 Zsófia Kohajda1,2†
Zsófia Kohajda1,2† Axel Loewe
Axel Loewe Noémi Tóth
Noémi Tóth András Varró
András Varró Norbert Nagy
Norbert Nagy- 1MTA-SZTE Research Group of Cardiovascular Pharmacology, Hungarian Academy of Sciences, Szeged, Hungary
- 2Department of Pharmacology and Pharmacotherapy, Faculty of Medicine, University of Szeged, Szeged, Hungary
- 3Institute of Biomedical Engineering, Karlsruhe Institute of Technology (KIT), Karlsruhe, Germany
The electrophysiological mechanism of the sinus node automaticity was previously considered exclusively regulated by the so-called “funny current”. However, parallel investigations increasingly emphasized the importance of the Ca2+-homeostasis and Na+/Ca2+ exchanger (NCX). Recently, increasing experimental evidence, as well as insight through mechanistic in silico modeling demonstrates the crucial role of the exchanger in sinus node pacemaking. NCX had a key role in the exciting story of discovery of sinus node pacemaking mechanisms, which recently settled with a consensus on the coupled-clock mechanism after decades of debate. This review focuses on the role of the Na+/Ca2+ exchanger from the early results and concepts to recent advances and attempts to give a balanced summary of the characteristics of the local, spontaneous, and rhythmic Ca2+ releases, the molecular control of the NCX and its role in the fight-or-flight response. Transgenic animal models and pharmacological manipulation of intracellular Ca2+ concentration and/or NCX demonstrate the pivotal function of the exchanger in sinus node automaticity. We also highlight where specific hypotheses regarding NCX function have been derived from computational modeling and require experimental validation. Nonselectivity of NCX inhibitors and the complex interplay of processes involved in Ca2+ handling render the design and interpretation of these experiments challenging.
Introduction
Few areas of cardiac cellular electrophysiology had more intense debate over the years than ‘what makes our hearts beat'—the electrophysiology of the sinus node (SN). In the last two decades, we could witness the struggle of two competing oscillator concepts explaining SN automaticity: in essence, the funny-current (If) and the Ca2+-induced depolarization of the membrane potential (Vm) via Na+/Ca2+ exchanger (NCX, INaCa). Year by year, several studies were published supporting both theories. Thus, the exciting exploration of SN pacemaking mechanisms has been full of debates for over a half century. Numerous mechanisms have been suggested, tested, refuted, accepted, tested, and retested again, updated and after intense point–counterpoint public debates finally settled at a delicate balance known as the coupled-clock mechanism that includes both membrane and sarcoplasmic reticulum (SR) linked contributions. The general field of SN pacemaking (Lakatta et al., 2010; Maltsev et al., 2014; DiFrancesco, 2020) and computational modeling thereof has been excellently reviewed elsewhere (Wilders, 2007; Li et al., 2013). In this review, we focus on the role of NCX function on SN pacemaking and aim to give a balanced view from the very early to recent experimental findings and accompanying computational models.
The SNCs show a characteristic slow diastolic depolarization (DD) starting from the maximal diastolic potential (MDP) and ending at the action potential (AP) threshold of about −40 mV. This threshold is called takeoff potential (Kharche et al., 2011). At the takeoff potential, the course of the AP changes qualitatively from slow DD to the AP upstroke (Figure 1A). If, NCX, and ICaT (T-type Ca2+-current) are mainly active before (at Vm more negative than the takeoff potential), whereas ICaL (L-type Ca2+-current) and IKr (rapid delayed rectifier potassium current) are mainly active thereafter (at more positive Vm), while NCX is active throughout the entire AP cycle. Early studies suggested a major role of a decaying delayed rectifier potassium current (IKr) (Noble, 1962) and the nonselective, cAMP-dependent and hyperpolarization activated current [funny-current, If, (DiFrancesco, 1991)] in the development of DD. This mechanism, governed by transmembrane ion channels with Hodgkin–Huxley kinetics was termed as “membrane-clock” (M-clock). Later studies identified rhythmic, spontaneous locally propagating subsarcolemmal Ca2+ releases (LCR) generated by the SR via ryanodine receptors (RYRs) during the DD (Figure 1B) (Huser et al., 2000; Bogdanov et al., 2001; Monfredi et al., 2013). The LCRs activate the forward mode of the Na+/Ca2+ exchanger generating inward current that contributes to the DD (Bogdanov et al., 2001). Under certain experimental conditions, and at least temporarily, this mechanism can operate independently of the M-clock, therefore it was termed as “Ca2+-clock” (Maltsev et al., 2006). Many of the effects of the LCRs can be represented by their influence on the Ca2+ concentrations in the different compartments of the cell as done in most computational models of SNCs. After intense and long debate, several studies provided evidence that the M-clock and Ca2+-clock are functionally coupled, converging to the concept of a coupled-clock system (Figure 1B). The coupling is based on numerous voltage-, time- and Ca2+-dependent mechanisms (Maltsev and Lakatta, 2009; Lakatta et al., 2010) including an important role of the L-type Ca2+ current that “resets” and “refuels” the Ca2+-clock (Maltsev and Lakatta, 2009) (Figure 1). Both ICaL and NCX have important roles in the two clock-like subsystems, i.e. they contribute to both the membrane voltage-clock and the Ca2+-clock (Figure 1B).
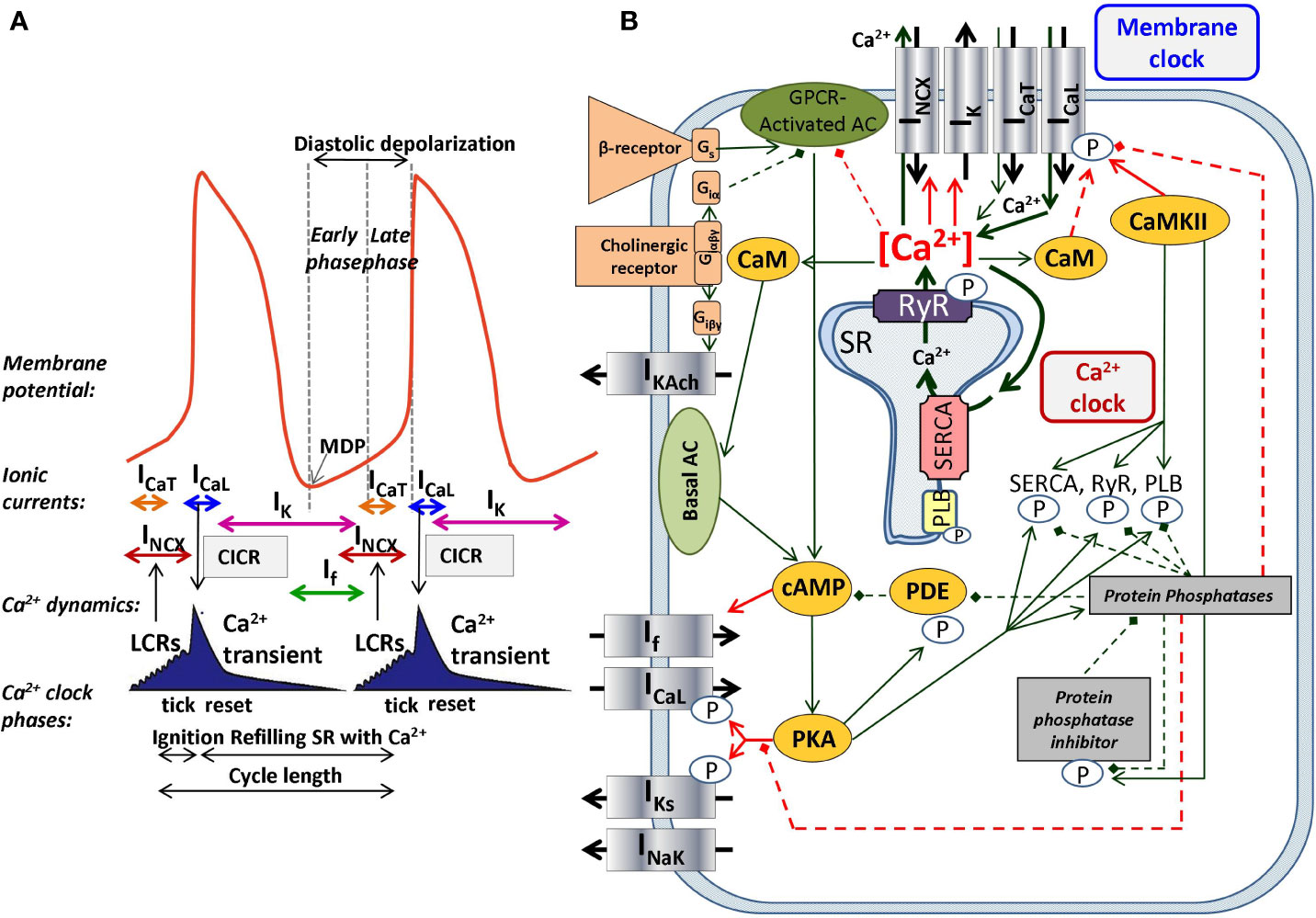
Figure 1 The coupled-clock pacemaker system of the sinoatrial node. Schematic illustration of the currents and functional interactions between the two oscillator subsystem contributing to the SN AP (A). The interplay of the regulatory molecules comprising the coupled-clock pacemaker system is illustrated in (B). Membrane clock (M-clock) of sinoatrial node pacemaker cells comprises the major surface membrane currents: L-type Ca2+ channels (ICaL), T-type Ca2+ channels (ICaT), delayed rectifier K+ channels (IK), hyperpolarization activated funny channels (If), Na+/Ca2+exchanger (NCX), and Na+/K+ ATPase (INaK). Regulatory factors that affect the function of proteins of both clocks are indicated with different colors: molecules with red letters couple the Ca2+ clock to the M-clock; factors in orange regulate both the M-clock and Ca2+ clock via the GPCR pathway. Interrupted lines refer to inhibitory effects. AC, adenylate-cyclase; CaM, calmodulin; cAMP, cyclic adenosine monophosphate; CaMKII, Ca2+/calmodulin-dependent protein kinase II; GPCR, G-protein coupled receptor; PDE, phosphodiesterase; PKA, protein kinase A; PLB, phospholamban; RyR, ryanodine receptor; SERCA, sarcoplasmic reticulum Ca2+ ATPase 2a.
The Role of NCX in the Ca2+ Flux Balance
Na+/Ca2+ exchangers were identified as members of the Ca2+/cation antiporters (CaCA) superfamily, as the exchanger carries three Na+ in exchange for one Ca2+. Thus, the exchanger is electrogenic and affects the transmembrane voltage. The first data regarding the exchange ratio of NCX was published by Reuter and Seitz describing the Na+/Ca2+ exchange as neutral, i.e., changing two Na+ ions for one Ca2+ (Reuter and Seitz, 1968). Later, in squid axon experiments, it became clear that the NCX function is electrogenic, with a suggested stoichiometry of 4:1 (Mullins and Requena, 1979). Chapman and Tunstall (1980) suggested a 3:1 ratio in the heart muscle (Chapman and Tunstall, 1980). Nowadays, the 3:1 Na+/Ca2+ stoichiometry is clearly supported by several experimental studies and generally accepted as the functional exchanger ratio (Bers and Ginsburg, 2007). The electrogenic operation of the Na+/Ca2+ exchanger results in an ion current with one net elementary charge in each transport cycle. The mode in which the exchanger operates is defined by the relationship of Vm and the NCX equilibrium potential (ENCX), where ENCX is determined by the transmembrane Na+ and Ca2+ gradients (ENCX = 3ENa − 2ECa). When the difference between Vm and the equilibrium potential is positive, the exchanger acts in the reverse mode; however Ca2+ extrusion is favored when Vm is more negative than the equilibrium potential (Bers, 2002). Depending on the mode of action (forward vs. reverse), the Na+/Ca2+ exchanger can generate either inward or outward current, it can depolarize or repolarize the cell membrane, respectively.
The activity of the exchanger has complex intra- and extracellular regulatory molecules: Among the intracellular factors, the Ca2+i, ATP, phosphatidylinositol-di-phosphate-2 (PIP2), NO and extracellular factors such as monovalent cations, PKC activators, and agents that induce proteolysis or protein phosphorylation can activate the exchanger (Ballard and Schaffer, 1996; Bers, 2001; Bers, 2008). Other intracellular factors like Na+i, H+, bivalent and trivalent cations, and protein dephosphorylation are inhibitory factors of NCX function (Ballard and Schaffer, 1996; Bers, 2001; Bers, 2008). Modulatory proteins of NCX are PKA, PKC, PP1, and PP2A (Ballard and Schaffer, 1996; Bers, 2001; Bers, 2008). Based on previous experimental results, the modulatory effect of the cAMP-PKA pathway on the NCX function is controversial. Several studies have shown PKA-related activation of the Na+/Ca2+ exchanger with forskolin and isoproterenol (Iwamoto et al., 1995; Iwamoto et al., 1996; Iwamoto et al., 1998); however, other studies do not demonstrate such effects. Latest studies concluded that the NCX function is not modified by PKA activation (Ginsburg and Bers, 2005).
Three mammalian NCX isoforms have been identified: NCX1 is expressed in the heart, kidney, and brain, having the most extended spectrum of expression among the isoforms, NCX2 is present in the brain, and NCX3 is expressed in the brain and skeletal muscle (Nicoll et al., 1990; Reilly and Shugrue, 1992; Kofuji et al., 1994; Li et al., 1994; Nicoll et al., 1996).
Pharmacology of the Na+/Ca2+ Exchanger
Since NCX plays a crucial role under physiological circumstances in the maintenance of Ca2+ flux balance and SN pacemaking as well as in pathological settings, e.g., in arrhythmia generation (delayed afterdepolarizations, Ca2+ overload), selective inhibition of the exchanger became one of the most intensive research areas demanding the development of properly selective agents. In this chapter, we summarize the NCX inhibitors from the early nonspecific substances to novel, highly selective agents.
Nonspecific Inhibitory Substances
Divalent (Co2+, Sr2+, Mg2+, Cd2+, Ba2+, Mn2+) and trivalent (La3+, Nd3+, Tm3+, Y3+) cations have a nonspecific inhibitory effect on the NCX current (Wakabayashi and Goshima, 1981; Trosper and Philipson, 1983) resulting from direct action on the NCX1 isoform or by replacing Ca2+ ions at the transport sites. It is important to note that these cations are nonselective blockers, even the most commonly used Ni2+ has nonspecific effects. Ni2+ is used in concentrations between 1 and 10 mM and inhibits the reverse mode rather than the forward mode of NCX.
“Selective” Benzyloxyphenyl Derivative Inhibitors
Some benzyloxyphenyl derivatives such as KB-R7943 (2-[2-[4-(4-nitrobenzyl-oxy)phenyl]ethyl]isothio-urea-methanesulfonate, carbamidothioic acid) were developed as novel NCX inhibitors with promising blocking potency; however experimental studies reported that KB-R7943 exerts complex nonspecific effects on other transmembrane ion currents including ICaL, INa, IKr. The IC50 values of KB-R7943 are 3.35 ± 0.82 µM on the forward mode and 4.74 ± 0.69 µM on the reverse mode (Birinyi et al., 2005). SEA-0400 (2-(4-(2,5-difluorobenzyloxy)phenoxy)-5-ethoxy aniline) has improved selectivity compared to KB-R7943. However, an ICaL inhibition of approximately 20% was reported (Birinyi et al., 2005). The reverse and forward modes were equally inhibited, having IC50 values of 108 ± 18 nM and 111 ± 43 nM, respectively (Birinyi et al., 2005).
Novel NCX Inhibitors With Increased Selectivity
Due to the lack of selective NCX inhibitors, the exact role of the exchanger in SN pacemaking, cardiac arrhythmogenesis, and Ca2+-handling could not be investigated directly. Efforts were made continuously to develop a potent, selective inhibitor to clarify the role of this highly complex transport system in cardiac function. Recently, two novel NCX blocking compounds were identified: ORM-10103 and ORM-10962. The selectivity of both agents was intensively tested by (Jost et al., 2013) and (Kohajda et al., 2016). While ORM-10103 has improved selectivity compared to previous blockers, it still exhibits minor nonspecific effects of inhibiting IKr at 3 μM. The estimated EC50 values of ORM-10103 at 1 μM concentration for the forward and reverse modes of the NCX are 800 and 960 nM, respectively. In contrast, the EC50 values of ORM-10962 on the inward and outward NCX current are 55 and 67 nM, respectively. The inhibitory effect of ORM-10962 is highly selective with high efficacy on the NCX current and no effect on the L-type Ca2+ current, peak and late sodium current, sodium-potassium pump, and all the repolarizing potassium currents (IKr, IKs, Ito (transient outward potassium current), IK1 (inward rectifier potassium current)) (Kohajda et al., 2016). The effect of selective NCX-inhibition on SN pacemaking by using 1 μM ORM-10962 was also investigated recently (Kohajda et al., 2020).
NCX Activators
Probably the most important activating factor for NCX is Ca2+ itself. The activation is strongly promoted by elevated Na+i and increased by higher pacing frequency (Ginsburg et al., 2013). Pharmacological selective activation of the forward NCX mode is expected to facilitate Ca2+ extrusion. The utility of a possible NCX activator compound is controversial, since the facilitation of the forward NCX function could be useful under Ca2+ overload; however the arrhythmogenic inward current would also be increased, especially in ventricular cells. A possible increase of the reverse mode, at the same time, could largely modulate this effect. In SNCs, activation of the forward NCX mode may accelerate the firing rate without β-adrenergic activation and may decrease intracellular Ca2+. However, currently there is no selective NCX activator compound available. Li+ is known to stimulate NCX, but its effect on NCX1 is markedly smaller than on NCX2 and NCX3 (Nicoll et al., 1990; Iwamoto et al., 1998). Several papers also claimed that NCX1 is sensitive to redox agents (Reeves et al., 1986) (Amoroso et al., 2000; Santacruz-Toloza et al., 2000). A combination of a reductant (GSH, DTT, Fe2+) and an oxidant (Fe3+, H2O2) factor is required to enhance NCX. Diethylpyrocarbonate increased Na+-dependent Ca2+ uptake (Ottolia et al., 2002). Several peptides (concanavalin-A and insulin) were also shown to activate the exchanger (Gupta et al., 1986; Makino et al., 1988).
Discovery and Characterization of the Role of NCX in SN Pacemaking
Pioneer Studies Regarding the Role of NCX in SN Pacemaking
The first experimental results regarding NCX function in SNCs were published by Brown et al. (Brown et al., 1984). They suggested that the slow inward current is composed of two different types of inward currents: a fast component, which they attributed to a channel-gated mechanism (ICa,f) and a slow component: NCX triggered by the release of stored intracellular Ca2+ (INaCa). Satoh et al. (Satoh, 1993) provided experimental evidence that caffeine applied in the bath solution at concentrations of 1 to 10 mM caused a frequency decrease and arrhythmias in isolated rabbit cells. This work did not discuss the ionic currents (e.g., NCX) in detail but demonstrated that cellular Ca2+ overload was induced during exposure of SNCs to caffeine (Satoh, 1993). However, considering the complex effects of caffeine especially if it is applied in the bath solution makes the interpretation extremely difficult. In the same year, Zhou and Lipsius strengthened the initial suggestions of NCX's role in SN automaticity by using latent pacemaker cells isolated from cat atrium (Zhou and Lipsius, 1993). They demonstrated that 1 µM ryanodine notably decreased the spontaneous pacemaking activity but did not stop automaticity, which led to the conclusion that a ryanodine sensitive component contributes to but is not essential for pacemaking. In line with these results, Rigg and Terrar (1996) found that 2 µM ryanodine and 100 µM CPA applied to guinea pig SNCs substantially decreased the spontaneous firing rate (29 ± 2% and 37 ± 6% respectively). They concluded that both agents markedly reduce the transient amplitude and the diastolic Ca2+ levels causing various changes in the Ca2+ dependent mechanisms and could affect the contribution of NCX. Similar conclusions were drawn one year later by Li et al. (1997) using ryanodine on rabbit isolated SNCs.
The first computational pacemaking model of mammalian SNCs including NCX was proposed by Noble and; Noble (1984) and later extended by Wilders et al. (1991) (Noble and Noble, 1984; Wilders et al., 1991). Another early SN model comprising NCX was proposed by Rasmusson et al. in 1990 (Rasmusson et al., 1990) for bullfrog SNCs. However, in their model parametrization, NCX was small and while modulating the MDP, it had little influence on the AP, which they attributed to Ca2+ buffering by myoplasmic proteins. In contrast, the Demir et al. (1994) rabbit model comprised a larger inward NCX (peak −96 pA) during the first third of the pacemaker potential, which then declined slowly through the remainder of the cycle (Demir et al., 1994). The model by Dokos et al. (1996) was mainly driven by an inward background Na+ current (Dokos et al., 1996). Upon complete block of their NCX formulation, which reproduced the saturation characteristics at high Ca2+i and negative potential low Ca2+i, automaticity ceased. The Dokos et al. NCX formulation was later on integrated in a rabbit SNC model by Kurata et al. (2002) in which it contributed significantly to DD (Kurata et al., 2002). However, NCX current amplitude decreases during DD in the Kurata et al. model (Figure 2E) in contrast to later models introduced below [e.g., (Maltsev and Lakatta, 2009)] where INaCa increases before ICaL is strongly activated. The NCX time course during the spontaneous APs of different early SNC models is shown in Figure 2. For the models available in the CellML repository, Figure 2 also shows their response to complete NCX block (red lines). The Demir et al. model exhibited CLs as during control after 5 s, i.e., continued to spontaneously generate APs in the absence of NCX (data not shown), the Dokos et al. and Kurata et al. models did not exhibit sustained pacemaking behavior upon complete NCX block. The effect of clamping the intracellular (and in the Kurata et al. model also the subspace) Ca2+ concentration to the initial value (i.e., value during early DD) is shown in Figure 2 as well (yellow lines). Under these conditions, pacemaking ceased in the Dokos et al. model; the CL was shortened in the Demir et al. model, whereas it was prolonged in the Kurata et al. model. Table 1 compares all SN models comprising a NCX formulation mentioned in this paper. Almost all NCX formulations originate from either DiFrancesco and Noble (1985) or Matsuoka and Hilgemann (1992).
