 Stephen A. Jannetti
Stephen A. Jannetti Brian M. Zeglis1,2,3,4
Brian M. Zeglis1,2,3,4 Michael R. Zalutsky
Michael R. Zalutsky- 1Department of Biochemistry, Hunter College, New York, NY, United States
- 2Ph.D. Program in Biochemistry, CUNY Graduate Center, New York, NY, United States
- 3Department of Radiology, Memorial Sloan Kettering Cancer Center, New York, NY, United States
- 4Ph.D. Program in Chemistry, CUNY Graduate Center, New York, NY, United States
- 5Department of Radiology, Duke University Medical Center, Durham, NC, United States
- 6Department of Radiology, Weill Cornell Medical College, New York, NY, United States
- 7Chemical Biology Program, Memorial Sloan Kettering Cancer Center, New York, NY, United States
Poly(ADP-ribose)polymerase-1 (PARP1) is a DNA repair enzyme highly expressed in the nuclei of mammalian cells, with a structure and function that have attracted interest since its discovery. PARP inhibitors, moreover, can be used to induce synthetic lethality in cells where the homologous recombination (HR) pathway is deficient. Several small molecule PARP inhibitors have been approved by the FDA for multiple cancers bearing this deficiency These PARP inhibitors also act as radiosensitizing agents by delaying single strand break (SSB) repair and causing subsequent double strand break (DSB) generation, a concept that has been leveraged in various preclinical models of combination therapy with PARP inhibitors and ionizing radiation. Researchers have determined the efficacy of various PARP inhibitors at sub-cytotoxic concentrations in radiosensitizing multiple human cancer cell lines to ionizing radiation. Furthermore, several groups have begun evaluating combination therapy strategies in mouse models of cancer, and a fluorescent imaging agent that allows for subcellular imaging in real time has been developed from a PARP inhibitor scaffold. Other PARP inhibitor scaffolds have been radiolabeled to create PET imaging agents, some of which have also entered clinical trials. Most recently, these highly targeted small molecules have been radiolabeled with therapeutic isotopes to create radiotherapeutics and radiotheranostics in cancers whose primary interventions are surgical resection and whole-body radiotherapy. In this review we discuss the utilization of these small molecules in combination therapies and in scaffolds for imaging agents, radiotherapeutics, and radiotheranostics. Development of these radiolabeled PARP inhibitors has presented promising results for new interventions in the fight against some of the most intractable cancers.
Introduction
Poly(ADP-ribose)polymerase-1 (PARP1) is a 116 kDa DNA repair enzyme with nuclear concentrations ranging from 2 × 105 to 1 × 106 enzymes/nucleus in eukaryotic cells (Ludwig et al., 1988; Herceg and Wang, 2001). Within 30 seconds of the advent of DNA damage, PARP PARylates itself, activating the enzyme and leading to a 500-fold increase in its activity over basal levels (Benjamin and Gill, 1980; Alvarez-Gonzalez and Althaus, 1989; Haince et al., 2008; Hassa and Hottiger, 2008; Langelier et al., 2012). Unsuccessful PARP1 mediated repair can result in cell death through multiple pathways, including apoptosis (Kaufmann et al., 1993), ATP depletion (Martin et al., 2000), parthanatos (David et al., 2009), and mitotic catastrophe (Schoonen and van Vugt, 2018).
Over the past decade, inhibitors of PARP have emerged as a common monotherapy for certain subtypes of breast and ovarian cancers (Tangutoori et al., 2015). Moreover, preclinical data has demonstrated that PARP inhibition can increase radiosensitivity in cancer cells (Wang et al., 2019). The efficacy of combination therapies employing PARP inhibitors and external beam radiation has been demonstrated in the clinic, and several phase I clinical trials based on this approach have been completed at the time of writing (NCT00770471, NCT00649207, NCT01264432, NCT01477489, NCT01514201, NCT01657799), with results being available for some of them (Russo et al., 2009; Tangutoori et al., 2015; Dréan et al., 2016). The use of PARP inhibitors as scaffolds for radiopharmaceuticals has also blossomed in recent years (Irwin et al., 2014; Salinas et al., 2015; Carney et al., 2016; Carney et al., 2017; Jannetti et al., 2018; Reilly et al., 2018; Makvandi et al., 2019; Pirovano et al., 2019). To wit, several clinical trials of PARP-inhibitor-based diagnostic imaging agents are currently in progress or have been completed ([18F]FluorThanatrace (Michel et al., 2017), PARPi-FL (Kossatz et al., 2019)), and [18F]PARPi (Schöder et al., 2019)) and a number of therapeutic radiopharmaceuticals based on PARP inhibitors have been employed in preclinical animal models (Kossatz et al., 2016; Michel et al., 2017; Sander Effron et al., 2017).
Mechanism of PARP Inhibition
DNA Binding
PARP1 is composed of six domains. Moving from the N-terminus to the C-terminus, the enzyme contains three zinc fingers (Zn1, Zn2, Zn3), one domain for auto-poly(ADP-ribose)ylation (autoPARylation; AD) that contains a breast cancer 1 protein (BRCT) motif on the c-terminus of the domain, one domain that interacts with open chromatin (WGR) (Altmeyer et al., 2009; Thomas et al., 2019), and one domain associated with the enzyme’s catalytic activity (CAT) comprised of a helical subdomain (HD) and a conserved ADP-ribosyl transferase subdomain (ART). Zn1 and Zn2 are homologous domains that recognize and bind DNA, though it has been shown that the enzyme can bind DNA with only one of these two domains (Langelier et al., 2011) (Figure 1). Taken together, the Zn fingers engage not specific sequences of DNA but rather structural motifs such as blunt ends, single strand breaks (SSBs), double strand breaks (DSBs), 3′ single-base overhangs, and long overhangs (D’Amours et al., 1999; D’Silva et al., 1999; Pion et al., 2003). It is important to note that in each of these cases, PARP1 binds to the irregular angle in the broken DNA strand, not the exposed nucleotides (Lonskaya et al., 2005). Furthermore, each Zn finger seems to play a particular role in a different aspect of the enzyme’s function. For example, Zn1 is responsible for binding DSBs, interacting with the catalytic domain, and activating PARP1. Zn2 seems to be responsible for the recognition of SSBs. (Eustermann et al., 2011). Zn3 has been shown to play a critical role in protein–protein interactions during DNA-dependent autoPARylation by initiating hydrolysis of the NAD+ substrate (Langelier et al., 2008; Venere et al., 2014). Unlike Zn1 and Zn2, Zn3 is not required for DNA activation, though it does mediate PARP1–chromatin interactions (Langelier et al., 2010; Venere et al., 2014).
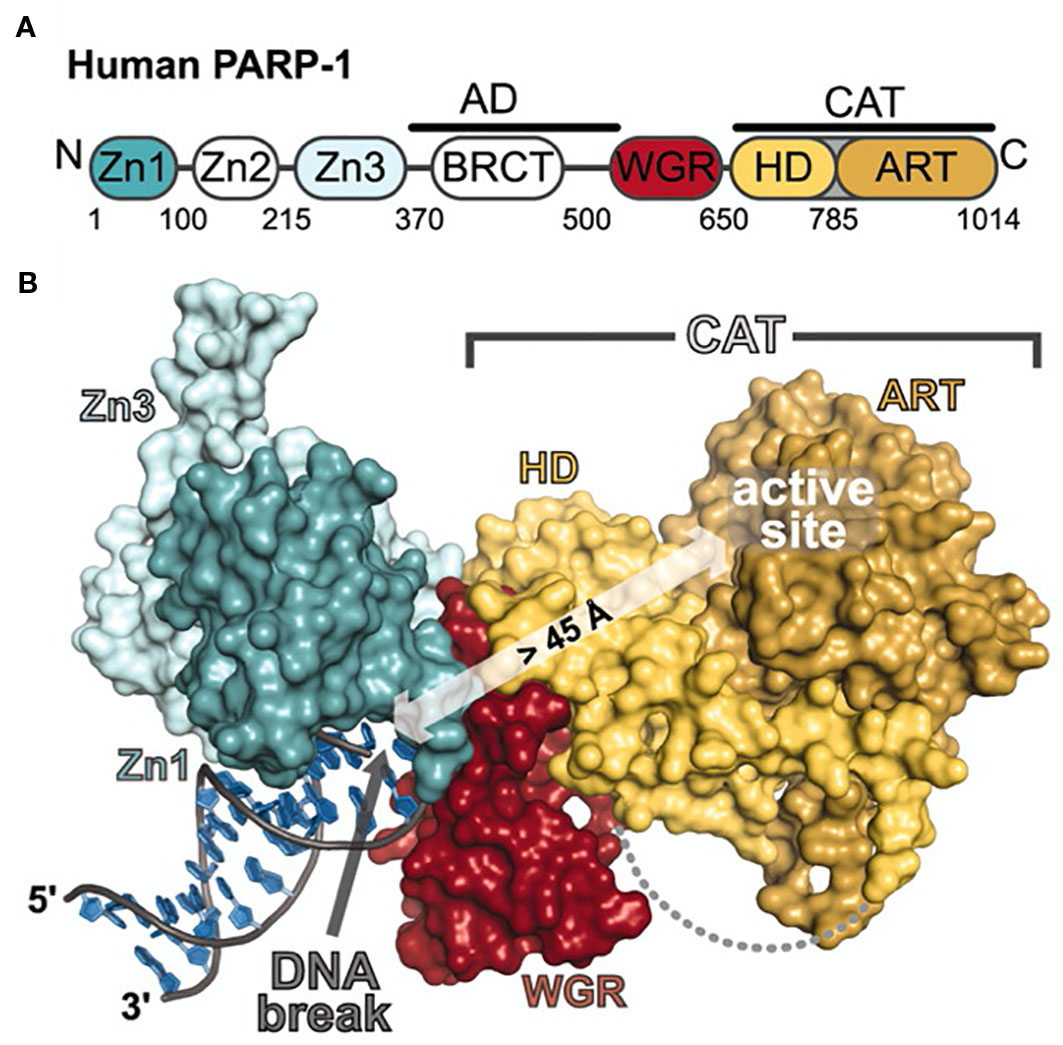
Figure 1 Schematic representation and crystal structure of PARP1 domains and subdomains. (A) Schematic representation of human PARP-1 domains and subdomains. A BRCA C-terminus (BRCT) fold is located within the region of PARP-1 that is primarily targeted for automodification. The catalytic domain is composed of an alphahelical subdomain (HD) and an ADP-ribosyl transferase subdomain (ART). (B) Crystal structure of the PARP-1/DNA structure. Zn1, Zn3, and WGR-CAT are shown as surfaces. The arrow indicates the location of the PARP-1 automodification region near the catalytic active site. From Langelier et al., 2012. Reprinted with permission from AAAS. [PDB code 4DQY, (Langelier et al., 2012)].
Single and Double Strand DNA Repair
The complete mechanisms of poly(ADP-ribosyl)ation by the PARP family of enzymes and its role in DNA repair and regulation warrants further investigation (Alemasova and Lavrik, 2019). One proposed hypothesis is the homodimerization of PARP1 after recruitment to the side of DNA damage to begin autoPARylation and the repair process (Bauer et al., 1986; Bauer et al., 1990; Mendoza-Alvarez and Alvarez-Gonzalez, 1993; Mendoza-Alvarez and Alvarez-Gonzalez, 1999; Mendoza-Alvarez and Alvarez-Gonzalez, 2004). PARP1 has been shown to dimerize in its active form in solution which, greatly increased its specific activity (pmol of product/min per pmol of enzyme) (Bauer et al., 1990). PARP1 dimerization was also confirmed by dynamic light scattering (Vasil’eva et al., 2019). The rate of automodification is a function of PARP concentration consistent with second order kinetics. The rate kinetics suggest a dimerized model in which two PARP1 enzymes initialize PARylation, each with an active NAD+ binding site (Mendoza-Alvarez and Alvarez-Gonzalez, 1993; Mendoza-Alvarez and Alvarez-Gonzalez, 1999; Mendoza-Alvarez and Alvarez-Gonzalez, 2004). Interestingly, an earlier study supports a model where optimal enzyme activity and PAR chain formation occur in a PARP1:DNA binding stoichiometry of 2:1 where one PARP1 enzyme is catalytically active and the other PARP1 acts as a receptor for PARylation (Panzeter and Althaus, 1994). This model is supported by the crystal structure of the Zn1 and Zn2 domains from separate PAPR1 enzymes in a dimer, complexing damaged DNA (Ali et al., 2012). Another study found the 5′-recessed structure of DNA will recruit PARP1 in a 2:1 PARP1:DNA stoichiometry (Pion et al., 2003). Once dimerized, PARylation and autoPARylation are initiated. AutoPARylation takes place on glutamate and lysine residues found in the AD (Altmeyer et al., 2009; Venere et al., 2014). This domain is also the site of protein–protein interactions (WGR) with the downstream protein targets of PARP1 (Venere et al., 2014). However, in the absence of an AD domain, PARP1 can still PARylate other proteins (Altmeyer et al., 2009).
