 Giovanna Cenini
Giovanna Cenini Wolfgang Voos
Wolfgang Voos
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 23 August 2019
Sec. Experimental Pharmacology and Drug Discovery
Volume 10 - 2019 | https://doi.org/10.3389/fphar.2019.00902
This article is part of the Research Topic Alzheimer’s Disease: Original Mechanisms and Translational Impact View all 19 articles
Alzheimer disease (AD) is a progressive and deleterious neurodegenerative disorder that affects mostly the elderly population. At the moment, no effective treatments are available in the market, making the whole situation a compelling challenge for societies worldwide. Recently, novel mechanisms have been proposed to explain the etiology of this disease leading to the new concept that AD is a multifactor pathology. Among others, the function of mitochondria has been considered as one of the intracellular processes severely compromised in AD since the early stages and likely represents a common feature of many neurodegenerative diseases. Many mitochondrial parameters decline already during the aging, reaching an extensive functional failure concomitant with the onset of neurodegenerative conditions, although the exact timeline of these events is still unclear. Thereby, it is not surprising that mitochondria have been already considered as therapeutic targets in neurodegenerative diseases including AD. Together with an overview of the role of mitochondrial dysfunction, this review examines the pros and cons of the tested therapeutic approaches targeting mitochondria in the context of AD. Since mitochondrial therapies in AD have shown different degrees of progress, it is imperative to perform a detailed analysis of the significance of mitochondrial deterioration in AD and of a pharmacological treatment at this level. This step would be very important for the field, as an effective drug treatment in AD is still missing and new therapeutic concepts are urgently needed.
Alzheimer disease (AD) is a complex and heterogeneous disorder strongly affecting the cognitive functions and the memory of seniors.
Many risk factors were proposed to be significant contributors for the AD onset such as senescence, autophagy defects, genetic factors [i.e., ApolipoproteinaE-allele4 (APOE4), Triggering receptor expressed on myeloid cells 2 (Trem2)], microbiota alterations, lifestyle choices, cardiovascular and traumatic brain injury, as well as environmental factors (level of education, hypertension, obesity, diabetes, smoking, hearing loss, depression, physical inactivity, social isolation) (Livingston et al., 2017). It is now well accepted that important cellular pathways are compromised in AD. Together with intraneuronal neurofibrillary tangles (NFT) made of hyperphosphorylated tau protein and the extraneuronal senile plaques (SP) made of beta-amyloid (Aβ) peptides, synaptic failure, vascular damage, increased oxidative stress, neuronal and axonal injury, microglia-regulated neuroinflammation, and mitochondrial dysfunction are hallmarks of the disease (Figure 1).
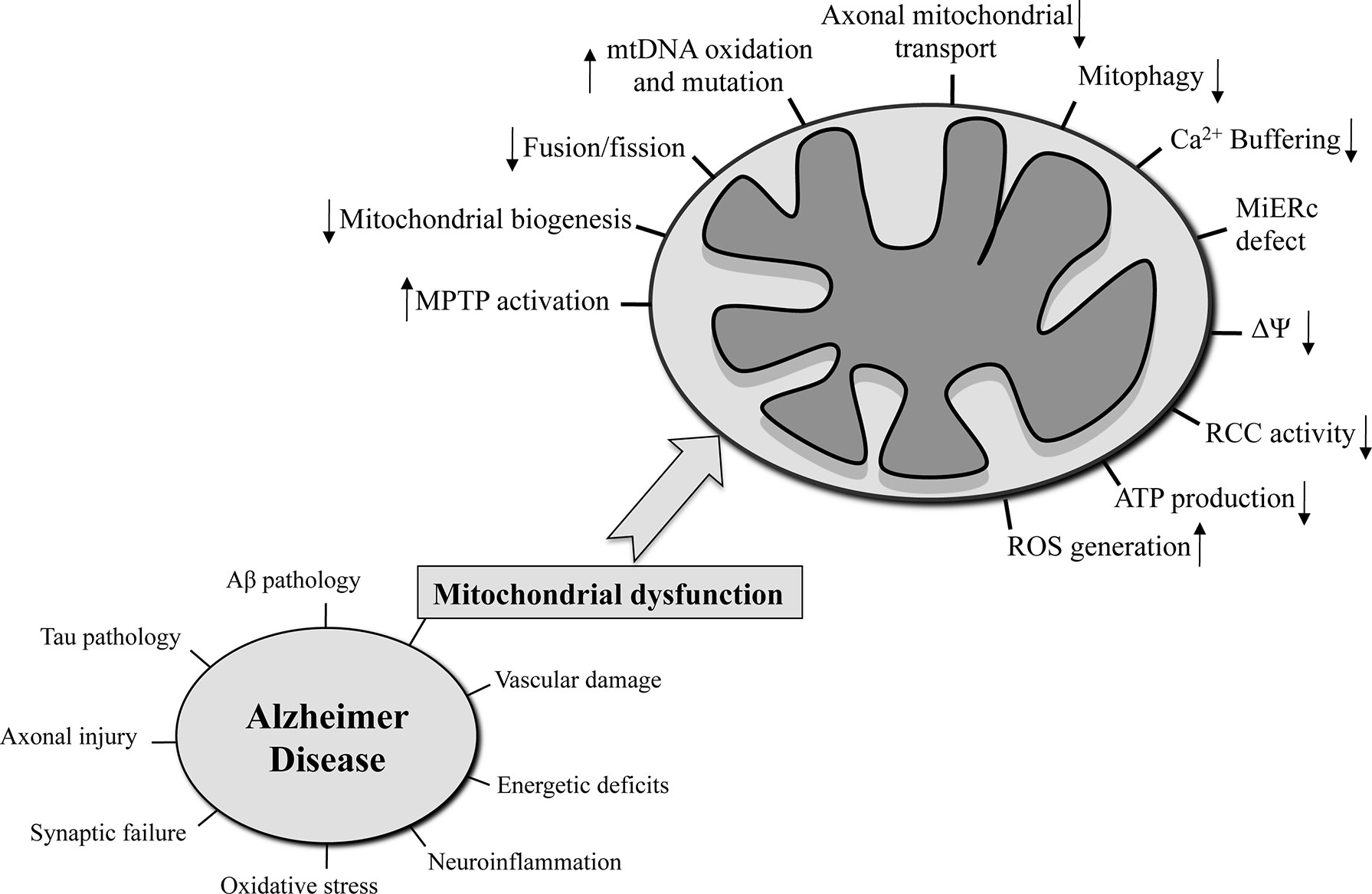
Figure 1 The hallmarks that characterized AD are reported in the left side of the figure. On the right side, the mitochondria-related functions that are seriously compromised in AD are on focus.
Along the past years, Aβ peptides have been considered one of the most promising therapeutic targets for AD. However, many clinical studies based on the Aβ cascade hypothesis failed, and the idea that Aβ pathology is not anymore the leading primary cause of AD has risen (Morris et al., 2018). Instead, nowadays the belief that AD is a multi-factorial disease is growing steadily, and mitochondrial dysfunction is one of the factors that may actively contribute to the disease onset and progression (Iturria-Medina et al., 2017; Veitch et al., 2019). Despite that, a logical temporal order of the events in AD, as well as a valid and effective therapy, is still missing. However, our society urgently requires medical interventions to counteract this deleterious disease because of the severe negative impact on the quality of lives of the afflicted patients as well as on the health system as a whole due to a rapidly aging population.
This review focuses on the description of the role of mitochondrial dysfunction and the status of mitochondrial therapy in AD. The main question addressed here is: could the mitochondrial organelle be a valid pharmacologic target to prevent or delay the AD onset or to block the AD progression?
The mitochondrion is a cellular organelle with a characteristic and unique structure formed by two membranes, respectively called outer mitochondrial membrane (OMM) and inner mitochondrial membrane (IMM) that surround the matrix. Mitochondria are defined as the powerhouse of the cell because every cell in the human body relies on the energy provided by these organelles to sustain their vital functions. Mitochondrial energy production via the so-called process of oxidative phosphorylation takes place at the IMM through the activity of respiratory chain complexes (RCC), generating an inner membrane potential (mtΔΨ) that is used by the ATP-synthase enzyme complex to synthesize adenosine triphosphate (ATP). This process depends on the supply of reducing equivalents by the end-oxidation of nutrients via the Krebs cycle or β-oxidation in the mitochondrial matrix compartment (Stock et al., 2000). Mitochondria contain their own DNA (mtDNA) located in the matrix that encodes mainly 13 protein subunits of the RCC. All other mitochondrial protein components are encoded in the nuclear DNA (nuDNA) and are imported into the organelle after the translation at cytosolic ribosomes. Hence, the maintenance of an entire and functional mitochondrial proteome requires a fine-tuned and well-coordinated sequence of many reactions and a close integration of organellar and cellular biogenesis processes (Pfanner et al., 2019).
Neurons are strictly dependent on the presence of mitochondria in particular at the synapses where these organelles produce ATP and buffer Ca2+-ion concentration, both fundamental processes for the implementation of neurotransmission and generation of membrane potential along the axon (Li et al., 2004; Verstreken et al., 2005; Gazit et al., 2016). This justifies the high amount of mitochondria at the synaptic area, higher than any other part of the neurons. Linked to that, a correct and efficient transport of neuronal mitochondria at the synaptic terminals is fundamental for their correct function. Both non-synaptic and synaptic mitochondria are usually synthesized in the neuronal soma and then transported in the other area of the neurons where they are required. The transport of mitochondria along the axons is guaranteed via microtubules and requires motor proteins such as kinesin, dynein, as well as the OMM protein Mitochondrial Rho GTPase (Miro). Axonal transport of mitochondria is also influenced by the metabolic demand and the Ca2+ status at the synaptic level (Yi et al., 2004; Glater et al., 2006; Russo et al., 2009; Sheng and Cai, 2012).
The enzymatic activity of the mitochondrial RCC results essentially in two “side effects.” First, the generation of the mtΔΨ along the IMM is essential also for the execution of mitochondrial import of nuclear-encoded proteins and overall it is a parameter that reflects the health status of mitochondria and cells (Shariff et al., 2004). Second, a leakage of electrons from the RCC contributes significantly to the formation of reactive oxygen species (ROS). Therefore, ROS are considered a typical by-product of bioenergetic pathways (Quinlan et al., 2013). However, under normal physiological conditions, ROS production is well balanced by the presence of adequate antioxidant systems, and the damage to the diverse cellular constituents is contained. However, during aging, as well as during several pathological conditions, in particular in neurodegenerative diseases, this equilibrium becomes unbalanced. Increased ROS concentrations result in molecular damage at the site where they are produced or, through diffusion, in surrounding areas, leading to the generation of the so-called oxidative stress condition. ROS targets essentially comprise all cellular macromolecules, ranging from proteins, lipids, carbohydrates, up to nucleic acids (Cipak Gasparovic et al., 2017). The hippocampus region, the cortex, and more generally the brain are particularly vulnerable to oxidative stress because of their high consumption of oxygen and dependence on mitochondrial energy production. This susceptibility is increased by low levels of antioxidant defenses and a high content of polyunsaturated fats, which are especially vulnerable to oxidative alterations (Cobley et al., 2018).
Mitochondria form a dynamic tubular network extended throughout the cytosol, a behavior that is often misrepresented by the cell biology textbooks. Two crucial processes, fusion and fission, regulate the entire morphology and structure of this mitochondrial network (Mishra and Chan, 2016). During the fission reaction, a part of the mitochondrial tubule is divided into fragments, a process that is regulated by a member of the dynamin family, Dynamin-1-like protein (Drp1), together with the OMM fission factors Mitochondrial fission 1 protein (Fis1) and Mitochondrial dynamics protein MID49 [Mitochondrial elongation factor 2 (MIEF2)]. Fusion, where two or more pieces of mitochondria are fused together to one structure, happens through joint activity of the proteins Dynamin-like 120 kDa protein [or Optic atrophy protein 1 (OPA1)] and Mitofusin 1 and 2 (Mfn1 and Mfn2). Fusion/fission processes together with the precursor proteins import and internal proteins translation are part of the mitochondrial biogenesis in which the cells increase their mitochondrial mass (Sanchis-Gomar et al., 2014). A master regulator of mitochondrial biogenesis is Peroxisome-proliferator-activated receptor γ coactivator-1α (PGC-1α) (Scarpulla, 2011) that activates a series of transcriptional factors, including the Mitochondrial transcription factor A (TFAM), which regulates transcription and replication of mtDNA (Kang et al., 2018), and Nuclear respiratory factor 1 (NFR-1) and 2 (NFR-2), which control the mitochondrial protein-encoded nuclear genes (Scarpulla, 2011).
The buffer of intracellular Ca2+ is mediated mainly by the cooperation between endoplasmic reticulum (ER) and mitochondria through the formation of contact sites (Krols et al., 2016) that permit the Ca2+ uptake from the cytosol and the exchange of the ion between the two organelles (Rizzuto and Pozzan, 2006). Ca2+ regulates important mitochondrial metabolic enzymes (McCormack et al., 1990). The mitochondria contain two types of Ca2+ channels: the Mitochondria calcium uniporter (MCU) with high selectivity for this ion and localized in the IMM (De Stefani et al., 2011) and the Voltage-dependent anion channel (VDAC) localized in the OMM that regulates the release of the Ca2+ from the mitochondria (Krols et al., 2016). Furthermore, VDAC cooperates with the adenine nucleotide transporter in the IMM and the cyclophin D (CypD) in the matrix on the formation of the mitochondrial permeability transition pore (mPTP) (Bernardi, 1999). An mPTP opening leads to activation of apoptosis and then cell death (Green and Kroemer, 2004). As already mentioned above, at the synaptic level, mitochondria regulate the amount of Ca2+ fundamental for neurotransmission and in general for the exertion of synaptic functions (Werth and Thayer, 1994; Billups and Forsythe, 2002).
Mitochondrial functions and eventually cellular homeostasis are guaranteed by a dedicated mitochondrial quality control system (mtQCS). The mtQCS comprises a multitude of different biochemical mechanisms that act at different levels, affecting individual polypeptides as well as the whole organelle. While the folding state and activities of mitochondrial proteins are controlled by endogenous chaperones and proteases (Voos, 2013), damaged mitochondria may be removed by a selective autophagy pathway, termed mitophagy (Youle and Narendra, 2011). The primary regulator of the mitophagy is a specialized signaling system consisting of the protein PTEN-induced kinase 1 (Pink1) and the ubiquitin ligase Parkin that is activated after the loss of mtΔΨ (Rüb et al., 2017). An accumulation of Pink1 at the OMM of damaged mitochondria is thought to recruit Parkin that leads to a labeling of the mitochondria for the subsequent mitophagy process. This is followed by the formation of an autophagosomal membrane engulfing the mitochondria followed by its fusion with the lysosomes where ultimately the digestion of the mitochondrial material takes place.
In AD brain, the alteration of energetic pathways, also linked to the reduction of glucose consumption, is a well-established feature of the disease (Gibson and Shi, 2010). The glucose uptake in the brain is usually measured with the positron emission tomography (PET) tracer 18-fluorodeoxyglucose (fDG). In subjects with AD, PET studies have consistently demonstrated a low rate of glucose metabolism (between 20% and 30% lower than healthy individuals) in brain regions involved in processing memory (e.g., the hippocampus, posterior cingulate, temporal, and parietal lobes) (Kapogiannis and Mattson, 2011). Furthermore, it was proposed that the metabolic changes appeared earlier than the onset of the histopathological markers and symptoms (Gibson and Shi, 2010). Although the real cause is still unclear, the defective metabolism that characterizes AD could be easily linked to mitochondrial dysfunction.
Since its formulation in 1992 (Hardy and Higgins, 1992), the “amyloid cascade hypothesis” has dominated the AD field in the past 30 years. This hypothesis was based on two clear evidences: Aβ peptides constitute the extraneuronal senile plaques and mutation of Aβ peptides precursor, amyloid-β precursor protein (APP), leads to an early onset of AD. However, due to the fails in all Phase III clinical trials in human AD, this hypothesis has substantially lost ground and needed to be strongly revised or integrated with other hypotheses (Karran et al., 2011). In 2004, a new hypothesis was proposed to explain the onset of sporadic AD. The hypothesis, called “mitochondrial cascade hypothesis,” described that each human genetic heritage influences mitochondrial functions with a primary repercussion on the onset of AD pathology. In other words, according to this hypothesis, the mitochondrial dysfunction is the primary process to trigger all the cascade of events that lead to sporadic late-onset AD (Swerdlow and Khan, 2004; Swerdlow et al., 2014).