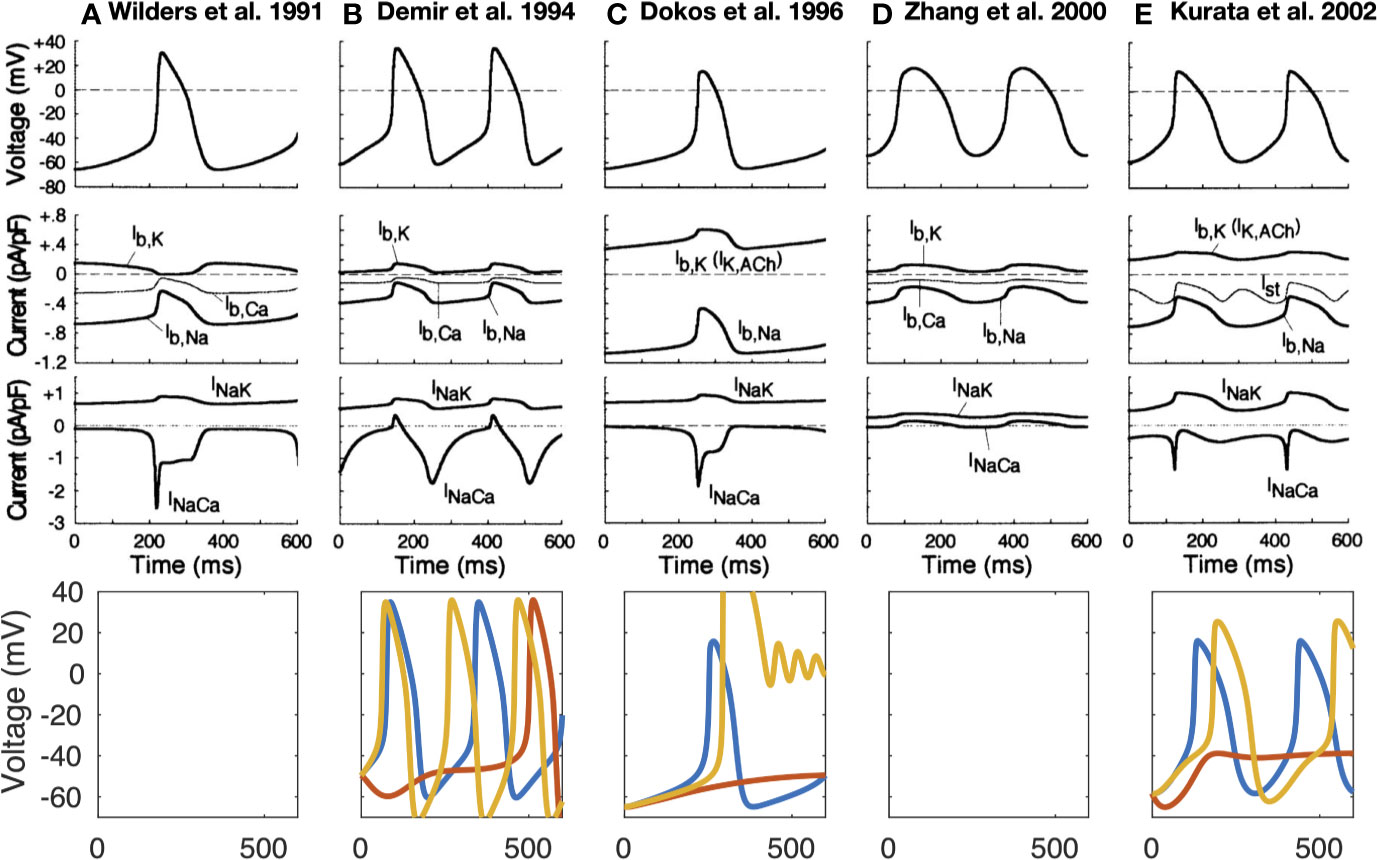
Figure 2 Time-independent background and transporter currents including NCX during spontaneous pacemaking in early SN models: (Wilders et al., 1991) (A), (Demir et al., 1994) (B), (Dokos et al., 1996) (C), (Zhang et al., 2000) [(D); central model], and (Kurata et al., 2002) (E). Top 3 rows reproduced from Kurata et al. (2002) with permission. Bottom row: effect of complete NCX block (red line) and clamping intracellular Ca2+ to the initial value (yellow line). As the Kurata et al. model comprised a separate subspace Ca2+ concentration, this was clamped as well. The Wilders et al. model was not available in the CellML repository, the Zhang model did not exhibit sustained pacemaking behavior in the version deposited in the CellML repository.
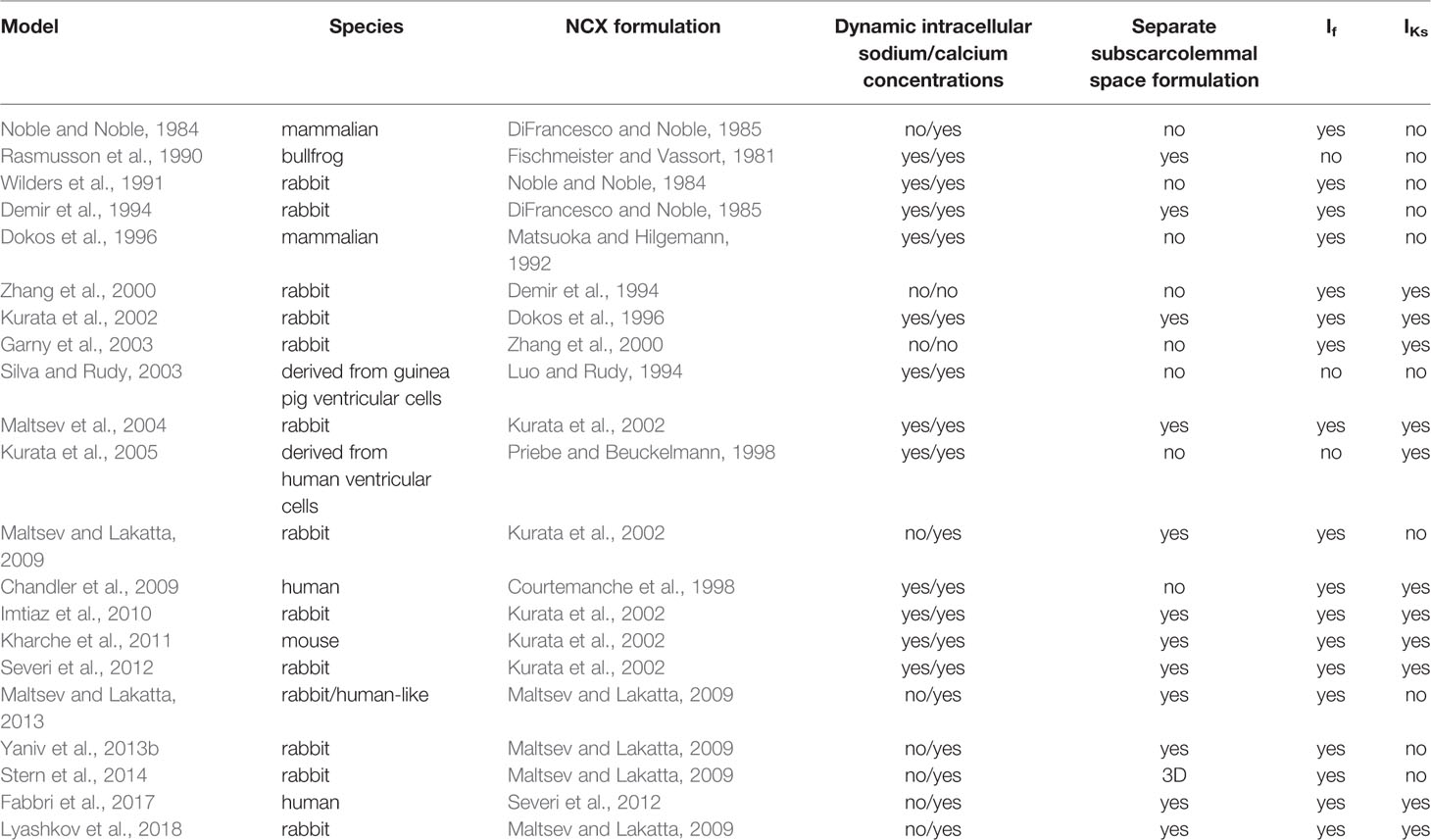
Table 1 Comparison of model features for the 21 SNC models described in this review. Only studies that introduced new models or markedly advanced existing models are listed.
In 1998, Ju and Allen (1998) studied the potential role of Ca;2+-dynamics in the spontaneous firing rate of APs in sinus venosus of cane toad and provided quantitative data regarding the magnitude of the NCX current during the SN AP. They measured 20–27 pA NCX current in early diastole (Ca2+i: 250–300 nM) and 12 pA (Ca2+i: 200 nM) in late diastole in toad isolated sinus venosus cells. The authors emphasized that the net inward current (i.e., total current) required during DD to generate a pacemaker potential is less than 1 pA (DiFrancesco, 1993):
Therefore, quite small relative changes of NCX current (as a response of Ca2+i change) can cause substantial changes in the firing rate. Hagiwara and Irisawa (1989) reported 1 pA/pF NCX current density at Vm of −40 mV in the presence of 500 nM Ca2+i (Hagiwara and Irisawa, 1989). Wilders et al. (1991), in a numerical model, demonstrated that the NCX current had an amplitude of 0.64 pA/pF under similar conditions (Wilders et al., 1991). However, one should keep in mind that the absolute value of NCX current amplitude per se does not exclusively define the contribution of the exchanger to DD. The current integral, i.e. the total charge transferred through the membrane and its relation to other, concurrently active currents, determines the role for pacemaking, meaning that larger NCX current amplitude does not necessarily imply larger contribution (cf. Figure 3).
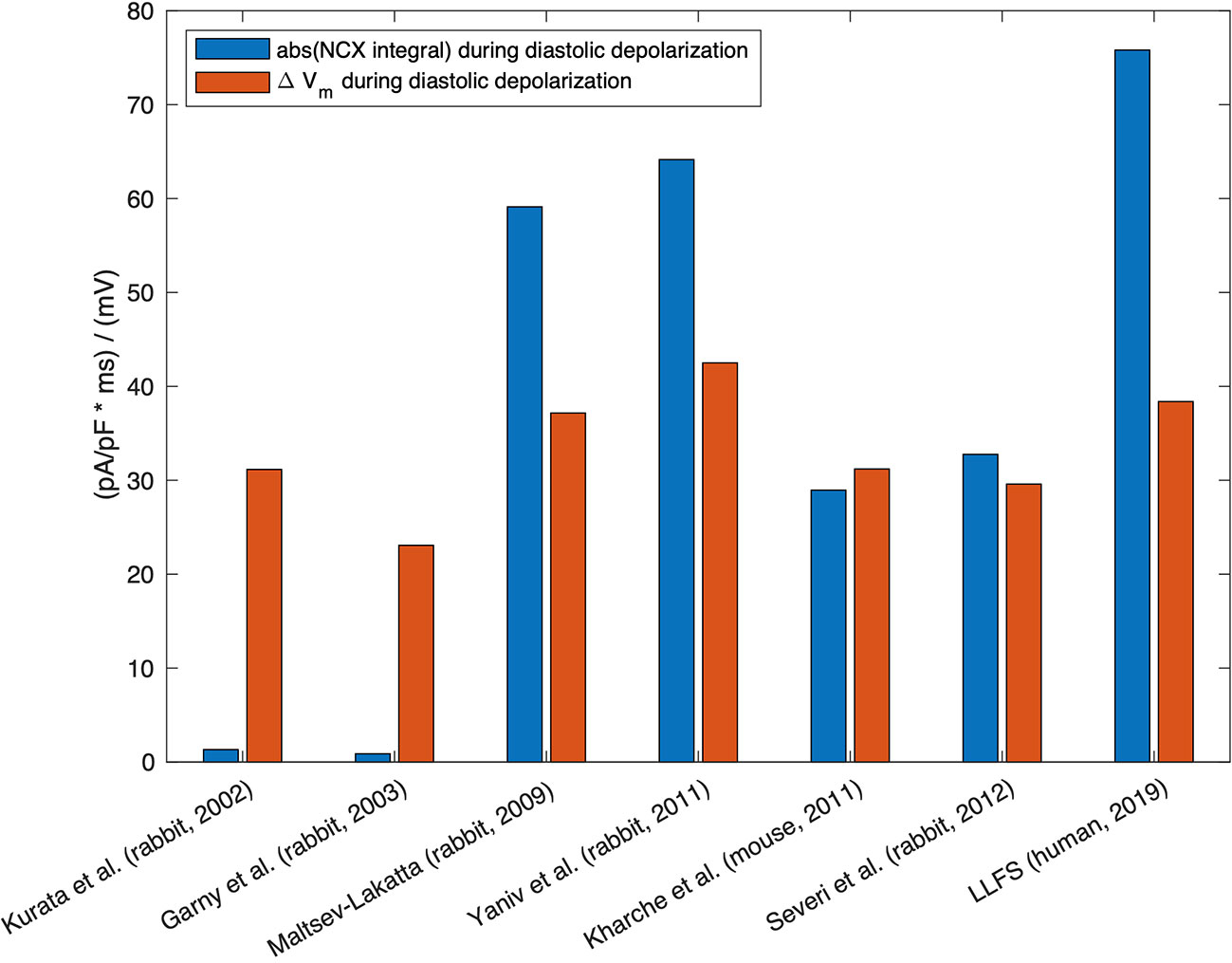
Figure 3 Absolute value of the integral of signed NCX during the DD (MDP to AP takeoff) (blue, unit: ms * pA/pF ≡ mV), in relation to the difference in maximum diastolic Vm and takeoff potential for computational models of different species: rabbit (Kurata et al., 2002; Garny et al., 2003; Maltsev and Lakatta, 2009; Yaniv et al., 2011; Severi et al., 2012), mouse (Kharche et al., 2011), and the human Loewe–Lutz–Fabbri–Severi (LLFS) model (Loewe et al., 2019a; Loewe et al., 2019b).
Ju and Allen (1998) also reported that BAPTA and ryanodine (2 µM) caused frequency reduction until the cells stopped beating. However, this happened after 30 min when other nonspecific actions and spontaneous current decline could not be ruled out. They also demonstrated that decrease of external Na+ caused a rise of Ca2+i due to reverse mode NCX. Application of Na+-free solution resulted in a rapid rise in Ca2+i that exceeded the preceding systolic Ca2+i level. This rapid rise of Ca2+i and the generation of an inward current when intracellular [Ca2+] is elevated are characteristics of the activation of Na+/Ca2+ exchange (Allen et al., 1983; Ju and Allen, 1998). The authors also demonstrated that the exchanger current seemed to be close to a linear function of Ca2+i. Bufo marinus pacemaker cells do not contain any If (Ju et al., 1995). Therefore, If decrease can be ruled out as a contamination of the experiments. They conclude that NCX has a crucial role in setting the resting heart rate, and the actual level of Ca2+i has great importance for indirectly setting the AP firing rate.
Discovery and Characterization of Local Ca2+ Release (LCR) Events: The Role of NCX in SN Automaticity
A seminal study was published by Huser et al. in 2000 (Huser et al., 2000). By using voltage clamp and confocal fluorescence microscopy in cat atrial spontaneously beating cells, they discovered an increase of Ca2+i in the last third of DD just before the action potential depolarization, due to local release of Ca2+ from the SR. As small dose (25–50 µM) of NiCl2 suppressed the local release, thus they suggested the T-type Ca2+ channels triggered subsarcolemmal Ca2+ sparks, which in turn stimulated the inward NCX to depolarize the pacemaker potential to threshold.
One year later, the Lakatta group (Bogdanov et al., 2001) further analyzed the spontaneous Ca2+ releases (local Ca2+ releases, LCR, Figure 4) in isolated rabbit sinoatrial cells. Their findings indicate that the pre-AP releases are locally propagating Ca2+ waves, resulting from ryanodine-sensitive Ca2+-induced Ca2+ release (CICR). In turn, the negative chronotropic effect of ryanodine is the result of disappearance of localized pre-AP Ca2+ releases. Substituting Na+ for Li+ to inhibit NCX caused sinus arrest leading to the conclusion that Ca2+ release-NCX cross-talk has crucial importance in pacemaking. The authors provided a possible scenario for the current interactions underlying spontaneous automaticity: after reaching the maximal diastolic potential, If activates to depolarize the membrane and activates ICaT and ICaL causing LCR from the SR, which in turn activates NCX and the inward current augments to activate the remaining L-type Ca2+ channels. These results were interpreted such that SR Ca2+ release has a pivotal impact in defining the actual firing rate and inactivation of either the RyR or NCX activity lead to frequency slowing or abolishment.
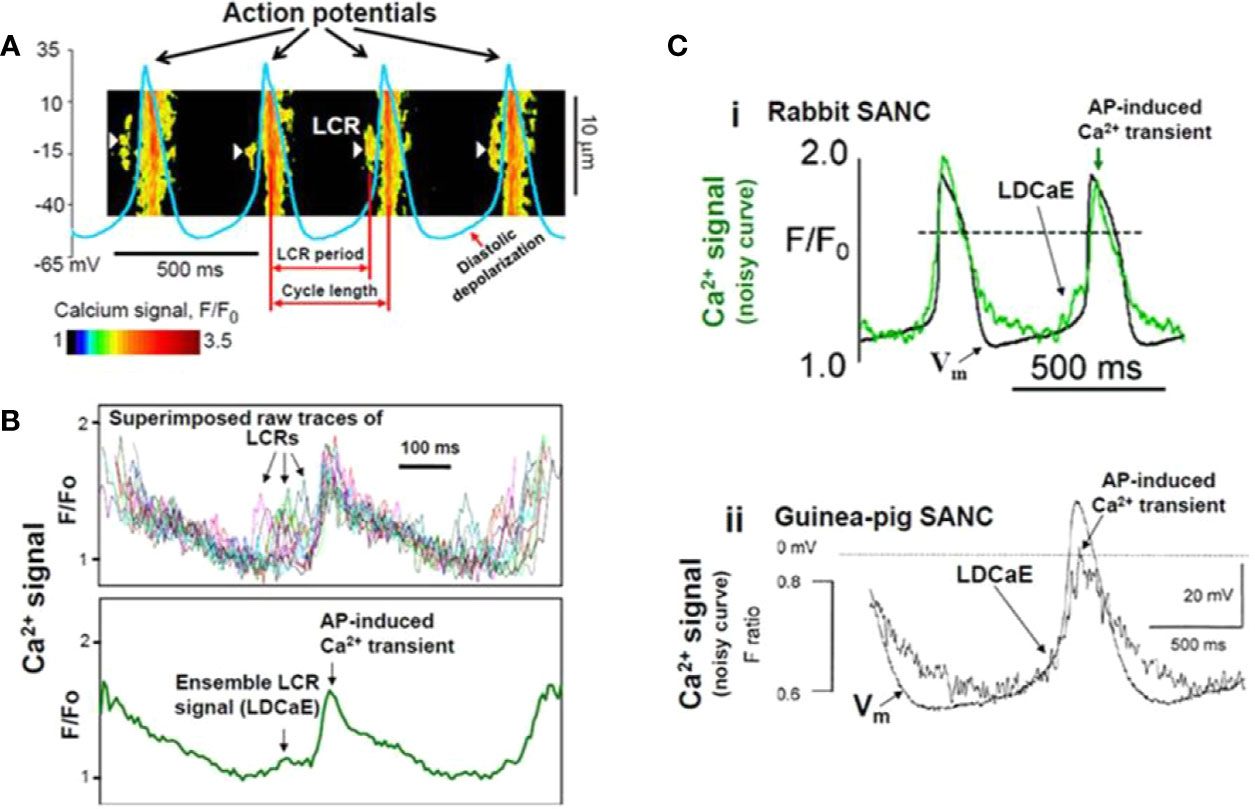
Figure 4 Confocal line scanning image of Ca2+-signal with superimposed spontaneous AP traces recorded in rabbit SNCs show DD, distribution of LCRs (white arrows), LCR period, and CL (A). Confocal microscopic image of LCRs are depicted in [(B), upper panel], ensemble LCR signal or Late Diastolic Ca2+ Elevation (LDCaE) that precedes Ca2+-transient are created from temporal average of LCRs [(B), lower panel]. LDCaE in single SNC of rabbit [(C), panel i] and guinea-pig [(C), panel ii] is shown. LCR: local Ca2+ release. From (Maltsev et al., 2014) with permission.
The pivotal role of the strong interaction between the Ca2+ concentration in the subspace and Vm via NCX for spontaneous depolarizations in SNCs was numerically reproduced and mechanistically underpinned by Maltsev et al. (2004) in a biophysical modeling study in 2004 based on an extension of the Kurata et al. (2002) model (Kurata et al., 2002). In 2009, Maltsev and Lakatta proposed a coupled oscillator model incorporating aspects of both a membrane and a Ca2+ clock, which yielded rhythmic spontaneous depolarization over a wider range of parameters of the currents involved in the M-clock (ICaL, If) (Maltsev and Lakatta, 2009). By modulating SR Ca2+ pumping rate, the coupled-clock yielded robust pacemaking in the range of 1.8 to 4.6 Hz, which is wider than could be achieved by modulation of M-clock properties alone. Kurata et al. (2012) performed stability and bifurcation analyses on this coupled-clock model and found that the most important model parameters for generation of cyclic depolarization (equilibrium point instability) are the SR Ca2+ pumping rate, NCX density as well as ICaL conductance (Kurata et al., 2012). However, the model system does not exhibit cyclic intracellular Ca2+ oscillations when Vm is clamped (independent of the clamped value), i.e., arguing against an independent Ca2+ clock, thus underlining the coupled nature of the pacemaking mechanism. In line with this finding, Maltsev and Lakatta (2009) observed damped oscillations when clamping the transmembrane voltage to −65 mV (at Pup = 20 mM/s) (Maltsev and Lakatta, 2009). However, sustained oscillations were observed at Pup = 40 mM/s. The NCX was a major contributor to the observed equilibrium point in the study by Kurata et al. (2012).
Vinogradova et al. explored in 2004 whether the LCRs require previous membrane depolarization as was suggested so far (Vinogradova et al., 2004). Experimental measurements on isolated rabbit SNCs as well as numerical modeling revealed three findings i) the LCRs during the DD do not require previous membrane depolarization; ii) the LCRs are rhythmic and generate rhythmic inward current movements; iii) the LCR periods are highly correlated with the spontaneous CLs.
In contrast to the increasing body of evidence, Sanders et al. claimed in 2006 that the contribution of NCX to spontaneous depolarization does not exclusively depend on the intact SR function in guinea pig SNCs (Sanders et al., 2006). They demonstrated that low Na+ solution abolished spontaneous firing in the presence of CPA, an inhibitor of SR uptake. On the other hand, if NCX would be entirely a consequence of SR Ca2+ release, the CPA should have a similar effect as a low Na+ solution. However, the SR inhibition only slowed the heart rate, while low Na+ abolished spontaneous pacemaking within one beat, with concomitant decrease of Ca2+i. However, considering the rather complex effects of reduced Na+ solution, the results could not unequivocally support the authors' hypothesis. The application of KB-R7943 to inhibit NCX has to be considered problematic because of its nonselectivity.