There are several proteins which can be modified with potentially large, negatively charged PAR chains, which have been found to be between 1 and 200 units long (Hakmé et al., 2008). It has been suggested that as the length of the PAR chain increases the affinity of PARP1 for DNA decreases, mostly due to its highly negative charge, allowing PARP1 to release the DNA and leave space for other DNA repair proteins to bind (Ogata et al., 1980; Poirier et al., 1982; Tulin and Spradling, 2003; Timinszky et al., 2009). The PAR chain can be hydrolyzed to shorter PAR chains, mono(ADP-ribose) by ADP-ribose hydrolase (ARH3) or PAR glycohydrolase (PARG) (Oka et al., 2006; Min and Wang, 2009). When ARH3 or PARG cleaves the first ADP-ribose in a PAR chain from PARP it reestablishes the enzyme’s ability to recognize and bind DNA damage, essentially “resetting” the PARP (Rouleau et al., 2010).
The most direct use of the PAR chain is as energy in the form of ATP when the cell is experiencing decreased levels of ATP (Petermann, 2003; Walker et al., 2006). It is known that DNA damage leads to rapid depletion of ATP reserves in the cell (Sims et al., 1983). This lends to the hypothesis that one role of PARP is to provide ATP from NAD+ for the ligation step in the BER pathway, which is favored in times of ATP depletion (Petermann, 2003; Walker et al., 2006).
The PAR chain has been reported to have the potential to recruit an array of different proteins (Rouleau et al., 2010). When interacting with acceptor proteins, the PAR chains can modulate localization, function, and structure (Kraus, 2008; Krishnakumar and Kraus, 2010). Aside from autoPARylation, target proteins for PARylation by PARP1 include other DNA repair proteins, transcription factors, histones, and chromatin modulators (D’Amours et al., 1999; Hassa and Hottiger, 2008). One of the PARylation targets during DNA damage repair is H1, whose targeting results in the relaxation of the chromatin super structure and recruitment of repair proteins such as XRCC1 (El-Khamisy, 2003; Okano et al., 2003). XRCC1 binds directly to the PAR chain, whereas other repair proteins interact with mediating proteins that in turn bind PAR (Rouleau et al., 2010).
Necessary, additional interactions between DNA repair proteins occur through a BRCA1 carboxy-terminal (BRCT) repeat motif found in many other proteins involved in DNA damage repair (Kameshita et al., 1984). This domain, containing a conserved ADP binding sequence comprised of 20 amino acids, has been identified and found to overlap with domains in many proteins associated with DNA binding, nuclear localization, nuclear export, protein degradation, and protein–protein interaction (Pleschke et al., 2000). Two enzymes essential to HR, ataxia telangiectasia-mutated (ATM) and mitotic recombination 11 (MRE11) are signaled through PAR as well (Haince et al., 2008; Sugimura et al., 2008).
Synthetic Lethality
Exploiting synthetic lethality—a relationship between two cellular mechanisms wherein the functional loss of one is survivable but the loss of both is lethal—was proposed as a treatment for cancer almost a quarter of a century ago (Hartwell et al., 1997). This strategy was validated during the phase I trial of the PARP1/2 inhibitor olaparib, when the majority of patients with BRCA1/2 mutations saw a benefit from PARPi intervention (Fong et al., 2009). In 2014, olaparib received FDA approval for treatment of advanced ovarian cancer (Kim et al., 2015).
When the homologous recombination (HR) pathway is compromised, inhibition of the remaining PARP-dependent BER pathway can be lethal to cells, although the exact mechanism is not entirely understood (Helleday, 2011). Presently, the model for PARPi-mediated synthetic lethality is that an inhibitor will bind a DNA-bound PARP enzyme, preventing it from PARylating proteins or dissociating from the DNA. These lesions, caused by trapped PARPs, accumulate and prevent DNA repair and replication, causing cytotoxicity (Murai et al., 2012; Murai et al., 2014; Pommier et al., 2016). Likely, the HR pathway fails in BRCA-deficient cells due to loss of function of BRCA1/2, causing the cells to rely upon BER to repair damaged DNA (Ström et al., 2011). The BRCA1 and BRCA2 genes code for the eponymous tumor-suppressing proteins essential to the HR repair pathway (Roy et al., 2011). Loss of function of one of these genes via mutation is associated with a high risk of breast and ovarian cancer (Miki et al., 1994; Wooster et al., 1995). These types of BRCA1/2 negative cancers are naturally sensitive to PARP inhibitors (Bryant et al., 2005; Farmer et al., 2005; Liu et al., 2008; Rottenberg et al., 2008; Fong et al., 2009; Evers et al., 2010).
While synthetic lethality via PARP inhibitors is frequently associated with BRCA1/2 mutations in the literature, many genes play crucial roles in various stages of the HR repair pathway (Cejka, 2015; Hoa et al., 2015). An example of how PARP inhibitors can be lethal to HR-deficient cells is their effect on XRCC1-deficient cells. An increased amount of SSBs were detected when XRCC1-deficient cells were treated with a PARP inhibitor (Ström et al., 2011). Certain tumors arising from hereditary cancers that share an HR-deficient phenotype, not just BRCA deficiency, are sometimes described by the term “BRCAness.” This includes any mutation that would affect replication fork stability, or any genes involved in the HR pathway, for example ATM, ATR, FANC, or PALB2 (Turner et al., 2004; McCabe et al., 2006; Lord and Ashworth, 2016). There is evidence of BRCAness and PARP inhibitors inducing synthetic lethality in cancers that are known to have HR-deficient pathways, such as high-grade serous ovarian cancer (HGS-OVCa), advanced prostate cancer, and pancreatic cancers (Bell et al., 2011; Carnevale and Ashworth, 2015; Mateo et al., 2015; Waddell et al., 2015; Lord and Ashworth, 2016).
The first evidence of PARP inhibition inducing lethality appeared in the 1970s when NAD+ analogs were used to bind PARP1 in combination with a genotoxic agent (Brightwell et al., 1975; Terada et al., 1979; Purnell and Whish, 1980). At the time of writing, four therapeutic PARP inhibitors have been approved by the FDA (olaparib, rucaparib, niraparib, and talazoparib) and four more are in various stages of clinical trials (veliparib, E7016, CEP-9722, BGB-290; NCT01827384, NCT01605162, NCT01345357, NCT03150810, respectively).
Increased Genomic Instability After PARP Inhibition
Originally, the mechanism proposed for PARP-inhibitor-mediated synthetic lethality was the accumulation of DSBs produced when a replication fork failed after encountering an inhibited PARP bound to an SSB (Farmer et al., 2005). There is evidence that cells undergoing PARP inhibition contain no significant increase in SSBs (Gottipati et al., 2010; Ström et al., 2011). PARP knockout cells and PARP knockdown cells contain no higher level of SSBs than wildtype cells (Fisher et al., 2007). These findings suggest alternate explanation of PARP inhibitor-mediated synthetic lethality.
PARP more directly affects the genome through PARylating histones and other nuclear proteins to unwind the chromatin structure (De Murcia et al., 1986; Althaus et al., 1994). An increased level of biomarkers of genomic instability, such as DNA strand breaks, gene amplification, DNA recombination, and SCE were found in cells with decreased PARP activity after treatment with DNA-damaging agents. These findings were made using PARP inhibitors, PARP knockout models, and asRNA models (Küpper et al., 1990; Waldman and Waldman, 1991; Ding and Smulson, 1994; Schreiber et al., 1995).
These results lead to the hypothesis that when HR and BER pathways are inaccessible to cells, they rely on non-homologous end joining (NHEJ). HR and BER are conservative DNA repair methods, maintaining the original DNA sequence that was damaged. When HR is not an option for the cell, it has to rely on BER and NHEJ. NHEJ is a non-conservative repair pathway because it will excise the damaged DNA, leading to genomic instability (Moynahan et al., 2001; Tutt et al., 2001). It has been shown that NHEJ is promoted in cells with defective HR pathways after treatment of PARP inhibitors. Also, resistance to PARP inhibitors is acquired when the NHEJ pathway is inhibited (Patel et al., 2011). These findings indicate that PARP-inhibitor-induced lethality can also be attributed to genomic instability as a result of the NHEJ pathway in non-irradiated situations.
PARP Trapping
PARP trapping is the formation of a PARP–DNA complex through inhibition of DNA-bound PARP. PARP–DNA complexes were detected in cell lines treated with olaparib and rucaparib (Murai et al., 2012; Murai et al., 2014). PARP inhibitors prevent PARP from synthesizing PAR chains by competitively binding PARP’s natural substrate, NAD+.
Interestingly, inhibiting PARP is more cytotoxic than the absence of PARP itself (Thomas et al., 2018). One hypothesis for this effect might be due to replication fork stalling and subsequent collapse, a mechanism shared with topoisomerase I (TOP1) and TOP1 inhibitors. Further evidence of PARP trapping and collapsing replication forks is PARP1’s role in restarting stalled replication forks, a task prevented by PARP inhibitors (Yang et al., 2004; Bryant et al., 2009). This mechanism sheds some light on the lethality of PARP inhibitors in cells with and without BRCA mutations (Strumberg et al., 2000).
The efficacy of PARP trapping was shown to be independent of the half-maximal inhibitory concentration (IC50) of the PARP inhibitors (Murai et al., 2012; Murai et al., 2014). Of the FDA-approved PARP inhibitors, veliparib is the least effective at PARP trapping, irrespective of the fact that its IC50 value is lower than that of niraparib (2 nM and 3.2 nM, respectively). Olaparib has a significantly more favorable IC50 than rucaparib (5 nM and 1.4 nM for olaparib and rucaparib, respectively), and they exhibit equal efficacy as PARP trapping agents. Talazoparib has the more favorable IC50 and functions as the best PARP trapping agent (Murai et al., 2012). Talazoparib’s ability to trap PARP is likely due to its bulky structure and rigidly which contributes to a slow rate of dissociation (Shen et al., 2013; Murai et al., 2014; Pommier et al., 2016). Recent combination trials have demonstrated new indications of PARP inhibitors in combination with other therapeutics, extending their use beyond cancers with BRCAness. One such example includes a combination therapy of rucaparib and temozolomide in metastatic melanoma, a cancer not typically associated with BRCA1/2 mutations (Plummer et al., 2013).
PARP Inhibition as a Radiosensitizer
While PARP inhibition in cancers with “BRCAness” can induce synthetic lethality, PARPi in other cell lines can radiosensitize them. The PARPi 3-aminobenzamide was able to radiosensitize two breast cancer cell lines, MDA-MB-231 and MDA-MB-436, one with and one without BRCA mutation, respectively (Zhao et al., 2019). The mechanism of PARPi induced radiosensitivity is most likely a DNA replication-dependent model in which replication forks collapse during delayed SSB repair, as demonstrated by Dungey et al. in which a replication-dependent increase in ɣH2AX foci in G2 cells was observed after treatment with olaparib and fractionated ionizing radiation (IR) in the T98G model of glioblastoma (Dungey et al., 2008). Noel et al. showed HeLa cells were radiosensitized by the PARPi 4-amino-1,8-naphthalimide during S phase. Irradiation of these cells produced an increase of DSBs hours after irradiation (Noel et al., 2006). Evidence supporting a cell-cycle dependent effect was provided earlier by Chalmers et al. when hamster fibroblast cell lines V79-379A and CHO-K1 and human glioma cell line T98G treated with PARPis were the most radiosensitive during periods of rapid growth. Once cells had been arrested in G1 phase, radiosensitivity was lost (Chalmers et al., 2004).
