 James R. Groome
James R. Groome Landon Bayless-Edwards
Landon Bayless-Edwards
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 28 February 2020
Sec. Pharmacology of Ion Channels and Channelopathies
Volume 11 - 2020 | https://doi.org/10.3389/fphar.2020.00160
This article is part of the Research Topic Molecular Mechanisms of Voltage-Gating in Ion Channels View all 11 articles
Voltage-gated ion channels share a common structure typified by peripheral, voltage sensor domains. Their S4 segments respond to alteration in membrane potential with translocation coupled to ion permeation through a central pore domain. The mechanisms of gating in these channels have been intensely studied using pioneering methods such as measurement of charge displacement across a membrane, sequencing of genes coding for voltage-gated ion channels, and the development of all-atom molecular dynamics simulations using structural information from prokaryotic and eukaryotic channel proteins. One aspect of this work has been the description of the role of conserved negative countercharges in S1, S2, and S3 transmembrane segments to promote sequential salt-bridge formation with positively charged residues in S4 segments. These interactions facilitate S4 translocation through the lipid bilayer. In this review, we describe functional and computational work investigating the role of these countercharges in S4 translocation, voltage sensor domain hydration, and in diseases resulting from countercharge mutations.
The seminal work of Hodgkin and Huxley (1952) described voltage-dependent gating particles that determine membrane permeability to sodium and potassium ions during an action potential. Their experimental work culminated in a mathematical description of the action potential based on activation gates for sodium (m) and potassium (n) ions, and an inactivation gate for sodium (h). This paradigm describing the ionic basis for the action potential has since been upheld and augmented with biophysical explanations of the molecular phenomena through which voltage-gated ion channels (VGICs) dictate membrane excitability.
The description of the structure of DNA and the discovery of the genetic code allowed molecular biology to become an integral part of research into VGIC function. Genes for ion channels and other proteins involved in electrical signaling were cloned using strategies of peptide purification from a diversity of electrically excitable tissues [reviewed by (Dolphin, 2018)], and by employing unique phenotypes of Drosophila mutants to generate probes for gene cloning [i.e., Shaker potassium channel (Papazian et al., 1987)], and para sodium channel [reviewed by (Papazian et al., 1988; Ganetzky, 2000)].
A striking pattern emerged in the amino acid sequences predicted from gene sequences for VGICs. Regularly spaced positively charged amino acid residues were observed within the fourth segment (S4) of hydrophobicity (Noda et al., 1984; Figure 1). Subsequent models of channel structure that were put forth included postulates regarding the interaction of positive and negative charges, and a helical screw mechanism of S4 translocation in channel activation (Greenblatt et al., 1985; Guy and Seetharamulu, 1986). With functional reconstitution of ion channel proteins in mammalian cells or Xenopus oocytes (Noda et al., 1986; Stuhmer et al., 1988; Timpe et al., 1988; Leonard et al., 1989), investigations of the structure to function relationships in VGICs focused first on the role of S4 segments in channel activation and fast inactivation (Stuhmer et al., 1989; Zagotta et al., 1989; Papazian et al., 1991). Progress in our understanding of the mechanisms of voltage-gating in ion channels has since been facilitated with advanced electrophysiological techniques, crystallography, and computational work. In this review, we examine the research efforts that describe the contributions made by negative countercharges of S1 to S3 segments to the voltage-gating of ion channels.

Figure 1 Multiple sequence alignments of voltage sensor domains (VSDs) (S1–S4) from a sample of voltage-gated ion channel (VGIC) alpha subunit domains. Prokaryotic sodium channels are NaVAb from Arcobacter butzleri (Payandeh et al., 2011), and the NaChBac orthologue NaVRh from alpha proteobacterium (Zhang et al., 2012). The remaining sequences are human. For each alignment, putative consensus negative charge regions are highlighted with boxes colored according to frequently observed residues (glutamate, yellow; asparagine, red; aspartate, blue). The first five positively charged residues in S4 are shown in green.
Voltage-gated channels show considerable homology in S1–S4 segments that comprise the voltage sensor domain (VSD). Alignments for a sample of voltage-gated sodium, calcium, and potassium channel VSDs are shown in Figure 1. Loci of conserved negatively charged (acidic) or polar residues are observed in S1–S3 segments. These residues represent putative countercharges to conserved, positively charged residues observed in the S4 segment.
An extensive analysis of VSDs from over 6,500 sequence alignments across taxa (Figure 2A; Palovcak et al., 2014) highlights the conservation of acidic or polar charges in S1–S3, positive charges in S4, and the degree of sequence identity. Figure 2B shows the location of conserved countercharges in relation to a central hydrophobic constriction site (HCS) that separates the outer and inner vestibules of the VSD. Countercharges above the HCS are located in an extracellular negatively charged region (ENC), with those nearer to the cytoplasmic region located in the intracellular negatively charged region (INC). Also shown is a conserved S2 aromatic residue located in a putative, gating charge transfer center (GCTC); this aromatic residue may act as a steric barrier to S4 translocation (Tao et al., 2010; Schwaiger et al., 2013). Current models of that S4 movement postulate that S4 arginine residues pass through an aqueous gating pore in the HCS region of the VSD [reviewed by (Groome et al., 2018)]. Side chains of S1–S3 countercharges face toward the guanidyl groups of the conserved S4 arginine residues (Figure 2C). The high degree of conservation of countercharge residues suggests that they play an important role or roles in channel function. Interactions of these countercharges with S4 residues have been investigated in a set of functional experiments described below, that support roles for countercharges in protein folding and S4 translocation.
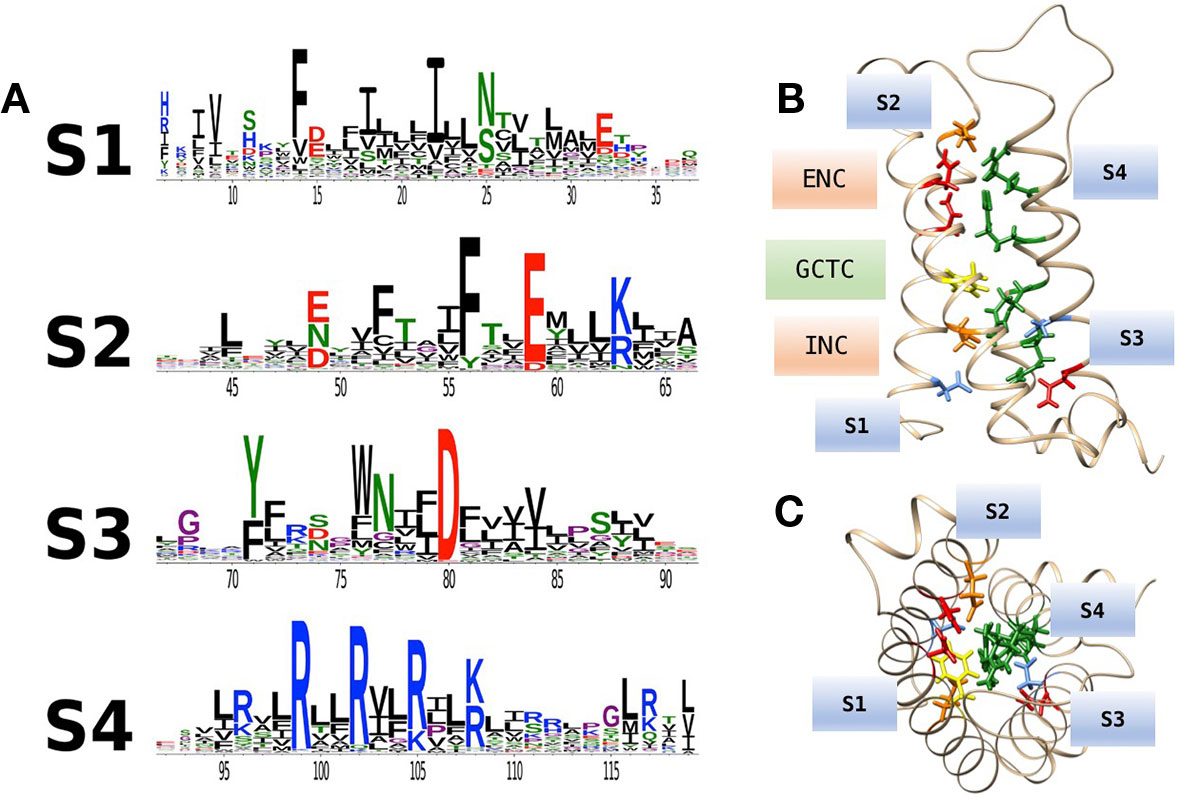
Figure 2 (A) Conservation of amino acid residues in voltage sensor domains (VSDs) across taxa, highlighting the evolutionary conservation of polar or acidic residues in S1–S3 (putative countercharges), positively charged arginine or lysine residues in S4, and conserved aromatic residues. With permission from J. Gen. Physiol. and first author (Palovcak et al., 2014). (B) Homology model of hNaV1.4 domain IV VSD based on prokaryotic structural information [3RVY.pdb, (Payandeh et al., 2011)] showing locations of consensus S1–S3 countercharges in the extracellular negatively charged (ENC) and intracellular negatively charged (INC) regions. A conserved aromatic in S2 (yellow) is also shown as part of the gating charge transfer center (GCTC). (C) Top view of the VSD showing side chains of S1–S3 countercharges facing the S4 arginine guanidyl groups.
Experiments to describe gating charge movements associated with sodium channel function in the squid giant axon (Armstrong and Bezanilla, 1973; Armstrong and Bezanilla, 1977) were followed by characterization of gating charge movement in Shaker potassium channels (Perozo et al., 1992; Perozo et al., 1994; Bezanilla et al., 1994). The biophysical basis for displacement of the gating charge, and thus voltage-sensitivity, is S4 translocation in response to altered membrane potential [reviewed by (Bezanilla, 2000; Bezanilla, 2018)]. Investigations of the role of the S4 segment as the voltage sensor have employed mutagenesis including mutant cycle analysis, toxins to trap resting or activated states, thiosulfonate reagents to determine S4 residue accessibility, fluorescence measurements, and molecular dynamics simulations [reviewed by (Groome, 2014)]. Pertinent to this review, a series of investigations have supported the premise that sequential, salt-bridge (countercharge/S4 residue) interactions facilitate transitions from the resting to fully activated state of VGICs. Here, we describe the functional experiments that provide support for that role of countercharges in voltage-gating.
Countercharge residues that interact with S4 arginines were first identified in potassium channels [Table 1, and reviewed by (Fedida and Hesketh, 2001; Papazian et al., 2002; Kuang et al., 2015)]. In a series of papers, Papazian and colleagues showed that electrostatic interactions between S4 residues and negative countercharges in S2 and S3 were crucial in the folding or maturation of the fully functional tetrameric Shaker channel (Papazian et al., 1995; Seoh et al., 1996; Tiwari-Woodruff et al., 1997; Schulteis et al., 1998). Protein maturation in Shaker was dependent on short-range electrostatic interactions between S2 ENC residue E283 and S4 residues R368 (R3) and R371 (R4), and between INC residues E293 (S2) and D316 (S3) and S4 residue K374 (K5).
