 Ran Li
Ran Li Yingguang Shan
Yingguang Shan Lu Gao
Lu Gao Xi Wang
Xi Wang Xule Wang
Xule Wang Fang Wang
Fang Wang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 05 June 2019
Sec. Cardiovascular and Smooth Muscle Pharmacology
Volume 10 - 2019 | https://doi.org/10.3389/fphar.2019.00537
The Glp-1 analog, liraglutide (Lir), has been shown to reduce infarct size and improve cardiac function after myocardial ischemia in rodents with or without diabetes. However, the effect of Lir on angiotensin II (AngII) and pressure overload induced cardiac hypertrophy in nondiabetic mice and the underlying mechanisms are unclear. The aim of this study was to investigate the effect of Lir on cardiac hypertrophy induced by AngII infusion and pressure overload and to explore its possible mechanism. Mice were subjected to AngII as well as thoracic aorta coarctation (TAC) to induce a cardiac hypertrophy model. Mice were daily injected with either liraglutide or saline for 2 weeks after AngII infusion. Mice were also subjected to either liraglutide or saline for 25 days after TAC surgery. Neonatal rat cardiomyocytes and human AC cell lines were stimulated with AngII to induce a cardiomyocytes hypertrophy model. The results indicated Lir significantly inhibited cardiac hypertrophy and fibrosis and improved cardiac function in both the AngII and pressure overload induced model. The in vitro study showed that Lir inhibits AngII induced cell hypertrophy. Mechanistically, Lir directly suppressing the activation of PI3K/Akt1 and stimulated AMPKα signaling pathways in cardiomyocytes, which was confirmed by use of an mTOR activator (MHY1485), overexpression of constitutively active Akt, and the knockdown of AMPKa2 expression. Moreover, the protective effects of Lir were lost in AMPKa2 knockout mice. Taken together, Lir inhibits AngII and pressure overload induced cardiac remodeling via regulating PI3K/Akt1 and AMPKα signaling.
Cardiac hypertrophy is an independent risk factor contributing to the development of and mortality in cases of cardiovascular diseases (Ferrario, 2016). It may also occur as a complication associated with hypertension, valvular heart diseases, acute myocardial infarction, and congenital heart diseases (Ma et al., 2018). Pathological cardiac remodeling is initially invoked to reduce ventricular wall stress and temporally preserve cardiac pump function, eventually evolving into a devastating spiral of maladaptive alterations that culminate in HF and death (Wu et al., 2017). The signaling mechanisms involved in the remodeling response are complex, including phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt), Ca2+/calcinerin, nuclear factor of activated T cells (NFAT), nuclear factor-kappa B (NF-κB), and mitogen-activated protein kinase (MAPKs) signaling. However, the molecular mechanisms have not been clearly elucidated, and its treatment is still facing a bottleneck. A better understanding of the factors that regulate remodeling pathways could reveal potential therapeutic targets for treating cardiac hypertrophy and heart failure.
Glucagon-likepeptide1 (GLP-1), an incretin hormone mainly derived from intestinal L-cells, stimulates insulin secretion, inhibits glucagon secretion, delays gastric emptying and reduces postprandial hyperglycemia (Dai et al., 2013; Kawatani et al., 2018). GLP-1 agonists and dipeptidyl peptidase-4 (DPP-4) inhibitors (that prevent the degradation of the GLP-1) have been approved for the treatment of type 2 diabetes (Trujillo and Nuffer, 2014). GLP1 and its analogs have also been demonstrated to exert cardioprotective actions in a few ischemia/reperfusion models (Ban et al., 2008; Noyan-Ashraf et al., 2009; Timmers et al., 2009). GLP1 analogs have been reported to reverse the molecular pathology and cardiovascular dysfunction in obese mice (Noyan-Ashraf et al., 2013). In 2009, Noyan-Ashraf et al. (2009) reported that the GLP-1R agonist liraglutide activates cytoprotective pathways and improves outcomes after experimental myocardial infarction in mice. Moreover, a Glp-1 analog, liraglutide, ameliorated hepatic steatosis, and cardiac hypertrophy in C57BL/6J mice fed a western diet (Mells et al., 2012). However, Kyhl et al. (2017) revealed that 4-week treatment with liraglutide did not affect cardiac remodeling following a nonreperfused myocardial infarction. These inconsistent results have caused confusion in regards to the usage of Lir in cardiovascular disease.