Preclinical Models of Combination Therapies
In Vitro—Clonogenic Assays
The Marples group out of Wayne State University has demonstrated that radiosensitivity can be increased through PARP inhibition in human glioma cell lines U373-MG and T98G. Clonogenicity was evaluated with increasing concentrations of PARP inhibitors (1–3 μM) that were found to be non-toxic in the absence of radiation. A 3 μM concentration of PARP inhibitor, the highest concentration of inhibitor that was non-toxic in the absence of IR, was then used in conjunction with low levels (0.05–0.3 Gy) of ionizing radiation (IR) to induce toxicity (Chalmers et al., 2004). Treatment with a small molecule PARP inhibitor, AG14361, followed by 8 Gy IR reduced survivability in colorectal cancer cell lines (LoVo) by 73% (Calabrese et al., 2004). Non-small cell lung cancer (NSCLC) cell lines A549 and Calu-6 were each treated with 1 μM and 5 μM of olaparib before being exposed to 0, 2, 4, and 6 Gy to find dose-dependent sensitization of both cell lines. For A549 and Calu-6 the Survival Enhancement Ratios (SER) values at 1 μM were found to be 1.3 and 1.5, respectively. These values increased to 1.6 μM and 1.8 at 5 μM (Senra et al., 2011). Veliparib was shown to have a limit on radiosensitization with concentrations above 2.5 μM no longer increasing radiosensitivity in a NSCLC cell line, H1299. Survival fractions were decreased when IR was supplemented by pretreatment of 2.5 μM veliparib. This effect was also observed in human prostate cancer cell lines (DU145 and 22Rv1) (Liu et al., 2008). The PARP inhibitor E7016 was able to increase radiosensitivity across multiple cancer cell lines as well. A dose enhance factor ≥1.4 was calculated for glioblastoma (U251) and pancreatic (MiaPaCa), and prostate cancer (DU145) cell lines when treated with E7016 prior to IR. Surviving fractions in all three cell lines were reduced to 0.1 in clonogenic assays (Russo et al., 2009). Veliparib demonstrated no effect on colony formation in PC-3 prostate cancer cells when incubated in 10 μM veliparib. The same treatment, followed by 2 Gy IR, reduced colony formation to 47% (Barreto-Andrade et al., 2011). 22Rv1 prostate cancer cells had PARylation reduced by 97–100% after incubation with the PARP inhibitor olaparib. The radiosensitization enhancement ratio was found to be ≥1.2 when combined with IR compared to PARP inhibitor alone. This result was found in acutely hypoxic, chronically hypoxic, and normoxic conditions (Gani et al., 2015). These works establish the efficacy of a variety of PARP inhibitors as radiosensitizers for multiple human cancer cell lines at low μM concentrations, often below cytotoxic concentrations of the PARP inhibitors themselves.
In Vivo—Tumor Growth Delay and Survival
Combination therapy is a more efficacious approach to treating H460 models of non-small cell lung cancer. A tumor growth delay assay using a five-fold increase in tumor volume as an endpoint saw a 1-day or 7-day delay using the PARP inhibitor veliparib or external beam radiation alone, respectively. When these therapies were combined, the five-fold increase in tumor volume was delayed by 13.5 days (Albert et al., 2007). A subcutaneous LoVo xenograft model of colorectal cancer exhibited tumor growth delay of 19 days with a fractionated regimen of IR that was increased to 37 days when combined with a low dose of AG14361, which did not delay tumor growth when administered alone (Calabrese et al., 2004). Tumor growth was significantly impeded in a dose-dependent trend of GPI-15427 and 2 Gy in mouse models of JHU006 and JHU012 HNSCC (Khan et al., 2009). A dose response dependency of veliparib was demonstrated in a human colon cancer mouse xenograft model, HCT116, when administered through a subcutaneously implanted osmotic pump in conjunction with IR compared to IR alone (Donawho et al., 2007). Calu-6 mouse xenograft models received a daily 50 mg/kg dose of olaparib for 5 days, 5 days of 2 Gy IR daily, or both therapies. The combination therapy cohort experienced a significant delay of 10 days to reach the endpoint compared to either monotherapy or control cohort (Senra et al., 2011).
Mouse models of HCT116 colorectal cancer receiving twice daily orally administered doses of 12.5 mg/kg of veliparib in conjunction with 2 Gy fractions of IR displayed significant tumor growth delay compared to control groups of IR alone (Shelton et al., 2013). Olaparib was also tested as a radiosensitizer in subcutaneous mouse models of glioblastoma-initiating cells. Daily treatment of olaparib over 7 days was administered concurrently with 3 Gy of IR every other day for three total doses to find greater tumor growth delay than vehicle, IR alone, or IR with vehicle (Venere et al., 2014). The effect of fractionated RT after sensitization by olaparib was evaluated in a 22Rv1 human prostate cancer mouse model. One cohort received a single 8 Gy dose on the third day of three consecutive daily doses of intraperitoneally administered PARP inhibitor. A second cohort was treated with seven consecutive days of olaparib, with 5 × 2 Gy doses every other day starting 3 days after the initial PARP inhibitor injection. The group receiving fractionated doses displaying a non-significant delay in tumor growth compared to the vehicle + fractionated IR control group (Gani et al., 2015). The above data suggests that a combination therapy between PARP inhibitors and RT is more effective in vitro and in vivo than either therapy alone and is summarized in Table 1.
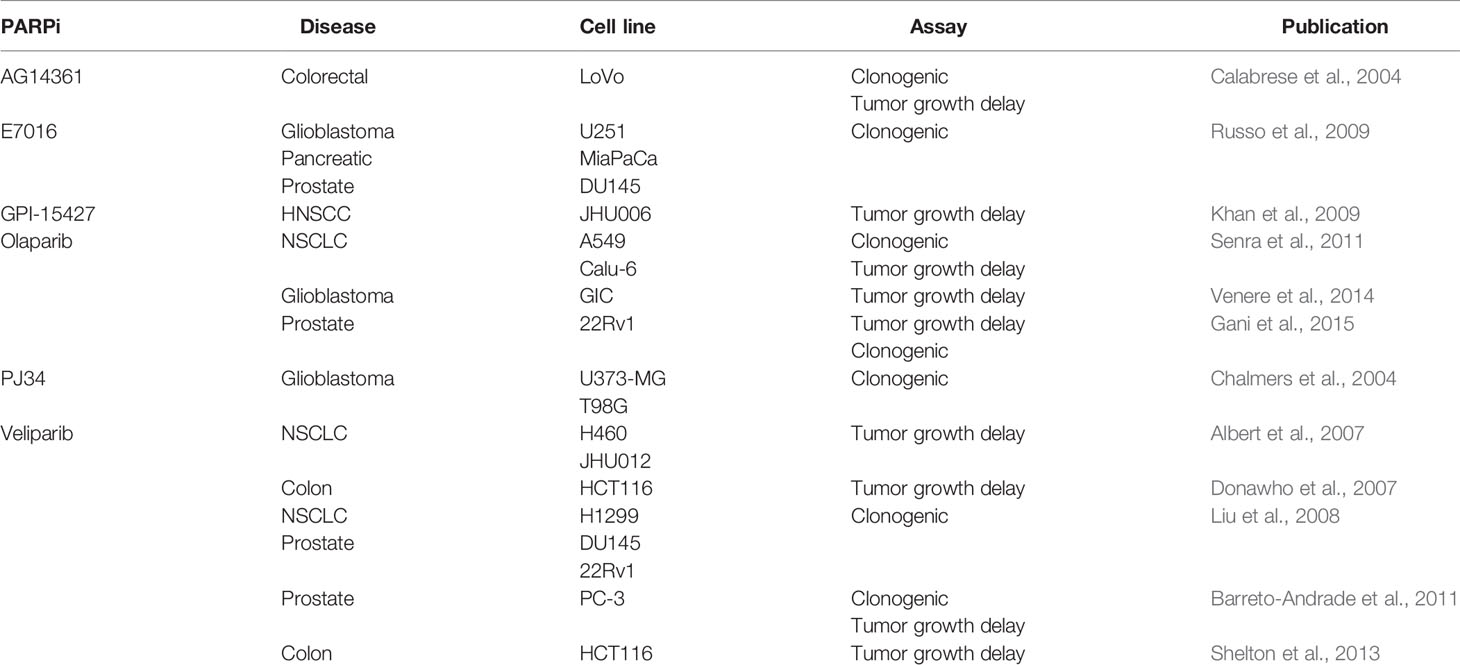
Table 1 Preclinical combination therapies with PARP inhibitors and ionizing radiation.
Combination Therapy Clinical Trials
Ionizing Radiation With Chemotherapy and PARPis
There are currently several completed clinical trials exploring the efficacy of combining PARP inhibitors, radiotherapy and chemotherapy, none of which have available results. In a phase I study of patients with phase II or III rectal cancer, patients were given 825 mg/m2 capecitabine twice daily and 1.8 Gy fractions daily for a total of 50.4 Gy over approximately 6 weeks in conjunction with escalating doses (20–400 mg) of veliparib orally twice daily. Maximum tolerated dose was not reached, and the study found 400 mg twice daily to be the appropriate dose of veliparib for the phase II study (NCT01589419, Czito et al., 2017). Two other phase I studies evaluating the combination of veliparib and temozolomide against diffuse pontine glioma and glioblastoma have also concluded, but results have not yet been posted (NCT01514201, NCT00770471). The PARADIGM-2 study is two parallel phase I studies in which one arm evaluated dose escalation of olaparib (50–150 mg/daily) is combined with 60 Gy in 30 fractions over 3 weeks of radiotherapy followed by four additional weeks of olaparib. The second arm included the same regimen with concomitant temozolomide at 75 mg/m2 daily throughout radiotherapy and again after the 4 weeks of olaparib (Fulton et al., 2018). One study aims to find the MTD of b 25–200 mg of olaparib twice daily beginning 3 days prior to the first dose of cetuximab. The initial 400 mg/m² dose of cetuximab will precede the start of radiation by 5–7 days. 69.3 Gy of radiation therapy will be administered in 33 fractions over 6.5 weeks (NCT01758731).
Ionizing Radiation With PARPis
The first clinical trial exploring combination therapy between PARP inhibitors and ionizing radiation to publish results combines veliparib and whole brain radiation therapy (WBRT) in adult patients with brain metastases from non-small cell lung cancer (NCT00649207). Patients were age >18 years with Karnofsky performance status (KPS) scores ≥70. One arm received WBRT administered daily in 2.5 Gy fractions over 15 sessions for 37.5 Gy total. A second arm was treated with 150 mg of veliparib twice daily with concurrent daily fractions of 3.0 Gy fractions over 2 weeks for 30 Gy.
All three arms of the study received a 30 Gy fractionated dose of 10 × 3 Gy doses, excluding weekends and holidays. The variable was the quantity of drug received twice daily: placebo, 50 mg veliparib, or 200 mg veliparib. The primary outcome was survival up to 36 months. While the patient tumor population was homogenous, 88–90% of patients in this trial had Graded Prognosis Assessment scores ≤2.5, amounting to an unfavorable prognosis, and the primary outcome was not met (Chabot et al., 2017). It is worth noting that even when a combination therapy significantly prolongs survival in patient populations with favorable prognoses (GPA 2.5–4), it falls short of significance in populations with unfavorable prognoses (Aoyama et al., 2015). This study progressed to phase II (NCT01657799), where no benefit was found in combining WBRT with veliparib compared to WBRT and a placebo (Chabot et al., 2017).
