
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol., 29 April 2020
Sec. Inflammation Pharmacology
Volume 11 - 2020 | https://doi.org/10.3389/fphar.2020.00533
This article is part of the Research TopicEicosanoids in CancerView all 12 articles
 Alicia M. Hidalgo-Estévez1†
Alicia M. Hidalgo-Estévez1† Konstantinos Stamatakis2,3†
Konstantinos Stamatakis2,3† Marta Jiménez-Martínez2
Marta Jiménez-Martínez2 Ricardo López-Pérez2
Ricardo López-Pérez2 Manuel Fresno2,3*
Manuel Fresno2,3*Colorectal cancer (CRC) is one of the most common and recurrent types of cancer, with high mortality rates. Several clinical trials and meta-analyses have determined that the use of pharmacological inhibitors of cyclooxygenase 2 (COX-2), the enzyme that catalyses the rate-limiting step in the synthesis of prostaglandins (PG) from arachidonic acid, can reduce the incidence of CRC as well as the risk of recurrence of this disease, when used together with commonly used chemotherapeutic agents. These observations suggest that inhibition of COX-2 may be useful in the treatment of CRC, although the current drugs targeting COX-2 are not widely used since they increase the risk of health complications. To overcome this difficulty, a possibility is to identify genes regulated by COX-2 activity that could give an advantage to the cells to form tumors and/or metastasize. The modulation of those genes as effectors of COX-2 may cancel the beneficial effects of COX-2 in tumor transformation and metastasis. A review of the available databases and literature and our own data have identified some interesting molecules induced by prostaglandins or COX-2 that have been also described to play a role in colon cancer, being thus potential pharmacological targets in colon cancer. Among those mPGES-1, DUSP4, and 10, Programmed cell death 4, Trop2, and many from the TGFβ and p53 pathways have been identified as genes upregulated in response to COX-2 overexpression or PGs in colon carcinoma lines and overexpressed in colon tumor tissue. Here, we review the available evidence of the potential roles of those molecules in colon cancer in the context of PG/COX signaling pathways that could be critical mediators of some of the tumor growth and metastasis advantage induced by COX-2. At the end, this may allow defining new therapeutic targets/drugs against CRC that could act specifically against tumor cells and would be effective in the prevention and treatment of CRC, lacking the unwanted side effects of COX-2 pharmacological inhibitors, providing alternative approaches in colon cancer.
Colorectal cancer (CRC) is the third most common type of cancer and the second leading cause of cancer-related death in the world, with around 900,000 deaths per year (Globocan, 2018; Keum and Giovannucci, 2019). Even in patients who have undergone tumor resection, 40–50% relapse and die of metastases, being the overall 5-year survival less than 60%. Despite this health burden, only few therapeutic options exist for this disease, including combination of chemotherapy and anti-angiogenic agents (Kuipers et al., 2015; Karpisheh et al., 2019). Thus, currently available therapeutic options do not achieve the desired efficiency and development of new strategies is required.
There are two types of CRC: hereditary or familial colon cancer and sporadic colon cancer (Sancho et al., 2004; Ma et al., 2018). Nonetheless, sporadic colon cancer is the most common with an incidence of 75–80% in diagnosed cases. The alterations generated in two main molecular pathways subdivide this type of cancer. In 80–85% of sporadic CRCs there are mutations or deletions in suppressor genes such as KRAS, APC, DCC, and p53, promoting chromosomal instability (Sancho et al., 2004; Moran et al., 2010). In 1990, Fearon and Vogelstein proposed that the mutation in three or four of the aforementioned oncogenes and/or suppressor genes is necessary to initiate the tumorigenesis process, where the final properties of the tumor are determined from the accumulation and order in which changes appear (Fearon and Vogelstein, 1990). Later, it was described how the epithelial cells of the CRC acquire the genetic alterations in a strict order and involve the Wnt and TGFβ signaling pathways in cancer promotion (Arends, 2000). On the other hand, the non-canonical route, also called “mutator”, comprises 15–20% of sporadic CRCs and depends mainly on the alteration due to the instability of microsatellite sequences (MSI, Microsatellite Sequences Instability) in the genome due to the accumulation of mutations in the genes for rupture repair (MMR, Mismatch Repair) produced during DNA replication (Imai and Yamamoto, 2008).
Many clinical trials and epidemiological studies have suggested that the use of non-steroidal anti-inflammatory drugs (NSAIDs), which are classic inhibitors of COX enzymatic activity reduce the risk of developing cancer in general, and more specifically in CRC (reviewed in (Thun et al., 2012; Rothwell, 2013; Piazuelo and Lanas, 2015; Patrignani and Patrono, 2016). Taking together all these data strongly suggest that Cox targeting is a mechanism for cancer prevention.
COX enzymes catalyse the rate limiting step in the conversion of arachidonic acid, derived from membrane phospholipids by phospholipase A2, into prostanoids (Williams and Dubois, 1996). Two main isoforms of this enzyme are known: COX-1 encoded by prostaglandin-endoperoxide synthase 1 (PTGS1) and COX-2 (encoded by PTGS2). Although both proteins show the same cyclooxygenase and peroxidase activity, their differences are found in substrates, cell expression, inhibition, and intracellular localization and induction (Garavito et al., 2002). COX-1 is constitutively expressed in most tissues and involved in physiological processes. However, COX-2 is not usually detectable in normal tissues but is induced by numerous cytokines, growth factors, hormones, and tumor promoters. Prostanoids modulate immune responses, renal function, blood clotting and play a role in many pathological conditions, such as inflammation, pain, fever, swelling, etc. (Iniguez et al., 2003; Iniguez et al., 2008; Sreeramkumar et al., 2012).
Considerable amounts of evidence in clinical settings further support a role of COX-2 in colorectal carcinogenesis and tumor progression [reviewed in (Subbaramaiah and Dannenberg, 2003; Wang and Dubois, 2010)]. Thus, COX-2 is expressed early during the adenoma-carcinoma sequence that occurs in CRC, suggesting an important role of this enzyme in colorectal carcinogenesis. COX-2 expression is upregulated in human colorectal adenocarcinomas when compared with normal adjacent colonic tissue. Moreover, polymorphisms of the PTGS2 gene were associated with risk of CRC (Cox et al., 2004; Agundez et al., 2015). The use of Min/+ mice as a CRC model showed elevated levels of COX-1 and COX-2 in sporadically formed adenomas (Williams et al., 1996) and inhibition of COX-2 resulted in a substantial decrease in intestinal polyp number and size (Jacoby et al., 2000). In the same way, Apc716 mice developed a smaller number and size of tumor polyps when the COX-2 gene was eliminated (Oshima et al., 1996).
In addition, many reports of colorectal tumor cells either overexpressing COX-2 or having it silenced have correlated increased COX-2 expression with their invasive and metastatic properties in xenografted tumors in mice (Tsujii and Dubois, 1995; Tsujii et al., 1997; O’mahony et al., 1999; Chen et al., 2001; Sun et al., 2002; Yoshimoto et al., 2002; Charames and Bapat, 2006; Strillacci et al., 2006; Stamatakis et al., 2015). However, the molecular mechanisms by which COX-2 expression in intestinal epithelial cells leads to that phenotype have not been fully elucidated yet.
In view of the abundant biological and phenotypic evidence, several clinical trials have been performed, aimed to evaluate the efficacy of specific inhibitors of COX-2 (COXIBs) (Fitzgerald and Patrono, 2001) to prevent or delay the onset (or recurrence) of tumors in high-risk patients, including those with prior removal of colon tumors. These studies indicate that specific inhibition of COX-2 prevents the (re)appearance of tumors but also show cardiovascular side-effects [reviewed in (Sinicrope, 2006; Bertagnolli, 2007)].
Recent studies, remark the role of COX-2 in constitutive IDO1 expression by human tumors and substantiate the use of COX-2 inhibitors to improve the efficacy of cancer immunotherapy, either by reducing constitutive IDO1 expression, which contributed to the lack of T-cell infiltration in tumors that fail to respond to immunotherapy (Hennequart et al., 2017), or by synergizing with anti-checkpoint antibodies (Zelenay et al., 2015).
For all of the above, the study of the expression of COX-2 in the different phases of tumor progression and metastasis and the finding of new signaling pathways triggered by this enzyme are essential in order to develop new drugs that inhibit the effects of COX-2 both in cancer prevention and therapy (Rizzo, 2011).
The activity of cyclooxygenases (COX) is coupled to several terminal synthases that produce the five primary different prostanoids: prostaglandin D2 (PGD2), prostaglandin E2 (PGE2), prostaglandin F2α (PGF2α), prostaglandin I2/prostacyclin (PGI2), and thromboxane A2 (TXA2) (Iniguez et al., 2003; Iniguez et al., 2008) being also some of them implicated in colon cancer.
Among the prostanoids, PGE2 has been proposed as the principal prostanoid promoting tumor growth and survival in CRC. PGE2 is present in the healthy colon but its levels are elevated in CRC (Pugh and Thomas, 1994; Cathcart et al., 2012; Zuo and Shureiqi, 2013) and correlate with tumor size (Pugh and Thomas, 1994; Yang et al., 1998) and disease progression (Karpisheh et al., 2019). The elevation of PGE2 levels may favor tumor growth and invasion via several mechanisms as stimulating cell proliferation, inducing local immunosuppression, inhibiting apoptosis, promoting angiogenesis, and increasing cell migration and invasion as well as drug resistance in colon cancer cells (Iniguez et al., 2003; Corral et al., 2007; Greenhough et al., 2009; Wang and Dubois, 2010; Karpisheh et al., 2019). Moreover, there is a positive feedback loop between COX-2 and PGE2, in which COX-2 induces PGE2 production, and that in turn increases further the expression of COX-2 in colon cancer cells (Stamatakis et al., 2015; Karpisheh et al., 2019).
Three PGE2 synthases from PGH2 have been described (Hara et al., 2010), two microsomal, mPGES1, mPGES-2, and the cytoplasmic cPGES, encoded by the PTGES, PTGES2, and PTGES3 genes, respectively. MPGES1 expression has been associated to CRC incidence and prognosis (Seo et al., 2009; Sasaki et al., 2012) and has been proposed to cooperate with COX-2 to enhance tumor growth (Kamei et al., 2003). cPGES is a non-inducible isoform, constitutively expressed in most tissues and associated with COX-1 activity while maintaining the production of PGE2 (Tanioka et al., 2000).
Notably, we found co-localization of COX-2 and MPGES1 in human CRC biopsies and that PTGS2 and PTGES gene expression levels strongly correlate in human microarray databases. Interestingly, we have described the joint induction of mPGES-1 and COX-2 by PGE2 due to the involvement of the transcription factor EGR1 (Early Growth Response Protein 1), representing a positive feedback loop between COX-2, mPGES-1, and PGE2 (Stamatakis et al., 2015). Moreover, mPGES-1 overexpressing carcinoma cell lines have increased tumorigenic capacity in vivo indicating that high levels of any of the two enzymes is sufficient to enhance CRC growth (Stamatakis et al., 2015). Our results demonstrate that mPGES1 is induced by COX-2 overexpression, via autocrine PGs release, likely PGF2α through an EGR1-dependent mechanism in colon carcinoma.
Thus, mPGES1 has gained attention recently as alternative target to COX-2 for CRC chemoprevention and chemotherapy (Sasaki et al., 2015). In this sense, mPGES1 inhibitors have demonstrated promising effects in inhibiting colon carcinoma growth (Dixon et al., 2013; Wang et al., 2018; Karpisheh et al., 2019), that may circumvent the in vivo cardiovascular toxicity associated with COX-2 inhibitors.
PGE2 acts through its binding to prostanoid-E (EP) receptors. Four subtypes of EP receptors (EP1, EP2, EP3, EP4) have been identified. Their tissue-specific expression and activation trigger different signaling pathways that favor, for example, the cellular proliferation of the intestinal epithelium (Takafuji et al., 2000). EPs belong to the family of G-protein coupled receptors (GPCRs) and transduce PGE2 signaling through different intracellular messengers. EP2 and EP4 preferably increase intracellular levels of cyclic adenosine monophosphate (cAMP) while signaling by EP3 mostly mobilizes cAMP and the activation of EP1 mobilizes calcium (Ca2+) (Regan, 2003; Alfranca et al., 2006). In different mice models of colon cancer, genetic deletion of the EP1 (Kawamori et al., 2005), EP2 (Ma et al., 2015), and EP4 receptors inhibits colonic tumorigenesis (Mutoh et al., 2002).