Despite the fact that the validity of the mitochondrial cascade hypothesis has yet to be demonstrated in different AD models as well as human patients, the following mitochondrial functions were found severely compromised in the AD context (Hauptmann et al., 2009): mitochondrial morphology (Johnson and Blum, 1970) and number (Hirai et al., 2001), oxidative phosphorylation, mtΔΨ, Ca2+ buffering, ROS production (Butterfield and Halliwell, 2019), mtDNA oxidation and mutation (Wang et al., 2006), mitochondrial-ER contact sites (Area-Gomez et al., 2018), mitochondrial biogenesis, mitochondrial transport along the neuronal axon (Calkins and Reddy, 2011), and mitophagy (Figure 1). In a neuronal context, any of these dysfunctional processes could lead to synaptic deficits and critical consequences not only for single neurons but also for a more complex structure like the brain (Cai and Tammineni, 2017).
In AD brains, the activities of the enzymes involved in mitochondrial energy production, such as complex IV cytochrome c oxidase (COX), pyruvate dehydrogenase complex, mitochondrial isocitrate dehydrogenase, α-ketoglutarate dehydrogenase (αKGDH), and ATP synthase complex were found decreased, while the succinate dehydrogenase (complex II) and malate dehydrogenase activities were increased (Maurer et al., 2000; Cardoso et al., 2004; Gibson and Shi, 2010; Wojsiat et al., 2015). This definitely compromises the maintenance of the mtΔΨ and eventually of the mitochondrial ATP production (Beck et al., 2016).
In line with that, the imbalance between ROS production and antioxidant power was observed in AD brains, cerebrospinal fluid (CSF), and blood (García-Blanco et al., 2017). Since the 1990s, the ROS-induced oxidative stress has received considerable attention as one of the main factors contributing to the AD pathogenesis (Mark et al., 1997). Already the mild cognitive impairment (MCI), an early stage in the AD chronology, is characterized by the significant increase of oxidative stress markers, such as lipid peroxidation and protein oxidation products, and the decrease of antioxidants in the brain and peripheral compartments (Praticò et al., 2002; Rinaldi et al., 2003; Butterfield et al., 2006).
The analysis of the samples from different AD experimental models and AD patients showed a strong link between the oxidative stress and mitochondrial dysfunction. In the transgenic mice over-expressing human APP (Tg mAPP mice), an early and progressive accumulation of Aβ peptide in synaptic mitochondria led to a mitochondrial synaptic dysfunction such as damaged mitochondrial respiratory activity, increased mPTP and oxidative stress, and impaired mitochondrial axonal transport (Du et al., 2010). Data from the 3xTg-AD mice showed that the compromised mitochondria bioenergetics together with elevated oxidative stress levels are early phenomena appearing before the development of observable Aβ plaques (Hauptmann et al., 2009; Yao et al., 2009). Oxidation of one of the mitochondrial enzymes involved in the oxidative phosphorylation, ATP synthase, was found in isolated lymphocytes from AD peripheral blood as well as in MCI and AD brains (Sultana et al., 2006; Reed et al., 2008; Tramutola et al., 2018). This may explain the compromised activity of the ATP synthase and the reduction of ATP levels in AD. Another paper showed a correlation between the reduction of the mitochondrial enzyme Aconitase (ACO2) activity and the plasma antioxidant levels in peripheral lymphocytes from MCI and AD patients proving again the strong association between the oxidative stress and the mitochondrial dysfunction in AD (Mangialasche et al., 2015). Interestingly, the new and innovative technology for AD modeling obtained with the human induced pluripotent stem cells (iPSCs) directly from AD patients demonstrated further that AD-relevant mitochondrial aberrations, including oxidative stress, have a causative role in the developments of the disease. Indeed, neurons and astrocytes from AD-iPSCs presented increased ROS production and RCC levels and enhanced susceptibility to the stressors (Ochalek et al., 2017; Oksanen et al., 2017; Birnbaum et al., 2018).
The mitochondrial dynamics such as fusion and fission processes were found unbalanced in AD, potentially leading to i) compromised distribution and morphology of mitochondria in the neurons (Hirai et al., 2001) and ii) fragmented mitochondria observed in fibroblasts and brains from AD patients (Wang et al., 2008a; Wang et al., 2009). The mitochondrial fusion and fission proteins were differentially expressed in AD hippocampus with an increase of the mitochondrial fission protein Fis1 alongside with a significant downregulation of Drp1 and fusion proteins Mfn1, Mfn2, and OPA1 (Wang et al., 2009). Similar results were found in a AD cybrids model, together with bleb like- and shorter mitochondria compared to control samples (Gan et al., 2014). Furthermore, increased phosphorylation at Ser 616 site and S-nitrosylation of Drp1, which both facilitate the mitochondrial fission (Taguchi et al., 2007; Cho et al., 2009), were higher in a AD brains compared to control (Wang et al., 2009). Beside that, the protein Drp1 was seen interacting with Aβ and phosphorylated tau in brain homogenates from AD patients (Manczak et al., 2011; Manczak and Reddy, 2012). A recent study performed in samples from AD and healthy control subjects showed the significant association between a specific polymorphism in MFN2 gene and AD suggesting that genetic polymorphism of fusion process regulation might be involved in the AD pathogenesis (Kim et al., 2017). In addition, mfn2 protein act as a tether between mitochondria and ER membranes (de Brito and Scorrano, 2008). In this regard, mfn2 influences the Presenilin 2 (PS2), whose mutation is linked to the familial AD (FAD), in the modulation of the mitochondria-ER contact sites (Filadi et al., 2016).
Several experimental AD models linked to APP overexpression or Aβ peptides treatments are characterized as well by mitochondrial fragmentation and abnormal mitochondrial distribution along the neurons due to an alteration of mitochondrial fusion and fission proteins levels (Wang et al., 2008b; Du et al., 2010; Zhao et al., 2010; Calkins and Reddy, 2011; Wang et al., 2017). All these results lead to two critical remarks: i) the altered balance between fusion and fission that interferes with mitochondrial transport contributes actively to the AD pathogenesis and ii) the mitochondrial dynamics impairment could be a new therapeutic target in AD.
Another key mitochondrial function, the mitochondrial biogenesis, was impaired in AD. The significant reduction of the number of mitochondria in AD human hippocampus and in cell culture models already suggests that the mitochondrial biogenesis is compromised (Hirai et al., 2001; Wang et al., 2008b). Furthermore, the level of protein regulating the mitochondrial biogenesis such as PGC-1α, NRF1 and 2, and TFAM was significantly reduced in human AD hippocampus and cellular models overexpressing APP Swedish mutation (Qin et al., 2009; Sheng et al., 2012). In the AD mouse model harboring mutant human transgenes of APP and Presenilin-1 (PS1), the mitochondrial biogenesis markers were found again declined in particular in the hippocampus region, and the use of melatonin brought beneficial effects (Song et al., 2018).
Interestingly, on one side, mitophagy was able to reverse the memory impairment, to prevent the cognitive deterioration and the Aβ peptide/tau pathology in several AD models (Fang et al., 2019). However, on the other side, mitophagy was also strongly affected in AD, leading to the accumulation of damaged mitochondria and consequently to dysfunctional neurons. One cause may be the impairment of the fusion between the autophagosome and lysosomes. This was observed in cultured cells overexpressing mutant APP, in AD mouse models, and also in neurons from AD patients’ brain (Boland et al., 2008; Lee et al., 2010; Coffey et al., 2014). In AD brains, the somatic mutations found in mtDNA are higher than in healthy brains, potentially triggering other neuropathological consequences such as the increased ROS production in neurons and the promotion of amyloidogenic processing of APP (Lin et al., 2002).
The two major and typical histopathological markers of AD, Aβ peptide and tau, harmfully accumulate in or interact non-specifically with mitochondria (Eckert et al., 2010). Aβ peptide and abnormal tau negatively affect axonal transport and consequently the transport of mitochondria along the axon from the neuronal soma to the synapses. AD mouse models, overexpressing Aβ peptides, have damaged mitochondria usually characterized by impaired axonal transport of mitochondria, a reduced mtΔΨ, and inhibited RCC with a compromised ATP production (Rui et al., 2006). The accumulation of Aβ peptides or of the precursor APP inside the mitochondria (Anandatheerthavarada et al., 2003; Hansson Petersen et al., 2008) and even the interaction of Aβ peptides with some component of the mitochondrial matrix (Lustbader et al., 2004) would be the most straightforward and rational explanations to justify the mitochondrial dysfunctions in the animal models of AD. However, mitochondria lack APP and the set of the enzymes required for Aβ peptide generation, making a mitochondria-localized production of Aβ peptides unlikely. Furthermore, a solid mechanism that explains the mitochondrial import of Aβ peptides and the direct negative effects of Aβ peptides on mitochondria is still missing, suggesting that the mitochondrial dysfunctions identified in all these AD models are indirect effects of Aβ peptides. In support of this point, a recent study showed that Aβ peptides impaired mitochondrial import of nuclear-encoded precursor proteins due to an extra mitochondrial co-aggregation process (Cenini et al., 2016).
Tauopathies including AD are also characterized by mitochondrial dysfunction. Tau influences, directly and indirectly, the mitochondrial transport along the neuronal axon and the mitochondrial functions. This leads to the reduction and impairment of mitochondria at the presynaptic terminals with obvious deleterious consequences (Dubey et al., 2008; DuBoff et al., 2012). In AD brains, phosphorylated tau was found interacting with VDAC1 leading as well to mitochondrial dysfunction (Manczak and Reddy, 2012). Hyperphosphorylation of tau negatively affects complex I activity with a decrease of ATP production, an increase of oxidative stress, dissipation of mtΔΨ, induction of the mitochondrial fission, and excessive mitochondrial fragmentation in postmortem brains from AD patients and in murine models (Manczak et al., 2011; Eckert et al., 2014). In addition, mitochondrial stress was shown to promote tau-hyperphosphorylation in a mouse model (Melov et al., 2007). These observations argue for a prominent role of tau pathology in the mitochondrial dysfunction of AD.
The Translocase of outer membrane 40 kDa submit homolog (Tomm40) is a mitochondrial channel localized in OMM that is fundamental for the import of nuclear-encoded mitochondrial preproteins (Chacinska et al., 2009). Aβ peptides affected directly or indirectly the mitochondrial import machinery including Tomm40, and this may also contribute to the mitochondrial dysfunction observed in AD (Devi et al., 2006; Anandatheerthavarada and Devi, 2007; Cenini et al., 2016). TOMM40 gene is contained in a tight gene cluster together with APOE gene in the chromosome 19 (Gottschalk et al., 2014; Subramanian et al., 2017). APOE is one of the most significant genetic risk factors for late-onset sporadic AD (LOAD) with the ε4/ε4 isoform linked to the highest risk (Saunders et al., 1993). It seems that also a variable-length, deoxythymidine homopolymer polymorphism in intron 6 of the TOMM40 gene represents a genetic risk for LOAD. However, different groups showed that TOMM40 SNPs (single-nucleotide polymorphisms) are associated with the LOAD (Martin et al., 2000; Takei et al., 2009; Kim et al., 2011; Davies et al., 2014). In a Caucasian ethnic group three variants of the TOMM40 polymorphisms were identified, and the variant rs10524523 has received particular attention since it lowered the age of LOAD onset by 7 years in APOE3/4 carriers (Roses et al., 2010). Furthermore, this variant was associated with impaired cognition and the gray matter volume in the brain area susceptible to AD (Johnson et al., 2011). Different groups also demonstrated the strong influence of TOMM40 “523” variant on TOMM40 and APOE genes transcription (Linnertz et al., 2014; Payton et al., 2016).
The integration of all these facts into a significant biological context like neuronal cells in AD, suggests that the accumulation of dysfunctional mitochondria at the synapses and the lack of their replacement would contribute substantially to the neurons degeneration and consequently to the worsening of the AD condition.
AD is still without a cure and also essentially lacks a rational understanding of the primary event triggering the disease. Nevertheless, an improved comprehension of this deleterious disorder and the development of effective treatments are essential not only to heal the disease but also eventually to prevent or postpone the onset of the symptoms in the patients.
The traditional cures used nowadays to treat the AD patients are so far the cholinesterase inhibitors (donepezil, rivastigmine, and galantamine) and memantine that block the N-methyl-D-aspartate (NMDA) receptor and the excess of glutamate activity. NMDA receptors and acetylcholin (Ach) are fundamental in memory and learning processes and their concentration and function are compromised in AD (Francis, 2005). However, these treatments improve the cognitive and memory functions, without really slowing down the progression of the disease.
As described above, mitochondrial dysfunctions and a compromised energetic metabolism are two prominent aspects of AD pathology. Therefore, mitochondria should be seriously considered as pharmacological targets. In the course of history, nevertheless, different compounds affecting mitochondria were already tested in AD without a successful outcome. However, as the idea of AD as a multifactorial disease gained more ground in the last years, a reconsideration of mitochondria as a valid therapeutic target together with other medications is strongly recommended.
Mitochondria could be targeted through two ways: i) by pharmacologic approaches acting on mitochondria directly or ii) by action on the lifestyle that indirectly hits this organelle (Figure 2). In the following section, we describe the most popular mitochondrial treatments that have been used until today on AD patients, and in Table 1, we summarize specifically the beneficial effects of these compounds on mitochondria in different experimental AD models. The table is also a proof that these treatments are able to act effectively and positively on mitochondria, and therefore a revision and improvement of their use in AD would be worthy.
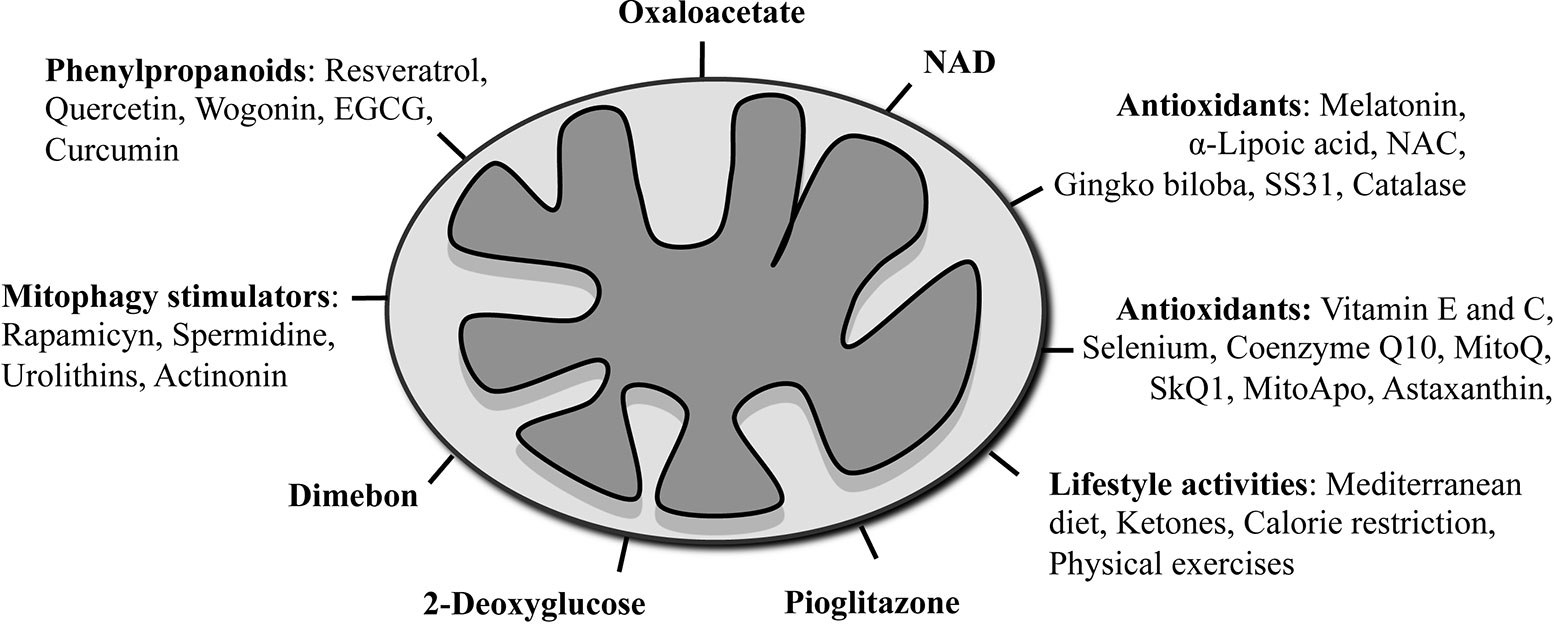
Figure 2 Schematic summary of mitochondrial-targeted therapies used in AD models and clinical trials.