New Frontiers—PARPi Diagnostics and Radiotherapies
PARPi-FL
PARPi-FL was first reported in 2012 by the Weissleder Lab at Massachusetts General Hospital in human pancreatic cancer cells (Reiner et al., 2012). It can be used for real-time visualization of intracellular kinetics of PARP inhibitors (Thurber et al., 2013). It was later shown to be a viable imaging agent in vivo in a mouse model of glioblastoma (Irwin et al., 2014). Composed of a BODIPY-FL dye conjugated to an olaparib scaffold, it retains a similar pharmacokinetic profile, including the low IC50 value of 12.2 nM compared to the 5.0 nM value of olaparib (Menear et al., 2008). It can also be blocked by pretreatment with olaparib. PARPi-FL uptake in tumors is rapid, with statistically significantly increased tumor-to-muscle and tumor-to-brain ratios of ≥10 in a mouse model of glioblastoma. Uptake of PARPi-FL was correlated to PARP1 expression, and increased after irradiation (Irwin et al., 2014; Kossatz et al., 2016). Retention persists for hours, with <50% metabolites present in the blood at peak uptake in tumors (Irwin et al., 2014). PARPi-FL has been used for real-time measurements of drug–target interaction in vitro and in vivo (Dubach et al., 2014; Dubach et al., 2017). The translational potential of PARPi-FL was highlighted when high tumor-to-organ ratios were observed in an orthotopic model of oral squamous cell carcinoma using clinical imaging instruments (Kossatz et al., 2016; Carney et al., 2017), and early clinical outcomes have been reported [NCT03085147 and (Kossatz et al., 2019)].
PARPi-Derived PET Tracers
The first radiolabeled PARP inhibitor for PET imaging was designed to monitor tissue necrosis. The Mach Group at Washington University labeled the small molecule PARP1 inhibitor PJ34 with carbon-11 and had good yields with increased uptake in target tissue in a rat model of Type I diabetes (Tu et al., 2005). The first reported fluorine-18-labeled PARP inhibitor was [18F]FE-LS-75 from the Roesch Group at Johannes Gutenberg-University, which showed high yields up to 80% (Riss et al., 2009) but did not report in vitro/vivo experiments. The first fluorinated PARP-targeted small molecule based on a later FDA-approved PARP inhibitor was 18F-BO (Keliher et al., 2011). Uptake was shown to correlate to PARP1 expression in breast cancer mouse models. A dose of olaparib prior to injection with 18F-BO was able to reduce uptake in vivo. Favorable uptake was also observed in pancreatic and ovarian cancer models (Reiner et al., 2012) (Figures 2A, B). 18F-PARPi-FL was developed as a dual modality PET/fluorescent imaging agent (Keliher et al., 2014). PARP1-specific uptake was demonstrated in glioblastoma xenografts. Both modalities showed similar tumor-to-brain uptake ratios (PET, 9:1; fluorescence, 7:1) as determined by autoradiography and fluorescence microscopy (Carlucci et al., 2015). 18F-PARPi is an olaparib-based PET imaging agent that exhibits high specificity for PARP1 in mouse models of orthotopic glioblastoma, diffuse intrinsic pontine glioma, and small-cell lung cancer (Carney et al., 2016; Kossatz et al., 2017; Carney et al., 2018) (Figures 2C, D). 18F-PARPi has potential to non-invasively monitor disease progression and is currently in phase I clinical trials [(Schöder et al., 2019), NCT03631017]. Wilson et al. has published the synthesis and in vivo biodistribution of a fluorine-18 isotopologue of olaparib. Pre-irradiation of the cells and tumors was shown to increase uptake of the compound in several pancreatic cancer cell lines (Wilson et al., 2019). 18F-FluorThanatrace (18F-FTT) is a rucaparib-based PET imaging agent first published in a human breast cancer mode (Zhou et al., 2014) (Figure 2E). It was the first PARP-targeted PET imaging agent to be tested in the clinic and is currently involved in several phase I clinical trials, evaluating uptake in different cancers (Michel et al., 2017; Makvandi et al., 2018).
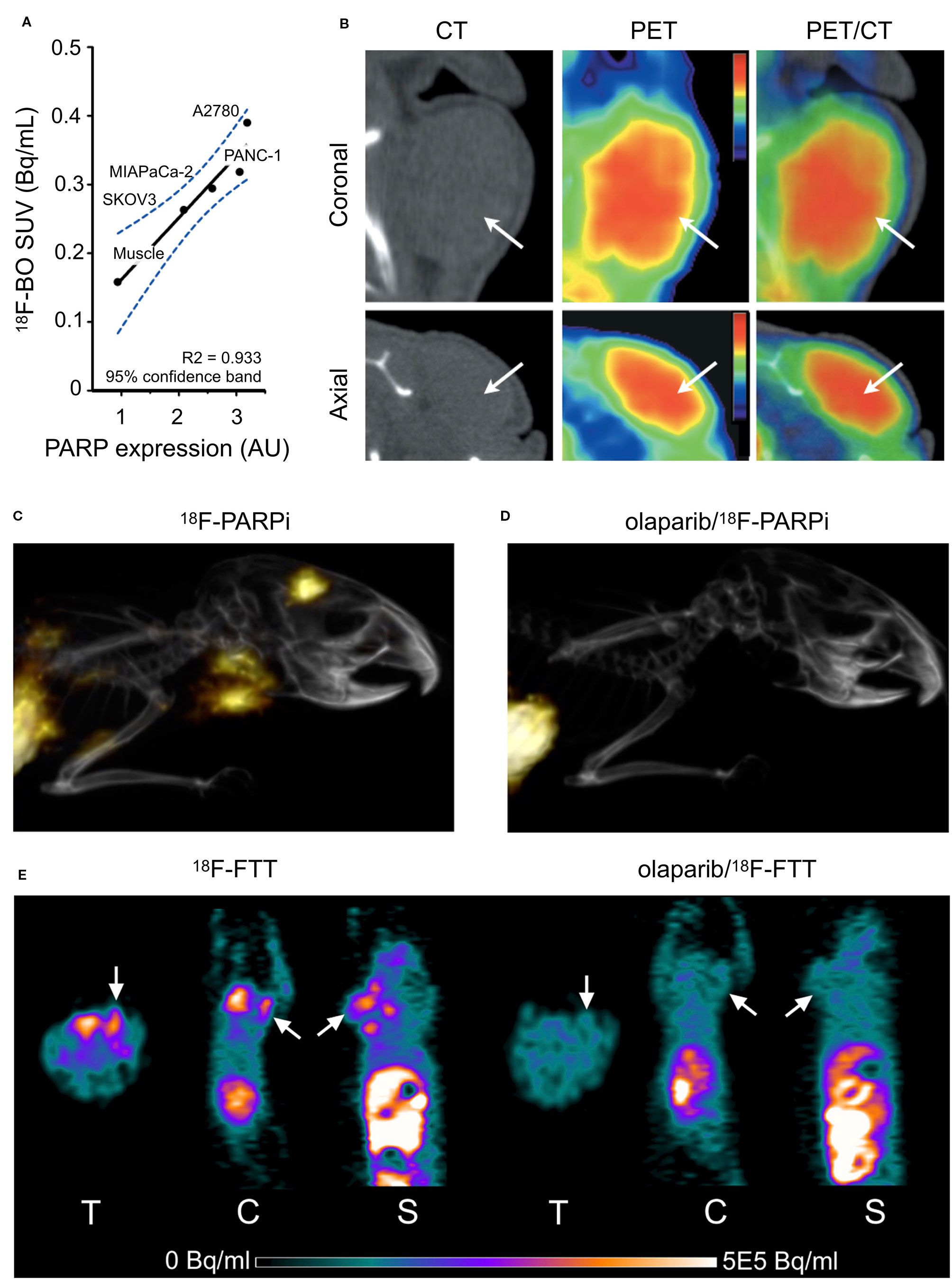
Figure 2 In vivo PET/CT Imaging. (A) Correlation of 18F-BO uptake and PARP expression in two ovarian and two pancreatic tumor types as determined by immunoblotting. (B) Coronal and axial PET/CT scans of a subcutaneous A2780 tumor-bearing mouse (Reiner et al., 2012). (C) Sagittal PET/CT images of orthotopic U251 MG tumor-bearing mice 2 h post-injection of 18F-PARPi. (D) PET/CT 2 h post-injection 18F-PARPi after 30 m pre-injection of 500-fold excess olaparib (Carney et al., 2016). (E) MicroPET images of MDA-MB-231 tumors in mice tumor at 60 min after 18F-FTT injection before and after treatment with olaparib (ip 50 mg/kg 20 min pretreatment; Zhou et al., 2014). Reprinted by permission from Springer Nature: Springer Molecular Imaging and Biology (Carney et al., 2016) (Non-invasive PET imaging of PARP1 expression in glioblastoma models, Carney, B, G Carlucci, B Salinas, V Di Gialleonardo, S Kossatz, A Vansteene, VA Longo, A Bolaender, G Chiosis, KR Keshari, WA Weber, and T Reiner), copyright (2016).
PARPi Radiotherapeutics
Several PARP inhibitors have also been labeled with therapeutic isotopes (Salinas et al., 2015; Jannetti et al., 2018; Reilly et al., 2018; Makvandi et al., 2019; Pirovano et al., 2019). The pharmacokinetic profiles of several iodinated PARP inhibitors based on olaparib were explored in human glioblastoma models in vitro and in vivo. Various length linkers were evaluated using an olaparib scaffold and a small library of iodobenzoic acids. The compounds with the best pharmacokinetics were radioiodinated and evaluated in culture and orthotopic mouse models of human glioblastoma for PARP1 specificity (Salinas et al., 2015). The rucaparib scaffold was also leveraged in the design and synthesis of alpha- and auger-emitting radiotherapeutics using copper-catalyzed halogenation of boronic esters (Reilly et al., 2018; Makvandi et al., 2019). The efficacy of PARP-targeted radiotherapeutics was first published in subcutaneous mouse models of glioblastoma, and later in orthotopic models of human glioblastoma (Jannetti et al., 2018; Pirovano et al., 2019). Intratumoral injections were implemented to mimic Convection Enhanced Delivery (CED). A reporter cell line transduced from U87 cells was designed to respond to p53 activation, as well as cellular density. This allowed imaging of cell death following treatment with the iodinated PARP inhibitor. Therapeutic efficacy was evaluated in a tumor growth delay experiment that found a fractionated dose of the drug could significantly delay the endpoint of the study (Figure 3A). Use of a CED-mimicking subcutaneous implant allowed approximately 9 Gy to be delivered to a brain-tumor-bearing mouse compared to 1 Gy in a healthy mouse (Jannetti et al., 2018) (Figures 3B, C). These results were leveraged in the synthesis and validation of an Auger-electron emitting isotopologue. Uptake of the drug can be decreased by pretreatment with olaparib and proved lethal to cells at concentrations lower than that of olaparib (EC50 = 69 nm). The radiotheranostic proved efficacious in prolonging survival of treated mice, and intratumoral administration of the drug in mice bearing human brain tumor significantly increased survival compared to vehicle alone (p = 0.0094). Application of CED-mimicking implants replicated this effect in the same model (p = 0.0361, Pirovano et al., 2019) (Figures 3D, E). High radiochemical yields (≥89%) were reported for halogenation of both olaparib and rucaparib scaffolds with astatine-211 and iodine-125 (Reilly et al., 2018). Antitumor effects were observed using an astatinated PARP inhibitor in a mouse model of neuroblastoma. Favorable uptake was observed in the tumor after 2 h. A single dose of the alpha-emitting drug was able to significantly delay tumor growth and prolong survival against a control group (Makvandi et al., 2019) (Figures 3F, G).