PGF2α is mainly produced by the activity of PGF synthase (PGFS) from PGH2 and specifically activates the GPCR type FP receptor (Breyer et al., 2001; Komoto et al., 2006). Signaling through the two FP isoforms leads to the activation of phospholipase C (PLC) and, consequently, an increase in intracellular Ca2+ and the activation of protein kinase C (PKC). However, PGF2α can be synthesized from other PGs such as PGE2 and PGD2, and can signal through other receptors such as EP1 and EP3, thus varying the intracellular signaling pathways (Breyer et al., 2001; Komoto et al., 2006). For example, induction of COX-2 through FP can occur through Rho activation and transcription mediated by β-catenin/Tcf (Fujino and Regan, 2003). Along those lines, elevated levels of cAMP have been detected in cells isolated from colonic crypts after treatment with PGF2α (Collins et al., 2009).
There is very little evidence of the possible role of PGF2α in CRC other than its higher levels, together with PGE2 (Nugent et al., 1996). Also, in the tumor tissue of patients with CRC, a differential regulation of the cancer-dependent FP receptor has been detected (Gustafsson et al., 2007). PGF2α also promotes the invasion and mobility of epithelial cells of adenomas and colon carcinomas in collaboration with PGE2 (Qualtrough et al., 2007).
Recently, we have shown that colorectal tumor cells produce PGF2α though COX-2 and have provided some evidence that PGF2α may also play a role in colorectal tumorigenesis (Stamatakis et al., 2015). Colorectal adenoma and carcinoma-derived cell lines secrete PGF2α while they also express FP indicating potential autocrine effects. Interestingly, this secreted PGF2α was able to increase mPGES1 but also COX-2 in colon carcinoma cells, via FP receptor and EGR1, further enhancing PGE2 levels indicating a positive feedback loop between COX-2/mPGES1/PGE2 and PGF2α (Stamatakis et al., 2015). However, PGF2α failed to directly induce cell proliferation in CRC cell lines (Cassano et al., 2000).
The roles of the other COX-derived prostanoids in CRC tumors, as well as in carcinoma cell lines remains poorly understood (Cathcart et al., 2011). It has been reported that the levels of some prostaglandin receptors, such as IP (PGI2 receptor) and DP (PGD2 receptor), were reduced in CRC cells and the downregulation of one of them, DP2, has been related to differentiation of healthy epithelium to tumor (Gustafsson et al., 2007). The expression of thromboxane synthase (TXS), the enzyme that catalyses the conversion of PGH2 to TXA2, was significantly increased in CRC tumors compared to normal tissue. Furthermore, genetic or pharmacological reduction of TXS diminishes proliferation in CRC cell lines (Ekambaram et al., 2011). In addition it has also been described that the lack of the enzyme 15-PGDH, responsible for the inactivation of prostaglandins and lipoxins, is associated with CRC (Backlund et al., 2005) and together with the increase in the PGE2 synthesis, induces tumor formation in Min (multiple intestinal neoplasia) CRC model in mice. (Myung et al., 2006). Besides the loss of 15‐PGDH and induction of this enzyme has been associated with the suppression of inflammation-driven colon carcinogenesis in mice (Choi et al., 2014).
The potential implication of the Cox-PG synthases-PG receptors in CRC can be also studied through expression levels of the pathway´s components using The Cancer Genome Atlas (TCGA) gene expression data. A summary of these findings is shown in Figure 1, based on the TCGA Colon Cancer dataset. These analyses, comparing tumor and normal colon epithelia, showed that gene expressions from several members of this pathway are upregulated in tumors, namely PTGS2, PTGES, and TBXAS1, indicating that the biosynthetic and signaling pathways of PGE2 and TXA2 are favored in established tumors. Conversely, expression downregulation in tumor tissue was observed for genes encoding components of the pathway leading to the signaling of other prostanoids, but most notably of HPGD that encodes the PGE2-inactivating enzyme 15-PGDH. Analysis with other different datasets, i.e., the OncoMine microarray database (Compendia Bioscience), resulted in very similar data regarding PTGS2, PTGES (Stamatakis et al., 2015). Together, those analyses confirm the important role of COX2-2 and PGE2 in colon cancer. Nonetheless, since COX is the rate-limiting enzyme in the synthesis of all prostanoids, an increased expresion of this enzyme could result in the elevation of other prostanoids besides PGE2.
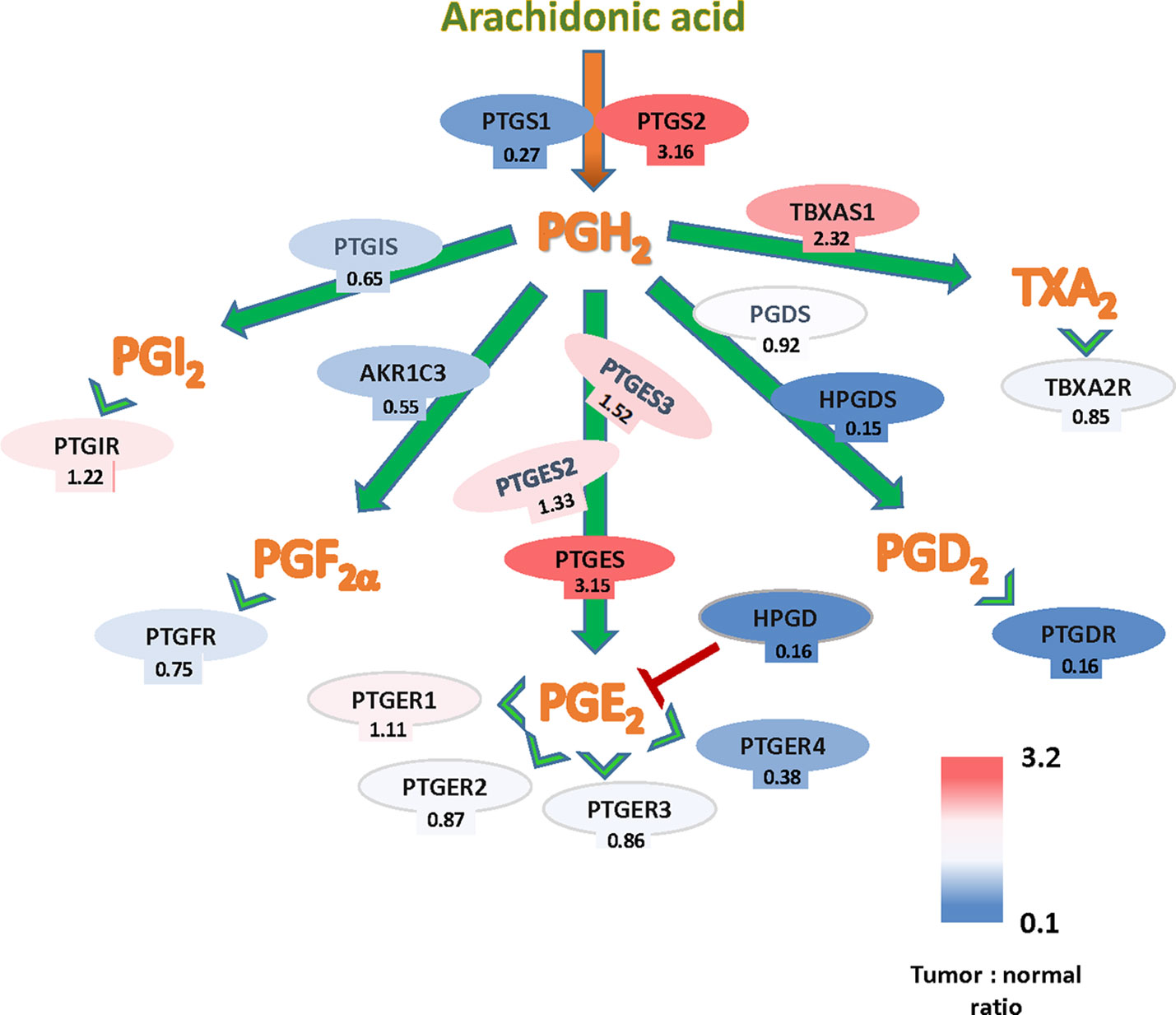
Figure 1 Arachidonate-prostanoids pathway gene expression in colorectal cancer (CRC) tumors. Shown are mRNA expressions in tumors of enzymes and receptors from the arachidonate-prostanoids pathway. Numbers indicate the fold differences with normal adjacent tissue based on the The Cancer Genome Atlas (TCGA) Colon Cancer dataset.
As mentioned above, the treatment of patients with NSAIDs or COXIBs greatly decreases tumor progression in a number of clinical trials in CRC. However, since general inhibitors of COXs, such as aspirin, can cause gastrointestinal bleeding problems and specific inhibitors of COX-2 increase the risk of cardiovascular disease, alternative treatments are needed targeting the downstream effector of COX-2 protumoral activity, likely minimizing side effects, and if possible aiming exclusively to cancer cells (Cathcart et al., 2011).
Although PGE2 blockade may represent a good therapeutic option, other PGs besides PGE2 could be responsible for COX-2 effect in colon carcinoma cells. Besides, although prostanoids may modulate the growth and invasiveness of colon carcinoma cells, they also modulate the immune responses and inflammation and their inhibition may also have unwanted side effects.
To address this issue, we have concentrated in the effects of COX-2 in colon carcinoma cells. COX-2 activity regulates gene expression, which confers cells advantages for growth, migration, invasion, and metastasis (Stamatakis et al., 2015). We found that stable COX-2 overexpression in carcinoma cell lines (HT-29, HCT116, and Caco2) significantly affects gene transcription. Analysis of genes modified by COX-2 overexpression indicates that many genes involved in transcription, growth, apoptosis, angiogenesis, and migration were elevated (ArrayExpress, E-MEXP-2343). Moreover, a review of the available databases and literature in other carcinoma cell lines overexpressing COX-2 or repressed siRNA also identify some COX-2 downstream effectors responsible for the pro-tumorigenic properties of COX-2, although none of them performed a comprehensive gene expression analysis. Altogether, those analyses identified some interesting molecules. We selected some genes and validated them by quantitative RT-PCR in colon carcinoma cell lines, either induced by PGE2 or by overexpression of COX-2 or mPGES1. The results of mRNA levels obtained for some of those genes are shown in Table 1. Besides, we performed analyses of mRNA expression and survival in colon adenocarcinomas using public databases. From all these analyses some expected genes emerged, but others are relatively new and amenable to drug targeting.