Table 1 List of compounds and lifestyle activities effects on mitochondria in experimental models for AD.
More information about the ongoing clinical trials concerning mitochondria in AD are summarized in Wilkins et al. and in Perez Ortiz et al. (Perez Ortiz and Swerdlow, 2019; Wilkins and Morris, 2017), and they can also be found in www.clinicaltrials.gov.
Since the increased oxidative stress accompanied by the reduction of the antioxidant power was measured in the brain, CSF, and blood from AD patients, treatments with antioxidant compounds were tested to counteract this oxidative unbalance and slow down the progression of the AD symptoms.
Typical antioxidants were the vitamins, E and C, but their effects in the context of AD remain questionable. For example, in two studies with vitamin E, some markers of lipid peroxidation were found decreased in AD patients’ CSF, with no consistent effect on or even a deterioration of cognitive functions (Arlt et al., 2012; Galasko et al., 2012). Vitamin E was also administered in combination with selenium. However, high levels of selenium were found toxic with a pro-oxidant effect, glial activation, and neuronal death (Vinceti et al., 2014). There is an important study called PREADViSE that was performed to see the long-term effect of anti-oxidant supplements (Vitamin E, selenium, Vitamin E + selenium or placebo) on dementia incidence among asymptomatic men. However, the supplement did not prevent dementia occurrence (Kryscio et al., 2017).
Targeting directly the mitochondria with antioxidant compounds was always one of the most considered therapeutic strategies in AD. In this regard, an antioxidant directed to mitochondria that has been tried was the coenzyme Q10 (CoQ10). CoQ10 has a quinone structure and is a component of the mitochondrial RCC. In a rat model for AD, CoQ10 prevented the cognitive decline (Dehghani Dolatabadi et al., 2012). Still, due to a low bioavailability in the brain (Kwong et al., 2002), CoQ10 has never been successful in humans. To overcome this issue, the mitoquinone mesylate (MitoQ) was optimized. MitoQ is an antioxidant compound made of ubiquinone conjugate with triphenylphosphonium (TPP). The TPP is necessary to target the molecule to the mitochondria because it helps to cross the lipid bilayers accumulating on the negative site of mitochondrial membranes (Kelso et al., 2001; Smith et al., 2003). MitoQ behaved as ROS scavenger and was tested in different AD model systems (see Table 1). Here, MitoQ shown to prevent oxidative damage, to protect RCC activity, to reduce Aβ peptide levels, synaptic loss, and astrogliosis, and to improve cognitive functions (McManus et al., 2011; Ng et al., 2014). As reported in the review from Ortiz (Perez Ortiz and Swerdlow, 2019), at the moment, MitoQ is tested in a small clinical trial to check its effect on cerebrovascular blood flow in AD. Similarly to MitoQ, other antioxidant compounds (SkQ1, MitoApo, astaxanthin) affect positively the mitochondrial functions (see Table 1) and could be potentially used to treat AD (Lobos et al., 2016; Stefanova et al., 2016; Brenza et al., 2017).
Another group of antioxidant molecules such as melatonin, α-lipoic acid (LA), N-Acetyl-cysteine (NAC), and Ginkgo biloba were tested in vivo and in vitro and showed protective effects on Aβ peptide accumulation and mitochondrial toxicity as well as on cognitive functions (Dong et al., 2010; Rosales-Corral et al., 2012a). Melatonin is a neurohormone produced by the pineal gland with neuroprotective functions in AD pathogenesis (Shukla et al., 2017). Melatonin is a ROS scavanger and showed some anti-amyloidogenic properties (Dong et al., 2010; Rosales-Corral et al., 2012a). At mitochondrial level, melatonin prevented the ROS production, the cardiolipin oxidation, and the mPTP opening, restored the Ca2+ balance, and reduced the caspase-3 and -9 levels (Feng and Zhang, 2004; Jou et al., 2004; Petrosillo et al., 2009; Espino et al., 2010). Treatments with α-lipoic acid, a cofactor for many RCC enzymes, exhibited a positive effect on cognitive functions in clinical trials on AD patients and in murine models of aging and AD, α-lipoic acid affected also the formation and the stabilization of Aβ peptide fibril as well as the protection against the Aβ peptide toxicity in cultured hippocampal neurons (Liu et al., 2002; Lovell et al., 2003; Ono et al., 2006; Hager et al., 2007; Quinn et al., 2007; Sancheti et al., 2013). N-Acetyl-cysteine (NAC) is the precursor of the endogenous antioxidant glutathione (GSH), a key molecule for the maintenance of mitochondrial functions (Traber et al., 1992). In vitro and in vivo, NAC had beneficial effects on Aβ peptide and phosphorylated tau levels with improvement of cognitive functions, protection against memory decline, and reduction of oxidative stress markers (see also Table 1) (Studer et al., 2001; Fu et al., 2006; Huang et al., 2010; Costa et al., 2016). In two clinical trials, subjects with MCI, AD, or early memory loss were treated for a long time with a nutraceutical formulation that also included NAC. Improvement of cognitive and behavioral functions was observed (Remington et al., 2015; Remington et al., 2016). G. biloba is a natural antioxidant already used in the Chinese traditional medicine. Table 1 shows all the effects of G. biloba on mitochondrial functions. Two clinical trials were performed to test the effect of G. biloba in the prevention against memory and cognitive decline in older adults and AD subjects. Unfortunately, no positive effects were observed in these tests (Snitz et al., 2009; Vellas et al., 2012).
The Szeto-Schiller (SS) tetrapeptides are a group of small peptides that due to their structure act as antioxidants and can reach the mitochondrial matrix and the IMM (Szeto, 2006). In one of AD murine models, the SS31 reduced Aβ peptide production, mitochondrial dysfunction, and enhanced mitochondrial biogenesis and synaptic activity (Calkins et al., 2011; Reddy et al., 2017). Recently, a combination of SS31 and the mitochondrial division inhibitor 1 (Mdivi1) was tested in cultured AD cells with positive effects, suggesting that a combined treatment of mitochondria-targeted antioxidants could have higher effectiveness (Reddy et al., 2018).
An interesting preclinical study proposed to target the antioxidant enzyme catalase to the mitochondria. Catalase catalyzes the decomposition of hydrogen peroxide (H2O2) in water (H2O) and oxygen (O2) and is typically localized in the peroxisome. A double transgenic mouse with mitochondria-targeted catalase (MCAT) and APP was created, and the protective effects against abnormal APP processing, Aβ peptide pathology, and lifespan extension were tested. Mitochondrial catalase showed beneficial outcomes in this highly artificial model. Although most of the antioxidant clinical trials were not entirely successful, this study proved that a direct target of an antioxidant to the mitochondria might still have a chance as a therapeutic approach in AD (Mao et al., 2012).
Despite the oxidative stress unbalance is an evident hallmark in AD and some mitochondrial-targeted antioxidant strategies showed promising effect on cognitive functions, none entered so far in the market as a valid AD treatment. There are different reasons to justify the failures (summarized in Persson et al. paper; Persson et al., 2014). The antioxidants at certain concentrations and conditions could behave as pro-oxidants and therefore they are more harmful than useful. The antioxidant administration during the clinical trials was probably started too late during the development of the disease suggesting that an early intervention could be more effective. Last, the antioxidant bioavailability in the brain could be low due to the difficulty of these molecules to cross the blood–brain barrier (BBB) requiring a rational modification of their structure to overpass this issue.
The phenylpropanoids are natural compounds that exert many physiological functions crucial for the survival of plants. In this heterogeneous group of substances, many subclasses have been identified such as stilbenoids, flavonoids, curcuminoids, phenolate esters, and lignans. These compounds showed an effect against the Aβ peptide and tau pathologies, on the activation of the inflammation response, on the oxidative stress, and also on the mitochondrial dysfunction (Kolaj et al., 2018). Between others, resveratrol, quercetin, wogonin, epigallocatechin-3-gallate (EGCG), and curcumin were already tested and showed to promote mitochondrial biogenesis, to impede apoptotic pathways through inhibition of DNA fragmentation, ROS formation, and caspase-3 activation, and to reduce perturbation of mtΔΨ and ATP levels (see also Table 1 for the effects of phenylpropanoids on mitochondria in AD models) (Lagouge et al., 2006; Davis et al., 2009; Im et al., 2012; Valenti et al., 2013; Reddy et al., 2016). Furthermore, these compounds were able to restore the mitochondrial functions in a transgenic mouse model of AD (Dragicevic et al., 2011b). In particular in an in vitro study, EGCG, a major flavonoid component of the green tea, accumulated in mitochondria and exerted a strong influence on the mitochondrial functions proposing it as pharmacological treatment in AD (Schroeder et al., 2009; Dragicevic et al., 2011b). However, phenylpropanoids have a dual effect on mitochondrial function, depending on the concentration. For example, EGCG could increase apoptosis in cultured neurons at specific concentrations, while quercetin protected cultured hippocampal cells against Aβ peptide-induced apoptosis only in low concentrations (Chung et al., 2007; Ansari et al., 2009). Curcumin is an antioxidant compound with massive potential for the prevention and treatment of AD. It showed beneficial effects on Tg2576 AD model mice, such as reduction of the brain oxidative stress and the neuroinflammation, but no effect in AD patients, probably due to a low bioavailability (Lim et al., 2001; Baum et al., 2008; Ringman et al., 2012). New strategies have been implemented to overpass this limitation and improve the curcumin pharmacokinetics, such as the nanotechnology-based delivery system, new pharmaceutical formulations, and the change in the way of administration (Reddy et al., 2014; Serafini et al., 2017).
Like the antioxidant, the use of the phenylpropanoids in AD treatment needs to be considered with caution and none of them has become a real therapy yet. The new AD clinical trials based on this group of molecules definitely require a broad design, a substantial revision, and a careful implementation.
Lifestyle activities, in particular exercise and diet, have been known to act at the mitochondrial level and should therefore be considered as possible interventions to treat AD. Table 1 reports the effects of the compounds and activities strictly related to the lifestyle on mitochondria from AD models.
A Mediterranean diet has been correlated to the reduction of the incidence of AD (Scarmeas et al., 2006; Karstens et al., 2019). The Mediterranean diet is mainly composed of fruits, vegetables, and omega-3 fatty acids, which are enriched in olive oil. It was observed that, for example, polyphenol-rich extra-virgin oil reduced mitochondria-generated oxidative stress and insulin resistance in high-fat diet fed rats (Lama et al., 2017). Another polyphenol component of olive oil called oleuropein aglycone (OLE) promoted autophagy, decreased aggregated proteins levels, and reduced the cognitive impairment in AD patients’ brain (Grossi et al., 2013; Cordero et al., 2018). Hydroxytyrosol (HT), another bioactive compound of olive oil, ameliorated mitochondrial dysfunction in an animal model of AD (Peng et al., 2016). On the other side, higher consumption of fructose affected negatively the mitochondrial function in hippocampus from adult rats, suggesting that fructose consumption should be actively avoided (Cigliano et al., 2018). Ketones are another source of energy for the brain when there is a limited amount of available glucose (Owen et al., 1967). The ketone ester diet in a model of AD (3xTgAD) had positive effects also on mitochondrial functions (Pawlosky et al., 2017). The therapeutic ketosis was suggested to reduce the AD brain pathology including the accumulation of Aβ plaques and NFT (Kashiwaya et al., 2013). Of course, the results obtained in AD murine models have to be proven in humans through clinical trials (Puchowicz and Seyfried, 2017). In this regard, there are experiments going on at the University of Kansas about the effect of a ketogenic diet (KD) on participants with AD, but no definitive results are available yet (Taylor et al., 2018; Taylor et al., 2019).
An extreme form of diet is represented by calorie restriction (CR). CR is a strong limitation on calorie intake without facing a lack of nutrients. It is well known that CR is an excellent way to extend lifespan, to increase insulin sensitivity, and to prevent age-related diseases (Mattison et al., 2017). At the mitochondrial level, CR showed positive effects by affecting mitochondrial biogenesis through the induction of NO synthetase (eNOS) (Nisoli et al., 2005). Newly synthesized mitochondria led to an increase of mitophagy, reduction of ROS, increased ATP levels, and overall improvement of the mitochondrial quality and cell bioenergetics (López-Lluch et al., 2006). Furthermore, CR affected the mtDNA content as well as the amount of TFAM-bound mtDNA in rats (Picca et al., 2013). There are ongoing clinical studies around the world concerning the effect of CR and dietary intervention on MCI (Wilkins and Morris, 2017).
Physical exercise (PE) has been demonstrated to generally benefit the health of the body and mind, affecting properties such as brain plasticity and cognitive function. Hence, it could be a good prevention for age-related diseases (Hernández et al., 2015; Paillard et al., 2015). It is well known that PE targets mitochondria and improved mitochondrial function (see Table 1 to check the effects of PE on mitochondria in AD models). A study showed that PE increased mtDNA repair, ameliorated mitochondria respiratory function through the increase of RCC activity, attenuated ROS generation capacity together with a reduction of Aβ1-42 peptide levels, and correlated with an amelioration of cognitive function in the hippocampus from the APP/PS1 transgenic mouse model of AD (Bo et al., 2014). However, data obtained in another AD mouse model (3xTg-AD) demonstrated that short-term exercise did not augment the critical gene expression of mitochondrial biogenesis, even if the glucose metabolism was overall improved (Do et al., 2018). Maternal exercise during pregnancy resulted in a positive effect on mitochondrial function concerning the onset of AD. In this study, a protective effect against Aβ oligomer-induced neurotoxicity in the adult offspring brain rats was shown (Klein et al., 2019). Clinical trials with PE were performed in older adults with healthy as well as impaired cognitive function. Aβ1-42 concentration in plasma and CSF was modified. In the brain, improvements of cognitive and executive functions, and even a change of hippocampal volume and memory, were observed, together with a reduced brain atrophy (Baker et al., 2010; Erickson et al., 2011; Vidoni et al., 2015; Yokoyama et al., 2015). Of course, in these human studies, neither a direct effect of PE on mitochondria nor the molecular mechanisms of PE benefits have been proved. However, all the studies performed in animal models positively supported the hypothesis that PE may have a beneficial effect on mitochondrial functions and glucose metabolism also in humans.
Diet, CR, and PE can also be combined to improve the quality of human aging and to prevent neurodegenerative disease (Rege et al., 2017). These approaches were shown to affect mitophagy, the cellular removal mechanism for damaged mitochondria, indicating the mitophagy as a new and promising therapeutic target to prevent the progression of the diseases. Experimental evidences from rodent studies showed that fasting and exercises could have a beneficial effect not only on mitophagy but also on mitochondrial biogenesis, reduction of oxidative stress, and overall neuronal plasticity (Alirezaei et al., 2010). Other strategies to boost mitophagy in order to delay AD are the use of compounds like 2-deoxyglucose, which protects neurons and enhances mitochondrial functions (Table 1) (Duan and Mattson, 1999; Yao et al., 2011). Additional molecules that promote autophagy/mitophagy are rapamycin, spermidine, urolithins, and the antibiotic actinonin (Spilman et al., 2010; Morselli et al., 2011; Ryu et al., 2016; Fang et al., 2019). The mTOR inhibitor rapamycin was already demonstrated to have beneficial effects on a mouse AD model (Spilman et al., 2010). Testing these molecules in clinical AD might be worth it.
Treatment with oxaloacetate (OOA), an intermediate of the Krebs cycle and gluconeogenesis, has been proposed as a new therapeutic approach for AD, and it was already tested in some AD subjects (Swerdlow et al., 2016). Studies involving OOA performed in mice showed positive effects on glycolysis, respiratory fluxes, mtDNA and mtDNA-encoded proteins, activation of mitochondrial biogenesis, hippocampal neurogenesis activity, neuroinflammation, and change in brain insulin signaling (Wilkins et al., 2014). Despite there are no studies about the direct efficacy of OOA treatment on mitochondria in AD models, clinical trials with OOA in AD are ongoing.
Nicotinamide adenine dinucleotide (NAD) is an intermediate common to several mitochondrial metabolic pathways such as glycolysis, TCA cycle, and oxidative phosphorylation. Studies on in vitro and in vivo AD models proved that NAD treatments acted directly on mitochondrial functions and were beneficial (Table 1). In the past, the effect of a stabilized oral NAD formulation on cognitive functions in AD patients was also tested. The rationale behind this testing was based on the enhancement of the cellular bioenergetic to improve brain performance in the fight against neurodegenerative diseases. Interestingly, after 6 months of treatment, the subjects with probable AD showed no cognitive deterioration suggesting that NAD could be an excellent method to prevent the AD progression (Demarin et al., 2004). However, further studies are needed to prove NAD as an effective treatment to slow down AD.