Using high-speed Ca2+ imaging, Yaniv et al. (2013) could show that the coupled clocks also control beat-to-beat regulation of the spontaneous beating rate (Yaniv et al., 2013b). Acute application of low concentrations of caffeine (2 to 4 mM) caused Ca2+ release from the SR and subsequently led to increased spontaneous beating rates via membrane depolarization caused by increased NCX activation. This effect could be reproduced mechanistically in an extended version of the Maltsev–Lakatta computational model (Yaniv et al., 2013b). Toward this end, Severi et al. (2012) proposed an update of the Maltsev–Lakatta rabbit SNC model incorporating a coupled-clock with Ca2+-handling (including NCX) and refined current formulations based on more recent experimental data (Severi et al., 2012). On the other hand, the Severi et al. model could not predict the effect of acute caffeine injection described above and complete block of If lead to cessation of spontaneous activity. Simulations including the effects of 1 µM isoproterenol [causing, among other, a 25% increase of SR Ca2+ pumping as in Maltsev and Lakatta (2010) (Maltsev and Lakatta, 2010)], yielded a 28.2% rate increase in the Severi et al. model in good agreement with the experimentally observed increase of 26.3 ± 5.4% by Bucchi et al. (2007b).
In 2013, Sirenko et al. confirmed the presence of LCR in SNCs (Sirenko et al., 2013). The authors compared the spontaneous Ca2+ releases in permeabilized (i.e., in the absence of functional ion channels) rabbit SN vs. ventricular cells. They found that in the presence of similar physiological Ca2+ concentrations, the SNCs generate large rhythmic spontaneous Ca2+ releases in contrast to ventricular cells in which they found Ca2+ sparks with small amplitude and stochastic nature, as was originally described (Cheng et al., 1993). This ability of SNCs was associated with more abundant SERCA, reduced extent of phospholamban (PLB), and increased Ca2+-dependent phosphorylation of PLB and RyR.
In 2013, Yaniv et al. found new evidence regarding the membrane and Ca2+ clock coupling (Yaniv et al., 2013a). Application of the If-inhibitor ivabradine (3 µM) reduced the AP firing rate, partially by inhibiting If and by decreasing the SR Ca2+ content in isolated SNCs. This means that despite the fact that ivabradine has no direct effect on intracellular Ca2+ cycling and does not exert effects on surface membrane channels other than If, it still suppresses the SR Ca2+ load and the LCR events. These novel findings further supported the hypothesis that cross-talk between the membrane and Ca2+ clock regulates SN automaticity.
Sirenko et al. (2016) analyzed in detail the electrochemical Na+/Ca2+ gradients in the pacemaker function of the NCX (Sirenko et al., 2016). They used a combination of numerical modeling and experimental rabbit approaches (by using the Na/K inhibitor digoxigenin) to determine how the coupling of Na+ and Ca2+ electrochemical gradients regulates pacemaking. Minimal increase of Na+i (5%) enhanced the LCRs, shortened the CL, which was interpreted as an extension of normal chronotropic response. Further increase of Na+i by up to 15 mM caused reduction of LCRs, large CL variability as a consequence of Vm-Ca2+ clock uncoupling. In parallel with the biphasic CL changes, the NCX current also showed biphasic alterations: initially the INCX increased due to the high amplitude LCRs, then declined as ENCX reduced. The authors claimed that ENa, ECa, and ENCX tightly regulate the SN automaticity via influencing several ion channels and components of the SN clocks.
Stern et al. (2014) proposed a computational model with subcellular spatial resolution that allows studying intracellular Ca2+ release propagation patterns that was employed in Maltsev et al. (2017) to classify LCRs and provide insight into mechanisms increasing robustness of pacemaking at various SERCA pumping rates. Consistent with previous studies, they found a major role of NCX in the fight-or-flight response: faster SR Ca2+ pumping prepared the cell for stronger (higher amplitude) and more synchronous LCRs (more subcellular Ca2+ wave collisions) in the next cycle, which elicited a higher NCX current.
Torrente et al. in 2016 studied the role of the predominant isoform of L-type Ca2+ channels (Cav1.3) in the generation of the LCRs and Ca2+i dynamics in SNCs from wild-type and Cav1.3 knockout mice (Torrente et al., 2016). This work challenged the results of previous experimental work (under voltage-clamp conditions with Vm below the activation of Cav1.3 (Vinogradova et al., 2004) and in membrane-permeabilized SNCs, without the influence of any plasma membrane ion channels (Sirenko et al., 2013) claiming that LCRs are clearly spontaneous in nature and entirely independent from the M-clock. Cav1.3 deficiency significantly impaired Ca2+i dynamics by reducing the frequency of LCR events and preventing synchronization. The results of Torrente et al. suggest that the local increase of Ca2+i generated by Cav1.3 Ca2+ current induces RyR opening, thus supporting the coupling between membrane depolarization and SR Ca2+ release. Thus Cav1.3 channels can be inducers of LCR generation in the late DD. Considering this mechanism, Cav1.3 could stimulate the NCX-mediated depolarizing current to reach the threshold potential necessary to trigger an AP. Although LCRs are likely triggered by Cav1.3-mediated ICaL release in mouse SNCs (Torrente et al., 2016), LCR generation has different mechanisms in pacemaker cells of various species. As mentioned before, Vinogradova et al. showed that LCRs in rabbit SNCs do not require a change of Vm (Vinogradova et al., 2004). Instead, LCRs are spontaneous in nature and occur in permeabilized SNCs. Takimoto et al. showed that the α1D Ca2+ channel mRNA is expressed in rat atrium (Takimoto et al., 1997). This observation was confirmed in mouse and human cells by Mangoni et al. (Mangoni et al., 2003). However, it remains unclear whether α1D Ca2+ channels are present and functional in rabbit SNCs since expression at the protein level has not been verified (Qu et al., 2005).
Huser et al. found that T-type Ca2+ channels can also have a role in the generation of LCRs since in cat atrial pacemaker cells, LCRs are triggered by voltage-dependent activation of T-type Ca2+ channels (Huser et al., 2000).
The Role of Phosphorylation in SN Pacemaking and the Fight-or-Flight Response
A crucial feature of the SN is the capability of adapting the firing rate to the momentary requirements of the body. β-adrenergic activation stimulates the cAMP-PKA cascade and phosphorylates several components of the Ca2+ handling machinery and transmembrane ion channels. As a consequence, higher frequency and magnitude of LCRs provide enhanced drive for NCX, thus accelerating DD (Vinogradova et al., 2005; Vinogradova and Lakatta, 2009).
CaMKII
In 2000, Vinogradova et al. reported a crucial role of CaMKII in SN pacemaking (Vinogradova et al., 2000). Selective inhibition of CaMKII activity (by autocamtide 2-inhibitory peptide, and KN-93/KN-92) reduced the pacemaker frequency in rabbit SNCs, furthermore, larger doses of the compounds completely terminated SN automaticity. The authors also demonstrated that CaMKII activity is driven by subsarcolemmal Ca2+ movements and located in the vicinity of L-type Ca2+ channels. Therefore, the vital role of CaMKII could be based on the modulation of ICaL via subsarcolemmal Ca2+ changes (Vinogradova et al., 2000).
In 2011, Gao and Anderson showed that CaMKII can support heart rate increase independent of β-adrenergic stimulation (Gao et al., 2011). Furthermore, CaMKII is required for β-adrenergic response and Ca2+-based mechanism of pacemaking.
Li et al. demonstrated that CaMKII inhibition reduced phosphorylation of RyR and PLB, decreased the LCR size, and increased the LCR period (Li et al., 2016). As a consequence, SN firing rate was reduced. These results led the authors to the conclusion that high basal CaMKII activation ultimately regulates the pacemaker function via phosphorylation of Ca2+ handling proteins.
cAMP-PKA-PDE
The Lakatta group in 2008 demonstrated considerably high basal activity of phosphodiesterase (PDE) enzyme, which restricts local RyR Ca2+ release during DD via reduction of cAMP PKA-dependent protein phosphorylation to keep the basal spontaneous SNC firing under control (Vinogradova et al., 2008). Also in 2008, Younes et al. demonstrated in isolated SNCs that the adenylate-cyclase is activated in the entire physiological concentration range of intracellular Ca2+ and the cAMP-PKA axis activation drives SR Ca2+ release, which in turn activates AC (Younes et al., 2008). This feed-forward “fail-safe” system has a crucial role in maintaining the normal rhythm of SNCs.
In 2011, Liu et al. studied the role of the AC-cAMP-PKA-Ca2+ signaling cascade in mouse SNCs, and they demonstrated that the Ca2+ handling proteins are abundantly expressed, and LCRs were also detected in skinned cells. They showed that inhibition of intrinsic PKA activity reduces PLB phosphorylation and prolongs the LCR period causing reduction in the SNCs firing rate. The PDE inhibition resulted in the opposite effects: increased PLB phosphorylation, shortened LCR period, and accelerated firing. In the same study the authors also demonstrated that β-adrenergic activation requires intact Ca2+ handling, indicating a pivotal role of AC-cAMP-PKA-Ca2+ signaling cascade in maintaining the normal automaticity in mouse SNCs (Liu et al., 2011).
Vinogradova et al. provided data regarding the synergistic role of PDE3/PDE4 activity in rabbit SNCs in 2018. Individual inhibition of PDE3 or PDE4 caused a moderate increase in the beating rate (20 vs. 5%, respectively) (Vinogradova et al., 2018a). However, parallel block of both caused a 45% increase of pacemaking rate, indicating a synergistic effect. Furthermore, dual inhibition enhanced the LCR numbers and size, reduced the SR refilling time and LCR period due to decrease of cAMP/PKA phosphorylation (Vinogradova et al., 2018b). An amplification of local RyR Ca2+ release activates augmented NCX current at earlier times leading to an increase in the DD rate and spontaneous SANC beating rate. When RyRs were disabled by ryanodine, PDE inhibition failed to amplify local Ca2+ releases and increase NCX current. As a result, there was no increase in the DDR and spontaneous SN beating rate (Vinogradova et al., 2018b). In this context, Vinogradova et al. showed in the same year that PDE3/PDE4 modulation of SNCs is self-adaptive, i.e., full functional effect is achieved only when both PDEs are inhibited. Such inhibition will lead to an elevation of the local level of cAMP and PKA phosphorylation. The authors claimed that local cAMP signals may have greater importance in the regulation of spontaneous automaticity in SNCs than the ‘global cAMP' (Vinogradova et al., 2018a).
Autonomic Modulation
In 2002, Vinogradova et al. elucidated the Ca2+ release during DD as the specific link between β-adrenergic stimulation and increased firing rate in rabbit SNCs (Vinogradova et al., 2002). They demonstrated that 3 µM ryanodine abolished the effect of 0.1 µM isoproterenol. In the presence of ryanodine, the β-adrenergic stimulation failed to alter the slope of DD, firing rate, and subsarcolemmal Ca2+ releases, whereas the ICaL amplitude exerted clear β-adrenergic stimulation-induced increase. In 2006, Bogdanov et al. described how application of agents affecting the timing or amplitude of LCRs (ryanodine, BAPTA, nifedipine or isoproterenol) caused immediate changes of spontaneous beating rate, which could be traced back to changes of local NCX using mechanistic computational modeling (Bogdanov et al., 2006). Similarly, PKA-dependent phosphorylation affected LCR spatiotemporal synchronization and therefore modulated beating rate mediated by altered NCX (Vinogradova et al., 2006).
Himeno et al. (2008) studied β1-adrenergic stimulation in a computational model of guinea pig SNCs with large IKs in which they described the role of NCX (Himeno et al., 2008). While activation of IKs during the AP preceding the β1-adrenergic stimulation was negligibly small, IKs counterbalanced the increase in ICaL and NCX, which otherwise compromised the positive chronotropic effect of the β1-adrenergic stimulation by elongating the APD in their model.
Lyashkov et al. (2009) investigated the mechanism of muscarinic-receptor stimulation on heart rate reduction (Lyashkov et al., 2009). They found that carbachol (at IC50) reduced the number and size of LCRs and lengthened the LCR period with concomitant decrease of the beating rate. Numerical modeling indicated that cholinergic-modulation of the beating rate is integrated into the Ca2+ cycling via LCR-mediated function of the NCX. The authors concluded that in the presence of low doses of carbachol, the muscarinic activation induced beating rate reduction is mediated by suppression of cAMP-PKA-Ca2+ signaling, while IK(Ach) activation contributes only under higher carbachol concentrations.
In the same year, Maltsev and Lakatta (2010) published a modeling study demonstrating how G protein-coupled receptor modulation of spontaneous SNC beating rate is not only mediated by effects on membrane currents but also by effects on the SR Ca2+ pumping rate, which in turn affect diastolic NCX (Maltsev and Lakatta, 2010). Also in 2010, Gao et al. demonstrated in healthy canine hearts that complete or almost complete If block attenuated but did not eliminate the positive chronotropy of isoproterenol suggesting that If is not the only target of the β-adrenergic response (Gao et al., 2010). Their results also support the notion of spontaneous Ca2+ releases during late DD, which was sensitive to isoproterenol. In their experiments, 2 µM ryanodine caused a 14% decrease in pacemaker frequency but dramatically decreased the effect of isoproterenol. They also provided evidence that ryanodine has no direct effect on If. The authors concluded that both voltage and Ca2+ dynamics have a role in the pacemaker mechanism.
In the same year, Vinogradova and Lakatta demonstrated that the LCR period and the spontaneous SN cycle is tightly regulated by the SR refilling time in rabbit SNCs (Vinogradova et al., 2010). Therefore, phosphorylation/dephosphorylation of the SERCA regulator protein PLB has also critical roles in setting the actual CL of the SN automaticity especially during β-adrenergic stimulation.
In a theoretical analysis of the relative importance of the membrane and Ca2+ oscillators, Imtiaz et al. (2010) could show that increased SR Ca2+ uptake leads to i) hyperpolarized MDP via a reduction of NCX due to lower Ca2+i (Imtiaz et al., 2010). This means that due to lower Ca2+ the smaller inward current through NCX enables more negative MDP. ii) The more negative Vm increases the magnitude of If and ICaT during early diastole. iii) The enhanced Ca2+ entry during DD increases intracellular Ca2+ and CICR causing a higher NCX current.
Transgenic Modifications of NCX
In 2013, several studies using transgenic NCX knockout mice were published. First, Gao et al. used cardiac specific, incomplete NCX1 knockout mice where the baseline sinus frequency was completely identical between control and NCX1-deleted mutants (Gao et al., 2013). However, they found that NCX1 deletion only partially impaired the isoproterenol effect indicating that If is also required. The authors concluded that the NCX has a minor role in maintaining the normal heart rate but NCX1 deletion dramatically enhanced the effects of ivabradine on isoproterenol-induced automaticity indicating that NCX and If mutually contribute in the fight-or-flight response. Since the genetic knockout of NCX was incomplete, one cannot rule out the possibility that the remaining NCX may be able to provide enough current to fulfill its role in maintaining the Ca2+ balance.
In response to that paper, Maltsev et al. in 2013 provided rabbit SN numerical simulations (Maltsev et al., 2013) to address some unanswered questions by Gao et al. (2013). The authors extended their simulation with new stabilization of NCX via local control of CICR. They argued that the Ca2+ released from the RyR is able to recruit the neighboring RyRs producing Ca2+ wavelets (LCRs) having amplitudes larger than the Ca2+ sparks and smaller than global Ca2+ transients. Since the NCX extrudes Ca2+ from the vicinity of RyRs it attenuates the spread of Ca2+ leading to weaker recruitment. However, when NCX is genetically decreased (i.e. incomplete NCX1 knockout), the Ca2+ spread and recruitment are enhanced providing larger Ca2+ and driving force for the remaining NCX molecules. These results indicate that NCX has a crucial role under the normal heart rate, and a small fraction (i.e. 20%) of NCX is able to produce sufficient current under DD. However, when NCX expression is low, its capacity is fully used for Ca2+ extrusion under rest and there is no more support for frequency increase under β-adrenergic response.
Herrmann et al. in 2013 used inducible and SN specific Cre transgenic mice lacking NCX1 selectively in SN pacemaking cells (Herrmann et al., 2013). The NCX1 was genetically ablated in a temporally controlled and tissue selective manner. The animals exerted severe bradycardia and large variability with arrhythmias leading to the conclusion that NCX has a role to maintain normal rhythm. The caffeine induced transient decay was markedly slowed (3.6% of control) in cpNCXKO cells indicating that in SNCs, almost the entire trans-sarcolemmal Ca2+ extrusion is carried by NCX. At the same time, it is an interesting finding that the Ca2+ transient amplitude was decreased and the decay was slowed while the SR Ca2+ content was unaffected. The authors claimed that the NCX1 deleted cells are not able to compensate the NCX1 deletion. They argue for a fundamental role of NCX in pacemaking even under resting conditions, and the normal pacemaking function is not possible without proper NCX function.
Groenke et al. found that in NCX1 knockout mice (Cre/LoxP system), there is no indication of spontaneous atrial depolarizations on the ECG and the animals exerted junctional escape rhythm (Groenke et al., 2013). Furthermore, isolated cells did not show spontaneous automaticity despite the presence of If and intact Ca2+ stores. They claim that NCX has a crucial role in normal pacemaking. They observed LCRs but no Ca2+ transients in NCX deleted cells. They also conclude that If is not sufficient to depolarize the cell membrane if NCX is completely lacking, interestingly not even if isoproterenol was added. Torrente et al. in 2015 created an atrial-specific NCX knockout mouse where NCX was totally eliminated from the atria including the SN (Torrente et al., 2015). These cells lacked spontaneous APs despite intact If. In contrast, the intact tissue showed spontaneous burst–pause type depolarizations. The authors conclude that in the absence of NCX-mediated depolarizations, ICaL and If are able to initiate AP burst where Ca2+ is gradually accumulated and finally terminated burst activity via Ca2+-dependent inactivation or by other Ca2+ activated processes such as small-conductance Ca2+-activated K+ currents (Torrente et al., 2017).
In 2016, Choi et al. observed regular oscillations of inward currents in voltage-clamped human embryonic stem cell-derived cardiomyocytes, which exhibit spontaneous APs when not being voltage clamped (Choi et al., 2016). The oscillatory Ca2+ releases could be eliminated by blocking NCX (with 1 µM SN-6 and 10 µM KB-R7943) but not by blocking If (with 3 mM ivabradine). Their observations suggest that in these hESC-CMs, the M-clock and the voltage clock act as two redundant pacemaker mechanisms which can work independently.
Kaese et al. (2017) used a homozygously overexpressed NCX mouse model and found that the basal heart rate is not affected, suggesting that NCX is not a crucial factor in setting the resting heart rate (Kaese et al., 2017). In contrast, β-adrenergic stimulation elicited higher heart rates in NCX-overexpressing mice, suggesting more inward NCX current which enhances the speed of DD.
Between 2003 and 2011 several genetically manipulated mice studies with deficient If function were published where autonomic rate modulation was preserved (Ludwig et al., 2003; Herrmann et al., 2007; Harzheim et al., 2008; Hoesl et al., 2008; Baruscotti et al., 2011). These results further indicate the lack of an essential and irreplaceable role of If in SN pacemaking and emphasize that If works together with its “teammates” within the coupled-clock system (Lakatta and Maltsev, 2012)
AP Ignition Model as the Most Current SN Pacemaking Mechanism
In 2018, a new model was proposed by the Lakatta group extending SN electrophysiology by an integrated regulatory mechanism including close interactions of LCRs, NCX, and ICaL coupled by a positive feedback via diastolic CICR and DD acceleration (Lyashkov et al., 2018). In contrast to previous theories arguing for a role of LCRs during the late DD, the AP ignition model suggests that early LCRs (ICaL-independent) activate inward NCX, which defines the ignition onset. If, and ICaT also contribute to early depolarization to reach the ICaL threshold. The physiological role of the low-voltage activation threshold L-type Ca2+ channel isoform (Cav1.3) may have more importance to regulate the early ignition phase. However, the details of the specific contribution of Cav1.3 require further investigation. The ICaL-mediated Ca2+-influx recruits more LCRs via diastolic CICR, and this positive feedback further increases NCX. It was also shown that ICaT also has a role in the diastolic CICR, similarly as early results suggested (Huser et al., 2000). However, previously ivabradine and ryanodine were considered to cause AP CL lengthening by different mechanisms (i.e. ivabradine by If inhibition and ryanodine by takeoff potential depolarization). The underlying mechanism of ryanodine induced decrease in the firing rate could be driven by the marked decrease in DDR, which was observed in isolated rabbit sinoatrial node cells by Vinogradova et al. (2002). The AP ignition model brings them common ground: regardless of whether the M-clock (in case of ivabradine) or the Ca2+-clock (in case of ryanodine) is perturbed, it will indirectly affect the other clock via the coupling mechanism. As a coupling, similar responses happened, the ignition potential become depolarized, and time-to-ignition phase becomes longer. This provides delay in clock-coupling by CICR delay.
The Role of NCX in Human SNCs in Comparison to Other Species
In contrast to the significant amount of experimental data on the role of NCX in pacemaking derived from animal experiments (mostly isolated rabbit SNCs), data from human SNCs or tissue are scarce. Considering the vast difference in the main quantity of interest, the beating rate, between commonly used laboratory animals and humans (mouse: 500 bpm, rabbit: 300 bpm, dog: 100 bpm, human: 60 bpm), it however cannot be assumed that the details of pacemaking mechanisms and most importantly the delicate balance of competing effects can be transferred from animal models to human in general. Indeed, it has been shown that computational models of SNCs of different species (mouse, rabbit, human) react markedly different to changes of e.g. extracellular Ca2+ concentration (Loewe et al., 2019b). Given the potential relevance of this alteration regarding sudden bradycardic death in dialysis patients (Loewe et al., 2019a), species-dependent investigations are desirable. As a particular example of interspecies differences, IKr has been reported to be absent in porcine SNCs and IKs to be negligibly small in rabbit SNCs during rest, while both are present in guinea pig SNCs (Satoh, 2003). Moreover, SN AP duration is markedly different between species (mouse: 80 ms, rabbit: 200 ms, human: 300 ms; approximate values) as are other electrophysiological parameters such as AP upstroke velocity, MDP, and DDR (Opthof, 2001). Regarding the role of NCX, its importance in terms of relative contribution to Ca2+ extrusion differs between species as well: 28% in rabbit compared to 7% in rat, i.e., a 4-fold difference (Bers, 2002).