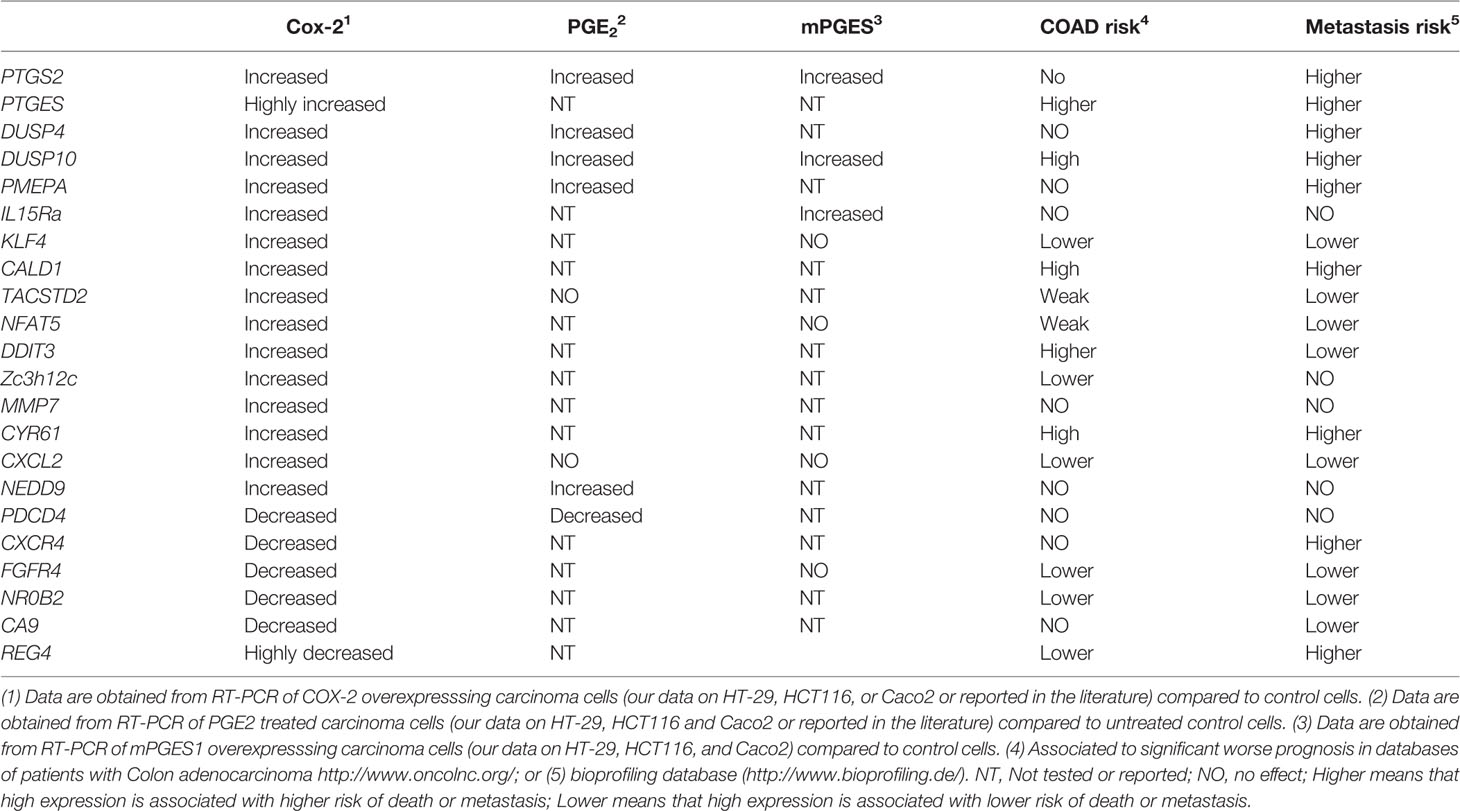
Table 1 Genes upregulated by COX-2, mPGES1, or PGE2.
Dual-specificity Mitogen Activated Protein Kinase (MAPK) phosphatase 10 (DUSP10), also named MPK5, was found altered by COX-2 overexpression in CRC cell lines in several data sets (Doherty et al., 2009) included our own. DUSP10 is able to dephosphorylate p38 and c-Jun NH2-terminal kinase (JNK). Until recently, there were few reports regarding the putative role of DUSP10 in cancer. Most of those studies have found increased DUSP10 mRNA in tumor tissue, thus suggesting a pro-tumorigenic role for this phosphatase (Jimenez-Martinez et al., 2019b). In CRC, the expression of certain DUSP10 polymorphisms are linked to an enhanced CRC risk (Duan et al., 2015).
We found that DUSP10 overexpression increased the growth of CRC cell lines and mouse xenografts, while the opposite phenotype was observed by DUSP10 silencing. Cancer progression is related to an uncontrolled cell division and DUSP10 overexpression produces the loss of cell-contact inhibition through the dephosphorylation of Yes-associated protein (YAP) at Ser397. This dephosphorylation retains YAP in the nucleus. In fact, we found that high amounts of DUSP10 and YAP1 are located in the nucleus of CRC cells (Jimenez-Martinez et al., 2019a). Additionally, the quantity of nuclear DUSP10 in CRC tumor biopsies is directly correlated with high tumor stage CRC and poor prognosis and survival in a large cohort of CRC patients, being also associated to high expression of nuclear YAP1. All these data point at DUSP10 as a downstream protein in COX-2 signaling and provide evidence of the role of DUSP10 in CRC progression via YAP1 regulation (Jimenez-Martinez et al., 2019a). Interestingly, an inhibitor of DUSP10 have been described (Hommo et al., 2015), supporting its use in CRC.
Another dual-specificity MAPK phosphatase, DUSP4/MKP-2, was also upregulated by COX-2. DUSP4 role in several cancers has been documented (Low and Zhang, 2016; Seternes et al., 2019). An altered expression of DUSP4 is related to colon tumorigenesis. Thus, elevated DUSP4 expression is associated with microsatellite instability in CRC patients (Gröschl et al., 2013) being also associated with liver and lung metastases of CRC (Saigusa et al., 2013). Moreover, DUSP4 overexpression in CRC cell lines produced upregulation of MAPK targets, such as EGR1, FOS, and MYC, and downregulation of the mismatch repair gene MSH2, all these effects leading to an increase in cell proliferation (Gröschl et al., 2013). In contrast, it has been more recently reported that the decrease in the expression of DUSP4 in CRC cell lines activated ERKs, causing cell proliferation and invasiveness (Ichimanda et al., 2018).
Interestingly, DUSP4 overexpression in CRC cell lines decreases their sensitivity to doxorubicin, a drug used to treat CRC (Kang et al., 2017). Moreover DUSP4 genetic inactivation increased the resistance to cetuximab, an epidermal growth factor receptor (EGFR)-blocking antibody, widely used and approved for treatment of metastatic CRC in cell lines and to another well‐known EGFR inhibitor, erlotinib (Park et al., 2019). Interestingly, those effects are also related to COX-2 expression, since DUSP4 was the highest induced gene in cetuximab-resistant CRC cells (Lu et al., 2016). All these data together point at DUSP4, enzyme regulated by COX-2, as a factor whose overexpression leads to CRC development and invasion, and which can be a promising therapeutic target. Moreover, DUSP4 specific inhibitors have been described (Park et al., 2014) supporting its clinical testing in CRC at least in some drug resistant tumors.
Trophoblast cell-surface antigen 2 (TROP2), also called Tumor-Associated Calcium Signal Transducer 2 (TACSTD2), is a cell-surface glycoprotein expressed during embryonic and fetal development which is involved in cell proliferation, cell binding, motility, and metastasis (Mcdougall et al., 2015). TROP2 overexpression can induce cancer growth and is associated with poor prognosis and drug resistance in cancer cells (Shvartsur and Bonavida, 2015).
Our COX-2, but not mPGES1, overexpressing colon carcinoma cells present high levels of TACSTD2. Besides, high expression of PTGS2 and TACSTD2 genes have been found also associated in lung cancer metastasis (Citterio et al., 2012). Previous studies in CRC patients, already showed the correlation between high expression of TROP2 and metastasis in CRC (Kapoor, 2013). The overexpression of TROP2 together with MMP7, another COX-2 induced gene (see below), is a predictor of worse prognosis and relapse in CRC (Fang et al., 2009). TROP2 expression enhances anchorage-independent growth in colon carcinoma cell lines (Wang et al., 2008) and its activation of TROP2 by Tumor Necrosis Factor (TNF)-alpha induces cell migration and invasion (Zhao and Zhang, 2018).
More importantly, an anti-TROP-2 antibody, sacituzumab govitecan, is a potential therapeutic drug for metastatic solid tumors (Starodub et al., 2015). Since TROP2 is consistently induced by COX-2 overexpression, this makes it an alternative target to COX-2 blockade therapy in CRC.
Matrix metalloproteinases (MMPs) are proteolytic enzymes which degrade and remodel the extracellular matrix (ECM) in physiological processes, such as in cell migration, and have also been involved in metastasis (Zucker and Vacirca, 2004). It has been reported that MMP7 is one of the most important MMPs in colorectal tumorigenesis, promoting angiogenesis, invasiveness, and tumor survival. Thus, MMP-7 may enhance CRC progression due to its proteolytic activity on several cell surface molecules as EGFR, Fas-L, etc. (Wang et al., 2006). MMP7 is abundantly synthesised by colon carcinoma cells (Zucker and Vacirca, 2004). A study of CRC patients demonstrated an increase in COX-2 and MMP-7 expression when compared to normal tissue and colon polyps sample (Bengi et al., 2015). High levels of MMP-7 together with PTEN down-regulation where detected in CRC and were related to tumor stage and progression (Bi et al., 2013). Moreover, COX-2 overexpression in carcinoma cells modulates the adhesive properties of MMPs (Tsujii and Dubois, 1995; Tsujii et al., 1997). PGE2 can transactivate EGFR thereby inducing the proliferation of CRC cell lines and exerts its functions in part through molecules such as MMP-7 (Pai et al., 2002; Karpisheh et al., 2019).
Krüppel-like factor 4 (KLF4) is a transcription factor that was firstly identified as a regulator of cell growth arrest being one of the most important factors in Cancer Stem Cells (CSC). It is expressed in differentiating cells and it is also known to have a role in apoptosis suppression (Ghaleb and Yang, 2017). KLF4 mRNA is consistently and strongly induced in our COX-2 but not in mPGES1 overexpressing colon carcinoma cells and it was shown that COX-2 and KLF4 colocalized in carcinoma cells of CRC tumor samples (Shao et al., 2008). Moreover, in HT-29 colon cancer cells, the expression of KLF4 was induced by 15-Deoxy-Delta-12, 14 PGJ2, a downstream product of COX-2 signaling pathway (Chen and Tseng, 2005).
It has been described that the levels of high KLF4 mRNA in normal tissues can be used as a prognostic indicator of survival in CRC patients (Lee et al., 2014), suggesting a regulatory role in CRC progression. In contrast, other reports have indicated a protumoral role of KLF4 in CRC. Thus, KLF4 was found in colon adenocarcinoma metastasis to the liver (Humphries et al., 2018) and low KLF4 expression was found in poorly differentiated CRC tissues (Hu et al., 2011). A study performed in cancer stem cell (CSC)-enriched spheroid CRC cell lines revealed a role of KLF4 in the invasiveness, migration, resistance to treatment, and ability to generate tumors as well as in the induction CSCs markers in those cells (Leng et al., 2013).
The expression of programmed cell death 4 (PDCD4), a tumor suppressor protein that inhibits tumor development in many cancer types, is decreased in CRC and this downregulation in colon tumors has been related to worse prognosis (Long et al., 2019). Several studies correlate PDCD4 overexpression with inhibition of cell proliferation and invasion in different tumor cancer cell types. In colon carcinoma cells, high levels of PDCD4 inhibited Akt signaling pathway, decreasing invasiveness (Wang et al., 2017). Moreover, a COX-2 inhibitor, NS-398, increases PDCD4 in HCA-7 cells, a colon cancer cell line (Zhang and Dubois, 2001), in agreement with our observation that is consistently downregulated by COX-2 overexpression in carcinoma cell lines. The overexpression of COX-2 in response to inflammation induces an increase in PGE2 production and PGE2 decreases PDCD4 protein levels through upregulation of miR-21 (Peacock et al., 2014). MiR-21 role in cancer has been extensively studied and its upregulation may explain the intrinsic resistance of some cancers to chemotherapy (Pfeffer et al., 2015), being miR-21 and PDCD4 inversely correlated in CRC (Chang et al., 2011; Horiuchi et al., 2012). Thus increasing PDCD4 levels through pharmacological manipulation of miR-21 could represent a novel therapeutic strategy in the treatment of CRC, as an alternative to COX-2/PGE2 blockade in CRC.
Nuclear Factor of Activated T cells- 5 (NFAT5) is a transcription factor belonging to the NFAT family whose expression is upregulated by osmotic stress to difference with the other four members of this family (Qin et al., 2014). Dysregulation of NFAT signaling has been linked to tumor progression in several cancers (Qin et al., 2014). Interestingly, NFAT is a key transcription factor regulating COX-2 expression in CRC cell lines (Duque et al., 2005), suggesting the existence of a positive feedback loop between NFATs and COX-2. However, little is know on NFAT5 and CRC. We found NFAT5 is induced by COX-2 overexpression and it has been described that promotes metastasis through the induction of alpha(6)beta(4) integrin in human breast and colon tumors (Jauliac et al., 2002), and besides some associations between NAFT5 SNPs and CRC risk has been found (Slattery et al., 2011). NFAT5 expression has also been associated to metastasis in other cancer types and to proliferation of lung adenocarcinoma cells (Guo and Jin, 2015).
