 Angélica P. Escobar1*
Angélica P. Escobar1* Andrés E. Chávez
Andrés E. Chávez Pablo R. Moya
Pablo R. Moya
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Pharmacol. , 15 November 2019
Sec. Translational Pharmacology
Volume 10 - 2019 | https://doi.org/10.3389/fphar.2019.01362
This article is part of the Research Topic Celebrating 40 Years of the Chilean Society of Pharmacology View all 34 articles
Obsessive compulsive disorder (OCD) is a heterogeneous psychiatric disorder affecting 1%–3% of the population worldwide. About half of OCD afflicted individuals do not respond to currently available pharmacotherapy, which is mainly based on serotonin reuptake inhibition. Therefore, there is a critical need to search novel and improved therapeutic targets to treat this devastating disorder. In recent years, accumulating evidence has supported the glutamatergic hypothesis of OCD, and particularly pointing a potential role for the neuronal glutamate transporter EAAT3. This mini-review summarizes recent findings regarding the neurobiological basis of OCD, with an emphasis on the glutamatergic neurotransmission and EAAT3 as a key player in OCD etiology.
Obsessive compulsive disorder (OCD) is a psychiatric disorder affecting 1%–3% of the population worldwide (Grabe et al., 2000; Angst et al., 2004). It is characterized by recurrent, intrusive worries, feelings or unwanted thoughts (obsessions), and repetitive, structured, ritualistic mental acts and/or behaviors (compulsions). OCD is clinically heterogeneous, displaying a wide range of symptomatic expression (Murphy et al., 2013). Most OCD patients also have high levels of anxiety, likely due to the inability to control or stop the appearance of obsessions (Pauls et al., 2014). Both obsessions and compulsions are time consuming, causing a high impairment in social and occupational areas (American Psychiatric Association, 2013).
Current OCD treatment involves cognitive-behavioral therapy alone or combined with pharmacotherapy mainly based on serotonin reuptake inhibitors (SRI), such as fluoxetine, clomipramine or paroxetine (Bouvard et al., 2004; Cottraux et al., 2005; Skapinakis et al., 2016). However, only about half of patients achieve an adequate decrease in the severity of symptoms (Hirschtritt et al., 2017), highlighting the need for new therapeutic options to treat this devastating disorder.
Since original reports, neuroimaging studies have consistently indicated that a dysfunction in the cortico-striato-thalamo-cortical (CSTC) loop is involved in OCD (Milad and Rauch, 2012; Nakao et al., 2014; Burguiere et al., 2015). In a brief and oversimplified manner, the CSTC loop is composed of glutamatergic pyramidal cortical neurons that project onto striatal subnuclei. From here, GABAergic neurons project to basal ganglia and the thalamus throughout both direct and indirect pathways; in turn, the thalamus sends recurrent projections back to cortical areas (Alexander and Crutcher, 1990). A normal CSTC loop function is required for regulating habitual and goal-directed behaviors (Gerfen and Surmeier, 2011). Neuroimaging studies, for example, indicated changes in the volume of the anterior cingulate cortex and the thalamus in OCD patients compared to healthy controls (Atmaca et al., 2007; Radua and Mataix-Cols, 2009), an alteration that was shown to be restored in responders to SRI treatment (Atmaca et al., 2006). At the functional level, hyperactivity of the anterior cingulate, orbitofrontal cortex, and caudate (Saxena et al., 2001; Maia et al., 2008), as well as altered functional connectivity between the medial frontal cortex and striatal regions have been reported in OCD (Fitzgerald et al., 2011; Posner et al., 2014).
Multiple neurotransmitter systems have been implicated in the etiology of OCD. Since clinically efficacious pharmacotherapy targets the serotonin system, impairments in this neurotransmitter were hypothesized to be disease-causing (Insel et al., 1985; March et al., 1989; Barr et al., 1993) and probably explain certain aspects of OCD pathophysiology. However, like other neuropsychiatric entities, OCD is multifactorial and thereby additional hypotheses have been proposed (Pauls et al., 2014). For instance, some SRI-refractory OCD patients can benefit from combined therapy with antipsychotic drugs targeting dopamine D2 receptors (Dold et al., 2013; Dold et al., 2015), consistent with changes in dopaminergic system reported in OCD patients (Denys et al., 2004), including a decrease in dopamine transporter (DAT) and dopamine D2 receptors levels in basal ganglia (Denys et al., 2004; Hesse et al., 2005). Both monoamine hypotheses are supported by reports of OCD-like behaviors in animal models where modifications of serotonin or dopamine neurotransmitter systems were achieved by genetic or pharmacological intervention (Yadin et al., 1991; Szechtman et al., 1998; Chou-Green et al., 2003; Ralph-Williams et al., 2003).
Over the last decades, increasing lines of evidence support a glutamatergic hypothesis in OCD. For instance, multiple trials have reported promising results in OCD using glutamate-targeting drugs. Memantine, ketamine, and rapastinel, all drugs acting on glutamate NMDA receptors (NMDAR) have shown beneficial effects in OCD treatment (Bakhla et al., 2013; Rodriguez et al., 2016; Modarresi et al., 2018). Moreover, Riluzole, a drug that reduces glutamate synaptic levels (by acting at both presynaptically and at glial glutamate transporters) has been shown to improve OCD symptomatology in about 50% of patients when given concomitantly with SRI (Coric et al., 2005; Grant et al., 2007; Pittenger et al., 2015). Other studies have shown that the adjunctive use of N-acetyl cysteine alleviates OCD symptomatology (Lafleur et al., 2006; Lalanne et al., 2014; Ghanizadeh et al., 2017; di Michele et al., 2018). N-acetyl cysteine is a cysteine donor, precursor for glutathione (a potent antioxidant), and also increases glutamate levels by activating the glial glutamate/cysteine antiporter.
Alterations in fluid or imaging biomarkers of glutamatergic neurotransmission have been reported in OCD, including increased glutamate content in cerebrospinal fluid of OCD untreated patients (Chakrabarty et al., 2005) and a positive correlation between compulsive behavior and glutamate concentration in the anterior cingulate cortex (Naaijen et al., 2017). Interestingly, a previous study performed in child and adolescent OCD patients reported decreased, rather than increased glutamate levels in this brain region (Rosenberg et al., 2004). Higher glutamate concentration was also found in the caudate nucleus in child OCD patients that was restored after treatment with paroxetine (Rosenberg et al., 2000). Such alterations in the levels of glutamate in distinct nuclei of the CSTC loop might lead to (or reflect) functional changes that ultimately, lead to abnormal neuronal circuit activity. Consistent with this idea, three recently developed animal models of OCD display glutamatergic alterations in cortico-striatal synapses (Welch et al., 2007; Shmelkov et al., 2010; Delgado-Acevedo et al., 2019).
Genetic studies also provide support for the glutamatergic hypothesis of OCD, including linkage and association studies. For instance, the 5072T/G variant of GRIN2B gene, which encodes for the NR2B subunit of NMDARs, was significantly associated with OCD in a family-based study (Arnold et al., 2004), whereas the rs1019385 polymorphism was associated with reduced glutamate levels in the anterior cingulate in drug-free pediatric OCD patients. Variants in the GRIK2 gene encoding the kainate receptor subunit 2 have been also reported in OCD (Delorme et al., 2004; Sampaio et al., 2011). Another glutamate-related gene proposed in OCD is DLGAP3, which encodes for the postsynaptic scaffolding protein SAPAP3 implicated in the anchoring of glutamate receptors. As discussed below, SAPAP3 knock-out (KO) mice display OCD relevant behaviors (Welch et al., 2007). These findings prompted genetic studies where some DLGAP3 gene variants were found to be more associated with grooming disorders than with OCD (Bienvenu et al., 2009; Zuchner et al., 2009; Boardman et al., 2011).