One of the few experiments using human SNCs was performed by Tsutsui et al. (2018). The authors show that similarly to animal models, spontaneous rhythmic LCRs generated by the Ca2+ clock are coupled to electrogenic surface membrane molecules to initiate rhythmic APs, and that Ca2+-cAMP-protein kinase A (PKA) signaling has a regulatory role in the clock coupling in human SNCs (Tsutsui et al., 2018). In the absence of strong coupling between the oscillator subsystems, it was shown that SNCs fail to trigger spontaneous APs and only exhibit disorganized LCRs that were unable to activate the M-clock (Tsutsui et al., 2018). In summary, the study by Tsutsui et al. provides evidence that also in humans it is the coupled-clock mechanism that fundamentally drives SNC pacemaking and provides hints for mechanisms underlying SN dysfunction (Tsutsui et al., 2018).
To highlight interspecies differences and provide an overview of the characteristics of the role of NCX in different models, basic in silico experiments were performed.
Figure 5 shows the time courses of Vm, Ca2+i, total membrane current and NCX for several computational models of different species. Magnitude and temporal dynamics of NCX vary markedly between models and species. While NCX does not play a major role in the Garny et al. and Kurata et al. rabbit models, there is some early diastolic NCX in the Severi et al., Kharche et al. and Loewe–Lutz–Fabbri–Severi (LLFS) models, which becomes more pronounced only slightly before AP ignition. In the Yaniv et al. and Maltsev–Lakatta models in contrast, NCX is apparent markedly earlier. A current integral reflects the total charge transported across the membrane during a specific period of time. For a current normalized to the membrane capacitance (pA/pF), the integral is identical to the potential difference caused by the charge accumulated on the capacitive membrane due to the current (units: ms * pA/pF ≡ mV) is a measure of how much of the DD (in terms of mV) can be attributed to NCX. To quantify the role of NCX to DD in the models of different species, we computed the NCX integral during DD:
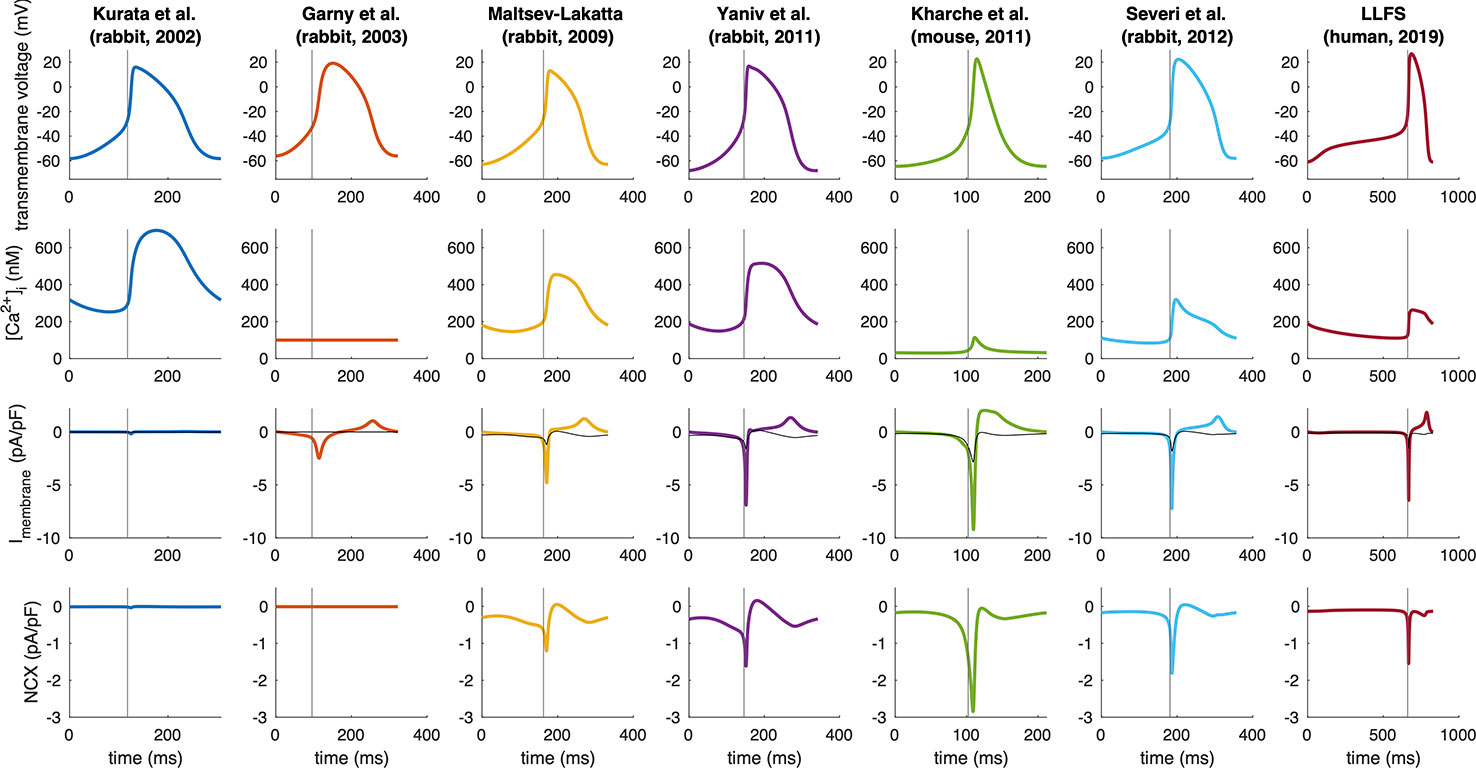
Figure 5 Spontaneous APs, Ca2+ transients, total membrane currents, and NCX in computational models of different species (compare Figure 3). Note the different time axes as a result of the markedly different spontaneous beating rates. The vertical gray line indicates the time of AP takeoff. For comparison, NCX is plotted in gray in the total membrane current panels.
where ttakeoff was defined as the first time step for which d2Vm/dt2 exceeded 15% of the maximum d2Vm/dt2 in the time interval between MDP and max(Vm). The curvature based measure was inspired by the shape of the SN APs with the threshold empirically chosen to strike a balance between a detection early in the upstroke and robustness to minor increases of d2Vm/dt2 during DD. Figure 3 compares the NCX integral in relation to the total rise in Vm during DD and gives an idea about how much of the total DD can be attributed to NCX in each of the models across different species. Under the simplifying assumption of independent currents, almost the entire DD can be attributed to NCX in the Severi et al. rabbit and Kharche et al. mouse model. In the Yaniv et al., LLFS, and Maltsev–Lakatta models, NCX alone would cause more DD than the net ΔVm observed, i.e., outward currents partly compensate the NCX-induced depolarization whereas the role of NCX is almost negligible in the Garny et al. and Kurata et al. rabbit models.
To further illustrate interspecies differences in the role of NCX in SN pacemaking, we evaluated how the models of different species respond to gradual block of NCX. Figure 6 shows that the Garny et al. model exhibits an almost linear relation between NCX block and CL prolongation, however with a limited overall effect of +29.6 ms upon complete elimination of NCX. All other models required a certain amount of remaining NCX for reliable pacemaking (between 10% and 50%). In the Yaniv et al. model, 60% NCX block already terminated stable pacemaking. Instead, blocks of two subsequent APs followed by low amplitude Vm oscillations for several seconds were observed (compare Figure 7). The Yaniv et al. and Maltsev–Lakatta models showed a monotonic relation with CL prolongation for all degrees of block. In contrast, the Severi et al., Kharche et al., and LLFS models exhibited a biphasic relation with CL shortening up to a certain degree of block (80%, 50%, 50% block, respectively) and pronounced CL prolongation beyond (up to +420 ms in the LLFS model).
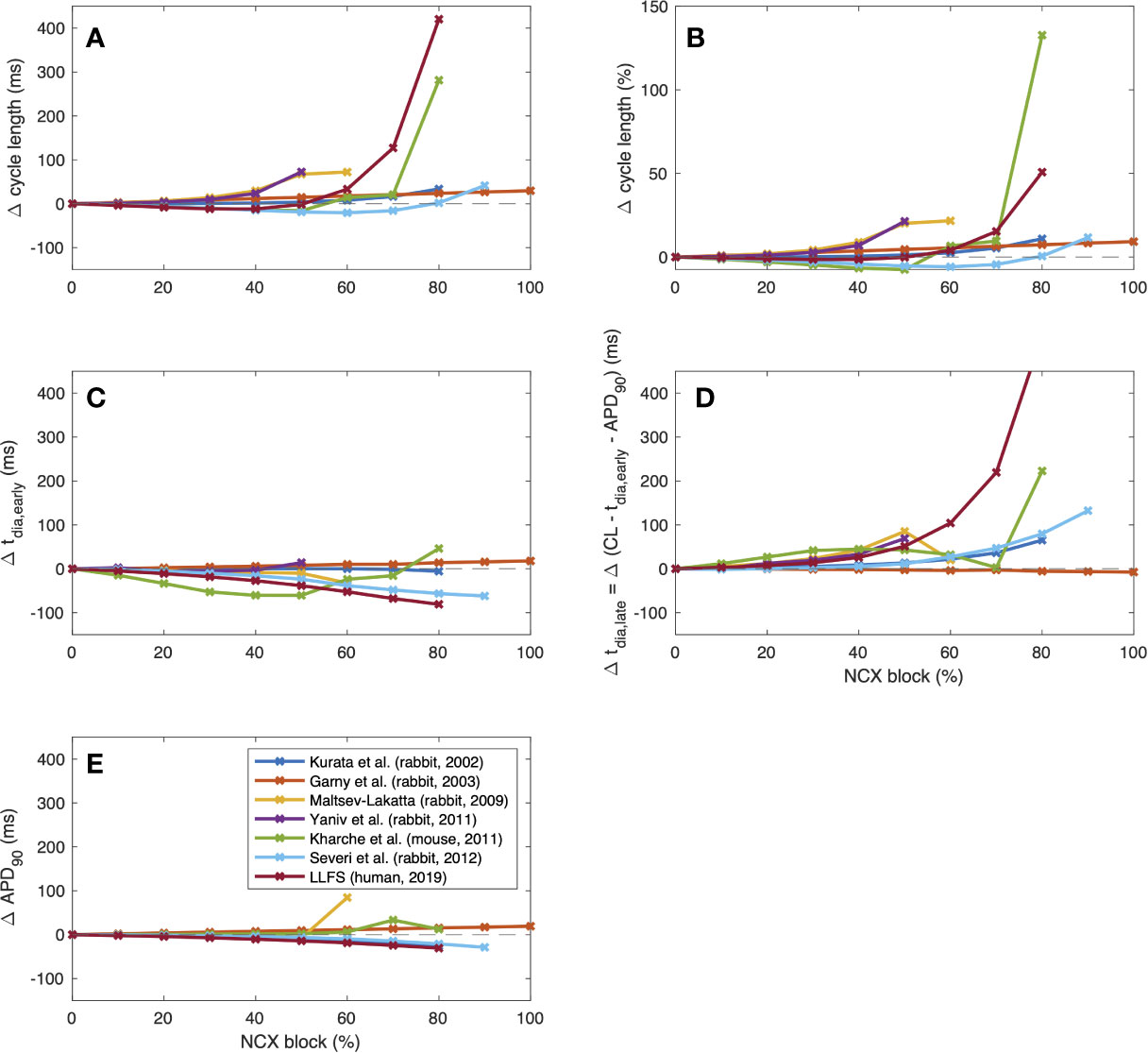
Figure 6 Change of CL [absolute (A) and relative (B)], tdia,early (C), tdia,late (D) and APD (E) of computational models of different species (compare Figure 4) in response to gradual NCX block.
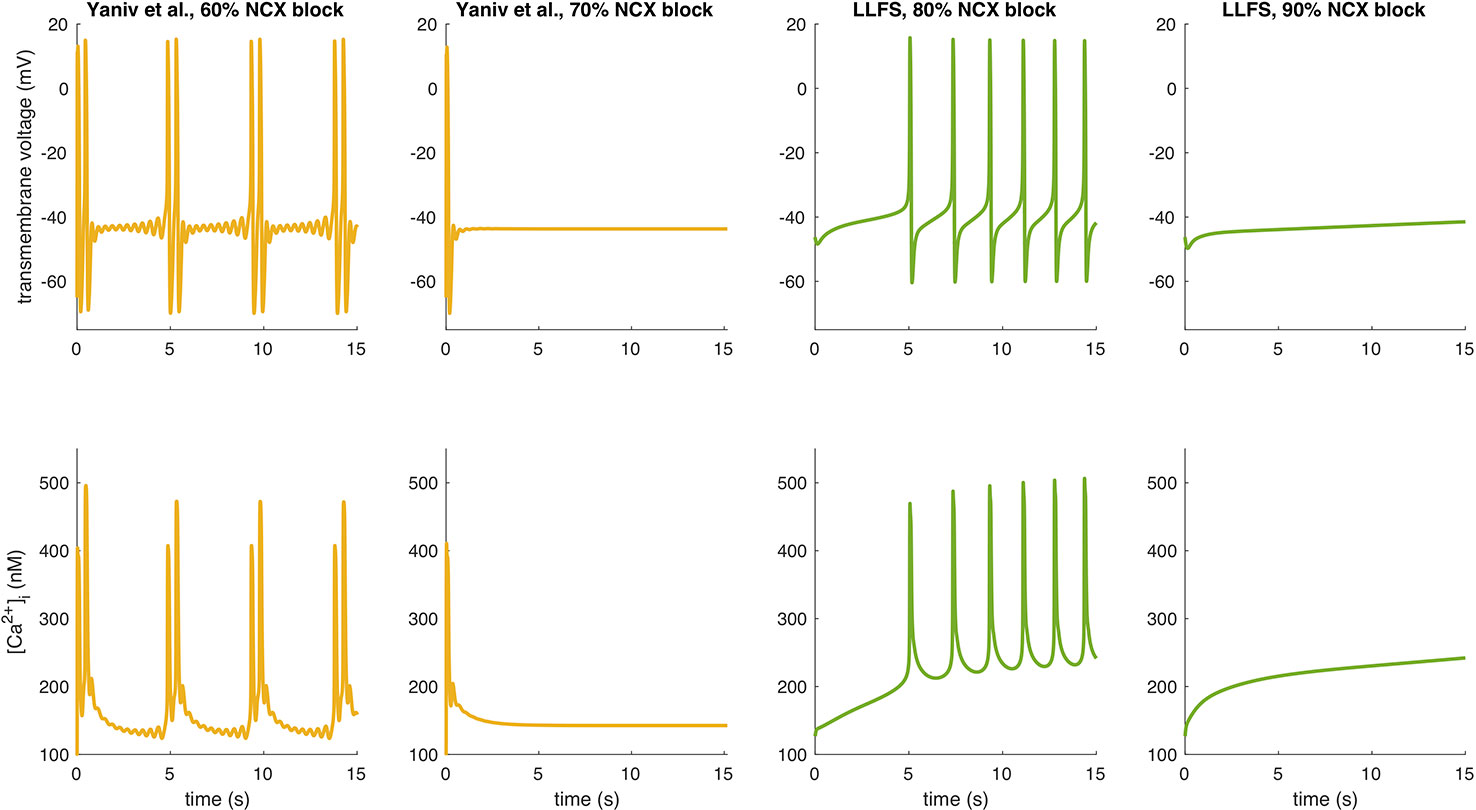
Figure 7 APs and Ca2+ transients of the Yaniv et al. and LLFS models for NCX block degrees slightly below and above the threshold for cessation of automaticity.
To identify the drivers for these CL changes across species, we evaluated the effects on APD as well as early and late depolarization with the following metrics:
where DDRearly was defined as a first order approximation of the DDR during the first 35 ms for the small animal models and the first 100 ms for the human LLFS model. APD shortening was responsible for the initial CL shortening in the Severi et al. rabbit and LLFS human models in combination with a reduction of tdia,early, which also affected the CL of the Kharche et al. model for degrees of block ≤70%. The superlinear CL prolongation for high degrees of block was mainly driven by changes in tdia,late, i.e., changes that could not be attributed to APD, Vtakeoff, or changes in early DDR. Absolute values of CL, overshoot potential, MDP, APD90, DDRearly, and dVm/dtmax upon NCX block of different degrees are shown in Figure 8. In addition to the effects included in the integral measures in Figure 6, a monotonic reduction of AP upstroke velocity could be observed in all but the Garny et al. rabbit model. As models of the same species (such as the Severi et al., Garny et al., and Yaniv et al. rabbit models presented here) sometimes exhibit different properties, it cannot be clearly pinned down which of the effects are truly species-dependent and which are simply model-dependent. Future studies should address this issue to identify which of the insights derived from animal experiments can be transferred to the human setting in which ways.
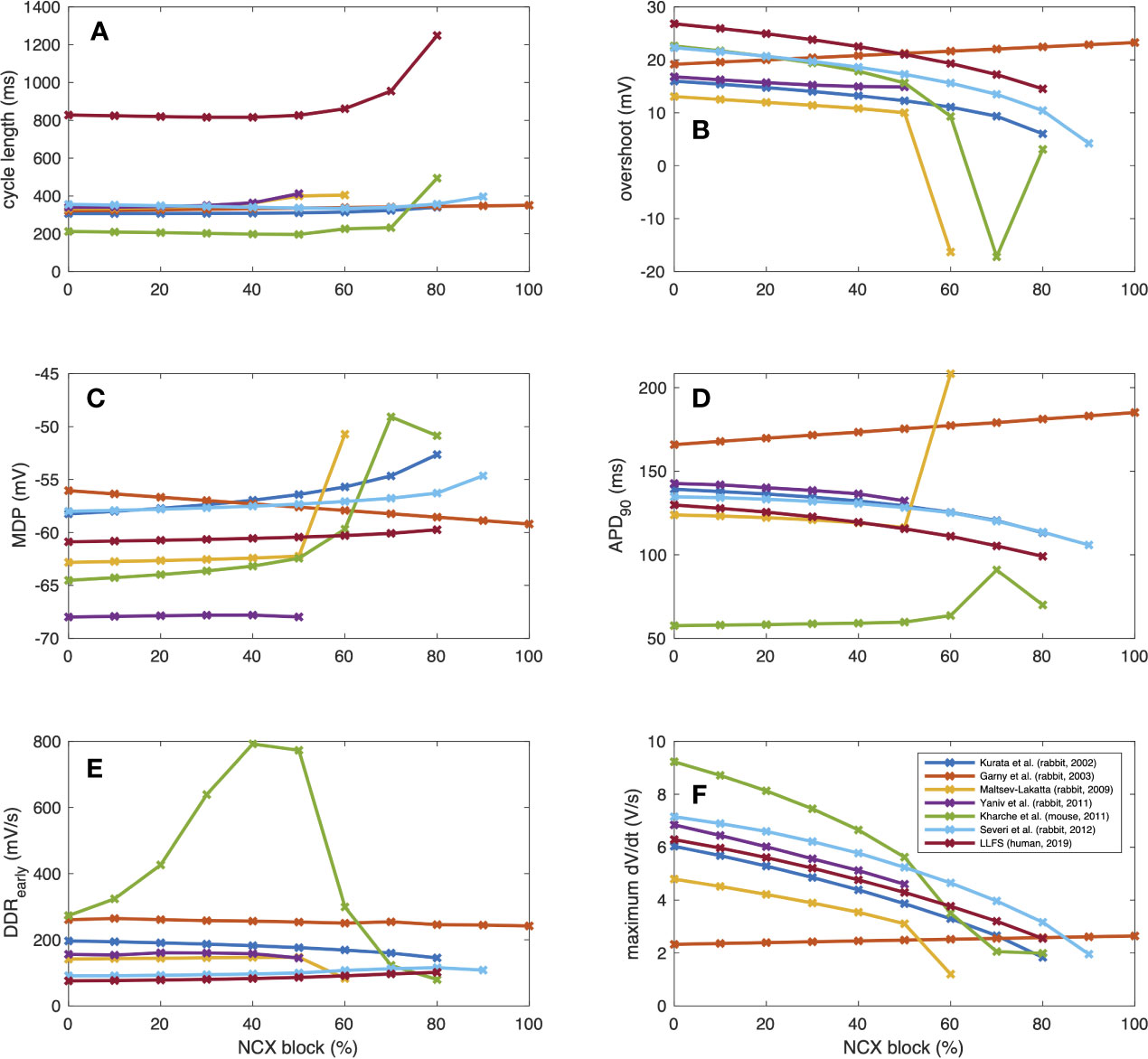
Figure 8 AP properties of computational models of different species (compare Figure 3) in response to gradual NCX block: CL (A), overshoot (B), MDP (C), APD (D), early DDR (E), (dVm/dt)max (F).
To study the biophysical identity of human SNCs, Chandler et al. (2009) performed an mRNA and protein expression analysis of human SNCs suggesting similar NCX1 in SN and atrial cells (Chandler et al., 2009). Based on their experimental findings, they derived one of the first computational models of human SNCs by adapting the atrial Courtemanche et al. (1998) model (Courtemanche et al., 1998) based on their experimental findings where NCX was scaled down by 26%. Allah et al. (2011) found rabbit SN NCX1 mRNA expression levels to be 78% of that of right atrial and 69% of that of left ventricular myocytes, respectively (Allah et al., 2011). Based on these two studies and If recordings in human SNCs, Verkerk et al. presented another early human SNC model (Verkerk et al., 2007; Verkerk et al., 2013). Compared to rabbit cells, their model of human SN cells had smaller absolute NCX, smaller absolute If, and a less pronounced Ca2+ transient. When comparing to the net diastolic membrane current, NCX was of similar magnitude between human and rabbit cells whereas If remained smaller in human cells.
The first human SNC model originating from a SNC model rather that an atrial model was presented by Fabbri et al. (2017). Their model is based on the Severi et al. rabbit model (Severi et al., 2012) and considered human data wherever available (Fabbri et al., 2017). The NCX formulation was adopted from the parent model and goes back to Kurata et al. (2002) (Kurata et al., 2002). As a result of the model parameter fitting to experimental data on the AP and Ca2+ transient level, the maximal NCX current was reduced by 53% (3.34 nA) compared to the parent model. Interestingly, NCX block experiments caused an increase in beating rate in the Fabbri et al. model. (50% block: 83 bpm [+12.2%]; 75% block: 93 bpm [+25.7%]) mediated by a synergistic acceleration of DD caused by elevated Ca2+i, shortening of APD and less negative maximum diastolic potential. Upon 90% NCX block, automaticity ceased. The β-adrenergic rate modulation experiments by Fabbri et al. did not include any direct effects on NCX but the SR Ca2+ pumping was modeled as a target of isoproterenol. The application of 1 µM isoproterenol yielded a 27.9% increase in beating rate. If only SR Ca2+ pumping was modeled as a target of isoproterenol, this increase was markedly attenuated and even slightly reversed with a remaining effect of −0.4%. Loewe et al. proposed the Loewe–Lutz–Fabbri–Severi model (LLFS) (Loewe et al., 2019b) as an extension of the Fabbri et al. model which considers, among other improvements, the dynamics and transients of the intracellular sodium and potassium concentrations in contrast to the previously fixed concentrations, thus further constraining the model physiologically by requiring homeostasis across time spans of minutes (Loewe et al., 2019a). In the LLFS model, the response to complete If block (CL +25.9%) was in accordance with the available experimental human data [+26% reported by Verkerk et al. (2007)] (Verkerk et al., 2007).
