme, the co-substrate acetyl-CoA, and the target protein. Following acetyl-CoA and substrate binding to the enzyme, the acetyl group is transferred to the lysine ϵ-amino group (Tanner et al., 2000).
Gcn5 and PCAF are closely related proteins, sharing 73% sequence similarity and having H3K14 as main in vitro substrate (Roth et al., 2001). Gcn5 acetylation activity has been shown to regulate cell cycle progression through the acetylation of CDC6 (cell division control protein 6 homolog), a critical factor for the assembly of the pre-replicative complex in G1 phase (Paolinelli et al., 2009). Similarly, PCAF is involved in cell cycle regulation, as well as transcriptional regulation and differentiation. For instance, under conditions of stress, PCAF acetylates H3 on p21 promoter, thus inhibiting cell growth (Love et al., 2012).
α-TAT1 is structurally similar to Gcn5 and acetylates Lys40 of α-tubulin (Kormendi et al., 2012; Taschner et al., 2012). Although the structural similarity to Gcn5, α-TAT1 contains a wider substrate binding pocket and further specific structural elements that play an essential role in α-tubulin specific acetylation (Friedmann et al., 2012). Given the specificity for α-tubulin, α-TAT1 activity is associated to processes such as cell adhesion, migration, and invasion (Castro-Castro et al., 2012; Aguilar et al., 2014).
The MYST family comprises the following enzymes: MOZ (monocytic leukemia zinc finger protein, KAT6A), MORF (MOZ related factor, KAT6B), MOF (males absent on the first, KAT8, Figure 2C), Tip60 (Tat-interacting protein, KAT5), and HBO1 (HAT bound to ORC1, KAT7). Enzymes belonging to this family possess a highly conserved catalytic region (the MYST domain) containing the acetyl-CoA binding site and zing finger motifs (Neuwald and Landsman, 1997; Sapountzi and Cote, 2011). A characteristic feature of MYST enzymes is the mechanism of catalysis consisting of a double displacement reaction (Yan et al., 2002; Wapenaar et al., 2015). In brief, the co-substrate acetyl-CoA donates the acetyl moiety to a cysteine residue, which in turn transfers it to the lysine of the substrate protein. A key role is played by a glutamate in the catalytic site that deprotonates the lysine of the substrate protein, thus facilitating the nucleophilic attack.
MOZ and MORF present high sequence homology and have the histone H3 as main target. They both form multi-protein complexes involved in signalling related to transcriptional activation (Avvakumov and Cote, 2007) and development processes such as haematopoiesis (Perez-Campo et al., 2009; Perez-Campo et al., 2013) and skeletogenesis (Crump et al., 2006).
Tip60 is the first described human member of MYST family and acetylates H4 at different Lys residues and transcription factors such as p53 (Tang et al., 2006), c-Myc (Patel et al., 2004), and E2F1 (Van Den Broeck et al., 2012). Given its wide range of substrates, Tip60 is involved in multiple pathways including transcriptional activation, apoptosis (Sykes et al., 2006) and DNA-damage response (Murr et al., 2006).
MOF is the latest discovered member of the MYST family and has H4K16 as main substrate, but it is also active towards H4K5, H4K8, TIP5, and p53. MOF is the acetylating subunit of two distinct complexes, MSL (male-specific lethal) and NSL (non-specific lethal), that modulate its target specificity and thus downstream effects (Ravens et al., 2014). Interestingly, evidence shows that MOF is also involved in the transcriptional regulation of mitochondrial DNA (Chatterjee et al., 2016).
As explained above, further human KATs are represented by proteins that are part of transcription factor complexes. For instance, TFIIIC90 acetylates H3 and is involved in the recruitment of RNA polymerase III for transcriptional activation (Hsieh et al., 1999).
CLOCK and NCoA-1 are co-activators of nuclear hormone receptors with substrate specificity towards H3 and H4. CLOCK is involved in circadian rhythm regulation (Doi et al., 2006), while NCoA-1- acetylation is a consequence of steroid-mediated transcriptional activation (Spencer et al., 1997).
Given their wide substrate specificity (including histones, transcriptional factors, kinases, and tumor suppressors) and involvement with key cellular processes, KATs play a pivotal role in cellular physiology and disease (Fiorentino et al., 2018). The dysregulation of KAT expression or enzymatic activity may lead to tumorigenesis and be the cause of diseases like inflammatory disorders, respiratory, cardiovascular, and neurological pathologies (Dekker and Haisma, 2009; Schneider et al., 2013; Huang et al., 2015; Sun et al., 2015).
For instance, p300/CBP may form chimeric proteins with the MYST family members MOZ and MOF, or the histone methyltransferase MLL which are the main cause of acute myeloid leukaemia (AML). These chimeras present aberrant catalytic activity given the presence of two active sites, thus leading to increased protein expression, particularly oncogenes (Chaffanet et al., 2000; Sun et al., 2015). Mutations and deletions of p300/CBP are related to a subset of cancer types such as lung, colon, and ovarian carcinomas (Gayther et al., 2000; Kishimoto et al., 2005), but also developmental disorders, like the Rubinstein-Taybi syndrome. This disease is characterised by mental and physical anomalies and increased susceptibility to cancer (Portela and Esteller, 2010). One of the main targets of p300/CBP is the transcription factor NF-κB. It has been shown that NF-κB acetylation is correlated to a deceased affinity for the inhibitory protein IκB, hence facilitating the expression of NF-κB target genes involved in the inflammation pathway (Schneider et al., 2013).
Gcn5 has been shown to be upregulated in a diverse range of cancer types. For instance, Gcn5 promotes the expression of cell cycle factors in non-small cell lung cancer (NSCLC) (Chen et al., 2013), as well as colon cancer, where the expression of the oncogene c-Myc was increased (Yin et al., 2015). Differently, PCAF may be up- or down-regulated depending on the cancer type. Its overexpression is associated with invasive urothelial carcinoma (Shiota et al., 2010), while PCAF was found to be down-regulated in oesophageal squamous cell carcinoma (Zhu et al., 2009), hepatocellular carcinoma (Li et al., 2016), ovarian and gastric cancer (Ying et al., 2010). Furthermore, Gcn5 and PCAF aberrant activity has been related to type 2 diabetes. Both proteins acetylate PGC-1α (peroxisome proliferator-activated receptor gamma co-activator 1 alpha), a co-activator of hepatic gluconeogenesis. Acetylation is a trigger for PGC-1α degradation, and an anomalous Gnc5/PCAF activity leads to a decrease in blood and hepatic glucose output (Sun et al., 2014).
The MYST family members MOZ and MORF form hybrid proteins with p300/CBP contributing to the insurgence of AML, as already discussed. Apart from p300/CBP, MOZ can also form chimeras with the nuclear receptor co-activator TIF2, and this event is also implicated in AML (Kindle et al., 2005). Similarly to PCAF, both MOF and Tip60 have tissue-specific roles in cancer. For instance, their expression is related to tumor growth in oral tongue squamous cell carcinoma (MOF) (Li et al., 2015), and prostate cancer (Tip60) (Halkidou et al., 2003). Conversely, both proteins are down-regulated in breast cancer (Pfister et al., 2008; Bassi et al., 2016). MYST family members have demonstrated to be important for neural development, indeed MORF is highly expressed in the brain and mutations are associated with neurodevelopmental disorders in humans such as the genitopatellar syndrome, a skeletal dysplasia with cerebral and genital anomalies (Campeau et al., 2012).
Given the extensive association between KATs abnormal activity and a variety of diseases, many molecules have been developed to interfere with their activity, as well as clarify their physiopathology (Fiorentino et al., 2018). To this end, many research groups have developed inhibitors of KATs (KATi) and a great amount of them are represented by natural products or their (semi)-synthetic derivatives. In recent years KATi discovery has finally succeeded in releasing highly potent and isoform selective inhibitors, along with their relative co-crystal structures. The p300 inhibitor A-485 (Lasko et al., 2017), and the MOZ/MORF inhibitors WM8014 and WM1119 (Baell et al., 2018) represent an important step forward in the field of KAT drug discovery. However, many other KAT subtypes still lack good inhibitors, which may be obtained using natural occurring products as scaffolds for focused drug design and development projects.
This review will focus on KATi obtained from natural sources describing their mechanisms of action, structure-activity relationships and (semi)-synthetic derivatives.
The connection between anomalous protein acetylation and the onset of different diseases has inspired the research on KATi with the final goal of developing therapeutics, as well as chemical probes to further investigate KAT function.
Interestingly, over last decades, a great number of drugs and biological tools have been developed from natural products with examples including antimicrobial drugs (beta lactam and glycopeptide antibiotics), but also analgesics (aspirin and opioids) and cancer chemotherapeutics (paclitaxel and derivatives) (Dias et al., 2012; Newman and Cragg, 2012). Therefore, it is not surprising that research groups exploit natural occurring molecules to develop biologically active compounds. In fact, natural products represent a valid alternative to synthetic molecules given their higher chemical complexity and the different chemical space they occupy. Natural compounds usually present a preference of oxygen more than sulphur or nitrogen as heteroatoms, as well as a higher number of stereogenic centres and fused rings (Cherblanc et al., 2013). On the other hand, they are usually poorly selective towards their target given the presence of electrophile or redox centres and polyphenol moieties. Nevertheless, natural products have always been crucial for the development of therapeutics, representing scaffolds for the development of semi-synthetic derivatives, like in the case of the different generations of penicillin derivatives. Moreover, natural products may be a start point for ligand-based drug discovery campaigns, where the initial lead compound may be modified through (bio)isosteric replacement or may undergo structural simplification, to remove unnecessary or undesirable substructures and modulate potency and target specificity. A classic example of structural simplification are the opioid analgesics such as levorphanol or fentanyl which represent two different degrees of simplification of the parent natural molecule morphine (Wang et al., 2019).
The above-described features also characterise natural occurring KAT modulators and, although they may show some drawbacks related to their physicochemical properties, current efforts are made towards the improvement of their activity and selectivity. In fact, their optimization into new (semi)-synthetic derivatives could represent a significant milestone towards the obtainment of novel selective and potent KATi.
Garcinol (1A) is a polyisoprenylated benzophenone extracted from Garcinia indica showing inhibitory activity in the low micromolar range towards p300 and PCAF (IC50 = 7 and 5 μM, respectively) (Balasubramanyam et al., 2004a). Isogarcinol (1B) is the result of the intramolecular cyclization of garcinol and can be extracted from Garcinia mangostana (Cen et al., 2013). Similarly, it shows nonspecific KATi activity towards p300 and PCAF in the same micromolar range (Mantelingu et al., 2007b). Different derivatives of 1B were synthesized in order to improve selectivity and potency, and to decrease toxicity: LTK-13 (1C) and LTK-14 (1D) are monoaliphatic substituted derivatives, while LTK-19 (1E) is a bis methyl sulfonyl derivative (Figure 3). The three compounds showed specific inhibition of p300 with IC50 values in the 5–7 μM range. Among these, 1D displayed higher efficiency in vivo, and impaired reproduction of HIV through the down regulation of p300-mediated acetylation of p53; moreover, it was nontoxic towards T-cells (Mantelingu et al., 2007b). Molecular simplification of 1A led to the benzylidene barbituric acid derivative 1F which showed selective p300 inhibition with an IC50 value of 2.1 μM. To overcome the susceptibly to hydrolysis of compound 1F, two methyl groups at the ortho positions of the benzylidene moiety were inserted, thus leading to EML425 (1G), a selective inhibitor of p300/CBP active in the low micromolar range [IC50 (p300) = 2.9 μM and IC50 (CBP) = 1.1 μM]. AlphaLISA homogeneous proximity immunoassays performed by varying either histone H3 or acetyl-CoA concentration and adding increasing concentrations of 1G, indicated a non-competitive mode of action. Despite the presence of an α,β-unsaturation would make 1G a good Michael acceptor with potential pleiotropic mechanisms of action, remarkably, tests in human leukaemia U937 cells showed a 1G-promoted reduction of H4K5 and H3K9 acetylation levels, and the induction of cell cycle arrest in the G0/G1 phase (Milite et al., 2015).