Although some of the studies have indicated GLP-1 agonists could reduce infarct size and improve cardiac function after myocardial ischemia in rodents with or without diabetes, the relative effects of these agents on cardiac remodeling in nondiabetic mice have not been studied. Furthermore, the underlying mechanisms of these agents in cardioprotection have not been completely delineated. Since Lir exerts cardioprotection in many other cardiovascular diseases, we assume Lir may be beneficial in cardiac hypertrophy, and thus, we used AngII and pressure loading to induce a cardiac remodeling model.
All of the studies were performed in accordance with the guidelines of the NIH (Guide for the Care and Use of Laboratory Animals, 1996) and were approved by the Animal Care and Use Committee of the First Affiliated Hospital of Zhenzhou University. A mouse cardiac hypertrophy model was induced by Ang II infusion. Ang II (1.4 mg/kg per day and dissolved in 0.9% NaCl) was subcutaneously infused for 4 weeks using an osmotic minipump (Alzet model 2004, Alza Corp.) implanted into each mouse. Saline-infused animals served as infusion controls and were subjected to the same procedures as the experimental animals, with the exception of the Ang II infusion. To investigate whether Lir can ameliorate cardiac hypertrophy, mice were randomized to receive intraperitoneal injections of Lir (200 μg/kg, dissolved in saline (0.9% NaCl), Novo Nordisk, Princeton, NJ, United States) or saline daily for 2 weeks after 2 weeks of AngII infusion when cardiac hypertrophy can be significantly documented. All of the mice (C57BL6J wild type mice) used in our studies were 8–10 week old males with a BW of 23.5–27.5 g, and the common diet for rodent intervention studies was used.
Adenosine 5′-monophosphate (AMP)-activated protein kinase (AMPKa)2 knockout mice (with a C57BL6J background) were purchased from Cyagen Biosciences Inc. (Guangzhou, China).
Thoracic aorta coarctation was performed as described in a previous study (Xiao et al., 2017). Mice were subjected to Lir (200 μg/kg, dissolved in saline, Novo Nordisk, Princeton, NJ, United States) from 3 days after TAC surgery until 28 days after surgery. The mice were sacrificed 4 weeks after TAC surgery.
After the mice were killed, their hearts were dissected and weighed to compare the heart weight/body weight (HW/BW, mg/g), heart weight/tibial length (HW/TL, mg/mm), and lung weight/body weight (LW/BW, mg/g) ratios.
Mice were anesthetized with pentobarbital sodium (6 mg/kg, i.p.) for cardiac echocardiography using an ultrasound machine (Vivid 7, GE Medical System, Milwaukee, WI, United States). The left ventricular ejection fraction (LVEF), interventricular septal thickness at diastole (IVSD), LVESd, and LVEDd were calculated from the M-mode recording. Hemodynamics were measured in anesthetized (1.5% isoflurane) mice using cardiac catheterization. A microtip catheter transducer (SPR-839; Millar Instruments, Houston, TX, United States) was inserted into the right carotid artery and then advanced into the LV. Fifteen minutes after stabilization, pressure signals and the heart rate were continuously recorded with a Millar Pressure-Volume System (MPVS-400; Millar Instruments) coupled with a Powerlab/4SP A/D converter and then stored and displayed on a personal computer. Data were processed using the PVAN data analysis software.
Total RNA was extracted using the TRIzol reagent as previously described (Liu et al., 2015) and reverse transcribed. The first-strand cDNA was used for quantitative real-time PCR to quantify mRNA expression using GAPDH as the normalized control. The promoters used are listed in Table 1.

Table 1. Primer sequences used for RT-PCR.
Heart tissues and cardiomyocytes were lysed in RIPA buffer. Proteins were isolated as previously described (Liu et al., 2015). Briefly, the left ventricles were polished and the cardiomyocytes were lysed in RIPA buffer, and then, the protein concentrations were measured using the BCA Protein Assay Kit (Thermo, 23227) and an ELISA Reader (Synergy HT, Bio-Tek). The cell lysates (50 mg) were loaded into each lane and subjected to SDS-PAGE, and the proteins were then transferred onto Immobilon-FL membranes (Millipore, IPFL00010). The membranes were incubated overnight at 4°C with primary antibodies against one of the following proteins: phosphorylated (p-) and total (T) PI3K, Akt1, AMPKα, AMPKα, mammalian target of rapamycin (mTOR), ribosomal protein S6 kinase beta-1 (70S6K), S6, extracellular regulated protein kinases (ERK)1/2, NFAT2 and GAPDH (all of the antibodies were purchased form Cell Signaling Technology and diluted at 1:1000). The blots were scanned using a two-color infrared imaging system (Odyssey, LI-COR). Specific protein expression levels were normalized to the GAPDH protein for the total cell lysates and cytosolic proteins.