Among the genes induced by COX2, we found in our arrays many genes of the TGF-β pathway, which we later confirmed by PCR (Table 2). Many of them are also induced by PGE2 treatment although in a much lower extent, suggesting that other COX2 derived products, as PGF2α, are also involved. Apart from those, some other genes induced by TGF-β were also induced by COX-2/PGs.
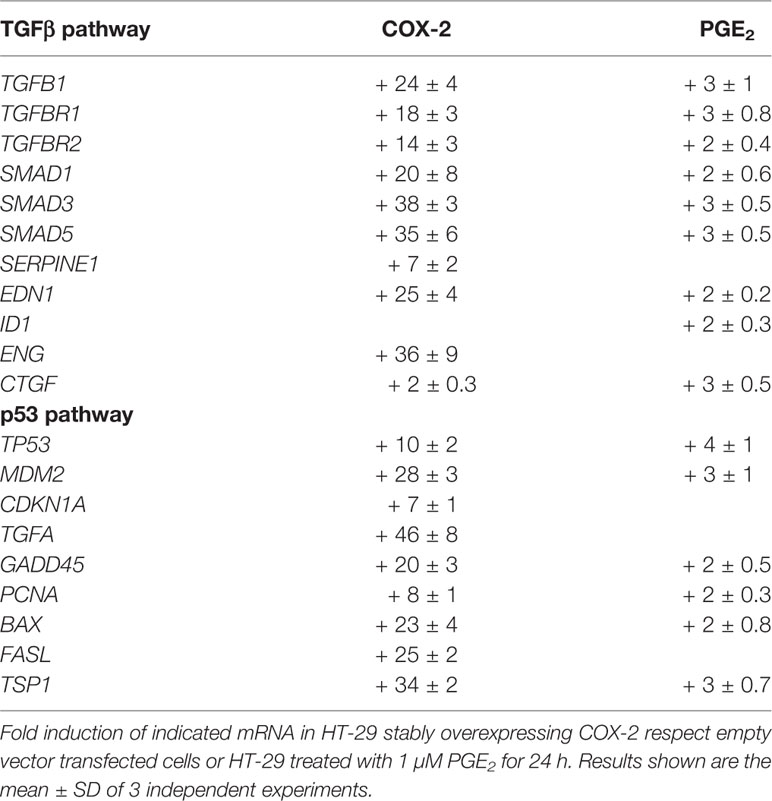
Table 2 Induction of TGFβ pathway or p53 pathway genes by COX-2 overexpression or PGE2 treatment in HT-29 colon carcinoma cells.
One of them was Prostate Transmembrane Protein, Androgen Induced 1 (PMEPA1, also known as TMEPA1, transmembrane prostate androgen induced 1). PMEPA1 was first identified as being upregulated in renal cell carcinoma and was designated as solid tumor associated gene 1 (STAG1) (Rae et al., 2001). PMEPA1 is known to be induced by TGF-β and modulates TGF-β signaling by competing with SARA (SMAD Anchor for Receptor Activation) for R-SMAD binding to sequester R-SMAD phosphorylation and promoting lysosomal degradation of TGF-β receptor (Watanabe et al., 2010). PMEPA1, through a negative feedback loop, is described as the responsible to convert TGF-β from a tumor suppressor to a tumor promoter in breast cancer (Singha et al., 2010).
We have demonstrated that PMEPA1 is a COX-2, PGE2, PGF2α, and calcium (through Ionophore treatment) -induced gene in colon and ovary cancer cells (Jimenez-Segovia et al., 2019). Furthermore, PMEPA1 is involved in differentiation of colon epithelial cells and high levels are also found in all phases of CRC development, including metastasis, in patients (Brunschwig et al., 2003). Furthermore, its high expression in CRC is related to poor prognosis and postulated as a predictor of relapse risk (Xu et al., 2017; Zhang et al., 2019). PMEPA1 is also overexpressed in intestinal tumors in Apc (Min) mice, which are prone to intestinal adenoma formation and is also dysregulated in human CRC adenomas (Reichling et al., 2005).
A recent study using colon cancer cell lines revealed the role of PMEPA1 in cell migration and invasion through bone morphogenetic proteins (BMP) signaling pathway activation and phosphorylation of the transcription factors Smad1 and Smad5 (Zhang et al., 2019), which also correlates with its described effect in ovarian cancer cells (Jimenez-Segovia et al., 2019).
NEDD9 (Neural Precursor Cell Expressed, Developmentally Down-Regulated-9) or HEF1 (Human enhancer of filamentation 1) is a structural protein mainly found in epithelial cells, which is upregulated by TGF-β and expressed transiently during embryonic life in mice. Many functions have been attributed to NEDD9, among them, the transmission of growth control signals between focal adhesions. It has also been involved in melanoma tumorigenesis and, most notably, in metastasis (O’neill et al., 2007; Tikhmyanova et al., 2010; Lee et al., 2011).
NEDD9 was also identified as one of key genes in CRC, being upregulated in CRC tumors (Li et al., 2011; Cui et al., 2017). Interestingly, two independent groups have shown that NEDD9 overexpression elicited the same effects as PGE2 treatment on cell proliferation, cell cycle progression, colony formation, migration, and xenograft tumor growth in colon carcinoma cell lines (Xia et al., 2010; Li et al., 2011). Interestingly, those PGE2 effects were reversed by inhibition of NEDD9 expression, indicating that NEDD9/HEF-1 could be an important downstream mediator in the pro-tumoral activity of PGE2 in CRC. In this sense, it would be possible to develop inhibitors to disrupt NEDD9 interactions with oncogenic partners (Tikhmyanova et al., 2010).
Cysteine Rich Angiogenic Inducer 61 (Cyr61/CNN1) is another TGF-β regulated gene (Han et al., 2016). It is a member of the CNN family of extracellular matrix associated proteins, which induces cell division and differentiation, chemotaxis, angiogenesis, and adhesion. In CRC, high levels of this protein induce migration and increase invasiveness (Chang et al., 2014). Cyr61/CNN1 has been described as potential serum marker for CRC (Song et al., 2017), whereas Cyr61 expression in the tumor tissue indicates poor prognosis in colon cancer patients being statistically associated with greater mortality (Jeong et al., 2014). Altered activation of CYR61 gene enhancers occurs during CRC development, being CYR61 expression induced by FOXA1 and CBP in colon cancer cells pointing at both transcription factors as targets for CRC treatment (Xie et al., 2019). Interestingly, PGF2α through FP receptors can upregulate the expression of CYR61 mRNA (Xu et al., 2009). We also found a significant positive correlation of COX2, DUSP10 with CYR61 in colon carcinoma cell lines (Jimenez-Martinez et al., 2019a).
Caldesmon (CaD), a major actin-associated protein, is found in smooth muscle and non-muscle cells with contractile function. In normal cells, CaD regulates contraction through the binding to two of the components of the thin filaments, actin, and tropomyosin, in the presence of Ca2+ (Wang and Coluccio, 2010). CaD expression was reported in both adenomas and adenocarcinomas (Porter et al., 1993). There are two alternatively spliced isoforms of CaD, high (h-CaD) and low (l-CaD), which differ in their molecular weight. H-CaD levels are lower in the late stages of colorectal adenocarcinoma while the low-molecular weight variant expression varies during tumor development and it is related to metastasis (Köhler, 2011; Kim et al., 2012). CaD is absent in the stroma in CRC but present in smooth muscle, thus it is a marker for desmoplasia and tumor invasiveness of the large intestine’s muscularis propria (Roberts et al., 2014). It has been also described that CaD mRNA levels are higher in colon carcinoma cell lines than in non-transformed normal colon epithelial cells (Nimmrich et al., 2000).
However, little is known about the role of the protein in the carcinogenic process. CaD is an important regulator of podosome formation which is involved in the degradation of the extracellular matrix (ECM), promoting in this way cell invasion, that is required for metastasis (Yoshio et al., 2007). L-CaD overexpression promotes expression of the regulatory p21 and c-PARP and suppresses NF-κB and phosphorylated mTOR expression, whereas its suppression increases sensitivity to chemo- and radiotherapy in colon carcinoma lines (Kim et al., 2012). TGF-β1 regulates the expression and phosphorylation of CaD in epithelial cells modulating epithelial-mesenchymal (EMT) transition (Nalluri et al., 2018).
The role of p53 in cancer in general and CRC in particular is well known [reviewed in (De Moraes et al., 2007; Watson and Collins, 2011)]. Mutations in the tumor suppressor gene TP53, which encodes the protein p53, are frequently found in human cancers. Mutations in K-ras, adenomatous polyposis coli (APC), and p53 induce the transition from healthy colonic epithelia to CRC (Dixon et al., 2013).
Among the genes induced by COX-2, we found in our arrays many genes of the p53 pathway, as it was found for the TGF pathway, that were also later confirmed by PCR (Table 2). Among those are TGFα, as well as others involved in cell cycle, apoptosis, DNA repair, and angiogenesis that my explain some of the diverse pro-tumoral activities of COX-2 in CRC. Again some of them were also induced by exogenous addition of PGE2 but in lesser extent, further suggesting that other prostaglandins, likely PGF2α, may be involved.
In addition, there is a well-known crosstalk between p53 and COX-2, in which COX-2 decreases p53 transcription, and p53 also regulates COX-2 expression (De Moraes et al., 2007). Furthermore, many COX-2 inducers, such as hypoxia, cytokines, oncogene activation, carcinogens, and inflammation, can also activate p53. The role of p53 in the regulation of COX-2 expression and activity has been extensively described. Thus, it has been reported the increase in COX-2 transcript in human tumor cells expressing p53 (Han et al., 2002; De Moraes et al., 2007). COX-2 upregulation by p53 has been attributed to an increase on the binding of NF-κB to COX-2 promoter in response to p53 overexpression (Benoit et al., 2006). Conversely, COX-2 avoids the transcriptional activity of p53 on target genes (De Moraes et al., 2007). COX-2 is not only regulating p53 function but also its subcellular localization, as it was demonstrated in human colon cell lines treated with celecoxib, a COX-2 inhibitor, in which p53 nuclear location was promoted (Swamy et al., 2003). Interestingly, joint nuclear expression of COX-2 and p53 was significantly associated with adenoma recurrence in CRC (Brand et al., 2013).
Nonetheless there is less reported evidence of COX-2 effects on the p53 pathway. Interestingly our analyses have defined many p53 related genes that are modulated by COX-2 overexpression in colon carcinoma cells. Many of them were not described before as modulated by COX-2 and can provide clues in new therapeutic approaches.
COX-2 has been identified through clinical trials, epidemiological meta-analyses, and preclinical models and as a key molecule in the development of CRC. However, the use of specific COX-2 inhibitors in the clinic has been hampered by their side effects. The identification of genes regulated by COX-2 activity has allowed us to identify some genes that have been already implicated in several aspects of CRC development and therapy. The relationships among those genes and with the PG pathways are summarized in Figure 2.