The pioglitazone is a peroxisome proliferator-activated receptor gamma (PPARγ) agonist. PPARγ is a ligand-activated nuclear transcription factor that has a role in regional transcriptional regulation of chr19q13.32 (Subramanian et al., 2017). This region contains the TOMM40-APOE-APOC1 genes and, as already mentioned, TOMM40 and APOE4 genes are risk factors for the LOAD development. Pioglitazone was able to decrease the transcription of TOMM40, APOE, and APOC1 genes making this molecule an interesting candidate in the AD therapy (Subramanian et al., 2017). In CHO cell line overexpressing APP695 isoform, pioglitazone lowered the Aβ1-42 level and restored the mitochondrial activity (Chang et al., 2015). These results were then confirmed in vivo in APP/PSEN1 mice (Table 1) (Chang et al., 2019).
Pioglitazone is usually used to treat diabetes mellitus type 2. Some years ago, the pharmaceutical company Takeda used this compound in a large and global Alzheimer’s prevention study called TOMMORROW to slow down the progression from MCI to AD. The people involved were selected based on their APOE and TOMM40 genotype without considering Aβ status. In 2018, phase III of this prevention trial, unfortunately, closed down because the results against symptomatic AD were negative, despite some improvement in brain metabolism.
Another compound that affects mitochondria but failed the AD clinical trial was dimebon (latrepirdine). Dimebon (latrepirdine) is an old antihistaminic drug (first generation of H1-antagonist) used against allergies that was selected in an AD clinical trial because it demonstrated cognition and memory-enhancing properties in rats treated with neurotoxin (Bachurin et al., 2001). Moreover, dimebon showed a substantial effect on mitochondria from different AD models (Table 1). Anyway, dimebon lacked reproducibility in the AD clinical trials and showed opposite effects on neuropsychiatric and cognitive symptoms, and daily activities (Bachurin et al., 2001; Doody et al., 2008). In a review from 2018, Eckert et al. asked the scientific community to reevaluate the drug dimebon as a potential treatment of AD since one of the clinical trials was able to show a slight improvement of mitochondrial functions after using dimebon in respect of the substantial effect on cognition and behavior (Eckert et al., 2018).
In a multitude of studies, mitochondrial dysfunction has been demonstrated to be a crucial feature of AD. Several experimental results suggested that a decline of mitochondrial activity happens during aging and may get worse at early stages of the disease, contributing to disease onset. However, more thorough investigations are needed to properly address this point. The suitability of the mitochondria as a target in AD treatment is still under discussion, considering that some pharmacological trials were not successful and others were more promising, but none led to a real marketable AD drug. Nevertheless, the current understanding of AD indicates that a complete cure may not be reachable yet. Future research efforts should be invested to i) understand the real chronology of events, ii) collocate correctly the mitochondrial dysfunction inside this temporal sequence, and iii) establish if the mitochondrial dysfunctions are a primary cause or a secondary event. Only when these three key points will be correctly settled, it will be easier to intervene pharmacologically and no more time and money will be wasted for futile therapeutic studies. The failures of the respective drugs or clinical trials often happened because the underlying scientific background was not always very robust or because the models and the tools used to prove the basal hypothesis were not always well defined or validated. Therefore, a more rational approach to a complex human disease like AD is needed as well as an improvement of communication between the different scientific disciplines in order to achieve a better understanding of the disease etiology and to develop new and more effective drugs.
GC conceived the idea and prepared the manuscript. WV reviewed the draft and provided important information for the completion of this manuscript.
The Deutsche Forschungsgemeinschaft (Grant No VO 657/5-2 to WV) supported the work in our laboratory.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ajith, T. A., Nima, N., Veena, R. K., Janardhanan, K. K., Antonawich, F. (2014). Effect of palladium α-lipoic acid complex on energy in the brain mitochondria of aged rats. Altern. Ther. Health Med. 20, 27–35.
Alirezaei, M., Kemball, C. C., Flynn, C. T., Wood, M. R., Whitton, J. L., Kiosses, W. B. (2010). Short-term fasting induces profound neuronal autophagy. Autophagy 6, 702–710. doi: 10.4161/auto.6.6.12376
Anandatheerthavarada, H. K., Biswas, G., Robin, M.-A., Avadhani, N. G. (2003). Mitochondrial targeting and a novel transmembrane arrest of Alzheimer’s amyloid precursor protein impairs mitochondrial function in neuronal cells. J. Cell. Biol. 161, 41–54. doi: 10.1083/jcb.200207030
Anandatheerthavarada, H. K., Devi, L. (2007). Mitochondrial translocation of amyloid precursor protein and its cleaved products: relevance to mitochondrial dysfunction in Alzheimer’s disease. Rev Neurosci. 18, 343–354. doi: 10.1515/revneuro.2007.18.5.343
Ansari, M. A., Abdul, H. M., Joshi, G., Opii, W. O., Butterfield, D. A. (2009). Protective effect of quercetin in primary neurons against Abeta(1-42): relevance to Alzheimer’s disease. J. Nutr. Biochem. 20, 269–275. doi: 10.1016/j.jnutbio.2008.03.002
Area-Gomez, E., de Groof, A., Bonilla, E., Montesinos, J., Tanji, K., Boldogh, I., et al. (2018). A key role for MAM in mediating mitochondrial dysfunction in Alzheimer disease. Cell. Death Dis. 9, 335. doi: 10.1038/s41419-017-0215-0
Arlt, S., Müller-Thomsen, T., Beisiegel, U., Kontush, A. (2012). Effect of one-year vitamin C- and E-supplementation on cerebrospinal fluid oxidation parameters and clinical course in Alzheimer’s disease. Neurochem. Res. 37, 2706–2714. doi: 10.1007/s11064-012-0860-8
Bachurin, S., Bukatina, E., Lermontova, N., Tkachenko, S., Afanasiev, A., Grigoriev, V., et al. (2001). Antihistamine agent Dimebon as a novel neuroprotector and a cognition enhancer. Ann. N. Y. Acad. Sci. 939, 425–435. doi: 10.1111/j.1749-6632.2001.tb03654.x
Baker, L. D., Frank, L. L., Foster-Schubert, K., Green, P. S., Wilkinson, C. W., McTiernan, A., et al. (2010). Aerobic exercise improves cognition for older adults with glucose intolerance, a risk factor for Alzheimer’s disease. J. Alzheimers Dis. 22, 569–579. doi: 10.3233/JAD-2010-100768
Balaban, H., Nazıroğlu, M., Demirci, K., Övey, İ. S. (2017). The protective role of selenium on scopolamine-induced memory impairment, oxidative stress, and apoptosis in aged rats: the involvement of TRPM2 and TRPV1 channels. Mol. Neurobiol. 54, 2852–2868. doi: 10.1007/s12035-016-9835-0
Baum, L., Lam, C. W. K., Cheung, S. K.-K., Kwok, T., Lui, V., Tsoh, J., et al. (2008). Six-month randomized, placebo-controlled, double-blind, pilot clinical trial of curcumin in patients with Alzheimer disease. J. Clin. Psychopharmacol. 28, 110–113. doi: 10.1097/jcp.0b013e318160862c
Beck, S. J., Guo, L., Phensy, A., Tian, J., Wang, L., Tandon, N., et al. (2016). Deregulation of mitochondrial F1FO-ATP synthase via OSCP in Alzheimer’s disease. Nat. Commun. 7, 11483. doi: 10.1038/ncomms11483
Bernardi, P. (1999). Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol. Rev. 79, 1127–1155. doi: 10.1152/physrev.1999.79.4.1127
Biasibetti, R., Tramontina, A. C., Costa, A. P., Dutra, M. F., Quincozes-Santos, A., Nardin, P., et al. (2013). Green tea (-)epigallocatechin-3-gallate reverses oxidative stress and reduces acetylcholinesterase activity in a streptozotocin-induced model of dementia. Behav. Brain Res. 236, 186–193. doi: 10.1016/j.bbr.2012.08.039
Billups, B., Forsythe, I. D. (2002). Presynaptic mitochondrial calcium sequestration influences transmission at mammalian central synapses. J. Neurosci. 22, 5840–5847. doi: 10.1523/JNEUROSCI.22-14-05840.2002
Birnbaum, J. H., Wanner, D., Gietl, A. F., Saake, A., Kündig, T. M., Hock, C., et al. (2018). Oxidative stress and altered mitochondrial protein expression in the absence of amyloid-β and tau pathology in iPSC-derived neurons from sporadic Alzheimer’s disease patients. Stem Cell. Res. 27, 121–130. doi: 10.1016/j.scr.2018.01.019
Bo, H., Kang, W., Jiang, N., Wang, X., Zhang, Y., Ji, L. L. (2014). Exercise-induced neuroprotection of hippocampus in APP/PS1 transgenic mice via upregulation of mitochondrial 8-oxoguanine DNA glycosylase. Oxid. Med. Cell. Longev. 2014, 834502. doi: 10.1155/2014/834502
Boland, B., Kumar, A., Lee, S., Platt, F. M., Wegiel, J., Yu, W. H., et al. (2008). Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. J. Neurosci. 28, 6926–6937. doi: 10.1523/JNEUROSCI.0800-08.2008
Brenza, T. M., Ghaisas, S., Ramirez, J. E. V., Harischandra, D., Anantharam, V., Kalyanaraman, B., et al. (2017). Neuronal protection against oxidative insult by polyanhydride nanoparticle-based mitochondria-targeted antioxidant therapy. Nanomedicine 13, 809–820. doi: 10.1016/j.nano.2016.10.004
Butterfield, D. A., Halliwell, B. (2019). Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 20, 148–160. doi: 10.1038/s41583-019-0132-6
Butterfield, D. A., Poon, H. F., St Clair, D., Keller, J. N., Pierce, W. M., Klein, J. B., et al. (2006). Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: insights into the development of Alzheimer’s disease. Neurobiol. Dis. 22, 223–232. doi: 10.1016/j.nbd.2005.11.002
Cai, Q., Tammineni, P. (2017). Mitochondrial aspects of synaptic dysfunction in Alzheimer’s disease. J. Alzheimers Dis. 57, 1087–1103. doi: 10.3233/JAD-160726
Calkins, M. J., Reddy, P. H. (2011). Amyloid beta impairs mitochondrial anterograde transport and degenerates synapses in Alzheimer’s disease neurons. Biochim. Biophys. Acta 1812, 507–513. doi: 10.1016/j.bbadis.2011.01.007
Calkins, M. J., Manczak, M., Mao, P., Shirendeb, U., Reddy, P. H. (2011). Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 20, 4515–4529. doi: 10.1093/hmg/ddr381
Cardoso, S. M., Proença, M. T., Santos, S., Santana, I., Oliveira, C. R. (2004). Cytochrome c oxidase is decreased in Alzheimer’s disease platelets. Neurobiol. Aging 25, 105–110. doi: 10.1016/S0197-4580(03)00033-2
Cenini, G., Rüb, C., Bruderek, M., Voos, W. (2016). Amyloid β-peptides interfere with mitochondrial preprotein import competence by a coaggregation process. Mol. Biol. Cell 27, 3257–3272. doi: 10.1091/mbc.E16-05-0313
Chacinska, A., Koehler, C. M., Milenkovic, D., Lithgow, T., Pfanner, N. (2009). Importing mitochondrial proteins: machineries and mechanisms. Cell 138, 628–644. doi: 10.1016/j.cell.2009.08.005
Chang, C.-H., Chen, H.-X., Yü, G., Peng, C.-C., Peng, R. Y. (2014). Curcumin-protected PC12 cells against glutamate-induced oxidative toxicity. Food Technol. Biotechnol. 52, 468–478. doi: 10.17113/ftb.52.04.14.3622
Chang, K. L., Pee, H. N., Tan, W. P., Dawe, G. S., Holmes, E., Nicholson, J. K., et al. (2015). Metabolic profiling of CHO-AβPP695 cells revealed mitochondrial dysfunction prior to amyloid-β pathology and potential therapeutic effects of both PPARγ and PPARα Agonisms for Alzheimer’s disease. J. Alzheimers Dis. 44, 215–231. doi: 10.3233/JAD-140429
Chang, K. L., Wong, L. R., Pee, H. N., Yang, S., Ho, P. C.-L. (2019). Reverting metabolic dysfunction in cortex and cerebellum of APP/PS1 mice, a model for Alzheimer’s disease by pioglitazone, a peroxisome proliferator-activated receptor gamma (PPARγ) agonist. Mol. Neurobiol. doi: 10.1007/s12035-019-1586-2
Chen, P., Wang, R.-R., Ma, X.-J., Liu, Q., Ni, J.-Z. (2013). Different forms of selenoprotein M differentially affect Aβ aggregation and ROS generation. Int. J. Mol. Sci. 14, 4385–4399. doi: 10.3390/ijms14034385
Cho, D.-H., Nakamura, T., Fang, J., Cieplak, P., Godzik, A., Gu, Z., et al. (2009). S-Nitrosylation of Drp1 mediates β-amyloid–related mitochondrial fission and neuronal injury. Science 324, 102. doi: 10.1126/science.1171091
Chung, W.-G., Miranda, C. L., Maier, C. S. (2007). Epigallocatechin gallate (EGCG) potentiates the cytotoxicity of rotenone in neuroblastoma SH-SY5Y cells. Brain Res. 1176, 133–142. doi: 10.1016/j.brainres.2007.07.083
Cigliano, L., Spagnuolo, M. S., Crescenzo, R., Cancelliere, R., Iannotta, L., Mazzoli, A., et al. (2018). Short-term fructose feeding induces inflammation and oxidative stress in the hippocampus of young and adult rats. Mol. Neurobiol. 55, 2869–2883. doi: 10.1007/s12035-017-0518-2
Cipak Gasparovic, A., Zarkovic, N., Zarkovic, K., Semen, K., Kaminskyy, D., Yelisyeyeva, O., et al. (2017). Biomarkers of oxidative and nitro-oxidative stress: conventional and novel approaches. Br. J. Pharmacol. 174, 1771–1783. doi: 10.1111/bph.13673
Cobley, J. N., Fiorello, M. L., Bailey, D. M. (2018). 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 15, 490–503. doi: 10.1016/j.redox.2018.01.008
Coffey, E. E., Beckel, J. M., Laties, A. M., Mitchell, C. H. (2014). Lysosomal alkalization and dysfunction in human fibroblasts with the Alzheimer’s disease-linked presenilin 1 A246E mutation can be reversed with cAMP. Neuroscience 263, 111–124. doi: 10.1016/j.neuroscience.2014.01.001
Cordero, J. G., García-Escudero, R., Avila, J., Gargini, R., García-Escudero, V. (2018). Benefit of oleuropein aglycone for Alzheimer’s disease by promoting autophagy. Oxid. Med. Cell. Longev. 2018, 5010741. doi: 10.1155/2018/5010741
Costa, M., Bernardi, J., Fiuza, T., Costa, L., Brandão, R., Pereira, M. E. (2016). N-acetylcysteine protects memory decline induced by streptozotocin in mice. Chem. Biol. Interact. 253, 10–17. doi: 10.1016/j.cbi.2016.04.026
Davies, G., Harris, S. E., Reynolds, C. A., Payton, A., Knight, H. M., Liewald, D.C., et al. (2014). A genome-wide association study implicates the APOE locus in nonpathological cognitive ageing. Mol. Psychiatry 19, 76–87. doi: 10.1038/mp.2012.159
Davis, J. M., Murphy, E. A., Carmichael, M. D., Davis, B. (2009). Quercetin increases brain and muscle mitochondrial biogenesis and exercise tolerance. Am. J. Physiol. Regul. Integr. Comp. Physiol. 296, R1071–R1077. doi: 10.1152/ajpregu.90925.2008
de Brito, O. M., Scorrano, L. (2008). Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605–610. doi: 10.1038/nature07534
De Stefani, D., Raffaello, A., Teardo, E., Szabò, I., Rizzuto, R. (2011). A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476, 336–340. doi: 10.1038/nature10230
Dehghani Dolatabadi, H. R., Reisi, P., Alaei, H., Azizi Malekabadi, H., Pilehvarian, A. A. (2012). Folic acid and coenzyme Q10 ameliorate cognitive dysfunction in the rats with intracerebroventricular injection of streptozotocin. Iran J. Basic Med. Sci. 15, 719–724.