Several heart sections (5 μm thick) were prepared as previously described (Liu et al., 2015). The cardiomyocyte histopathology was detected using HE. Single myocytes were measured using a quantitative digital image analysis system (Image-Pro Plus, version 6.0). Between 100 and 200 LV myocytes were outlined in each group. Collagen production was detected using PSR. Between 10 and 20 LV fields were outlined in each heart. All of the results were visualized and photographed via light microscopy (Eclipse8, Nikon, Tokyo, Japan) and the images were analyzed with ImagePro Plus software (Media Cybernetics, Bethesda, MD, United States) to quantify the cardiomyocyte area and the collagen percentage.
The 1- to 2-day-old Sprague-Dawley rats were killed by swift decapitation. Their hearts were quickly removed and only the ventricles were kept. They were washed with PBS three times and incubated with 0.125% trypsin-EDTA (Gibco, 2520-072) for 15 min. They were then enzymatically digested four times for 15 min in 0.125% trypsin-EDTA in PBS. Digestion was stopped by the addition of FBS at a final concentration of 10%. The cells were then centrifuged at 250 g for 8 min and then resuspended in DMEM/F12 (Gibco, C11330) supplemented with 10% FBS. The cells were then incubated for 1–2 h in 100-mm dishes to allow noncardiac myocytes (mainly cardiac fibroblasts) to adhere to the plastic. They were then plated in six well plates at a density of 5 × 105 cells per well with 1% bromodeoxyuridine for 48 h. All of the antibodies used in the Western blotting are the same as those used in the in vivo studies. For immunofluorescence staining, an α-actin antibody (Abcam, diluted at 1:100) was used.
AC16 cells (ATCC, Bethesda, MD, United States) were cultured in DMEM supplemented with 12.5% fetal bovine serum and 1% penicillin-streptomycin. Cells in exponential growth were dissociated with 0.25% trypsin (GIBCO, 25200) and were seeded in six-well culture plates at a seeding density of 1 × 106/well before being incubated for 24 h. Then, the cells were cultured with serum-free DMEM for another 12 h before treatment.
Cells were treated with Lir (25, 50, 100 nM) and AngII (1 μM) for 24 h. Cell viability was detected using CCK-8 assays. The expression levels of ANP, B-type natriuretic peptide (BNP), β-MHC, and α-MHC genes were detected by RT-PCR. The cardiomyocyte cross-sectional area in each group was evaluated by immunofluorescence staining for α-actin. After treatment with Ang II and/or Lir (25, 50, 100 nM) for 24 h, protein levels were detected by using WB assays. For cell transfection, replication-defective adenoviral (Ad) vectors were used to overexpress constitutively active Akt or to knock down AMPKa. The constitutively active Akt1 and NC adenoviral (Ad) vectors used in our study were generated by the Hanbio Biotechnology Co. (Shanghai, China). At 48 h after plating, the cardiomyocytes were infected with adenovirus diluted in DMEM/F12 at 100 MOI. After infection, cardiomyocyte cells were starved with FBS-free medium for 16 h and treated with Ang II and/or Lir for 24 h.
SPSS software, version 13.0, was used for data analysis. All data are expressed as the means ± SE. The Kolmogorov-Smirnov test was used to analyze the data distribution. All data were normally distributed. The groups were compared by one-way analysis of variance (ANOVA) followed by the post hoc LSD test when ANOVA found a significant value of F and no variance in homogeneity; otherwise, Tamhane’s T2 post hoc test was used. Comparisons between the two groups were performed using Student’s unpaired t-test. p < 0.05 indicates a statistical difference.
Mice were subjected to Ang II infusion to induce a cardiac hypertrophy model. Four weeks after the infusion, a significant hypertrophic response was observed as evidenced by an increased heart weight to body weight ratio (HW/BW), HW to tibial length (HW/TL) ratio, increased cross section area, increased transcription levels of hypertrophic markers [ANP, BNP, β-MHC] and a decreased transcription level of α-MHC. After 2 weeks of Lir treatment, these hypertrophic responses were lower than in the Ang II group as shown by decreased HW/BW and HW/TL ratios, a decreased cross section area, decreased transcription levels of hypertrophic markers (ANP, BNP, β-MHC) and an increased transcription level of α-MHC (Figures 1A–D).