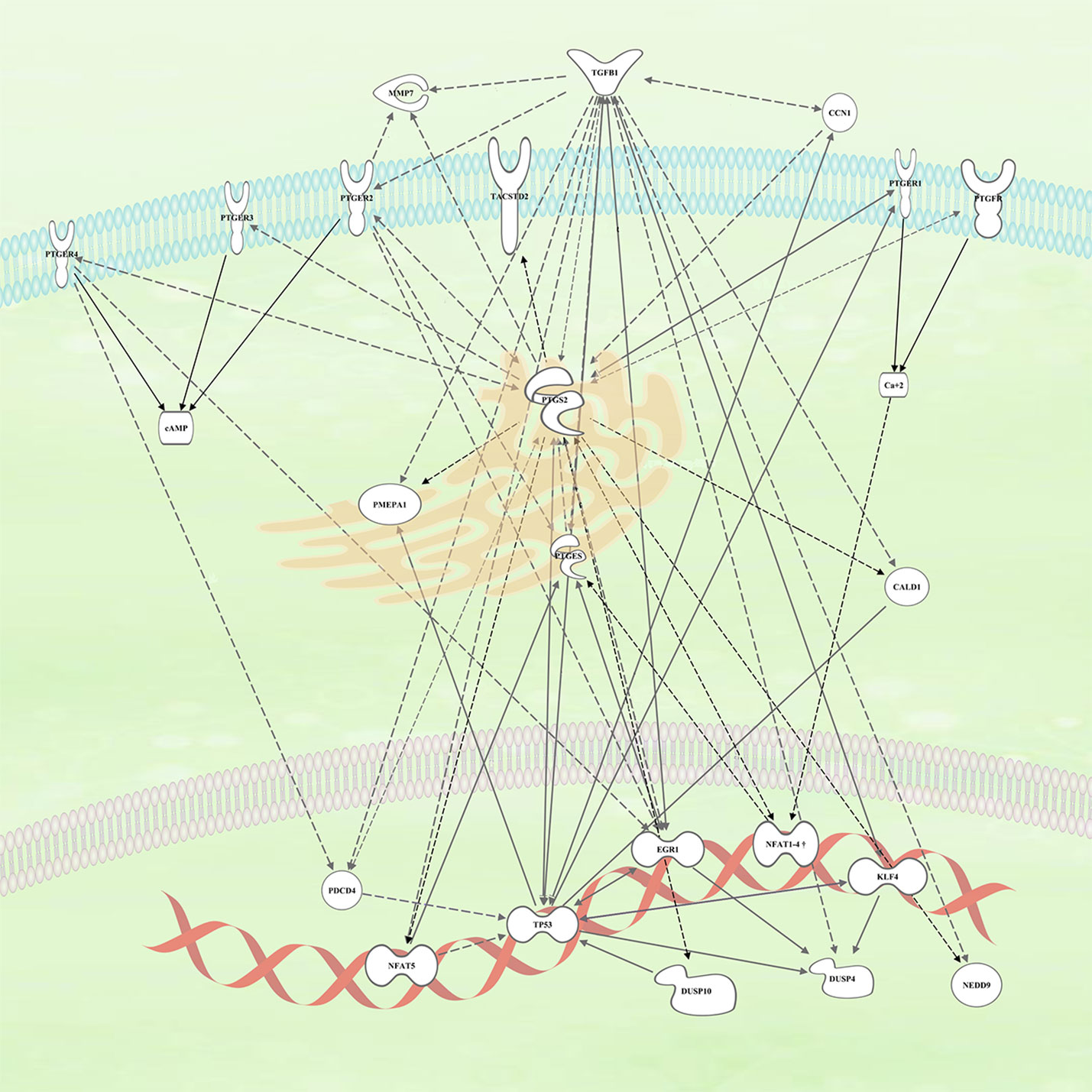
Figure 2 Connection within the different genes upregulated by COX2 and likely involved in colorectal cancer (CRC) tumors. The continuous arrows are of direct interaction/activation while the discontinuous ones are indirect activation in which intermediaries participate.
Of all molecules described here, some of them are more amenable to drug discovery. Besides some molecules had already specific inhibitors developed, as the phosphatases DUSP10 and DUSP4, as well as monoclonal antibodies to TROP2. This, together with the clinical evidence available in CRC, pointed out to these 3 molecules as the most suitable to be considered as putative drug targets and to engage in clinical trials especially in drug resistant settings or advanced CRC stages most likely in combination therapy. Also the interaction of the TGF-β and COX2 pathway deserve further clinical exploration. In summary, drugs targeting the COX-2 downstream molecules described will likely lack the unwanted side effects of COX-2 pharmacological inhibitors, providing alternative approaches in colon cancer.
Writing—review and editing: MF, AH-E, KS, MJ-M, and RL-P. Review of published literature: MF, AH-E, KS, MJ-M, and RL-P. Analysis of the data in figures and tables: KS.
This research was funded by grants from “Ministerio de Ciencia e Innovación” (SAF2013-42850-R and SAF2016-75988-R) “Comunidad de Madrid (S2017/BMD-3671. INFLAMUNE-CM), Fondo de Investigaciones Sanitarias” (BIOIMID) to MF and Institutional grants from “Fundación Ramón Areces” and “Banco de Santander”. KS was the recipient of a Spanish Association Against Cancer Oncology Investigator grant (AECC AIO).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Agundez, J. A., Blanca, M., Cornejo-Garcia, J. A., Garcia-Martin, E. (2015). Pharmacogenomics of cyclooxygenases. Pharmacogenomics 16, 501–522. doi: 10.2217/pgs.15.6
Alfranca, A., Iniguez, M. A., Fresno, M., Redondo, J. M. (2006). Prostanoid signal transduction and gene expression in the endothelium: role in cardiovascular diseases. Cardiovasc. Res. 70, 446–456. doi: 10.1016/j.cardiores.2005.12.020
Arends, J. W. (2000). Molecular interactions in the Vogelstein model of colorectal carcinoma. J. Pathol. 190, 412–416. doi: 10.1002/(SICI)1096-9896(200003)190:4<412::AID-PATH533>3.0.CO;2-P
Backlund, M. G., Mann, J. R., Holla, V. R., Buchanan, F. G., Tai, H. H., Musiek, E. S., et al. (2005). 15-Hydroxyprostaglandin dehydrogenase is down-regulated in colorectal cancer. J. Biol. Chem. 280, 3217–3223. doi: 10.1074/jbc.M411221200
Bengi, G., Keles, D., Topalak, Ö., Yalçin, M., Kiyak, R., Oktay, G. (2015). Expressions of TIMP-1, COX-2 and MMP-7 in Colon Polyp and Colon Cancer. Euroasian J. Hepato-gastroenterol. 5, 74–79. doi: 10.5005/jp-journals-10018-1138
Benoit, V., De Moraes, E., Dar, N. A., Taranchon, E., Bours, V., Hautefeuille, A., et al. (2006). Transcriptional activation of cyclooxygenase-2 by tumor suppressor p53 requires nuclear factor-kappaB. Oncogene 25, 5708–5718. doi: 10.1038/sj.onc.1209579
Bertagnolli, M. M. (2007). Chemoprevention of colorectal cancer with cyclooxygenase-2 inhibitors: two steps forward, one step back. Lancet Oncol. 8, 439–443. doi: 10.1016/S1470-2045(07)70139-0
Bi, Z., Dong, L.-D., Gu, X.-M. (2013). Clinical significance of MMP-7 and PTEN expression in colorectal cancer. Hepato-gastroenterology 60, 32–36. doi: 10.5754/hge12537
Brand, L., Munding, J., Pox, C. P., Ziebarth, W., Reiser, M., Hüppe, D., et al. (2013). ß-Catenin, Cox-2 and p53 immunostaining in colorectal adenomas to predict recurrence after endoscopic polypectomy. Int. J. Colorectal Dis. 28, 1091–1098. doi: 10.1007/s00384-013-1667-z
Breyer, R. M., Bagdassarian, C. K., Myers, S. A., Breyer, M. D. (2001). Prostanoid receptors: subtypes and signaling. Annu. Rev. Pharmacol. Toxicol. 41, 661–690. doi: 10.1146/annurev.pharmtox.41.1.661
Brunschwig, E. B., Wilson, K., Mack, D., Dawson, D., Lawrence, E., Willson, J. K., et al. (2003). PMEPA1, a transforming growth factor-beta-induced marker of terminal colonocyte differentiation whose expression is maintained in primary and metastatic colon cancer. Cancer Res. 63, 1568–1575.
Cassano, G., Gasparre, G., Susca, F., Lippe, C., Guanti, G. (2000). Lack of effect by prostaglandin F2alpha on the proliferation of the HCT-8 and HT-29 human adenocarcinoma cell lines. Oncol. Rep. 7, 183–186. doi: 10.3892/or.7.1.183
Cathcart, M. C., Lysaght, J., Pidgeon, G. P. (2011). Eicosanoid signalling pathways in the development and progression of colorectal cancer: novel approaches for prevention/intervention. Cancer Metastasis Rev. 30, 363–385. doi: 10.1007/s10555-011-9324-x
Cathcart, M.-C., O’byrne, K. J., Reynolds, J. V., O’sullivan, J., Pidgeon, G. P. (2012). COX-derived prostanoid pathways in gastrointestinal cancer development and progression: Novel targets for prevention and intervention. Biochim. Biophys. Acta (BBA) - Rev. Cancer 1825, 49–63. doi: 10.1016/j.bbcan.2011.09.004
Chang, K. H., Miller, N., Kheirelseid, E. A., Ingoldsby, H., Hennessy, E., Curran, C. E., et al. (2011). MicroRNA-21 and PDCD4 expression in colorectal cancer. Eur. J. Surg. Oncol. 37, 597–603. doi: 10.1016/j.ejso.2011.04.001
Chang, C. C., Lin, B. R., Wu, T. S., Jeng, Y. M., Kuo, M. L. (2014). Input of microenvironmental regulation on colorectal cancer: role of the CCN family. World J. Gastroenterol. 20, 6826–6831. doi: 10.3748/wjg.v20.i22.6826
Charames, G. S., Bapat, B. (2006). Cyclooxygenase-2 knockdown by RNA interference in colon cancer. Int. J. Oncol. 28, 543–549. doi: 10.3892/ijo.28.2.543
Chen, Z. Y., Tseng, C. C. (2005). 15-deoxy-Delta12,14 prostaglandin J2 up-regulates Kruppel-like factor 4 expression independently of peroxisome proliferator-activated receptor gamma by activating the mitogen-activated protein kinase kinase/extracellular signal-regulated kinase signal transduction pathway in HT-29 colon cancer cells. Mol. Pharmacol. 68, 1203–1213. doi: 10.1124/mol.105.014944
Chen, W. S., Wei, S. J., Liu, J. M., Hsiao, M., Kou-Lin, J., Yang, W. K. (2001). Tumor invasiveness and liver metastasis of colon cancer cells correlated with cyclooxygenase-2 (COX-2) expression and inhibited by a COX-2-selective inhibitor, etodolac. Int. J. Cancer 91, 894–899. doi: 10.1002/1097-0215(200102)9999:9999<894::AID-IJC1146>3.0.CO;2-#
Choi, S. H., Kim, B. G., Robinson, J., Fink, S., Yan, M., Sporn, M. B., et al. (2014). Synthetic triterpenoid induces 15-PGDH expression and suppresses inflammation-driven colon carcinogenesis. J. Clin. Invest. 124, 2472–2482. doi: 10.1172/JCI69672
Citterio, C., Menacho-Márquez, M., García-Escudero, R., Larive, R. M., Barreiro, O., Sánchez-Madrid, F., et al. (2012). The rho exchange factors vav2 and vav3 control a lung metastasis-specific transcriptional program in breast cancer cells. Sci. Signaling 5, ra71–ra71. doi: 10.1126/scisignal.2002962
Collins, D., Hogan, A. M., Skelly, M. M., Baird, A. W., Winter, D. C. (2009). Cyclic AMP-mediated chloride secretion is induced by prostaglandin F2alpha in human isolated colon. Br. J. Pharmacol. 158, 1771–1776. doi: 10.1111/j.1476-5381.2009.00464.x
Corral, R. S., Iniguez, M. A., Duque, J., Lopez-Perez, R., Fresno, M. (2007). Bombesin induces cyclooxygenase-2 expression through the activation of the nuclear factor of activated T cells and enhances cell migration in Caco-2 colon carcinoma cells. Oncogene 26, 958–969. doi: 10.1038/sj.onc.1209856
Cox, D. G., Pontes, C., Guino, E., Navarro, M., Osorio, A., Canzian, F., et al. (2004). Polymorphisms in prostaglandin synthase 2/cyclooxygenase 2 (PTGS2/COX2) and risk of colorectal cancer. Br. J. Cancer 91, 339–343. doi: 10.1038/sj.bjc.6601906
Cui, X., Shen, K., Xie, Z., Liu, T., Zhang, H. (2017). Identification of key genes in colorectal cancer using random walk with restart. Mol. Med. Rep. 15, 867–872. doi: 10.3892/mmr.2016.6058
De Moraes, E., Dar, N. A., De Moura Gallo, C. V., Hainaut, P. (2007). Cross-talks between cyclooxygenase-2 and tumor suppressor protein p53: Balancing life and death during inflammatory stress and carcinogenesis. Int. J. Cancer 121, 929–937. doi: 10.1002/ijc.22899
Dixon, D. A., Blanco, F. F., Bruno, A., Patrignani, P. (2013). Mechanistic aspects of COX-2 expression in colorectal neoplasia. Recent Results Cancer Res. 191, 7–37. doi: 10.1007/978-3-642-30331-9_2
Doherty, G. A., Byrne, S. M., Austin, S. C., Scully, G. M., Sadlier, D. M., Neilan, T. G., et al. (2009). Regulation of the apoptosis-inducing kinase DRAK2 by cyclooxygenase-2 in colorectal cancer. Br. J. Cancer 101, 483–491. doi: 10.1038/sj.bjc.6605144
Duan, X., Gao, Y., Yang, H., Feng, T., Jin, T., Long, Y., et al. (2015). Polymorphisms in the DUSP10 gene are associated with sex-specific colorectal cancer risk in a Han population. Int. J. Clin. Exp. Pathol. 8, 2018–2025.