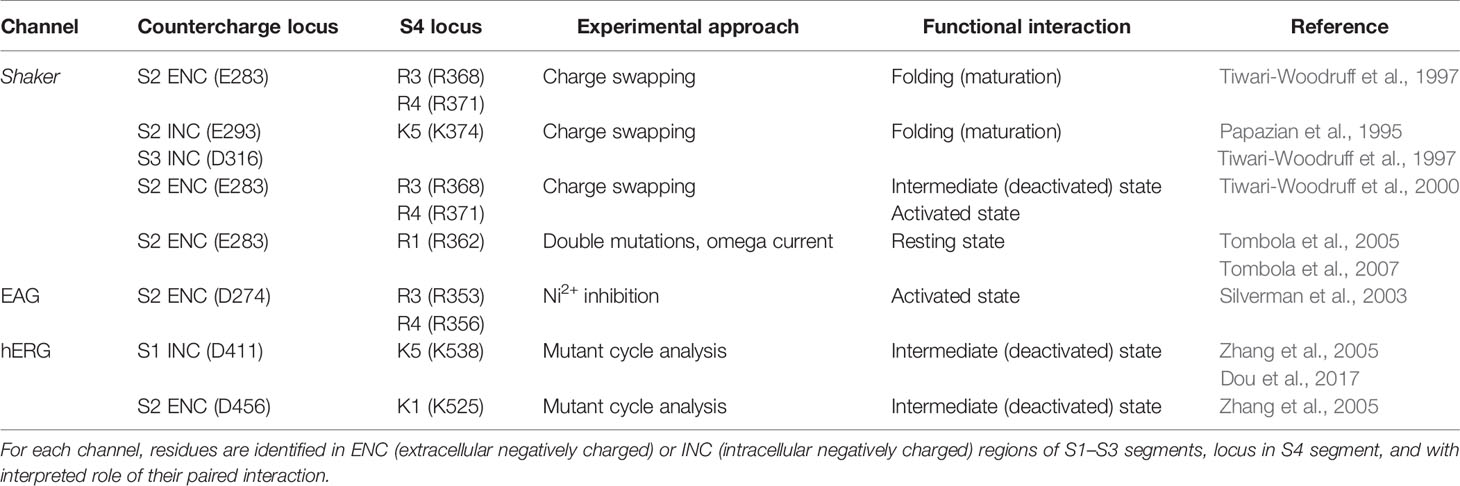
Table 1 Specific countercharge/S4 residue interactions identified in potassium channels.
Comparison of the effects of single (charge-reversing) versus double (charge-swapping) mutations reveled pairwise interactions in the functional Shaker channel between E283 and R368 (intermediate, closed state activation) and between E283 and R371 [open-state activation; (Tiwari-Woodruff et al., 2000)]. A closed, resting state interaction was suggested between E283 and R362 (R1) by comparing the amplitude of omega current in single and double mutations using an R1S construct (Tombola et al., 2005; Tombola et al., 2007). Studies of EAG potassium channels confirmed interactions of countercharge and S4 arginine residues during activation. Silverman et al. (2003) showed that a unique, inhibitory effect of divalent ion (Ni2+) on the S2 ENC mutant D274A was reversed by pairing that mutant with R353Q (R3). Pairing D274A with R356Q (R4) did not reverse Ni2+ inhibition. Finally, the observation that Ni2+ inhibition was reversed in the triple D274A/R353Q/R356Q mutation suggested stepwise translocation of R353 and R356 to promote their interaction with D274. These results supported a two-stage model of S4 activation mediated by interaction of an S2 ENC residue with S4 arginine residues R3 and R4, as in Shaker channels.
In studies of human EAG channels (hERG), mutant cycle analysis showed that the S1 INC countercharge D411 and S2 ENC countercharge D456 interact with S4 residues K538 (K5) and K525 (K1), respectively, dictating early closed-state transitions (Zhang et al., 2005; Dou et al., 2017). Results using a similar approach suggested that S2 and S3 countercharges D460 and D509 may influence S4 translocation later in the activation pathway (Liu et al., 2003; Zhang et al., 2005). Taken together, results from these studies on potassium channels were consistent with a model of voltage-gating in which, from a resting conformation with R1 in the GCTC (Lin et al., 2011), sequential pairwise interactions of countercharge residues with S4 residues mediate channel activation.
Cloning of the prokaryotic sodium channel NaChBac (Ren et al., 2001) provided an advantage to study specific electrostatic interactions in the VSD of sodium channels. Like potassium channels, functional NaChBac is comprised of a homotetramer from the channel gene coding for one domain, providing robust effect with mutagenesis and more direct interpretation of the effect of these mutations on channel function.
In studies with NaChBac, cysteine substitution of countercharge and S4 residue pairs allowed for interpretation of their putative interaction (Table 2). For cysteine substitutions in close proximity, disulfide bond formation resulted in loss of channel availability. A key element of these experiments was the observation that reducing agents such as beta-mercaptoethanol or TCEP (tris 2-carboxyethyl phosphine) were able to rescue channel function by breaking the disulfide linkage. For mutations in which partial loss of channel opening was observed, interactions were determined from results showing that oxidizing agents such as hydrogen peroxide more fully abolished sodium currents, and with subsequent treatment with reducing agents that restored channel function. Double-mutant cycle analysis of the free energy change required for activation was used in these experiments to corroborate interactions [for review see (Bosshard et al., 2004)].
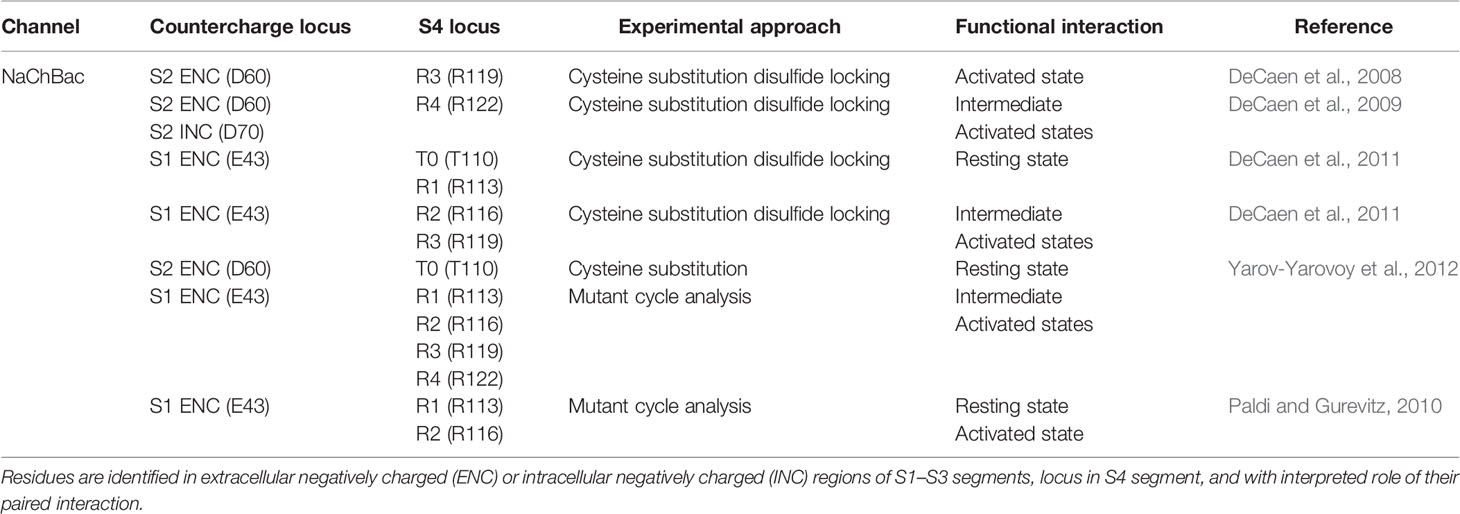
Table 2 Specific countercharge/S4 residue interactions identified in NaChBac sodium channels.
These experiments showed that NaChBac S2 residues D60 (ENC) and D70 (INC) interact with R119 (R3) and/or R122 (R4) during activation (DeCaen et al., 2008; DeCaen et al., 2009). Subsequent studies employing disulfide locking (DeCaen et al., 2011), and/or mutant cycle analysis (Paldi and Gurevitz, 2010; Yarov-Yarovoy et al., 2012) revealed interactions between a highly conserved ENC S1 residue (E43) and S4 residues T110 (T0), R113 (R1) in the resting state, and with R116 (R2), R119 (R3), and R122 (R4) during intermediate to fully activated states of the channel. A combined set of results was compared in Rosetta models illustrating the pairwise interaction of countercharge/S4 residue interactions from resting to activated states (Yarov-Yarovoy et al., 2012), with an interpretation of VSD gating similar to that suggested by the aforementioned experiments in potassium channels.
With the reports of crystal structures for prokaryotic sodium channels (Payandeh et al., 2011; Payandeh et al., 2012; Zhang et al., 2012) supporting the functional characterization of countercharge/S4 residue interactions in NaChBac, research attention was given to eukaryotic sodium channels. Here, both molecular dynamics simulations (Gosselin-Badaroudine et al., 2012) and scanning mutagenesis (Groome and Winston, 2013; Pless et al., 2014) probed each of the four domains of the skeletal muscle sodium channel (NaV1.4) for domain-specific roles of countercharges in activation and fast inactivation. Groome and Winston (2013) found that effects on activation probability were notable for mutations in domain II, while effects on fast inactivation kinetics were characteristic in domain IV (Figure 3). Using unnatural amino acid substitutions to precisely target the relative impact of charge content versus structure for gating contributions of countercharge residues, Pless et al. (2014) showed that mutations of ENC residues in domains I and II produce effects dependent on charge, whereas the effects of INC mutations are independent of charge alteration. These results were similar to those observed for Shaker channel mutations at ENC versus INC loci (Pless et al. (2011), and these authors presented the possibility that INC charges influence S4 movement through hydration of the inner vestibule of the VSD. Clearly, the significant differences observed in the functional effects of S1 to S3 mutations across domains, or at different loci (ENC versus INC) should be considered in the interpretation of the role(s) of countercharges in voltage-gating.
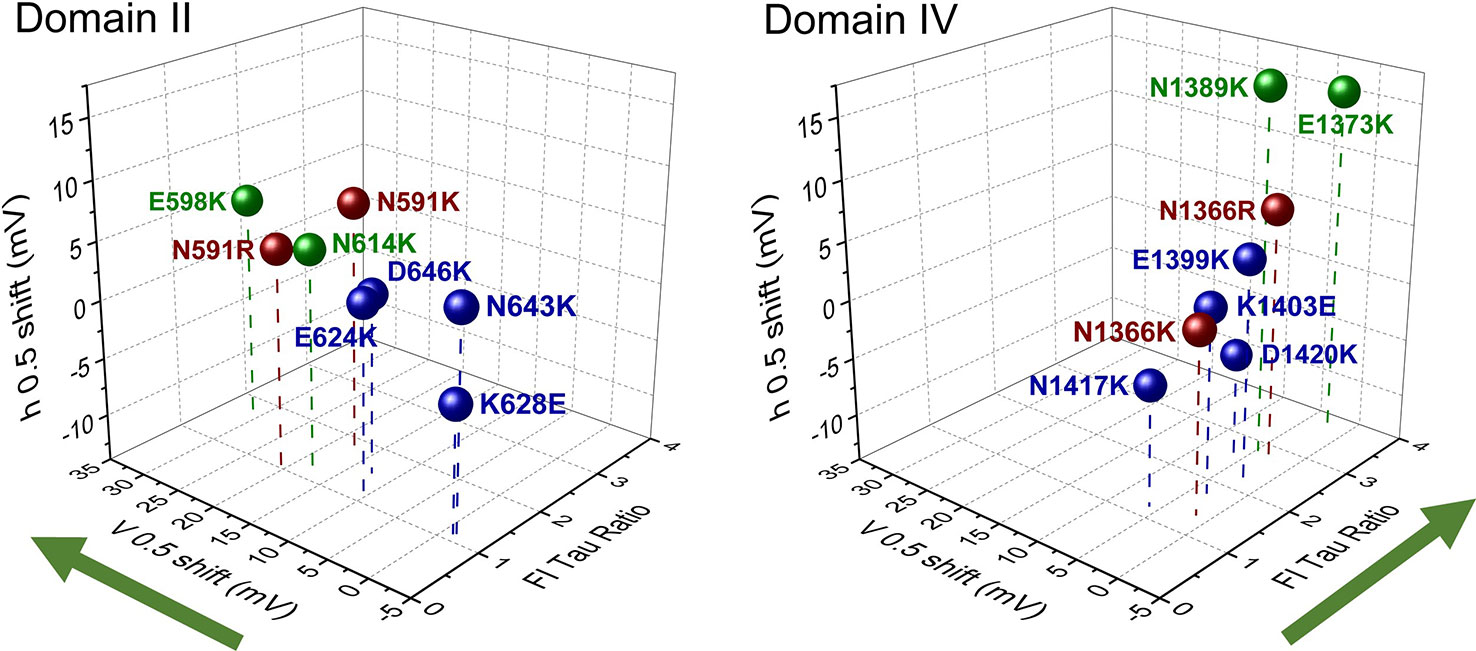
Figure 3 Scanning mutagenesis of putative countercharges in domains II and IV of the skeletal muscle sodium channel NaV1.4. Charge-reversing mutations in domain II decrease activation probability (indicated by shift in V0.5), while those in domain IV slow the entry of channels into a fast-inactivated state (indicated by the ratio of time constant (tau) of mutation with respect to that of wild type, at 20 mV). These effects are consistent with the hypothesis that countercharges facilitate certain domain-specific functions highlighted by green arrows (DII activation, DIV fast inactivation). Adapted from Groome and Winston (2013). Colors indicate countercharge loci as extracellular negatively charged (ENC) (green, red), or intracellular negatively charged (INC) (blue).