Local Control of NCX
Several studies suggested that in cardiac muscle (Lipp et al., 1990) and other cells such as squid axon (Mullins and Requena, 1979), pancreatic acinar cells (Osipchuk et al., 1990), and smooth muscle (Stehno-Bittel and Sturek, 1992) ionic concentrations close to the intracellular surface of the plasma membrane may be different from those in the cytoplasm. These concentration gradients are thought to result from transient net fluxes across the surface membrane and may crucially influence the transmembrane ion channels. In atrial cells, which lack a well-developed t-tubule system, a non-uniform distribution of Ca2+ within the cell following release from the SR has been found and the results were interpreted in terms of distinct release sites under different control mechanisms (Lipp et al., 1990). Trafford et al. in rat ventricular myocytes also demonstrated the existence of the submembrane “area” by using the NCX current as a reporter and found that Ca2+i changes near the surface membrane can be considerably larger than those averaged over the cell. The differences can be explained by a diffusion barrier between the release site and the bulk cytosol (Trafford et al., 1995).
In SNCs, the diastolic NCX is not only regulated by Vm but also by the LCRs, which in turn depend on local cross-talk including Ca2+-dependent functions of NCX, RyR, and SERCA (Figure 1B). This feedback mechanism between LCR and NCX can rapidly change NCX. It is known that RyRs are tightly organized in clusters, which are coupled with the L-type Ca2+ channels to form Ca2+ release units (CRU). RyRs are activated by an increase of local Ca2+i resulting in Ca2+-induced Ca2+ release (CICR). Each CRU generates small, temporal Ca2+ release events, called Ca2+ sparks which have a radius of 1.5 µm (Stern et al., 2013; Hoang-Trong et al., 2015). Therefore, a LCR (4–12 µm) involves several sparks fired by neighboring CRUs via fire-diffuse-fire propagation (Maltsev et al., 2011a; Stern et al., 2014). Spark characteristics can be modulated by β-adrenergic response via variation of single CRU Ca2+ currents (Ispark). The LCR controls SN chronotropy (acceleration and slowing) via changing the LCR size, amplitude, and number per cell surface area.
In 2011, Maltsev et al. demonstrated, by two-dimensional Ca2+ measurements on rabbit SNCs and via numerical modeling, that increasing the Ispark amplitude results in local synchronization of CRU firing yielding increasing LCR size, rhythmicity, and occurrence rate (Maltsev et al., 2011a). Ispark regulates both the time shift and the amplitude of the spontaneous release. Since the LCRs are directly coupled with NCX, this means that Ispark amplitude indirectly influences NCX during DD (Figure 9). In 2013, Maltsev et al. discovered a novel stabilization mechanism via local control of CICR (Maltsev et al., 2013). The Ca2+ released from the RyRs during DD is able to recruit the neighboring RyRs to release more Ca2+. This local recruitment depends on the extent of released Ca2+ and its diffusion to the neighboring RyRs. Since NCX extrudes Ca2+ from the vicinity of RyRs, it restrains CICR because the Ca2+ occurrence to diffuse and activate neighboring RyRs is reduced. The diastolic NCX increase is described by:
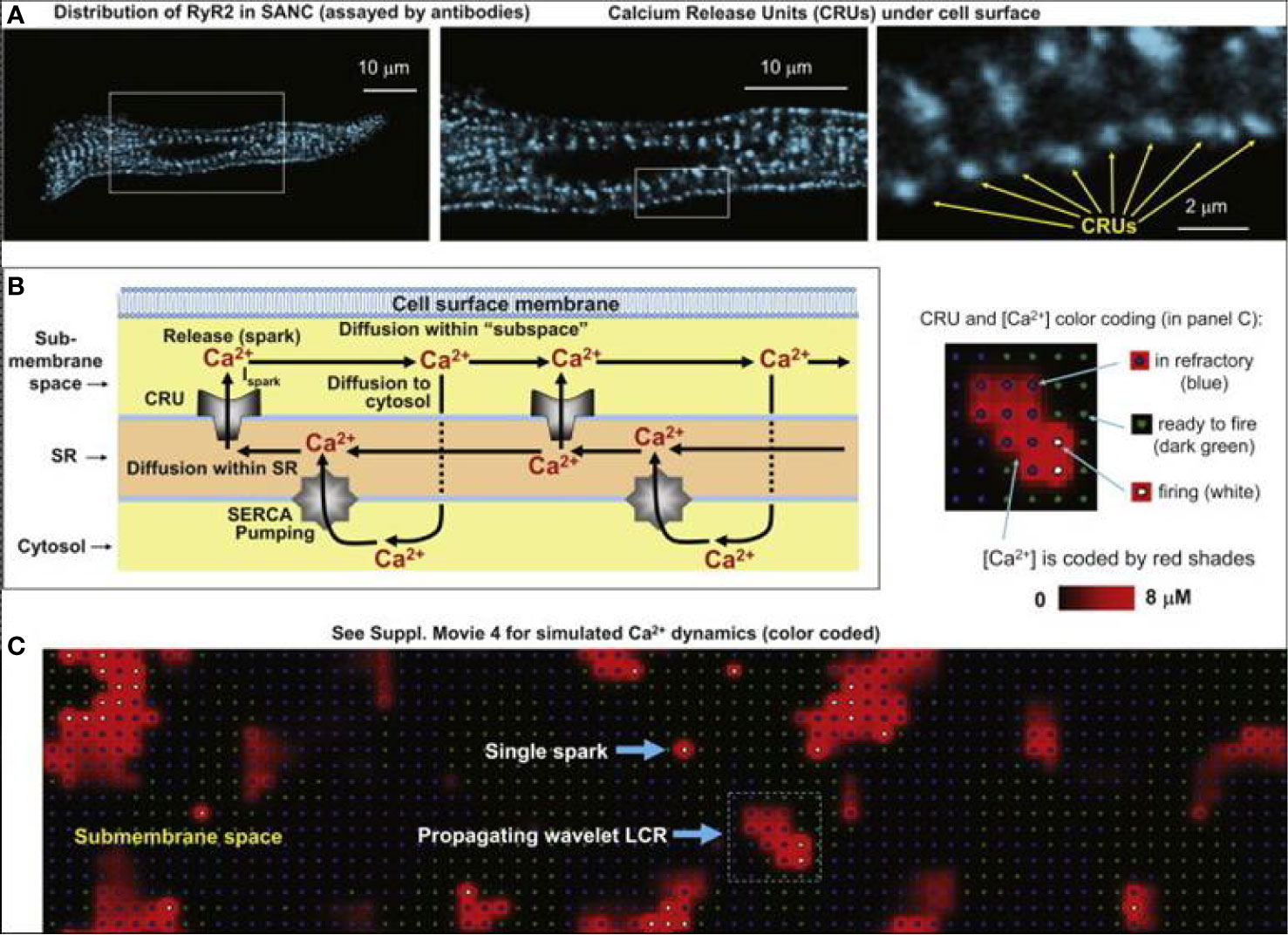
Figure 9 Development of the numerical model of local Ca2+ dynamics in rabbit SNCs: Ca2+ release units (CRUs) and Ca2+ cycling. RyR clusters, i.e., CRUs, are visualized in confocal images of immuno-fluorescence in rabbit SNCs under cell surface membrane (A). Schematic drawing of local Ca2+ cycling through different cell compartments (submembrane space, SR, and cytosol) (B). Local Ca2+ fluxes are depicted with arrows, Ca2+ waves travel from left to right. (C) Instantaneous distributions of local [Ca2+] in the submembrane space predicted by our model: single Ca2+ spark and a wavelet LCRs (propagating from left to right). Inset illustrates the color-coding for CRU states and [Ca2+] (from (Maltsev et al., 2011a) with permission).
where nsparks is the number of Ca2+ sparks and NCXspark is the NCX current generated by one spark. This formula predicts that during NCX suppression the decreased NCX current (NCXspark) in turn increases the number of sparks (nsparks) since there is more Ca2+ available for diffusion and activation of additional RyRs. As the above formula predicts, the larger number of sparks tends to increase the NCX current, and finally, these opposite changes are able to largely compensate each other providing marginal change in the NCX. Stern et al. in 2014 provided three-dimensional SNC computational modeling including diffusion and buffering of Ca2+ in the cytosol and SR but omitted the submembrane space and inactivation of RyRs because of the lack of experimental validation data (Stern et al., 2014). Immunostaining experiments revealed that bridging RyR groups exist between large RyR clusters, organized in irregular networks. This architecture provides the basis for propagating LCR events. When a structure without bridges was incorporated in the model, the separated RyR clusters produced only isolated sparks, without LCRs and NCX activity. The model indicates that this hierarchical RyR clustering provides a pivotal adaptive mechanism and contributes to the β-adrenergic adaptation. Maltsev et al. (2017) characterized the complex spatiotemporal structure of LCRs in cardiac pacemaker cells (Maltsev et al., 2017). Using automatic classification of LCRs, they demonstrated that SR pumping controls LCRs and pacemaker rate via timely synchronized occurrence of LCRs creating a powerful ensemble signal to activate NCX. This also means that when SR pumping rate is low, the LCRs exert smaller amplitudes due to the lower SR Ca2+ load. However, the system compensates and maintains the NCX signal by increasing LCR size and duration. This mechanism ensures fail-safe pacemaker function within a wide range of pacing frequencies.
Heterogeneity of SNCs
Functionally and electrophysiologically, the SN has a heterogeneous structure [as excellently reviewed by Boyett et al. (2000) and Lancaster et al. (2004)]. During normal cardiac activation, the spontaneous AP is initiated from the center, the leading pacemaker site of the SN, and then the AP is conducted through the periphery to the surrounding atrial muscle of the crista terminalis (Bleeker et al., 1980). In response to different interventions, the leading pacemaker site can shift to the peripheral region causing major importance in the pacemaker function. In the center the cells are described being smaller compared to those in the periphery and contain fewer myofilaments (Bleeker et al., 1980; Masson-Pevet et al., 1984; Boyett et al., 2000). The possible differences in the AP characteristics and the expression of ion channels were intensively studied, and the question whether the expression levels of proteins depend on the cell size of the SNCs remains equivocal (Honjo et al., 1996; Boyett et al., 1998; Lyashkov et al., 2007). The majority of the studies report that the expression of the Ca2+ regulatory proteins differs between regions of the SN, which results in heterogeneity of intracellular Ca2+ handling and pacemaker activity between cells across the nodal tissue. Musa et al. showed that the density of ICaL was significantly (p < 0.001) correlated with cell capacitance in rabbit sinoatrial node, and larger cells showed greater density (Musa et al., 2002). The authors also performed immunocytochemical labeling of L-type Ca2+ channel, RYR2, and SERCA2, showing in significantly higher density in cells from the periphery compared to the center of the SN (Musa et al., 2002). In line with this, larger cells exhibited greater systolic Ca2+, diastolic Ca2+, Ca2+ transient amplitude, and spontaneous rate compared with smaller, probably central cells (Lancaster et al., 2004). Sarcolemmal Ca2+ ATPase and NCX have a lower activity in central cells, and the exchanger is responsible for a larger proportion of sarcolemmal Ca2+ extrusion in those cells compared with larger, peripheral cells (Lancaster et al., 2004). In 2007, Lyashkov et al. determined the density and distribution of cRyR, NCX1, and SERCA2 in SNCs of different sizes from the rabbit SN tissue (Lyashkov et al., 2007). They found that both bigger and smaller SN cells have identical AP-induced Ca2+ transients and spontaneous localized Ca2+ release characteristics (Lyashkov et al., 2007). They provided evidence on robust NCX1 as well as SERCA2 and cRyR cellular labeling densities in SNCs and they found that the entire sinoatrial node—including the Cx43 negative primary pacemaker area—exhibits positive labeling for NCX1, cRyR, and SERCA2 (Lyashkov et al., 2007). Furthermore, submembrane colocalization of NCX1 and cardiac RyR was shown, which exceeds the degree observed in atrial or ventricular cells (Lyashkov et al., 2007). However, Tellez et al. examined the molecular basis of the ion currents in rabbit SN with measuring the abundance of messenger RNAs coding for ion channels and regulatory proteins of the SN tissue. Ca2+ handling proteins abundance showed variability between tissues from different parts of the SN (Tellez et al., 2006). The abundance of NCX1 mRNA was significantly lower in the periphery of the SN compared with other regions. In the transcript profile, an apparent isoform switch from the atrial muscle to the center of the SN was found: RYR2 to RYR3, Nav1.5 to Nav1.1, Cav1.2 to Cav1.3 and Kv1.4 to Kv4.2 (Tellez et al., 2006).
Maltsev and Lakatta in 2013, by using a numerical model, explored large variation of ensembles of electrogenic molecules to identify the minimum set of proteins that confer a robust (i.e. fail-safe) and flexible (i.e. adaptation to a given vegetative stimulus) pacemaker function (Maltsev and Lakatta, 2013). The minimal model was found to contain: ICaL+ IKr+ NCX+ Ca2+ clock. The further addition of ICaT and If decreased flexibility but increased robustness of the system. Therefore the higher balance of flexibility and robustness contains five parameters: ICaL + IKr + NCX + Ca2+ clock + If. It is important to note that in the four parameter models, all model sets without NCX failed to generate rhythmic APs in at least one of the tests, confirming the fundamental importance of the exchanger in the pacemaking.
Monfredi et al. examined the major ionic currents in intercaval pacemaker cells (IPCs) obtained from rabbit heart and found marked electrophysiological heterogeneity in IPCs (Monfredi et al., 2018). Experiments and numerical simulations indicated that there is an IPC cell population with minimal or zero If, without correlation between If density and cell size. In the absence of If, the diastolic NCX in response to Ca2+ release (i.e., the Ca2+ clock) could drive the pacemaking. Wide ranges of ICaL and IK densities were found in these cells. Functional heterogeneity of SNCs was recently described by Kim et al. (2018). Their experiments performed in guinea pigs demonstrated three types of SNCs: rhythmically firing, dysrhythmically beating, and dormant cells exerting no spontaneous activation. LCRs were present in all three types; however in the dysrhythmic and dormant cells only smaller, stochastic LCRs without spatial and temporal synchronization were observed. β-adrenergic stimulation synchronized LCRs in all dysrhythmic cells and in the 1/3 of the dormant cells. Furthermore, the dormant cells developed automaticity in response to β-adrenergic stimulation. These results indicate partial or complete uncoupling between Ca2+ and M-clocks in the dysrhythmic and dormant cells.
What is the Role of NCX in Pacemaking Under Resting Heart Rate?
When active in forward mode, NCX generates inward current and at the same time represents the most important pathway for Ca2+ extrusion to maintain the Ca2+ flux balance (Bers, 2001). Since these two functions are not separable, profound (but incomplete) NCX inhibition may always have secondary effects by altering the intracellular Ca2+ levels. Considering the published results and the nature of NCX inhibition, it could be that pharmacological NCX inhibition (partial for all available agents) or incomplete transgenic knockout mutants are not able to unequivocally pin down NCX's function in pacemaking. Theoretically, partial NCX suppression (acute pharmacological, or incomplete transgenic models) may lead to 3 different outcomes, characterized by the intracellular Ca2+ concentration since NCX is the major pathway for Ca2+ extrusion:
i) NCX inhibition does not increase Ca2+i because of activation of other compensatory mechanisms (such as plasma membrane ATPase). In this fortunate case, all NCX inhibition is pure current inhibition with minimal secondary, indirect mechanisms through changes of Ca2+i. However, this scenario may have the lowest probability.
ii) NCX inhibition does not increase Ca2+i because the (potentially small) uninhibited fraction is able to remove the Ca2+. This suggests a non-linear relationship between the degree of NCX inhibition and reduction of NCX function in terms of Ca2+ extrusion during the DD, making the interpretation difficult.
iii) NCX inhibition elevates Ca2+i. In this case, the Ca2+ gain will enhance the Ca2+-dependent inactivation of the ICaL, which may shorten the AP. However, also the AP threshold is expected to be higher, requiring more time to be reached. According to the ignition theory (Lyashkov et al., 2018), the decreased contribution of ICaL also reduces the ICaL-LCR-NCX interactions. Furthermore, the Ca2+ increase may activate other Ca2+-dependent mechanisms such as IK(Ca). These indirect mechanisms may counteract the effect of NCX inhibition.
Groenke et al. demonstrated that NCX1 KO mice show no spontaneous atrial depolarization (Groenke et al., 2013); only junctional escape rhythm was observed. The cells did not show spontaneous automaticity despite the presence of If and intact Ca2+ stores. Current mechanistic models of both human (Loewe et al., 2019a) and rabbit (Yaniv et al., 2011) SNCs also support no spontaneous automaticity when NCX function is completely absent. Considering pharmacological data, it can be firmly stated that different maneuvers aiming to decrease the intracellular Ca2+ level such as ryanodine, CPA, BAPTA in all cases lead to a decrease of SN frequency to varying extents. However, the kinetics of Ca2+ currents also changes due to these interventions. Therefore, taking together the published results, it is doubtless that if NCX is largely inhibited or completely absent, the normal Ca2+ flux balance is considerably impaired which per se may influence spontaneous automaticity. These secondary shifts of Ca2+ balance make the experimental evaluation of the total NCX contribution during DD more difficult. At the same time, the available experimental data with manipulation of Ca2+i, modifications of second messengers (such as cAMP, PKA, PDE, CaMKII), transgenic animals and numerical modeling clearly indicate the fundamental role of NCX in maintaining the resting heart rate.
What is the Role of NCX in the Fight-or-Flight Response?
The β-adrenergic response involves the increase of intracellular cAMP and PKA, increases CaMKII activity and increases the intracellular Ca2+ load via phosphorylation of various channels (Figure 1B). A large body of evidence indicates that LCRs and therefore also NCX are under the influence and delicate control of several intracellular signaling molecules (such as CaMKII, PKA, cAMP, PDE), which are sensitive to β-adrenergic activation (see The Role of Phosphorylation in SN Pacemaking and the Fight-or-Flight Response). These results indicate a pivotal role of NCX not only in maintaining the basal SN automaticity but also in the sympathetic stimulus-mediated heart rate acceleration.
Does the Reverse Mode of NCX Influence SN Pacemaking?
Currently, a transport-selective NCX inhibitor is not available. However, the KB-R7943 preferentially inhibits the reverse mode of NCX (Birinyi et al., 2005); it markedly influences ICaL (Birinyi et al., 2005) making the interpretation difficult. The electrophysiological properties of the NCX predict functioning reverse mode when Vm is depolarized (more than 0 mV) and Na+i is high. Experimental results as well as modeling data demonstrate that SN APs peak around 20 mV, which could be considered high enough to “switch on” reverse mode; however the time at depolarized levels is very short. During normal Na+i, mechanistic modeling predicts only marginal reverse mode at the beginning of the AP, thus the NCX current is mainly inward throughout the entire AP. The Yaniv et al. (Yaniv et al., 2013b) and LFSS model (Loewe et al., 2019a) predicted termination of beating when intracellular Na+i is elevated, suggesting that this is not a way to augment reverse NCX during SN automaticity.
Maltsev and Lakatta in 2013 (Maltsev and Lakatta, 2013) reported a notable reverse mode of the NCX that was lacked in the previously published models. Numerical calculations revealed 2.45 pC of Ca2+ influx via reverse mode that is comparable with ICaL-mediated Ca2+ influx for one AP cycle. This means that reverse NCX contributes in “refueling” the Ca2+ clock almost equal to ICaL.
Criticisms of CA2+-Theory
The role of Ca2+i in the SN pacemaker function has been challenged from the beginning. However, the importance of intact Ca2+ handling in normal SN automaticity was accepted in a relatively short time. In contrast, the dominance of If over NCX and vice versa was a recurrent and often appearing question (Lakatta and DiFrancesco, 2009). Since the funny-current was the first proposed mechanism underlying pacemaking, it is hardly surprising that the Ca2+ theory was challenged many times experimentally.
Ryanodine Effect
Serious criticisms have emerged regarding the frequency modulating effect of ryanodine. Authors often found a 15–30% frequency decrease after application of ryanodine which was attributed to the effect of indirectly reduced NCX activity. However, Bucchi and DiFrancesco in 2003 showed that the ryanodine mediated increase of CL is not the consequence of decreased DD slope but the positive shift of AP threshold (Bucchi et al., 2003). A further consistent finding was that ryanodine considerably reduces the frequency-increasing effect of β-adrenergic activation (Bucchi et al., 2003). The DiFrancesco group (Bucchi et al., 2003) demonstrated that ryanodine disrupts the normal signal transduction pathway of of β-adrenergic response, and the intracellularly used cAMP analogues were able to increase the frequency in the presence of 3 µM ryanodine to a similar extent as during control. The authors claim that the SR Ca2+ release may represent only a “safety mechanism” for the DD to reach the threshold level. Another paper of DiFrancesco group (Bucchi et al., 2007b) investigated “signatures” of rate changes caused by different pharmacological maneuvers. They found that agents modifying If such as ivabradine, isoproterenol, cAMP, and acetylcholine affect only the slope of DD while ryanodine-mediated bradycardia is caused by an increase of the take-off potential. They conclude that while other mechanisms could be important If appears to be the simplest and most direct contributor to rate modulation. While the reduction of pacing rate after ryanodine application is an unequivocal finding, the underlying mechanisms (change of take-off potential vs. slowed DDR) are a matter of debate. Vinogradova et al. showed that ryanodine has a marked effect on the DD by decreasing DDR, thus causing significant CL prolongation (Vinogradova et al., 2002). In another study, they also observed that in the presence of ryanodine, the ability of the cAMP analogue CPT-cAMP to increase the SNC beating rate was markedly reduced from ~35% (in control) to ~10% (Vinogradova et al., 2006). The different findings of this study and the previously mentioned study by Bucchi et al. (2003) could be partly due to different experimental approaches. DiFrancesco's group used clusters of SNCs with whole cell recordings of APs, while Lakatta's group used single SNCs with the perforated patch technique.