Duque, J., Fresno, M., Iniguez, M. A. (2005). Expression and function of the nuclear factor of activated T cells in colon carcinoma cells: involvement in the regulation of cyclooxygenase-2. J. Biol. Chem. 280, 8686–8693. doi: 10.1074/jbc.M413076200
Ekambaram, P., Lambiv, W., Cazzolli, R., Ashton, A. W., Honn, K. V. (2011). The thromboxane synthase and receptor signaling pathway in cancer: an emerging paradigm in cancer progression and metastasis. Cancer Metastasis Rev. 30, 397–408. doi: 10.1007/s10555-011-9297-9
Fang, Y. J., Lu, Z. H., Wang, G. Q., Pan, Z. Z., Zhou, Z. W., Yun, J. P., et al. (2009). Elevated expressions of MMP7, TROP2, and survivin are associated with survival, disease recurrence, and liver metastasis of colon cancer. Int. J. Colorectal Dis. 24, 875–884. doi: 10.1007/s00384-009-0725-z
Fearon, E. R., Vogelstein, B. (1990). A genetic model for colorectal tumorigenesis. Cell 61, 759–767. doi: 10.1016/0092-8674(90)90186-I
Fitzgerald, G. A., Patrono, C. (2001). The coxibs, selective inhibitors of cyclooxygenase-2. N Engl. J. Med. 345, 433–442. doi: 10.1056/NEJM200108093450607
Fujino, H., Regan, J. W. (2003). Prostaglandin F(2alpha) stimulation of cyclooxygenase-2 promoter activity by the FP(B) prostanoid receptor. Eur. J. Pharmacol. 465, 39–41. doi: 10.1016/S0014-2999(03)01485-7
Garavito, R. M., Malkowski, M. G., Dewitt, D. L. (2002). The structures of prostaglandin endoperoxide H synthases-1 and -2. Prostaglandins Other Lipid Mediat. 68-69, 129–152. doi: 10.1016/S0090-6980(02)00026-6
Ghaleb, A. M., Yang, V. W. (2017). Krüppel-like factor 4 (KLF4): What we currently know. Gene 611, 27–37. doi: 10.1016/j.gene.2017.02.025
Globocan (2018). https://gco.iarc.fr/today/data/factsheets/cancers/10_8_9-Colorectum-fact-sheet.pdf.
Greenhough, A., Smartt, H. J., Moore, A. E., Roberts, H. R., Williams, A. C., Paraskeva, C., et al. (2009). The COX-2/PGE2 pathway: key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis 30, 377–386. doi: 10.1093/carcin/bgp014
Gröschl, B., Bettstetter, M., Giedl, C., Woenckhaus, M., Edmonston, T., Hofstädter, F., et al. (2013). Expression of the MAP kinase phosphatase DUSP4 is associated with microsatellite instability in colorectal cancer (CRC) and causes increased cell proliferation. Int. J. Cancer 132, 1537–1546. doi: 10.1002/ijc.27834
Guo, K., Jin, F. (2015). NFAT5 promotes proliferation and migration of lung adenocarcinoma cells in part through regulating AQP5 expression. Biochem. Biophys. Res. Commun. 465, 644–649. doi: 10.1016/j.bbrc.2015.08.078
Gustafsson, A., Hansson, E., Kressner, U., Nordgren, S., Andersson, M., Lonnroth, C., et al. (2007). Prostanoid receptor expression in colorectal cancer related to tumor stage, differentiation and progression. Acta Oncol. 46, 1107–1112. doi: 10.1080/02841860701403061
Han, J. A., Kim, J. I., Ongusaha, P. P., Hwang, D. H., Ballou, L. R., Mahale, A., et al. (2002). P53-mediated induction of Cox-2 counteracts p53- or genotoxic stress-induced apoptosis. EMBO J. 21, 5635–5644. doi: 10.1093/emboj/cdf591
Han, S., Bui, N. T., Ho, M. T., Kim, Y. M., Cho, M., Shin, D. B. (2016). Dexamethasone Inhibits TGF-β1-Induced Cell Migration by Regulating the ERK and AKT Pathways in Human Colon Cancer Cells Via CYR61. Cancer Res. Treat 48, 1141–1153. doi: 10.4143/crt.2015.209
Hara, S., Kamei, D., Sasaki, Y., Tanemoto, A., Nakatani, Y., Murakami, M. (2010). Prostaglandin E synthases: Understanding their pathophysiological roles through mouse genetic models. Biochimie 92, 651–659. doi: 10.1016/j.biochi.2010.02.007
Hennequart, M., Pilotte, L., Cane, S., Hoffmann, D., Stroobant, V., Plaen, E., et al. (2017). Constitutive IDO1 Expression in Human Tumors Is Driven by Cyclooxygenase-2 and Mediates Intrinsic Immune Resistance. Cancer Immunol. Res. 5, 695–709. doi: 10.1158/2326-6066.CIR-16-0400
Hommo, T., Pesu, M., Moilanen, E., Korhonen, R. (2015). Regulation of Inflammatory Cytokine Production by MKP-5 in Macrophages. Basic Clin. Pharmacol. Toxicol. 117, 96–104. doi: 10.1111/bcpt.12380
Horiuchi, A., Iinuma, H., Akahane, T., Shimada, R., Watanabe, T. (2012). Prognostic significance of PDCD4 expression and association with microRNA-21 in each Dukes’ stage of colorectal cancer patients. Oncol. Rep. 27, 1384–1392. doi: 10.3892/or.2012.1648
Hu, R., Zuo, Y., Zuo, L., Liu, C., Zhang, S., Wu, Q., et al. (2011). KLF4 Expression Correlates with the Degree of Differentiation in Colorectal Cancer. Gut liver 5, 154–159. doi: 10.5009/gnl.2011.5.2.154
Humphries, H. N., Wickremesekera, S. K., Marsh, R. W., Brasch, H. D., Mehrotra, S., Tan, S. T., et al. (2018). Characterization of Cancer Stem Cells in Colon Adenocarcinoma Metastasis to the Liver. Front. Surg. 4, 76–76. doi: 10.3389/fsurg.2017.00076
Ichimanda, M., Hijiya, N., Tsukamoto, Y., Uchida, T., Nakada, C., Akagi, T., et al. (2018). Downregulation of dual-specificity phosphatase 4 enhances cell proliferation and invasiveness in colorectal carcinomas. Cancer Sci. 109, 250–258. doi: 10.1111/cas.13444
Imai, K., Yamamoto, H. (2008). Carcinogenesis and microsatellite instability: the interrelationship between genetics and epigenetics. Carcinogenesis 29, 673–680. doi: 10.1093/carcin/bgm228
Iniguez, M. A., Rodriguez, A., Volpert, O. V., Fresno, M., Redondo, J. M. (2003). Cyclooxygenase-2: a therapeutic target in angiogenesis. Trends Mol. Med. 9, 73–78. doi: 10.1016/S1471-4914(02)00011-4
Iniguez, M. A., Cacheiro-Llaguno, C., Cuesta, N., Diaz-Munoz, M. D., Fresno, M. (2008). Prostanoid function and cardiovascular disease. Arch. Physiol. Biochem. 114, 201–209. doi: 10.1080/13813450802180882
Jacoby, R. F., Seibert, K., Cole, C. E., Kelloff, G., Lubet, R. A. (2000). The cyclooxygenase-2 inhibitor celecoxib is a potent preventive and therapeutic agent in the min mouse model of adenomatous polyposis. Cancer Res. 60, 5040–5044.
Jauliac, S., López-Rodriguez, C., Shaw, L. M., Brown, L. F., Rao, A., Toker, A. (2002). The role of NFAT transcription factors in integrin-mediated carcinoma invasion. Nat. Cell Biol. 4, 540–544. doi: 10.1038/ncb816
Jeong, D., Heo, S., Sung Ahn, T., Lee, S., Park, S., Kim, H., et al. (2014). Cyr61 expression is associated with prognosis in patients with colorectal cancer. BMC Cancer 14, 164–164. doi: 10.1186/1471-2407-14-164
Jimenez-Martinez, M., Ostale, C. M., Van Der Burg, L. R., Galan-Martinez, J., Hardwick, J. C. H., Lopez-Perez, R., et al. (2019a). DUSP10 Is a Regulator of YAP1 Activity Promoting Cell Proliferation and Colorectal Cancer Progression. Cancers (Basel) 11 (11), 1767. doi: 10.3390/cancers11111767
Jimenez-Martinez, M., Stamatakis, K., Fresno, M. (2019b). The Dual-Specificity Phosphatase 10 (DUSP10): Its Role in Cancer, Inflammation, and Immunity. Int. J. Mol. Sci. 20 (7), 1626. doi: 10.3390/ijms20071626
Jimenez-Segovia, A., Mota, A., Rojo-Sebastian, A., Barrocal, B., Rynne-Vidal, A., Garcia-Bermejo, M. L., et al. (2019). Prostaglandin F2alpha-induced Prostate Transmembrane Protein, Androgen Induced 1 mediates ovarian cancer progression increasing epithelial plasticity. Neoplasia 21, 1073–1084. doi: 10.1016/j.neo.2019.10.001
Kamei, D., Murakami, M., Nakatani, Y., Ishikawa, Y., Ishii, T., Kudo, I. (2003). Potential role of microsomal prostaglandin E synthase-1 in tumorigenesis. J. Biol. Chem. 278, 19396–19405. doi: 10.1074/jbc.M213290200
Kang, X., Li, M., Zhu, H., Lu, X., Miao, J., Du, S., et al. (2017). DUSP4 promotes doxorubicin resistance in gastric cancer through epithelial-mesenchymal transition. Oncotarget 8, 94028–94039. doi: 10.18632/oncotarget.21522
Kapoor, S. (2013). TROP2 expression and its evolving role in tumor pathogenesis in systemic tumors. Tumour. Biol. 34, 1967–1968. doi: 10.1007/s13277-012-0586-x
Karpisheh, V., Nikkhoo, A., Hojjat-Farsangi, M., Namdar, A., Azizi, G., Ghalamfarsa, G., et al. (2019). Prostaglandin E2 as a potent therapeutic target for treatment of colon cancer. Prostaglandins Other Lipid Mediat. 144, 106338. doi: 10.1016/j.prostaglandins.2019.106338
Kawamori, T., Kitamura, T., Watanabe, K., Uchiya, N., Maruyama, T., Narumiya, S., et al. (2005). Prostaglandin E receptor subtype EP(1) deficiency inhibits colon cancer development. Carcinogenesis 26, 353–357. doi: 10.1093/carcin/bgh322
Keum, N., Giovannucci, E. (2019). Global burden of colorectal cancer: emerging trends, risk factors and prevention strategies. Nat. Rev. Gastroenterol. Hepatol. 16, 713–732. doi: 10.1038/s41575-019-0189-8
Kim, K.-H., Yeo, S.-G., Kim, W. K., Kim, D. Y., Yeo, H. Y., Hong, J. P., et al. (2012). Up-regulated expression of l-caldesmon associated with malignancy of colorectal cancer. BMC Cancer 12, 601–601. doi: 10.1186/1471-2407-12-601
Köhler, C. (2011). Histochemical localization of caldesmon isoforms in colon adenocarcinoma and lymph node metastases. Virchows Archiv.: an Int. J. Pathol. 459, 81–89. doi: 10.1007/s00428-011-1091-0
Komoto, J., Yamada, T., Watanabe, K., Woodward, D. F., Takusagawa, F. (2006). Prostaglandin F2alpha formation from prostaglandin H2 by prostaglandin F synthase (PGFS): crystal structure of PGFS containing bimatoprost. Biochemistry 45, 1987–1996. doi: 10.1021/bi051861t
Kuipers, E. J., Grady, W. M., Lieberman, D., Seufferlein, T., Sung, J. J., Boelens, P. G., et al. (2015). Colorectal cancer. Nat. Rev. Dis. Primers 1, 15065. doi: 10.1038/nrdp.2015.65
Lee, T. K., Murthy, S. R. K., Cawley, N. X., Dhanvantari, S., Hewitt, S. M., Lou, H., et al. (2011). An N-terminal truncated carboxypeptidase E splice isoform induces tumor growth and is a biomarker for predicting future metastasis in human cancers. J. Clin. Invest. 121, 880–892. doi: 10.1172/JCI40433
Lee, H.-Y., Ahn, J. B., Rha, S. Y., Chung, H. C., Park, K. H., Kim, T. S., et al. (2014). High KLF4 level in normal tissue predicts poor survival in colorectal cancer patients. World J. Surg. Oncol. 12, 232–232. doi: 10.1186/1477-7819-12-232