Domain specific roles for voltage sensors in calcium channels have also been identified (Pantazis et al., 2014; Tuluc et al., 2016a). One source of functional variability across domains in CaV channels may be related to an outer countercharge observed in DIIS3 and DIVS3 segments (Coste de Bagneaux et al., 2018). In CaV1.1, aspartate D1196, identified as D4 in the extracellular region of DIVS3, interacts with DIVS4 arginines R1 and R2, albeit differentially in distinct splice variants (Tuluc et al., 2016b). Specifically, the alternative splice variant CaV1.1e excluding 19 amino acids in the DIVS3-S4 linker enhances activation, suggesting that in CaV1.1a, the DIVS3-S4 linker disrupts D4 interaction with R1 and R2. Differential effects of DIVS3 D4 mutations in CaV1.2 and CaV1.3 are not solely dependent on the intact linker (Coste de Bagneaux et al., 2018) but also support a role for this countercharge in channel gating, and provide insight into domain-specific voltage-sensitivity in these channels.
VSD S1–S3 negative charges have roles in gating and selectivity in the voltage-gated proton channel HV1. Countercharge/S4 residue interactions during HV1 gating are suggested by mutant cycle analysis of the effects of single and double cysteine mutations (Chamberlain et al., 2014), and molecular dynamics simulations have defined salt-bridge interactions between countercharge and S4 residues during HV1 activation (Gianti et al., 2016). Interestingly, mutation of negative charge D112 in S1 results in channels that are non-functional (D112V), that display a shift in permeability to anions (i.e., D112H), or that maintain proton selectivity (D112E; Musset et al., 2012). The proton selectivity function of D112 might be based on its salt-bridge formation with S4 residue R211 (Chamberlain et al., 2015; Dudev et al., 2015). D112 and other countercharges in Hv1 S1–S3 appear to facilitate proton conduction through a mechanism in which proton binding sites are comprised of three pairs of acidic countercharges, with conformational change in the VSD promoting exchange of protons between sites (van Keulen et al., 2017). Proton-mediated disruption of the cation-pi interaction of S2 aromatic residue F150 with R211 has been proposed as a key determinant in the closed to open transition of the channel, facilitating the S2 E153 to S1 D112 proton exchange across the VSD permeation pathway.
Following the successful crystallization of the KcSA potassium channel (Doyle et al., 1998), efforts to determine the atomic structure of VGICs intensified, and led to alternative hypotheses about the mechanism through which S4 voltage-sensing segments promote channel activation. The first full length structure of a voltage-gated potassium channel (KVAP) was solved with crystallography at a resolution of 3.2 Å (Jiang et al., 2003). In addition, an isolated VSD was solved at a resolution of 1.9 Å and whose structure revealed a salt-bridge between the S4 residue R133 (R5) with the S2 ENC countercharge D62. Importantly, the authors hypothesized that the second helical region in S3, along with the S4 helix, formed a voltage-sensor paddle that moved through the lipid membrane in response to membrane depolarization. Crystallization of the eukaryotic potassium channel KV1.2 (Long et al., 2005a; Long et al., 2005b) and the chimera KV1.2/2.1 (Long et al., 2007) partially reconciled the paddle hypothesis of charge displacement with functional data that supported vertical displacement of the S4 helix [reviewed by (Horn, 2002; Gandhi and Isacoff, 2002; Elinder et al., 2007)]. As described later, structural data for KV VSDs were utilized in molecular dynamics simulations (Delemotte et al., 2011; Jensen et al., 2012) that supported a mechanism of voltage-gating dependent on multiple salt-bridge interactions facilitating intermediate states of channel activation.
The crystal structure of the prokaryotic sodium channel NaVAb (Payandeh et al., 2011) revealed the proximity of ENC and INC countercharges with specific S4 residues. Importantly, Kv1.2/2.1 and NaVAb structures showed the S4 segment in nearly identical positions (Long et al., 2007; Payandeh et al., 2011), confirming the vertical position of the S4 segment in the activated state. Countercharge to S4 arginine residue interactions have been revealed with crystallographic data from NaVAb, NaVRh, and NaVMs (Payandeh et al., 2011; Payandeh et al., 2012; Zhang et al., 2012; Sula et al., 2017; Wisedchaisri et al., 2019). It has also been hypothesized that negatively charged residues tune the hydrophilicity of the inner and outer vestibules of the VSD (Palovcak et al., 2014), while the central, hydrophobic region separates these vestibules and focuses the electric field (Starace and Bezanilla, 2004; Ahern and Horn, 2005; Chanda and Bezanilla, 2008; Lacroix et al., 2014).
Recently, improvements in cryo-electron microscopy (cryo-EM) have allowed for rapid advances in structure determination of large membrane proteins, including eukaryotic sodium and calcium channels. Shen et al. (2017) used cryo-EM to determine the structure of NaVPaS, the cockroach isoform of NaV1.4, at 3.8 Å. Shortly thereafter, the structure of EeNaV1.4, the electric eel construct of NaV1.4, was solved to 4.0 Å and in complex with the accessory β1 subunit (Yan et al., 2017). Many of the countercharge/S4 residue interactions described in the structures of prokaryotic sodium channels are reiterated in these eukaryotic structures, and in the cryo-EM structure of CaV1.1 (Wu et al., 2016). When the structure of the human NaV1.4 channel/β1 subunit was solved to 3.2 Å (Pan et al., 2018), an expanded set of interactions between S1 and S3 countercharges and S4 residues was presented, and suggested a role for polar residues in stabilizing VSDs across domains. Together, these cryo-EM structures have provided a structural framework to complement functional investigations of domain-specific functions of eukaryotic VSDs (Chanda and Bezanilla, 2002; Goldschen-Ohm et al., 2013; Tuluc et al., 2016a) and of domain-specific effects of S1–S3 countercharge mutations (Groome and Winston, 2013; Pless et al., 2014).
Molecular dynamics simulations have been used extensively in computational approaches to investigate ion channel function, by providing a view of time-dependent interactions within the VSD in response to applied membrane potential on the system [for reviews see (Delemotte et al., 2009; Cournia et al., 2015; Howard et al., 2018)]. These mathematical simulations solve the equations for motion in three dimensions for atoms with a defined mass and charge. As parameters such as membrane potential and amino acid sequence can be easily manipulated, these simulations allow researchers to test specific hypotheses about countercharge interactions with S4 residues, as the S4 segment vertically transverses the electric field. However, these simulations are computationally limited to short timescales, making it difficult to simulate an entire conformational transition.
Molecular dynamics simulations of VSDs conducted following the publication of voltage-gated potassium channel structures (Jiang et al., 2003; Long et al., 2005a; Long et al., 2005b; Long et al., 2007) probed the activated state of the voltage sensor. Several research groups were interested in hydration of the intracellular and extracellular vestibules, as well an interaction between the voltage sensor and surrounding lipid environment. Treptow and Tarek (2006) found that the outer S4 arginine residues are accessible to water in the activated state, and Jogini and Roux (2007) proposed that S4 arginine residues interact with both negatively charged lipid phosphate groups and S2 countercharges. While their continuum electrostatic calculations showed that the membrane potential varies greatly across the membrane, the greatest membrane potential was focused on the center of the voltage sensor, a hypothesis that has been supported functionally (Starace and Bezanilla, 2004; Ahern and Horn, 2005). Simulations of S4 dynamics in response to an applied membrane potential suggested that the S4 segment transverses the membrane in a screw-like motion (Nishizawa and Nishizawa, 2008), with salt-bridges (Treptow et al., 2009) and hydrogen bonding networks (Bjelkmar et al., 2009) stabilizing intermediate states. These simulations supported a model of vertical translocation of the S4 helix through the membrane, and groups began focusing on defining the intermediate states of the voltage sensor and interactions that occurred within each state.
Due to computational limitations, simulating each intermediate state of the VSD required biasing simulations based on initial starting structure (Yarov-Yarovoy et al., 2006; Wood et al., 2012), a directed path of S4 helix motion through the membrane (Delemotte et al., 2011; Schwaiger et al., 2011) or enhanced sampling techniques (Delemotte et al., 2015). Resting state models have been built by multiple groups. Yarov-Yarovoy et al. (2006); Pathak et al. (2007), and Henrion et al. (2012) used a de novo and homology modeling approach, Delemotte et al. (2011) used a combination of harmonic restraints and steered molecular dynamics, Vargas et al. (2011) computed an average structure over multiple simulations, and Jensen et al. (2012) ran a long time-scale simulation to reach a resting state structure. Although the methodologies to produce them were unique, these models showed a high degree of similarity, with the S4 helix rotated and translated inwardly, and R1 located between the S2 ENC and INC countercharges, and near the conserved S2 aromatic residue of the GCTC. A consensus model of the resting state was then proposed (Vargas et al., 2012). While simulations have suggested that the S4 segment adopts a 310 helical secondary structure to favor the resting state and align S4 arginine side chains with those of negative countercharges (Villalba-Galea et al., 2008; Khalili-Araghi et al., 2010; Schow et al., 2010; Schwaiger et al., 2011), the functional contribution of 310 helical structure to VGIC gating remains to be clearly elucidated (Kubota et al., 2014; Bassetto et al., 2019).
Computational approaches to VSD activation have confirmed and refined the description of intermediate states. Nishizawa and Nishizawa (2008) initially hypothesized that the free energy landscape between intermediate states would be “ragged,” indicating multiple pathways of transition between states due to the large number of electrostatic interactions taking place. This hypothesis was confirmed by Delemotte et al. (2015), who characterized the transition between the resting and a partially activated state using well-tempered metadynamics. They showed that R1 must transit across the hydrophobic septum prior to the transition of R3 engaged with intracellular lipid phosphate groups, to R3 engaged with the INC S2 countercharge. Although this single most favorable pathway between a resting and intermediate state was found, their results did show a rough free energy landscape, as previously hypothesized. However, when the free energy was plotted as a function of gating charge, a smooth free energy landscape was produced, with two well-defined energy wells corresponding to the resting and intermediate states. It is important to note that the bulk of these simulations were completed using the Kv1.2 channel as a model. Thus, although many of these findings can be translated to other channel types, it is yet unknown how the differences between VSDs affect dynamics. For example, the cryo-EM structure of hNaV1.4 (Pan et al., 2018) suggests unique electrostatic interactions within each VSD, and differences in the extent of voltage sensor activation across domains.
To further explore the differences in interactions between domains of hNaV1.4, we have conducted a network analysis from short time-scale molecular dynamics simulations (Bayless-Edwards, 2019). Figure 4 shows each S4 segment translocated intracellularly from an initial activated structure (Pan et al., 2018) to produce both an intermediate state and a potential resting state structure, using methods similar to those of Wood et al. (2012). gRINN (get Residue Interaction eNergies and Networks) was used to complete the network analysis (Sercinoglu and Ozbek, 2018). Our results support the presence of unique electrostatic interactions in each domain over multiple conformations. The strongest interactions are between acidic countercharges with S4 residues, consistent with other simulations (Gosselin-Badaroudine et al., 2012) and functional experiments in sodium channels (Groome and Winston, 2013; Pless et al., 2014).