However, ryanodine may have a direct inhibitory effect on ICaT channels, which would complicate the interpretation of such experimental data (Li et al., 1997).
Bucchi et al. and Vinogradova et al. (Vinogradova et al., 2002; Bucchi et al., 2003; Vinogradova et al., 2006) were not the only ones investigating the effects of RyR Ca2+ release suppression by ryanodine in the response of β-adrenergic stimulation. The influence of ryanodine on the modulation of the chronotropic effect of β-adrenoceptor stimulation has been extensively studied in a variety of different species. In guinea-pig SNCs exposed to 100 nmol/L isoprenaline, a decrease in firing rate and decrease in the amplitude of Ca2+ transients were observed after ryanodine application, supporting that ryanodine decreases the positive chronotropic effect of isoprenaline (Rigg et al., 2000). In isolated mouse SNCs, in the presence of ryanodine, the chronotropic effect of β-adrenoceptor stimulation was entirely suppressed and a reduction of spontaneous AP frequency was observed after isoprenaline application (Wu et al., 2009). Consistent with this finding, Joung et al. showed that the isoprenaline dose-dependent increase of heart rate was suppressed by ryanodine infusion and SR Ca2+ depletion with ryanodine prevented isoproterenol-induced LDCaE and blunted sinus rate acceleration in canine right atrium (Joung et al., 2009).
It is important to note that the ryanodine effect highly depends on the concentration and experiment time (Rousseau et al., 1987; Smith et al., 1988; Buck et al., 1992; Zimanyi et al., 1992; Fill and Copello, 2002). In low doses, it locks the RyR in a subconductance open state (Buck et al., 1992). Before the SR depletion, the Ca2+ flux increases the firing rate indicating the direct role of Ca2+ and NCX in pacemaking (Rubenstein and Lipsius, 1989). The AP ignition model (Lyashkov et al., 2018) predicts that ryanodine substantially depolarizes the ignition potential and prolongs both CL and time-to-ignition, but does not significantly affect the MDP. Additional simulations with Ca2+ dynamics showed that Ca2+ release continues in the presence of SR Ca2+ leak (the leaky SR model mimics ryanodine-dependent lock of RyRs in the subconductance state) but it becomes persistent. In this case, the diffusional resistance between the network and junctional SR limits intra-SR flux and preserves the Ca2+-load of the network SR. This maintains SR Ca2+ load and drives persistent release flux (Lyashkov et al., 2018).
Ca2+-Dependence of If
A possible Ca2+-dependence of the funny current would raise serious questions since the maneuvers to alter Ca2+i levels were generally considered to influence the SR-NCX axis only, without interfering with If. Hagiwara and Irisawa (1989) demonstrated Ca2+ dependent changes of If (Hagiwara and Irisawa, 1989). Later inside-out patch clamp experiments revealed that Ca2+ ions do not directly influence the If current (Zaza et al., 1991). However, a Ca2+-activated adenylate-cyclase isoform was found in SNCs (Mattick et al., 2007), indicating that a Ca2+ rise after NCX inhibition will induce augmented funny current. This effect is also proposed to counterbalance the effect of NCX inhibition on the CL. Furthermore, it may be another link between Ca2+ homeostasis and Vm. The actual value of the MDP is mainly defined by the potassium conductance during repolarization. Therefore, potassium channels indirectly influence automaticity. The small conductance Ca2+ activated potassium current may provide a direct link between Ca2+ handling and repolarization. Even though no effect was found in ventricular myocardium (Nagy et al., 2009), a possible contribution for pacemaking was reported (Torrente et al., 2017; Saeki et al., 2019).
Intracellular Buffering
The forward mode of the NCX extrudes Ca2+ from the intracellular space while generating inward current that contributes to DD. Since the primary driver of NCX during DD is the Ca2+ concentration, buffering of intracellular Ca2+ affects the role of NCX and potentially attenuates or terminates spontaneous automaticity. Several authors challenged the role of SR release in setting the actual frequency of SN pacemaking by application of the rapid Ca2+ buffer BAPTA. The results are summarized in Table 2. The data clearly demonstrate the significant role of Ca2+i in the SN pacemaking, i.e., the function of the inward NCX during DD. A contradictory result was published by Himeno et al., where unaffected AP firing rate was demonstrated in the presence of high Ca2+i buffering, thus challenging the Ca2+ hypothesis (Himeno et al., 2011). Their results were later interpreted by the Lakatta group (Maltsev et al., 2011b) as methodological problems, i.e. incomplete seal resistance. When rabbit cells were loaded with the caged Ca2+ buffer NP-EGTA, the firing rate was slow and dysrhythmic with low SR Ca2+ levels. Rapid photolysis induced Ca2+ increase, in turn, markedly increased the frequency (by about 50%), accelerated the DD, augmented the LCRs, and reduced the CL variability (Yaniv et al., 2011).
Table 2 Effects of BAPTA on spontaneous firing rate of SNCs.
Pharmacological Problems
Important pharmacological problems arose regarding If and NCX inhibitors. The contribution of If was traditionally investigated using caesium or ivabradine. The heart rate lowering effects of these compounds varied within a large range (7–31%) and even 20 mM caesium was unable to terminate spontaneous SN beating (Sohn and Vassalle, 1995). Therefore, the pacemaking role of If was regularly challenged by considering its I–V relationship (small current in the DD range), slow activation kinetics, and moderate effects of inhibitors (DiFrancesco, 1995; Vassalle, 1995; Bucchi et al., 2007a; Lakatta and DiFrancesco, 2009). However, both CsCl2 and ivabradine cause incomplete and voltage-dependent block. A further issue with ivabradine is the significant IKr block even at the widely used concentration of 3 μM (Koncz et al., 2011). Similar situations can be found with selective NCX inhibitors. Both ORM-10103 and ORM-10962 however, exert no influence on other currents and partially inhibit NCX, may also depend on Ca2+. As discussed above (What is the Role of NCX in Pacemaking Under Resting Heart Rate)?, increased Ca2+ limits the effect of NCX inhibition via secondary effects and covers the exact role of NCX during DD. Taking together these results, it seems likely that complete pharmacological inhibition of If or NCX is currently not feasible using the available compounds, impeding the identification of the role of NCX and If in SN pacemaking.
An indirect experiment was also published in 2015 by Szepesi et al. (2015) using 10 µM ORM-10103 as a selective NCX inhibitor (Jost et al., 2013) in Langendorff perfused rat hearts. The results exerted identical R-R intervals after application of selective NCX blockade.
In 2008, Farkas et al. investigated the possible antiarrhythmic effect of selective NCX inhibition against dofetilide-induced Torsades de pointes arrhythmia by using 1 µM SEA0400, a nonselective NCX inhibitor in rabbit and rat Langendorff-perfused hearts (Farkas et al., 2008). However, the measured R-R intervals indicated no effect on sinus function of NCX inhibition in both species. In contrast, by using the most selective available NCX inhibitor ORM-10962, Kohajda et al. showed a tendency to decrease the heart rate in guinea pig hearts in vivo, however within the experimental variance (Kohajda et al., 2016). 1 µM ORM-10962 exerted statistically significant, however marginal (8%) decrease in rabbit atrial tissue, while Ca2+i was significantly increased in SNCs (Kohajda et al., 2020).
Applications
Besides its fundamental role for pacemaking under physiological conditions, various applications of engineered NCX and potential roles in pathogenesis have been put forward. Silva and Rudy (2003) numerically replicated biological pacemakers derived from guinea pig ventricular cells (Silva and Rudy, 2003). Suppression of IK1 by 81% elicited rhythmic spontaneous depolarizations at a CL of 594 ms caused by NCX as the main pacemaking current. The role of NCX was underlined by a bifurcation analysis of biological pacemaking driven by an unstable equilibrium point via a saddle-node bifurcation by Kurata et al. (2005) using a modified Priebe-Beuckelmann model in which NCX was identified as the primary pacemaker current (Kurata et al., 2005). However, they found that NCX is not necessarily required for rhythmic spontaneous depolarizations and that equilibrium point instability as a prerequisite for stable pacemaking is caused by ICaL but not NCX. Jones et al. (2012) identified NCX as one of the key determinants of pulmonary vein automaticity, i.e. pacemaking in non-SN cells, which is considered as one of the most important triggers initiating atrial fibrillation (Jones et al., 2012). Youm et al. (2006) modeled pacemaker activity in mouse small intestine, i.e., noncardiac, cells and found a role of NCX for automaticity in these cells as well (Youm et al., 2006).
Loewe et al. investigated the effect of extracellular Ca2+ concentration changes on SN pacemaking in the LLFS computational model motivated by the observation that the heart rate of dialysis patients, who experience marked shifts of extracellular ion concentrations, drops to very low values before they suffer from sudden cardiac death with an unexplained high incidence (Loewe et al., 2019a). They found that Ca2+ changes markedly affected the beating rate (46 bpm/mM ionized Ca2+ without autonomic control). While this pronounced bradycardic effect of hypocalcemia was mediated primarily by ICaL, NCX was subsequently attenuated due to the reduction of Ca2+i. Wolf et al. (2013) proposed a computational model of ankyrin-B syndrome SNCs in which alterations of ICaL and NCX as well as INaK due to ankyrin-B dysfunction increase variability in SN automaticity (Wolf et al., 2013). Choudhury et al. (2018) overexpressed NCX1 in bradycardic rat subsidiary atrial pacemaker tissue and found that it did not accelerate the rate of spontaneous depolarizations in contrast to overexpression of TBX18, which increased rate as well as stability and rescued isoproterenol response (Choudhury et al., 2018).
Concluding Remarks
A large body of experimental evidence indicates and mechanistic in silico modeling supports the crucial role of NCX in SN automaticity both under normal heart rate as well as during β-adrenergic stimulation. However, SN automaticity probably could be one of the areas of experimental cardiology where concise hypotheses have been derived from computational modeling that call for experimental testing which is not yet on the horizon, especially in context of NCX function. The complex effects of drugs modulating the Ca2+ handling (CPA, ryanodine, isoproterenol), the shortcomings of transgenic animal models, and the nonselectivity and/or indirect effects of parallel Ca2+ increase of selective inhibitors indicate a clear demand of further experimental research to fully reveal the role of NCX in SN pacemaking.
Author Contributions
AL—preparing the manuscript: in silico mechanistic modeling, preparing the figures. AV—supervising the study. NN—preparing the manuscript: experimental results of the NCX function in SN. ZK, NT—searching of relevant publications, preparing subchapters of the manuscript.
Funding
We declare that all sources of funding received is submitted.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the National Research Development and Innovation Office (NKFIH PD-125402 (for NN), FK-129117 (for NN), GINOP-2.3.2-15-2016-00006 and GINOP-2.3.2-15-2016-00012), the LIVE LONGER EFOP-3.6.2-16-2017-00006 project, the János Bolyai Research Scholarship of the Hungarian Academy of Sciences (for NN), the UNKP-18-4-SZTE-76 New National Excellence Program of the Ministry for Innovation and Technology (for NN), the EFOP 3.6.3 VEKOP-16-2017-00009 (for NT). AL gratefully acknowledges financial support by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—Project-ID 399 258734477—SFB 1173 and through Project-ID 391128822-LO 2093/1-1. University of Szeged Open Access Fund, No.: 4314.
Abbreviations
AP, action potential; APD, action potential duration; ATP, adenosine triphosphate; Ca2+/CA, Ca2+-cation antiporter; Ca2+i, intracellular Ca2+ concentration; CaM, calmodulin; CaMKII, Ca2+/calmodulin-dependent protein kinase; cAMP, cyclic adenosine monophosphate; CICR, Ca2+-induced Ca2+ release; CL, cycle length; CRU, Ca2+- release unit; DD, diastolic depolarization; DDR, diastolic depolarization rate; EC50, half maximal effective concentration; ENCX, NCX equilibrium potential; GPCR, G-protein coupled receptor; hESC-CM, human embryonic stem cell-derived cardiomyocyte; IC50, half maximal inhibitory concentration; ICaL, L-type Ca2+-current; ICaT, T-type Ca2+-current; If, funny-current; IK1, inward rectifier potassium current; IK(Ca): small-conductance Ca2+-activated K+ current; IKr, rapid delayed rectifier potassium current; IKs, slow delayed rectifier potassium current; Ito, transient outward potassium current; LCR, local Ca2+ releases; LDCaE, late diastolic Ca2+ elevation; LLFS model, Loewe–Lutz–Fabbri–Severi model; M-clock, membrane clock; MDP, maximal diastolic potential; Na+i, intracellular Na+ concentration; NCX, Na+/Ca2+ exchanger; NO, nitrogen-monoxide; PDE, phosphodiesterase; PKA, protein kinase A; PKC, protein kinase C; PLB, phospholamban; PP1, protein phosphatase 1; PP2A, protein phosphatase 2; RyR, ryanodine receptor; SERCA, sarcoplasmic reticulum Ca2+ ATPase; SN, sinus node; SNC, sinus node cell; SR, sarcoplasmic reticulum; Vm, transmembrane potential.
References
Allah, E. A., Tellez, J. O., Yanni, J., Nelson, T., Monfredi, O., Boyett, M. R., et al. (2011). Changes in the expression of ion channels, connexins and Ca2+-handling proteins in the sino-atrial node during postnatal development. Exp. Physiol. 96 (4), 426–438. doi: 10.1113/expphysiol.2010.055780
Allen, D. G., Eisner, D. A., Lab, M. J., Orchard, C. H. (1983). The effects of low sodium solutions on intracellular calcium concentration and tension in ferret ventricular muscle. J. Physiol. 345, 391–407. doi: 10.1113/jphysiol.1983.sp014984
Amoroso, S., Tortiglione, A., Secondo, A., Catalano, A., Montagnani, S., Di Renzo, G., et al. (2000). Sodium nitroprusside prevents chemical hypoxia-induced cell death through iron ions stimulating the activity of the Na+-Ca2+ exchanger in C6 glioma cells. J. Neurochem. 74 (4), 1505–1513. doi: 10.1046/j.1471-4159.2000.0741505.x
Ballard, C., Schaffer, S. (1996). Stimulation of the Na+/Ca2+ exchanger by phenylephrine, angiotensin II and endothelin 1. J. Mol. Cell Cardiol. 28 (1), 11–17. doi: 10.1006/jmcc.1996.0002
Baruscotti, M., Bucchi, A., Viscomi, C., Mandelli, G., Consalez, G., Gnecchi-Rusconi, T., et al. (2011). Deep bradycardia and heart block caused by inducible cardiac-specific knockout of the pacemaker channel gene Hcn4. Proc. Natl. Acad. Sci. U. S. A 108 (4), 1705–1710. doi: 10.1073/pnas.1010122108
Bers, D. M., Ginsburg, K. S. (2007). Na:Ca stoichiometry and cytosolic Ca-dependent activation of NCX in intact cardiomyocytes. Ann. N. Y. Acad. Sci. 1099, 326–338. doi: 10.1196/annals.1387.060
Bers, D. M. (2001). Excitation-Contraction Coupling and Cardiac Contractile Force (Netherlands: Springer). doi: 10.1007/978-94-010-0658-3
Bers, D. M. (2002). Cardiac excitation-contraction coupling. Nature 415 (6868), 198–205. doi: 10.1038/415198a
Bers, D. M. (2008). Calcium cycling and signaling in cardiac myocytes. Annu. Rev. Physiol. 70, 23–49. doi: 10.1146/annurev.physiol.70.113006.100455
Birinyi, P., Acsai, K., Banyasz, T., Toth, A., Horvath, B., Virag, L., et al. (2005). Effects of SEA0400 and KB-R7943 on Na+/Ca2+ exchange current and L-type Ca2+ current in canine ventricular cardiomyocytes. Naunyn Schmiedebergs Arch. Pharmacol. 372 (1), 63–70. doi: 10.1007/s00210-005-1079-x
Bleeker, W. K., Mackaay, A. J., Masson-Pevet, M., Bouman, L. N., Becker, A. E. (1980). Functional and morphological organization of the rabbit sinus node. Circ. Res. 46 (1), 11–22. doi: 10.1161/01.res.46.1.11
Bogdanov, K. Y., Vinogradova, T. M., Lakatta, E. G. (2001). Sinoatrial nodal cell ryanodine receptor and Na(+)-Ca(2+) exchanger: molecular partners in pacemaker regulation. Circ. Res. 88 (12), 1254–1258. doi: 10.1161/hh1201.092095
Bogdanov, K. Y., Maltsev, V. A., Vinogradova, T. M., Lyashkov, A. E., Spurgeon, H. A., Stern, M. D., et al. (2006). Membrane potential fluctuations resulting from submembrane Ca2+ releases in rabbit sinoatrial nodal cells impart an exponential phase to the late diastolic depolarization that controls their chronotropic state. Circ. Res. 99 (9), 979–987. doi: 10.1161/01.RES.0000247933.66532.0b
Boyett, M. R., Honjo, H., Yamamoto, M., Nikmaram, M. R., Niwa, R., Kodama, I. (1998). Regional differences in effects of 4-aminopyridine within the sinoatrial node. Am. J. Physiol. 275 (4), H1158–H1168. doi: 10.1152/ajpheart.1998.275.4.H1158
Boyett, M. R., Honjo, H., Kodama, I. (2000). The sinoatrial node, a heterogeneous pacemaker structure. Cardiovasc. Res. 47 (4), 658–687. doi: 10.1016/s0008-6363(00)00135-8
Brown, H. F., Kimura, J., Noble, D., Noble, S. J., Taupignon, A. (1984). The slow inward current, isi, in the rabbit sino-atrial node investigated by voltage clamp and computer simulation. Proc. R. Soc. Lond. B. Biol. Sci. 222 (1228), 305–328. doi: 10.1098/rspb.1984.0066
Bucchi, A., Baruscotti, M., Robinson, R. B., DiFrancesco, D. (2003). I(f)-dependent modulation of pacemaker rate mediated by cAMP in the presence of ryanodine in rabbit sino-atrial node cells. J. Mol. Cell Cardiol. 35 (8), 905–913. doi: 10.1016/S0022-2828(03)00150-0
Bucchi, A., Barbuti, A., Baruscotti, M., DiFrancesco, D. (2007a). Heart rate reduction via selective ‘funny' channel blockers. Curr. Opin. Pharmacol. 7 (2), 208–213. doi: 10.1016/j.coph.2006.09.005
Bucchi, A., Baruscotti, M., Robinson, R. B., DiFrancesco, D. (2007b). Modulation of rate by autonomic agonists in SAN cells involves changes in diastolic depolarization and the pacemaker current. J. Mol. Cell Cardiol. 43 (1), 39–48. doi: 10.1016/j.yjmcc.2007.04.017
Buck, E., Zimanyi, I., Abramson, J. J., Pessah, I. N. (1992). Ryanodine stabilizes multiple conformational states of the skeletal muscle calcium release channel. J. Biol. Chem. 267 (33), 23560–23567.
Chandler, N. J., Greener, I. D., Tellez, J. O., Inada, S., Musa, H., Molenaar, P., et al. (2009). Molecular architecture of the human sinus node: insights into the function of the cardiac pacemaker. Circulation 119 (12), 1562–1575. doi: 10.1161/CIRCULATIONAHA.108.804369
Chapman, R. A., Tunstall, J. (1980). The interaction of sodium and calcium ions at the cell membrane and the control of contractile strength in frog atrial muscle. J. Physiol. 305, 109–123. doi: 10.1113/jphysiol.1980.sp013353
Cheng, H., Lederer, W. J., Cannell, M. B. (1993). Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science 262 (5134), 740–744. doi: 10.1126/science.8235594
Choi, S. W., Lee, H. A., Moon, S. H., Park, S. J., Kim, H. J., Kim, K. S., et al. (2016). Spontaneous inward currents reflecting oscillatory activation of Na(+)/Ca(2)(+) exchangers in human embryonic stem cell-derived cardiomyocytes. Pflugers Arch. 468 (4), 609–622. doi: 10.1007/s00424-015-1769-2
Choudhury, M., Black, N., Alghamdi, A., D'Souza, A., Wang, R., Yanni, J., et al. (2018). TBX18 overexpression enhances pacemaker function in a rat subsidiary atrial pacemaker model of sick sinus syndrome. J. Physiol. 596 (24), 6141–6155. doi: 10.1113/JP276508
Courtemanche, M., Ramirez, R. J., Nattel, S. (1998). Ionic mechanisms underlying human atrial action potential properties: insights from a mathematical model. Am. J. Physiol. 275 (1), H301–H321. doi: 10.1152/ajpheart.1998.275.1.H301
Demir, S. S., Clark, J. W., Murphey, C. R., Giles, W. R. (1994). A mathematical model of a rabbit sinoatrial node cell. Am. J. Physiol. 266 (3 Pt 1), C832–C852. doi: 10.1152/ajpcell.1994.266.3.C832
DiFrancesco, D., Noble, D. (1985). A model of cardiac electrical activity incorporating ionic pumps and concentration changes. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 307 (1133), 353–398. doi: 10.1098/rstb.1985.0001
DiFrancesco, D. (1991). The contribution of the ‘pacemaker' current (if) to generation of spontaneous activity in rabbit sino-atrial node myocytes. J. Physiol. 434, 23–40. doi: 10.1113/jphysiol.1991.sp018457
DiFrancesco, D. (1993). Pacemaker mechanisms in cardiac tissue. Annu. Rev. Physiol. 55, 455–472. doi: 10.1146/annurev.ph.55.030193.002323
DiFrancesco, D. (1995). Cesium and the pacemaker current. J. Cardiovasc. Electrophysiol. 6 (12), 1152–1155. doi: 10.1111/j.1540-8167.1995.tb00394.x
DiFrancesco, D. (2020). A Brief History of Pacemaking. Front. Physiol. 10, 1599. doi: 10.3389/fphys.2019.01599
Dokos, S., Celler, B., Lovell, N. (1996). Ion currents underlying sinoatrial node pacemaker activity: a new single cell mathematical model. J. Theor. Biol. 181 (3), 245–272. doi: 10.1006/jtbi.1996.0129
Fabbri, A., Fantini, M., Wilders, R., Severi, S. (2017). Computational analysis of the human sinus node action potential: model development and effects of mutations. J. Physiol. 595 (7), 2365–2396. doi: 10.1113/JP273259
Farkas, A. S., Acsai, K., Nagy, N., Toth, A., Fulop, F., Seprenyi, G., et al. (2008). Na(+)/Ca(2+) exchanger inhibition exerts a positive inotropic effect in the rat heart, but fails to influence the contractility of the rabbit heart. Br. J. Pharmacol. 154 (1), 93–104. doi: 10.1038/bjp.2008.83
Fill, M., Copello, J. A. (2002). Ryanodine receptor calcium release channels. Physiol. Rev. 82 (4), 893–922. doi: 10.1152/physrev.00013.2002
Fischmeister, R., Vassort, G. (1981). The electrogenic Na-Ca exchange and the cardiac electrical activity. I--Simulation on Purkinje fibre action potential. J Physiol (Paris) 77 (6–7), 705–709.