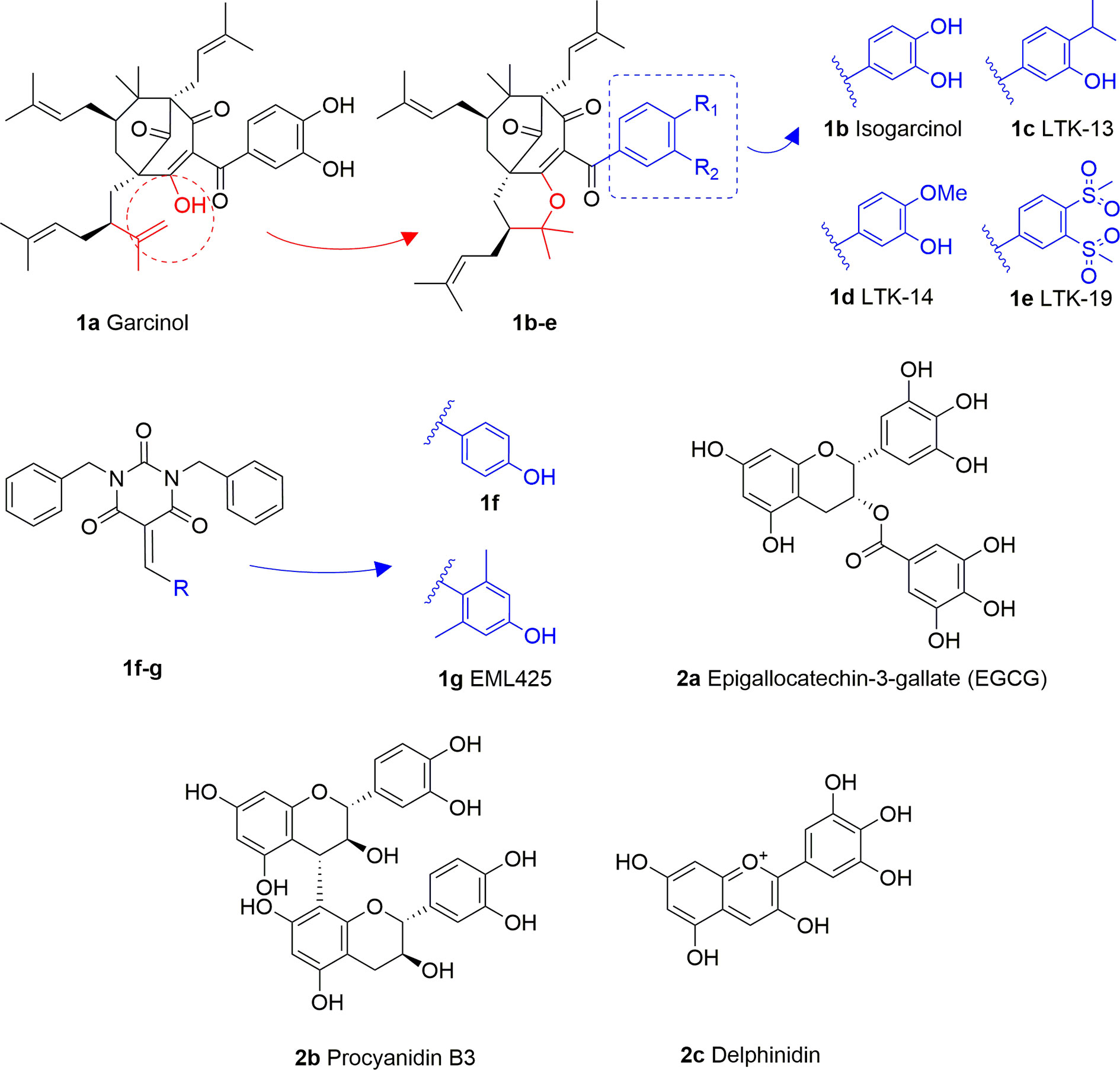
Figure 3 Structures and modifications of garcinol (1a-g), EGCG and related derivatives (2a-b), and delphinidin (2c).
Epigallocatechin-3-gallate (EGCG, 2A), is a non-selective KATi active against p300, CBP, PCAF and Tip60 with IC50 values of 30, 50, 60, and 70 μM, respectively (Choi et al., 2009). However, polyphenols like 2A are able to interact with an extensive range of proteins and the optimization of their activity for every single target is quite challenging (Mai et al., 2008). Procyanidin B3 (2B, Figure 3) showed up to 90% KAT activity inhibition in a dose-dependent fashion, as well as 60% p300 activity following a non-competitive mode of action. In addition, it inhibited p300-mediated androgen receptor acetylation and acetylation-dependent prostate cancer cell proliferation (Choi et al., 2011).
Delphinidin (2C, Figure 3), is an anthocyanidin extracted from pomegranate (Punica granatum), showing specific p300/CBP inhibition (IC50 = 30 μM). This compound has shown potential as anti-inflammatory agent as it showed suppression of inflammatory cytokines expression in MH7A cell through hypoacetylation of NF-κB, as well as inhibition of cytokine release in Jurkat T lymphocyte cell lines (Seong et al., 2011).
Curcumin (3A) is a natural product extracted from the rhizome of turmeric (Curcuma longa), a common preparation used in both traditional Indian and Chinese medicines. Curcumin is a non-competitive p300 inhibitor (IC50 = 25 μM) (Balasubramanyam et al., 2004b) presenting a mode of action likely relying on the covalent binding to the target enzyme through its cinnamoyl moieties serving as Michael acceptors (Marcu et al., 2006). Interestingly, curcumin induced down-regulation of mGlu2 receptor in mouse spinal cord after systemic administration and decreased the acetylation levels of histones H3 and H4 in dorsal root ganglia (DRG) (Zammataro et al., 2014). On the other hand, given its covalent mode of action and its polyphenolic nature, curcumin is not a selective KATi and several additional targets have been identified so far, including COX2, protein kinase C, thioredoxin reductase, tubulin, and 5-lipoxygenase (Anand et al., 2008). Because of its pleiotropic nature, despite numerous studies indicated the positive effects of curcumin in various pathologies such as diabetes (Maradana et al., 2013), cardiovascular diseases (Srivastava and Mehta, 2009) and others, it is hard to attribute them to KAT inhibition. Curcumin was also shown to have membrane disruption properties, thus that some of its cellular effects may be the result of this additional feature (Ingólfsson et al., 2014). Curcumin has been evaluated in more than 120 clinical trials for various conditions, however the outcomes of the completed double blinded, placebo controlled studies (including trials for conditions such as pancreatic and colon cancer or Alzheimer’s disease) have highlighted the in vivo inefficacy of curcumin. Indeed, features like chemical instability, unspecific cross-reactivity, and low oral bioavailability hampered the translation of in vitro activity into in vivo effects (Nelson et al., 2017). However, in order to improve its potency and selectivity, many research groups developed several synthetic derivatives of curcumin such as compounds 3B and 3C, where the methoxy residues were replaced by a carboxylic acid or a bromine atom, respectively (Figure 4). Both derivatives showed improved inhibitory activity towards p300, in fact the IC50 values were 33 and 21 μM, respectively, while curcumin showed an IC50 value higher than 400 μM in the same biophysical assay. Compound 3D (RC56) is a cyclic ketone derivative of 3C showing improved potency towards p300 (IC50 = 5 μM) and represents the scaffold for the development of various analogues, including 3E and 3F, the iodinated and dibrominated derivatives of 3D, respectively (Figure 4). While the iodinated analogue 3E displayed slightly lower potency (IC50 = 8.1 μM), the dibrominated derivative 3F had an increased p300 inhibitory activity (IC50 = 2.3 μM) (Costi et al., 2007; Mai et al., 2008). These data suggest that the presence of electron-withdrawing substituents in the phenyl ring is favourable for protein-ligand interactions. Finally, the sodium salt of phenylpyrazolocurcumin CTK7A (3G, Figure 4) is a water-soluble inhibitor of p300 and PCAF displaying a mixed type p300 inhibition for acetyl-CoA and histones. It suppressed H3 acetylation in KB cells (oral squamous cell carcinoma), being more active towards H3K14 than H3K9 and inhibited tumor proliferation in a related mouse xenograft model (Arif et al., 2010).
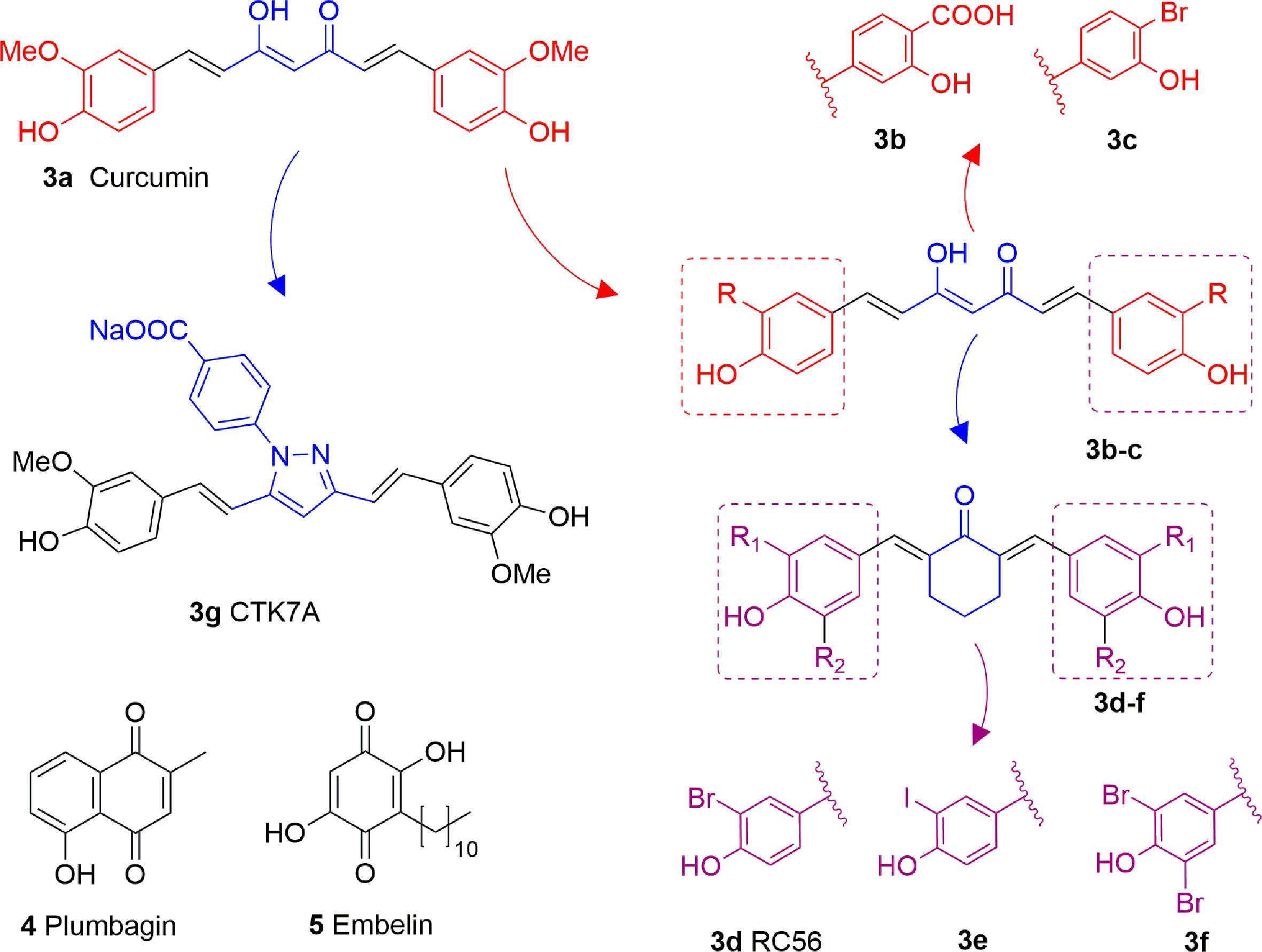
Figure 4 Structures and modifications of curcumin and its derivatives (3a-g), plumbagin (4) and embelin (5).