Genetic studies have also suggested a role for the SLC1A1 gene, encoding the neuronal glutamate transporter 3 (EAAT3) in OCD. This gene was originally proposed in two independent genome-wide linkage OCD studies (Hanna et al., 2002; Willour et al., 2004). Subsequent family-based association and case-control studies found SLC1A1 gene variants that are associated with OCD (Arnold et al., 2006; Dickel et al., 2006; Stewart et al., 2007; Shugart et al., 2009; Wendland et al., 2009; Samuels et al., 2011). In addition, an association between an SLC1A1 haplotype and the appearance of atypical antipsychotic-induced OCD symptoms has been also reported (Kwon et al., 2009), reinforcing the idea that modifications of SLC1A1 gene might underlie the generation of compulsive behavior. A recent study found that some SLC1A1 variants are associated with white matter microstructure modifications in child and adolescent OCD patients (Gasso et al., 2015), suggesting that they might also underlie the anatomical alterations seen in OCD. Nevertheless, a meta-analysis study found just a weak association between the SLC1A1 variant rs301443 and OCD, while rs12682807 was modestly associated in male subjects (Stewart et al., 2013). The lack of stronger association might be attributed to inadequate sample size, distinct clinical subtypes of OCD and genetic/phenotypic heterogeneity of the subjects (Stewart et al., 2013; Rajendram et al., 2017). Two genome-wide association studies reported no SLC1A1 variants reaching genome wide significance, potentially due to statistical power limitations given low sample size (Stewart et al., 2013; Mattheisen et al., 2015).
To date, three studies have addressed the impact of SLC1A1 gene variants on EAAT3 expression/function. Veenstra-VanderWeele and colleagues characterized the rare SLC1A1 coding variant T164A found in an OCD family, which was shown to have modest effects on both EAAT3 Vmax and Km parameters, suggesting a decrease in the number of EAAT3 available and its affinity for glutamate, respectively, which might be involved in the disorder (Veenstra-VanderWeele et al., 2012). However, no protein quantification was carried out in this work. In a second study, Bailey and colleagues characterized the EAAT3 loss-of-function variants R445W and I395del found in human dicarboxylic aminoaciduria; both mutants dramatically reduced or abolished glutamate and cysteine transport by EAAT3 and led to almost absent EAAT3 surface expression in a cell line model (Rodenas-Ruano et al., 2012). Although no psychiatric assessment was available for the subjects in this study, authors reported that one of the patients carrying R445W variants exhibited features consistent with a diagnosis of OCD (Bailey et al., 2011). In a third study, Wendland and colleagues described SLC1A1 variants that affect mRNA expression in human dorsolateral prefrontal cortex tissue and that were associated with OCD in a large case-control study (Wendland et al., 2009). Collectively, these data suggest that gene variants impacting EAAT3 expression might underlie the pathogenesis of OCD. A more definitive answer will hopefully be provided in the near future through adequately-powered, large genome-wide association or sequencing studies that can yield statistically robust insights into the role of SLC1A1 and other glutamatergic system genes in OCD.
EAAT3 belongs to the excitatory amino acid transporters family (EAAT1-5) that regulates the extracellular levels of glutamate. Although its expression is approximately 100-fold lower than other EAAT in brain (Holmseth et al., 2012), enriched EAAT3 content is found in CSTC loop, including the cerebral cortex, hippocampus, striatum, and basal ganglia (Rothstein et al., 1994; Furuta et al., 1997; Shashidharan et al., 1997; Kanai and Hediger, 2004; Holmseth et al., 2012), in glutamatergic, GABAergic, and dopaminergic neurons (Coco et al., 1997; Conti et al., 1998; Sidiropoulou et al., 2001; Underhill et al., 2014). EAAT3 is localized peri-synaptically on the postsynaptic spine (Coco et al., 1997; He et al., 2000), and several studies have indicated that its contribution to the overall glutamate uptake is much lesser compared to that of the astrocytic transporters EAAT1 and EAAT2 (Tong and Jahr, 1994; Rothstein et al., 1996; Diamond and Jahr, 1997; Tanaka et al., 1997). Nonetheless, increasing evidence indicate that EAAT3 contributes to buffer glutamate released during synaptic events and prolong the time course of astrocytic clearance in the hippocampus (Scimemi et al., 2009), and to regulate the responses of extra-synaptic glutamate NMDA and AMPA receptors (Scimemi et al., 2009; Jarzylo and Man, 2012; Underhill et al., 2014; Delgado-Acevedo et al., 2019) as well as metabotropic glutamate receptors (Otis et al., 2004; Wadiche and Jahr, 2005). As changes in EAAT3 activity can modify the trafficking of AMPARs and function of NMDARs, it is not surprising that EAAT3 has been shown to be involved in excitatory synaptic plasticity (Cao et al., 2014; Bjorn-Yoshimoto and Underhill, 2016; Delgado-Acevedo et al., 2019). EAAT3 can also control neuronal activity through a negative feedback of excitatory neurotransmission by regulating the synthesis of GABA at inhibitory terminals (Sepkuty et al., 2002; Mathews and Diamond, 2003) and by regulating the expression of D1 dopamine receptors (Bellini et al., 2018). Collectively, the evidence strongly supports the notion that EAAT3 regulates glutamate levels in the synaptic cleft and thus the function of postsynaptic receptors. Therefore, changes in EAAT3 expression and/or function at glutamatergic synapses within the CSTC loop might underlie the neurobiological basis of OCD.
EAAT3 has been also suggested to play a role on the neuronal redox balance as they also transport cysteine, the rate limiting substrate for glutathione synthesis (Himi et al., 2003; Aoyama et al., 2006; Watabe et al., 2007). Indeed, EAAT3 KO mice have reduced glutathione brain levels, which correlates with the brain atrophy and hippocampal neurodegeneration in aged mice and impaired spatial memory reported in these mice (Aoyama et al., 2006; Berman et al., 2011). Increased oxidative stress and a reduced number of dopaminergic neurons were also found in EAAT3 KO mice (Berman et al., 2011). Correspondingly, mice lacking GTRAP3-18, a reticulum protein that interacts with and retains EAAT3 on cellular compartment (Lin et al., 2001) not only showed increased EAAT3 levels at the plasma membrane, but also increased cysteine and glutathione brain content as well as improved performance in spatial memory tasks (Aoyama et al., 2012). In contrast, mice overexpressing EAAT3 have higher glutathione brain levels (Delgado-Acevedo et al., 2019), raising the possibility that alterations in redox balance might be also involved in OCD.
Three different genetic models, namely, the Sapap3 KO (Welch et al., 2007) mice, the Slitrk5 KO mice (Shmelkov et al., 2010), and the EAAT3 overexpressing mice (Delgado-Acevedo et al., 2019), have been reported to recapitulate OCD hallmarks including cortico-striatal glutamatergic alterations, particularly changes in NMDAR function, and displaying OCD-like behaviors that could be rescued by chronic administration of SSRI (See Table 1). Mice lacking SAPAP3 (a member of SAP90/PSD-95-associated protein family (SAPAPs)) exhibit an aberrantly increased self-grooming leading to skin wounds (Welch et al., 2007). SAPAP3 KO mice also showed increased anxiety-like behavior in the open field, zero-maze, and light-dark emergence tests; both increased anxiety and hyper-grooming behaviors were alleviated by fluoxetine administration for 6 days (Welch et al., 2007). SAPAP3 KO mice also show abnormalities at cortico-striatal synapses, including a decrease in AMPAR-mediated transmission and an increase in NMDAR-mediated transmission. Such alterations were specific for cortico-striatal synapses, as they were not observed in the hippocampus (Welch et al., 2007) neither in thalamo-striatal synapses (Wan et al., 2014) and were rescued by striatal injections of a lentivirus expressing SAPAP3 (Welch et al., 2007). While the reduction in AMPAR-mediated transmission is likely due to AMPAR endocytosis, an increased activity of mGluR5 receptors was also reported and might be involved in the increase of silent synapses (Wan et al., 2011). This increased mGluR5 activity seemed not only to accompany the OCD-like phenotype but could also be a causative agent, since acute mGluR5 activation recapitulated OCD-like behavior in wildtype mice, and mGluR5 antagonism reverted the phenotype on SAPAP3 KO mice (Ade et al., 2016). Additional dysregulation in serotonin and dopamine release and/or metabolism (Wood et al., 2018) as well as deficits in behavioral inhibition have been also reported in SAPAP3 KO mice (Burguiere et al., 2013). While this evidence links SAPAP3 to compulsive behaviors, human DLGAP3/SAPAP3 gene variants seem to be more related to disorders belonging to the obsessive-compulsive spectrum rather than directly to OCD, as discussed above (Bienvenu et al., 2009; Zuchner et al., 2009; Boardman et al., 2011).