Delic, V., Brownlow, M., Joly-Amado, A., Zivkovic, S., Noble, K., Phan, T.-A., et al. (2015). Calorie restriction does not restore brain mitochondrial function in P301L tau mice, but it does decrease mitochondrial F0F1-ATPase activity. Mol. Cell. Neurosci. 67, 46–54. doi: 10.1016/j.mcn.2015.06.001
Demarin, V., Podobnik, S. S., Storga-Tomic, D., Kay, G. (2004). Treatment of Alzheimer’s disease with stabilized oral nicotinamide adenine dinucleotide: a randomized, double-blind study. Drugs Exp. Clin. Res. 30, 27–33.
Deng, H., Mi, M.-T. (2016). Resveratrol attenuates Aβ25-35 caused neurotoxicity by inducing autophagy through the TyrRS-PARP1-SIRT1 signaling pathway. Neurochem. Res. 41, 2367–2379. doi: 10.1007/s11064-016-1950-9
Devi, L., Prabhu, B. M., Galati, D. F., Avadhani, N. G., Anandatheerthavarada, H. K. (2006). Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J. Neurosci. 26, 9057–9068. doi: 10.1523/JNEUROSCI.1469-06.2006
Dixit, S., Fessel, J. P., Harrison, F. E. (2017). Mitochondrial dysfunction in the APP/PSEN1 mouse model of Alzheimer’s disease and a novel protective role for ascorbate. Free Radic. Biol. Med. 112, 515–523. doi: 10.1016/j.freeradbiomed.2017.08.021
Do, K., Laing, B. T., Landry, T., Bunner, W., Mersaud, N., Matsubara, T., et al. (2018). The effects of exercise on hypothalamic neurodegeneration of Alzheimer’s disease mouse model. PLoS ONE 13, e0190205. doi: 10.1371/journal.pone.0190205
Dong, W., Huang, F., Fan, W., Cheng, S., Chen, Y., Zhang, W., et al. (2010). Differential effects of melatonin on amyloid-beta peptide 25-35-induced mitochondrial dysfunction in hippocampal neurons at different stages of culture. J. Pineal. Res. 48, 117–125. doi: 10.1111/j.1600-079X.2009.00734.x
Doody, R. S., Gavrilova, S. I., Sano, M., Thomas, R. G., Aisen, P. S., Bachurin, S. O., et al. (2008). Effect of dimebon on cognition, activities of daily living, behaviour, and global function in patients with mild-to-moderate Alzheimer’s disease: a randomised, double-blind, placebo-controlled study. Lancet 372, 207–215. doi: 10.1016/S0140-6736(08)61074-0
Dragicevic, N., Copes, N., O’Neal-Moffitt, G., Jin, J., Buzzeo, R., Mamcarz, M., et al. (2011a). Melatonin treatment restores mitochondrial function in Alzheimer’s mice: a mitochondrial protective role of melatonin membrane receptor signaling. J. Pineal. Res. 51, 75–86. doi: 10.1111/j.1600-079X.2011.00864.x
Dragicevic, N., Delic, V., Cao, C., Copes, N., Lin, X., Mamcarz, M., et al. (2012). Caffeine increases mitochondrial function and blocks melatonin signaling to mitochondria in Alzheimer’s mice and cells. Neuropharmacology 63, 1368–1379. doi: 10.1016/j.neuropharm.2012.08.018
Dragicevic, N., Smith, A., Lin, X., Yuan, F., Copes, N., Delic, V., et al. (2011b). Green tea epigallocatechin-3-gallate (EGCG) and other flavonoids reduce Alzheimer’s amyloid-induced mitochondrial dysfunction. J. Alzheimers Dis. 26, 507–521. doi: 10.3233/JAD-2011-101629
Du, H., Guo, L., Yan, S., Sosunov, A. A., McKhann, G. M., Yan, S. S. (2010). Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc. Natl. Acad. Sci. U.S.A. 107, 18670–18675. doi: 10.1073/pnas.1006586107
Duan, W., Mattson, M. P. (1999). Dietary restriction and 2-deoxyglucose administration improve behavioral outcome and reduce degeneration of dopaminergic neurons in models of Parkinson’s disease. J. Neurosci. Res. 57, 195–206. doi: 10.1002/(SICI)1097-4547(19990715)57:2<195::AID-JNR5>3.0.CO;2-P
Dubey, M., Chaudhury, P., Kabiru, H., Shea, T. B. (2008). Tau inhibits anterograde axonal transport and perturbs stability in growing axonal neurites in part by displacing kinesin cargo: neurofilaments attenuate tau-mediated neurite instability. Cell Motil. Cytoskeleton 65, 89–99. doi: 10.1002/cm.20243
DuBoff, B., Götz, J., Feany, M. B. (2012). Tau promotes neurodegeneration via DRP1 mislocalization in vivo. Neuron 75, 618–632. doi: 10.1016/j.neuron.2012.06.026
Dumont, M., Kipiani, K., Yu, F., Wille, E., Katz, M., Calingasan, N. Y., et al. (2011). Coenzyme Q10 decreases amyloid pathology and improves behavior in a transgenic mouse model of Alzheimer’s disease. J. Alzheimers Dis 27, 211–223. doi: 10.3233/JAD-2011-110209
Durán-Prado, M., Frontiñán, J., Santiago-Mora, R., Peinado, J. R., Parrado-Fernández, C., Gómez-Almagro, M. V., et al. (2014). Coenzyme Q10 protects human endothelial cells from β-amyloid uptake and oxidative stress-induced injury. PLoS ONE 9, e109223. doi: 10.1371/journal.pone.0109223
Eckert, A., Keil, U., Kressmann, S., Schindowski, K., Leutner, S., Leutz, S., et al. (2003). Effects of EGb 761 Ginkgo biloba extract on mitochondrial function and oxidative stress. Pharmacopsychiatry 36 Suppl 1, S15–S23. doi: 10.1055/s-2003-40449
Eckert, A., Keil, U., Scherping, I., Hauptmann, S., Müller, W. E. (2005). Stabilization of mitochondrial membrane potential and improvement of neuronal energy metabolism by Ginkgo biloba extract EGb 761. Ann. N. Y. Acad. Sci. 1056, 474–485. doi: 10.1196/annals.1352.023
Eckert, A., Nisbet, R., Grimm, A., Götz, J. (2014). March separate, strike together—Role of phosphorylated TAU in mitochondrial dysfunction in Alzheimer’s disease. (BBA) - Mol. Basis Dis. 1842, 1258–1266. doi: 10.1016/j.bbadis.2013.08.013
Eckert, A., Schulz, K. L., Rhein, V., Götz, J. (2010). Convergence of amyloid-β and Tau pathologies on mitochondria in vivo. Mol. Neurobiol. 41, 107–114. doi: 10.1007/s12035-010-8109-5
Eckert, S. H., Eckmann, J., Renner, K., Eckert, G. P., Leuner, K., Muller, W. E. (2012). Dimebon ameliorates amyloid-β induced impairments of mitochondrial form and function. J. Alzheimers Dis. 31, 21–32. doi: 10.3233/JAD-2012-120310
Eckert, S. H., Gaca, J., Kolesova, N., Friedland, K., Eckert, G. P., Muller, W. E. (2018). Mitochondrial pharmacology of dimebon (latrepirdine) calls for a new look at its possible therapeutic potential in Alzheimer’s disease. Aging Dis. 9, 729–744. doi: 10.14336/AD.2017.1014
Elipenahli, C., Stack, C., Jainuddin, S., Gerges, M., Yang, L., Starkov, A., et al. (2012). Behavioral improvement after chronic administration of coenzyme Q10 in P301S transgenic mice. J. Alzheimers Dis. 28, 173–182. doi: 10.3233/JAD-2011-111190
Erickson, K. I., Voss, M. W., Prakash, R. S., Basak, C., Szabo, A., Chaddock, L., et al. (2011). Exercise training increases size of hippocampus and improves memory. Proc. Natl. Acad. Sci. U.S.A. 108, 3017–3022. doi: 10.1073/pnas.1015950108
Espino, J., Bejarano, I., Redondo, P. C., Rosado, J. A., Barriga, C., Reiter, R. J., et al. (2010). Melatonin reduces apoptosis induced by calcium signaling in human leukocytes: evidence for the involvement of mitochondria and Bax activation. J. Membr. Biol. 233, 105–118. doi: 10.1007/s00232-010-9230-0
Fang, E. F., Hou, Y., Palikaras, K., Adriaanse, B. A., Kerr, J. S., Yang, B., et al. (2019). Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 22, 401–412. doi: 10.1038/s41593-018-0332-9
Feng, Z., Zhang, J.-T. (2004). Protective effect of melatonin on beta-amyloid-induced apoptosis in rat astroglioma C6 cells and its mechanism. Free Radic. Biol. Med. 37, 1790–1801. doi: 10.1016/j.freeradbiomed.2004.08.023
Filadi, R., Greotti, E., Turacchio, G., Luini, A., Pozzan, T., Pizzo, P. (2016). Presenilin 2 modulates endoplasmic reticulum–mitochondria coupling by tuning the antagonistic effect of mitofusin 2. Cell. Rep. 15, 2226–2238. doi: 10.1016/j.celrep.2016.05.013
Francis, P. T. (2005). The interplay of neurotransmitters in Alzheimer’s disease. CNS Spectr. 10, 6–9. doi: 10.1017/S1092852900014164
Fu, A.-L., Dong, Z.-H., Sun, M.-J. (2006). Protective effect of N-acetyl-L-cysteine on amyloid beta-peptide-induced learning and memory deficits in mice. Brain Res. 1109, 201–206. doi: 10.1016/j.brainres.2006.06.042
Galasko, D. R., Peskind, E., Clark, C. M., Quinn, J. F., Ringman, J. M., Jicha, G. A., et al. (2012). Antioxidants for Alzheimer disease: a randomized clinical trial with cerebrospinal fluid biomarker measures. Arch. Neurol. 69, 836–841. doi: 10.1001/archneurol.2012.85
Gan, X., Huang, S., Wu, L., Wang, Y., Hu, G., Li, G., et al. (2014). Inhibition of ERK-DLP1 signaling and mitochondrial division alleviates mitochondrial dysfunction in Alzheimer’s disease cybrid cell. Biochim. Biophys. Acta 1842, 220–231. doi: 10.1016/j.bbadis.2013.11.009
García-Blanco, A., Baquero, M., Vento, M., Gil, E., Bataller, L., Cháfer-Pericás, C. (2017). Potential oxidative stress biomarkers of mild cognitive impairment due to Alzheimer disease. J. Neurol. Sci. 373, 295–302. doi: 10.1016/j.jns.2017.01.020
Gazit, N., Vertkin, I., Shapira, I., Helm, M., Slomowitz, E., Sheiba, M., et al. (2016). IGF-1 Receptor differentially regulates spontaneous and evoked transmission via mitochondria at hippocampal synapses. Neuron 89, 583–597. doi: 10.1016/j.neuron.2015.12.034
Gerenu, G., Liu, K., Chojnacki, J. E., Saathoff, J. M., Martínez-Martín, P., Perry, G., et al. (2015). Curcumin/melatonin hybrid 5-(4-hydroxy-phenyl)-3-oxo-pentanoic acid [2-(5-methoxy-1H-indol-3-yl)-ethyl]-amide ameliorates AD-like pathology in the APP/PS1 mouse model. ACS Chem. Neurosci. 6, 1393–1399. doi: 10.1021/acschemneuro.5b00082
Gibson, G. E., Shi, Q. (2010). A mitocentric view of Alzheimer’s disease suggests multi-faceted treatments. J. Alzheimers Dis. 20 Suppl 2, S591–S607. doi: 10.3233/JAD-2010-100336
Glater, E. E., Megeath, L. J., Stowers, R. S., Schwarz, T. L. (2006). Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J. Cell Biol. 173, 545–557. doi: 10.1083/jcb.200601067
Godoy, J. A., Lindsay, C. B., Quintanilla, R. A., Carvajal, F. J., Cerpa, W., Inestrosa, N. C. (2017). Quercetin exerts differential neuroprotective effects against H2O2 and Aβ aggregates in hippocampal neurons: the role of mitochondria. Mol. Neurobiol. 54, 7116–7128. doi: 10.1007/s12035-016-0203-x
Gottschalk, W. K., Lutz, M. W., He, Y. T., Saunders, A. M., Burns, D. K., Roses, A. D., et al. (2014). The broad impact of TOM40 on neurodegenerative diseases in aging. J. Parkinsons Dis Alzheimers Dis. 1, 1–25. doi: 10.13188/2376-922X.1000003
Green, D. R., Kroemer, G. (2004). The pathophysiology of mitochondrial cell death. Science 305, 626–629. doi: 10.1126/science.1099320
Grossi, C., Rigacci, S., Ambrosini, S., Ed Dami, T., Luccarini, I., Traini, C., et al. (2013). The polyphenol oleuropein aglycone protects TgCRND8 mice against Aß plaque pathology. PLoS ONE 8, e71702. doi: 10.1371/journal.pone.0071702
Guo, Z. H., Mattson, M. P. (2000). In vivo 2-deoxyglucose administration preserves glucose and glutamate transport and mitochondrial function in cortical synaptic terminals after exposure to amyloid beta-peptide and iron: evidence for a stress response. Exp. Neurol. 166, 173–179. doi: 10.1006/exnr.2000.7497
Hager, K., Kenklies, M., McAfoose, J., Engel, J., Münch, G. (2007). Alpha-lipoic acid as a new treatment option for Alzheimer’s disease—a 48 months follow-up analysis. J. Neural Transm. Suppl. 72, 189–193. doi: 10.1007/978-3-211-73574-9_24
Hagl, S., Heinrich, M., Kocher, A., Schiborr, C., Frank, J., Eckert, G. P. (2014). Curcumin micelles improve mitochondrial function in a mouse model of Alzheimer’s disease. J. Prev. Alzheimers Dis. 1, 80–83. doi: 10.14283/jpad.2014.2
Hansson Petersen, C. A., Alikhani, N., Behbahani, H., Wiehager, B., Pavlov, P. F., Alafuzoff, I., et al. (2008). The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. U.S.A. 105, 13145–13150. doi: 10.1073/pnas.0806192105
Hardy, J. A., Higgins, G. A. (1992). Alzheimer’s disease: the amyloid cascade hypothesis. Science 256, 184–185. doi: 10.1126/science.1566067