Plumbagin (4, Figure 4) is a hydroxynaphthoquinone extracted from Plumbago rosea roots, which has been shown to inhibit KAT activity in vitro and in cells with IC50 values for p300 and PCAF of 20 and 50 μM, respectively, with a non-competitive mode of action (Vasudevarao et al., 2014). Docking investigations combined with site-directed mutagenesis of p300 catalytic domain suggested that the hydroxyl group of 4 forms a crucial hydrogen bond with Lys1358 in the active site of the enzyme, thus it is crucial for inhibition, as confirmed by the loss of inhibitory activity of derivatives where the hydroxyl group was substituted with other moieties (Ravindra et al., 2009). Compound 4 suffers from high thiol reactivity due to the presence of a Michael acceptor, which can be overcome through methylation on C3, still retaining the KAT non-competitive inhibition (Vasudevarao et al., 2014).
Embelin (5, Figure 4) is a hydroxybenzoquinone derivative obtained from Embelia ribes berries. It displayed H3K9 acetylation suppression in mouse models and PCAF in vitro inhibitory activity (IC50 = 7.2 μM). Molecular modelling studies have postulated that the hydroxyl group and the undecyl chain are necessary for ligand-protein interaction. Indeed, the hydroxyl group interacts with the amide backbone through hydrogen bonds, while the alkyl chain is inserted into a narrow hydrophobic groove near the binding site of the β-mercaptoethylamine portion of CoA. Experimental works further confirmed these findings as the conversion of one hydroxyl group to methylamine or the shortening of the alkyl chain to a 10-carbon tail completely eliminates PCAF binding (Modak et al., 2013).
Anacardic acid (6A) is a 6-pentadecylsalicylic acid extracted from the cashew nut shells and has been indicated as a non-competitive and non-selective inhibitor of KAT enzymes with a broad range of IC50 values, depending on the reports. Indeed, IC50 values span from 8.5 μM (Balasubramanyam et al., 2003) and over 1,000 μM (Wu et al., 2009) in the case of p300, and from 5 μM (Balasubramanyam et al., 2003) to 667.1 μM (Wu et al., 2009) in the case of PCAF. Anacardic acid also showed inhibitory activity towards MYST family members, in particular Tip60 (IC50s from 64 to 347.6 μM) (Wu et al., 2009; Ghizzoni et al., 2012) and MOF (Ki and IC50 values of 64 and 43 μM, respectively) (Ghizzoni et al., 2012; Wapenaar et al., 2015). At cellular level, administration of 6A induced suppression of NF-κB signalling as a consequence of the inhibition of the acetylation of the p65 subunit (Sung et al., 2008). However, similarly to curcumin, 6A inhibits multiple proteins (Hemshekhar et al., 2012), thus its cellular effect might be a consequence of interactions with targets other than KAT enzymes. Furthermore, 6A presents low cell permeability, thus different derivatives have been synthesized to reduce the lipophilicity, as well as to improve the inhibitory potency and isoform selectivity.
Changes in the alkyl chain length and regiochemistry led to compounds 6B and 6C, which exhibited inhibition towards MOF (IC50 = 37 and 57 μM, respectively). Compound 6B possesses a decyl aliphatic chain instead of the pentadecyl chain of 6A and, while 6C is a 6B derivative where the alkyl chain position has been switched from ortho to meta with respect to the carboxyl group (Figure 5). Furthermore, derivatives bearing shorter alkyl chain lengths completely lost inhibitory activity, indicating the importance of the hydrophobic contacts played by the aliphatic chain for the ligand-protein interactions (Wapenaar et al., 2015). Replacement of the alkyl chain at 6-position with a substituted phenethyl moiety led to compounds 6D-6H (Figure 5), which were tested against p300, PCAF, and Tip60, displaying more than 75% inhibition of Tip60 at 200 μM (Ghizzoni et al., 2010; Ghizzoni et al., 2012). Amongst them, derivative 6F (MG149) was the most potent [IC50 (Tip60) = 74 μM, IC50 (MOF) = 47 μM], showing competitive inhibition towards acetyl-CoA in Tip60 and uncompetitive inhibition towards acetyl-CoA in MOF (Ghizzoni et al., 2012). Furthermore, it displayed KAT inhibitory activity in cell based assays where the acetyltransferase activity was measured in hippocampus, amygdala and prefrontal cortex nuclear extracts (Ghizzoni et al., 2012), and inhibited the expression of pro-inflammatory genes in murine precision-cut lung slices (van den Bosch et al., 2017).
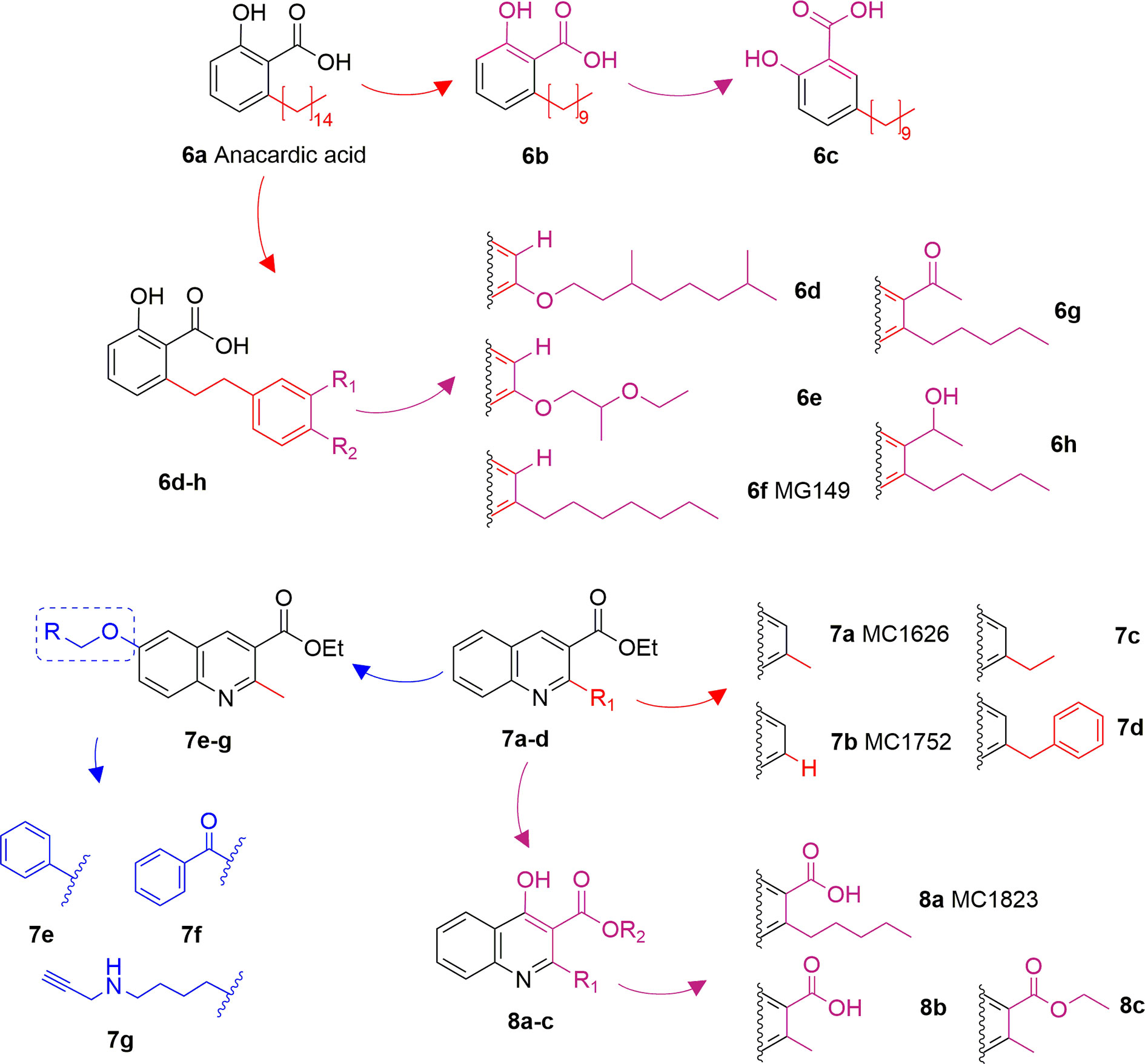
Figure 5 Structures and modifications of anacardic acid and its derivatives (6a-h, 7a-g, and 8a-c).
Compounds 7A (MC1626) and 7B (MC1752) can be regarded as quinoline analogues of anacardic acid that were identified through a phenotypic screening in S. cerevisiae. Indeed, both molecules impaired yeast cell growth resembling the effects of Gcn5 deletion mutants. In particular, 7A also suppressed acetylation and gene transcription mediated by Gcn5 (Ornaghi et al., 2005). To improve inhibitory activity, novel 7A derivatives presenting alternative aliphatic/aromatic chains at the C2-quinoline or additional residues at the C6-quinoline positions have been designed and synthesized. In compounds 7C and 7D the 2-methyl group is replaced by n-propyl (7C) or benzyl (7D) groups, while molecules 7E-G are derivatives of 7A bearing side chains at C6 (Figure 5). These changes improved p300 inhibitory activity and selectivity compared to 7A. Indeed, whilst 7A presented 22.6% p300 inhibition when tested at 100 μM, 7E-G showed 58.5–69.3% of p300 inhibition when evaluated at the same concentration. Among the three compounds, the C6 substituted molecule 7G was the most potent (69.3% p300 inhibition at 100 μM, and IC50 = 57.5 μM). Moreover, both 7C and 7G caused a massive reduction in H3 and H4 acetylation levels in human leukaemia U937 cells (Lenoci et al., 2014).
Further quinoline analogues of 6A are compounds 8A (MC1823), 8B, and 8C. In this case, the three substituents of the quinoline core retain the same relative positions as in 6A. In compound 8A a linear pentyl tail replaces the pentadecyl chain (Mai et al., 2006), while 8B is a 2-methyl derivative, and 8C is the ethyl ester of 8B (Figure 5). Compound 8A displayed 81% CBP inhibition at 50 μM, while 8B and 8C caused more than 80% inhibition of both p300 and CBP at 50 μM (Mai et al., 2009). When compared to 6A in a nuclear extract KAT activity inhibition assay at the concentration of 50 μM, compound 8A decreased the total KAT activity by 30%, while 6A by just 15% (Mai et al., 2006).
Long chain alkylidenmalonates (LoCAMs) are a class of KAT modulators derived from the molecular simplification of anacardic acid. SPV106 (9A) is the parent compound of this series showing p300/CBP inhibition comparable to 6A, as well as a peculiar PCAF activation (Sbardella et al., 2008). SPV106 has been subjected to various modifications leading to two interesting series of compounds (Figure 6): bicarboxylic (9B and 9C) and acetoacetic derivatives (9D and 9E). Bicarboxylic derivatives are obtained from the hydrolysis of SPV106 ethyl esters and differ in the length of the aliphatic tail (14 and 15 carbons for 9B and 9C, respectively). They are both low micromolar p300 inhibitors (IC50 = 1.3 and 1.1 μM for 9B and 9C, respectively) and weak PCAF inhibitors. On the other hand, the acetoacetic derivatives are p300 inhibitors (IC50 = 2.4 and 4.7 μM for 9D and 9E, respectively), but also potent PCAF activators (Castellano et al., 2015). Considering the activities of LoCAM compounds towards p300 and PCAF we can conclude that the ester hydrolysis improves p300 inhibitory activity, but decreases affinity towards PCAF, whilst replacement of carboxylic moiety with an acetyl group is favourable for PCAF activation (Sbardella et al., 2008; Castellano et al., 2015). Furthermore, variations to the length of the alkyl chain or introduction of a heteroatom is detrimental for the interaction with both enzymes (Castellano et al., 2015).