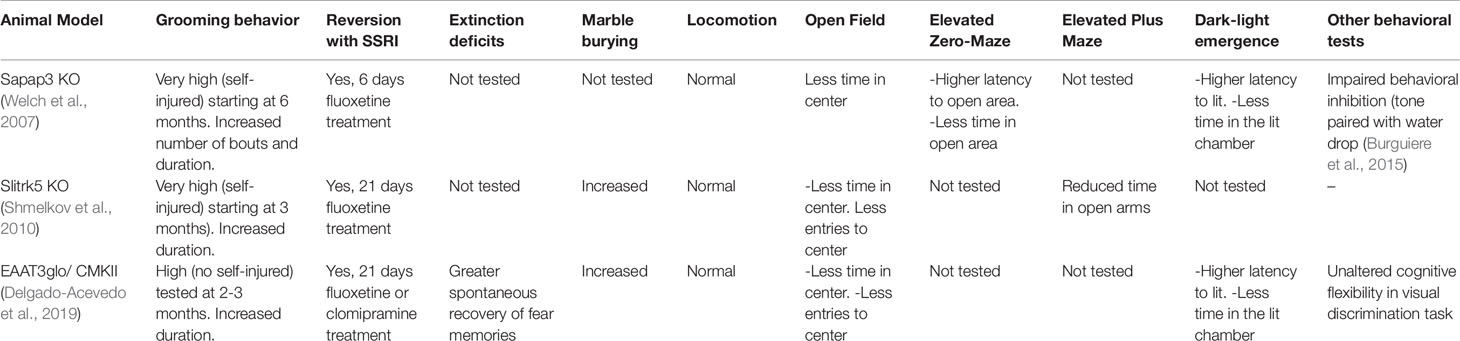
Table 1 Summary of the behavioral alterations relevant to obsessive-compulsive disorder reported in the SAPAP3 KO mouse, Slitrk5 KO mouse, and EAAT3glo/CMKII mouse.
Similarly, the deletion of SlitrK5 gene was found to induce compulsive and anxiety-like behaviors in mice (Shmelkov et al., 2010). The N-terminal region of SlitrK5, a transmembrane protein, is similar to Slit proteins conformed by two leucine-rich domains, whereas the C-terminal is similar to Trk neurotrophin receptor (Aruga and Mikoshiba, 2003). Like SAPAP3 KO mice, increased self-grooming leading to skin lesions and facial hair loss has been reported in SlitrK5 KO mice (Shmelkov et al., 2010). They also display compulsive-like behavior in the marble burying test and increased anxiety-like behavior in the open field and elevated plus maze tests, behavioral impairments that were restored by chronic (18 days) treatment with fluoxetine (Shmelkov et al., 2010). Importantly, SlitrK5 KO mice have selective over-activation of the orbitofrontal cortex and abnormalities in striatal glutamatergic signaling, including a reduction in the level of glutamate receptor subunits GluR1, GluR2, NR2A, and NR2B and a decrease in the amplitude of spikes in response to the cortico-striatal stimulation (Shmelkov et al., 2010). While the exact mechanism by which Slitrk5 deletion impacts glutamatergic signaling remains unclear, Slitrk5 might play an important role in neuronal development as a rare synaptogenesis-impairing mutation in the SLITRK5 gene has been recently associated with OCD (Song et al., 2017), suggesting that changes in the development and/or maturation of excitatory neurons in the CSTC circuit might underlie OCD pathophysiology. Consistent with this idea, SlitrK5 KO mice display decreased arbor complexity in striatal medium spiny neurons (Shmelkov et al., 2010), which might explain the decreased number of postsynaptic glutamate receptors and glutamatergic function, two hallmarks of OCD disorders.
Mouse models with altered EAAT3 expression have been useful to study OCD neurobiology. The first EAAT3 KO mouse model was reported in 1997 and indicated reduced spontaneous locomotor activity, but no other neurological dysfunctions nor OCD relevant behaviors (Peghini et al., 1997). Later, it was found that EAAT3 KO mice also display an age-dependent decrease of dopaminergic neurons number in the substantia nigra, which might explain the impaired locomotor activity (Berman et al., 2011). Similarly, changes in the volume of the hippocampal CA1 region, associated with an age-progressive decline in swim velocity and in spatial memory performance were also reported (Aoyama et al., 2006). Moreover, neurons from EAAT3 KO mice have been found to be more sensitive to oxidative stress (Aoyama et al., 2006), ischemia (Jang et al., 2012; Choi et al., 2014), and traumatic brain injury (Choi et al., 2016). Since some of the alterations in EAAT3 KO mice can be rescued by the treatment with N-acetyl cysteine (Berman et al., 2011), it is plausible to suggest that EAAT3 has a role in neuroprotection due to its cysteine donor function. In this context, increased oxidative status in OCD patients as well as a positive correlation between the severity of OCD symptoms and the level of lipid peroxidation have been reported (Chakraborty et al., 2009). Increased glutathione peroxidase levels in the plasma serum of treatment-naïve children with OCD (Simsek et al., 2016) and lower levels of glutathione in the posterior cingulate cortex have been also reported (Brennan et al., 2016). While animal and human studies indicate that SRIs can restore neuron oxidative status observed in mood disorders, to the best of our knowledge, only one study has suggested that fluoxetine treatment restores the oxidative status in OCD patients (Chakraborty et al., 2009).
After two decades of research since the first publication of an EAAT3 KO mouse model (Peghini et al., 1997), only one study has reported some behaviors reminiscent of compulsive-like behavior in EAAT3 KO mice (Bellini et al., 2018). A second EAAT3 KO mouse model was recently developed by a different research group and, in agreement with the majority of the studies, they found unaltered anxiety or compulsive-like behaviors in this model (Zike et al., 2017). Indeed, a decrease in grooming behavior and amphetamine-induced locomotor activity, as well as in D1 dopamine receptor agonist-induced stereotypy (an effect correlated with reduced dopamine release in the striatum) was reported in this second EAAT3 KO model (Quinlan et al., 1999). While genetic background (C57BL/6 vs CD1 strain) and/or the age of the animals might explain some of the differences observed in the repetitive behavior in EAAT3 KO mouse, both studies highlight the impact of EAAT3 ablation on dopamine neurotransmission (Zike et al., 2017; Bellini et al., 2018). Altogether, these data suggest that OCD susceptibility might not be related to a reduced (or absent) EAAT3 expression/function. Consistent with this idea, one of the SLC1A1 gene variants highly replicated in OCD genetic studies is associated with increased rather than decreased EAAT3 mRNA levels in human brain tissue (Wendland et al., 2009). To explore this EAAT3 gain-of-function hypothesis, a conditional transgenic mouse line, in which EAAT3 was overexpressed under CamKIIα driver (EAAT3glo/CMKII) was recently reported (Delgado-Acevedo et al., 2019). This model exhibits increased anxiety and compulsive-like behaviors that were restored by chronic, but not acute, fluoxetine, or clomipramine treatment. Moreover, EAAT3glo/CMKII mice displayed alterations in the glutamatergic system in the striatum, including changes in NMDARs subunit composition (increased NR2B/NR2A ratio), and impaired NMDA-dependent synaptic plasticity (Delgado-Acevedo et al., 2019). Interestingly, EAAT3glo/CMKII mice also displayed higher spontaneous recovery of fear memory, suggesting impairment in long-term extinction, which has also been described in OCD patients (Milad et al., 2013) and, to the best of our knowledge, for the first time in an OCD mouse model (Delgado-Acevedo et al., 2019). In view of their high density in extinction-related brain areas, the NMDAR has been suggested to be a candidate for modulating extinction learning (for review see (Myers and Davis, 2002; Davis, 2011). However, whether changes in NMDAR subunits composition observed in the striatum of EAAT3glo/CMKII mice (Delgado-Acevedo et al., 2019) also occur in extinction-related areas remain to be determined.