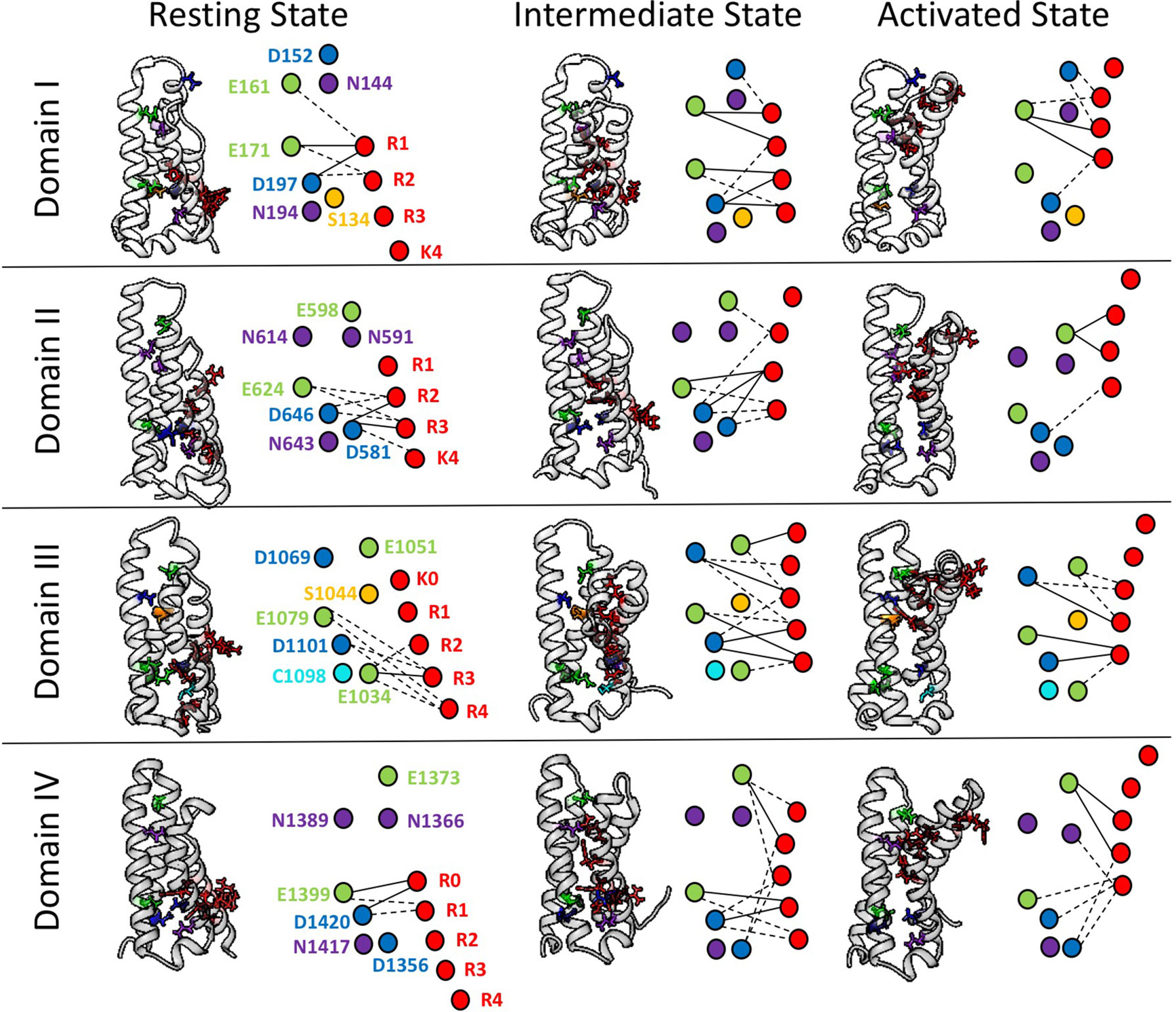
Figure 4 Electrostatic interactions between countercharges (aspartate, blue; glutamate, green; asparagine, purple; serine, yellow, cysteine, cyan) and each of four to five positively charged residues in S4 segments (arginine or lysine, red) in domains I–IV of the skeletal muscle sodium channel NaV1.4. Nodes indicate charged and polar residues within the voltage sensor, while edge width indicates strength of interaction. The profile of interactions is given for a presumptive resting state, intermediate closed state, and activated state. Reproduced with permission from Idaho State University Libraries (Bayless-Edwards, 2019).
Solvation of the VSD is an essential component in models of voltage-gating in which positively charged S4 arginine residues traverse the membrane through an aqueous environment (Freites and Tobias, 2015). Water permeation within the VSD is often measured by calculating either the solvent-accessible surface or the simulated average water occupancy along the z-axis of the membrane. As is expected by the hourglass shape of the VSD comprising inner and outer vestibules, water occupancy is highest at the edges of the membrane and decreases to the hydrophobic septum (Freites et al., 2006; Schow et al., 2010; Gosselin-Badaroudine et al., 2012). Chakrapani et al. (2010) used site-directed spin-labeling to quantify the hydrophilicity of outer and inner vestibules of the VSD and incorporated these environmental restraints into molecular dynamics simulations of NaChBac. Their results showed that S4 arginines R1 and R2 move through a hydrophilic environment toward their interaction with phospholipid heads of the bilayer as the S2 ENC (D60)/S4 R3 salt-bridge interaction is formed with activation of the channel. The significance of hydration in voltage-gating is further emphasized with the finding that S4 aspartate substitutions in the Shaker channel are remarkably tolerated with respect to gating function (Diaz-Franulic et al., 2018), and by the corollary observation that mutation of S1–S3 residues comprising the hydrophobic plug have dramatic effects on gating charge movement (Lacroix et al., 2014, and reviewed by Bezanilla, 2018).
In diseases such as hypokalemic periodic paralysis and dilated cardiomyopathy, mutation of S4 arginines can lead to permeation of protons or cations between the inner and outer vestibules of the VSD, a finding supported using both molecular dynamics simulations (Jensen et al., 2012; Monteleone et al., 2017) and cryo-EM (Jiang et al., 2018). This omega current is a characteristic effect of mutations in these diseases, and is a significant factor in pathogenesis [reviewed by Groome et al., 2018)]. Interestingly, the width of the hydrophobic region varies between sodium channel domains (Gosselin-Badaroudine et al., 2012), and the large hydrophobic septum in DIV may explain the lack of observed omega current associated with DIVS4 channelopathy mutations (Francis et al., 2011), even with periodic paralysis as a contributing element to the phenotype (Poulin et al., 2018).
It has been hypothesized that mutations near the hydrophobic septum disrupt not only steric inhibition of water molecules and ions (Jiang et al., 2018), but also hydrogen bonding (Monteleone et al., 2017; Poulin et al., 2018) and electrostatic interactions (Bayless-Edwards et al., 2018). This hypothesis is consistent with the premise that charged and polar residues play an important role in the hydration of ion channel vestibules in VSDs (Pless et al., 2011; Palovcak et al., 2014). An interesting set of questions remain as to the nature and extent of contributions of polar, countercharge residues to specific aspects of voltage-gating. For example, mutations of VSD polar residues do elicit substantial effects on channel activation, fast inactivation, and slow inactivation (Gamal El-Din et al., 2013; Groome and Winston, 2013; Pless et al., 2014; Groome et al., 2019). The domain-specific VSD conformations underlying these channel functions are an important target for investigation (Yarov-Yarovoy and DeCaen, 2019) and a determination of the impact of hydration provided by polar countercharges in voltage-gating may provide some unique insight.
Inherited mutations in the genes for VGICs comprise a set of channelopathies including epilepsy and migraine syndromes, pain, cardiac arrhythmia syndromes, myotonia, periodic paralysis, and congenital myasthenia (for reviews see Catterall, 2012; Bezzina et al., 2015; Cannon, 2015). These mutations may result in a truncated protein, disrupt trafficking or assembly with accessory subunits, or produce gating defects. Channelopathy mutations have been studied in heterologous expression to characterize their biophysical impact on membrane excitability, and in transgenics to test their impact on physiology and behavior [reviewed by (Cannon, 2018; Catterall, 2018)].
A number of variants predicting VSD countercharge mutations associated with disease phenotypes have been identified, with missense mutations for these channelopathies given in Table 3. Noteworthy on this list are the prevalence of SCN1A mutations identified in genetic screening of patients with intractable epilepsy syndromes. These countercharge mutations have yet to be functionally characterized in heterologous expression (Brunklaus et al., 2020). However, several countercharge mutations identified in cardiac arrhythmia or skeletal muscle syndromes have been characterized, and are described here.

Table 3 Voltage-gated ion channel (VGIC) countercharge S1–S3 mutations identified in human disease phenotypes.
Channelopathy mutations identified in patients with Brugada syndrome and/or overlapping phenotypes such as dilated cardiomyopathy confer loss of function in the cardiac sodium channel hNaV1.5 (SCN5A; for reviews see Garcia-Elias and Benito, 2018; Asatryan, 2019). A few of these mutations are at countercharge loci, and for which biophysical characterization has been reported. Loss of function may be explained to some extent with observed reduction in current density, as in DIS2 ENC mutation E161Q (Meregalli et al., 2009). In contrast, N1541D (DIVS1 ENC) elicits specific gating defects including enhanced probability of fast inactivation, and slowed recovery from open-state fast inactivation (Dharmawan et al., 2019). Interestingly, each of these gating defects are reiterated in the homologous NaV1.4 mutation N1366D (Groome and Winston, 2013).
The SCN5A DIIIS3 INC mutation D1275N produces mild, loss of function gating defects including impaired activation and enhanced fast inactivation when studied in heterologous expression (Groenewegen et al., 2003; Hayano et al., 2017). However, D1275N promotes significant cardiac defects such as slowed conduction, heart block, atrial fibrillation, ventricular tachycardia, and dilated cardiomyopathy when inserted in transgenic mice (Watanabe et al., 2011) or zebrafish (Huttner et al., 2013). Two SCN5A channelopathy mutations have been characterized at D1595 (DIVS3 INC). First, D1595N has been linked to atrioventricular block (Wang et al., 2002). The mutation reduces current density and enhances fast and slow inactivation consistent with the cardiac muscle phenotype as shown in action potential modeling. Histidine substitution at this locus (D1595H) is identified in patients with arrhythmic dilated cardiomyopathy (Olson et al., 2005), and enhances fast inactivation (Nguyen et al., 2008).
In the skeletal muscle sodium channel hNaV1.4 (SCN4A), the mutation D1069N (DIIIS2 ENC) has been identified in family members presenting with congenital myopathy (Zaharieva et al., 2016). The mutation produces a right shift in the midpoint of the activation curve, consistent with loss of function (hypoexcitability). In contrast, N1366S (DIVS1 ENC), identified in a family diagnosed with paramyotonia congenita, produces gain of function effects including enhanced activation, slowed entry into the fast-inactivated state, and accelerated recovery (Ke et al., 2017). These effects are consistent with a hyperexcitable skeletal muscle fiber phenotype that characterizes the disease. Thus, functional characterizations of countercharge mutations have extended the genotype to phenotype correlation for loss or gain of function in SCN5A and SCN4A channelopathies.