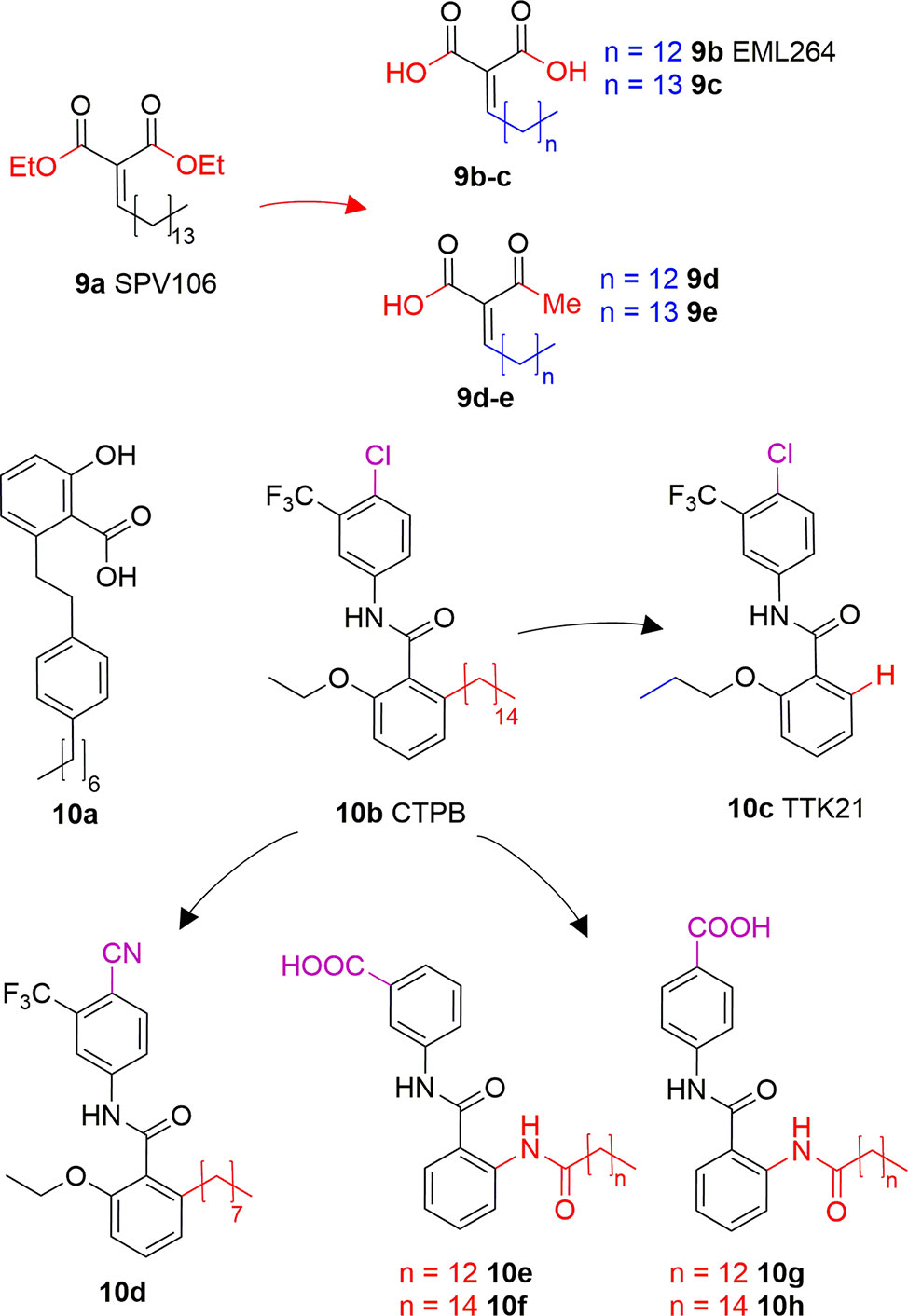
Figure 6 Structures and modifications of anacardic acid derivatives with mixed KAT inhibiting/activating properties: LoCAMs (9a-e) and compounds 10a-h.
Another reported anacardic acid derivative presenting both KAT inhibitory and activating properties is compound 10A which inhibits MOF and Tip60 with IC50 values of 47 and 64 μM, respectively, and exhibits PCAF activation (Ghizzoni et al., 2012). Differently, the anacardic acid benzamide derivative 10B (Figure 6) displays p300 activation as confirmed by the observed p300-dependent transcriptional activation (Balasubramanyam et al., 2003; Mantelingu et al., 2007a). The removal of CTPB long alkyl chain (compound 10C, TTK21, Figure 6) does not impair the p300/CBP activating ability (Chatterjee et al., 2013). Remarkably, compound 10D, presenting an octyl chain and a nitrile group replacing the chlorine has a reversed activity, showing 50% p300 inhibition at 100 μM in vitro. It also inhibited p300 KAT activity and induced apoptosis in immortalized HEK cells (Souto et al., 2008). Further derivatives replacing the alkyl chain with a long chain inverted amide (10E-H, Figure 6) displayed inhibitory activity against PCAF and were active in several human cancer cell lines (Park and Ma, 2012). Compound 10E showed 79% PCAF inhibition at 100 μM, which was higher than 6A, used as a positive control for the experiment. When translated to cytotoxic assays, IC50 values of compounds 10E-H were ranging from 25 to 80 μM in HCT, A549, HT-29, Hep3B, MDA- 231, and HeLa cancer cell lines (Park and Ma, 2012).
Alkaloids are plant-derived compounds containing multiple cycles and nitrogen atoms. A structure-based virtual screening identified four alkaloids (11A-D, Figure 7) possessing inhibitory activity against p300 in the low micromolar range, and against PCAF in the medium micromolar range [p300: IC50 (11A) = 0.69 μM, IC50 (11B) = 1.05 μM, IC50 (11C) = 0.58 μM IC50 (11D) = 4.85 μM; PCAF: IC50 (11A) = 14.13 μM, IC50 (11B) = 10.0 μM, IC50 (11C) = 27.1 μM IC50 (11D) = 7.16 μM]. When docked on p300, these compounds share a similar binding mode, consisting on the formation of hydrogen bonds with key residues Arg1410, Thr1411, and Trp1466, similarly to acetyl-CoA. However, none of these compounds showed activity on cancer cell lines, apart from 11D, although the authors of the paper do not exclude off-target effects in this case (Guo-Bo et al., 2016).
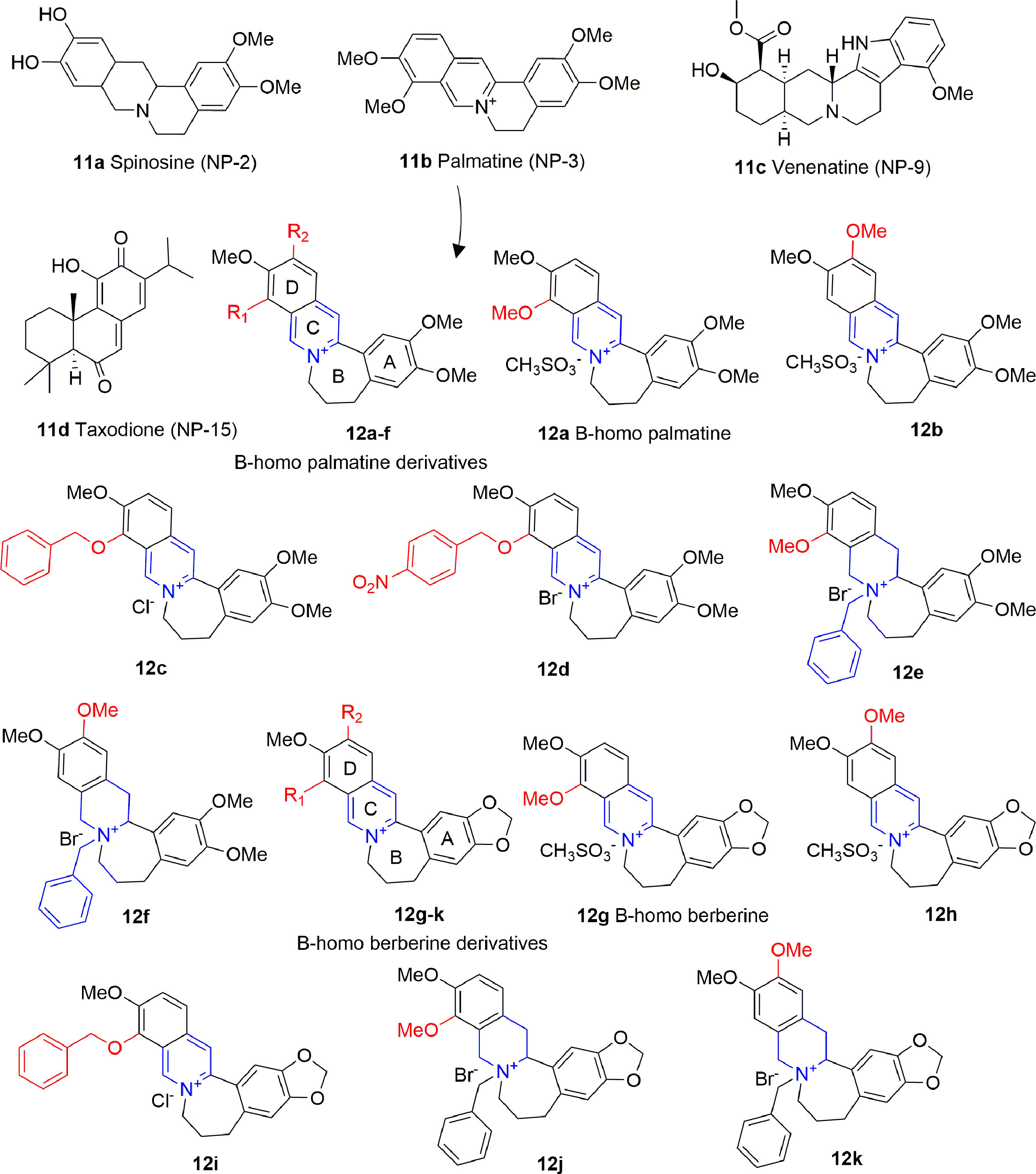
Figure 7 Structures and (semi)-synthetic derivatives of alkaloids with KAT inhibitory activity (11a-d and 12a-k).
Compound 11B (also indicated as Palmatine) has been used as source of inspiration to design B-homo palmatine (12A-F) and B-homo berberine derivatives (12G-K) all endowed with a central dihydroazepine B ring (Figure 7). Amongst them, B-homo palmatine derivative 12B showed the best potency towards p300 (IC50 of 0.42 μM) with a 10-fold increase compared to its parent compound 12A (IC50 = 4.5 μM). Similarly, the B-homo berberine derivative 12H showed a better inhibitory activity than its parent compound 12G (IC50 = 9.2 and 1.8 μM for 12G and 12H, respectively). The SAR evaluation indicates that generally B-homo berberine derivatives are less active than the palmatine ones, and the shift of the methoxy group from R1 to R2 position improves the inhibitory activity. Moreover, the replacement of methoxy groups with bulky substituents such as benzyloxy moieties does not affect or even decreases the activity of these compounds against p300 [IC50 (12C) = 4.7 μM, IC50 (12D) = 9.4 μM, IC50 (12I) = 2.5 μM], and the addition of a benzyl group to the quaternary amine massively decreases the inhibitory activity in both series [IC50 (12E) = 4.7 μM, IC50 (12F) = 7.4 μM, IC50 (12J) = 34 μM, IC50 (12K) = 44 μM] (Yang et al., 2018).