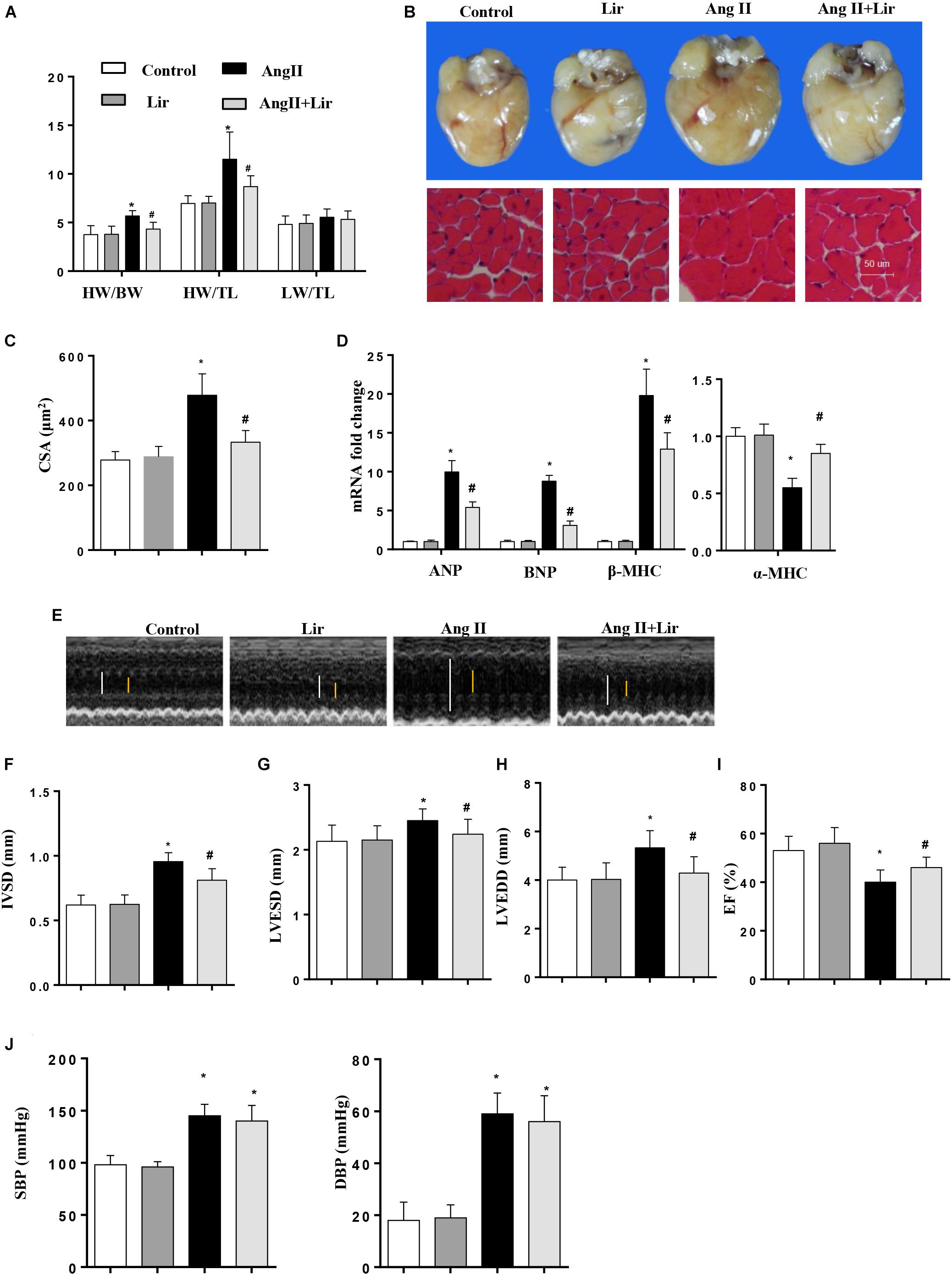
Figure 1. The effect of Lir on AngII-induced cardiac hypertrophy and dysfunction. Statistical results of the HW/BW, HW/TL, and LW/BW ratios (n = 10) at 4 weeks after AngII and/or Lir treatment (A); the gross heart, HE staining (B) and CSA (n = 100 cells per section) (C) at 4 weeks after AngII and/or Lir treatment; RT-PCR analysis of hypertrophic markers including ANP, BNP, β-MHC, and α-MHC from the heart of different groups (n = 9) (D); echocardiography results 4 weeks after AngII and/or Lir treatment (n = 10, white line: LVEDD, yellow line: LVESD) (E–I). Pressure loop measurements of systolic (SBP) and diastolic blood pressure (DBP) in different groups (n = 10 mice/group, J). ∗P < 0.05, compared to the control group; #P < 0.05, compared to the AngII group.