Converging evidence from genetic, neuroanatomical, pharmacological, and preclinical studies in both humans and animal models support that glutamate dysregulations may contribute to the pathophysiology of OCD. As discussed above, the findings from genetic mouse models that are relevant of OCD not only highlight glutamatergic alterations, and particularly altered NMDAR function in the OCD etiology, but also establish EAAT3 overexpression rather than an EAAT3 reduction as an attractive model to further explore in this and other related disorders. While the exact mechanism by which altered EAAT3 levels impact NMDARs subunit composition remains unknown, the fact that they can change in the order of minutes to hours in response to neuronal activity (Barria and Malinow, 2002; Bellone and Nicoll, 2007; Matta et al., 2011), as well as in a long-lasting manner by early life experience (Quinlan et al., 1999; Rodenas-Ruano et al., 2012) strongly suggests that EAAT3 not only limits NMDARs function by rapidly binding synaptically released glutamate (Scimemi et al., 2009), but also might lead to a cascade of events that, ultimately, can modify the relative levels of NR2B to NR2A subunits composition of NMDARs in a long-lasting manner during brain development to impair function. Since redox balance also alters NMDAR function (Choi and Lipton, 2000), the EAAT3-dependent cysteine uptake might also impact glutamatergic neurotransmission by modifying the neuronal oxidative status. Further studies are warranted to better dissect the interplay between of NMDAR and redox balance in the etiology of OCD.
PM conceived the work. AE, JW, AC, PM drafted the work, critically revised the work and provided important intellectual content. AE, JW, AC, PM provided approval for publication of the content and agreed to be accountable for all aspects of the work.
This work was supported by grants Fondecyt N° 1190833, Fondecyt N° 3190843, and ICM-MINECOM N° P09-022-F CINV.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a shared affiliation, though no other collaboration, with the authors AE, JW, AC, PM at time of review.
Ade, K. K., Wan, Y., Hamann, H. C., O’Hare, J. K., Guo, W., Quian, A., et al. (2016). Increased metabotropic glutamate receptor 5 signaling underlies obsessive-compulsive disorder-like behavioral and striatal circuit abnormalities in mice. Biol. Psychiatry 80, 522–533. doi: 10.1016/j.biopsych.2016.04.023
Alexander, G. E., Crutcher, M. D. (1990). Functional architecture of basal ganglia circuits: neural substrates of parallel processing. Trends Neurosci. 13, 266–271. doi: 10.1016/0166-2236(90)90107-L
Angst, J., Gamma, A., Endrass, J., Goodwin, R., Ajdacic, V., Eich, D., et al. (2004). Obsessive-compulsive severity spectrum in the community: prevalence, comorbidity, and course. Eur. Arch. Psychiatry Clin. Neurosci. 254, 156–164. doi: 10.1007/s00406-004-0459-4
Aoyama, K., Suh, S. W., Hamby, A. M., Liu, J., Chan, W. Y., Chen, Y., et al. (2006). Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat. Neurosci. 9, 119–126. doi: 10.1038/nn1609
Aoyama, K., Wang, F., Matsumura, N., Kiyonari, H., Shioi, G., Tanaka, K., et al. (2012). Increased neuronal glutathione and neuroprotection in GTRAP3-18-deficient mice. Neurobiol. Dis. 45, 973–982. doi: 10.1016/j.nbd.2011.12.016
Arnold, P. D., Rosenberg, D. R., Mundo, E., Tharmalingam, S., Kennedy, J. L., Richter, M. A. (2004). Association of a glutamate (NMDA) subunit receptor gene (GRIN2B) with obsessive-compulsive disorder: a preliminary study. Psychopharmacol. (Berl.) 174, 530–538. doi: 10.1007/s00213-004-1847-1
Arnold, P. D., Sicard, T., Burroughs, E., Richter, M. A., Kennedy, J. L. (2006). Glutamate transporter gene SLC1A1 associated with obsessive-compulsive disorder. Arch. Gen. Psychiatry 63, 769–776. doi: 10.1001/archpsyc.63.7.769
Aruga, J., Mikoshiba, K. (2003). Identification and characterization of Slitrk, a novel neuronal transmembrane protein family controlling neurite outgrowth. Mol. Cell Neurosci. 24, 117–129. doi: 10.1016/S1044-7431(03)00129-5
American Psychiatric Association. (2013). Diagnostic and statistical manual of mental disorders (5th ed.). Arlington, VA
Atmaca, M., Yildirim, B. H., Ozdemir, B. H., Aydin, B. A., Tezcan, A. E., Ozler, A. S. (2006). Volumetric MRI assessment of brain regions in patients with refractory obsessive-compulsive disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry 30, 1051–1057. doi: 10.1016/j.pnpbp.2006.03.033
Atmaca, M., Yildirim, H., Ozdemir, H., Tezcan, E., Poyraz, A. K. (2007). Volumetric MRI study of key brain regions implicated in obsessive-compulsive disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry 31, 46–52. doi: 10.1016/j.pnpbp.2006.06.008
Bailey, C. G., Ryan, R. M., Thoeng, A. D., Ng, C., King, K., Vanslambrouck, J. M., et al. (2011). Loss-of-function mutations in the glutamate transporter SLC1A1 cause human dicarboxylic aminoaciduria. J. Clin. Invest. 121, 446–453. doi: 10.1172/JCI44474
Bakhla, A. K., Verma, V., Soren, S., Sarkhel, S., Chaudhury, S. (2013). An open-label trial of memantine in treatment-resistant obsessive-compulsive disorder. Ind. Psychiatry J. 22, 149–152. doi: 10.4103/0972-6748.132930
Barr, L. C., Goodman, W. K., Price, L. H. (1993). The serotonin hypothesis of obsessive compulsive disorder. Int. Clin. Psychopharmacol. 8 Suppl 2, 79–82. doi: 10.1097/00004850-199311002-00011
Barria, A., Malinow, R. (2002). Subunit-specific NMDA receptor trafficking to synapses. Neuron. 35, 345–353. doi: 10.1016/S0896-6273(02)00776-6
Bellini, S., Fleming, K. E., De, M., McCauley, J. P., Petroccione, M. A., D’Brant, L. Y., et al. (2018). Neuronal glutamate transporters control dopaminergic signaling and compulsive behaviors. J. Neurosci. 38, 937–961. doi: 10.1523/JNEUROSCI.1906-17.2017
Bellone, C., Nicoll, R. A. (2007). Rapid bidirectional switching of synaptic NMDA receptors. Neuron. 55, 779–785. doi: 10.1016/j.neuron.2007.07.035
Berman, A. E., Chan, W. Y., Brennan, A. M., Reyes, R. C., Adler, B. L., Suh, S. W., et al. (2011). N-acetylcysteine prevents loss of dopaminergic neurons in the EAAC1-/- mouse. Ann. Neurol. 69, 509–520. doi: 10.1002/ana.22162
Bienvenu, O. J., Wang, Y., Shugart, Y. Y., Welch, J. M., Grados, M. A., Fyer, A. J., et al. (2009). Sapap3 and pathological grooming in humans: Results from the OCD collaborative genetics study. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 150B, 710–720. doi: 10.1002/ajmg.b.30897
Bjorn-Yoshimoto, W. E., Underhill, S. M. (2016). The importance of the excitatory amino acid transporter 3 (EAAT3). Neurochem. Int. 98, 4–18. doi: 10.1016/j.neuint.2016.05.007
Boardman, L., van der Merwe, L., Lochner, C., Kinnear, C. J., Seedat, S., Stein, D. J., et al. (2011). Investigating SAPAP3 variants in the etiology of obsessive-compulsive disorder and trichotillomania in the South African white population. Compr. Psychiatry 52, 181–187. doi: 10.1016/j.comppsych.2010.05.007
Bouvard, M. A., Milliery, M., Cottraux, J. (2004). Management of obsessive compulsive disorder. Psychother. Psychosom. 73, 149–157. doi: 10.1159/000076452