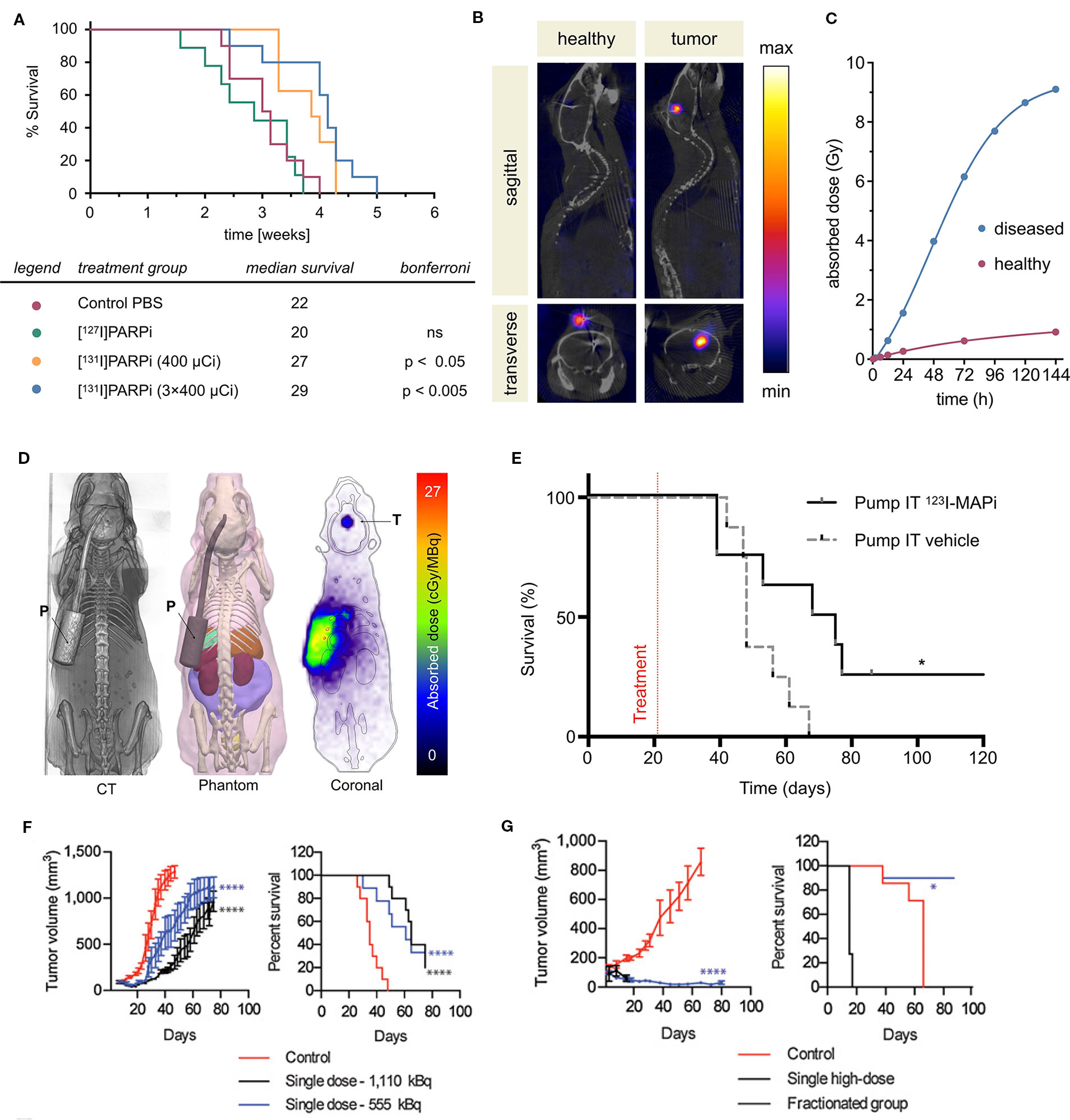
Figure 3 PARP targeted radiotherapies. Radiolabeled PARP inhibitors in mouse models of cancer. (A) Survival plot and table of treatment groups with median survival of subcutaneous U87-p53 tumor-bearing mice. P = 0.0001. (B) SPECT/CT of orthotopic U87-p53 tumors during osmotic pump treatment of [131I]PARPi at 72 hours. (C) Calculated absorbed dose to brain* during treatment. *Brain and tumor have been considered together as a single organ in organ level dose calculation. (Jannetti et al., 2018). (D) Dosimetry of the subcutaneous pump model showing CT, phantom, and Monte Carlo simulation of dose accumulation in the tumor. (E) Kaplan–Meier survival study of pump implanted mice shows an improvement of survival of 123I-MAPi treated mice (n = 8) when compared to control (n = 8). *P < 0.05 (Pirovano et al., 2019). (F) Tumor growth and Kaplan–Meier curves for IMR-05 tumor-bearing mice (F) treated with single dose of 555 or 1,110 kBq of [211At]MM4 (control vs. 555 kBq and 1,110 kBq mixed linear model p < 0.0001; control vs. 555 and 1,110 kBq survival Mantel–Cox test p < 0.0001, 555 vs. 1,110 kBq not significant, (G) and single high dose of 1,480 kBq vs. a fractionated dose of 370 kBq twice weekly for a cumulative dose of 1,480 kBq (control vs. fractionated mixed linear model p < 0.0001, fractionated vs. high dose not significant; survival Mantel–Cox test high dose vs. control p < 0.0001, fractionated vs. control p < 0.03 (Makvandi et al., 2019).
Summary and Outlook
Since its discovery we have elucidated the multiple roles PARP1 plays in the cell. PARP inhibitors have given rise to promising new cancer therapies and treatment strategies. We have recently witnessed PARPis receive approval as monotherapies for several cancers, and are waiting on the next generation of these small molecules. Many research groups are already evaluating the potential of PARPis as radiosensitizing agents in preclinical models of combination therapies. PARPis are currently being applied in the clinic as radiosensitizing agents in addition to clinical trials using combination PARPis with chemotherapies and radiation. A handful of these small molecules have been labeled to create a new class of diagnostic and radiotherapeutic agents, several of which are currently in clinical trials. The broad versatility and applications of these PARPis are providing the research community with a new set of tools for diagnosis, patient stratification, and therapy in some of the most lethal cancers.
Author Contributions
All authors have read and approved the manuscript, and the authors disclose no conflict of interests. The contributions of said authors can be broken down as follows: Drafting the manuscript, or critically contributing to or revising the manuscript, or enhancing its intellectual content: SJ, BZ, MZ, TR. Approving the final content of the manuscript: SJ, BZ, MZ, TR.
Conflict of Interest
SP, CB, JSL, and TR are shareholders of Summit Biomedical Imaging, LLC. SP and TR are co-inventors on filed U.S. patent (WO2016164771) that covers methods of use for PARPi-FL. TR is a co-inventor on U.S. patents (WO2012074840 and WO2016033293), covering the compositions of matter for PARPi-FL and 18F-PARPi, respectively. TR is a paid consultant for and has received grant support from Theragnostics, Inc., which has licensed 18F-PARPi. This arrangement has been reviewed and approved by Memorial Sloan Kettering Cancer Center in accordance with its conflict of interest policies.
References
Albert, J. M., Cao, C., Kim, K. W., Willey, C. D., Geng, L., Xiao, D., et al. (2007). Inhibition of poly(ADP-ribose) polymerase enhances cell death and improves tumor growth delay in irradiated lung cancer models. Clin. Cancer Res. 13, 3033–3042. doi: 10.1158/1078-0432.CCR-06-2872
Alemasova, E. E., Lavrik, O. I. (2019). Poly (ADP-ribosyl)ation by PARP1: reaction mechanism and regulatory proteins. Nucleic Acids Res. 47, 3811–3827. doi: 10.1093/nar/gkz120
Ali, A. A. E., Timinszky, G., Arribas-Bosacoma, R., Kozlowski, M., Hassa, P. O., Hassler, M., et al. (2012). The zinc-finger domains of PARP1 cooperate to recognize DNA strand breaks. Nat. Struct. Mol. Biol. 19, 685. doi: 10.1038/nsmb.2335
Althaus, F. R., Höfferer, L., Kleczkowska, H. E., Malanga, M., Naegeli, H., Panzeter, P. L., et al. (1994). Histone shuttling by poly ADP-ribosylation. Mol. Cell Biochem. 138, 53–59. doi: 10.1007/BF00928443
Altmeyer, M., Messner, S., Hassa, P. O., Fey, M., Hottiger, M. O. (2009). Molecular mechanism of poly(ADP-ribosyl)ation by PARP1 and identification of lysine residues as ADP-ribose acceptor sites. Nucleic Acids Res. 37, 3723–3738. doi: 10.1093/nar/gkp229
Alvarez-Gonzalez, R., Althaus, F. R. (1989). Poly(ADP-ribose) catabolism in mammalian cells exposed to DNA-damaging agents. Mutat. Res. 218, 67–74. doi: 10.1016/0921-8777(89)90012-8
Aoyama, H., Tago, M., Shirato, H., Radiation Oncology Study Group 99-1 (JROSG 99-1) Investigators Japanese. (2015). Stereotactic radiosurgery with or without whole-brain radiotherapy for brain metastases: secondary analysis of the JROSG 99-1 randomized clinical trial. JAMA Oncol. 1, 457–464. doi: 10.1001/jamaoncol.2015.1145
Barreto-Andrade, J. C., Efimova, E. V., Mauceri, H. J., Beckett, M. A., Sutton, H. G., Darga, T. E., et al. (2011). Response of human prostate cancer cells and tumors to combining PARP inhibition with ionizing radiation. Mol. Cancer Ther. 10, 1185–1193. doi: 10.1158/1535-7163.MCT-11-0061
Bauer, P. I., Hakam, A., Kun, E. (1986). Mechanisms of poly (ADP-ribose) polymerase catalysis; mono-ADP-ribosylation of poly (ADP-ribose) polymerase at nanomolar concentrations of NAD. FEBS Lett. 195, 331–338. doi: 10.1016/0014-5793(86)80188-0
Bauer, P. I., Buki, K. G., Hakam, A., Kun, E. (1990). Macromolecular association of ADP-ribosyltransferase and its correlation with enzymic activity. Biochem. J. 270, 17–26. doi: 10.1042/bj2700017
Bell, O., Tiwari, V. K., Thomä, N. H., Schübeler, D. (2011). Determinants and dynamics of genome accessibility. Nat. Rev. Genet. 12, 554. doi: 10.1038/nrg3017
Benjamin, R. C., Gill, D.M. (1980). Poly (ADP-ribose) synthesis in vitro programmed by damaged DNA. A comparison of DNA molecules containing different types of strand breaks. J. Biol. Chem. 255, 10502–10508.