We also detected the cardiac function by echocardiography. Four weeks after AngII infusion, the mice revealed ventricular wall hypertrophy and expansion as well as cardiac dysfunction as evidenced by an increased IVSd, LVEDd and LVESd, and decreased LVEF. Lir treatment ameliorated these changes with reduced IVSD, LVEDD and LVESD, and increases in LVEF compared with the Ang II group (Figures 1E–I). The systolic and diastolic blood pressure (SBP/DBP) were detected after 4 weeks of AngII infusion. SBP and DBP were found to be elevated in the AngII group, and Lir slightly reduced the SBP and DBP (P > 0.05) (Figure 1J). Collectively, Lir attenuates AngII induced cardiac hypertrophy and relieves cardiac dysfunction.
Cardiac fibrosis is one of the main features of cardiac hypertrophy. Thus, we determined the effects of Lir on cardiac fibrosis. PSR staining revealed a significant perivascular and interstitial fibrosis in the AngII infused mice heart while Lir attenuated this fibrosis level (Figures 2A,B). Subsequent analysis of mRNA and protein expression levels of TGF-β1, collagen I, collagen III, and CTGF, which are responsible for cardiac fibrosis, showed similar results (Figures 2C–E). These data suggested that Lir attenuated the cardiac fibrosis induced by Ang II.
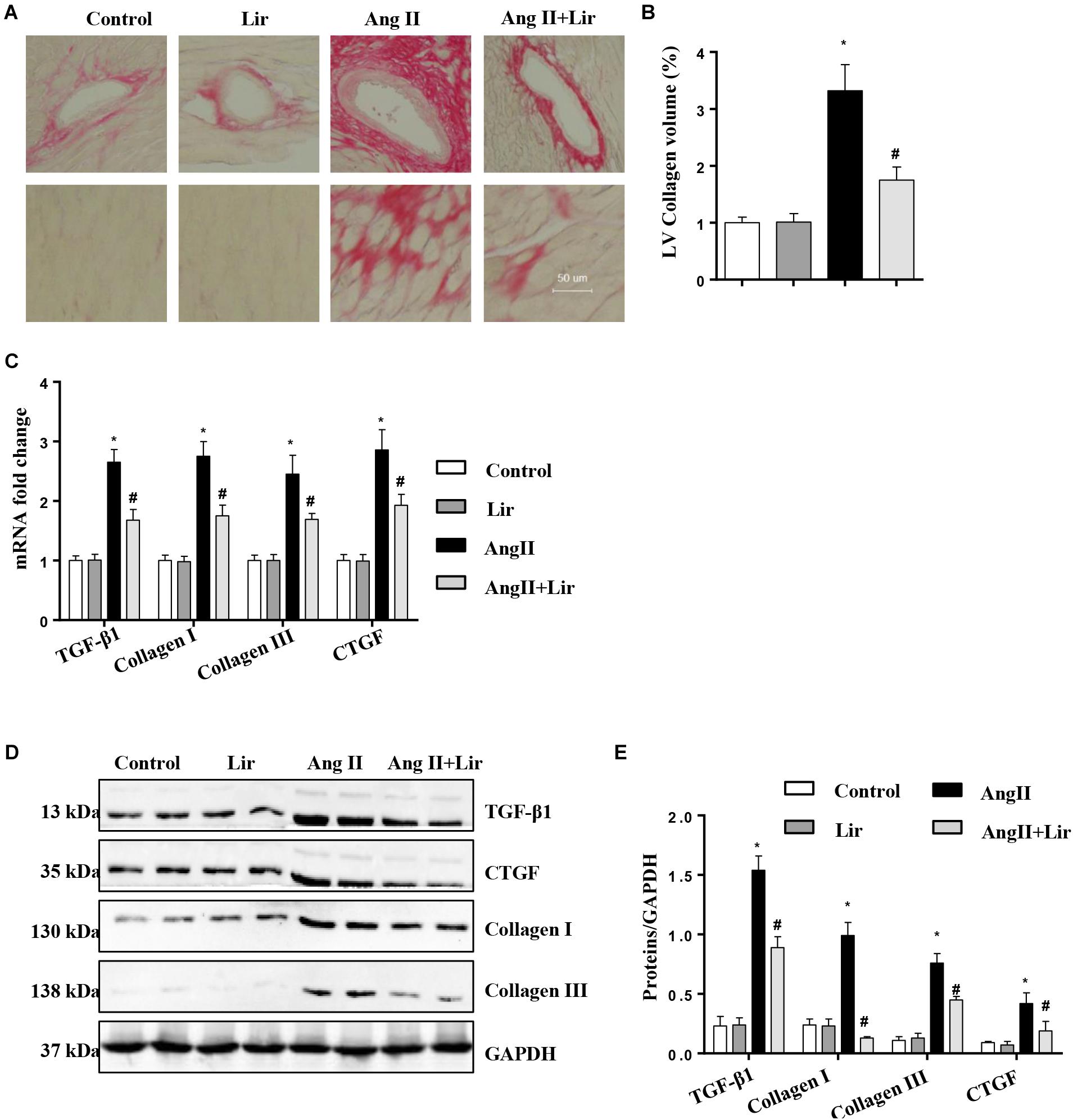
Figure 2. The effect of Lir on AngII-induced cardiac fibrosis. Histological sections of the heart were stained for PSR in the indicated groups at 4 weeks after AngII and/or Lir treatment. Left ventricular interstitial collagen volume fraction in the indicated groups was quantified using an image-analyzing system (A,B, the