Gao, Z., Chen, B., Joiner, M. L., Wu, Y., Guan, X., Koval, O. M., et al. (2010). I(f) and SR Ca(2+) release both contribute to pacemaker activity in canine sinoatrial node cells. J. Mol. Cell Cardiol. 49 (1), 33–40. doi: 10.1016/j.yjmcc.2010.03.019
Gao, Z., Singh, M. V., Hall, D. D., Koval, O. M., Luczak, E. D., Joiner, M. L., et al. (2011). Catecholamine-independent heart rate increases require Ca2+/calmodulin-dependent protein kinase II. Circ. Arrhythm. Electrophysiol. 4 (3), 379–387. doi: 10.1161/CIRCEP.110.961771
Gao, Z., Rasmussen, T. P., Li, Y., Kutschke, W., Koval, O. M., Wu, Y., et al. (2013). Genetic inhibition of Na+-Ca2+ exchanger current disables fight or flight sinoatrial node activity without affecting resting heart rate. Circ. Res. 112 (2), 309–317. doi: 10.1161/CIRCRESAHA.111.300193
Garny, A., Kohl, P., Hunter, P. J., Boyett, M. R., Noble, D. (2003). One-dimensional rabbit sinoatrial node models: benefits and limitations. J. Cardiovasc. Electrophysiol. 14 (10 Suppl.), S121–S132. doi: 10.1046/j.1540.8167.90301.x
Ginsburg, K. S., Bers, D. M. (2005). Isoproterenol does not enhance Ca-dependent Na/Ca exchange current in intact rabbit ventricular myocytes. J. Mol. Cell Cardiol. 39 (6), 972–981. doi: 10.1016/j.yjmcc.2005.09.005
Ginsburg, K. S., Weber, C. R., Bers, D. M. (2013). Cardiac Na+-Ca2+ exchanger: dynamics of Ca2+-dependent activation and deactivation in intact myocytes. J. Physiol. 591 (8), 2067–2086. doi: 10.1113/jphysiol.2013.252080
Groenke, S., Larson, E. D., Alber, S., Zhang, R., Lamp, S. T., Ren, X., et al. (2013). Complete atrial-specific knockout of sodium-calcium exchange eliminates sinoatrial node pacemaker activity. PLoS One 8 (11), e81633. doi: 10.1371/journal.pone.0081633
Gupta, M. P., Makino, N., Khatter, K., Dhalla, N. S. (1986). Stimulation of Na+-Ca2+ exchange in heart sarcolemma by insulin. Life Sci. 39 (12), 1077–1083. doi: 10.1016/0024-3205(86)90199-2
Hagiwara, N., Irisawa, H. (1989). Modulation by intracellular Ca2+ of the hyperpolarization-activated inward current in rabbit single sino-atrial node cells. J. Physiol. 409, 121–141. doi: 10.1113/jphysiol.1989.sp017488
Harzheim, D., Pfeiffer, K. H., Fabritz, L., Kremmer, E., Buch, T., Waisman, A., et al. (2008). Cardiac pacemaker function of HCN4 channels in mice is confined to embryonic development and requires cyclic AMP. EMBO J. 27 (4), 692–703. doi: 10.1038/emboj.2008.3
Herrmann, S., Stieber, J., Stockl, G., Hofmann, F., Ludwig, A. (2007). HCN4 provides a ‘depolarization reserve' and is not required for heart rate acceleration in mice. EMBO J. 26 (21), 4423–4432. doi: 10.1038/sj.emboj.7601868
Herrmann, S., Lipp, P., Wiesen, K., Stieber, J., Nguyen, H., Kaiser, E., et al. (2013). The cardiac sodium-calcium exchanger NCX1 is a key player in the initiation and maintenance of a stable heart rhythm. Cardiovasc. Res. 99 (4), 780–788. doi: 10.1093/cvr/cvt154
Himeno, Y., Sarai, N., Matsuoka, S., Noma, A. (2008). Ionic mechanisms underlying the positive chronotropy induced by beta1-adrenergic stimulation in guinea pig sinoatrial node cells: a simulation study. J. Physiol. Sci. 58 (1), 53–65. doi: 10.2170/physiolsci.RP015207
Himeno, Y., Toyoda, F., Satoh, H., Amano, A., Cha, C. Y., Matsuura, H., et al. (2011). Minor contribution of cytosolic Ca2+ transients to the pacemaker rhythm in guinea pig sinoatrial node cells. Am. J. Physiol. Heart Circ. Physiol. 300 (1), H251–H261. doi: 10.1152/ajpheart.00764.2010
Hoang-Trong, T. M., Ullah, A., Jafri, M. S. (2015). Calcium Sparks in the Heart: Dynamics and Regulation. Res. Rep. Biol. 6, 203–214. doi: 10.2147/RRB.S61495
Hoesl, E., Stieber, J., Herrmann, S., Feil, S., Tybl, E., Hofmann, F., et al. (2008). Tamoxifen-inducible gene deletion in the cardiac conduction system. J. Mol. Cell Cardiol. 45 (1), 62–69. doi: 10.1016/j.yjmcc.2008.04.008
Honjo, H., Boyett, M. R., Kodama, I., Toyama, J. (1996). Correlation between electrical activity and the size of rabbit sino-atrial node cells. J. Physiol. 496 ( Pt 3), 795–808. doi: 10.1113/jphysiol.1996.sp021728
Huser, J., Blatter, L. A., Lipsius, S. L. (2000). Intracellular Ca2+ release contributes to automaticity in cat atrial pacemaker cells. J. Physiol. 524 Pt 2, 415–422. doi: 10.1111/j.1469-7793.2000.00415.x
Imtiaz, M. S., von der Weid, P. Y., Laver, D. R., van Helden, D. F. (2010). SR Ca2+ store refill–a key factor in cardiac pacemaking. J. Mol. Cell Cardiol. 49 (3), 412–426. doi: 10.1016/j.yjmcc.2010.03.015
Iwamoto, T., Wakabayashi, S., Shigekawa, M. (1995). Growth factor-induced phosphorylation and activation of aortic smooth muscle Na+/Ca2+ exchanger. J. Biol. Chem. 270 (15), 8996–9001. doi: 10.1074/jbc.270.15.8996
Iwamoto, T., Watano, T., Shigekawa, M. (1996). A novel isothiourea derivative selectively inhibits the reverse mode of Na+/Ca2+ exchange in cells expressing NCX1. J. Biol. Chem. 271 (37), 22391–22397. doi: 10.1074/jbc.271.37.22391
Iwamoto, T., Pan, Y., Nakamura, T. Y., Wakabayashi, S., Shigekawa, M. (1998). Protein kinase C-dependent regulation of Na+/Ca2+ exchanger isoforms NCX1 and NCX3 does not require their direct phosphorylation. Biochemistry 37 (49), 17230–17238. doi: 10.1021/bi981521q
Jones, G., Spencer, B. D., Adeniran, I., Zhang, H. (2012). Development of biophysically detailed electrophysiological models for pacemaking and non-pacemaking human pulmonary vein cardiomyocytes. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2012, 199–202. doi: 10.1109/EMBC.2012.6345905
Jost, N., Nagy, N., Corici, C., Kohajda, Z., Horvath, A., Acsai, K., et al. (2013). ORM-10103, a novel specific inhibitor of the Na+/Ca2+ exchanger, decreases early and delayed afterdepolarizations in the canine heart. Br. J. Pharmacol. 170 (4), 768–778. doi: 10.1111/bph.12228
Joung, B., Tang, L., Maruyama, M., Han, S., Chen, Z., Stucky, M., et al. (2009). Intracellular calcium dynamics and acceleration of sinus rhythm by beta-adrenergic stimulation. Circulation 119 (6), 788–796. doi: 10.1161/CIRCULATIONAHA.108.817379
Ju, Y. K., Allen, D. G. (1998). Intracellular calcium and Na+-Ca2+ exchange current in isolated toad pacemaker cells. J. Physiol. 508 ( Pt 1), 153–166. doi: 10.1111/j.1469-7793.1998.153br.x
Ju, Y. K., Saint, D. A., Hirst, G. D., Gage, P. W. (1995). Sodium currents in toad cardiac pacemaker cells. J. Membr. Biol. 145 (2), 119–128. doi: 10.1007/bf00237370
Kaese, S., Bogeholz, N., Pauls, P., Dechering, D., Olligs, J., Kolker, K., et al. (2017). Increased sodium/calcium exchanger activity enhances beta-adrenergic-mediated increase in heart rate: Whole-heart study in a homozygous sodium/calcium exchanger overexpressor mouse model. Heart Rhythm. 14 (8), 1247–1253. doi: 10.1016/j.hrthm.2017.05.001
Kharche, S., Yu, J., Lei, M., Zhang, H. (2011). A mathematical model of action potentials of mouse sinoatrial node cells with molecular bases. Am. J. Physiol. Heart Circ. Physiol. 301 (3), H945–H963. doi: 10.1152/ajpheart.00143.2010
Kim, M. S., Maltsev, A. V., Monfredi, O., Maltseva, L. A., Wirth, A., Florio, M. C., et al. (2018). Heterogeneity of calcium clock functions in dormant, dysrhythmically and rhythmically firing single pacemaker cells isolated from SA node. Cell Calcium 74, 168–179. doi: 10.1016/j.ceca.2018.07.002
Kofuji, P., Lederer, W. J., Schulze, D. H. (1994). Mutually exclusive and cassette exons underlie alternatively spliced isoforms of the Na/Ca exchanger. J. Biol. Chem. 269 (7), 5145–5149.
Kohajda, Z., Farkas-Morvay, N., Jost, N., Nagy, N., Geramipour, A., Horvath, A., et al. (2016). The Effect of a Novel Highly Selective Inhibitor of the Sodium/Calcium Exchanger (NCX) on Cardiac Arrhythmias in In Vitro and In Vivo Experiments. PLoS One 11 (11), e0166041. doi: 10.1371/journal.pone.0166041
Kohajda, Z., Tóth, N., Szlovák, J., Loewe, A., Bitay, G., Gazdag, P., et al. (2020). Novel Na+/Ca2+ Exchanger Inhibitor ORM-10962 Supports Coupled Function of Funny-Current and Na+/Ca2+ Exchanger in Pacemaking of Rabbit Sinus Node Tissue. Front. Pharmacol. 10, 1632. doi: 10.3389/fphar.2019.01632
Koncz, I., Szel, T., Bitay, M., Cerbai, E., Jaeger, K., Fulop, F., et al. (2011). Electrophysiological effects of ivabradine in dog and human cardiac preparations: potential antiarrhythmic actions. Eur. J. Pharmacol. 668 (3), 419–426. doi: 10.1016/j.ejphar.2011.07.025
Kurata, Y., Hisatome, I., Imanishi, S., Shibamoto, T. (2002). Dynamical description of sinoatrial node pacemaking: improved mathematical model for primary pacemaker cell. Am. J. Physiol. Heart Circ. Physiol. 283 (5), H2074–H2101. doi: 10.1152/ajpheart.00900.2001
Kurata, Y., Hisatome, I., Matsuda, H., Shibamoto, T. (2005). Dynamical mechanisms of pacemaker generation in IK1-downregulated human ventricular myocytes: insights from bifurcation analyses of a mathematical model. Biophys. J. 89 (4), 2865–2887. doi: 10.1529/biophysj.105.060830
Kurata, Y., Hisatome, I., Shibamoto, T. (2012). Roles of sarcoplasmic reticulum Ca2+ cycling and Na+/Ca2+ exchanger in sinoatrial node pacemaking: insights from bifurcation analysis of mathematical models. Am. J. Physiol. Heart Circ. Physiol. 302 (11), H2285–H2300. doi: 10.1152/ajpheart.00221.2011
Lakatta, E. G., DiFrancesco, D. (2009). What keeps us ticking: a funny current, a calcium clock, or both? J. Mol. Cell Cardiol. 47 (2), 157–170. doi: 10.1016/j.yjmcc.2009.03.022
Lakatta, E. G., Maltsev, V. A. (2012). Rebuttal: what I(f) the shoe doesn't fit? “The funny current has a major pacemaking role in the sinus node”. Heart Rhythm. 9 (3), 459–460. doi: 10.1016/j.hrthm.2011.09.024
Lakatta, E. G., Maltsev, V. A., Vinogradova, T. M. (2010). A coupled SYSTEM of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart's pacemaker. Circ. Res. 106 (4), 659–673. doi: 10.1161/CIRCRESAHA.109.206078
Lancaster, M. K., Jones, S. A., Harrison, S. M., Boyett, M. R. (2004). Intracellular Ca2+ and pacemaking within the rabbit sinoatrial node: heterogeneity of role and control. J. Physiol. 556 (Pt 2), 481–494. doi: 10.1113/jphysiol.2003.057372
Li, Z., Matsuoka, S., Hryshko, L. V., Nicoll, D. A., Bersohn, M. M., Burke, E. P., et al. (1994). Cloning of the NCX2 isoform of the plasma membrane Na(+)-Ca2+ exchanger. J. Biol. Chem. 269 (26), 17434–17439.
Li, J., Qu, J., Nathan, R. D. (1997). Ionic basis of ryanodine's negative chronotropic effect on pacemaker cells isolated from the sinoatrial node. Am. J. Physiol. 273 (5), H2481–H2489. doi: 10.1152/ajpheart.1997.273.5.H2481
Li, P., Lines, G., Maleckar, M., Tveito, A. (2013). Mathematical models of cardiac pacemaking function. Front. Phys. 1, 20. doi: 10.3389/fphy.2013.00020
Li, Y., Sirenko, S., Riordon, D. R., Yang, D., Spurgeon, H., Lakatta, E. G., et al. (2016). CaMKII-dependent phosphorylation regulates basal cardiac pacemaker function via modulation of local Ca2+ releases. Am. J. Physiol. Heart Circ. Physiol. 311 (3), H532–H544. doi: 10.1152/ajpheart.00765.2015
Lipp, P., Pott, L., Callewaert, G., Carmeliet, E. (1990). Simultaneous recording of Indo-1 fluorescence and Na+/Ca2+ exchange current reveals two components of Ca2(+)-release from sarcoplasmic reticulum of cardiac atrial myocytes. FEBS Lett. 275 (1-2), 181–184. doi: 10.1016/0014-5793(90)81467-3
Liu, J., Sirenko, S., Juhaszova, M., Ziman, B., Shetty, V., Rain, S., et al. (2011). A full range of mouse sinoatrial node AP firing rates requires protein kinase A-dependent calcium signaling. J. Mol. Cell Cardiol. 51 (5), 730–739. doi: 10.1016/j.yjmcc.2011.07.028
Loewe, A., Lutz, Y., Fabbri, A., Severi, S. (2019a). Sinus Bradycardia Due to Electrolyte Changes as a Potential Pathomechanism of Sudden Cardiac Death in Hemodialysis Patients. Biophys. J. 116, 231a. doi: 10.1016/j.bpj.2018.11.1271
Loewe, A., Lutz, Y., Nagy, N., Fabbri, A., Schweda, C., Varró, A., et al. (2019b). Inter-Species Differences in the Response of Sinus Node Cellular Pacemaking to Changes of Extracellular Calcium, in: 2019 41st Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC). 1875–1878. doi: 10.1109/EMBC.2019.8857573
Ludwig, ,. A., Budde, T., Stieber, J., Moosmang, S., Wahl, C., Holthoff, K., et al. (2003). Absence epilepsy and sinus dysrhythmia in mice lacking the pacemaker channel HCN2. EMBO J. 22 (2), 216–224. doi: 10.1093/emboj/cdg032
Luo, C. H., Rudy, Y. (1994). A dynamic model of the cardiac ventricular action potential. I. Simulations of ionic currents and concentration changes. Circ. Res. 74 (6), 1071–96.
Lyashkov, A. E., Juhaszova, M., Dobrzynski, H., Vinogradova, T. M., Maltsev, V. A., Juhasz, O., et al. (2007). Calcium cycling protein density and functional importance to automaticity of isolated sinoatrial nodal cells are independent of cell size. Circ. Res. 100 (12), 1723–1731. doi: 10.1161/CIRCRESAHA.107.153676
Lyashkov, A. E., Vinogradova, T. M., Zahanich, I., Li, Y., Younes, A., Nuss, H. B., et al. (2009). Cholinergic receptor signaling modulates spontaneous firing of sinoatrial nodal cells via integrated effects on PKA-dependent Ca(2+) cycling and I(KACh). Am. J. Physiol. Heart Circ. Physiol. 297 (3), H949–H959. doi: 10.1152/ajpheart.01340.2008
Lyashkov, A. E., Behar, J., Lakatta, E. G., Yaniv, Y., Maltsev, V. A. (2018). Positive Feedback Mechanisms among Local Ca Releases, NCX, and ICaL Ignite Pacemaker Action Potentials. Biophys. J. 114 (5), 1176–1189. doi: 10.1016/j.bpj.2017.12.043
Makino, N., Zhao, D., Dhalla, N. S. (1988). Stimulation of heart sarcolemmal Na+-Ca2+ exchange by concanavalin A. Biochem. Biophys. Res. Commun. 154 (1), 245–251. doi: 10.1016/0006-291x(88)90676-6
Maltsev, V. A., Lakatta, E. G. (2009). Synergism of coupled subsarcolemmal Ca2+ clocks and sarcolemmal voltage clocks confers robust and flexible pacemaker function in a novel pacemaker cell model. Am. J. Physiol. Heart Circ. Physiol. 296 (3), H594–H615. doi: 10.1152/ajpheart.01118.2008
Maltsev, V. A., Lakatta, E. G. (2010). A novel quantitative explanation for the autonomic modulation of cardiac pacemaker cell automaticity via a dynamic system of sarcolemmal and intracellular proteins. Am. J. Physiol. Heart Circ. Physiol. 298 (6), H2010–H2023. doi: 10.1152/ajpheart.00783.2009
Maltsev, V. A., Lakatta, E. G. (2013). Numerical models based on a minimal set of sarcolemmal electrogenic proteins and an intracellular Ca(2+) clock generate robust, flexible, and energy-efficient cardiac pacemaking. J. Mol. Cell Cardiol. 59, 181–195. doi: 10.1016/j.yjmcc.2013.03.004
Maltsev, V. A., Vinogradova, T. M., Bogdanov, K. Y., Lakatta, E. G., Stern, M. D. (2004). Diastolic calcium release controls the beating rate of rabbit sinoatrial node cells: numerical modeling of the coupling process. Biophys. J. 86 (4), 2596–2605. doi: 10.1016/S0006-3495(04)74314-3
Maltsev, V. A., Vinogradova, T. M., Lakatta, E. G. (2006). The emergence of a general theory of the initiation and strength of the heartbeat. J. Pharmacol. Sci. 100 (5), 338–369. doi: 10.1254/jphs.cr0060018
Maltsev, A. V., Maltsev, V. A., Mikheev, M., Maltseva, L. A., Sirenko, S. G., Lakatta, E. G., et al. (2011a). Synchronization of stochastic Ca(2)(+) release units creates a rhythmic Ca(2)(+) clock in cardiac pacemaker cells. Biophys. J. 100 (2), 271–283. doi: 10.1016/j.bpj.2010.11.081
Maltsev, V. A., Vinogradova, T. M., Stern, M. D., Lakatta, E. G. (2011b). Letter to the editor: “Validating the requirement for beat-to-beat coupling of the Ca2+ clock and M clock in pacemaker cell normal automaticity”. Am. J. Physiol. Heart Circ. Physiol. 300 (6), H2323–2324; author reply H2325-2326. doi: 10.1152/ajpheart.00110.2011
Maltsev, A. V., Yaniv, Y., Stern, M. D., Lakatta, E. G., Maltsev, V. A. (2013). RyR-NCX-SERCA local cross-talk ensures pacemaker cell function at rest and during the fight-or-flight reflex. Circ. Res. 113 (10), e94–e100. doi: 10.1161/CIRCRESAHA.113.302465
Maltsev, V. A., Yaniv, Y., Maltsev, A. V., Stern, M. D., Lakatta, E. G. (2014). Modern perspectives on numerical modeling of cardiac pacemaker cell. J. Pharmacol. Sci. 125 (1), 6–38. doi: 10.1254/jphs.13r04cr
Maltsev, A. V., Maltsev, V. A., Stern, M. D. (2017). Stabilization of diastolic calcium signal via calcium pump regulation of complex local calcium releases and transient decay in a computational model of cardiac pacemaker cell with individual release channels. PloS Comput. Biol. 13 (8), e1005675. doi: 10.1371/journal.pcbi.1005675
Mangoni, M. E., Couette, B., Bourinet, E., Platzer, J., Reimer, D., Striessnig, J., et al. (2003). Functional role of L-type Cav1.3 Ca2+ channels in cardiac pacemaker activity. Proc. Natl. Acad. Sci. U. S. A 100 (9), 5543–5548. doi: 10.1073/pnas.0935295100
Masson-Pevet, M. A., Bleeker, W. K., Besselsen, E., Treytel, B. W., Jongsma, H. J., Bouman, L. N. (1984). Pacemaker cell types in the rabbit sinus node: a correlative ultrastructural and electrophysiological study. J. Mol. Cell Cardiol. 16 (1), 53–63. doi: 10.1016/s0022-2828(84)80714-2
Matsuoka, S., Hilgemann, D. W. (1992). Steady-state and dynamic properties of cardiac sodium-calcium exchange. Ion and voltage dependencies of the transport cycle. J. Gen. Physiol. 100 (6), 963–1001. doi: 10.1085/jgp.100.6.963
Mattick, P., Parrington, J., Odia, E., Simpson, A., Collins, T., Terrar, D. (2007). Ca2+-stimulated adenylyl cyclase isoform AC1 is preferentially expressed in guinea-pig sino-atrial node cells and modulates the I(f) pacemaker current. J. Physiol. 582 (Pt 3), 1195–1203. doi: 10.1113/jphysiol.2007.133439
Monfredi, O., Maltseva, L. A., Spurgeon, H. A., Boyett, M. R., Lakatta, E. G., Maltsev, V. A. (2013). Beat-to-Beat Variation in Periodicity of Local Calcium Releases Contributes to Intrinsic Variations of Spontaneous Cycle Length in Isolated Single Sinoatrial Node Cells. PLoS One 8 (6), e67247. doi: 10.1371/journal.pone.0067247
Monfredi, O., Tsutsui, K., Ziman, B., Stern, M. D., Lakatta, E. G., Maltsev, V. A. (2018). Electrophysiological heterogeneity of pacemaker cells in the rabbit intercaval region, including the SA node: insights from recording multiple ion currents in each cell. Am. J. Physiol. Heart Circ. Physiol. 314 (3), H403–H414. doi: 10.1152/ajpheart.00253.2016
Mullins, L. J., Requena, J. (1979). Calcium measurement in the periphery of an axon. J. Gen. Physiol. 74 (3), 393–413. doi: 10.1085/jgp.74.3.393
Musa, H., Lei, M., Honjo, H., Jones, S. A., Dobrzynski, H., Lancaster, M. K., et al. (2002). Heterogeneous expression of Ca(2+) handling proteins in rabbit sinoatrial node. J. Histochem. Cytochem. 50 (3), 311–324. doi: 10.1177/002215540205000303
Nagy, N., Szuts, V., Horvath, Z., Seprenyi, G., Farkas, A. S., Acsai, K., et al. (2009). Does small-conductance calcium-activated potassium channel contribute to cardiac repolarization? J. Mol. Cell Cardiol. 47 (5), 656–663. doi: 10.1016/j.yjmcc.2009.07.019
Nicoll, D. A., Longoni, S., Philipson, K. D. (1990). Molecular cloning and functional expression of the cardiac sarcolemmal Na(+)-Ca2+ exchanger. Science 250 (4980), 562–565. doi: 10.1126/science.1700476
Nicoll, D. A., Quednau, B. D., Qui, Z., Xia, Y. R., Lusis, A. J., Philipson, K. D. (1996). Cloning of a third mammalian Na+-Ca2+ exchanger, NCX3. J. Biol. Chem. 271 (40), 24914–24921. doi: 10.1074/jbc.271.40.24914
Noble, D., Noble, S. J. (1984). A model of sino-atrial node electrical activity based on a modification of the DiFrancesco-Noble, (1984) equations. Proc. R. Soc. Lond. B. Biol. Sci. 222 (1228), 295–304. doi: 10.1098/rspb.1984.0065
Noble, D. (1962). A modification of the Hodgkin–Huxley equations applicable to Purkinje fibre action and pace-maker potentials. J. Physiol. 160, 317–352. doi: 10.1113/jphysiol.1962.sp006849
Opthof, T. (2001). Function and structure of the mouse sinus node: nothing you can see that isn't shown. Cardiovasc. Res. 52 (1), 1–4. doi: 10.1016/s0008-6363(01)00417-5
Osipchuk, Y. V., Wakui, M., Yule, D. I., Gallacher, D. V., Petersen, O. H. (1990). Cytoplasmic Ca2+ oscillations evoked by receptor stimulation, G-protein activation, internal application of inositol trisphosphate or Ca2+: simultaneous microfluorimetry and Ca2+ dependent Cl- current recording in single pancreatic acinar cells. EMBO J. 9 (3), 697–704. doi: 10.1002/j.1460-2075.1990.tb08162.x
Ottolia, M., Schumann, S., Nicoll, D. A., Philipson, K. D. (2002). Activation of the cardiac Na+/Ca2+ exchanger by DEPC. Ann. N. Y. Acad. Sci. 976, 85–88. doi: 10.1111/j.1749-6632.2002.tb04720.x
Priebe, L., Beuckelmann, D. J. (1998). Simulation study of cellular electric properties in heart failure. Circ. Res. 82 (11), 1206–1223.