Common sequence motifs in VSDs include the positively charged S4 region, and conserved aromatic, acidic, or polar amino acid residues in S1–S3 segments. Investigations of the mechanism of voltage-gating in VGICs have supported the hypothesis that VSD negatively charged residues act as countercharges to facilitate S4 translocation in an energetically unfavorable membrane environment. Functional experiments have revealed pairwise interactions between countercharge/S4 residues that play an important role in protein folding, the resting state of the channel, and in outward movement of the S4 segment in the steps leading to channel opening. Determinations of protein structure at atomic resolution supports such countercharge/S4 residue interactions, and have facilitated in-depth analyses of their choreography, using molecular dynamics simulations. These simulations have also been instrumental in present efforts to elucidate the mechanisms by which countercharge residues regulate VSD hydration and to explore the role of hydration in ion channel function. An expanding genetic database has revealed that countercharge mutations comprise a portion of published variants of ion channel genes in patients presenting with neural, cardiac, or skeletal muscle disorders. Genotype to phenotype correlation for countercharge mutations is an important aspect of continued research of the biophysical mechanisms of channelopathies.
JG and LB-E contributed equally to the writing of this review article.
This work was supported by the NIH under 1R15NS093579-01A1 to JG and Idaho INBRE (through an Institutional Development Award from the National Institute of General Medical Sciences) under Grant P20GM103408.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The research presented in Figure 4 (Bayless-Edwards, 2019) made use of the Idaho National Laboratory High Performance Computing Center, which is supported by the Office of Nuclear Energy of the U.S. DOE and the Nuclear Science User Facilities under Contract No. DE-AC07-05ID14517.
Ahern, C. A., Horn, R. (2005). Focused electric field across the voltage sensor of potassium channels. Neuron 48 (1), 25–29. doi: 10.1016/j.neuron.22056.08.020
Armstrong, C. M., Bezanilla, F. (1973). Currents related to movement of the gating particles of the sodium channels. Nature 242 (5398), 459–461. doi: 10.1038/242459a0
Armstrong, C. M., Bezanilla, F. (1977). Inactivation of the sodium channel. II. Gating current experiments. J. Gen. Physiol. 70 (5), 567–590. doi: 10.1085/jgp.70.5.567
Asatryan, B. (2019). Cardiac sodium channel dysfunction and dilated cardiomyopathy: a contemporary reappraisal of pathophysiological concepts. J. Clin. Med. 8 (7), E1029. doi: 10.3390/jcm8071029
Azmanov, D. N., Zhelyaskova, S., Dimova, P. S., Radionova, M., Bojinova, V., Florez, L., et al. (2010). Mosaicism of a missense SCN1A mutation and Dravet syndrome in a Roma/Gypsy family. Epileptic Disord. 12 (2), 117–124. doi: 10.1684/epd.2010.0311
Bassetto, C. A. Z., Jr., Carvalho-de-Souza, J. L., Bezanilla, F. (2019). Metal bridge in S4 segment supports helix transition in Shaker channel. Biophys. J. 117, 1–12. doi: 10.1016/j.bpj.2019.08.035.
Bayless-Edwards, L., Winston, V., Lehmann-Horn, F., Arinze, P., Groome, J. R., Jurkat-Rott, K. (2018). NaV1.4 DI-S4 periodic paralysis mutation R222W enhances inactivation and promotes leak current to attenuate action potentials and depolarize muscle fibers. Sci. Rep. 8, 10372. doi: 10.1038/s41598-018-28594-5
Bayless-Edwards, L. (2019). From molecule to cell: using electrophysiology and multiscale modelling to understand how NaV1.4 controls electrical excitability. [Master’s thesis]. (Pocatello, ID: Idaho State University).
Bezanilla, F., Perozo, E., Stefani, E. (1994). Gating of Shaker K+ channels: II. The components of gating currents and a model of channel inactivation. Biophys. J. 66 (4), 1011–1021. doi: 10.1016/S0006-3495(94)80882-3
Bezanilla, F. (2000). The voltage sensor in voltage-dependent ion channels. Physiol. Rev. 80 (2), 555–592. doi: 10.1151/physrev.2000.80.2.555
Bezanilla, F. (2018). Gating currents. J. Gen. Physiol. 150 (7), 911–932. doi: 10.1085/jgp.201812157
Bezzina, C. R., Lahrouchi, N., Priori, S. G. (2015). Genetics of sudden cardiac death. Circ. Res. 116 (12), 1919–1936. doi: 10.1161/CIRCRESAHA.116.304030
Bjelkmar, P., Niemela, P. S., Vattulainen, I., Lindahl, E. (2009). Conformational changes and slow dynamics through microsecond polarized atomistic molecular simulation of an integral Kv1.2 ion channel. PLoS Computat. Biol. 5 (2), e1000289. doi: 10.1371/journal.pcbi.1000289
Bosshard, H. R., Marti, D. N., Jelesarov, I. (2004). Protein stabilization of salt bridges: concepts, experimental approaches and clarification of some misunderstandings. J. Mol. Recognit. 17 (1), 1–16. doi: 10.1002/jmr.657
Brunklaus, A., Schorge, S., Smith, A. D., Ghanty, I., Stewart, K., Gardiner, S., et al. (2020). SCN1A variants from bench to bedside – improved prediction from functional characterization. Hum. Mutat. 41, 363–374. doi: 10.1002/humu.23943. in press.
Cannon, S. C. (2015). Channelopathies of skeletal muscle excitability. Compr. Physiol. 5 (2), 761–790. doi: 10.1002/cphy.c140062
Cannon, S. C. (2018). “Sodium channelopathies of skeletal muscle,” in Voltage-gated sodium channels: structure, function and channelopathies, vol. 246 . Ed. Chahine, M. Handbook of Experimental Pharmacology (Switzerland: Springer International Publishing AG), 309–330. doi: 10.10017/164_2017_52
Catterall, W. A. (2012). Voltage-gated sodium channels at 60: structure, function and pathophysiology. J. Physiol. 590 (11), 2577–2589. doi: 10.1113/jphysiol.2011.224204
Catterall, W. A. (2018). Dravet syndrome: a sodium channel interneuronopathy. Curr. Opin. Physiol. 2, 42–50. doi: 10.1016/j.cophys.2017.12.007
Chakrapani, S., Sompornpisut, P., Intharathep, P., Roux, B., Perozo, E. (2010). The activated state of a sodium channel voltage sensor in a membrane environment. Proc. Natl. Acad. Sci. U. S. A. 107 (12), 5435–5440. doi: 10.1073/pnas.0914109107
Chamberlain, A., Qiu, F., Rebolledo, S., Wang, Y., Noskov, S. Y., Larsson, H. P. (2014). Hydrophobic plug functions as a gate in voltage-gated proton channels. Proc. Natl. Acad. Sci. U. S. A. 111 (2), E273–E282. doi: 10.1073/pnas.1318018111
Chamberlain, A., Qiu, F., Wang, Y., Noskov, S. Y., Larsson, H. P. (2015). Mapping the gating and permeation pathways in the voltage-gated proton channel Hv1. J. Mol. Biol. 427 (1), 131–145. doi: 10.1016/j.jmb.2014.11.018
Chanda, B., Bezanilla, F. (2002). Tracking voltage-dependent conformational changes in skeletal muscle sodium channel during activation. J. Gen. Physiol. 120 (5), 629–645. doi: 10.1085/jgp.20028679
Chanda, B., Bezanilla, F. (2008). A common pathway for charge transport through voltage-sensing domains. Neuron 57 (3), 345–351. doi: 10.1016/j.neuron.2008.01.015
Coste de Bagneaux, P., Campiglio, M., Benedetti, B., Tuluc, P., Flucher, B. E. (2018). Role of putative voltage-sensor countercharge D4 in regulating gating properties of Cav1.2 and Cav1.3 calcium channels. Channels 12 (1), 249–261. doi: 10.1080/19336950.2018.1482183
Cournia, Z., Allen, T. W., Andricioaei, I., Antonny, B., Baum, D., Brannigan, G., et al. (2015). Membrane protein structure, function and dynamics: a perspective from experiments and theory. J. Mem. Biol. 248 (4), 611–640. doi: 10.1007/s00232-015-9802-0
DeCaen, P. G., Yarov-Yarovoy, V., Zhao, Y., Scheuer, T., Catterall, W. A. (2008). Disulfide-locking of a sodium channel voltage sensor reveals ion pair formation during activation. Proc. Natl. Acad. Sci. U. S. A. 105 (39), 15142–15147. doi: 10.1073/pnas.0806486105
DeCaen, P. G., Yarov-Yarovoy, V., Sharp, E. M., Scheuer, T., Catterall, W. A. (2009). Sequential formation of ion pairs during activation of a sodium channel voltage sensor. Proc. Natl. Acad. Sci. U. S. A. 106 (52), 22498–22503. doi: 10.1073/pnas.0912307106
DeCaen, P. G., Yarov-Yarovoy, V., Scheuer, T., Catterall, W. A. (2011). Gating charge interactions with the S1 segment during activation of a Na+ channel voltage sensor. Proc. Natl. Acad. Sci. U. S. A. 108 (46), 18825–18830. doi: 10.1073/pnas.1116449108
Delemotte, L., Dehez, F., Treptow, W., Tarek, M. (2009). Modeling membranes under a transmembrane potential. J. Phys. Chem. B. 112 (18), 5547–5550. doi: 10.1021/jp710846y
Delemotte, L., Tarek, M., Klein, M. L., Amaral, C., Treptow, W. (2011). Intermediate states of the Kv1.2 voltage sensor from atomistic molecular dynamics simulations. Proc. Natl. Acad. Sci. U. S. A. 108 (15), 6019–6114. doi: 10.1073/pnas.1102724108
Delemotte, L., Kasimova, M. A., Klein, M. L., Tarek, M., Carnevale, V. (2015). Free-energy landscape of ion channel voltage-sensor domain activation. Proc. Natl. Acad. Sci. U. S. A. 112 (1), 124–129. doi: 10.1073/pnas.1416959112
Depienne, C., Trouillard, O., Saint-Martin, C., Gourfinkel-An, I., Bouteiller, D., Carpentier, W., et al. (2009). Spectrum of SCN1A gene mutations associated with Dravet syndrome: analysis of 333 patients. J. Med. Genet. 46 (3), 183–191. doi: 10.1136/jmg.2008.062323
Depienne, C., Trouillard, O., Gourfinkel-An, I., Saint-Martin, C., Bouteiller, D., Barthez-Carpentier, M. A., et al. (2010). Mechanisms for variable expressivity of inherited SCN1A mutations causing Dravet syndrome. J. Med. Genet. 47 (6), 404–410. doi: 10.1136/jmg.2009.074328
Dharmawan, T., Nakajima, T., Iizuka, T., Tamura, S., Matsui, H., Kaneko, Y., et al. (2019). Enhanced closed-state inactivation of mutant cardiac sodium channels (SCN5A N1541D and R1632C) through different mechanisms. J. Mol. Cell. Cardiol. 130, 88–95. doi: 10.1016/j.yjmcc.2019.02.023
Diaz-Franulic, I., Gonzalez-Perez, V., Moldenhauer, H., Navarro-Quezada, N., Naranjo, D. (2018). Gating-induced large aqueous volumetric remodeling and aspartate tolerance in the voltage sensor domain of Shaker K+ channels. Proc. Natl. Acad. Sci. U. S. A. 115 (32), 8203–8208. doi: 10.1073/pnas.1806578115