Some cyclopentenone prostaglandins (CyPGs) were shown to possess p300 inhibitory activity (Ravindra et al., 2012). In particular, Δ12-PGJ2 (13A) and PGJ2 (13B) (Figure 8) displayed IC50 values of 0.75 μM and > 2 μM, respectively. Docking studies suggest that 13A assumes a conformation such that the electrophilic carbon of the α,β-unsaturation in the cyclopentenone ring is close enough to Cys1438 of p300 to form a covalent Michael adduct. Further experiments involving site-directed mutagenesis of the p300 KAT domain, peptide competition assays, and mass spectrometric analysis validated the hypothesis of the covalent interaction of Δ12-PGJ2 with Cys1438. Both Δ12-PGJ2 and PGJ2 were found to inhibit H3 histone acetylation in cell-based assays. In addition, Δ12-PGJ2 also inhibited acetylation of the HIV-1 Tat by recombinant p300 in vitro. This effect was translated in U1/HIV cells (human monocytic cells chronically infected with HIV-1), contributing to the reduction of HIV viral gene expression (Ravindra et al., 2012).
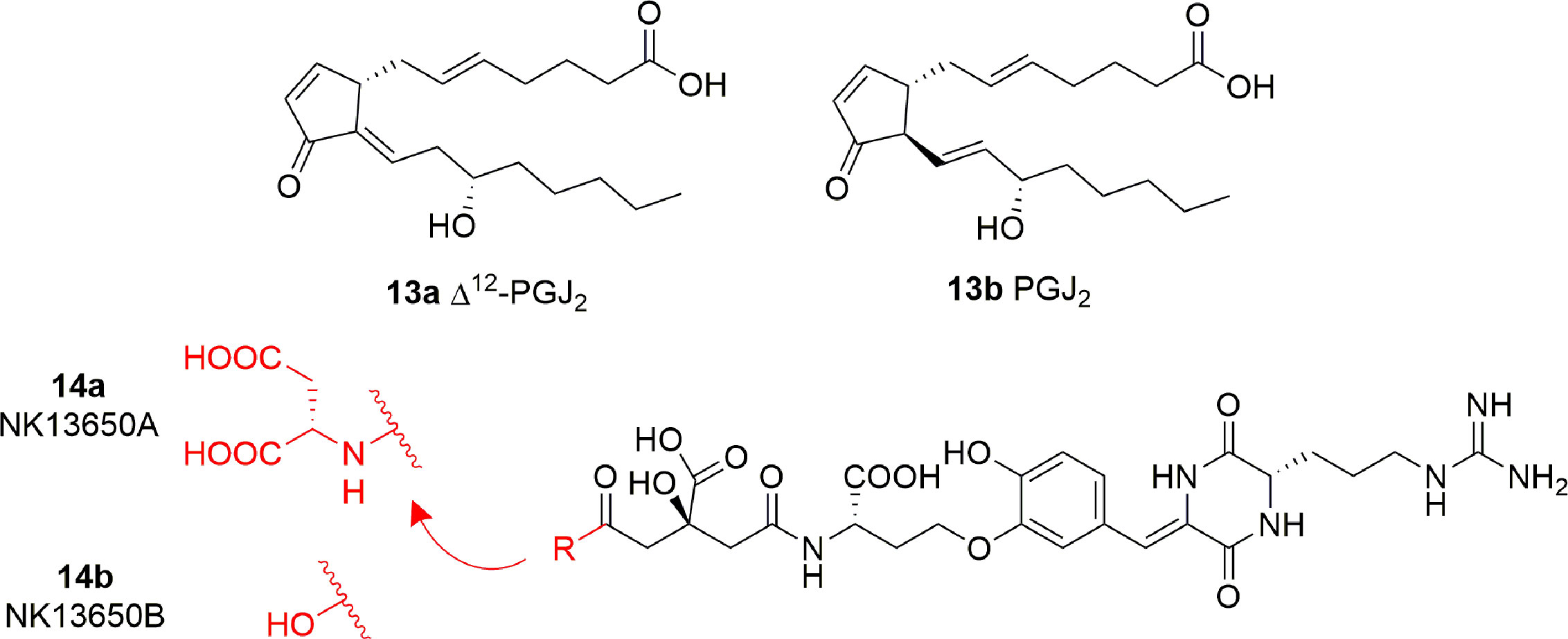
Figure 8 Structures of prostaglandins (CyPGs) and Penicillium peptide metabolites with KAT inhibitory activity (13a-b and 14a-b).
The peptide metabolites of Penicillium species 14A (NK13650A) and 14B (NK13650B) (Figure 8) are potent and selective inhibitors p300 with IC50 values of 11 and 22 nM, respectively. Remarkably, they suppress the transcriptional activation mediated by the oestrogen and androgen receptors and reduce the cell viability in tumor cell lines, including prostate cancer cells (Tohyama et al., 2012). Their peptide nature is the main cause of low cell absorption and metabolic instability, however given their high potency and selectivity these compounds represent a good starting point for the development of optimized peptidomimetic drugs.
Over the past decades, KAT aberrant activity has been connected to a vast range of diseases, particularly cancer. Thus, it is not surprising that great efforts have been made towards the discovery of KAT modulators. Natural compounds have always been a valid starting point for the development of small molecule drugs and, as we have described in this review, a great number of KATi have been obtained from or are indeed (semi)-synthetic derivatives of plant extracts or other natural sources (Table 1).
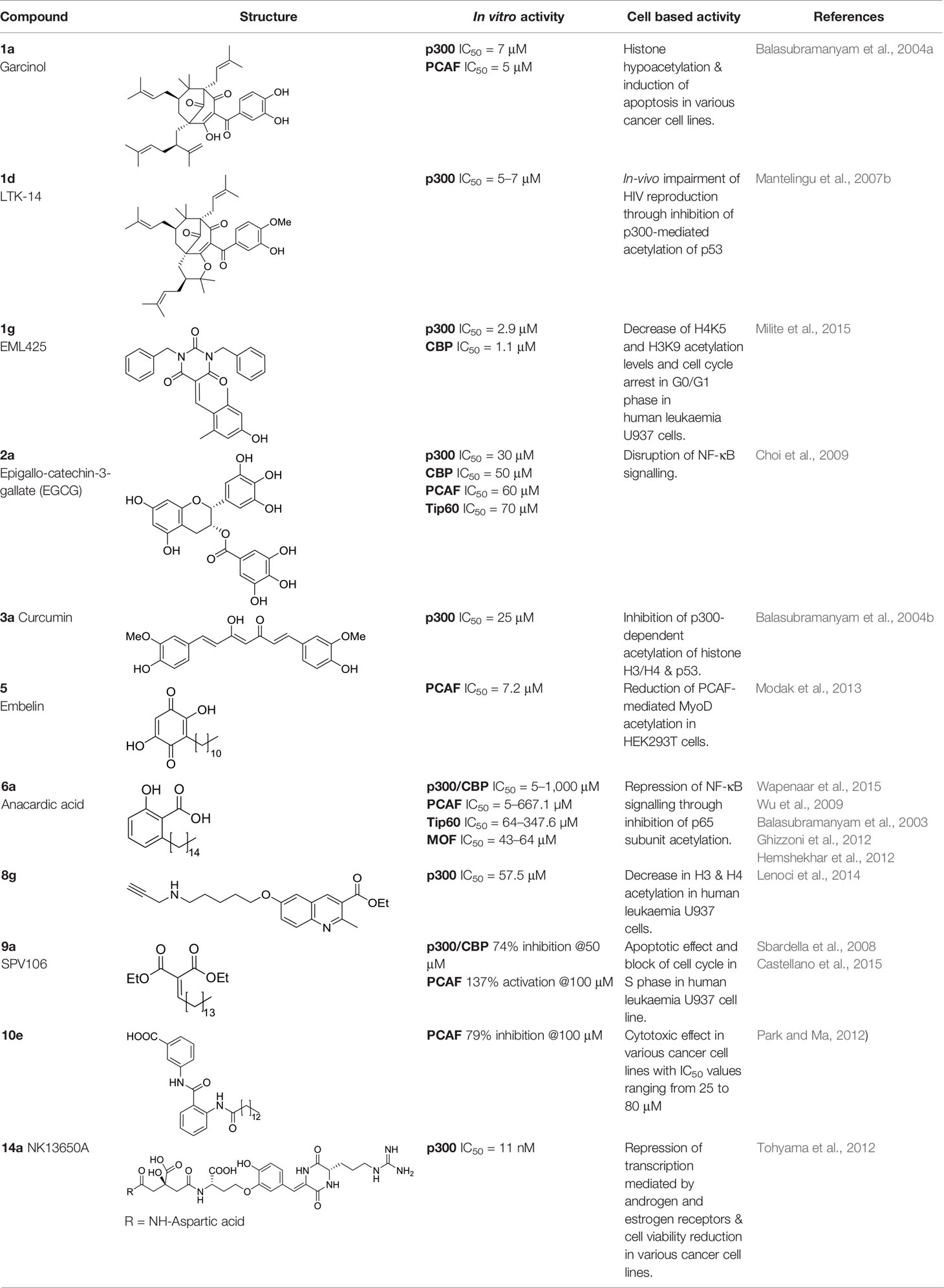
Table 1 Most significant KAT modulators from natural sources.
However, many reported naturally derived KATi present poorly understood mechanism of action and have many off-target effects. Therefore, often further structure-activity optimization efforts are necessary to obtain the desired potency, selectivity, and in cell activity. This would be particularly beneficial not only for the obtainment of clinically active drugs, but also for the development of biological tools to further understand the intricate and multifaceted roles of the various KAT isoforms in the context of cellular homeostasis. In fact, while targeting different proteins may be useful to obtain the desired phenotypic effect (for instance, decreased expression of oncoproteins), especially in the epigenetics field (Tomaselli et al., 2020), a biological probe must have a specific target in order to correlate the observed phenotypic effect to a specific molecular interaction.
The release of the co-crystal structures of the recently discovered p300 inhibitor A-485 (Lasko et al., 2017), and the MOZ/MORF inhibitors WM8014 and WM1119 (Baell et al., 2018) have paved the way for the development of novel highly potent and selective KATi. Nonetheless, may other KAT isoforms remain untargeted and natural products represent an ideal starting point for the development of novel therapeutics using both structure-based and ligand-based drug discovery approaches.
We envisage that the impact of natural products in the development of KATi, and more in general in drug discovery, will continue to be central. The continuous improvements in protein purification, bioanalytical technologies (Johnson et al., 2011), and structural biology approaches such as cryo-electron microscopy (Poepsel et al., 2018) and structural mass spectrometry (Mehmood et al., 2015; Fiorentino et al., 2019), will further facilitate the evaluation and the employment of these molecules in research.
FF, AM, and DR contributed to conception, manuscript writing, and proof reading. FF and DR searched the literature. FF and DR prepared the figures and the table. DR supervised and coordinated the whole writing work. All authors contributed to the article and approved the submitted version.