Leng, Z., Tao, K., Xia, Q., Tan, J., Yue, Z., Chen, J., et al. (2013). Krüppel-like factor 4 acts as an oncogene in colon cancer stem cell-enriched spheroid cells. PloS One 8, e56082. doi: 10.1371/journal.pone.0056082
Li, Y., Bavarva, J. H., Wang, Z., Guo, J., Qian, C., Thibodeau, S. N., et al. (2011). HEF1, a novel target of Wnt signaling, promotes colonic cell migration and cancer progression. Oncogene 30, 2633–2643. doi: 10.1038/onc.2010.632
Long, J., Yin, Y., Guo, H., Li, S., Sun, Y., Zeng, C., et al. (2019). The mechanisms and clinical significance of PDCD4 in colorectal cancer. Gene 680, 59–64. doi: 10.1016/j.gene.2018.09.034
Low, H. B., Zhang, Y. (2016). Regulatory Roles of MAPK Phosphatases in Cancer. Immune Netw. 16, 85–98. doi: 10.4110/in.2016.16.2.85
Lu, Y., Shi, C., Qiu, S., Fan, Z. (2016). Identification and validation of COX-2 as a co-target for overcoming cetuximab resistance in colorectal cancer cells. Oncotarget 7, 64766–64777. doi: 10.18632/oncotarget.8649
Ma, X., Aoki, T., Tsuruyama, T., Narumiya, S. (2015). Definition of Prostaglandin E2-EP2 Signals in the Colon Tumor Microenvironment That Amplify Inflammation and Tumor Growth. Cancer Res. 75, 2822–2832. doi: 10.1158/0008-5472.CAN-15-0125
Ma, H., Brosens, L. A. A., Offerhaus, G. J. A., Giardiello, F. M., De Leng, W. W. J., Montgomery, E. A. (2018). Pathology and genetics of hereditary colorectal cancer. Pathology 50, 49–59. doi: 10.1016/j.pathol.2017.09.004
Mcdougall, A. R. A., Tolcos, M., Hooper, S. B., Cole, T. J., Wallace, M. J. (2015). Trop2: from development to disease. Dev. Dynamics 244, 99–109. doi: 10.1002/dvdy.24242
Moran, A., Ortega, P., De Juan, C., Fernandez-Marcelo, T., Frias, C., Sanchez-Pernaute, A., et al. (2010). Differential colorectal carcinogenesis: Molecular basis and clinical relevance. World J. Gastrointest Oncol. 2, 151–158. doi: 10.4251/wjgo.v2.i3.151
Mutoh, M., Watanabe, K., Kitamura, T., Shoji, Y., Takahashi, M., Kawamori, T., et al. (2002). Involvement of prostaglandin E receptor subtype EP(4) in colon carcinogenesis. Cancer Res. 62, 28–32.
Myung, S. J., Rerko, R. M., Yan, M., Platzer, P., Guda, K., Dotson, A., et al. (2006). 15-Hydroxyprostaglandin dehydrogenase is an in vivo suppressor of colon tumorigenesis. Proc. Natl. Acad. Sci. U. S. A 103, 12098–12102. doi: 10.1073/pnas.0603235103
Nalluri, S. M., O’connor, J. W., Virgi, G. A., Stewart, S. E., Ye, D., Gomez, E. W. (2018). TGFbeta1-induced expression of caldesmon mediates epithelial-mesenchymal transition. Cytoskeleton (Hoboken) 75, 201–212. doi: 10.1002/cm.21437
Nimmrich, I., Erdmann, S., Melchers, U., Finke, U., Hentsch, S., Moyer, M. P., et al. (2000). Seven genes that are differentially transcribed in colorectal tumor cell lines. Cancer Lett. 160, 37–43. doi: 10.1016/S0304-3835(00)00553-X
Nugent, K. P., Spigelman, A. D., Phillips, R. K. (1996). Tissue prostaglandin levels in familial adenomatous polyposis patients treated with sulindac. Dis. Colon Rectum 39, 659–662. doi: 10.1007/BF02056946
O’mahony, C. A., Beauchamp, R. D., Albo, D., Tsujii, M., Sheng, H. M., Shao, J., et al. (1999). Cyclooxygenase-2 alters transforming growth factor-beta 1 response during intestinal tumorigenesis. Surgery 126, 364–370. doi: 10.1016/S0039-6060(99)70178-9
O’neill, G. M., Seo, S., Serebriiskii, I. G., Lessin, S. R., Golemis, E. A. (2007). A new central scaffold for metastasis: parsing HEF1/Cas-L/NEDD9. Cancer Res. 67, 8975–8979. doi: 10.1158/0008-5472.CAN-07-1328
Oshima, M., Dinchuk, J. E., Kargman, S. L., Oshima, H., Hancock, B., Kwong, E., et al. (1996). Suppression of intestinal polyposis in Apc delta716 knockout mice by inhibition of cyclooxygenase 2 (COX-2). Cell 87, 803–809. doi: 10.1016/S0092-8674(00)81988-1
Pai, R., Soreghan, B., Szabo, I. L., Pavelka, M., Baatar, D., Tarnawski, A. S. (2002). Prostaglandin E2 transactivates EGF receptor: a novel mechanism for promoting colon cancer growth and gastrointestinal hypertrophy. Nat. Med. 8, 289–293. doi: 10.1038/nm0302-289
Park, H., Jeon, T., Chien, P., Park, S., Oh, S., Kim, S., et al. (2014). Discovery of Novel DUSP4 Inhibitors through the Virtual Screening with Docking Simulations. Bull. Korean Chem. Soc. 35, 2655–2659. doi: 10.5012/bkcs.2014.35.9.2655
Park, S.-M., Hwang, C. Y., Cho, S.-H., Lee, D., Gong, J.-R., Lee, S., et al. (2019). Systems analysis identifies potential target genes to overcome cetuximab resistance in colorectal cancer cells. FEBS J. 286, 1305–1318. doi: 10.1111/febs.14773
Patrignani, P., Patrono, C. (2016). Aspirin and Cancer. J. Am. Coll. Cardiol. 68, 967–976. doi: 10.1016/j.jacc.2016.05.083
Peacock, O., Lee, A. C., Cameron, F., Tarbox, R., Vafadar-Isfahani, N., Tufarelli, C., et al. (2014). Inflammation and MiR-21 pathways functionally interact to downregulate PDCD4 in colorectal cancer. PloS One 9, e110267. doi: 10.1371/journal.pone.0110267
Pfeffer, S. R., Yang, C. H., Pfeffer, L. M. (2015). The Role of miR-21 in Cancer. Drug Dev. Res. 76, 270–277. doi: 10.1002/ddr.21257
Piazuelo, E., Lanas, A. (2015). NSAIDS and gastrointestinal cancer. Prostaglandins Lipid Mediat. 120, 91–96. doi: 10.1016/j.prostaglandins.2015.06.001
Porter, R. M., Holme, T. C., Newman, E. L., Hopwood, D., Wilkinson, J. M., Cuschieri, A. (1993). Monoclonal antibodies to cytoskeletal proteins: an immunohistochemical investigation of human colon cancer. J. Pathol. 170, 435–440. doi: 10.1002/path.1711700406
Pugh, S., Thomas, G. A. (1994). Patients with adenomatous polyps and carcinomas have increased colonic mucosal prostaglandin E2. Gut 35, 675–678. doi: 10.1136/gut.35.5.675
Qin, J. J., Nag, S., Wang, W., Zhou, J., Zhang, W. D., Wang, H., et al. (2014). NFAT as cancer target: mission possible? Biochim. Biophys. Acta 1846, 297–311. doi: 10.1016/j.bbcan.2014.07.009
Qualtrough, D., Kaidi, A., Chell, S., Jabbour, H. N., Williams, A. C., Paraskeva, C. (2007). Prostaglandin F(2alpha) stimulates motility and invasion in colorectal tumor cells. Int. J. Cancer 121, 734–740. doi: 10.1002/ijc.22755
Rae, F. K., Hooper, J. D., Nicol, D. L., Clements, J. A. (2001). Characterization of a novel gene, STAG1/PMEPA1, upregulated in renal cell carcinoma and other solid tumors. Mol. Carcinog 32, 44–53. doi: 10.1002/mc.1063
Regan, J. W. (2003). EP2 and EP4 prostanoid receptor signaling. Life Sci. 74, 143–153. doi: 10.1016/j.lfs.2003.09.031
Reichling, T., Goss, K. H., Carson, D. J., Holdcraft, R. W., Ley-Ebert, C., Witte, D., et al. (2005). Transcriptional profiles of intestinal tumors in Apc(Min) mice are unique from those of embryonic intestine and identify novel gene targets dysregulated in human colorectal tumors. Cancer Res. 65, 166–176.
Rizzo, M. T. (2011). Cyclooxygenase-2 in oncogenesis. Clin. Chim Acta 412, 671–687. doi: 10.1016/j.cca.2010.12.026
Roberts, J. A., Waters, L., Ro, J. Y., Zhai, Q. J. (2014). Smoothelin and caldesmon are reliable markers for distinguishing muscularis propria from desmoplasia: a critical distinction for accurate staging colorectal adenocarcinoma. Int. J. Clin. Exp. Pathol. 7, 792–796.
Rothwell, P. M. (2013). Aspirin in prevention of sporadic colorectal cancer: current clinical evidence and overall balance of risks and benefits. Recent Results Cancer Res. 191, 121–142. doi: 10.1007/978-3-642-30331-9_7
Saigusa, S., Inoue, Y., Tanaka, K., Toiyama, Y., Okugawa, Y., Shimura, T., et al. (2013). Decreased expression of DUSP4 is associated with liver and lung metastases in colorectal cancer. Med. Oncol. (Northwood London Eng.) 30, 620–620. doi: 10.1007/s12032-013-0620-x
Sancho, E., Batlle, E., Clevers, H. (2004). Signaling pathways in intestinal development and cancer. Annu. Rev. Cell Dev. Biol. 20, 695–723. doi: 10.1146/annurev.cellbio.20.010403.092805
Sasaki, Y., Kamei, D., Ishikawa, Y., Ishii, T., Uematsu, S., Akira, S., et al. (2012). Microsomal prostaglandin E synthase-1 is involved in multiple steps of colon carcinogenesis. Oncogene 31, 2943–2952. doi: 10.1038/onc.2011.472
Sasaki, Y., Nakatani, Y., Hara, S. (2015). Role of microsomal prostaglandin E synthase-1 (mPGES-1)-derived prostaglandin E2 in colon carcinogenesis. Prostaglandins Other Lipid Mediat. 121, 42–45. doi: 10.1016/j.prostaglandins.2015.06.006
Seo, T., Tatsuguchi, A., Shinji, S., Yonezawa, M., Mitsui, K., Tanaka, S., et al. (2009). Microsomal prostaglandin E synthase protein levels correlate with prognosis in colorectal cancer patients. Virchows Arch. 454, 667–676. doi: 10.1007/s00428-009-0777-z
Seternes, O. M., Kidger, A. M., Keyse, S. M. (2019). Dual-specificity MAP kinase phosphatases in health and disease. Biochim. Biophys. Acta Mol. Cell Res. 1866, 124–143. doi: 10.1016/j.bbamcr.2018.09.002
Shao, J., Yang, V. W., Sheng, H. (2008). Prostaglandin E2 and Krüppel-like transcription factors synergistically induce the expression of decay-accelerating factor in intestinal epithelial cells. Immunology 125, 397–407. doi: 10.1111/j.1365-2567.2008.02847.x
Shvartsur, A., Bonavida, B. (2015). Trop2 and its overexpression in cancers: regulation and clinical/therapeutic implications. Genes Cancer 6, 84–105. doi: 10.18632/genesandcancer.40
Singha, P. K., Yeh, I. T., Venkatachalam, M. A., Saikumar, P. (2010). Transforming growth factor-beta (TGF-beta)-inducible gene TMEPAI converts TGF-beta from a tumor suppressor to a tumor promoter in breast cancer. Cancer Res. 70, 6377–6383. doi: 10.1158/0008-5472.CAN-10-1180
Sinicrope, F. A. (2006). Targeting cyclooxygenase-2 for prevention and therapy of colorectal cancer. Mol. Carcinog 45, 447–454. doi: 10.1002/mc.20232
Slattery, M. L., Lundgreen, A., Bondurant, K. L., Wolff, R. K. (2011). Tumor necrosis factor-related genes and colon and rectal cancer. Int. J. Mol. Epidemiol. Genet. 2, 328–338.