Dolphin, A. C. (2018). Voltage-gated calcium channels: their discovery, function and importance as drug targets. Brain Neurosci. Adv. 2, 1–8. doi: 10.1177/2398212818794805
Dou, Y., Macdonald, L. C., Wu, Y., Fedida, D. (2017). The fast component of hERG gating charge: an interaction between D411 in the S1 and S4 residues. Biophys. J. 113 (9), 1979–1991. doi: 10.1016/j.bpj.2017.09.004
Doyle, D. A., Morais Cabral, J., Pfuetzner, R. A., Kuo, A., Gulbis, J. M., Cohen, S. L., et al. (1998). The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science 280 (5360), 69–77. doi: 10.1126/science.280.5360.69
Dudev, T., Musset, B., Morgan, D., Cherny, V. V., Smith, S. M., Mazmanian, K., et al. (2015). Selectivity mechanism of the voltage-gated proton channel, HV1. Sci. Rep. 5, 10320. doi: 10.1038/srpe10320
Elinder, F., Nilsson, J., Arhem, P. (2007). On the opening of voltage-gated ion channels. Physiol. Behav. 92 (1-2), 1–7. doi: 10.1016/j.physbeh.2007.05.058
Fedida, D., Hesketh, J. C. (2001). Gating of voltage-gated potassium channels. Progr. Biophys. Mol. Biol. 75 (3), 165–199. doi: 10.1016/S0079-6107(01)00006-2
Francis, D. G., Rybalchenko, V., Struyk, A., Cannon, S. C. (2011). Leaky sodium channels from voltage sensor mutations in periodic paralysis, but not paramyotonia. Neurol 76 (19), 1635–1641. doi: 10.1212/WNL.0b013e318219fb57
Freites, J. A., Tobias, D. J. (2015). Voltage-sensing in membranes: from macroscopic currents to molecular motions. J. Membr. Biol. 248 (3), 419–430. doi: 10.1007/s00232-015-0805-x
Freites, J. A., Tobias, D. J., White, S. H. (2006). A voltage-sensor water pore. Biophys. J. 91 (11), L90–L92. doi: 10.1529/biophysj.106.096065
Fujiwara, T., Sugawara, T., Mazaki-Miyazaki, E., Takahashi, Y., Fukushima, K., Watanabe, M., et al. (2003). Mutations of sodium channel a subunit type I (SCN1A) in intractable childhood epilepsies with frequent generalized tonic-clonic seizures. Brain 126, 531–546. doi: 10.1093/brain/awg053
Gamal El-Din, T. M., Martinez, G. Q., Payandeh, J., Scheuer, T., Catterall, W. A. (2013). A gating charge interaction required for late slow inactivation of the bacterial sodium channel NaVAb. J. Gen. Physiol. 142 (3), 181–190. doi: 10.1085/jgp.201311012
Gandhi, C. S., Isacoff, E. Y. (2002). Molecular models of voltage sensing. J. Gen. Physiol. 120 (4), 455. doi: 10.1085/jgp.20028678
Ganetzky, B. (2000). Genetic analysis of ion channel dysfunction in Drosophila. Kidney Int. 57 (3), 766–771. doi: 10.1016/j.1523-1755.2000.00913.x
Garcia-Elias, A., Benito, B. (2018). Ion channel disorders and sudden cardiac death. Int. J. Mol. Sci. 19 (3), E692. doi: 10.3390/ijms19030692
Gianti, E., Delemotte, L., Klein, M., Carvnevale, V. (2016). On the role of water density fluctuations in the inhibition of a proton channel. Proc. Natl. Acad. Sci. U. S. A. 113 (52), E8359–E8368. doi: 10.1073/pna.1609964114
Glaudemans, B., van der Wijst, J., Scola, R. H., Lorenzoni, P. J., Heister, A., van der Kemp, A. W., et al. (2009). A missense mutation in the Kv1.1 voltage-gated potassium channel-encoding gene KCNA1 is linked to human autosomal dominant hypomagnesemia. J. Clin. Invest. 119 (4), 936–942. doi: 10.1172/JCI36948
Goldschen-Ohm, M. P., Capes, D. L., Oelstrom, K. M., Chanda, B. (2013). Multiple pore conformations driven by asynchronous movements of voltage sensors in a eukaryotic sodium channel. Nat. Commun. 4, 1350. doi: 10.1038/ncomms2356
Gosselin-Badaroudine, P., Delemotte, L., Moreau, A., Klein, M. L., Chahine, M. (2012). Gating pore currents and the resting state of NaV1.4 voltage sensor domains. Proc. Natl. Acad. Sci. U. S. A. 109 (47), 19250–19255. doi: 10.1073/pnas.1217990109
Greenblatt, R. E., Blatt, Y., Montal, M. (1985). The structure of the voltage-sensitive sodium channel: inferences from computer-aided analysis of the Electrophorus electricus channel primary structure. FEBS 193 (2), 125–134. doi: 10.1016/0014-5793(85)80136-8
Groenewegen, W. A., Firouzi, M., Bezzina, C. R., Vliex, S., van Langen, I. M., Sandkuijl, L., et al. (2003). A cardiac sodium channel mutation cosegregates with a rare connexin40 genotype in familial atrial standstill. Circ. Res. 92 (1), 14–22. doi: 10.1161/01.RES.0000050585.07097.D7
Groome, J. R., Winston, V. (2013). S1-S3 counter charges in the voltage sensor module of a mammalian sodium channel regulate fast inactivation. J. Gen. Physiol. 141 (5), 601–618. doi: 10.1085/jgp.201210935
Groome, J. R., Moreau, A., Delemotte, L. (2018). “Gating pore currents in sodium channels,” in Handbook of Experimental Pharmacology, vol. 246 . Ed. Chahine, M. Voltage-gated sodium channels: structure, function and channelopathies (Switzerland: Springer International Publishing AG), 371–399. doi: 10.1007/164_2017_54
Groome, J. R., Bayless-Edwards, L., Wheeler, A., Camp, R. (2019). Domain I countercharges limit slow inactivation in hNaV1.4 channels. Biophys. J. 116 (Supplement Issue3), 390a. doi: 10.1016/j.bpj.2018.11.2113
Groome, J. R. (2014). “The voltage sensor module in sodium channels,” in Handbook of Experimental Pharmacology, vol. 221 . Ed. Ruben, P. Voltage gated sodium channels (Berlin, Heidelberg: Springer International Publishing AG), 7–31. doi: 10.1007/978-3.642-41588-3_2
Guy, H. R., Seetharamulu, P. (1986). Molecular model of the action potential sodium channel. Proc. Natl. Acad. Sci. U. S. A. 83, 508–512. doi: 10.1073/pnas.83.2.508
Harkin, L. A., McMahon, J. M., Iona, X., Dibbens, L., Pelekanos, J. T., Zuberi, S. M., et al. (2007). The spectrum of SNC1A-related infantile epileptic encephalopathies. Brain 130 (3), 843–852. doi: 10.1093/brain/awm002
Hayano, M., Makiyama, T., Kamakura, T., Watanabe, H., Sasaki, K., Funakoshi, S., et al. (2017). Development of a patient-derived induced pluripotent stem cell model for the investigation of SCN5A-D1275N-related cardiac sodium channelopathy. Circ. J. 81 (12), 1783–1791. doi: 10.1253/circj.CJ-17-0064
Henrion, U., Renhorn, J., Borjesson, S. I., Nelson, E. M., Schwaiger, C. S., Bjelkmar, P., et al. (2012). Tracking a complete voltage-sensor cycle with metal-ion bridges. Proc. Natl. Acad. Sci. U. S. A. 109 (22), 8552–8557. doi: 10.1073/pnas.1116938109
Hodgkin, A. L., Huxley, A. F. (1952). A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol. 117, 500–544. doi: 10.1113/jphysiol.1952.sp004764
Horn, R. (2002). Coupled movements in voltage-gated ion channels. J. Gen. Physiol. 120 (4), 449–453. doi: 10.1085/jgp.20028658
Howard, R. J., Carnevale, V., Delemotte, L., Hellmich, U. A., Rothberg, B. S. (2018). Permeating disciplines: overcoming barriers between molecular simulations and classical structure-function approaches in biological transport. Biochim. Biophys. Acta Biomembr. 1860 (4), 927–942. doi: 10.1016/j.bbamem.2017.12.013
Huang, W., Liu, M., Yan, S. F., Yan, N. (2017). Structure-based assessment of disease-related mutations in human voltage-gated sodium channels. Protein Cell 8 (6), 401–438. doi: 10.1007/s13238-017-0372-z
Huttner, I. G., Trivedi, G., Jacoby, A., Mann, S. A., Vandenberg, J. I., Fatkin, D. (2013). A transgenic zebrafish model of a human cardiac sodium channel mutation exhibits bradycardia, conduction-system abnormalities and early death. J. Mol. Cell. Cardiol. 61, 123–132. doi: 10.1016/j.yjmcc.2013.06.005
Jensen, M. Ø., Jojini, V., Borhani, D. W., Leffler, A. E., Dror, R. O., Shaw, D. E. (2012). Mechanism of voltage gating in potassium channels. Science 336 (6078), 229–233. doi: 10.1126/science.1216533
Jiang, Y., Lee, A., Chen, J., Ruta, V., Cadene, M., Chait, B. T., et al. (2003). X-ray structure of a voltage-gated K+ channel. Nature 423 (6935), 33–41. doi: 10.1038/nature01580
Jiang, D., Gamal El-Din, T. M., Ing, C., Lu, P., Pomes, R., Zheng, N., et al. (2018). Structural basis for gating pore current in periodic paralysis. Nature 557 (7706), 590–594. doi: 10.1038/s41586-018-0120-4
Jogini, V., Roux, B. (2007). Dynamics of the Kv1.2 voltage-gated potassium channel in a membrane environment. Biophys. J. 93 (9), 3070–3082. doi: 10.1529/biophysj.107.112540
Jongbloed, R., Marcelis, C., Velter, C., Doevendans, P., Geraedts, J., Smeets, H. (2002). DHPLC analysis of potassium ion channel genes in congenital long QT syndrome. Hum. Mutat. 20 (5), 382–391. doi: 10.1002/humu.10131
Kapplinger, J. D., Tester, D. J., Salisbury, B. A., Carr, J. L., Harris-Kerr, C., Pollevick, G. D., et al. (2009). Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the FAMILION® long QT syndrome genetic test. Heart Rhythm. 6 (9), 1297–1303. doi: 10.1016/j.hrthm.2009.05.021
Kapplinger, J. D., Tester, D. J., Alders, M., Benito, B., Berthet, M., Brugada, J., et al. (2010). An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm. 7 (1), 33–46. doi: 10.1016/j.hrthm.2009.090.069
Ke, Q., Ye, J., Tang, S., Wang, J., Luo, B., Ji, F., et al. (2017). N1366S mutation of human skeletal muscle sodium channel causes paramyotonia congenita. J. Physiol. 595 (22), 6837–6850. doi: 10.1113/JP274877
Kearney, J. A., Wiste, A. K., Stephani, U., Trudeau, M. S., Siegel, A., RamachandranNair, R., et al. (2006). Recurrent de novo mutations of SCN1A in severe myoclonic epilepsy of infancy. Ped. Neurol. 34 (2), 116–120. doi: 10.1016/j.pediatrneurol.2005.07.009
Khalili-Araghi, F., Jogini, V., Yarov-Yarovoy, V., Tajkhorshid, E., Roux, B., Schulten, K. (2010). Calculation of the gating charge for KV1.2 voltage-activated potassium channel. Biophys. J. 98 (10), 2189–2198. doi: 10.1016/bpj.2010.02.056
Kodera, H., Kato, M., Nord, A. S., Walsh, T., Lee, M., Yamanaka, G., et al. (2013). Targeted capture and sequencing for detection of mutations causing early onset epileptic encephalopathy. Epilepsia 54 (7), 1262–1269. doi: 10.1111/epi.12203
Kuang, Q., Purhonen, P., Hebert, H. (2015). Structure of potassium channels. Cell. Mol. Life Sci. 72 (19), 3677–3693. doi: 10.1007/s00018-015-1948-5
Kubota, T., Lacroix, J. J., Bezanilla, F., Correa, A. M. (2014). Probing α-310 transitions in a voltage-sensing S4 helix. Biophys. J. 107 (5), 1117–1128. doi: 10.1016/j.bpj.2014.07.042