collagen in LV are shown in red); RT-PCR analysis of the mRNA expression of TGF-β1, collagen I, collagen III, and CTGF in the myocardium obtained from the indicated groups at 4 weeks after AngII and/or Lir treatment (n = 9) (C). Western blot analysis of the expression of TGF-β1, CTGF, collagen I, and collagen III in the myocardium (n = 6) (D,E). ∗P < 0.05, compared to the control group; #P < 0.05, compared to the AngII group.
The molecular mechanism of the anti-hypertrophic effect of Lir was investigated. The main signaling pathway that contributes to cardiac hypertrophy was determined. We found that compared with the control mice, AngII-infusion significantly increased the phosphorylation level of PI3K as well as the phosphorylated level of downstream molecules Akt1, mTOR, p70S6K, and S6 (Figures 3A–C). We also detected the activation of AMPKa was increased in the AngII infused mice heart. After Lir treatment, the activated PI3K/Akt pathway was inhibited compared with that in the AngII group. Furthermore, the increased AMPKa activation induced by AngII was augmented after Lir treatment following AngII treatment (Figures 3A–C).
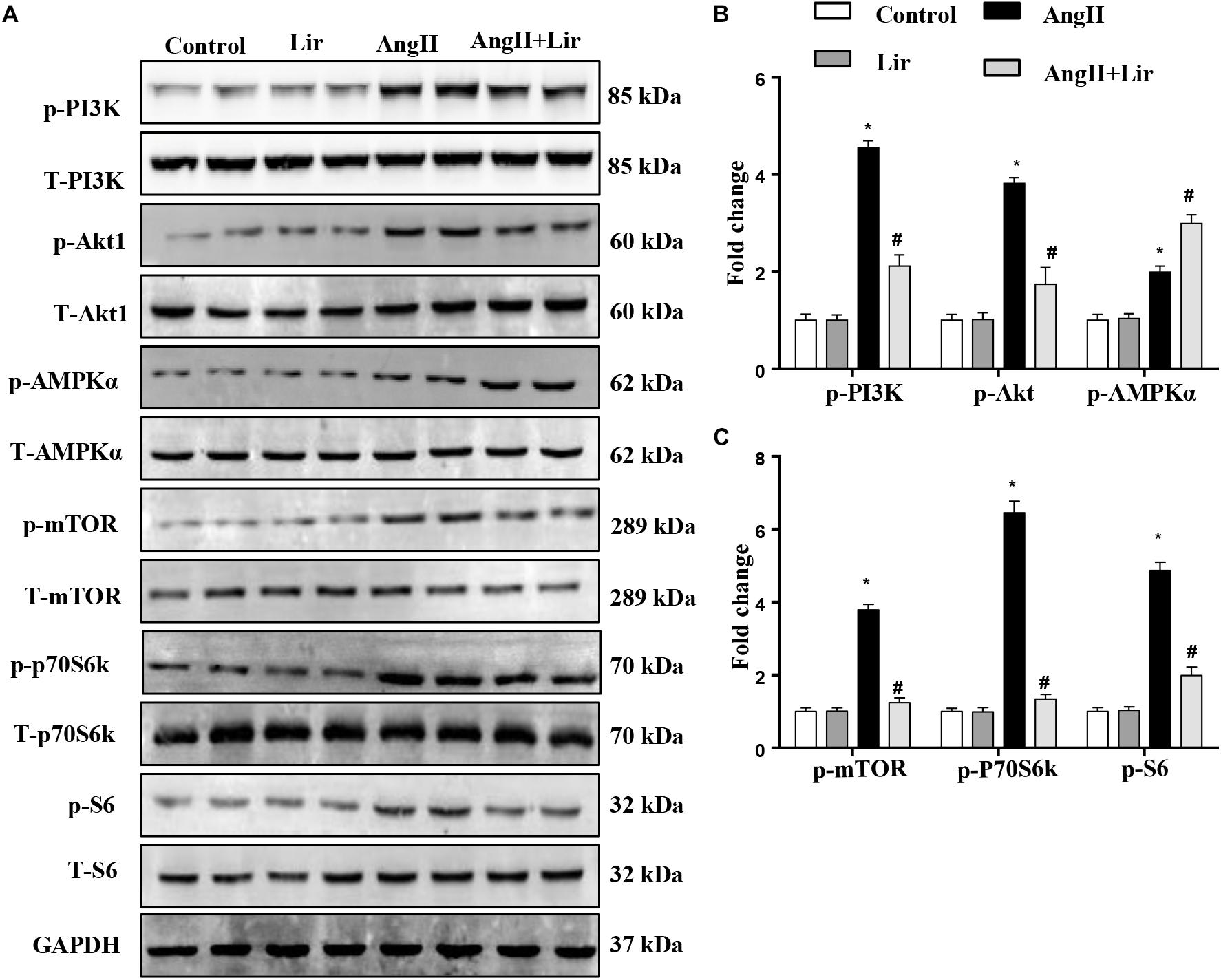
Figure 3. The effect of Lir on the AMPKα and Akt/mTOR/70S6K/S6 signaling pathways in vivo. The protein expression of phosphorylated and total PI3K, Akt, AMPKα, mTOR, 70S6K, and S6 in mice from the four groups. Representative WBs (A) and quantitative results (B,C) (n = 6). ∗P < 0.05, compared to the control group; #P < 0.05, compared to the AngII group.