Brennan, B. P., Jensen, J. E., Perriello, C., Pope, H. G., Jr., Jenike, M. A., Hudson, J. I., et al. (2016). Lower posterior cingulate cortex glutathione levels in obsessive-compulsive disorder. Biol. Psychiatry Cognit. Neurosci. Neuroimaging 1, 116–124. doi: 10.1016/j.bpsc.2015.12.003
Burguiere, E., Monteiro, P., Feng, G., Graybiel, A. M. (2013). Optogenetic stimulation of lateral orbitofronto-striatal pathway suppresses compulsive behaviors. Science 340, 1243–1246. doi: 10.1126/science.1232380
Burguiere, E., Monteiro, P., Mallet, L., Feng, G., Graybiel, A. M. (2015). Striatal circuits, habits, and implications for obsessive-compulsive disorder. Curr. Opin. Neurobiol. 30, 59–65. doi: 10.1016/j.conb.2014.08.008
Cao, J., Tan, H., Mi, W., Zuo, Z. (2014). Glutamate transporter type 3 regulates mouse hippocampal GluR1 trafficking. Biochim. Biophys. Acta 1840, 1640–1645. doi: 10.1016/j.bbagen.2014.01.006
Chakrabarty, K., Bhattacharyya, S., Christopher, R., Khanna, S. (2005). Glutamatergic dysfunction in OCD. Neuropsychopharmacology 30, 1735–1740. doi: 10.1038/sj.npp.1300733
Chakraborty, S., Singh, O. P., Dasgupta, A., Mandal, N., Nath Das, H. (2009). Correlation between lipid peroxidation-induced TBARS level and disease severity in obsessive-compulsive disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry 33, 363–366. doi: 10.1016/j.pnpbp.2009.01.001
Choi, Y. B., Lipton, S. A. (2000). Redox modulation of the NMDA receptor. Cell Mol. Life Sci. 57, 1535–1541. doi: 10.1007/PL00000638
Choi, B. Y., Kim, J. H., Kim, H. J., Lee, B. E., Kim, I. Y., Sohn, M., et al. (2014). EAAC1 gene deletion increases neuronal death and blood brain barrier disruption after transient cerebral ischemia in female mice. Int. J. Mol. Sci. 15, 19444–19457. doi: 10.3390/ijms151119444
Choi, B. Y., Kim, I. Y., Kim, J. H., Lee, B. E., Lee, S. H., Kho, A. R., et al. (2016). Decreased cysteine uptake by EAAC1 gene deletion exacerbates neuronal oxidative stress and neuronal death after traumatic brain injury. Amino Acids 48, 1619–1629. doi: 10.1007/s00726-016-2221-4
Chou-Green, J. M., Holscher, T. D., Dallman, M. F., Akana, S. F. (2003). Compulsive behavior in the 5-HT2C receptor knockout mouse. Physiol. Behav. 78, 641–649. doi: 10.1016/S0031-9384(03)00047-7
Coco, S., Verderio, C., Trotti, D., Rothstein, J. D., Volterra, A., Matteoli, M. (1997). Non-synaptic localization of the glutamate transporter EAAC1 in cultured hippocampal neurons. Eur. J. Neurosci. 9, 1902–1910. doi: 10.1111/j.1460-9568.1997.tb00757.x
Conti, F., DeBiasi, S., Minelli, A., Rothstein, J. D., Melone, M. (1998). EAAC1, a high-affinity glutamate tranporter, is localized to astrocytes and gabaergic neurons besides pyramidal cells in the rat cerebral cortex. Cereb. Cortex 8, 108–116. doi: 10.1093/cercor/8.2.108
Coric, V., Taskiran, S., Pittenger, C., Wasylink, S., Mathalon, D. H., Valentine, G., et al. (2005). Riluzole augmentation in treatment-resistant obsessive-compulsive disorder: an open-label trial. Biol. Psychiatry 58, 424–428. doi: 10.1016/j.biopsych.2005.04.043
Cottraux, J., Bouvard, M. A., Milliery, M. (2005). Combining Pharmacotherapy with cognitive-behavioral interventions for obsessive-compulsive disorder. Cognit. Behav. Ther. 34, 185–192. doi: 10.1080/16506070510043750
Davis, M. (2011). NMDA receptors and fear extinction: implications for cognitive behavioral therapy. Dialogues Clin. Neurosci. 13, 463–474.
Delgado-Acevedo, C., Estay, S. F., Radke, A. K., Sengupta, A., Escobar, A. P., Henriquez-Belmar, F., et al. (2019). Behavioral and synaptic alterations relevant to obsessive-compulsive disorder in mice with increased EAAT3 expression. Neuropsychopharmacology 44, 1163–1173. doi: 10.1038/s41386-018-0302-7
Delorme, R., Krebs, M. O., Chabane, N., Roy, I., Millet, B., Mouren-Simeoni, M. C., et al. (2004). Frequency and transmission of glutamate receptors GRIK2 and GRIK3 polymorphisms in patients with obsessive compulsive disorder. Neuroreport 15, 699–702. doi: 10.1097/00001756-200403220-00025
Denys, D., Zohar, J., Westenberg, H. G. (2004). The role of dopamine in obsessive-compulsive disorder: preclinical and clinical evidence. J. Clin. Psychiatry 65 Suppl 14, 11–17.
Denys, D., van der Wee, N., Janssen, J., De Geus, F., Westenberg, H. G. (2004). Low level of dopaminergic D2 receptor binding in obsessive-compulsive disorder. Biol. Psychiatry 55, 1041–1045. doi: 10.1016/j.biopsych.2004.01.023
di Michele, F., Siracusano, A., Talamo, A., Niolu, C. (2018). N-Acetyl cysteine and vitamin d supplementation in treatment resistant obsessive-compulsive disorder patients: a general review. Curr. Pharm. Des. 24, 1832–1838. doi: 10.2174/1381612824666180417124919
Diamond, J. S., Jahr, C. E. (1997). Transporters buffer synaptically released glutamate on a submillisecond time scale. J. Neurosci. 17, 4672–4687. doi: 10.1523/JNEUROSCI.17-12-04672.1997
Dickel, D. E., Veenstra-VanderWeele, J., Cox, N. J., Wu, X., Fischer, D. J., Van Etten-Lee, M., et al. (2006). Association testing of the positional and functional candidate gene SLC1A1/EAAC1 in early-onset obsessive-compulsive disorder. Arch. Gen. Psychiatry 63, 778–785. doi: 10.1001/archpsyc.63.7.778
Dold, M., Aigner, M., Lanzenberger, R., Kasper, S. (2013). Antipsychotic augmentation of serotonin reuptake inhibitors in treatment-resistant obsessive-compulsive disorder: a meta-analysis of double-blind, randomized, placebo-controlled trials. Int. J. Neuropsychopharmacol. 16, 557–574. doi: 10.1017/S1461145712000740
Dold, M., Aigner, M., Lanzenberger, R., Kasper, S. (2015). Antipsychotic augmentation of serotonin reuptake inhibitors in treatment-resistant obsessive-compulsive disorder: an update meta-analysis of double-blind, randomized, placebo-controlled trials. Int. J. Neuropsychopharmacol. 18:1-11. doi: 10.1093/ijnp/pyv047
Fitzgerald, K. D., Welsh, R. C., Stern, E. R., Angstadt, M., Hanna, G. L., Abelson, J. L., et al. (2011). Developmental alterations of frontal-striatal-thalamic connectivity in obsessive-compulsive disorder. J. Am. Acad. Child Adolesc. Psychiatry 50, 938–948 e3. doi: 10.1016/j.jaac.2011.06.011
Furuta, A., Rothstein, J. D., Martin, L. J. (1997). Glutamate transporter protein subtypes are expressed differentially during rat CNS development. J. Neurosci. 17, 8363–8375. doi: 10.1523/JNEUROSCI.17-21-08363.1997
Gasso, P., Ortiz, A. E., Mas, S., Morer, A., Calvo, A., Bargallo, N., et al. (2015). Association between genetic variants related to glutamatergic, dopaminergic and neurodevelopment pathways and white matter microstructure in child and adolescent patients with obsessive-compulsive disorder. J. Affect. Disord. 186, 284–292. doi: 10.1016/j.jad.2015.07.035
Gerfen, C. R., Surmeier, D. J. (2011). Modulation of striatal projection systems by dopamine. Annu. Rev. Neurosci. 34, 441–466. doi: 10.1146/annurev-neuro-061010-113641
Ghanizadeh, A., Mohammadi, M. R., Bahraini, S., Keshavarzi, Z., Firoozabadi, A., Alavi Shoshtari, A. (2017). Efficacy of N-Acetylcysteine augmentation on obsessive compulsive disorder: a multicenter randomized double blind placebo controlled clinical trial. Iran J. Psychiatry 12, 134–141.