Lacroix, J. J., Hyde, H. C., Campos, F. V., Bezanilla, F. (2014). Moving gating charges through the gating pore in a Kv channel voltage sensor. Proc. Natl. Acad. Sci. U. S. A. 111 (19), E1950–E1959. doi: 10.1073/pnas.1406161111
Leonard, R. J., Karschin, A., Jayashree-Aiyar, S., Davidson, N., Tanouye, M. A., Thomas, L., et al. (1989). Expression of Drosophila Shaker potassium channels in mammalian cells infected with recombinant vaccina virus. Proc. Natl. Acad. Sci. U. S. A. 86 (19), 7629–7633. doi: 10.1073/pnas.86.19.7629
Lin, M.-C. A., Hsieh, J.-Y., Mock, A. F., Papazian, D. M. (2011). R1 in the Shaker S4 occupies the gating charge transfer center in the resting state. J. Gen. Physiol. 138 (2), 155–163. doi: 10.1085/jgp.201110642
Liu, J., Zhang, M., Jiang, M., Tseng, G.-N. (2003). Negative charges in the transmembrane domains of the HERG K channel are involved in the activation- and deactivation-gating processes. J. Gen. Physiol. 121 (6), 599–614. doi: 10.1085/jgp.200308788
Long, S. B., Campbell, E. B., MacKinnon, R. (2005a). Voltage sensor of KV1.2: structural basis of electromechanical coupling. Science 309 (5736), 903–908. doi: 10.1126/science.1116270
Long, S. B., Campbell, E. B., MacKinnon, R. (2005b). Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science 309 (5736), 897–903. doi: 10.1126/science.1116269
Long, S. B., Tao, X., Campbell, E. B., MacKinnon, R. (2007). Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature 450 (7168), 376–382. doi: 10.1038/nature06265
Marini, C., Mei, D., Temudo, T., Ferrari, A. R., Buti, D., Dravet, C., et al. (2007). Idiopathic epilepsies with seizures precipitated by fever and SCN1A abnormalities. Epilepsia 48 (9), 1678–1685. doi: 10.1111/j.1528-1167.2007.01122x
Matalon, D., Goldberg, E., Medne, L., Marsh, E. D. (2014). Confirming an expanded spectrum of SCN2A mutations: a case series. Epileptic Disord. 16 (1), 13–18. doi: 10.1684/epd.2014.0641
McNair, W. P., Ku, L., Taylor, M. R. G., Fain, P. R., Dao, D., Wolfel, E., et al. (2004). SCN5A mutation associated with dilated cardiomyopathy, conduction disorder, and arrhythmia. Circulation 110, 2163–2167. doi: 10.1161/01.CIR.0000144458_58660.BB
Meregalli, P. G., Tan, H. L., Probst, V., Koopmann, T. T., Tanck, M. W., Bhuiyan, Z. A., et al. (2009). Type of SCN5A-mutation determines clinical severity and degree of conduction slowing in loss-of-function sodium channelopathies. Heart Rhythm. 6 (3), 341–348. doi: 10.1016/j.hrthm.2008.11.009
Monteleone, S., Lieb, A., Pinggera, A., Negro, G., Fuchs, J. E., Hofer, F., et al. (2017). Mechanisms responsible for w-pore currents in Cav calcium channel voltage-sensing domains. Biophys. J. 113 (7), 1485–1495. doi: 10.1016/j.bpj.2017.08.010
Musset, B., Smith, S. M. E., Rajan, S., Morgan, D., Cherny, V. V., DeCoursey, T. E. (2012). Aspartate is the selectivity filter of the human voltage gated proton channel. Nature 480 (7376), 273–277. doi: 10.1038/nature10557
Nakamura, K., Kato, M., Osaka, H., Yamashita, S., Nakagawa, E., Haginoya, K., et al. (2013). Clinical spectrum of SCN2A mutations expanding to Ohtahara syndrome. Neurol 81 (11), 992–998. doi: 10.1212/WNL.0b013e3182a43e57
Napolitano, C., Priori, S. G., Schwartz, P. J., Bloise, R., Ronchetti, E., Nastoli, J., et al. (2005). Genetic testing in the long QT syndrome: development and validation of an efficient approach to genotyping in clinical practice. JAMA 294 (23), 2975–2980. doi: 10.1001/jama.294.23.2975
Nguyen, T. P., Wang, D. W., Rhodes, T. H., George, A. L., Jr. (2008). Divergent biophysical defects caused by mutant sodium channels in dilated cardiomyopathy with arrhythmia. Circ. Res. 102 (3), 364–371. doi: 10.1161/CIRCRESAHA.107.164673
Nishizawa, M., Nishizawa, K. (2008). Molecular dynamics simulations of Kv channel voltage sensor helix in a lipid membrane with applied electric field. Biophys. J. 95 (4), L1729–L1744. doi: 10.1529/biophysj.108.130658
Noda, M., Shimizu, S., Tanabe, T., Takai, T., Kayano, T., Ikeda, T., et al. (1984). Primary structure of Electrophorus electricus sodium channel deduced from cDNA sequence. Nature 312 (5990), 121–127. doi: 10.1038/312121a0
Noda, M., Ikeda, T., Suzuki, H., Takeshima, H., Takahashi, T., Kuno, M., et al. (1986). Expression of functional sodium channels from cloned DNA. Nature 322 (6082), 826–828. doi: 10.1038/322826a0
Ogiwara, I., Ito, K., Sawaishi, Y., Osaka, H., Mazaki, E., Inoue, I., et al. (2009). De novo mutations of voltage-gated sodium channel alphaII gene SCN2A in intractable epilepsies. Neurol 73 (13), 1046–1053. doi: 10.1212/WNL.0b013e3181b0cebc
Olson, T. M., Michels, W., Ballew, J. D., Reyna, S. P., Karst, M. L., Herron, K. J., et al. (2005). Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA 293 (4), 447–454. doi: 10.1001/jama.293.4.447
Paldi, T., Gurevitz, M. (2010). Coupling between residues on S4 and S1 defines the voltage-sensor resting conformation in NaChBac. Biophys. J. 99 (2), 456–463. doi: 10.1016/bpj.2010.04.053
Palovcak, E., Delemotte, L., Klein, M. L., Carnevale, V. (2014). Evolutionary imprint of activation: the design principles of VSDs. J. Gen. Physiol. 143 (2), 145–156. doi: 10.1085/jgp.201311103
Pan, X., Li, Z., Zhou, Q., Shen, H., Wu, K., Huang, X., et al. (2018). Structure of the human voltage-gated sodium channel NaV1.4 in complex with beta1. Science 362 (6412), eaau2486. doi: 10.1126/science.aau2486
Pantazis, A., Savalli, N., Sigg, D., Neely, A., Olcese, R. (2014). Functional heterogeneity of the four voltage sensors of a human L-type calcium channel. Proc. Natl. Acad. Sci. U. S. A. 111 (51), 18381–18386. doi: 10.1073/pnas.1411127112
Papazian, D. M., Schwarz, T. L., Tempel, B. L., Jan, Y. N., Jan, L. Y. (1987). Cloning of genomic and complementary DNA from Shaker, a putative potassium gene from Drosophila. Science 237 (4816), 749–753. doi: 10.1126/science.2441470
Papazian, D. M., Schwarz, T. L., Tempel, B. L., Timpe, L. C., Jan, L. Y. (1988). Ion channels in Drosophila. Ann. Rev. Physiol. 50, 379–394. doi: 10.1146/annurev.ph.50.030188.002115
Papazian, D. M., Timpe, L. C., Jan, Y. N., Jan, L. Y. (1991). Alteration of voltage-dependence of Shaker potassium channel by mutations in the S4 sequence. Nature 349 (6307), 305–310. doi: 10.1038/349305a0
Papazian, D. M., Shao, X. M., Seoh, S.-A., Mock, A. F., Huang, Y., Wainstock, D. H. (1995). Electrostatic interactions of S4 voltage sensor in Shaker K+ channel. Neuron 14 (6), 1293–1301. doi: 10.1016/0896-6273(95)90276-7
Papazian, D. M., Silverman, W. R., Lin, M. C., Tiwari-Woodruff, S. K., Tang, C. Y. (2002). Structural organization of the voltage sensor in voltage-dependent K+ channels. Noavartis Found. Symp. 245, 178–190. doi: 10.1002/0470868759.ch13
Parrini, E., Marini, C., Mei, D., Galuppi, A., Cellini, E., Pucatti, D., et al. (2017). Diagnostic targeted resequencing in 349 patients with drug-resistant pediatric epilepsies identifies causative mutations in 30 different genes. Hum. Mutat. 38, 216–225. doi: 10.1002/humu.23149
Pathak, M. M., Yarov-Yarovoy, V., Agarwal, G., Roux, B., Barth, P., Kohout, S., et al. (2007). Closing in on the resting state of the shaker K+ channel. Neuron 56 (4), 124–140. doi: 10.1016/j.neuron.2007.09.023
Payandeh, J., Scheuer, T., Zheng, N., Catterall, W. A. (2011). The crystal structure of a voltage-gated sodium channel. Nature 475 (7356), 353–358. doi: 10.1038/nature10238
Payandeh, J., Gamal El-Din, T. M., Scheuer, T., Zheng, N., Catterall, W. A. (2012). Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature 486 (7401), 135–139. doi: 10.1038/nature11077
Perozo, E., Papazian, D. M., Stefani, E., Bezanilla, F. (1992). Gating currents in Shaker K+ channels. Implications for activation and inactivation models. Biophys. J. 62 (1), 160–168. doi: 10.1016/S0006-3495(92)81802-7
Perozo, E., Santacruz-Toloza, L., Stefani, E., Bezanilla, F., Papazian, D. M. (1994). S4 mutations alter gating currents of Shaker K channels. Biophys. J. 66, 345–354. doi: 10.1016/s0006-3495-(94)80783-0
Pless, S. A., Galpin, J. D., Niciforovic, A. P., Ahern, C. A. (2011). Contributions of counter-charge in a potassium channel voltage-sensor domain. Nat. Chem. Biol. 7 (9), 617–623. doi: 10.1038/nchembio.622
Pless, S. A., Elstone, F. D., Niciforovic, A. P., Galpin, J. D., Yang, R., Kurata, H. T., et al. (2014). Asymmetric functional contributions of acidic and aromatic side chains in sodium channel voltage-sensor domains. J. Gen. Physiol. 143 (5), 645–656. doi: 10.1085/jgp.201311036
Poulin, H., Gosselin-Badaroudine, P., Vicart, S., Habbout, K., Sternberg, D., Giuliano, S., et al. (2018). Substitutions of the S4DIV R2 residue (R1451) in NaV1.4 lead to complex forms of paramyotonia congenita and periodic paralyses. Sci. Rep. 8, 2041. doi: 10.1038/s41598-018-20468-0
Ren, D., Navarro, B., Xu, H., Yue, L., Shi, Q., Clapham, D. E. (2001). A prokaryotic voltage-gated sodium channel. Science 294 (5550), 2372–2375. doi: 10.1126/science.1065635
Schow, E. V., Frietes, J. A., Gogna, K., White, S. H., Tobias, D. J. (2010). Down-state model of the voltage-sensing domain of a potassium channel. Biophys. J. 98 (112), 2857–2866. doi: 10.1016/j.bpj.2010.03.031
Schulteis, C. T., Nagaya, N., Papazian, D. M. (1998). Subunit folding and assembly steps are interspersed during Shaker potassium channel biogenesis. J. Biol. Chem. 273 (40), 26210–26217. doi: 10.1074/jbc.273.40.26210
Schwaiger, C. S., Bjelkmar, P., Hess, B., Lindahl, E. (2011). 310-helix conformation facilitates the transition of a voltage sensor S4 segment toward the down state. Biophys. J. 100 (6), 1446–1454. doi: 10.1016/j.bpj.2011.02.003
Schwaiger, C. S., Liin, S. I., Elinder, F., Lindahl, E. (2013). The conserved phenylalanine in the K+ channel voltage-sensor domain creates a barrier with unidirectional effects. Biophys. J. 104 (1), 75–84. doi: 10.1016/j.bpj.2012.11.3827
Seoh, S.-A., Sigg, D., Papazian, D. M., Bezanilla, F. (1996). Voltage-sensing residues in the S2 and S4 segments of the Shaker K+ channel. Neuron 16, 1159–1167. doi: 10.1016/s0896-6273(00)80142-7