Song, Y. F., Xu, Z. B., Zhu, X. J., Tao, X., Liu, J. L., Gao, F. L., et al. (2017). Serum Cyr61 as a potential biomarker for diagnosis of colorectal cancer. Clin. Trans. Oncol. 19, 519–524. doi: 10.1007/s12094-016-1560-7
Sreeramkumar, V., Fresno, M., Cuesta, N. (2012). Prostaglandin E(2) and T cells: friends or foes? Immunol. Cell Biol. 90, 579–586. doi: 10.1038/icb.2011.75
Stamatakis, K., Jimenez-Martinez, M., Jimenez-Segovia, A., Chico-Calero, I., Conde, E., Galan-Martinez, J., et al. (2015). Prostaglandins induce early growth response 1 transcription factor mediated microsomal prostaglandin E2 synthase up-regulation for colorectal cancer progression. Oncotarget 6, 39941–39959. doi: 10.18632/oncotarget.5402
Starodub, A. N., Ocean, A. J., Shah, M. A., Guarino, M. J., Picozzi, V. J., Jr., Vahdat, L. T., et al. (2015). First-in-Human Trial of a Novel Anti-Trop-2 Antibody-SN-38 Conjugate, Sacituzumab Govitecan, for the Treatment of Diverse Metastatic Solid Tumors. Clin. Cancer Res. 21, 3870–3878. doi: 10.1158/1078-0432.CCR-14-3321
Strillacci, A., Griffoni, C., Spisni, E., Manara, M. C., Tomasi, V. (2006). RNA interference as a key to knockdown overexpressed cyclooxygenase-2 gene in tumour cells. Br. J. Cancer 94, 1300–1310. doi: 10.1038/sj.bjc.6603094
Subbaramaiah, K., Dannenberg, A. J. (2003). Cyclooxygenase 2: a molecular target for cancer prevention and treatment. Trends Pharmacol. Sci. 24, 96–102. doi: 10.1016/S0165-6147(02)00043-3
Sun, Y., Tang, X. M., Half, E., Kuo, M. T., Sinicrope, F. A. (2002). Cyclooxygenase-2 overexpression reduces apoptotic susceptibility by inhibiting the cytochrome c-dependent apoptotic pathway in human colon cancer cells. Cancer Res. 62, 6323–6328.
Swamy, M. V., Herzog, C. R., Rao, C. V. (2003). Inhibition of COX-2 in colon cancer cell lines by celecoxib increases the nuclear localization of active p53. Cancer Res. 63, 5239–5242.
Takafuji, V., Cosme, R., Lublin, D., Lynch, K., Roche, J. K. (2000). Prostanoid receptors in intestinal epithelium: selective expression, function, and change with inflammation. Prostaglandins Leukot. Essent. Fatty Acids 63, 223–235. doi: 10.1054/plef.2000.0144
Tanioka, T., Nakatani, Y., Semmyo, N., Murakami, M., Kudo, I. (2000). Molecular identification of cytosolic prostaglandin E2 synthase that is functionally coupled with cyclooxygenase-1 in immediate prostaglandin E2 biosynthesis. J. Biol. Chem. 275, 32775–32782. doi: 10.1074/jbc.M003504200
Thun, M. J., Jacobs, E. J., Patrono, C. (2012). The role of aspirin in cancer prevention. Nat. Rev. Clin. Oncol. 9, 259–267. doi: 10.1038/nrclinonc.2011.199
Tikhmyanova, N., Little, J. L., Golemis, E. A. (2010). CAS proteins in normal and pathological cell growth control. Cell Mol. Life Sci. 67, 1025–1048. doi: 10.1007/s00018-009-0213-1
Tsujii, M., Dubois, R. N. (1995). Alterations in cellular adhesion and apoptosis in epithelial cells overexpressing prostaglandin endoperoxide synthase 2. Cell 83, 493–501. doi: 10.1016/0092-8674(95)90127-2
Tsujii, M., Kawano, S., Dubois, R. N. (1997). Cyclooxygenase-2 expression in human colon cancer cells increases metastatic potential. Proc. Natl. Acad. Sci. U. S. A. 94, 3336–3340. doi: 10.1073/pnas.94.7.3336
Wang, C. L., Coluccio, L. M. (2010). New insights into the regulation of the actin cytoskeleton by tropomyosin. Int. Rev. Cell Mol. Biol. 281, 91–128. doi: 10.1016/S1937-6448(10)81003-2
Wang, D., Dubois, R. N. (2010). The role of COX-2 in intestinal inflammation and colorectal cancer. Oncogene 29, 781–788. doi: 10.1038/onc.2009.421
Wang, W. S., Chen, P. M., Su, Y. (2006). Colorectal carcinoma: from tumorigenesis to treatment. Cell. Mol. Life Sci.: CMLS 63, 663–671. doi: 10.1007/s00018-005-5425-4
Wang, J., Day, R., Dong, Y., Weintraub, S. J., Michel, L. (2008). Identification of Trop-2 as an oncogene and an attractive therapeutic target in colon cancers. Mol. Cancer Ther. 7, 280–285. doi: 10.1158/1535-7163.MCT-07-2003
Wang, Q., Zhu, J., Wang, Y. W., Dai, Y., Wang, Y. L., Wang, C., et al. (2017). Tumor suppressor Pdcd4 attenuates Sin1 translation to inhibit invasion in colon carcinoma. Oncogene 36, 6225–6234. doi: 10.1038/onc.2017.228
Wang, Y., Wang, W., Sanidad, K. Z., Shih, P. A., Zhao, X., Zhang, G. (2018). Eicosanoid signaling in carcinogenesis of colorectal cancer. Cancer Metastasis Rev. 37, 257–267. doi: 10.1007/s10555-018-9739-8
Watanabe, Y., Itoh, S., Goto, T., Ohnishi, E., Inamitsu, M., Itoh, F., et al. (2010). TMEPAI, a transmembrane TGF-beta-inducible protein, sequesters Smad proteins from active participation in TGF-beta signaling. Mol. Cell 37, 123–134. doi: 10.1016/j.molcel.2009.10.028
Watson, A. J. M., Collins, P. D. (2011). Colon cancer: a civilization disorder. Digest. Dis. (Basel Switzerland) 29, 222–228. doi: 10.1159/000323926
Williams, C. S., Dubois, R. N. (1996). Prostaglandin endoperoxide synthase: why two isoforms? Am. J. Physiol. 270, G393–G400. doi: 10.1152/ajpgi.1996.270.3.G393
Williams, C. S., Luongo, C., Radhika, A., Zhang, T., Lamps, L. W., Nanney, L. B., et al. (1996). Elevated cyclooxygenase-2 levels in Min mouse adenomas. Gastroenterology 111, 1134–1140. doi: 10.1016/S0016-5085(96)70083-5
Xia, D., Holla, V. R., Wang, D., Menter, D. G., Dubois, R. N. (2010). HEF1 is a crucial mediator of the proliferative effects of prostaglandin E(2) on colon cancer cells. Cancer Res. 70, 824–831. doi: 10.1158/0008-5472.CAN-09-2105
Xie, L., Song, X., Lin, H., Chen, Z., Li, Q., Guo, T., et al. (2019). Aberrant activation of CYR61 enhancers in colorectal cancer development. J. Exp. Clin. Cancer Res.: CR 38, 213–213. doi: 10.1186/s13046-019-1217-9
Xu, W., Chou, C.-L., Israel, D. D., Hutchinson, A. J., Regan, J. W. (2009). PGF(2alpha) stimulates FP prostanoid receptor mediated crosstalk between Ras/Raf signaling and Tcf transcriptional activation. Biochem. Biophys. Res. Commun. 381, 625–629. doi: 10.1016/j.bbrc.2009.02.102
Xu, G., Zhang, M., Zhu, H., Xu, J. (2017). A 15-gene signature for prediction of colon cancer recurrence and prognosis based on SVM. Gene 604, 33–40. doi: 10.1016/j.gene.2016.12.016
Yang, V. W., Shields, J. M., Hamilton, S. R., Spannhake, E. W., Hubbard, W. C., Hylind, L. M., et al. (1998). Size-dependent increase in prostanoid levels in adenomas of patients with familial adenomatous polyposis. Cancer Res. 58, 1750–1753.
Yoshimoto, T., Takahashi, Y., Kinoshita, T., Sakashita, T., Inoue, H., Tanabe, T. (2002). Growth stimulation and epidermal growth factor receptor induction in cyclooxygenase-overexpressing human colon carcinoma cells. Adv. Exp. Med. Biol. 507, 403–407. doi: 10.1007/978-1-4615-0193-0_62
Yoshio, T., Morita, T., Kimura, Y., Tsujii, M., Hayashi, N., Sobue, K. (2007). Caldesmon suppresses cancer cell invasion by regulating podosome/invadopodium formation. FEBS Lett. 581, 3777–3782. doi: 10.1016/j.febslet.2007.06.073
Zelenay, S., Van Der Veen, A. G., Bottcher, J. P., Snelgrove, K. J., Rogers, N., Acton, S. E., et al. (2015). Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell 162, 1257–1270. doi: 10.1016/j.cell.2015.08.015
Zhang, Z., Dubois, R. N. (2001). Detection of differentially expressed genes in human colon carcinoma cells treated with a selective COX-2 inhibitor. Oncogene 20, 4450–4456. doi: 10.1038/sj.onc.1204588
Zhang, L., Wang, X., Lai, C., Zhang, H., Lai, M. (2019). PMEPA1 induces EMT via a non-canonical TGF-β signalling in colorectal cancer. J. Cell. Mol. Med. 23, 3603–3615. doi: 10.1111/jcmm.14261
Zhao, P., Zhang, Z. (2018). TNF-alpha promotes colon cancer cell migration and invasion by upregulating TROP-2. Oncol. Lett. 15, 3820–3827. doi: 10.3892/ol.2018.7735
Zucker, S., Vacirca, J. (2004). Role of matrix metalloproteinases (MMPs) in colorectal cancer. Cancer Metastasis Rev. 23, 101–117. doi: 10.1023/A:1025867130437
Keywords: cyclooxygenase, prostaglandin, Colon cancer, therapy, tumor development, metastasis, effector genes/proteins
Citation: Hidalgo-Estévez AM, Stamatakis K, Jiménez-Martínez M, López-Pérez R and Fresno M (2020) Cyclooxygenase 2-Regulated Genes an Alternative Avenue to the Development of New Therapeutic Drugs for Colorectal Cancer. Front. Pharmacol. 11:533. doi: 10.3389/fphar.2020.00533
Received: 26 January 2020; Accepted: 06 April 2020;
Published: 29 April 2020.
Edited by:
Paola Patrignani, Università degli Studi G. d’Annunzio Chieti e Pescara, ItalyReviewed by:
Melania Dovizio, Università degli Studi G. d’Annunzio Chieti e Pescara, ItalyCopyright © 2020 Hidalgo-Estévez, Stamatakis, Jiménez-Martínez, López-Pérez and Fresno. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manuel Fresno, bWZyZXNub0BjYm0uY3NpYy5lcw==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.