Brightwell, M. D., Leech, C. E., O’Farrell, M. K., Whish, W. J., Shall, S. (1975). Poly(adenosine diphosphate ribose) polymerase in Physarum polycephalum. Biochem. J. 147, 119–129. doi: 10.1042/bj1470119
Bryant, H. E., Schultz, N., Thomas, H. D., Parker, K. M., Flower, D., Lopez, E., et al. (2005). Specific killing of BRCA2-deficient tumours with inhibitors of poly (ADP-ribose) polymerase. Nature 434, 913. doi: 10.1038/nature03443
Bryant, H. E., Petermann, E., Schultz, N., Jemth, A.-S., Loseva, O., Issaeva, N., et al. (2009). PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 28, 2601–2615. doi: 10.1038/emboj.2009.206
Calabrese, C. R., Almassy, R., Barton, S., Batey, M. A., Calvert, A. H., Canan-Koch, S., et al. (2004). Anticancer chemosensitization and radiosensitization by the novel poly(ADP-ribose) polymerase-1 inhibitor Ag14361. JNCI J. Natl. Cancer Inst. 96, 56–67. doi: 10.1093/jnci/djh005
Carlucci, G., Carney, B., Brand, C., Kossatz, S., Irwin, C. P., Carlin, S. D., et al. (2015). Dual-modality optical/PET imaging of PARP1 in glioblastoma. Mol. Imaging Biol. 17, 848–855. doi: 10.1007/s11307-015-0858-0
Carnevale, J., Ashworth, A. (2015). Assessing the significance of BRCA1 and BRCA2 mutations in pancreatic cancer. J. Clin. Oncol. 33 (28), 3080–3081. doi: 10.1200/JCO.2015.61.6961
Carney, B., Carlucci, G., Salinas, B., Di Gialleonardo, V., Kossatz, S., Vansteene, A., et al. (2016). Non-invasive PET imaging of PARP1 expression in glioblastoma models. Mol. Imaging Biol. 18, 386–392. doi: 10.1007/s11307-015-0904-y
Carney, B., Kossatz, S., Reiner, T. (2017). Molecular imaging of PARP. J. Nucl. Med. 58, 1025–1030. doi: 10.2967/jnumed.117.189936
Carney, B., Kossatz, S., Lok, B. H., Schneeberger, V., Gangangari, K. K., Pillarsetty, N. V. K., et al. (2018). Target engagement imaging of PARP inhibitors in small-cell lung cancer. Nat. Commun. 9, 176. doi: 10.1038/s41467-017-02096-w
Cejka, P. (2015). DNA end resection: nucleases team up with the right partners to initiate homologous recombination. J. Biol. Chem. 290, 22931–22938. doi: 10.1074/jbc.R115.675942
Chabot, P., Hsia, T.-C., Ryu, J.-S., Gorbunova, V., Belda-Iniesta, C., Ball, D., et al. (2017). Veliparib in combination with whole-brain radiation therapy for patients with brain metastases from non-small cell lung cancer: results of a randomized, global, placebo-controlled study. J. Neuro-oncol. 131, 105–115. doi: 10.1007/s11060-016-2275-x
Chalmers, A., Johnston, P., Woodcock, M., Joiner, M., Marples, B. (2004). PARP-1, PARP-2, and the cellular response to low doses of ionizing radiation. Int. J. Radiat. Oncol. Biol. Phys. 58, 410–419. doi: 10.1016/j.ijrobp.2003.09.053
Czito, B. G., Deming, D. A., Jameson, G. S., Mulcahy, M. F., Vaghefi, H., Dudley, M. W., et al. (2017). Safety and tolerability of veliparib combined with capecitabine plus radiotherapy in patients with locally advanced rectal cancer: a phase 1b study. Lancet Gastroenterol. Hepatol. 2, 418–426. doi: 10.1016/S2468-1253(17)30012-2
D’Amours, D., Desnoyers, S., D’Silva, I., Poirier, G. G. (1999). Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 342, 249–268. doi: 10.1042/bj3420249
D’Silva, I., Pelletier, J. D., Lagueux, J., D’Amours, D., Chaudhry, M. A., Weinfeld, M., et al. (1999). Relative affinities of poly(ADP-ribose) polymerase and DNA-dependent protein kinase for DNA strand interruptions. Biochim. Biophys. Acta 1430, 119–126. doi: 10.1016/S0167-4838(98)00278-7
David, K. K., Andrabi, S. A., Dawson, T. M., Dawson, V. L. (2009). “Parthanatos, a messenger of death. ” Front. In Biosci. (Landmark edition) 14, 1116. doi: 10.2741/3297
De Murcia, G., Huletsky, A., Lamarre, D., Gaudreau, A., Pouyet, J., Daune, M., et al. (1986). Modulation of chromatin superstructure induced by poly (ADP-ribose) synthesis and degradation. J. Biol. Chem. 261, 7011–7017.
Ding, R., Smulson, M. (1994). Depletion of nuclear poly(ADP-ribose) polymerase by antisense RNA expression: influences on genomic stability, chromatin organization, and carcinogen cytotoxicity. Cancer Res. 54, 4627–4634.
Donawho, C. K., Luo, Y., Luo, Y., Penning, T. D., Bauch, J. L., Bouska, J. J., et al. (2007). Abt-888, an orally active poly (ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin. Cancer Res. 13, 2728–2737. doi: 10.1158/1078-0432.CCR-06-3039
Dréan, A., Lord, C. J., Ashworth, A. (2016). PARP inhibitor combination therapy. Crit. Rev. Oncol. Hematol. 108, 73–85. doi: 10.1016/j.critrevonc.2016.10.010
Dubach, J. M., Vinegoni, C., Mazitschek, R., Fumene Feruglio, P., Cameron, L. A., Weissleder, R. (2014). In vivo imaging of specific drug–target binding at subcellular resolution. Nat. Commun. 5, 3946. doi: 10.1038/ncomms4946
Dubach, J. M., Kim, E., Yang, K., Cuccarese, M., Giedt, R. J., Meimetis, L. G., et al. (2017). “Quantitating drug–target engagement in single cells in vitro and in vivo.” Nat. Chem. Biol. 13, 168–173. doi: 10.1038/nchembio.2248
Dungey, F. A., Löser, D. A., Chalmers, A. J. (2008). Replication-dependent radiosensitization of human glioma cells by inhibition of poly (ADP-ribose) polymerase: mechanisms and therapeutic potential. Int. J. Radiat. Oncol. Biol. Phys. 72(4), 1188–1197. doi: 10.1016/j.ijrobp.2008.07.031
El-Khamisy, S. F. (2003). A requirement for PARP-1 for the assembly or stability of Xrcc1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 31, 5526–5533. doi: 10.1093/nar/gkg761
Eustermann, S., Videler, H., Yang, J.-C., Cole, P. T., Gruszka, D., Veprintsev, D., et al. (2011). The DNA-binding domain of human PARP-1 interacts with DNA single-strand breaks as a monomer through its second zinc finger. J. Mol. Biol. 407, 149–170. doi: 10.1016/j.jmb.2011.01.034
Evers, B., Helleday, T., Jonkers, J. (2010). Targeting homologous recombination repair defects in cancer. Trends Pharmacol. Sci. 31, 372–380. doi: 10.1016/j.tips.2010.06.001
Farmer, H., McCabe, N., Lord, C. J., Tutt, A. N., Johnson, D. A., Richardson, T. B., et al. (2005). Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921. doi: 10.1038/nature03445
Fisher, A. E., Hochegger, H., Takeda, S., Caldecott, K. W. (2007). Poly(ADP-ribose) polymerase 1 accelerates single-strand break repair in concert with poly(ADP-ribose) glycohydrolase. Mol. Cell Biol. 27, 5597–5605. doi: 10.1128/MCB.02248-06
Fong, P. C., Boss, D. S., Yap, T. A., Tutt, A., Wu, P., Mergui-Roelvink, M., et al. (2009). Inhibition of poly (ADP-ribose) polymerase in tumors from BRCA mutation carriers. New Engl. J. Med. 361, 123–134. doi: 10.1056/NEJMoa0900212
Fulton, B., Short, S. C., James, A., Nowicki, S., McBain, C., Jefferies, S., et al. (2018). PARADIGM-2: two parallel phase I studies of olaparib and radiotherapy or olaparib and radiotherapy plus temozolomide in patients with newly diagnosed glioblastoma, with treatment stratified by MGMT status. Clin. Trans. Radiat. Oncol. 8, 12–165. doi: 10.1016/j.ctro.2017.11.003
Gani, C., Coackley, C., Kumareswaran, R., Schütze, C., Krause, M., Zafarana, G., et al. (2015). In vivo studies of the PARP inhibitor, AZD-2281, in combination with fractionated radiotherapy: an exploration of the therapeutic ratio. Radiother. Oncol. 116, 486–494. doi: 10.1016/j.radonc.2015.08.003
Gottipati, P., Vischioni, B., Schultz, N., Solomons, J., Bryant, H. E., Djureinovic, T., et al. (2010). Poly (ADP-ribose) polymerase is hyperactivated in homologous recombination–defective cells. Cancer Res. 70, 5389–5398. doi: 10.1158/0008-5472.CAN-09-4716
Haince, J.-F, McDonald, D., Rodrigue, A., Déry, U., Masson, J.-Y., Hendzel, M. J., et al. (2008). PARP1-dependent kinetics of recruitment of Mre11 and Nbs1 proteins to multiple DNA damage sites. J. Biol. Chem. 283, 1197–1208. doi: 10.1074/jbc.M706734200
Hakmé, A., Wong, H.-K., Dantzer, F., Schreiber, V. (2008). The expanding field of poly (ADP-ribosyl)ation reactions. EMBO Rep. 9, 1094–1100. doi: 10.1038/embor.2008.191
Hartwell, L. H., Szankasi, P., Roberts, C. J., Murray, A. W., Friend, S. H. (1997). Integrating genetic approaches into the discovery of anticancer drugs. Science 278, 1064–1068. doi: 10.1126/science.278.5340.1064
Hassa, P. O., Hottiger, M. O. (2008). The diverse biological roles of mammalian PARPS, a small but powerful family of poly-ADP-ribose polymerases. Front. Biosci. 13, 3046–3082. doi: 10.2741/2909
Helleday, T. (2011). The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol. Oncol. 5, 387–393. doi: 10.1016/j.molonc.2011.07.001
Herceg, Z., Wang, Z. Q. (2001). Functions of poly(ADP-ribose) polymerase (PARP) in DNA repair, genomic integrity and cell death. Mutat. Res. 477, 97–110. doi: 10.1016/S0027-5107(01)00111-7
Hoa, N. N., Akagawa, R., Yamasaki, T., Hirota, K., Sasa, K., Natsume, T., et al. (2015). Relative contribution of four nucleases, CtIP, Dna2, Exo1 and Mre11, to the initial step of DNA double-strand break repair by homologous recombination in both the chicken DT40 and human TK6 cell lines. Genes Cells 20, 1059–1076. doi: 10.1111/gtc.12310
Irwin, C. P., Portorreal, Y., Brand, C., Zhang, Y., Desai, P., Salinas, B., et al. (2014). PARPi-FL—a fluorescent PARP1 inhibitor for glioblastoma imaging. Neoplasia 16, 432–440. doi: 10.1016/j.neo.2014.05.005
Jannetti, S. A., Carlucci, G., Carney, B., Kossatz, S., Shenker, L., Carter, L. M., et al. (2018). PARP1-targeted radiotherapy in mouse models of glioblastoma. J. Nucl. Med. 59, 1225–1233 doi: 10.2967/jnumed.117.205054
Küpper, J. H., De Murcia, G., Bürkle, A. (1990). Inhibition of poly (ADP-ribosyl) ation by overexpressing the poly (ADP-ribose) polymerase DNA-binding domain in mammalian cells. J. Biol. Chem. 265, 18721–18724.
Kameshita, I., Matsuda, Z., Taniguchi, T., Shizuta, Y. (1984). Poly (ADP-ribose) synthetase. Separation and identification of three proteolytic fragments as the substrate-binding domain, the DNA-binding domain, and the automodification domain. J. Biol. Chem. 259, 4770–4776.
Kaufmann, S. H., Desnoyers, S., Ottaviano, Y., Davidson, N. E., Poirier, G. G. (1993). Specific proteolytic cleavage of poly (ADP-ribose) polymerase: an early marker of chemotherapy-induced apoptosis. Cancer Res. 53, 3976–3985.