We also evaluated other hypertrophic signaling pathways such as ERK1/2 and NFAT. The levels of phosphorylated ERK and NFAT2 were increased in the AngII infused mice heart, but Lir did not affect the activation of ERK1/2 and NFAT2 (Supplementary Figures S1A,B). To detect whether the effects of liraglutide were mediated by activation of the GLP-1 receptor, the expression of GLP-1R was detected. As shown in Supplementary Figures S1C,D, GLP-1R expression was increased after stimulation with AngII but was not affected by liraglutide treatment in both NRCMs and ACs. These data suggest that Lir protects against cardiac hypertrophy through regulating PI3K/Akt1 and AMPKα.
We then evaluated whether Lir directly affected cardiomyocytes. Cells were cultured and stimulated with Lir. The cell viability detected by the CCK-8 assay indicated that Lir (25, 50, 100 nM) did not affect cell viability compared with the control group (Figure 4A). AngII (1 μM) treatment induced significant cardiomyocytes hypertrophy as assessed by an increased CSA and increased transcription levels of hypertrophic markers (ANP, β-MHC) compared with that in the control group. However, 50 and 100 nM Lir but not 25 nM Lir greatly inhibited the cardiomyocytes hypertrophic response (a decreased CSA and reduced transcription levels of hypertrophic markers) (Figures 4B–D). These data indicated that Lir protects against cardiac hypertrophy via directly affecting cardiomyocytes.
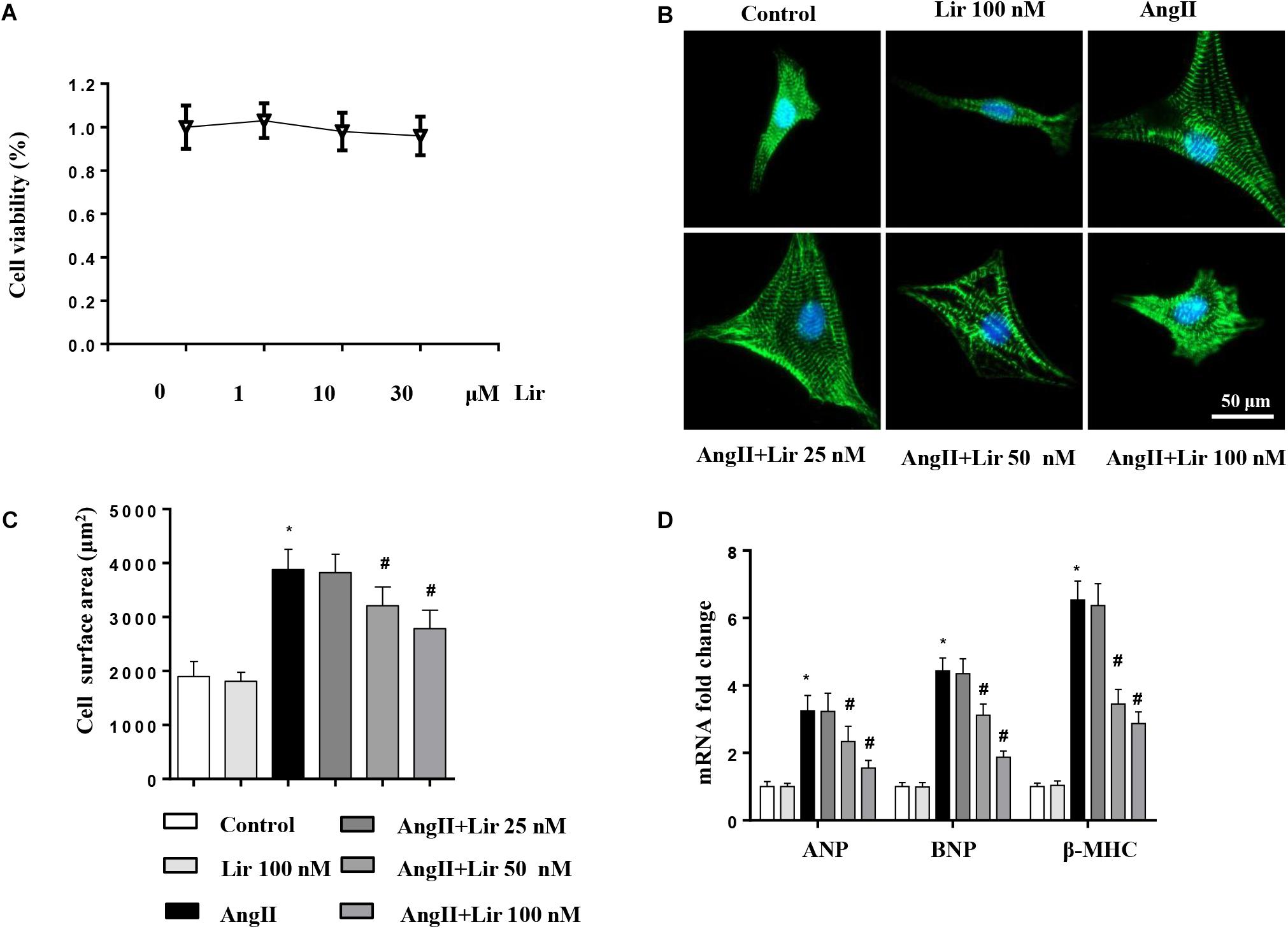
Figure 4. The effect of Lir on cardiomyocytes hypertrophy in vitro. Cardiomyocytes were treated with AngII and/or Lir (25, 50, 100 nM) for 24 h. (A) Cell viability of cardiomyocytes (n = 6). (B) Representative immunofluorescent staining of α-actin (n = 6). (C) Quantitative analysis of CSA (n > 50 cells per group). (D) RT-PCR analysis of the mRNA levels of ANP, BNP, and β-MHC in the indicated groups (n = 6). ∗P < 0.05, compared to the control group; #P < 0.05, compared to the AngII group.