Grabe, H. J., Meyer, C., Hapke, U., Rumpf, H. J., Freyberger, H. J., Dilling, H., et al. (2000). Prevalence, quality of life and psychosocial function in obsessive-compulsive disorder and subclinical obsessive-compulsive disorder in northern Germany. Eur. Arch. Psychiatry Clin. Neurosci. 250, 262–268. doi: 10.1007/s004060070017
Grant, P., Lougee, L., Hirschtritt, M., Swedo, S. E. (2007). An open-label trial of riluzole, a glutamate antagonist, in children with treatment-resistant obsessive-compulsive disorder. J. Child Adolesc. Psychopharmacol. 17, 761–767. doi: 10.1089/cap.2007.0021
Hanna, G. L., Veenstra-VanderWeele, J., Cox, N. J., Boehnke, M., Himle, J. A., Curtis, G. C., et al. (2002). Genome-wide linkage analysis of families with obsessive-compulsive disorder ascertained through pediatric probands. Am. J. Med. Genet. 114, 541–552. doi: 10.1002/ajmg.10519
He, Y., Janssen, W. G., Rothstein, J. D., Morrison, J. H. (2000). Differential synaptic localization of the glutamate transporter EAAC1 and glutamate receptor subunit GluR2 in the rat hippocampus. J. Comp. Neurol. 418, 255–269. doi: 10.1002/(SICI)1096-9861(20000313)418:3<255::AID-CNE2>3.0.CO;2-6
Hesse, S., Muller, U., Lincke, T., Barthel, H., Villmann, T., Angermeyer, M. C., et al. (2005). Serotonin and dopamine transporter imaging in patients with obsessive-compulsive disorder. Psychiatry Res. 140, 63–72. doi: 10.1016/j.pscychresns.2005.07.002
Himi, T., Ikeda, M., Yasuhara, T., Nishida, M., Morita, I. (2003). Role of neuronal glutamate transporter in the cysteine uptake and intracellular glutathione levels in cultured cortical neurons. J. Neural Transm. (Vienna) 110, 1337–1348. doi: 10.1007/s00702-003-0049-z
Hirschtritt, M. E., Bloch, M. H., Mathews, C. A. (2017). Obsessive-compulsive disorder: advances in diagnosis and treatment. JAMA 317, 1358–1367. doi: 10.1001/jama.2017.2200
Holmseth, S., Dehnes, Y., Huang, Y. H., Follin-Arbelet, V. V., Grutle, N. J., Mylonakou, M. N., et al. (2012). The density of EAAC1 (EAAT3) glutamate transporters expressed by neurons in the mammalian CNS. J. Neurosci. 32, 6000–6013. doi: 10.1523/JNEUROSCI.5347-11.2012
Insel, T. R., Mueller, E. A., Alterman, I., Linnoila, M., Murphy, D. L. (1985). Obsessive-compulsive disorder and serotonin: is there a connection?. Biol. Psychiatry 20, 1174–1188. doi: 10.1016/0006-3223(85)90176-3
Jang, B. G., Won, S. J., Kim, J. H., Choi, B. Y., Lee, M. W., Sohn, M., et al. (2012). EAAC1 gene deletion alters zinc homeostasis and enhances cortical neuronal injury after transient cerebral ischemia in mice. J. Trace Elem. Med. Biol. 26, 85–88. doi: 10.1016/j.jtemb.2012.04.010
Jarzylo, L. A., Man, H. Y. (2012). Parasynaptic NMDA receptor signaling couples neuronal glutamate transporter function to AMPA receptor synaptic distribution and stability. J. Neurosci. 32, 2552–2563. doi: 10.1523/JNEUROSCI.3237-11.2012
Kanai, Y., Hediger, M. A. (2004). The glutamate/neutral amino acid transporter family SLC1: molecular, physiological and pharmacological aspects. Pflugers Arch. 447, 469–479. doi: 10.1007/s00424-003-1146-4
Kwon, J. S., Joo, Y. H., Nam, H. J., Lim, M., Cho, E. Y., Jung, M. H., et al. (2009). Association of the glutamate transporter gene SLC1A1 with atypical antipsychotics-induced obsessive-compulsive symptoms. Arch. Gen. Psychiatry 66, 1233–1241. doi: 10.1001/archgenpsychiatry.2009.155
Lafleur, D. L., Pittenger, C., Kelmendi, B., Gardner, T., Wasylink, S., Malison, R. T., et al. (2006). N-acetylcysteine augmentation in serotonin reuptake inhibitor refractory obsessive-compulsive disorder. Psychopharmacol. (Berl.) 184, 254–256. doi: 10.1007/s00213-005-0246-6
Lalanne, L., Ayranci, G., Kieffer, B. L., Lutz, P. E. (2014). The kappa opioid receptor: from addiction to depression, and back. Front. Psychiatry 5, 170. doi: 10.3389/fpsyt.2014.00170
Lin, C. I., Orlov, I., Ruggiero, A. M., Dykes-Hoberg, M., Lee, A., Jackson, M., et al. (2001). Modulation of the neuronal glutamate transporter EAAC1 by the interacting protein GTRAP3-18. Nature 410, 84–88. doi: 10.1038/35065084
Maia, T. V., Cooney, R. E., Peterson, B. S. (2008). The neural bases of obsessive-compulsive disorder in children and adults. Dev. Psychopathol. 20, 1251–1283. doi: 10.1017/S0954579408000606
March, J. S., Gutzman, L. D., Jefferson, J. W., Greist, J. H. (1989). Serotonin and treatment in obsessive-compulsive disorder. Psychiatr. Dev. 7, 1–18.