This paper was funded by PRIN 2016 (prot. 20152TE5PK) (AM), Ricerca Finalizzata 2013 PE-2013-02355271 (AM), AIRC 2016 (n. 19162) (AM), Progetto di Ateneo ‘Sapienza’ 2017 n. RM11715C7CA6CE53 (DR), and NIH (n. R01GM114306) (AM). FF holds a SABS CDT Studentship supported by the EPSRC and the MRC (EP/L016044/1).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Aguilar, A., Becker, L., Tedeschi, T., Heller, S., Iomini, C., Nachury, M. V. (2014). Alpha-tubulin K40 acetylation is required for contact inhibition of proliferation and cell-substrate adhesion. Mol. Biol. Cell 25, 1854–1866. doi: 10.1091/mbc.E13-10-0609
Ali, I., Conrad, R. J., Verdin, E., Ott, M. (2018). Lysine Acetylation Goes Global: From Epigenetics to Metabolism and Therapeutics. Chem. Rev. 118, 1216–1252. doi: 10.1021/acs.chemrev.7b00181
Anand, P., Thomas, S. G., Kunnumakkara, A. B., Sundaram, C., Harikumar, K. B., Sung, B., et al. (2008). Biological activities of curcumin and its analogues (Congeners) made by man and Mother Nature. Biochem. Pharmacol. 76, 1590–1611. doi: 10.1016/j.bcp.2008.08.008
Arif, M., Vedamurthy, B. M., Choudhari, R., Ostwal, Y. B., Mantelingu, K., Kodaganur, G. S., et al. (2010). Nitric oxide-mediated histone hyperacetylation in oral cancer: target for a water-soluble HAT inhibitor, CTK7A. Chem. Biol. 17, 903–913. doi: 10.1016/j.chembiol.2010.06.014
Avvakumov, N., Cote, J. (2007). The MYST family of histone acetyltransferases and their intimate links to cancer. Oncogene 26, 5395–5407. doi: 10.1038/sj.onc.1210608
Baell, J. B., Leaver, D. J., Hermans, S. J., Kelly, G. L., Brennan, M. S., Downer, N. L., et al. (2018). Inhibitors of histone acetyltransferases KAT6A/B induce senescence and arrest tumour growth. Nature 560, 253–257. doi: 10.1038/s41586-018-0387-5
Balasubramanyam, K., Swaminathan, V., Ranganathan, A., Kundu, T. K. (2003). Small molecule modulators of histone acetyltransferase p300. J. Biol. Chem. 278, 19134–19140. doi: 10.1074/jbc.M301580200
Balasubramanyam, K., Altaf, M., Varier, R. A., Swaminathan, V., Ravindran, A., Sadhale, P. P., et al. (2004a). Polyisoprenylated benzophenone, garcinol, a natural histone acetyltransferase inhibitor, represses chromatin transcription and alters global gene expression. J. Biol. Chem. 279, 33716–33726. doi: 10.1074/jbc.M402839200
Balasubramanyam, K., Varier, R. A., Altaf, M., Swaminathan, V., Siddappa, N. B., Ranga, U. (2004b). Curcumin, a novel p300/CREB-binding protein-specific inhibitor of acetyltransferase, represses the acetylation of histone/nonhistone proteins and histone acetyltransferase-dependent chromatin transcription. J. Biol. Chem. 279, 51163–51167. doi: 10.1074/jbc.M409024200
Bassi, C., Li, Y. T., Khu, K., Mateo, F., Baniasadi, P. S., Elia, A., et al. (2016). The acetyltransferase Tip60 contributes to mammary tumorigenesis by modulating DNA repair. Cell Death Differ. 23, 1198–1208. doi: 10.1038/cdd.2015.173
Brownell, J. E., Allis, C. D. (1995). An activity gel assay detects a single, catalytically active histone acetyltransferase subunit in Tetrahymena macronuclei. Proc. Natl. Acad. Sci. U. S. A. 92, 6364–6368. doi: 10.1073/pnas.92.14.6364
Campeau, P. M., Kim, J. C., Lu, J. T., Schwartzentruber, J. A., Abdul-Rahman, O. A., Schlaubitz, S., et al. (2012). Mutations in KAT6B, encoding a histone acetyltransferase, cause Genitopatellar syndrome. Am. J. Hum. Genet. 90, 282–289. doi: 10.1016/j.ajhg.2011.11.023
Carafa, V., Rotili, D., Forgione, M., Cuomo, F., Serretiello, E., Hailu, G. S., et al. (2016). Sirtuin functions and modulation: from chemistry to the clinic. Clin. Epigent. 8, 61. doi: 10.1186/s13148-016-0224-3
Castellano, S., Milite, C., Feoli, A., Viviano, M., Mai, A., Novellino, E., et al. (2015). Identification of structural features of 2-alkylidene-1,3-dicarbonyl derivatives that induce inhibition and/or activation of histone acetyltransferases KAT3B/p300 and KAT2B/PCAF. ChemMedChem 10, 144–157. doi: 10.1002/cmdc.201402371
Castro-Castro, A., Janke, C., Montagnac, G., Paul-Gilloteaux, P., Chavrier, P. (2012). ATAT1/MEC-17 acetyltransferase and HDAC6 deacetylase control a balance of acetylation of alpha-tubulin and cortactin and regulate MT1-MMP trafficking and breast tumor cell invasion. Eur. J. Cell Biol. 91, 950–960. doi: 10.1016/j.ejcb.2012.07.001
Cen, J., Shi, M., Yang, Y., Fu, Y., Zhou, H., Wang, M., et al. (2013). Isogarcinol is a new immunosuppressant. PloS One 8, e66503. doi: 10.1371/journal.pone.0066503
Chaffanet, M., Gressin, L., Preudhomme, C., Soenen-Cornu, V., Birnbaum, D., Pebusque, M. J. (2000). MOZ is fused to p300 in an acute monocytic leukemia with t(8;22). Genes Chromosomes Cancer 28, 138–144. doi: 10.1002/(SICI)1098-2264(200006)28:2<138::AID-GCC2>3.0.CO;2-2
Chan, H. M., La Thangue, N. B. (2001). p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J. Cell. Sci. 114, 2363–2373.
Chatterjee, S., Mizar, P., Cassel, R., Neidl, R., Selvi, B. R., Mohankrishna, D. V., et al. (2013). A novel activator of CBP/p300 acetyltransferases promotes neurogenesis and extends memory duration in adult mice. J. Neurosci. 33, 10698–106712. doi: 10.1523/jneurosci.5772-12.2013
Chatterjee, A., Seyfferth, J., Lucci, J., Gilsbach, R., Preissl, S., Bottinger, L., et al. (2016). MOF Acetyl Transferase Regulates Transcription and Respiration in Mitochondria. Cell 167, 722–738. doi: 10.1016/j.cell.2016.09.052
Chen, L., Wei, T., Si, X., Wang, Q., Li, Y., Leng, Y., et al. (2013). Lysine acetyltransferase GCN5 potentiates the growth of non-small cell lung cancer via promotion of E2F1, cyclin D1, and cyclin E1 expression. J. Biol. Chem. 288, 14510–14521. doi: 10.1074/jbc.M113.458737
Cherblanc, F. L., Davidson, R. W., Di Fruscia, P., Srimongkolpithak, N., Fuchter, M. J. (2013). Perspectives on natural product epigenetic modulators in chemical biology and medicine. Nat. Prod. Rep. 30, 605–624. doi: 10.1039/c3np20097c
Choi, K. C., Jung, M. G., Lee, Y. H., Yoon, J. C., Kwon, S. H., Kang, H. B., et al. (2009). Epigallocatechin-3-gallate, a histone acetyltransferase inhibitor, inhibits EBV-induced B lymphocyte transformation via suppression of RelA acetylation. Cancer Res. 69, 583–592. doi: 10.1158/0008-5472.CAN-08-244
Choi, K. C., Park, S., Lim, B. J., Seong, A. R., Lee, Y. H., Shiota, M., et al. (2011). Procyanidin B3, an inhibitor of histone acetyltransferase, enhances the action of antagonist for prostate cancer cells via inhibition of p300-dependent acetylation of androgen receptor. Biochem. J. 433, 235–244. doi: 10.1042/bj20100980
Choudhary, C., Weinert, B. T., Nishida, Y., Verdin, E., Mann, M. (2014). The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 15, 536. doi: 10.1038/nrm3841
Costi, R., Di Santo, R., Artico, M., Miele, G., Valentini, P., Novellino, E., et al. (2007). Cinnamoyl compounds as simple molecules that inhibit p300 histone acetyltransferase. J. Med. Chem. 50, 1973–1977. doi: 10.1021/jm060943s
Crump, J. G., Swartz, M. E., Eberhart, J. K., Kimmel, C. B. (2006). Moz-dependent Hox expression controls segment-specific fate maps of skeletal precursors in the face. Development 133, 2661–2669. doi: 10.1242/dev.02435
Dancy, B. M., Cole, P. A. (2015). Protein lysine acetylation by p300/CBP. Chem. Rev. 115, 2419–2452. doi: 10.1021/cr500452k
Dekker, F. J., Haisma, H. J. (2009). Histone acetyl transferases as emerging drug targets. Drug Discovery Today 14, 942–948. doi: 10.1016/j.drudis.2009.06.008
Dias, D. A., Urban, S., Roessner, U. (2012). A historical overview of natural products in drug discovery. Metabolites 2, 303–336. doi: 10.3390/metabo2020303
Doi, M., Hirayama, J., Sassone-Corsi, P. (2006). Circadian regulator CLOCK is a histone acetyltransferase. Cell 125, 497–508. doi: 10.1016/j.cell.2006.03.033
Dyda, F., Klein, D. C., Hickman, A. B. (2000). GCN5-related N-acetyltransferases: a structural overview. Annu. Rev. Biophys. Biomol. Struct. 29, 81–103. doi: 10.1146/annurev.biophys.29.1.81
Falkenberg, K. J., Johnstone, R. W. (2014). Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discovery 13 (9), 673–691. doi: 10.1038/nrd4360
Filippakopoulos, P., Knapp, S. (2014). Targeting bromodomains: epigenetic readers of lysine acetylation. Nat. Rev. Drug Discovery 13, 337–356. doi: 10.1038/nrd4286
Fiorentino, F., Mai, A., Rotili, D. (2018). Lysine acetyltransferase inhibitors: structure-activity relationships and potential therapeutic implications. Future Med. Chem. 10, 1067–1091. doi: 10.4155/fmc-2017-0244
Fiorentino, F., Bolla, J. R., Mehmood, S., Robinson, C. V. (2019). The Different Effects of Substrates and Nucleotides on the Complex Formation of ABC Transporters. Structure 27, 651–659. doi: 10.1016/j.str.2019.01.010
Friedmann, D. R., Aguilar, A., Fan, J., Nachury, M. V., Marmorstein, R. (2012). Structure of the alpha-tubulin acetyltransferase, alphaTAT1, and implications for tubulin-specific acetylation. Proc. Natl. Acad. Sci. U.S.A. 109, 19655–19660. doi: 10.1073/pnas.1209357109
Gayther, S. A., Batley, S. J., Linger, L., Bannister, A., Thorpe, K., Chin, S. F., et al. (2000). Mutations truncating the EP300 acetylase in human cancers. Nat. Genet. 24, 300–303. doi: 10.1038/73536
Ghizzoni, M., Boltjes, A., Graaf, C., Haisma, H. J., Dekker, F. J. (2010). Improved inhibition of the histone acetyltransferase PCAF by an anacardic acid derivative. Bioorg. Med. Chem. 18, 5826. doi: 10.1016/j.bmc.2010.06.089
Ghizzoni, M., Wu, J., Gao, T., Haisma, H. J., Dekker, F. J., George Zheng, Y. (2012). 6-alkylsalicylates are selective Tip60 inhibitors and target the acetyl-CoA binding site. Eur. J. Med. Chem. 47, 337–344. doi: 10.1016/j.ejmech.2011.11.001
Guo-Bo, L., Lu-Yi, H., Hui, L., Sen, J., Lin-Li, L., Sheng-Yong, Y. (2016). Identification of new p300 histone acetyltransferase inhibitors from natural products by a customized virtual screening method. RSC Advances 6, 61137–61140. doi: 10.1039/C6RA11240D
Halkidou, K., Gnanapragasam, V. J., Mehta, P. B., Logan, I. R., Brady, M. E., Cook, S., et al. (2003). Expression of Tip60, an androgen receptor coactivator, and its role in prostate cancer development. Oncogene 22, 2466–2477. doi: 10.1038/sj.onc.1206342
Hemshekhar, M., Sebastin Santhosh, M., Kemparaju, K., Girish, K. S. (2012). Emerging roles of anacardic acid and its derivatives: a pharmacological overview. Basic Clin. Pharmacol. Toxicol. 110, 122–132. doi: 10.1111/j.1742-7843.2011.00833.x
Hsieh, Y. J., Kundu, T. K., Wang, Z., Kovelman, R., Roeder, R. G. (1999). The TFIIIC90 subunit of TFIIIC interacts with multiple components of the RNA polymerase III machinery and contains a histone-specific acetyltransferase activity. Mol. Cell. Biol. 19, 7697–7704. doi: 10.1128/MCB.19.11.7697
Huang, J., Wan, D., Li, J., Chen, H., Huang, K., Zheng, L. (2015). Histone acetyltransferase PCAF regulates inflammatory molecules in the development of renal injury. Epigenetics 10, 62–72. doi: 10.4161/15592294.2014.990780
Ingólfsson, H. I., Thakur, P., Herold, K. F., Hobart, E. A., Ramsey, N. B., Periole, X., et al. (2014). Phytochemicals perturb membranes and promiscuously alter protein function. ACS Chem. Biol. 9, 1788–1798. doi: 10.1021/cb500086e
Johnson, T. A., Sohn, J., Inman, W. D., Estee, S. A., Loveridge, S. T., Vervoort, H. C., et al. (2011). Natural product libraries to accelerate the high-throughput discovery of therapeutic leads. J. Nat. Prod. 74, 2545–2555. doi: 10.1021/np200673b
Kalkhoven, E. (2004). CBP and p300: HATs for different occasions. Biochem. Pharmacol. 68, 1145–1155. doi: 10.1016/j.bcp.2004.03.045
Kindle, K. B., Troke, P. J., Collins, H. M., Matsuda, S., Bossi, D., Bellodi, C., et al. (2005). MOZ-TIF2 inhibits transcription by nuclear receptors and p53 by impairment of CBP function. Mol. Cell. Biol. 25:988. doi: 10.1128/MCB.25.3.988-1002.2005
Kishimoto, M., Kohno, T., Okudela, K., Otsuka, A., Sasaki, H., Tanabe, C., et al. (2005). Mutations and deletions of the CBP gene in human lung cancer. Clin. Cancer Res. 11, 512–519.