Hauptmann, S., Scherping, I., Dröse, S., Brandt, U., Schulz, K. L., Jendrach, M., et al. (2009). Mitochondrial dysfunction: an early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol. Aging 30, 1574–1586. doi: 10.1016/j.neurobiolaging.2007.12.005
Hernández, S. S. S., Sandreschi, P. F., da Silva, F. C., Arancibia, B. A. V., da Silva, R., Gutierres, P. J. B., et al. (2015). What are the benefits of exercise for Alzheimer’s disease? A systematic review of the past 10 years. J. Aging Phys. Act. 23, 659–668. doi: 10.1123/japa.2014-0180
Hirai, K., Aliev, G., Nunomura, A., Fujioka, H., Russell, R. L., Atwood, C. S., et al. (2001). Mitochondrial abnormalities in Alzheimer’s disease. J. Neurosci. 21, 3017–3023. doi: 10.1523/JNEUROSCI.21-09-03017.2001
Huang, D.-S., Yu, Y.-C., Wu, C.-H., Lin, J.-Y. (2017). Protective effects of wogonin against Alzheimer’s disease by inhibition of amyloidogenic pathway. Evid Based Complement Alternat. Med. 2017, 3545169. doi: 10.1155/2017/3545169
Huang, Q., Aluise, C. D., Joshi, G., Sultana, R., St Clair, D. K., Markesbery, W. R., et al. (2010). Potential in vivo amelioration by N-acetyl-L-cysteine of oxidative stress in brain in human double mutant APP/PS-1 knock-in mice: toward therapeutic modulation of mild cognitive impairment. J. Neurosci. Res. 88, 2618–2629. doi: 10.1002/jnr.22422
Im, A.-R., Kim, Y.-H., Uddin, M. R., Lee, H. W., Chae, S. W., Kim, Y. H., et al. (2012). Scutellaria baicalensis extracts and flavonoids protect rat L6 cells from antimycin A-induced mitochondrial dysfunction. Evid Based Complement Alternat. Med. 2012, 517965. doi: 10.1155/2012/517965
Iturria-Medina, Y., Carbonell, F. M., Sotero, R. C., Chouinard-Decorte, F., Evans, A. C., Alzheimer’s Disease Neuroimaging Initiative (2017). Multifactorial causal model of brain (dis)organization and therapeutic intervention: application to Alzheimer’s disease. Neuroimage 152, 60–77. doi: 10.1016/j.neuroimage.2017.02.058
Jang, J.-H., Surh, Y.-J. (2003). Protective effect of resveratrol on beta-amyloid-induced oxidative PC12 cell death. Free Radic. Biol. Med. 34, 1100–1110. doi: 10.1016/S0891-5849(03)00062-5
Jiang, W., Luo, T., Li, S., Zhou, Y., Shen, X.-Y., He, F., et al. (2016). Quercetin protects against okadaic acid-induced injury via MAPK and PI3K/Akt/GSK3β signaling pathways in HT22 hippocampal neurons. PLoS ONE 11, e0152371. doi: 10.1371/journal.pone.0152371
Johnson, A. B., Blum, N. R. (1970). Nucleoside phosphatase activities associated with the tangles and plaques of Alzheimer’s disease: a histochemical study of natural and experimental neurofibrillary tangles. J. Neuropathol. Exp. Neurol. 29, 463–478. doi: 10.1097/00005072-197007000-00009
Johnson, S. C., La Rue, A., Hermann, B. P., Xu, G., Koscik, R. L., Jonaitis, E. M., et al. (2011). The effect of TOMM40 poly-T length on gray matter volume and cognition in middle-aged persons with APOE ε3/ε3 genotype. Alzheimers Dement. 7, 456–465. doi: 10.1016/j.jalz.2010.11.012
Jou, M.-J., Peng, T.-I., Reiter, R. J., Jou, S.-B., Wu, H.-Y., Wen, S.-T. (2004). Visualization of the antioxidative effects of melatonin at the mitochondrial level during oxidative stress-induced apoptosis of rat brain astrocytes. J. Pineal. Res. 37, 55–70. doi: 10.1111/j.1600-079X.2004.00140.x
Kang, I., Chu, C. T., Kaufman, B. A. (2018). The mitochondrial transcription factor TFAM in neurodegeneration: emerging evidence and mechanisms. FEBS Lett. 592, 793–811. doi: 10.1002/1873-3468.12989
Kapogiannis, D., Mattson, M. P. (2011). Disrupted energy metabolism and neuronal circuit dysfunction in cognitive impairment and Alzheimer’s disease. Lancet Neurol. 10, 187–198. doi: 10.1016/S1474-4422(10)70277-5
Karran, E., Mercken, M., De Strooper, B. (2011). The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 10, 698–712. doi: 10.1038/nrd3505
Karstens, A. J., Tussing-Humphreys, L., Zhan, L., Rajendran, N., Cohen, J., Dion, C., et al. (2019). Associations of the Mediterranean diet with cognitive and neuroimaging phenotypes of dementia in healthy older adults. Am. J. Clin. Nutr. 109, 361–368. doi: 10.1093/ajcn/nqy275
Kashiwaya, Y., Bergman, C., Lee, J.-H., Wan, R., King, M. T., Mughal, M. R., et al. (2013). A ketone ester diet exhibits anxiolytic and cognition-sparing properties, and lessens amyloid and tau pathologies in a mouse model of Alzheimer’s disease. Neurobiol. Aging 34, 1530–1539. doi: 10.1016/j.neurobiolaging.2012.11.023
Kaur, N., Dhiman, M., Perez-Polo, J. R., Mantha, A. K. (2015). Ginkgolide B revamps neuroprotective role of apurinic/apyrimidinic endonuclease 1 and mitochondrial oxidative phosphorylation against Aβ25-35 -induced neurotoxicity in human neuroblastoma cells. J. Neurosci. Res. 93, 938–947. doi: 10.1002/jnr.23565
Kelso, G. F., Porteous, C. M., Coulter, C. V., Hughes, G., Porteous, W. K., Ledgerwood, E. C., et al. (2001). Selective targeting of a redox-active ubiquinone to mitochondria within cells: antioxidant and antiapoptotic properties. J. Biol. Chem. 276, 4588–4596. doi: 10.1074/jbc.M009093200
Kim, S., Swaminathan, S., Shen, L., Risacher, S. L., Nho, K., Foroud, T., et al. (2011). Genome-wide association study of CSF biomarkers Abeta1-42, t-tau, and p-tau181p in the ADNI cohort. Neurology 76, 69–79. doi: 10.1212/WNL.0b013e318204a397
Kim, Y. J., Park, J. K., Kang, W. S., Kim, S. K., Han, C., Na, H. R., et al. (2017). Association between mitofusin 2 gene polymorphisms and late-onset Alzheimer’s disease in the Korean population. Psychiatry Investig. 14, 81–85. doi: 10.4306/pi.2017.14.1.81
Klein, C. P., Hoppe, J. B., Saccomori, A. B., Dos Santos, B. G., Sagini, J. P., Crestani, M. S., et al. (2019). Physical exercise during pregnancy prevents cognitive impairment induced by amyloid-β in adult offspring rats. Mol. Neurobiol. 56, 2022–2038. doi: 10.1007/s12035-018-1210-x
Kolaj, I., Imindu Liyanage, S., Weaver, D. F. (2018). Phenylpropanoids and Alzheimer’s disease: a potential therapeutic platform. Neurochem. Int. 120, 99–111. doi: 10.1016/j.neuint.2018.08.001
Kolosova, N. G., Tyumentsev, M. A., Muraleva, N. A., Kiseleva, E., Vitovtov, A. O., Stefanova, N. A. (2017). Antioxidant SkQ1 alleviates signs of Alzheimer’s disease-like pathology in old OXYS rats by reversing mitochondrial deterioration. Curr Alzheimer Res. 14, 1283–1292. doi: 10.2174/1567205014666170621111033
Kook, S.-Y., Lee, K.-M., Kim, Y., Cha, M.-Y., Kang, S., Baik, S. H., et al. (2014). High-dose of vitamin C supplementation reduces amyloid plaque burden and ameliorates pathological changes in the brain of 5XFAD mice. Cell. Death Dis. 5, e1083. doi: 10.1038/cddis.2014.26
Krols, M., Bultynck, G., Janssens, S. (2016). ER–mitochondria contact sites: a new regulator of cellular calcium flux comes into play. J. Cell. Biol. 214, 367–370. doi: 10.1083/jcb.201607124
Kryscio, R. J., Abner, E. L., Caban-Holt, A., Lovell, M., Goodman, P., Darke, A. K., et al. (2017). Association of antioxidant supplement use and dementia in the prevention of Alzheimer’s disease by vitamin E and selenium trial (PREADViSE). JAMA Neurol. 74, 567–573. doi: 10.1001/jamaneurol.2016.5778
Kwong, L. K., Kamzalov, S., Rebrin, I., Bayne, A.-C. V., Jana, C. K., Morris, P., et al. (2002). Effects of coenzyme Q(10) administration on its tissue concentrations, mitochondrial oxidant generation, and oxidative stress in the rat. Free Radic. Biol. Med. 33, 627–638. doi: 10.1016/S0891-5849(02)00916-4
Lagouge, M., Argmann, C., Gerhart-Hines, Z., Meziane, H., Lerin, C., Daussin, F., et al. (2006). Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 127, 1109–1122. doi: 10.1016/j.cell.2006.11.013
Lama, A., Pirozzi, C., Mollica, M. P., Trinchese, G., Di Guida, F., Cavaliere, G., et al. (2017). Polyphenol-rich virgin olive oil reduces insulin resistance and liver inflammation and improves mitochondrial dysfunction in high-fat diet fed rats. Mol. Nutr. Food Res. 61. doi: 10.1002/mnfr.201600418
Lee, J.-H., Yu, W. H., Kumar, A., Lee, S., Mohan, P. S., Peterhoff, C. M., et al. (2010). Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 141, 1146–1158. doi: 10.1016/j.cell.2010.05.008
Li, Z., Okamoto, K.-I., Hayashi, Y., Sheng, M. (2004). The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 119, 873–887. doi: 10.1016/j.cell.2004.11.003
Lim, G. P., Chu, T., Yang, F., Beech, W., Frautschy, S. A., Cole, G. M. (2001). The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. J. Neurosci. 21, 8370–8377. doi: 10.1523/JNEUROSCI.21-21-08370.2001
Lin, M. T., Simon, D. K., Ahn, C. H., Kim, L. M., Beal, M. F. (2002). High aggregate burden of somatic mtDNA point mutations in aging and Alzheimer’s disease brain. Hum. Mol. Genet. 11, 133–145. doi: 10.1093/hmg/11.2.133
Linnertz, C., Anderson, L., Gottschalk, W., Crenshaw, D., Lutz, M. W., Allen, J., et al. (2014). The cis-regulatory effect of an Alzheimer’s disease-associated poly-T locus on expression of TOMM40 and apolipoprotein E genes. Alzheimers Dement. 10, 541–551. doi: 10.1016/j.jalz.2013.08.280
Liu, J., Head, E., Gharib, A. M., Yuan, W., Ingersoll, R. T., Hagen, T. M., et al. (2002). Memory loss in old rats is associated with brain mitochondrial decay and RNA/DNA oxidation: partial reversal by feeding acetyl-L-carnitine and/or R-alpha -lipoic acid. Proc. Natl. Acad. Sci. U.S.A. 99, 2356–2361. doi: 10.1073/pnas.261709299
Livingston, G., Sommerlad, A., Orgeta, V., Costafreda, S. G., Huntley, J., Ames, D., et al. (2017). Dementia prevention, intervention, and care. Lancet 390, 2673–2734. doi: 10.1016/S0140-6736(17)31363-6
Lobos, P., Bruna, B., Cordova, A., Barattini, P., Galáz, J. L., Adasme, T., et al. (2016). Astaxanthin protects primary hippocampal neurons against noxious effects of Aβ-oligomers. Neural Plast. 2016, 3456783. doi: 10.1155/2016/3456783
Long, A. N., Owens, K., Schlappal, A. E., Kristian, T., Fishman, P. S., Schuh, R. A. (2015). Effect of nicotinamide mononucleotide on brain mitochondrial respiratory deficits in an Alzheimer’s disease-relevant murine model. BMC Neurol 15, 19. doi: 10.1186/s12883-015-0272-x
López-Lluch, G., Hunt, N., Jones, B., Zhu, M., Jamieson, H., Hilmer, S., et al. (2006). Calorie restriction induces mitochondrial biogenesis and bioenergetic efficiency. Proc. Natl. Acad. Sci. U.S.A. 103, 1768–1773. doi: 10.1073/pnas.0510452103
Loshchenova, P. S., Sinitsyna, O. I., Fedoseeva, L. A., Stefanova, N. A., Kolosova, N. G. (2015). Influence of antioxidant SkQ1 on accumulation of mitochondrial DNA deletions in the hippocampus of senescence-accelerated OXYS rats. Biochem. Mosc. 80, 596–603. doi: 10.1134/S0006297915050120
Lovell, M. A., Xie, C., Xiong, S., Markesbery, W. R. (2003). Protection against amyloid beta peptide and iron/hydrogen peroxide toxicity by alpha lipoic acid. J. Alzheimers Dis. 5, 229–239. doi: 10.3233/JAD-2003-5306
Lustbader, J. W., Cirilli, M., Lin, C., Xu, H. W., Takuma, K., Wang, N., et al. (2004). ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science 304, 448–452. doi: 10.1126/science.1091230
Manczak, M., Calkins, M. J., Reddy, P. H. (2011). Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: implications for neuronal damage. Hum. Mol. Genet. 20, 2495–2509. doi: 10.1093/hmg/ddr139
Manczak, M., Mao, P., Calkins, M. J., Cornea, A., Reddy, A. P., Murphy, M. P., et al. (2010). Mitochondria-targeted antioxidants protect against amyloid-beta toxicity in Alzheimer’s disease neurons. J. Alzheimers Dis. 20 Suppl 2, S609–S631. doi: 10.3233/JAD-2010-100564
Manczak, M., Reddy, P. H. (2012). Abnormal interaction of VDAC1 with amyloid beta and phosphorylated tau causes mitochondrial dysfunction in Alzheimer's disease. Hum. Mol. Genet. 21 (23), 5131–46. doi: 10.1093/hmg/dds360.