Sercinoglu, O., Ozbek, P. (2018). gRINN: a tool for calculation of residue interaction energies and protein energy network analysis of molecular dynamics simulations. Nucleic Acids Res. 46 (W1), W554–W526. doi: 10.1093/nar/gky381
Shen, H., Zhou, Q., Pan, X., Li, Z., Wu, J., Yan, N. (2017). Structure of a eukaryotic voltage-gated sodium channel at near-atomic resolution. Science 355 (6328), eaa14326. doi: 10.1126/scienceaa14326
Silverman, W. R., Roux, B., Papazian, D. M. (2003). Structural basis of two-stage voltage dependent activation in K+ channels. Proc. Natl. Acad. Sci. U. S. A. 100 (5), 2935–2940. doi: 10.1073/pnas.0636603100
Smits, J. P., Eckardt, L., Probst, V., Bezzina, C. R., Schott, J. J., Remme, C. A., et al. (2002). Genotype-phenotype relationship in Brugada syndrome: electrocardiographic features differentiate SCN5A-related patients from non-SCN5A-related patients. J. Am. Coll. Cardiol. 40 (2), 350–356. doi: 10.1016/s0735-1097(02)01962-9
Smits, J. P., Koopmann, T. T., Wilders, R., Veldkamp, M. W., Opthof, T., Bhuiyan, Z. A., et al. (2005). A mutation in the human cardiac sodium channel E161K contributes to sick sinus syndrome, conduction disease, and Brugada syndrome in two families. J. Mol. Cell. Cardiol. 38 (6), 969–981. doi: 10.1016/j.yjmcc.2005.02.024
Splawski, I., Shen, J., Timothy, K. W., Lehmann, M. H., Priori, S., Robinson, J. L., et al. (2000). Spectrum of mutations in long-QT syndrome genes KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circ 102, 1178–1185. doi: 10.1161/01.cir.102.10.1178
Starace, D. M., Bezanilla, F. (2004). A proton pore in a potassium channel voltage sensor reveals a focused electric field. Nature 427 (6974), 548–553. doi: 10.1038/nature02270
Stuhmer, W., Stocker, M., Sakmann, B., Seeburg, P., Baumann, A., Grupe, A., et al. (1988). Potassium channels expressed from rat brain DNA have delayed rectifier properties. FEBS Lettr. 242 (1), 199–206. doi: 10.1016/0014-5793(88)8105-9
Stuhmer, W., Conti, F., Suzuki, H., Wang, X. D., Noda, M., Yahagi, N., et al. (1989). Structural parts involved in the activation and inactivation of the sodium channel. Nature 339 (6226), 597–603. doi: 10.1038/339597a0
Sula, A., Booker, J., Ng, L. C. T., Naylor, C. E., DeCaen, P. G., Wallace, B. A. (2017). The complete structure of an activated open sodium channel. Nat. Commun. 8, 14205. doi: 10.1038/ncomms14205
Tao, X., Lee, A., Limapichat, W., Dougherty, D. A., MacKinnon, R. (2010). A gating charge transfer center in voltage sensors. Science 328 (5974), 67–73. doi: 10.1126/science.1185954
Tester, D. J., Will, M. L., Haglund, C. M., Ackermann, M. J. (2005). Compendium of cardiac sodium channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm. 2 (5), 507–517. doi: 10.1016/j.hrthm.2005.01.020
Timpe, L. C., Schwarz, T. L., Tempel, B. L., Papazian, D. M., Jan, Y. N., Jan, L. Y. (1988). Expression of functional potassium channels from Shaker cDNA in Xenopus oocytes. Nature 331 (6152), 143–145. doi: 10.1038/331143a0
Tiwari-Woodruff, S. K., Schulteis, C. T., Mock, A. F., Papazian, D. M. (1997). Electrostatic interactions between transmembrane segments mediate folding of Shaker K+ channel subunits. Biophys. J. 72 (4), 1489–1500. doi: 10.1016/S0006-3495(97)78797-6
Tiwari-Woodruff, S. K., Lin, M.-A., Schulteis, C. T., Papazian, D. M. (2000). Voltage-dependent structural interactions in the Shaker K+ channel. J. Gen. Physiol. 115 (2), 123–138. doi: 10.1085/jgp.115.2.123
Tombola, F., Pathak, M. M., Isacoff, E. Y. (2005). Voltage-sensing arginines in a potassium channel permeate and occlude cation-selective pores. Neuron 45 (3), 379–388. doi: 10.1016/j.neuron.2004.12.047
Tombola, F., Pathak, M. M., Gorostiza, P., Isacoff, E. Y. (2007). The twisted ion-permeation pathway of a resting voltage-sensing domain. Nature 445 (7127), 546–549. doi: 10.1038/nature05396
Treptow, W., Tarek, M. (2006). Environment of the gating charges in the Kv1.2 shaker potassium channel. Biophys. J. 90 (9), L64–L66. doi: 10.1529/biophysj.106.080754
Treptow, W., Tarek, M., Klein, M. L. (2009). Initial response of the potassium channel voltage sensor to a transmembrane potential. J. Am. Chem. Soc 131 (6), 2107–2109. doi: 10.1021/ja807330g
Tuluc, P., Benedetti, B., Coste de Bagneaux, P., Grabner, M., Flucher, B. E. (2016a). Two distinct voltage-sensing domains control voltage-sensitivity and kinetics of current activation in Cav1.1 calcium channels. J. Gen. Physiol. 147 (6), 437–439. doi: 10.1085/jgp.201511568
Tuluc, P., Yarov-Yarovoy, V., Benedetti, B., Flucher, B. E. (2016b). Molecular interactions in the voltage sensor controlling gating properties of CaV calcium channels. Structure 24 (2), 262–271. doi: 10.1016/j.str.2015.11.011
van Keulen, S. C., Gianti, E., Carnevale, V., Klein, M. L., Rothlisberger, U., Delemotte, L. (2017). Does proton conduction in the voltage-gated H+ channel hHv1 involve Grotthuss-like hopping via acidic residues? J. Phys. Chem. B 121, 3340–3351. doi: 10.1021/acs.jpcb.6b08339
Vargas, E., Bezanilla, F., Roux, B. (2011). In search of a consensus model of the resting state of a voltage-sensing domain. Neuron 72 (5), 713–720. doi: 10.1016/j.neuron.2011.09.024
Vargas, E., Yarov-Yarovoy, V., Khalili-Araghi, F., Catterall, W. A., Klein, M. L., Tarek, M., et al. (2012). An emerging consensus on voltage-dependent gating from computational modeling and molecular dynamics simulations. J. Gen. Physiol. 140 (6), 587–594. doi: 10.1085/jgp.201210873
Villalba-Galea, C. A., Sandtner, W., Starace, D. M., Bezanilla, F. (2008). S4-based voltage sensors have three major conformations. Proc. Natl. Acad. Sci. U. S. A. 105 (46), 17600–17607. doi: 10.1073/pnas.0807387105
Wang, D. W., Viswanathan, P. C., Balser, J. R., George, A. L., Jr., Benson, D. W. (2002). Clinical, genetic, and biophysical characterization of SCN5A mutations associated with atrioventricular conduction block. Circ 105 (3), 341–346. doi: 10.1161/hc0302.102592
Wang, J.-W., Shi, X.-Y., Kurahashi, H., Hwang, S.-K., Ishii, A., Higurashi, N., et al. (2012). Prevalence of SCN1A mutations in children with suspected Dravet syndrome and intractable childhood epilepsy. Epilepsy Res. 102 (3), 195–200. doi: 10.1016/j.eplepsyres.2012.06.006
Watanabe, H., Yang, T., Stroud, D. M., Lowe, J. S., Harris, L., Atack, T. C., et al. (2011). Striking in vivo phenotype of a disease-associated human SCN5A mutation producing minimal changes in vitro. Circ 124 (9), 1001–1011. doi: 10.1161/CIRCULATIONAHA.110.987248
Wisedchaisri, G., Tonggu, L., McCord, E., Gamal El-Din, T. M., Wang, L., Zheng, N., et al. (2019). Resting-state structure and gating mechanism of a voltage-gated sodium channel. Cell 178 (4), 993–1003. doi: 10.1016/j.cell.2019.06.031
Wong, V. C., Fung, C. W., Kwong, A. K. (2015). SCN2A mutation in a Chinese boy with infantile spasm – response to modified Atkins diet. Brain Dev. 37 (7), 729–732. doi: 10.1016/j.braindev.2014.10.008
Wood, M. L., Schow, E. V., Freites, J. A., White, S. H., Tombola, F., Tobias, D. J. (2012). Water wires in atomistic models of the Hv1 proton channel. Biochim. Biophys. Acta 1818 (2), 286–293. doi: 10.1016/j.bbamem.2011.07.045
Wu, J., Yan, Z., Li, Z., Qian, X., Lu, S., Dong, M., et al. (2016). Structure of the voltage-gated calcium channel Ca(v)1.1 at 3.6 Å resolution. Nature 537 (7619), 191–196. doi: 10.1038/nature19321
Yan, Z., Zhou, Q., Wang, L., Wu, J., Zhao, Y., Huang, G., et al. (2017). Structure of the NaV1.4-beta1 complex from electric eel. Cell 170 (3), 470–482. doi: 10.1016/j.cell.2017.06.039
Yarov-Yarovoy, V., DeCaen, P. G. (2019). The sodium channel voltage sensor slides to rest. Trends Pharmacol. Sci. 40 (10), 718–720. doi: 10.1016/j.tips.2019.08.009
Yarov-Yarovoy, V., Baker, D., Catterall, W. A. (2006). Voltage sensor conformations in the open and closed states in ROSETTA structural models of K(+) channels. Proc. Natl. Acad. Sci. U. S. A. 103 (19), 7292–7297. doi: 10.1073/pnas.0602350103
Yarov-Yarovoy, V., DeCaen, P. G., Westenbroek, R. E., Pan, C. Y., Scheuer, T., Baker, D., et al. (2012). Structural basis for gating charge movement in the voltage sensor of a sodium channel. Proc. Natl. Acad. Sci. U. S. A. 109 (2), E93–E102. doi: 10.1073/pnas.1118434109
Zagotta, W. N., Toshi, T., Aldrich, R. W. (1989). Gating of single Shaker potassium channels in Drosophila muscle and in Xenopus oocytes injected with Shaker mRNA. Proc. Natl. Acad. Sci. U. S. A. 86 (18), 7243–7247. doi: 10.1073/pnas.86.18.7243
Zaharieva, I. T., Thor, M. G., Oates, E. C., van Karnebeek, C., Hendson, G., Blom, E., et al. (2016). Loss-of-function mutations in SCN4A cause severe foetal hypokinesia or ‘classical’ congenital myopathy. Brain 139 (3), 674–691. doi: 10.1093/brain/awv352
Zhang, M., Liu, J., Jiang, M., Wu, D.-M., Sonawane, K., Guy, H. R., et al. (2005). Interactions between charged residues in the transmembrane segments of the voltage-sensing domain in the hERG channel. J. Mem. Biol. 207 (3), 169–181. doi: 10.1007/s00232-005-0812-1
Zhang, X., Ren, W., DeCaen, P., Yan, C., Tao, X., Tang., L., et al. (2012). Crystal structure of an orthologue of the NaChBac voltage-gated sodium channel. Nature 486 (7401), 130–134. doi: 10.1038/nature11054
Keywords: countercharge, crystallography, electrostatic, ion channel, molecular dynamics, channelopathy, sliding helix model, voltage sensor domain
Citation: Groome JR and Bayless-Edwards L (2020) Roles for Countercharge in the Voltage Sensor Domain of Ion Channels. Front. Pharmacol. 11:160. doi: 10.3389/fphar.2020.00160
Received: 22 November 2019; Accepted: 07 February 2020;
Published: 28 February 2020.
Edited by:
Gildas Loussouarn, Université de Nantes, FranceReviewed by:
Fredrik Elinder, Linköping University, SwedenCopyright © 2020 Groome and Bayless-Edwards. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James R. Groome, Z3Jvb2phbWVAaXN1LmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.