Kormendi, V., Szyk, A., Piszczek, G., Roll-Mecak, A. (2012). Crystal structures of tubulin acetyltransferase reveal a conserved catalytic core and the plasticity of the essential N terminus. J. Biol. Chem. 287, 41569–41575. doi: 10.1074/jbc.C112.421222
Lasko, L. M., Jakob, C. G., Edalji, R. P., Qiu, W., Montgomery, D., Digiammarino, E. L., et al. (2017). Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 550, 128–132. doi: 10.1038/nature24028
Lee, K. K., Workman, J. L. (2007). Histone acetyltransferase complexes: one size doesn’t fit all. Nat. Rev. Mol. Cell. Biol. 8, 284–295. doi: 10.1038/nrm2145
Lenoci, A., Tomassi, S., Conte, M., Benedetti, R., Rodriguez, V., Carradori, S., et al. (2014). Quinoline-based p300 histone acetyltransferase inhibitors with pro-apoptotic activity in human leukemia U937 cells. ChemMedChem 9, 542–548. doi: 10.1002/cmdc.201300536
Li, Q., Sun, H., Shu, Y., Zou, X., Zhao, Y., Ge, C. (2015). hMOF (human males absent on the first), an oncogenic protein of human oral tongue squamous cell carcinoma, targeting EZH2 (enhancer of zeste homolog 2). Cell Prolif. 48, 436–442. doi: 10.1111/cpr.12177
Li, Q., Liu, Z., Xu, M., Xue, Y., Yao, B., Dou, C., et al. (2016). PCAF inhibits hepatocellular carcinoma metastasis by inhibition of epithelial-mesenchymal transition by targeting Gli-1. Cancer Lett. 375, 190–198. doi: 10.1016/j.canlet.2016.02.053
Liu, X., Wang, L., Zhao, K., Thompson, P. R., Hwang, Y., Marmorstein, R., et al. (2008). The structural basis of protein acetylation by the p300/CBP transcriptional coactivator. Nature 451, 846–850. doi: 10.1038/nature06546
Lorch, Y., Beve, J., Gustafsson, C. M., Myers, L. C., Kornberg, R. D. (2000). Mediator-nucleosome interaction. Mol. Cell 6, 197–201. doi: 10.1016/S1097-2765(05)00007-9
Love, I. M., Sekaric, P., Shi, D., Grossman, S. R., Androphy, E. J. (2012). The histone acetyltransferase PCAF regulates p21 transcription through stress-induced acetylation of histone H3. Cell Cycle 11, 2458–2466. doi: 10.4161/cc.20864
Mai, A., Rotili, D., Tarantino, D., Ornaghi, P., Tosi, F., Vicidomini, C., et al. (2006). Small-molecule inhibitors of histone acetyltransferase activity: identification and biological properties. J. Med. Chem. 49, 6897–6907. doi: 10.1021/jm060601m
Mai, A., Cheng, D., Bedford, M. T., Valente, S., Nebbioso, A., Perrone, A., et al. (2008). Epigenetic multiple ligands: mixed histone/protein methyltransferase, acetyltransferase, and class III deacetylase (sirtuin) inhibitors. J. Med. Chem. 51, 2279–2290. doi: 10.1021/jm701595q
Mai, A., Rotili, D., Tarantino, D., Nebbioso, A., Castellano, S., Sbardella, G., et al. (2009). Identification of 4-hydroxyquinolines inhibitors of p300/CBP histone acetyltransferases. Bioorg. Med. Chem. Lett. 19, 1132–1135. doi: 10.1016/j.bmcl.2008.12.097
Mantelingu, K., Kishore, A. H., Balasubramanyam, K., Kumar, G. V., Altaf, M., Swamy, S. N., et al. (2007a). Activation of p300 histone acetyltransferase by small molecules altering enzyme structure: probed by surface-enhanced Raman spectroscopy. J. Phys. Chem. B. 111, 4527–4534. doi: 10.1021/jp067655s
Mantelingu, K., Reddy, B. A., Swaminathan, V., Kishore, A. H., Siddappa, N. B., Kumar, G. V., et al. (2007b). Specific inhibition of p300-HAT alters global gene expression and represses HIV replication. Chem. Biol. 14, 645–657. doi: 10.1016/j.chembiol.2007.04.011
Maradana, M. R., Thomas, R., O’sullivan, B. J. (2013). Targeted delivery of curcumin for treating type 2 diabetes. Mol. Nutr. Food Res. 57, 1550–1556. doi: 10.1002/mnfr.201200791
Marcu, M. G., Jung, Y. J., Lee, S., Chung, E. J., Lee, M. J., Trepel, J., et al. (2006). Curcumin is an inhibitor of p300 histone acetylatransferase. Med. Chem. 2, 169–174. doi: 10.2174/157340606776056133
Mehmood, S., Allison, T. M., Robinson, C. V. (2015). Mass spectrometry of protein complexes: from origins to applications. Annu. Rev. Phys. Chem. 66, 453–474. doi: 10.1146/annurev-physchem-040214-121732
Milite, C., Feoli, A., Sasaki, K., La Pietra, V., Balzano, A. L., Marinelli, L., et al. (2015). A novel cell-permeable, selective, and noncompetitive inhibitor of KAT3 histone acetyltransferases from a combined molecular pruning/classical isosterism approach. J. Med. Chem. 58, 2779–2798. doi: 10.1021/jm5019687
Modak, R., Basha, J., Bharathy, N., Maity, K., Mizar, P., Bhat, A. V., et al. (2013). Probing p300/CBP associated factor (PCAF)-dependent pathways with a small molecule inhibitor. ACS Chem. Biol. 8, 1311–1323. doi: 10.1021/cb4000597
6Murr, R., Loizou, J. I., Yang, Y. G., Cuenin, C., Li, H., Wang, Z. Q., et al. (2006). Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat. Cell Biol. 8, 91–99. doi: 10.1038/ncb1343
Nelson, K. M., Dahlin, J. L., Bisson, J., Graham, J., Pauli, G. F., Walters, M. A. (2017). The Essential Medicinal Chemistry of Curcumin. J. Med. Chem. 60, 1620–1637. doi: 10.1021/acs.jmedchem.6b00975
Neuwald, A. F., Landsman, D. (1997). GCN5-related histone N-acetyltransferases belong to a diverse superfamily that includes the yeast SPT10 protein. Trends Biochem. Sci. 22, 154–155. doi: 10.1016/S0968-0004(97)01034-7
Newman, D. J., Cragg, G. M. (2012). Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 75, 311–335. doi: 10.1021/np200906s
Ornaghi, P., Rotili, D., Sbardella, G., Mai, A., Filetici, P. (2005). A novel Gcn5p inhibitor represses cell growth, gene transcription and histone acetylation in budding yeast. Biochem. Pharmacol. 70, 911–917. doi: 10.1016/j.bcp.2005.06.013
Paolinelli, R., Mendoza-Maldonado, R., Cereseto, A., Giacca, M. (2009). Acetylation by GCN5 regulates CDC6 phosphorylation in the S phase of the cell cycle. Nat. Struct. Mol. Biol. 16, 412–420. doi: 10.1038/nsmb.1583
Park, W. J., Ma, E. (2012). Inhibition of PCAF histone acetyltransferase, cytotoxicity and cell permeability of 2-acylamino-1-(3- or 4-carboxy-phenyl)benzamides. Molecules 17, 13116–13131. doi: 10.3390/molecules171113116
Patel, J. H., Du, Y., Ard, P. G., Phillips, C., Carella, B., Chen, C. J. (2004). The c-MYC oncoprotein is a substrate of the acetyltransferases hGCN5/PCAF and TIP60. Mol. Cell Biol. 24, 10826–10834. doi: 10.1128/mcb.24.24.10826-10834.2004
Perez-Campo, F. M., Borrow, J., Kouskoff, V., Lacaud, G. (2009). The histone acetyl transferase activity of monocytic leukemia zinc finger is critical for the proliferation of hematopoietic precursors. Blood 113, 4866–4874. doi: 10.1182/blood-2008-04-152017
Perez-Campo, F. M., Costa, G., Lie-a-Ling, M., Kouskoff, V., Lacaud, G. (2013). The MYSTerious MOZ, a histone acetyltransferase with a key role in haematopoiesis. Immunology 139, 161–165. doi: 10.1111/imm.12072
Pfister, S., Rea, S., Taipale, M., Mendrzyk, F., Straub, B., Ittrich, C., et al. (2008). The histone acetyltransferase hMOF is frequently downregulated in primary breast carcinoma and medulloblastoma and constitutes a biomarker for clinical outcome in medulloblastoma. Int. J. Cancer 122, 1207–1213. doi: 10.1002/ijc.23283
Poepsel, S., Kasinath, V., Nogales, E. (2018). Cryo-EM structures of PRC2 simultaneously engaged with two functionally distinct nucleosomes. Nat. Struct. Mol. Biol. 25, 154–162. doi: 10.1038/s41594-018-0023-y
Portela, A., Esteller, M. (2010). Epigenetic modifications and human disease. Nat. Biotechnol. 28, 1057–1068. doi: 10.1038/nbt.1685
Ravens, S., Fournier, M., Ye, T., Stierle, M., Dembele, D., Chavant, V., et al. (2014). Mof-associated complexes have overlapping and unique roles in regulating pluripotency in embryonic stem cells and during differentiation. Elife 3, e02104. doi: 10.7554/eLife.02104
Ravindra, K. C., Selvi, B. R., Arif, M., Reddy, B. A., Thanuja, G. R., Agrawal, S., et al. (2009). Inhibition of lysine acetyltransferase KAT3B/p300 activity by a naturally occurring hydroxynaphthoquinone, plumbagin. J. Biol. Chem. 284, 24453–24464. doi: 10.1074/jbc.M109.023861
Ravindra, K. C., Narayan, V., Lushington, G. H., Peterson, B. R., Prabhu, K. S. (2012). Targeting of histone acetyltransferase p300 by cyclopentenone prostaglandin Delta(12)-PGJ(2) through covalent binding to Cys(1438). Chem. Res. Toxicol. 25, 337–347. doi: 10.1021/tx200383c
Roth, S. Y., Denu, J. M., Allis, C. D. (2001). Histone acetyltransferases. Annu. Rev. Biochem. 70, 81–120. doi: 10.1146/annurev.biochem.70.1.81
Sampath, V., Liu, B., Tafrov, S., Srinivasan, M., Rieger, R., Chen, E. I., et al. (2013). Biochemical characterization of Hpa2 and Hpa3, two small closely related acetyltransferases from Saccharomyces cerevisiae. J. Biol. Chem. 288, 21506–21513. doi: 10.1074/jbc.M113.486274s