Qu, Y., Baroudi, G., Yue, Y., El-Sherif, N., Boutjdir, M. (2005). Localization and modulation of {alpha}1D (Cav1.3) L-type Ca channel by protein kinase A. Am. J. Physiol. Heart Circ. Physiol. 288 (5), H2123–H2130. doi: 10.1152/ajpheart.01023.2004
Rasmusson, R. L., Clark, J. W., Giles, W. R., Robinson, K., Clark, R. B., Shibata, E. F., et al. (1990). A mathematical model of electrophysiological activity in a bullfrog atrial cell. Am. J. Physiol. 259 (2 Pt 2), H370–H389. doi: 10.1152/ajpheart.1990.259.2.H370
Reeves, J. P., Bailey, C. A., Hale, C. C. (1986). Redox modification of sodium-calcium exchange activity in cardiac sarcolemmal vesicles. J. Biol. Chem. 261 (11), 4948–4955.
Reilly, R. F., Shugrue, C. A. (1992). cDNA cloning of a renal Na(+)-Ca2+ exchanger. Am. J. Physiol. 262 (6 Pt 2), F1105–F1109. doi: 10.1152/ajprenal.1992.262.6.F1105
Reuter, H., Seitz, N. (1968). The dependence of calcium efflux from cardiac muscle on temperature and external ion composition. J. Physiol. 195 (2), 451–470. doi: 10.1113/jphysiol.1968.sp008467
Rigg, L., Terrar, D. A. (1996). Possible role of calcium release from the sarcoplasmic reticulum in pacemaking in guinea-pig sino-atrial node. Exp. Physiol. 81 (5), 877–880. doi: 10.1113/expphysiol.1996.sp003983
Rigg, L., Heath, B. M., Cui, Y., Terrar, D. A. (2000). Localisation and functional significance of ryanodine receptors during beta-adrenoceptor stimulation in the guinea-pig sino-atrial node. Cardiovasc. Res. 48 (2), 254–264. doi: 10.1016/s0008-6363(00)00153-x
Rousseau, E., Smith, J. S., Meissner, G. (1987). Ryanodine modifies conductance and gating behavior of single Ca2+ release channel. Am. J. Physiol. 253 (3 Pt 1), C364–C368. doi: 10.1152/ajpcell.1987.253.3.C364
Rubenstein, D. S., Lipsius, S. L. (1989). Mechanisms of automaticity in subsidiary pacemakers from cat right atrium. Circ. Res. 64 (4), 648–657. doi: 10.1161/01.res.64.4.648
Saeki, T., Kimura, T., Hashidume, K., Murayama, T., Yamamura, H., Ohya, S., et al. (2019). Conversion of Ca(2+) oscillation into propagative electrical signals by Ca(2+)-activated ion channels and connexin as a reconstituted Ca(2+) clock model for the pacemaker activity. Biochem. Biophys. Res. Commun. 510 (2), 242–247. doi: 10.1016/j.bbrc.2019.01.080
Sanders, L., Rakovic, S., Lowe, M., Mattick, P. A., Terrar, D. A. (2006). Fundamental importance of Na+-Ca2+ exchange for the pacemaking mechanism in guinea-pig sino-atrial node. J. Physiol. 571 (Pt 3), 639–649. doi: 10.1113/jphysiol.2005.100305
Santacruz-Toloza, L., Ottolia, M., Nicoll, D. A., Philipson, K. D. (2000). Functional analysis of a disulfide bond in the cardiac Na(+)-Ca(2+) exchanger. J. Biol. Chem. 275 (1), 182–188. doi: 10.1074/jbc.275.1.182
Satoh, H. (1993). Caffeine depression of spontaneous activity in rabbit sino-atrial node cells. Gen. Pharmacol. 24 (3), 555–563. doi: 10.1016/0306-3623(93)90212-G
Satoh, H. (2003). Sino-Atrial Nodal Cells of Mammalian Hearts: Ionic Currents and Gene Expression of Pacemaker Ionic Channels. J. Smooth Muscle Res. 39 (5), 175–193. doi: 10.1540/jsmr.39.175
Severi, S., Fantini, M., Charawi, L. A., DiFrancesco, D. (2012). An updated computational model of rabbit sinoatrial action potential to investigate the mechanisms of heart rate modulation. J. Physiol. 590 (18), 4483–4499. doi: 10.1113/jphysiol.2012.229435
Silva, J., Rudy, Y. (2003). Mechanism of pacemaking in I(K1)-downregulated myocytes. Circ. Res. 92 (3), 261–263. doi: 10.1161/01.res.0000057996.20414.c6
Sirenko, S., Yang, D., Li, Y., Lyashkov, A. E., Lukyanenko, Y. O., Lakatta, E. G., et al. (2013). Ca(2)(+)-dependent phosphorylation of Ca(2)(+) cycling proteins generates robust rhythmic local Ca(2)(+) releases in cardiac pacemaker cells. Sci. Signal 6 (260), ra6. doi: 10.1126/scisignal.2003391
Sirenko, S. G., Maltsev, V. A., Yaniv, Y., Bychkov, R., Yaeger, D., Vinogradova, T., et al. (2016). Electrochemical Na+ and Ca2+ gradients drive coupled-clock regulation of automaticity of isolated rabbit sinoatrial nodal pacemaker cells. Am. J. Physiol. Heart Circ. Physiol. 311 (1), H251–H267. doi: 10.1152/ajpheart.00667.2015
Smith, J. S., Imagawa, T., Ma, J., Fill, M., Campbell, K. P., Coronado, R. (1988). Purified ryanodine receptor from rabbit skeletal muscle is the calcium-release channel of sarcoplasmic reticulum. J. Gen. Physiol. 92 (1), 1–26. doi: 10.1085/jgp.92.1.1
Sohn, H. G., Vassalle, M. (1995). Cesium effects on dual pacemaker mechanisms in guinea pig sinoatrial node. J. Mol. Cell Cardiol. 27 (1), 563–577. doi: 10.1016/s0022-2828(08)80051-x
Stehno-Bittel, L., Sturek, M. (1992). Spontaneous sarcoplasmic reticulum calcium release and extrusion from bovine, not porcine, coronary artery smooth muscle. J. Physiol. 451, 49–78. doi: 10.1113/jphysiol.1992.sp019153
Stern, M. D., Rios, E., Maltsev, V. A. (2013). Life and death of a cardiac calcium spark. J. Gen. Physiol. 142 (3), 257–274. doi: 10.1085/jgp.201311034
Stern, M. D., Maltseva, L. A., Juhaszova, M., Sollott, S. J., Lakatta, E. G., Maltsev, V. A. (2014). Hierarchical clustering of ryanodine receptors enables emergence of a calcium clock in sinoatrial node cells. J. Gen. Physiol. 143 (5), 577–604. doi: 10.1085/jgp.201311123
Szepesi, J., Acsai, K., Sebok, Z., Prorok, J., Pollesello, P., Levijoki, J., et al. (2015). Comparison of the efficiency of Na+/Ca2+ exchanger or Na+/H+ exchanger inhibition and their combination in reducing coronary reperfusion-induced arrhythmias. J. Physiol. Pharmacol. 66 (2), 215–226.
Takimoto, K., Li, D., Nerbonne, J. M., Levitan, E. S. (1997). Distribution, splicing and glucocorticoid-induced expression of cardiac alpha 1C and alpha 1D voltage-gated Ca2+ channel mRNAs. J. Mol. Cell Cardiol. 29 (11), 3035–3042. doi: 10.1006/jmcc.1997.0532
Tellez, J. O., Dobrzynski, H., Greener, I. D., Graham, G. M., Laing, E., Honjo, H., et al. (2006). Differential expression of ion channel transcripts in atrial muscle and sinoatrial node in rabbit. Circ. Res. 99 (12), 1384–1393. doi: 10.1161/01.RES.0000251717.98379.69
Torrente, A. G., Zhang, R., Zaini, A., Giani, J. F., Kang, J., Lamp, S. T., et al. (2015). Burst pacemaker activity of the sinoatrial node in sodium-calcium exchanger knockout mice. Proc. Natl. Acad. Sci. U. S. A 112 (31), 9769–9774. doi: 10.1073/pnas.1505670112
Torrente, A. G., Mesirca, P., Neco, P., Rizzetto, R., Dubel, S., Barrere, C., et al. (2016). L-type Cav1.3 channels regulate ryanodine receptor-dependent Ca2+ release during sino-atrial node pacemaker activity. Cardiovasc. Res. 109 (3), 451–461. doi: 10.1093/cvr/cvw006
Torrente, A. G., Zhang, R., Wang, H., Zaini, A., Kim, B., Yue, X., et al. (2017). Contribution of small conductance K(+) channels to sinoatrial node pacemaker activity: insights from atrial-specific Na(+) /Ca(2+) exchange knockout mice. J. Physiol. 595 (12), 3847–3865. doi: 10.1113/JP274249
Trafford, A. W., Diaz, M. E., O'Neill, S. C., Eisner, D. A. (1995). Comparison of subsarcolemmal and bulk calcium concentration during spontaneous calcium release in rat ventricular myocytes. J. Physiol. 488 ( Pt 3), 577–586. doi: 10.1113/jphysiol.1995.sp020991
Trosper, T. L., Philipson, K. D. (1983). Effects of divalent and trivalent cations on Na+-Ca2+ exchange in cardiac sarcolemmal vesicles. Biochim. Biophys. Acta 731 (1), 63–68. doi: 10.1016/0005-2736(83)90398-x
Tsutsui, K., Monfredi, O. J., Sirenko-Tagirova, S. G., Maltseva, L. A., Bychkov, R., Kim, M. S., et al. (2018). A coupled-clock system drives the automaticity of human sinoatrial nodal pacemaker cells. Sci. Signal 11 (534), eaap7608. doi: 10.1126/scisignal.aap7608
Vassalle, M. (1995). The pacemaker current (I(f)) does not play an important role in regulating SA node pacemaker activity. Cardiovasc. Res. 30 (2), 309–310. doi: 10.1016/S0008-6363(95)00028-3
Verkerk, A. O., Wilders, R., van Borren, M. M., Peters, R. J., Broekhuis, E., Lam, K., et al. (2007). Pacemaker current (I(f)) in the human sinoatrial node. Eur. Heart J. 28 (20), 2472–2478. doi: 10.1093/eurheartj/ehm339
Verkerk, A. O., van Borren, M. M., Wilders, R. (2013). Calcium transient and sodium-calcium exchange current in human versus rabbit sinoatrial node pacemaker cells. ScientificWorldJournal 2013, 507872. doi: 10.1155/2013/507872
Vinogradova, T. M., Lakatta, E. G. (2009). Regulation of basal and reserve cardiac pacemaker function by interactions of cAMP-mediated PKA-dependent Ca2+ cycling with surface membrane channels. J. Mol. Cell Cardiol. 47 (4), 456–474. doi: 10.1016/j.yjmcc.2009.06.014
Vinogradova, T. M., Zhou, Y. Y., Bogdanov, K. Y., Yang, D., Kuschel, M., Cheng, H., et al. (2000). Sinoatrial node pacemaker activity requires Ca(2+)/calmodulin-dependent protein kinase II activation. Circ. Res. 87 (9), 760–767. doi: 10.1161/01.res.87.9.760
Vinogradova, T. M., Bogdanov, K. Y., Lakatta, E. G. (2002). beta-Adrenergic stimulation modulates ryanodine receptor Ca(2+) release during diastolic depolarization to accelerate pacemaker activity in rabbit sinoatrial nodal cells. Circ. Res. 90 (1), 73–79. doi: 10.1161/hh0102.102271
Vinogradova, T. M., Zhou, Y. Y., Maltsev, V., Lyashkov, A., Stern, M., Lakatta, E. G. (2004). Rhythmic ryanodine receptor Ca2+ releases during diastolic depolarization of sinoatrial pacemaker cells do not require membrane depolarization. Circ. Res. 94 (6), 802–809. doi: 10.1161/01.RES.0000122045.55331.0F
Vinogradova, T. M., Maltsev, V. A., Bogdanov, K. Y., Lyashkov, A. E., Lakatta, E. G. (2005). Rhythmic Ca2+ oscillations drive sinoatrial nodal cell pacemaker function to make the heart tick. Ann. N. Y. Acad. Sci. 1047, 138–156. doi: 10.1196/annals.1341.013
Vinogradova, T. M., Lyashkov, A. E., Zhu, W., Ruknudin, A. M., Sirenko, S., Yang, D., et al. (2006). High basal protein kinase A-dependent phosphorylation drives rhythmic internal Ca2+ store oscillations and spontaneous beating of cardiac pacemaker cells. Circ. Res. 98 (4), 505–514. doi: 10.1161/01.RES.0000204575.94040.d1
Vinogradova, T. M., Sirenko, S., Lyashkov, A. E., Younes, A., Li, Y., Zhu, W., et al. (2008). Constitutive phosphodiesterase activity restricts spontaneous beating rate of cardiac pacemaker cells by suppressing local Ca2+ releases. Circ. Res. 102 (7), 761–769. doi: 10.1161/CIRCRESAHA.107.161679
Vinogradova, T. M., Brochet, D. X., Sirenko, S., Li, Y., Spurgeon, H., Lakatta, E. G. (2010). Sarcoplasmic reticulum Ca2+ pumping kinetics regulates timing of local Ca2+ releases and spontaneous beating rate of rabbit sinoatrial node pacemaker cells. Circ. Res. 107 (6), 767–775. doi: 10.1161/CIRCRESAHA.110.220517
Vinogradova, T. M., Kobrinsky, E., Lakatta, E. G. (2018a). Dual Activation of Phosphodiesterases 3 and 4 Regulates Basal Spontaneous Beating Rate of Cardiac Pacemaker Cells: Role of Compartmentalization? Front. Physiol. 9, 1301. doi: 10.3389/fphys.2018.01301
Vinogradova, T. M., Sirenko, S., Lukyanenko, Y. O., Yang, D., Tarasov, K. V., Lyashkov, A. E., et al. (2018b). Basal Spontaneous Firing of Rabbit Sinoatrial Node Cells Is Regulated by Dual Activation of PDEs (Phosphodiesterases) 3 and 4. Circ. Arrhythm. Electrophysiol. 11 (6), e005896. doi: 10.1161/CIRCEP.117.005896
Wakabayashi, S., Goshima, K. (1981). Comparison of kinetic characteristics of Na+-Ca2+ exchange in sarcolemma vesicles and cultured cells from chick heart. Biochim. Biophys. Acta 645 (2), 311–317. doi: 10.1016/0005-2736(81)90202-9
Wilders, R., Jongsma, H. J., van Ginneken, A. C. (1991). Pacemaker activity of the rabbit sinoatrial node. A comparison of mathematical models. Biophys. J. 60 (5), 1202–1216. doi: 10.1016/S0006-3495(91)82155-5
Wilders, R. (2007). Computer modelling of the sinoatrial node. Med. Biol. Eng. Comput. 45 (2), 189–207. doi: 10.1007/s11517-006-0127-0
Wolf, R. M., Glynn, P., Hashemi, S., Zarei, K., Mitchell, C. C., Anderson, M. E., et al. (2013). Atrial fibrillation and sinus node dysfunction in human ankyrin-B syndrome: a computational analysis. Am. J. Physiol. Heart Circ. Physiol. 304 (9), H1253–H1266. doi: 10.1152/ajpheart.00734.2012
Wu, Y., Gao, Z., Chen, B., Koval, O. M., Singh, M. V., Guan, X., et al. (2009). Calmodulin kinase II is required for fight or flight sinoatrial node physiology. Proc. Natl. Acad. Sci. U. S. A 106 (14), 5972–5977. doi: 10.1073/pnas.0806422106
Yaniv, Y., Maltsev, V. A., Escobar, A. L., Spurgeon, H. A., Ziman, B. D., Stern, M. D., et al. (2011). Beat-to-beat Ca(2+)-dependent regulation of sinoatrial nodal pacemaker cell rate and rhythm. J. Mol. Cell Cardiol. 51 (6), 902–905. doi: 10.1016/j.yjmcc.2011.08.029
Yaniv, Y., Sirenko, S., Ziman, B. D., Spurgeon, H. A., Maltsev, V. A., Lakatta, E. G. (2013a). New evidence for coupled clock regulation of the normal automaticity of sinoatrial nodal pacemaker cells: bradycardic effects of ivabradine are linked to suppression of intracellular Ca(2)(+) cycling. J. Mol. Cell Cardiol. 62, 80–89. doi: 10.1016/j.yjmcc.2013.04.026
Yaniv, Y., Stern, M. D., Lakatta, E. G., Maltsev, V. A. (2013b). Mechanisms of beat-to-beat regulation of cardiac pacemaker cell function by Ca(2)(+) cycling dynamics. Biophys. J. 105 (7), 1551–1561. doi: 10.1016/j.bpj.2013.08.024
Youm, J. B., Kim, N., Han, J., Kim, E., Joo, H., Leem, C. H., et al. (2006). A mathematical model of pacemaker activity recorded from mouse small intestine. Philos. Trans. A Math. Phys. Eng. Sci. 364 (1842), 1135–1154. doi: 10.1098/rsta.2006.1759
Younes, A., Lyashkov, A. E., Graham, D., Sheydina, A., Volkova, M. V., Mitsak, M., et al. (2008). Ca(2+) -stimulated basal adenylyl cyclase activity localization in membrane lipid microdomains of cardiac sinoatrial nodal pacemaker cells. J. Biol. Chem. 283 (21), 14461–14468. doi: 10.1074/jbc.M707540200
Zaza, A., Maccaferri, G., Mangoni, M., DiFrancesco, D. (1991). Intracellular calcium does not directly modulate cardiac pacemaker (if) channels. Pflugers Arch. 419 (6), 662–664. doi: 10.1007/bf00370312
Zhang, H., Holden, A. V., Kodama, I., Honjo, H., Lei, M., Varghese, T., et al. (2000). Mathematical models of action potentials in the periphery and center of the rabbit sinoatrial node. Am. J. Physiol. Heart Circ. Physiol. 279 (1), H397–H421. doi: 10.1152/ajpheart.2000.279.1.H397
Zhou, Z., Lipsius, S. L. (1993). Na(+)-Ca2+ exchange current in latent pacemaker cells isolated from cat right atrium. J. Physiol. 466, 263–285. doi: 10.1113/jphysiol.1993.sp019720
Keywords: Na+/Ca2+ exchanger, pacemaking, sinus node, automaticity, Ca2+-handling
Citation: Kohajda Z, Loewe A, Tóth N, Varró A and Nagy N (2020) The Cardiac Pacemaker Story—Fundamental Role of the Na+/Ca2+ Exchanger in Spontaneous Automaticity. Front. Pharmacol. 11:516. doi: 10.3389/fphar.2020.00516
Received: 01 October 2019; Accepted: 01 April 2020;
Published: 28 April 2020.
Edited by:
Esther Pueyo, University of Zaragoza, SpainReviewed by:
Sanjay Ram Kharche, University of Western Ontario, CanadaEdward Lakatta, National Institutes of Health (NIH), United States
Tatiana M. Vinogradova, National Institutes of Health (NIH), United States
Matteo Elia Mangoni, Centre National de la Recherche Scientifique (CNRS), France
Copyright © 2020 Kohajda, Loewe, Tóth, Varró and Nagy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: András Varró, dmFycm8uYW5kcmFzQG1lZC51LXN6ZWdlZC5odQ==
†These authors have contributed equally to this work