Mangialasche, F., Baglioni, M., Cecchetti, R., Kivipelto, M., Ruggiero, C., Piobbico, D., et al. (2015). Lymphocytic mitochondrial aconitase activity is reduced in Alzheimer’s disease and mild cognitive impairment. J. Alzheimers Dis. 44, 649–660. doi: 10.3233/JAD-142052
Mao, P., Manczak, M., Calkins, M. J., Truong, Q., Reddy, T. P., Reddy, A. P., et al. (2012). Mitochondria-targeted catalase reduces abnormal APP processing, amyloid β production and BACE1 in a mouse model of Alzheimer’s disease: implications for neuroprotection and lifespan extension. Hum. Mol. Genet. 21, 2973–2990. doi: 10.1093/hmg/dds128
Marinelli, L., Fornasari, E., Di Stefano, A., Turkez, H., Arslan, M. E., Eusepi, P., et al. (2017). (R)-α-Lipoyl-Gly-l-Pro-l-Glu dimethyl ester as dual acting agent for the treatment of Alzheimer’s disease. Neuropeptides 66, 52–58. doi: 10.1016/j.npep.2017.09.001
Mark, R. J., Keller, J. N., Kruman, I., Mattson, M. P. (1997). Basic FGF attenuates amyloid beta-peptide-induced oxidative stress, mitochondrial dysfunction, and impairment of Na+/K+-ATPase activity in hippocampal neurons. Brain Res. 756, 205–214. doi: 10.1016/S0006-8993(97)00196-0
Martin, E. R., Lai, E. H., Gilbert, J. R., Rogala, A. R., Afshari, A. J., Riley, J., et al. (2000). SNPing away at complex diseases: analysis of single-nucleotide polymorphisms around APOE in Alzheimer disease. Am. J. Hum. Genet. 67, 383–394. doi: 10.1086/303003
Mattison, J. A., Colman, R. J., Beasley, T. M., Allison, D. B., Kemnitz, J. W., Roth, G.S., et al. (2017). Caloric restriction improves health and survival of rhesus monkeys. Nat. Commun. 8, 14063. doi: 10.1038/ncomms14063
Maurer, I., Zierz, S., Möller, H. J. (2000). A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients. Neurobiol. Aging 21, 455–462. doi: 10.1016/S0197-4580(00)00112-3
McCormack, J. G., Halestrap, A. P., Denton, R. M. (1990). Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol. Rev. 70, 391–425. doi: 10.1152/physrev.1990.70.2.391
McManus, M. J., Murphy, M. P., Franklin, J. L. (2011). The mitochondria-targeted antioxidant MitoQ prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer’s disease. J. Neurosci. 31, 15703–15715. doi: 10.1523/JNEUROSCI.0552-11.2011
Medina, S., Martínez, M., Hernanz, A. (2002). Antioxidants inhibit the human cortical neuron apoptosis induced by hydrogen peroxide, tumor necrosis factor alpha, dopamine and beta-amyloid peptide 1-42. Free Radic. Res. 36, 1179–1184. doi: 10.1080/107157602100006445
Melov, S., Adlard, P. A., Morten, K., Johnson, F., Golden, T. R., Hinerfeld, D., et al. (2007). Mitochondrial oxidative stress causes hyperphosphorylation of tau. PLoS ONE 2, e536. doi: 10.1371/journal.pone.0000536
Mishra, P., Chan, D. C. (2016). Metabolic regulation of mitochondrial dynamics. J. Cell Biol. 212, 379–387. doi: 10.1083/jcb.201511036
Moreira, P. I., Harris, P. L. R., Zhu, X., Santos, M. S., Oliveira, C. R., Smith, M. A., et al. (2007). Lipoic acid and N-acetyl cysteine decrease mitochondrial-related oxidative stress in Alzheimer disease patient fibroblasts. J. Alzheimers Dis. 12, 195–206. doi: 10.3233/JAD-2007-12210
Moreira, P. I., Santos, M. S., Sena, C., Nunes, E., Seiça, R., Oliveira, C. R. (2005). CoQ10 therapy attenuates amyloid beta-peptide toxicity in brain mitochondria isolated from aged diabetic rats. Exp. Neurol. 196, 112–119. doi: 10.1016/j.expneurol.2005.07.012
Morris, G. P., Clark, I. A., Vissel, B. (2018). Questions concerning the role of amyloid-β in the definition, aetiology and diagnosis of Alzheimer’s disease. Acta Neuropathol. 136, 663–689. doi: 10.1007/s00401-018-1918-8
Morselli, E., Mariño, G., Bennetzen, M. V., Eisenberg, T., Megalou, E., Schroeder, S., et al. (2011). Spermidine and resveratrol induce autophagy by distinct pathways converging on the acetylproteome. J. Cell Biol. 192, 615–629. doi: 10.1083/jcb.201008167
Naga, K. K., Geddes, J. W. (2011). Dimebon inhibits calcium-induced swelling of rat brain mitochondria but does not alter calcium retention or cytochrome C release. Neuromolecular Med. 13, 31–36. doi: 10.1007/s12017-010-8130-x
Ng, L. F., Gruber, J., Cheah, I. K., Goo, C. K., Cheong, W. F., Shui, G., et al. (2014). The mitochondria-targeted antioxidant MitoQ extends lifespan and improves healthspan of a transgenic Caenorhabditis elegans model of Alzheimer disease. Free Radic. Biol. Med. 71, 390–401. doi: 10.1016/j.freeradbiomed.2014.03.003
Nisoli, E., Tonello, C., Cardile, A., Cozzi, V., Bracale, R., Tedesco, L., et al. (2005). Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science 310, 314–317. doi: 10.1126/science.1117728
Ochalek, A., Mihalik, B., Avci, H. X., Chandrasekaran, A., Téglási, A., Bock, I., et al. (2017). Neurons derived from sporadic Alzheimer’s disease iPSCs reveal elevated TAU hyperphosphorylation, increased amyloid levels, and GSK3B activation. Alzheimers Res. Ther. 9, 90. doi: 10.1186/s13195-017-0317-z
Oksanen, M., Petersen, A. J., Naumenko, N., Puttonen, K., Lehtonen, Š., Gubert Olivé, M., et al. (2017). PSEN1 mutant iPSC-derived model reveals severe astrocyte pathology in Alzheimer’s disease. Stem Cell. Rep. 9, 1885–1897. doi: 10.1016/j.stemcr.2017.10.016
Omar, S. H., Kerr, P. G., Scott, C. J., Hamlin, A. S., Obied, H. K. (2017). Olive (Olea europaea L.) biophenols: a nutriceutical against oxidative stress in SH-SY5Y cells. Molecules 22, 1858. doi: 10.3390/molecules22111858
Ono, K., Hirohata, M., Yamada, M. (2006). Alpha-lipoic acid exhibits anti-amyloidogenicity for beta-amyloid fibrils in vitro. Biochem. Biophys. Res. Commun. 341, 1046–1052. doi: 10.1016/j.bbrc.2006.01.063
Owen, O. E., Morgan, A. P., Kemp, H. G., Sullivan, J. M., Herrera, M. G., Cahill, G. F. (1967). Brain metabolism during fasting. J. Clin. Invest. 46, 1589–1595. doi: 10.1172/JCI105650
Paillard, T., Rolland, Y., de Souto Barreto, P. (2015). Protective effects of physical exercise in Alzheimer’s disease and Parkinson’s disease: a narrative review. J. Clin. Neurol. 11, 212–219. doi: 10.3988/jcn.2015.11.3.212
Pantano, D., Luccarini, I., Nardiello, P., Servili, M., Stefani, M., Casamenti, F. (2017). Oleuropein aglycone and polyphenols from olive mill waste water ameliorate cognitive deficits and neuropathology. Br. J. Clin. Pharmacol. 83, 54–62. doi: 10.1111/bcp.12993
Pawlosky, R. J., Kemper, M. F., Kashiwaya, Y., King, M. T., Mattson, M. P., Veech, R. L. (2017). Effects of a dietary ketone ester on hippocampal glycolytic and tricarboxylic acid cycle intermediates and amino acids in a 3xTgAD mouse model of Alzheimer’s disease. J. Neurochem. 141, 195–207. doi: 10.1111/jnc.13958
Payton, A., Sindrewicz, P., Pessoa, V., Platt, H., Horan, M., Ollier, W., et al. (2016). A TOMM40 poly-T variant modulates gene expression and is associated with vocabulary ability and decline in nonpathologic aging. Neurobiol. Aging 39, 217.e1–217.e7. doi: 10.1016/j.neurobiolaging.2015.11.017
Peng, T.-I., Hsiao, C.-W., Reiter, R. J., Tanaka, M., Lai, Y.-K., Jou, M.-J. (2012). mtDNA T8993G mutation-induced mitochondrial complex V inhibition augments cardiolipin-dependent alterations in mitochondrial dynamics during oxidative, Ca(2+), and lipid insults in NARP cybrids: a potential therapeutic target for melatonin. J. Pineal. Res. 52, 93–106. doi: 10.1111/j.1600-079X.2011.00923.x
Peng, Y., Hou, C., Yang, Z., Li, C., Jia, L., Liu, J., et al. (2016). Hydroxytyrosol mildly improve cognitive function independent of APP processing in APP/PS1 mice. Mol. Nutr. Food Res. 60, 2331–2342. doi: 10.1002/mnfr.201600332
Perez Ortiz, J. M., Swerdlow, R. H. (2019). Mitochondrial dysfunction in Alzheimer’s disease: role in pathogenesis and novel therapeutic opportunities. Br. J. Pharmacol. 1–19 doi: 10.1111/bph.14585
Persson, T., Popescu, B. O., Cedazo-Minguez, A. (2014). Oxidative stress in Alzheimer’s disease: why did antioxidant therapy fail? Oxid. Med. Cell. Longev. 2014, 427318. doi: 10.1155/2014/427318
Petrosillo, G., Moro, N., Ruggiero, F. M., Paradies, G. (2009). Melatonin inhibits cardiolipin peroxidation in mitochondria and prevents the mitochondrial permeability transition and cytochrome c release. Free Radic. Biol. Med. 47, 969–974. doi: 10.1016/j.freeradbiomed.2009.06.032
Pfanner, N., Warscheid, B., Wiedemann, N. (2019). Mitochondrial proteins: from biogenesis to functional networks. Nat. Rev. Mol. Cell Biol. 20, 267–284. doi: 10.1038/s41580-018-0092-0
Picca, A., Fracasso, F., Pesce, V., Cantatore, P., Joseph, A.-M., Leeuwenburgh, C., et al. (2013). Age- and calorie restriction-related changes in rat brain mitochondrial DNA and TFAM binding. Age (Dordr) 35, 1607–1620. doi: 10.1007/s11357-012-9465-z
Porquet, D., Griñán-Ferré, C., Ferrer, I., Camins, A., Sanfeliu, C., Del Valle, J., et al. (2014). Neuroprotective role of trans-resveratrol in a murine model of familial Alzheimer’s disease. J. Alzheimers Dis. 42, 1209–1220. doi: 10.3233/JAD-140444
Praticò, D., Clark, C. M., Liun, F., Rokach, J., Lee, V. Y.-M., Trojanowski, J. Q. (2002). Increase of brain oxidative stress in mild cognitive impairment: a possible predictor of Alzheimer disease. Arch. Neurol. 59, 972–976. doi: 10.1001/archneur.59.6.972
Puchowicz, M. A., Seyfried, T. N. (2017). Novel ketone body therapy for managing Alzheimer’s disease: an editorial highlight for effects of a dietary ketone ester on hippocampal glycolytic and tricarboxylic acid cycle intermediates and amino acids in a 3xTgAD mouse model of Alzheimer’s disease. J. Neurochem. 141, 162–164. doi: 10.1111/jnc.13979
Qin, W., Haroutunian, V., Katsel, P., Cardozo, C. P., Ho, L., Buxbaum, J. D., et al. (2009). PGC-1alpha expression decreases in the Alzheimer disease brain as a function of dementia. Arch. Neurol. 66, 352–361. doi: 10.1001/archneurol.2008.588
Quinlan, C. L., Perevoshchikova, I. V., Hey-Mogensen, M., Orr, A. L., Brand, M. D. (2013). Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 1, 304–312. doi: 10.1016/j.redox.2013.04.005
Quinn, J. F., Bussiere, J. R., Hammond, R. S., Montine, T. J., Henson, E., Jones, R. E., et al. (2007). Chronic dietary alpha-lipoic acid reduces deficits in hippocampal memory of aged Tg2576 mice. Neurobiol. Aging 28, 213–225. doi: 10.1016/j.neurobiolaging.2005.12.014
Reddy, C. A., Somepalli, V., Golakoti, T., Kanugula, A. K., Karnewar, S., Rajendiran, K., et al. (2014). Mitochondrial-targeted curcuminoids: a strategy to enhance bioavailability and anticancer efficacy of curcumin. PLoS ONE 9, e89351. doi: 10.1371/journal.pone.0089351
Reddy, P. H., Manczak, M., Kandimalla, R. (2017). Mitochondria-targeted small molecule SS31: a potential candidate for the treatment of Alzheimer’s disease. Hum. Mol. Genet. 26, 1597. doi: 10.1093/hmg/ddx129
Reddy, P. H., Manczak, M., Yin, X., Grady, M. C., Mitchell, A., Kandimalla, R., et al. (2016). Protective effects of a natural product, curcumin, against amyloid β induced mitochondrial and synaptic toxicities in Alzheimer’s disease. J. Investig. Med. 64, 1220–1234. doi: 10.1136/jim-2016-000240
Reddy, P. H., Manczak, M., Yin, X., Reddy, A. P. (2018). Synergistic protective effects of mitochondrial division inhibitor 1 and mitochondria-targeted small peptide SS31 in Alzheimer’s disease. J. Alzheimers Dis. 62, 1549–1565. doi: 10.3233/JAD-170988
Reed, T., Perluigi, M., Sultana, R., Pierce, W. M., Klein, J. B., Turner, D. M., et al. (2008). Redox proteomic identification of 4-hydroxy-2-nonenal-modified brain proteins in amnestic mild cognitive impairment: insight into the role of lipid peroxidation in the progression and pathogenesis of Alzheimer’s disease. Neurobiol. Dis. 30, 107–120. doi: 10.1016/j.nbd.2007.12.007
Rege, S. D., Geetha, T., Broderick, T. L., Babu, J. R. (2017). Can diet and physical activity limit Alzheimer’s disease risk? Curr Alzheimer Res. 14, 76–93. doi: 10.2174/1567205013666160314145700
Remington, R., Bechtel, C., Larsen, D., Samar, A., Doshanjh, L., Fishman, P., et al. (2015). A phase II randomized clinical trial of a nutritional formulation for cognition and mood in Alzheimer’s disease. J. Alzheimers Dis. 45, 395–405. doi: 10.3233/JAD-142499
Remington, R., Bechtel, C., Larsen, D., Samar, A., Page, R., Morrell, C., et al. (2016). Maintenance of cognitive performance and mood for individuals with Alzheimer’s disease following consumption of a nutraceutical formulation: a one-year, open-label study. J. Alzheimers Dis. 51, 991–995. doi: 10.3233/JAD-151098
Rhein, V., Giese, M., Baysang, G., Meier, F., Rao, S., Schulz, K. L., et al. (2010). Ginkgo biloba extract ameliorates oxidative phosphorylation performance and rescues abeta-induced failure. PLoS ONE 5, e12359. doi: 10.1371/journal.pone.0012359
Rinaldi, P., Polidori, M. C., Metastasio, A., Mariani, E., Mattioli, P., Cherubini, A., et al. (2003). Plasma antioxidants are similarly depleted in mild cognitive impairment and in Alzheimer’s disease. Neurobiol. Aging 24, 915–919. doi: 10.1016/S0197-4580(03)00031-9
Ringman, J. M., Frautschy, S. A., Teng, E., Begum, A. N., Bardens, J., Beigi, M., et al. (2012). Oral curcumin for Alzheimer’s disease: tolerability and efficacy in a 24-week randomized, double blind, placebo-controlled study. Alzheimers Res. Ther. 4, 43. doi: 10.1186/alzrt146
Rizzuto, R., Pozzan, T. (2006). Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol. Rev. 86, 369–408. doi: 10.1152/physrev.00004.2005
Rosales-Corral, S. A., Lopez-Armas, G., Cruz-Ramos, J., Melnikov, V. G., Tan, D.-X., Manchester, L. C., et al. (2012a). Alterations in lipid levels of mitochondrial membranes induced by amyloid-β: a protective role of melatonin. Int J. Alzheimers Dis. 2012, 459806. doi: 10.1155/2012/459806
Rosales-Corral, S., Acuna-Castroviejo, D., Tan, D. X., López-Armas, G., Cruz-Ramos, J., Munoz, R., et al. (2012b). Accumulation of exogenous amyloid-beta peptide in hippocampal mitochondria causes their dysfunction: a protective role for melatonin. Oxid. Med. Cell. Longev. 2012, 843649. doi: 10.1155/2012/843649
Roses, A. D., Lutz, M. W., Amrine-Madsen, H., Saunders, A. M., Crenshaw, D. G., Sundseth, S. S., et al. (2010). A TOMM40 variable-length polymorphism predicts the age of late-onset Alzheimer’s disease. Pharmacogenomics J. 10, 375–384. doi: 10.1038/tpj.2009.69
Rüb, C., Wilkening, A., Voos, W. (2017). Mitochondrial quality control by the Pink1/Parkin system. Cell. Tissue Res. 367, 111–123. doi: 10.1007/s00441-016-2485-8
Rudnitskaya, E. A., Muraleva, N. A., Maksimova, K. Y., Kiseleva, E., Kolosova, N. G., Stefanova, N. A. (2015). Melatonin attenuates memory impairment, amyloid-β accumulation, and neurodegeneration in a rat model of sporadic Alzheimer’s disease. J. Alzheimers Dis. 47, 103–116. doi: 10.3233/JAD-150161
Rui, Y., Tiwari, P., Xie, Z., Zheng, J. Q. (2006). Acute impairment of mitochondrial trafficking by beta-amyloid peptides in hippocampal neurons. J. Neurosci. 26, 10480–10487. doi: 10.1523/JNEUROSCI.3231-06.2006
Russo, G. J., Louie, K., Wellington, A., Macleod, G. T., Hu, F., Panchumarthi, S., et al. (2009). Drosophila Miro is required for both anterograde and retrograde axonal mitochondrial transport. J. Neurosci. 29, 5443–5455. doi: 10.1523/JNEUROSCI.5417-08.2009
Ryu, D., Mouchiroud, L., Andreux, P. A., Katsyuba, E., Moullan, N., Nicolet-Dit-Félix, A. A., et al. (2016). Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat. Med. 22, 879–888. doi: 10.1038/nm.4132
Sadli, N., Barrow, C. J., McGee, S., Suphioglu, C. (2013). Effect of DHA and coenzymeQ10 against Aβ- and zinc-induced mitochondrial dysfunction in human neuronal cells. Cell. Physiol. Biochem. 32, 243–252. doi: 10.1159/000354433