Sano, Y., Ishii, S. (2001). Increased affinity of c-Myb for CREB-binding protein (CBP) after CBP-induced acetylation. J. Biol. Chem. 276, 3674–3682. doi: 10.1074/jbc.M006896200
Sapountzi, V., Cote, J. (2011). MYST-family histone acetyltransferases: beyond chromatin. Cell Mol. Life Sci. 68, 1147–1156. doi: 10.1007/s00018-010-0599-9
Sbardella, G., Castellano, S., Vicidomini, C., Rotili, D., Nebbioso, A., Miceli, M., et al. (2008). Identification of long chain alkylidenemalonates as novel small molecule modulators of histone acetyltransferases. Bioorg. Med. Chem. Lett. 18, 2788–2792. doi: 10.1016/j.bmcl.2008.04.017
Schneider, A., Chatterjee, S., Bousiges, O., Selvi, B. R., Swaminathan, A., Cassel, R., et al. (2013). Acetyltransferases (HATs) as targets for neurological therapeutics. Neurotherapeutics 10, 568–588. doi: 10.1007/s13311-013-0204-7
Seong, A. R., Yoo, J. Y., Choi, K., Lee, M. H., Lee, Y. H., Lee, J., et al. (2011). Delphinidin, a specific inhibitor of histone acetyltransferase, suppresses inflammatory signaling via prevention of NF-kappaB acetylation in fibroblast-like synoviocyte MH7A cells. Biochem. Biophys. Res. Commun. 410, 581–586. doi: 10.1016/j.bbrc.2011.06.029
Shiota, M., Yokomizo, A., Tada, Y., Uchiumi, T., Inokuchi, J., Tatsugami, K., et al. (2010). P300/CBP-associated factor regulates Y-box binding protein-1 expression and promotes cancer cell growth, cancer invasion and drug resistance. Cancer Sci. 101, 1797–1806. doi: 10.1111/j.1349-7006.2010.01598.x
Smith, B. C., Denu, J. M. (2009). Chemical mechanisms of histone lysine and arginine modifications. Biochim. Biophys. Acta 1789, 45–47. doi: 10.1016/j.bbagrm.2008.06.005
Souto, J. A., Conte, M., Alvarez, R., Nebbioso, A., Carafa, V., Altucci, L., et al. (2008). Synthesis of Benzamides Related to Anacardic Acid and Their Histone Acetyltransferase (HAT) Inhibitory Activities. ChemMedChem 3, 1435–1442. doi: 10.1002/cmdc.200800096
Spencer, T. E., Jenster, G., Burcin, M. M., Allis, C. D., Zhou, J., Mizzen, C. A., et al. (1997). Steroid receptor coactivator-1 is a histone acetyltransferase. Nature 389, 194–198. doi: 10.1038/38304
Srivastava, G., Mehta, J. L. (2009). Currying the heart: curcumin and cardioprotection. J. Cardiovasc. Pharmacol. Ther. 14, 22–27. doi: 10.1177/1074248408329608
Sun, C., Wang, M., Liu, X., Luo, L., Li, K., Zhang, S., et al. (2014). PCAF improves glucose homeostasis by suppressing the gluconeogenic activity of PGC-1alpha. Cell Rep. 9, 2250–2262. doi: 10.1016/j.celrep.2014.11.029
Sun, X. J., Man, N., Tan, Y., Nimer, S. D., Wang, L. (2015). The role of histone acetyltransferases in normal and malignant hematopoiesis. Front. Oncol. 5:108. doi: 10.3389/fonc.2015.00108
Sung, B., Pandey, M. K., Ahn, K. S., Yi, T., Chaturvedi, M. M., Liu, M., et al. (2008). Anacardic acid (6-nonadecyl salicylic acid), an inhibitor of histone acetyltransferase, suppresses expression of nuclear factor-kappaB-regulated gene products involved in cell survival, proliferation, invasion, and inflammation through inhibition of the inhibitory subunit of nuclear factor-kappaBalpha kinase, leading to potentiation of apoptosis. Blood 111, 4880–4891. doi: 10.1182/blood-2007-10-117994
Sykes, S. M., Mellert, H. S., Holbert, M. A., Li, K., Marmorstein, R., Lane, W. S., et al. (2006). Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol. Cell 24, 841–851. doi: 10.1016/j.molcel.2006.11.026
Tang, Y., Luo, J., Zhang, W., Gu, W. (2006). Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol. Cell 24, 827–839. doi: 10.1016/j.molcel.2006.11.021
Tanner, K. G., Langer, M. R., Kim, Y., Denu, J. M. (2000). Kinetic mechanism of the histone acetyltransferase GCN5 from yeast. J. Biol. Chem. 275, 22048–22055. doi: 10.1074/jbc.M002893200
Taschner, M., Vetter, M., Lorentzen, E. (2012). Atomic resolution structure of human alpha-tubulin acetyltransferase bound to acetyl-CoA. Proc. Natl. Acad. Sci. U.S.A. 109, 19649–19654. doi: 10.1073/pnas.1209343109
Tohyama, S., Tomura, A., Ikeda, N., Hatano, M., Odanaka, J., Kubota, Y., et al. (2012). Discovery and characterization of NK13650s, naturally occurring p300-selective histone acetyltransferase inhibitors. J. Org. Chem. 77, 9044–9052. doi: 10.1021/jo301534b
Tomaselli, D., Lucidi, A., Rotili, D., Mai, A. (2020). Epigenetic polypharmacology: A new frontier for epi-drug discovery. Med. Res. Rev. 40 (1), 190–244. doi: 10.1002/med.21600
Van Den Bosch, T., Leus, N. G. J., Wapenaar, H., Boichenko, A., Hermans, J., Bischoff, R., et al. (2017). A 6-alkylsalicylate histone acetyltransferase inhibitor inhibits histone acetylation and pro-inflammatory gene expression in murine precision-cut lung slices. Pulm. Pharmacol. Ther. 44, 88–95. doi: 10.1016/j.pupt.2017.03.006
Van Den Broeck, A., Nissou, D., Brambilla, E., Eymin, B., Gazzeri, S. (2012). Activation of a Tip60/E2F1/ERCC1 network in human lung adenocarcinoma cells exposed to cisplatin. Carcinogenesis 33, 320–325. doi: 10.1093/carcin/bgr292
Vasudevarao, M. D., Mizar, P., Kumari, S., Mandal, S., Siddhanta, S., Swamy, M. M., et al. (2014). Naphthoquinone-mediated inhibition of lysine acetyltransferase KAT3B/p300, basis for non-toxic inhibitor synthesis. J. Biol. Chem. 289, 7702–7717. doi: 10.1074/jbc.M113.486522
Wang, R., Cherukuri, P., Luo, J. (2005). Activation of Stat3 sequence-specific DNA binding and transcription by p300/CREB-binding protein-mediated acetylation. J. Biol. Chem. 280, 11528–11534. doi: 10.1074/jbc.M413930200
Wang, S., Dong, G., Sheng, C. (2019). Structural Simplification of Natural Products. Chem. Rev. 119, 4180–4220. doi: 10.1021/acs.chemrev.8b00504
Wapenaar, H., Van Der Wouden, P. E., Groves, M. R., Rotili, D., Mai, A., Dekker, F. J. (2015). Enzyme kinetics and inhibition of histone acetyltransferase KAT8. Eur. J. Med. Chem. 105, 289–296. doi: 10.1016/j.ejmech.2015.10.016
Wittschieben, B. O., Otero, G., De Bizemont, T., Fellows, J., Erdjument-Bromage, H., Ohba, R., et al. (1999). A novel histone acetyltransferase is an integral subunit of elongating RNA polymerase II holoenzyme. Mol. Cell 4, 123–128. doi: 10.1016/S1097-2765(00)80194-X
Wu, J., Xie, N., Wu, Z., Zhang, Y., Zheng, Y. G. (2009). Bisubstrate inhibitors of the MYST HATs Esa1 and Tip60. Bioorg. Med. Chem. 17, 1381–1386. doi: 10.1016/j.bmc.2008
Yan, Y., Harper, S., Speicher, D. W., Marmorstein, R. (2002). The catalytic mechanism of the ESA1 histone acetyltransferase involves a self-acetylated intermediate. Nat. Struct. Biol. 9, 862–869. doi: 10.1038/nsb849
Yang, X. J., Ogryzko, V. V., Nishikawa, J., Howard, B. H., Nakatani, Y. (1996). A p300/CBP-associated factor that competes with the adenoviral oncoprotein E1A. Nature 382, 319–324. doi: 10.1038/382319a0
Yang, Z., Zhang, Y., Chen, X., Li, W., Li, G.-B., Wu, Y. (2018). Total Synthesis and Evaluation of B-Homo Palmatine and Berberine Derivatives as p300 Histone Acetyltransferase Inhibitors - Yang - 2018 - European Journal of Organic Chemistry - Wiley Online Library. Eur. J. Org. Chem. 2018, 1041–1052. doi: 10.1002/ejoc.201701693
Yin, Y. W., Jin, H. J., Zhao, W., Gao, B., Fang, J., Wei, J., et al. (2015). The Histone Acetyltransferase GCN5 Expression Is Elevated and Regulated by c-Myc and E2F1 Transcription Factors in Human Colon Cancer. Gene Expr. 16, 187–196. doi: 10.3727/105221615x14399878166230
Ying, M. Z., Wang, J. J., Li, D. W., Yu, G., Wang, X., Pan, J., et al. (2010). The p300/CBP associated factor is frequently downregulated in intestinal-type gastric carcinoma and constitutes a biomarker for clinical outcome. Cancer Biol. Ther. 9, 312–320. doi: 10.4161/cbt.9.4.10748
Zammataro, M., Sortino, M. A., Parenti, C., Gereau, R. W. T., Chiechio, S. (2014). HDAC and HAT inhibitors differently affect analgesia mediated by group II metabotropic glutamate receptors. Mol. Pain 10, 68. doi: 10.1186/1744-8069-10-68
Keywords: lysine acetyltransferases, protein acetylation, natural products, enzyme inhibitors, epigenetics
Citation: Fiorentino F, Mai A and Rotili D (2020) Lysine Acetyltransferase Inhibitors From Natural Sources. Front. Pharmacol. 11:1243. doi: 10.3389/fphar.2020.01243
Received: 13 April 2020; Accepted: 29 July 2020;
Published: 12 August 2020.
Edited by:
Fidele Ntie-Kang, Martin Luther University of Halle-Wittenberg, GermanyReviewed by:
Wolfgang Sippl, Martin Luther University of Halle-Wittenberg, GermanyCopyright © 2020 Fiorentino, Mai and Rotili. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antonello Mai, YW50b25lbGxvLm1haUB1bmlyb21hMS5pdA==; Dante Rotili, ZGFudGUucm90aWxpQHVuaXJvbWExLml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.