Mathews, G. C., Diamond, J. S. (2003). Neuronal glutamate uptake Contributes to GABA synthesis and inhibitory synaptic strength. J. Neurosci. 23, 2040–2048. doi: 10.1523/JNEUROSCI.23-06-02040.2003
Matta, J. A., Ashby, M. C., Sanz-Clemente, A., Roche, K. W., Isaac, J. T. (2011). mGluR5 and NMDA receptors drive the experience- and activity-dependent NMDA receptor NR2B to NR2A subunit switch. Neuron. 70, 339–351. doi: 10.1016/j.neuron.2011.02.045
Mattheisen, M., Samuels, J. F., Wang, Y., Greenberg, B. D., Fyer, A. J., McCracken, J. T., et al. (2015). Genome-wide association study in obsessive-compulsive disorder: results from the OCGAS. Mol. Psychiatry 20, 337–344. doi: 10.1038/mp.2014.43
Milad, M. R., Rauch, S. L. (2012). Obsessive-compulsive disorder: beyond segregated cortico-striatal pathways. Trends Cognit. Sci. 16, 43–51. doi: 10.1016/j.tics.2011.11.003
Milad, M. R., Furtak, S. C., Greenberg, J. L., Keshaviah, A., Im, J. J., Falkenstein, M. J., et al. (2013). Deficits in conditioned fear extinction in obsessive-compulsive disorder and neurobiological changes in the fear circuit. JAMA Psychiatry 70, 608–618. doi: 10.1001/jamapsychiatry.2013.914
Modarresi, A., Sayyah, M., Razooghi, S., Eslami, K., Javadi, M., Kouti, L. (2018). Memantine augmentation improves symptoms in serotonin reuptake inhibitor-refractory obsessive-compulsive disorder: a randomized controlled trial. Pharmacopsychiatry 51, 263–269. doi: 10.1055/s-0043-120268
Murphy, D. L., Moya, P. R., Fox, M. A., Rubenstein, L. M., Wendland, J. R., Timpano, K. R. (2013). Anxiety and affective disorder comorbidity related to serotonin and other neurotransmitter systems: obsessive-compulsive disorder as an example of overlapping clinical and genetic heterogeneity. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 368, 20120435. doi: 10.1098/rstb.2012.0435
Myers, K. M., Davis, M. (2002). Behavioral and neural analysis of extinction. Neuron. 36, 567–584. doi: 10.1016/S0896-6273(02)01064-4
Naaijen, J., Zwiers, M. P., Amiri, H., Williams, S. C. R., Durston, S., Oranje, B., et al. (2017). Fronto-striatal glutamate in autism spectrum disorder and obsessive compulsive disorder. Neuropsychopharmacology 42, 2456–2465. doi: 10.1038/npp.2016.260
Nakao, T., Okada, K., Kanba, S. (2014). Neurobiological model of obsessive-compulsive disorder: evidence from recent neuropsychological and neuroimaging findings. Psychiatry Clin. Neurosci. 68, 587–605. doi: 10.1111/pcn.12195
Otis, T. S., Brasnjo, G., Dzubay, J. A., Pratap, M. (2004). Interactions between glutamate transporters and metabotropic glutamate receptors at excitatory synapses in the cerebellar cortex. Neurochem. Int. 45, 537–544. doi: 10.1016/j.neuint.2003.11.007
Pauls, D. L., Abramovitch, A., Rauch, S. L., Geller, D. A. (2014). Obsessive-compulsive disorder: an integrative genetic and neurobiological perspective. Nat. Rev. Neurosci. 15, 410–424. doi: 10.1038/nrn3746
Peghini, P., Janzen, J., Stoffel, W. (1997). Glutamate transporter EAAC-1-deficient mice develop dicarboxylic aminoaciduria and behavioral abnormalities but no neurodegeneration. EMBO J. 16, 3822–3832. doi: 10.1093/emboj/16.13.3822
Pittenger, C., Bloch, M. H., Wasylink, S., Billingslea, E., Simpson, R., Jakubovski, E., et al. (2015). Riluzole augmentation in treatment-refractory obsessive-compulsive disorder: a pilot randomized placebo-controlled trial. J. Clin. Psychiatry 76, 1075–1084. doi: 10.4088/JCP.14m09123
Posner, J., Marsh, R., Maia, T. V., Peterson, B. S., Gruber, A., Simpson, H. B. (2014). Reduced functional connectivity within the limbic cortico-striato-thalamo-cortical loop in unmedicated adults with obsessive-compulsive disorder. Hum. Brain Mapp. 35, 2852–2860. doi: 10.1002/hbm.22371
Quinlan, E. M., Olstein, D. H., Bear, M. F. (1999). Bidirectional, experience-dependent regulation of N-methyl-D-aspartate receptor subunit composition in the rat visual cortex during postnatal development. Proc. Natl. Acad. Sci. U.S.A. 96, 12876–12880. doi: 10.1073/pnas.96.22.12876
Radua, J., Mataix-Cols, D. (2009). Voxel-wise meta-analysis of grey matter changes in obsessive-compulsive disorder. Br. J. Psychiatry 195, 393–402. doi: 10.1192/bjp.bp.108.055046
Rajendram, R., Kronenberg, S., Burton, C. L., Arnold, P. D. (2017). Glutamate genetics in obsessive-compulsive disorder: a review. J. Can. Acad. Child Adolesc. Psychiatry 26, 205–213.
Ralph-Williams, R. J., Paulus, M. P., Zhuang, X., Hen, R., Geyer, M. A. (2003). Valproate attenuates hyperactive and perseverative behaviors in mutant mice with a dysregulated dopamine system. Biol. Psychiatry 53, 352–359. doi: 10.1016/S0006-3223(02)01489-0
Rodenas-Ruano, A., Chavez, A. E., Cossio, M. J., Castillo, P. E., Zukin, R. S. (2012). REST-dependent epigenetic remodeling promotes the developmental switch in synaptic NMDA receptors. Nat. Neurosci. 15, 1382–1390. doi: 10.1038/nn.3214
Rodriguez, C. I., Zwerling, J., Kalanthroff, E., Shen, H., Filippou, M., Jo, B., et al. (2016). Effect of a Novel NMDA receptor modulator, rapastinel (Formerly GLYX-13), in OCD: proof of concept. Am. J. Psychiatry 173, 1239–1241. doi: 10.1176/appi.ajp.2016.16080868
Rosenberg, D. R., MacMaster, F. P., Keshavan, M. S., Fitzgerald, K. D., Stewart, C. M., Moore, G. J. (2000). Decrease in caudate glutamatergic concentrations in pediatric obsessive-compulsive disorder patients taking paroxetine. J. Am. Acad. Child Adolesc. Psychiatry 39, 1096–1103. doi: 10.1097/00004583-200009000-00008
Rosenberg, D. R., Mirza, Y., Russell, A., Tang, J., Smith, J. M., Banerjee, S. P., et al. (2004). Reduced anterior cingulate glutamatergic concentrations in childhood OCD and major depression versus healthy controls. J. Am. Acad. Child Adolesc. Psychiatry 43, 1146–1153. doi: 10.1097/01.chi.0000132812.44664.2d
Rothstein, J. D., Martin, L., Levey, A. I., Dykes-Hoberg, M., Jin, L., Wu, D., et al. (1994). Localization of neuronal and glial glutamate transporters. Neuron. 13, 713–725. doi: 10.1016/0896-6273(94)90038-8
Rothstein, J. D., Dykes-Hoberg, M., Pardo, C. A., Bristol, L. A., Jin, L., Kuncl, R. W., et al. (1996). Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 16, 675–686. doi: 10.1016/S0896-6273(00)80086-0
Sampaio, A. S., Fagerness, J., Crane, J., Leboyer, M., Delorme, R., Pauls, D. L., et al. (2011). Association between polymorphisms in GRIK2 gene and obsessive-compulsive disorder: a family-based study. CNS Neurosci. Ther. 17, 141–147. doi: 10.1111/j.1755-5949.2009.00130.x
Samuels, J., Wang, Y., Riddle, M. A., Greenberg, B. D., Fyer, A. J., McCracken, J. T., et al. (2011). Comprehensive family-based association study of the glutamate transporter gene SLC1A1 in obsessive-compulsive disorder. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 156B, 472–477. doi: 10.1002/ajmg.b.31184
Saxena, S., Bota, R. G., Brody, A. L. (2001). Brain-behavior relationships in obsessive-compulsive disorder. Semin. Clin. Neuropsychiatry 6, 82–101. doi: 10.1053/scnp.2001.21833
Scimemi, A., Tian, H., Diamond, J. S. (2009). Neuronal transporters regulate glutamate clearance, NMDA receptor activation, and synaptic plasticity in the hippocampus. J. Neurosci. 29, 14581–14595. doi: 10.1523/JNEUROSCI.4845-09.2009
Sepkuty, J. P., Cohen, A. S., Eccles, C., Rafiq, A., Behar, K., Ganel, R., et al. (2002). A neuronal glutamate transporter contributes to neurotransmitter GABA synthesis and epilepsy. J. Neurosci. 22, 6372–6379. doi: 10.1523/JNEUROSCI.22-15-06372.2002