Sancheti, H., Akopian, G., Yin, F., Brinton, R. D., Walsh, J. P., Cadenas, E. (2013). Age-dependent modulation of synaptic plasticity and insulin mimetic effect of lipoic acid on a mouse model of Alzheimer’s disease. PLoS ONE 8, e69830. doi: 10.1371/journal.pone.0069830
Sanchis-Gomar, F., García-Giménez, J. L., Gómez-Cabrera, M. C., Pallardó, F. V. (2014). Mitochondrial biogenesis in health and disease. molecular and therapeutic approaches. Curr. Pharm. Des. 20, 5619–5633. doi: 10.2174/1381612820666140306095106
Saunders, A. M., Strittmatter, W. J., Schmechel, D., George-Hyslop, P. H., Pericak-Vance, M. A., Joo, S. H., et al. (1993). Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 43, 1467–1472. doi: 10.1212/WNL.43.8.1467
Scarmeas, N., Stern, Y., Tang, M.-X., Mayeux, R., Luchsinger, J. A. (2006). Mediterranean diet and risk for Alzheimer’s disease. Ann. Neurol. 59, 912–921. doi: 10.1002/ana.20854
Scarpulla, R. C. (2011). Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 1813, 1269–1278. doi: 10.1016/j.bbamcr.2010.09.019
Schloesser, A., Esatbeyoglu, T., Piegholdt, S., Dose, J., Ikuta, N., Okamoto, H., et al. (2015). Dietary tocotrienol/γ-cyclodextrin complex increases mitochondrial membrane potential and ATP concentrations in the brains of aged mice. Oxid. Med. Cell. Longev. 2015, 789710. doi: 10.1155/2015/789710
Schroeder, E. K., Kelsey, N. A., Doyle, J., Breed, E., Bouchard, R. J., Loucks, F. A., et al. (2009). Green tea epigallocatechin 3-gallate accumulates in mitochondria and displays a selective antiapoptotic effect against inducers of mitochondrial oxidative stress in neurons. Antioxid. Redox Signal 11, 469–480. doi: 10.1089/ars.2008.2215
Selvaraju, T. R., Khaza’ai, H., Vidyadaran, S., Abd Mutalib, M. S., Vasudevan, R. (2014). The neuroprotective effects of tocotrienol rich fraction and alpha tocopherol against glutamate injury in astrocytes. Bosn. J. Basic Med. Sci. 14, 195–204. doi: 10.17305/bjbms.2014.4.91
Serafini, M. M., Catanzaro, M., Rosini, M., Racchi, M., Lanni, C. (2017). Curcumin in Alzheimer’s disease: can we think to new strategies and perspectives for this molecule? Pharmacol. Res. 124, 146–155. doi: 10.1016/j.phrs.2017.08.004
Shariff, K., Ghosal, S., Matouschek, A. (2004). The force exerted by the membrane potential during protein import into the mitochondrial matrix. Biophys. J. 86, 3647–3652. doi: 10.1529/biophysj.104.040865
Sharma, D. R., Wani, W. Y., Sunkaria, A., Kandimalla, R. J., Sharma, R. K., Verma, D., et al. (2016). Quercetin attenuates neuronal death against aluminum-induced neurodegeneration in the rat hippocampus. Neuroscience 324, 163–176. doi: 10.1016/j.neuroscience.2016.02.055
Sheng, B., Wang, X., Su, B., Lee, H., Casadesus, G., Perry, G., et al. (2012). Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. J. Neurochem. 120, 419–429. doi: 10.1111/j.1471-4159.2011.07581.x
Sheng, Z.-H., Cai, Q. (2012). Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 13, 77–93. doi: 10.1038/nrn3156
Shukla, M., Govitrapong, P., Boontem, P., Reiter, R. J., Satayavivad, J. (2017). Mechanisms of melatonin in alleviating Alzheimer’s disease. Curr. Neuropharmacol. 15, 1010–1031. doi: 10.2174/1570159X15666170313123454
Smith, R. A. J., Porteous, C. M., Gane, A. M., Murphy, M. P. (2003). Delivery of bioactive molecules to mitochondria in vivo. Proc. Natl. Acad. Sci. U.S.A. 100, 5407–5412. doi: 10.1073/pnas.0931245100
Snitz, B. E., O’Meara, E. S., Carlson, M. C., Arnold, A. M., Ives, D. G., Rapp, S. R., et al. (2009). Ginkgo biloba for preventing cognitive decline in older adults: a randomized trial. JAMA 302, 2663–2670. doi: 10.1001/jama.2009.1913
Song, C., Li, M., Xu, L., Shen, Y., Yang, H., Ding, M., et al. (2018). Mitochondrial biogenesis mediated by melatonin in an APPswe/PS1dE9 transgenic mice model. Neuroreport 29, 1517–1524. doi: 10.1097/WNR.0000000000001139
Sood, P. K., Nahar, U., Nehru, B. (2011). Curcumin attenuates aluminum-induced oxidative stress and mitochondrial dysfunction in rat brain. Neurotox Res. 20, 351–361. doi: 10.1007/s12640-011-9249-8
Spilman, P., Podlutskaya, N., Hart, M. J., Debnath, J., Gorostiza, O., Bredesen, D., et al. (2010). Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PLoS ONE 5, e9979. doi: 10.1371/journal.pone.0009979
Stefanova, N. A., Muraleva, N. A., Maksimova, K. Y., Rudnitskaya, E. A., Kiseleva, E., Telegina, D. V., et al. (2016). An antioxidant specifically targeting mitochondria delays progression of Alzheimer’s disease-like pathology. Aging (Albany NY) 8, 2713–2733. doi: 10.18632/aging.101054
Stock, D., Gibbons, C., Arechaga, I., Leslie, A. G., Walker, J. E. (2000). The rotary mechanism of ATP synthase. Curr. Opin. Struct. Biol. 10, 672–679. doi: 10.1016/S0959-440X(00)00147-0
Studer, R., Baysang, G., Brack, C. (2001). N-Acetyl-L-cystein downregulates beta-amyloid precursor protein gene transcription in human neuroblastoma cells. Biogerontology 2, 55–60. doi: 10.1023/A:1010065103073
Subramanian, S., Gottschalk, W. K., Kim, S. Y., Roses, A. D., Chiba-Falek, O. (2017). The effects of PPARγ on the regulation of the TOMM40-APOE-C1 genes cluster. Biochim Biophys Acta Mol Basis Dis 1863, 810–816. doi: 10.1016/j.bbadis.2017.01.004
Sultana, R., Poon, H. F., Cai, J., Pierce, W. M., Merchant, M., Klein, J. B., et al. (2006). Identification of nitrated proteins in Alzheimer’s disease brain using a redox proteomics approach. Neurobiol. Dis. 22, 76–87. doi: 10.1016/j.nbd.2005.10.004
Swerdlow, R. H., Bothwell, R., Hutfles, L., Burns, J. M., Reed, G. A. (2016). Tolerability and pharmacokinetics of oxaloacetate 100 mg capsules in Alzheimer’s subjects. BBA Clin. 5, 120–123. doi: 10.1016/j.bbacli.2016.03.005
Swerdlow, R. H., Burns, J. M., Khan, S. M. (2014). The Alzheimer’s disease mitochondrial cascade hypothesis: progress and perspectives. Biochim. Biophys. Acta 1842, 1219–1231. doi: 10.1016/j.bbadis.2013.09.010
Swerdlow, R. H., Khan, S. M. (2004). A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med. Hypotheses 63, 8–20. doi: 10.1016/j.mehy.2003.12.045
Szeto, H. H. (2006). Cell-permeable, mitochondrial-targeted, peptide antioxidants. AAPS J. 8, E277–E283. doi: 10.1208/aapsj080232
Taguchi, N., Ishihara, N., Jofuku, A., Oka, T., Mihara, K. (2007). Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 282, 11521–11529. doi: 10.1074/jbc.M607279200
Takei, N., Miyashita, A., Tsukie, T., Arai, H., Asada, T., Imagawa, M., et al. (2009). Genetic association study on in and around the APOE in late-onset Alzheimer disease in Japanese. Genomics 93, 441–448. doi: 10.1016/j.ygeno.2009.01.003
Taylor, M. K., Sullivan, D. K., Mahnken, J. D., Burns, J. M., Swerdlow, R. H. (2018). Feasibility and efficacy data from a ketogenic diet intervention in Alzheimer’s disease. Alzheimers Dement. (N Y) 4, 28–36. doi: 10.1016/j.trci.2017.11.002
Taylor, M. K., Swerdlow, R. H., Burns, J. M., Sullivan, D. K. (2019). An experimental ketogenic diet for Alzheimer disease was nutritionally dense and rich in vegetables and avocado. Curr. Dev. Nutr. 3, nzz003. doi: 10.1093/cdn/nzz003
Tian, X., Zhang, L., Wang, J., Dai, J., Shen, S., Yang, L., et al. (2013). The protective effect of hyperbaric oxygen and Ginkgo biloba extract on Aβ25-35-induced oxidative stress and neuronal apoptosis in rats. Behav. Brain Res. 242, 1–8. doi: 10.1016/j.bbr.2012.12.026
Traber, J., Suter, M., Walter, P., Richter, C. (1992). In vivo modulation of total and mitochondrial glutathione in rat liver. Depletion by phorone and rescue by N-acetylcysteine. Biochem. Pharmacol. 43, 961–964. doi: 10.1016/0006-2952(92)90599-E
Tramutola, A., Abate, G., Lanzillotta, C., Triani, F., Barone, E., Iavarone, F., et al. (2018). Protein nitration profile of CD3+ lymphocytes from Alzheimer disease patients: novel hints on immunosenescence and biomarker detection. Free Radic. Biol. Med. 129, 430–439. doi: 10.1016/j.freeradbiomed.2018.10.414
Valenti, D., De Rasmo, D., Signorile, A., Rossi, L., de Bari, L., Scala, I., et al. (2013). Epigallocatechin-3-gallate prevents oxidative phosphorylation deficit and promotes mitochondrial biogenesis in human cells from subjects with Down’s syndrome. Biochim. Biophys. Acta 1832, 542–552. doi: 10.1016/j.bbadis.2012.12.011
Veitch, D. P., Weiner, M. W., Aisen, P. S., Beckett, L. A., Cairns, N. J., Green, R. C., et al. (2019). Understanding disease progression and improving Alzheimer’s disease clinical trials: recent highlights from the Alzheimer’s disease neuroimaging initiative. Alzheimers Dement. 15, 106–152. doi: 10.1016/j.jalz.2018.08.005
Vellas, B., Coley, N., Ousset, P.-J., Berrut, G., Dartigues, J.-F., Dubois, B., et al. (2012). Long-term use of standardised Ginkgo biloba extract for the prevention of Alzheimer’s disease (GuidAge): a randomised placebo-controlled trial. Lancet Neurol. 11, 851–859. doi: 10.1016/S1474-4422(12)70206-5
Verstreken, P., Ly, C. V., Venken, K. J. T., Koh, T.-W., Zhou, Y., Bellen, H. J. (2005). Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron 47, 365–378. doi: 10.1016/j.neuron.2005.06.018
Vidoni, E. D., Johnson, D. K., Morris, J. K., Van Sciver, A., Greer, C. S., Billinger, S. A., et al. (2015). Dose-response of aerobic exercise on cognition: a community-based, pilot randomized controlled trial. PLoS ONE 10, e0131647. doi: 10.1371/journal.pone.0131647
Vinceti, M., Mandrioli, J., Borella, P., Michalke, B., Tsatsakis, A., Finkelstein, Y. (2014). Selenium neurotoxicity in humans: bridging laboratory and epidemiologic studies. Toxicol. Lett. 230, 295–303. doi: 10.1016/j.toxlet.2013.11.016
Voos, W. (2013). Chaperone-protease networks in mitochondrial protein homeostasis. Biochim. Biophys. Acta 1833, 388–399. doi: 10.1016/j.bbamcr.2012.06.005
Wang, C.-F., Song, C.-Y., Wang, X., Huang, L.-Y., Ding, M., Yang, H., et al. (2019). Protective effects of melatonin on mitochondrial biogenesis and mitochondrial structure and function in the HEK293-APPswe cell model of Alzheimer’s disease. Eur. Rev. Med. Pharmacol. Sci. 23, 3542–3550. doi: 10.26355/eurrev_201904_17723
Wang, D.-M., Li, S.-Q., Wu, W.-L., Zhu, X.-Y., Wang, Y., Yuan, H.-Y. (2014). Effects of long-term treatment with quercetin on cognition and mitochondrial function in a mouse model of Alzheimer’s disease. Neurochem. Res. 39, 1533–1543. doi: 10.1007/s11064-014-1343-x
Wang, H., Jiang, T., Li, W., Gao, N., Zhang, T. (2018). Resveratrol attenuates oxidative damage through activating mitophagy in an in vitro model of Alzheimer’s disease. Toxicol. Lett. 282, 100–108. doi: 10.1016/j.toxlet.2017.10.021
Wang, J., Markesbery, W. R., Lovell, M. A. (2006). Increased oxidative damage in nuclear and mitochondrial DNA in mild cognitive impairment. J. Neurochem. 96, 825–832. doi: 10.1111/j.1471-4159.2005.03615.x
Wang, W., Yin, J., Ma, X., Zhao, F., Siedlak, S. L., Wang, Z., et al. (2017). Inhibition of mitochondrial fragmentation protects against Alzheimer’s disease in rodent model. Hum. Mol. Genet. 26, 4118–4131. doi: 10.1093/hmg/ddx299
Wang, X., Hu, X., Yang, Y., Takata, T., Sakurai, T. (2016). Nicotinamide mononucleotide protects against β-amyloid oligomer-induced cognitive impairment and neuronal death. Brain Res. 1643, 1–9. doi: 10.1016/j.brainres.2016.04.060
Wang, X., Su, B., Fujioka, H., Zhu, X. (2008a). Dynamin-like protein 1 reduction underlies mitochondrial morphology and distribution abnormalities in fibroblasts from sporadic Alzheimer’s disease patients. Am. J. Pathol. 173, 470–482. doi: 10.2353/ajpath.2008.071208
Wang, X., Su, B., Siedlak, S. L., Moreira, P. I., Fujioka, H., Wang, Y., et al. (2008b). Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc. Natl. Acad. Sci. U.S.A. 105, 19318–19323. doi: 10.1073/pnas.0804871105
Wang, X., Su, B., Lee, H., Li, X., Perry, G., Smith, M. A., et al. (2009). Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 29, 9090–9103. doi: 10.1523/JNEUROSCI.1357-09.2009
Weisová, P., Alvarez, S. P., Kilbride, S. M., Anilkumar, U., Baumann, B., Jordán, J., et al. (2013). Latrepirdine is a potent activator of AMP-activated protein kinase and reduces neuronal excitability. Transl Psychiatry 3, e317. doi: 10.1038/tp.2013.92
Werth, J. L., Thayer, S. A. (1994). Mitochondria buffer physiological calcium loads in cultured rat dorsal root ganglion neurons. J. Neurosci. 14, 348–356. doi: 10.1523/JNEUROSCI.14-01-00348.1994
Wilkins, H. M., Morris, J. K. (2017). New therapeutics to modulate mitochondrial function in neurodegenerative disorders. Curr. Pharm. Des. 23, 731–752. doi: 10.2174/1381612822666161230144517
Wilkins, H. M., Harris, J. L., Carl, S. M., L, E., Lu, J., Eva Selfridge, J., et al. (2014). Oxaloacetate activates brain mitochondrial biogenesis, enhances the insulin pathway, reduces inflammation and stimulates neurogenesis. Hum. Mol. Genet. 23, 6528–6541. doi: 10.1093/hmg/ddu371
Wojsiat, J., Prandelli, C., Laskowska-Kaszub, K., Martín-Requero, A., Wojda, U. (2015). Oxidative stress and aberrant cell cycle in Alzheimer’s disease lymphocytes: diagnostic prospects. J. Alzheimers Dis. 46, 329–350. doi: 10.3233/JAD-141977
Xue, Z., Guo, Y., Fang, Y. (2016). Moderate activation of autophagy regulates the intracellular calcium ion concentration and mitochondrial membrane potential in beta-amyloid-treated PC12 cells. Neurosci. Lett. 618, 50–57. doi: 10.1016/j.neulet.2016.02.044
Yao, J., Chen, S., Mao, Z., Cadenas, E., Brinton, R. D. (2011). 2-Deoxy-D-glucose treatment induces ketogenesis, sustains mitochondrial function, and reduces pathology in female mouse model of Alzheimer’s disease. PLoS ONE 6, e21788. doi: 10.1371/journal.pone.0021788
Yao, J., Irwin, R. W., Zhao, L., Nilsen, J., Hamilton, R. T., Brinton, R. D. (2009). Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 106, 14670–14675. doi: 10.1073/pnas.0903563106
Yi, M., Weaver, D., Hajnóczky, G. (2004). Control of mitochondrial motility and distribution by the calcium signal: a homeostatic circuit. J. Cell Biol. 167, 661–672. doi: 10.1083/jcb.200406038
Yokoyama, H., Okazaki, K., Imai, D., Yamashina, Y., Takeda, R., Naghavi, N., et al. (2015). The effect of cognitive-motor dual-task training on cognitive function and plasma amyloid β peptide 42/40 ratio in healthy elderly persons: a randomized controlled trial. BMC Geriatr 15, 60. doi: 10.1186/s12877-015-0058-4
Youle, R. J., Narendra, D. P. (2011). Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 12, 9–14. doi: 10.1038/nrm3028
Zhang, S., Hedskog, L., Petersen, C. A. H., Winblad, B., Ankarcrona, M. (2010). Dimebon (latrepirdine) enhances mitochondrial function and protects neuronal cells from death. J. Alzheimers Dis. 21, 389–402. doi: 10.3233/JAD-2010-100174
Zhang, Z.-X., Li, Y.-B., Zhao, R.-P. (2017). Epigallocatechin gallate attenuates β-amyloid generation and oxidative stress involvement of PPARγ in N2a/APP695 cells. Neurochem. Res. 42, 468–480. doi: 10.1007/s11064-016-2093-8
Keywords: Alzheimer disease, therapeutic strategy, mitochondria, mitochondrial dysfunction, mitochondrial therapy
Citation: Cenini G and Voos W (2019) Mitochondria as Potential Targets in Alzheimer Disease Therapy: An Update. Front. Pharmacol. 10:902. doi: 10.3389/fphar.2019.00902
Received: 10 April 2019; Accepted: 18 July 2019;
Published: 23 August 2019.
Edited by:
Cesare Mancuso, Catholic University of the Sacred Heart, ItalyReviewed by:
Amandine Grimm, University of Basel, SwitzerlandCopyright © 2019 Cenini and Voos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giovanna Cenini, Z2NlbmluaUBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.