Keliher, E. J., Reiner, T., Turetsky, A., Hilderbrand, S. A., Weissleder, R. (2011). High-yielding, two-step 18F labeling strategy for 18F-PARP1 inhibitors. ChemMedChem 6, 424–427. doi: 10.1002/cmdc.201000426
Keliher, E. J., Klubnick, J. A., Reiner, T., Mazitschek, R., Weissleder, R. (2014). Efficient acid-catalyzed (18) F/ (19) F fluoride exchange of BODIPY dyes. ChemMedChem 9, 1368–1373. doi: 10.1002/cmdc.201300506
Khan, K., Araki, K., Wang, D., Li, G., Li, X., Zhang, J., et al. (2009). Head and neck cancer radiosensitization by the novel poly (ADP-ribose) polymerase inhibitor GPI-15427. Head Neck NA–NA. 32, 381–391 doi: 10.1002/hed.21195
Kim, G., Ison, G., McKee, A. E., Zhang, H., Tang, S., Gwise, T., et al. (2015). FDA approval summary: olaparib monotherapy in patients with deleterious germline BRCA-mutated advanced ovarian cancer treated with three or more lines of chemotherapy. Clin. Cancer Res. 21, 4257–4261. doi: 10.1158/1078-0432.CCR-15-0887
Kossatz, S., Brand, C., Gutiontov, S., Liu, J. T., Lee, N. Y., Gönen, M., et al. (2016). Detection and delineation of oral cancer with a PARP1 targeted optical imaging agent. Sci. Rep. 6, 21371. doi: 10.1038/srep21371
Kossatz, S., Carney, B., Schweitzer, M., Carlucci, G., Miloushev, V. Z., Maachani, U. B., et al. (2017). Biomarker-based PET imaging of diffuse intrinsic pontine glioma in mouse models. Cancer Res. 77, 2112–2123. doi: 10.1158/0008-5472.CAN-16-2850
Kossatz, S., Pirovano, G., De Souza França, P. D., Strome, A. L., Sunny, S. P., Karassawa Zanoni, D., et al. (2019). PARP1 as a biomarker for early detection and intraoperative tumor delineation in epithelial cancers—first-in-human results. bioRxiv 663385 doi: 10.1101/663385
Kraus, W. L. (2008). Transcriptional control by PARP-1: chromatin modulation, enhancer-binding, coregulation, and insulation. Curr. Opin. In Cell Biol. 20, 294–302. doi: 10.1016/j.ceb.2008.03.006
Krishnakumar, R., Kraus, W.L. (2010). The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Mol. Cell 39, 8–24. doi: 10.1016/j.molcel.2010.06.017
Langelier, M.-F., Servent, K. M., Rogers, E. E., Pascal, J. M. (2008). A third zinc-binding domain of human poly(ADP-ribose) polymerase-1 coordinates DNA-dependent enzyme activation. J. Biol. Chem. 283, 4105–4114. doi: 10.1074/jbc.M708558200
Langelier, M. F., Ruhl, D. D., Planck, J. L., Kraus, W. L., Pascal, J. M. (2010). The Zn3 domain of human poly (ADP-ribose) polymerase-1 (PARP-1) functions in both DNA-dependent poly (ADP-ribose) synthesis activity and chromatin compaction. J. Biol. Chem. 285, 18877–18887. doi: 10.1074/jbc.M110.105668
Langelier, M. F., Planck, J. L., Roy, S., Pascal, J. M. (2011). Crystal structures of poly(ADP-ribose) polymerase-1 (PARP-1) zinc fingers bound to DNA: structural and functional insights into DNA-dependent PARP-1 activity. J. Biol. Chem. 286, 10690–10701. doi: 10.1074/jbc.M110.202507
Langelier, M. F., Planck, J. L., Roy, S., Pascal, J. M. (2012). Structural basis for DNA damage-dependent poly (ADP-ribosyl)ation by human PARP-1. Science 336, 728–732. doi: 10.1126/science.1216338
Liu, S. K., Coackley, C., Krause, M., Jalali, F., Chan, N., Bristow, R. G. (2008). A novel poly (ADP-ribose) polymerase inhibitor, Abt-888, radiosensitizes malignant human cell lines under hypoxia. Radiother. Oncol. 88, 258–268. doi: 10.1016/j.radonc.2008.04.005
Lonskaya, I., Potaman, V. N., Shlyakhtenko, L. S., Oussatcheva, E. A., Lyubchenko, Y. L., Soldatenkov, V. A. (2005). Regulation of poly(ADP-ribose) polymerase-1 by DNA structure-specific binding. J. Biol. Chem. 280, 17076–17083. doi: 10.1074/jbc.M413483200
Lord, C. J., Ashworth, A. (2016). BRCAness revisited. Nat. Rev. Cancer 16 (2), 110 doi: 10.1038/nrc.2015.21
Ludwig, A., Behnke, B., Holtlund, J., Hilz, H. (1988). Immunoquantitation and size determination of intrinsic poly (ADP-ribose) polymerase from acid precipitates. An analysis of the in vivo status in mammalian species and in lower eukaryotes. J. Biol. Chem. 263, 6993–6999.
Makvandi, M., Pantel, A., Schwartz, L., Schubert, E., Xu, K., Hsieh, C.-J., et al. (2018). A PET imaging agent for evaluating PARP-1 expression in ovarian cancer. J. Clin. Invest. 128(5), 2116–2126. doi: 10.1172/JCI97992
Makvandi, M., Lee, H., Puentes, L. N., Reilly, S. W., Rathi, K. S., Weng, C. C., et al. (2019). Targeting PARP-1 with alpha-particles is potently cytotoxic to human neuroblastoma in preclinical models. Mol. Cancer Ther. 18, 1195–1204. doi: 10.1158/1535-7163.MCT-18-0837
Martin, D. S., Bertino, J. R., Koutcher, J. A. (2000). ATP depletion + pyrimidine depletion can markedly enhance cancer therapy: fresh insight for a new approach. Cancer Res. 60, 6776–6783.
Mateo, J., Carreira, S., Sandhu, S., Miranda, S., Mossop, H., Perez-Lopez, R., et al. (2015). DNA-repair defects and olaparib in metastatic prostate cancer. New Engl. J. Med. 373, 1697–1708. doi: 10.1056/NEJMoa1506859
McCabe, N., Turner, N. C., Lord, C. J., Kluzek, K., Bialkowska, A., Swift, S., et al. (2006). Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 66, 8109–8115. doi: 10.1158/0008-5472.CAN-06-0140
Mendoza-Alvarez, H., Alvarez-Gonzalez, R. (1993). Poly(ADP-ribose) polymerase is a catalytic dimer and the automodification reaction is intermolecular. J. Biol. Chem. 268, 22575–22580.
Mendoza-Alvarez, H., Alvarez-Gonzalez, R. (1999). Biochemical characterization of mono (ADP-ribosyl) ated poly (ADP-ribose) polymerase. Biochemistry 38, 3948–3953. doi: 10.1021/bi982148p
Mendoza-Alvarez, H., Alvarez-Gonzalez, R. (2004). The 40 kDa carboxy-terminal domain of poly (ADP-ribose) polymerase-1 forms catalytically competent homo- and heterodimers in the absence of DNA. J. Mol. Biol. 336, 105–114. doi: 10.1016/j.jmb.2003.11.055
Menear, K. A., Adcock, C., Boulter, R., Cockcroft, X.-l., Copsey, L., Cranston, A., et al. (2008). 4-[3-(4-cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2h-phthalazin-1-one: a novel bioavailable inhibitor of poly (ADP-ribose) polymerase-1. J. Med. Chem. 51, 6581–6591. doi: 10.1021/jm8001263
Michel, L. S., Dyroff, S., Brooks, F. J., Spayd, K. J., Lim, S., Engle, J. T., et al. (2017). Pet of poly (ADP-ribose) polymerase activity in cancer: preclinical assessment and first in-human studies. Radiology 282, 453–463. doi: 10.1148/radiol.2016161929
Miki, Y., Swensen, J., Shattuck-Eidens, D., Futreal, P.A., Harshman, K., Tavtigian, S., et al. (1994). A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 66–71. doi: 10.1126/science.7545954
Min, W., Wang, Z. Q. (2009). Poly (ADP-ribose) glycohydrolase (PARG) and its therapeutic potential. Front. Biosci. (Landmark Ed) 14, 1619–1626. doi: 10.2741/3329
Moynahan, M. E., Cui, T. Y., Jasin, M. (2001). Homology-directed DNA repair, mitomycin-C resistance, and chromosome stability is restored with correction of a BRCA1 mutation. Cancer Res. 61, 4842–4850.
Murai, J., Huang, S. Y., Das, B. B., Renaud, A., Zhang, Y., Doroshow, J. H., et al. (2012). Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 72, 5588–5599. doi: 10.1158/0008-5472.CAN-12-2753
Murai, J., Huang, S.-Y. N., Renaud, A., Zhang, Y., Ji, J., Takeda, S., et al. (2014). Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol. Cancer Ther. 13, 433–443. doi: 10.1158/1535-7163.MCT-13-0803
Noel, G., Godon, C., Fernet, M., Giocanti, N., Megnin-Chanet, F., Favaudon, V. (2006). Radiosensitization by the poly (ADP-ribose) polymerase inhibitor 4-amino-1, 8-naphthalimide is specific of the S phase of the cell cycle and involves arrest of DNA synthesis. Mol. Cancer Ther. 5(3), 564–574. doi: 10.1158/1535-7163.MCT-05-0418
Ogata, N., Kawaichi, M., Ueda, K., Hayaishi, O. (1980). Poly (ADP-ribosyl)ation of 110,000 Dalton protein in human-lymphocytes treated with N-methyl-n′-nitro-n-nitrosoguanidine. Biochem. Int. 1, 229–236.
Oka, S., Kato, J., Moss, J. (2006). Identification and characterization of a mammalian 39-kDa poly(ADP-ribose) glycohydrolase. J. Biol. Chem. 281, 705–713. doi: 10.1074/jbc.M510290200
Okano, S., Lan, L., Caldecott, K. W., Mori, T., Yasui, A. (2003). Spatial and temporal cellular responses to single-strand breaks in human cells. Mol. Cell. Biol. 23, 3974–3981. doi: 10.1128/MCB.23.11.3974-3981.2003
Panzeter, P. L., Althaus, F. R. (1994). DNA strand break-mediated partitioning of poly (ADP-ribose) polymerase function. Biochemistry 33, 9600–9605. doi: 10.1021/bi00198a028
Patel, A. G., Sarkaria, J. N., Kaufmann, S. H. (2011). Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc. Natl. Acad. Sci. U. S. A 108, 3406–3411. doi: 10.1073/pnas.1013715108
Petermann, E. (2003). ATP-dependent selection between single nucleotide and long patch base excision repair. DNA Repair 2, 1101–1114. doi: 10.1016/S1568-7864(03)00117-4
Pion, E., Bombarda, E., Stiegler, P., Matthias Ullmann, G., Mély, Y., de Murcia, G., et al. (2003). Poly (ADP-ribose) polymerase-1 dimerizes at a 5′ recessed DNA end in vitro: a fluorescence study. Biochemistry 42, 12409–12417. doi: 10.1021/bi0344432
Pirovano, G., Jannetti, S. A., Carter, L. M., Sadique, A., Kossatz, S., Guru, N., et al. (2019). Targeted brain tumor radiotherapy using an Auger emitter. J. Labelled Compounds Radiopharmaceut. n/a–n/a. doi: 10.1101/649764
Pleschke, J. M., Kleczkowska, H. E., Strohm, M., Althaus, F. R. (2000). Poly(ADP-ribose) binds to specific domains in DNA damage checkpoint proteins. J. Biol. Chem. 275, 40974–40980. doi: 10.1074/jbc.M006520200
Plummer, R., Lorigan, P., Steven, N., Scott, L., Middleton, M. R., Wilson, R. H., et al. (2013). A phase II study of the potent PARP inhibitor, rucaparib (PF-01367338, AG014699), with temozolomide in patients with metastatic melanoma demonstrating evidence of chemopotentiation. Cancer Chemother. Pharmacol. 71, 1191–1199. doi: 10.1007/s00280-013-2113-1
Poirier, G. G., Niedergang, C., Madeleine, C., Alice, M., Mandel, P. (1982). Adenosine diphosphate ribosylation of chicken-erythrocyte histones H1, H5 and high-mobility-group proteins by purified calf-thymus poly (adenosinediphosphate-ribose) polymerase. Eur. J. Biochem. 127, 437–442. doi: 10.1111/j.1432-1033.1982.tb06891.x
Pommier, Y., O’Connor, M. J., de Bono, J. (2016). Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med. 8, 362ps17. doi: 10.1126/scitranslmed.aaf9246
Purnell, M. R., Whish, W. J. D. (1980). The effect of phenones on poly (adenosine diphosphate ribose) synthetase from porcine thymus. Biochem. J. 185 (3), 775. doi: 10.1042/bst0080175