Shashidharan, P., Huntley, G. W., Murray, J. M., Buku, A., Moran, T., Walsh, M., et al. (1997). Immunohistochemical localization of the neuron-specific glutamate transporter EAAC1 (EAAT3) in rat brain and spinal cord revealed by a novel monoclonal antibody. Brain Res. 773, 139–148. doi: 10.1016/S0006-8993(97)00921-9
Shmelkov, S. V., Hormigo, A., Jing, D., Proenca, C. C., Bath, K. G., Milde, T., et al. (2010). Slitrk5 deficiency impairs corticostriatal circuitry and leads to obsessive-compulsive-like behaviors in mice. Nat. Med. 16, 598–602. doi: 10.1038/nm.2125
Shugart, Y. Y., Wang, Y., Samuels, J. F., Grados, M. A., Greenberg, B. D., Knowles, J. A., et al. (2009). A family-based association study of the glutamate transporter gene SLC1A1 in obsessive-compulsive disorder in 378 families. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 150B, 886–892. doi: 10.1002/ajmg.b.30914
Sidiropoulou, K., Chao, S., Lu, W., Wolf, M. E. (2001). Amphetamine administration does not alter protein levels of the GLT-1 and EAAC1 glutamate transporter subtypes in rat midbrain, nucleus accumbens, striatum, or prefrontal cortex. Brain Res. Mol. Brain Res. 90, 187–192. doi: 10.1016/S0169-328X(01)00110-3
Simsek, S., Gencoglan, S., Yuksel, T. (2016). DNA damage and antioxidants in treatment naive children with obsessive-compulsive disorder. Psychiatry Res. 237, 133–137. doi: 10.1016/j.psychres.2016.01.054
Skapinakis, P., Caldwell, D. M., Hollingworth, W., Bryden, P., Fineberg, N. A., Salkovskis, P., et al. (2016). Pharmacological and psychotherapeutic interventions for management of obsessive-compulsive disorder in adults: a systematic review and network meta-analysis. Lancet Psychiatry 3, 730–739. doi: 10.1016/S2215-0366(16)30069-4
Song, M., Mathews, C. A., Stewart, S. E., Shmelkov, S. V., Mezey, J. G., Rodriguez-Flores, J. L., et al. (2017). Rare synaptogenesis-impairing mutations in slitrk5 are associated with obsessive compulsive disorder. PloS One 12, e0169994. doi: 10.1371/journal.pone.0169994
Stewart, S. E., Fagerness, J. A., Platko, J., Smoller, J. W., Scharf, J. M., Illmann, C., et al. (2007). Association of the SLC1A1 glutamate transporter gene and obsessive-compulsive disorder. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 144B, 1027–1033. doi: 10.1002/ajmg.b.30533
Stewart, S. E., Mayerfeld, C., Arnold, P. D., Crane, J. R., O’Dushlaine, C., Fagerness, J. A., et al. (2013). Meta-analysis of association between obsessive-compulsive disorder and the 3’ region of neuronal glutamate transporter gene SLC1A1. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 162B, 367–379. doi: 10.1002/ajmg.b.32137
Stewart, S. E., Yu, D., Scharf, J. M., Neale, B. M., Fagerness, J. A., Mathews, C. A., et al. (2013). Genome-wide association study of obsessive-compulsive disorder. Mol. Psychiatry 18, 788–798. doi: 10.1038/mp.2012.85
Szechtman, H., Sulis, W., Eilam, D. (1998). Quinpirole induces compulsive checking behavior in rats: a potential animal model of obsessive-compulsive disorder (OCD). Behav. Neurosci. 112, 1475–1485. doi: 10.1037//0735-7044.112.6.1475
Tanaka, K., Watase, K., Manabe, T., Yamada, K., Watanabe, M., Takahashi, K., et al. (1997). Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 276, 1699–1702. doi: 10.1126/science.276.5319.1699
Tong, G., Jahr, C. E. (1994). Block of glutamate transporters potentiates postsynaptic excitation. Neuron. 13, 1195–1203. doi: 10.1016/0896-6273(94)90057-4
Underhill, S. M., Wheeler, D. S., Li, M., Watts, S. D., Ingram, S. L., Amara, S. G. (2014). Amphetamine modulates excitatory neurotransmission through endocytosis of the glutamate transporter EAAT3 in dopamine neurons. Neuron. 83, 404–416. doi: 10.1016/j.neuron.2014.05.043
Veenstra-VanderWeele, J., Xu, T., Ruggiero, A. M., Anderson, L. R., Jones, S. T., Himle, J. A., et al. (2012). Functional studies and rare variant screening of SLC1A1/EAAC1 in males with obsessive-compulsive disorder. Psychiatr. Genet. 22, 256–260. doi: 10.1097/YPG.0b013e328353fb63
Wadiche, J. I., Jahr, C. E. (2005). Patterned expression of Purkinje cell glutamate transporters controls synaptic plasticity. Nat. Neurosci. 8, 1329–1334. doi: 10.1038/nn1539
Wan, Y., Feng, G., Calakos, N. (2011). Sapap3 deletion causes mGluR5-dependent silencing of AMPAR synapses. J. Neurosci. 31, 16685–16691. doi: 10.1523/JNEUROSCI.2533-11.2011
Wan, Y., Ade, K. K., Caffall, Z., Ilcim Ozlu, M., Eroglu, C., Feng, G., et al. (2014). Circuit-selective striatal synaptic dysfunction in the Sapap3 knockout mouse model of obsessive-compulsive disorder. Biol. Psychiatry 75, 623–630. doi: 10.1016/j.biopsych.2013.01.008
Watabe, M., Aoyama, K., Nakaki, T. (2007). Regulation of glutathione synthesis via interaction between glutamate transport-associated protein 3-18 (GTRAP3-18) and excitatory amino acid carrier-1 (EAAC1) at plasma membrane. Mol. Pharmacol. 72, 1103–1110. doi: 10.1124/mol.107.039461
Welch, J. M., Lu, J., Rodriguiz, R. M., Trotta, N. C., Peca, J., Ding, J. D., et al. (2007). Cortico-striatal synaptic defects and OCD-like behaviours in Sapap3-mutant mice. Nature 448, 894–900. doi: 10.1038/nature06104
Wendland, J. R., Moya, P. R., Timpano, K. R., Anavitarte, A. P., Kruse, M. R., Wheaton, M. G., et al. (2009). A haplotype containing quantitative trait loci for SLC1A1 gene expression and its association with obsessive-compulsive disorder. Arch. Gen. Psychiatry 66, 408–416. doi: 10.1001/archgenpsychiatry.2009.6
Willour, V. L., Yao Shugart, Y., Samuels, J., Grados, M., Cullen, B., Bienvenu, O. J., et al. (2004). Replication study supports evidence for linkage to 9p24 in obsessive-compulsive disorder. Am. J. Hum. Genet. 75, 508–513. doi: 10.1086/423899
Wood, J., LaPalombara, Z., Ahmari, S. E. (2018). Monoamine abnormalities in the SAPAP3 knockout model of obsessive-compulsive disorder-related behaviour. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 373 (1742). doi: 10.1098/rstb.2017.0023
Yadin, E., Friedman, E., Bridger, W. H. (1991). Spontaneous alternation behavior: an animal model for obsessive-compulsive disorder?. Pharmacol. Biochem. Behav. 40, 311–315. doi: 10.1016/0091-3057(91)90559-K
Zike, I. D., Chohan, M. O., Kopelman, J. M., Krasnow, E. N., Flicker, D., Nautiyal, K. M., et al. (2017). OCD candidate gene SLC1A1/EAAT3 impacts basal ganglia-mediated activity and stereotypic behavior. Proc. Natl. Acad. Sci. U.S.A. 114, 5719–5724. doi: 10.1073/pnas.1701736114
Keywords: EAAT3, OCD, obsessive-compulsive disorder, glutamate transporter, synaptic function, NMDAR, animal model, SLC1A1
Citation: Escobar AP, Wendland JR, Chávez AE and Moya PR (2019) The Neuronal Glutamate Transporter EAAT3 in Obsessive-Compulsive Disorder. Front. Pharmacol. 10:1362. doi: 10.3389/fphar.2019.01362
Received: 08 August 2019; Accepted: 28 October 2019;
Published: 15 November 2019.
Edited by:
Ramón Sotomayor-Zárate, University of Valparaíso, ChileReviewed by:
Marcela Julio-Pieper, Pontificia Universidad Católica de Valparaíso, ChileCopyright © 2019 Escobar, Wendland, Chávez and Moya. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Angélica P. Escobar, YW5nZWxpY2EuZXNjb2JhckBjaW52LmNs; Pablo R. Moya, cGFibG8ubW95YUB1di5jbA==
†Present address: Jens R. Wendlend, Takeda Pharmaceuticals, Cambridge, MA, United States
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.