Reilly, S. W., Makvandi, M., Xu, K., Mach, R. H. (2018). Rapid Cu-catalyzed [211At]astatination and [125I]iodination of boronic esters at room temperature. Org. Lett. 20, 1752–1755. doi: 10.1021/acs.orglett.8b00232
Reiner, T., Lacy, J., Keliher, E. J., Yang, K. S., Ullal, A., Kohler, R. H., et al. (2012). Imaging therapeutic PARP inhibition in vivo through bioorthogonally developed companion imaging agents. Neoplasia 14, 169–177. doi: 10.1593/neo.12414
Riss, P. J., Soskic, V., Schrattenholz, A., Roesch, F. (2009). Synthesis and radiosynthesis of N5-[18F] fluoroethyl-pirenzepine and its metabolite N5-[18F] fluoroethyl-LS 75. J. Labelled Compounds Radiopharmaceut. Off. J. Int. Isotope Soc. 52, 576–579. doi: 10.1002/jlcr.1687
Rottenberg, S., Jaspers, J. E., Kersbergen, A., van der Burg, E., Nygren, A. O. H., Zander, S. A. L., et al. (2008). High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc. Natl. Acad. Sci. 105, 17079–17084. doi: 10.1073/pnas.0806092105
Rouleau, M., Patel, A., Hendzel, M. J., Kaufmann, S. H., Poirier, G. G. (2010). PARP inhibition: PARP1 and beyond. Nat. Rev. Cancer 10, 293–301. doi: 10.1038/nrc2812
Roy, R., Chun, J., Powell, S. N. (2011). BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat. Rev. Cancer 12, 68–78. doi: 10.1038/nrc3181
Russo, A. L., Kwon, H. C., Burgan, W. E., Carter, D., Beam, K., Weizheng, X., et al. (2009). In vitro and in vivo radiosensitization of glioblastoma cells by the poly (ADP-ribose) polymerase inhibitor E7016. Clin. Cancer Res. 15, 607–612. doi: 10.1158/1078-0432.CCR-08-2079
Salinas, B., Irwin, C. P., Kossatz, S., Bolaender, A., Chiosis, G., Pillarsetty, N., et al. (2015). Radioiodinated PARP1 tracers for glioblastoma imaging. EJNMMI Res. 5, 123. doi: 10.1186/s13550-015-0123-1
Sander Effron, S., Makvandi, M., Lin, L., Xu, K., Li, S., Lee, H., et al. (2017). PARP-1 expression quantified by [18F]fluorthanatrace: a biomarker of response to PARP inhibition adjuvant to radiation therapy. Cancer Biother. Radiopharm. 32, 9–15. doi: 10.1089/cbr.2016.2133
Schöder, H., De Souza Franca, P. D., Nakajima, R., Burnazi, E., Roberts, S., Brand, C., et al. (2019). PARP1/2 imaging with 18F-PARPi in patients with head and neck cancer. medRxiv. doi: 10.1101/19009381
Schoonen, P. M., van Vugt, M. A. T. M. (2018). Never tear us a-PARP: dealing with DNA lesions during mitosis. Mol. Cell. Oncol. 5(1), e1382670. doi: 10.1080/23723556.2017.1382670
Schreiber, V., Hunting, D., Trucco, C., Gowans, B., Grunwald, D., De Murcia, G., et al. (1995). A dominant-negative mutant of human poly(ADP-ribose) polymerase affects cell recovery, apoptosis, and sister chromatid exchange following DNA damage. Proc. Natl. Acad. Sci. U. S. A. 92, 4753–4757. doi: 10.1073/pnas.92.11.4753
Senra, J. M., Telfer, B. A., Cherry, K. E., McCrudden, C. M., Hirst, D. G., O’Connor, M. J., et al. (2011). Inhibition of PARP-1 by olaparib (AZD2281) increases the radiosensitivity of a lung tumor xenograft. Mol. Cancer Ther. 10, 1949–1958. doi: 10.1158/1535-7163.MCT-11-0278
Shelton, J. W., Waxweiler, T. V., Landry, J., Gao, H., Xu, Y., Wang, L., et al. (2013). In vitro and in vivo enhancement of chemoradiation using the oral PARP inhibitor ABT-888 in colorectal cancer cells. Int. J. Radiat. Oncol. Biol. Phys. 86, 469–476. doi: 10.1016/j.ijrobp.2013.02.015
Shen, Y., Rehman, F. L., Feng, Y., Boshuizen, J., Bajrami, I., Elliott, R., et al. (2013). BMN 673, a novel and highly potent PARP1/2 inhibitor for the treatment of human cancers with DNA repair deficiency. Clin. Cancer Res. 19(18), 5003–5015. doi: 10.1158/1078-0432.CCR-13-1391
Sims, J. L., Berger, S. J., Berger, N. A. (1983). Poly(ADP-ribose) polymerase inhibitors preserve nicotinamide adenine dinucleotide and adenosine 5′-triphosphate pools in DNA-damaged cells: mechanism of stimulation of unscheduled DNA synthesis. Biochemistry 22, 5188–5194. doi: 10.1021/bi00291a019
Ström, C. E., Johansson, F., Uhlén, M., Szigyarto, C. A., Erixon, K., Helleday, T. (2011). Poly (ADP-ribose) polymerase (PARP) is not involved in base excision repair but PARP inhibition traps a single-strand intermediate. Nucleic Acids Res. 39, 3166–3175. doi: 10.1093/nar/gkq1241
Strumberg, D., Pilon, A. A., Smith, M., Hickey, R., Malkas, L., Pommier, Y. (2000). Conversion of topoisomerase I cleavage complexes on the leading strand of ribosomal DNA into 5′-phosphorylated DNA double-strand breaks by replication runoff. Mol. Cell. Biol. 20, 3977–3987. doi: 10.1128/MCB.20.11.3977-3987.2000
Sugimura, K., Takebayashi, S.-i., Taguchi, H., Takeda, S., Okumura, K. (2008). PARP-1 ensures regulation of replication fork progression by homologous recombination on damaged DNA. J. Cell Biol. 183, 1203–1212. doi: 10.1083/jcb.200806068
Tangutoori, S., Baldwin, P., Sridhar, S. (2015). PARP inhibitors: a new era of targeted therapy. Maturitas 81, 5–9. doi: 10.1016/j.maturitas.2015.01.015
Terada, M., Fujiki, H., Marks, P. A., Sugimura, T. (1979). Induction of erythroid differentiation of murine erythroleukemia cells by nicotinamide and related compounds. Proc. Natl. Acad. Sci. U. S. A. 76, 6411–6414. doi: 10.1073/pnas.76.12.6411
Thomas, A., Murai, J., Pommier, Y. (2018). The evolving landscape of predictive biomarkers of response to PARP inhibitors. J. Clin. Invest. 128, 1727–1730. doi: 10.1172/JCI120388
Thomas, C., Ji, Y., Wu, C., Datz, H., Boyle, C., MacLeod, B., et al. (2019). Hit and run versus long-term activation of PARP-1 by its different domains fine-tunes nuclear processes. Proc. Natl. Acad. Sci. U. S. A. 116, 9941–9946. doi: 10.1073/pnas.1901183116
Thurber, G. M., Yang, K. S., Reiner, T., Kohler, R. H., Sorger, P., Mitchison, T., et al. (2013). Single-cell and subcellular pharmacokinetic imaging allows insight into drug action in vivo. Nat. Commun. 4, 1504. doi: 10.1038/ncomms2506
Timinszky, G., Till, S., Hassa, P. O., Hothorn, M., Kustatscher, G., Nijmeijer, B., et al. (2009). A macrodomain-containing histone rearranges chromatin upon sensing PARP1 activation. Nat. Struct. Mol. Biol. 16, 923. doi: 10.1038/nsmb.1664
Tu, Z., Chu, W., Zhang, J., Dence, C. S., Welch, M. J., Mach, R. H. (2005). Synthesis and in vivo evaluation of [11C]PJ34, a potential radiotracer for imaging the role of PARP-1 in necrosis. Nucl. Med. Biol. 32, 437–443. doi: 10.1016/j.nucmedbio.2005.03.001
Tulin, A., Spradling, A. (2003). Chromatin loosening by poly(ADP)-ribose polymerase (PARP) at Drosophila puff loci. Science 299, 560–562. doi: 10.1126/science.1078764
Turner, N., Tutt, A., Ashworth, A. (2004). Hallmarks of ‘BRCAness’ in sporadic cancers. Nat. Rev. Cancer 4, 814. doi: 10.1038/nrc1457
Tutt, A., Bertwistle, D., Valentine, J., Gabriel, A., Swift, S., Ross, G., et al. (2001). Mutation in BRCA2 stimulates error-prone homology-directed repair of DNA double-strand breaks occurring between repeated sequences. EMBO J. 20, 4704–4716. doi: 10.1093/emboj/20.17.4704
Vasil’eva, I. A., Anarbaev, R. O., Moor, N. A., Lavrik, O. I. (2019). Dynamic light scattering study of base excision DNA repair proteins and their complexes. Biochim. Biophys. Acta (BBA)-Proteins Proteomics 1867, 297–305. doi: 10.1016/j.bbapap.2018.10.009
Venere, M., Hamerlik, P., Wu, Q., Rasmussen, R. D., Song, L. A., Vasanji, A., et al. (2014). Therapeutic targeting of constitutive PARP activation compromises stem cell phenotype and survival of glioblastoma-initiating cells. Cell Death Differ. 21, 258–269. doi: 10.1038/cdd.2013.136
Waddell, N., Pajic, M., Patch, A. M., Chang, D. K., Kassahn, K. S., Bailey, P., et al (2015). Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 518 (7540), 495–501 doi: 10.1038/nature14169
Waldman, A. S., Waldman, B. C. (1991). Stimulation of intrachromosomal homologous recombination in mammalian cells by an inhibitor of poly(ADP-ribosylation). Nucleic Acids Res. 19, 5943–5947. doi: 10.1093/nar/19.21.5943
Walker, J. W., Jijon, H. B., Madsen, K. L. (2006). AMP-activated protein kinase is a positive regulator of poly(ADP-ribose) polymerase. Biochem. Biophys. Res. Commun. 342, 336–341. doi: 10.1016/j.bbrc.2006.01.145
Wang, F., Zhu, S., Fisher, L. A., Wang, L., Eurek, N. J., Wahl, J. K., et al. (2019). Phosphatase 1 nuclear targeting subunit mediates recruitment and function of poly (ADP-ribose) polymerase 1 in DNA repair. Cancer Res. 79, 2526–2535. doi: 10.1158/0008-5472.CAN-18-1673
Wilson, T. C., Xavier, M. A., Knight, J., Verhoog, S., Torres, J. B., Mosley, M., et al. (2019). PET imaging of PARP expression using 18F-olaparib. J. Nucl. Med. 60, 504–510. doi: 10.2967/jnumed.118.213223
Wooster, R., Bignell, G., Lancaster, J., Swift, S., Seal, S., Mangion, J., et al. (1995). Identification of the breast cancer susceptibility gene BRCA2. Nature 378, 789. doi: 10.1038/378789a0
Yang, Y. G., Cortes, U., Patnaik, S., Jasin, M., Wang, Z. Q. (2004). Ablation of PARP-1 does not interfere with the repair of DNA double-strand breaks, but compromises the reactivation of stalled replication forks. Oncogene 23, 3872–3882. doi: 10.1038/sj.onc.1207491
Zhao, W., Hu, H., Mo, Q., Guan, Y., Li, Y., Du, Y., et al. (2019). Function and mechanism of combined PARP-1 and BRCA genes in regulating the radiosensitivity of breast cancer cells. Int. J. Clin. Exp. Pathol. 12(10), 3915.
Zhou, D., Chu, W., Xu, J., Jones, L. A., Peng, X., Li, S., et al. (2014). Synthesis, [18F] radiolabeling, and evaluation of poly (ADP-ribose) polymerase-1 (PARP-1) inhibitors for in vivo imaging of PARP-1 using positron emission tomography. Bioorg. Med. Chem. 22, 1700–1707. doi: 10.1016/j.bmc.2014.01.019
Keywords: PARP [poly(ADP-ribose) polymerase], radiotheranostic, molecular imaging, targeted radiotherapy (TRT), combination therapy
Citation: Jannetti SA, Zeglis BM, Zalutsky MR and Reiner T (2020) Poly(ADP-Ribose)Polymerase (PARP) Inhibitors and Radiation Therapy. Front. Pharmacol. 11:170. doi: 10.3389/fphar.2020.00170
Received: 13 November 2019; Accepted: 07 February 2020;
Published: 03 March 2020.
Edited by:
Salvatore Salomone, University of Catania, ItalyReviewed by:
Ross Carruthers, University of Glasgow, United KingdomRobert Henry Mach, University of Pennsylvania, United States
Copyright © 2020 Jannetti, Zeglis, Zalutsky and Reiner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas Reiner, reinert@mskcc.org