Previous studies have reported that Lir decreased angiotensin type 1 receptor (AT1R) expression and increased angiotensin type 2 receptor (AT2R) expression, thus attenuating AngII-induced tissue fibrosis in rats (Zhang et al., 2015). Thus, we detected the expression patterns of AT1R and AT2R. As shown in Supplementary Figures S1E,F, AngII infusion increased AT1R expression and reduced AT2R expression but Lir did not affect the AT1R/AT2R expression pattern in both the AngII infused heart tissue and the cardiomyocytes. These data indicate that Lir exerts an anti-hypertrophy effect in mice that is not dependent on AT1R/AT2R.
To confirm the PI3K/Akt1 and AMPKa pathways mediated the protective effects of Lir in cardiomyocytes, these signaling pathways were studied in vitro. The activation of PI3K and its downstream targets Akt1, mTOR, p70S6K, and S6 were significantly activated in cardiomyocytes treated with Ang II (1 μM). However, 50 and 100 nM Lir reduced the activation levels of PI3K, Akt1, mTOR, p70S6K, and S6 (Figures 5A–C). We also examined AMPKa signaling. Consistent with the in vivo studies, AMPKa was activated after 30 min of Ang II stimulation, while 50 and 100 nM Lir further increased the activation of AMPKa (Figures 5A–C). These findings indicated that Lir attenuated cell hypertrophy predominantly through inhibiting the Akt1/mTOR signaling pathways and stimulating the AMPKα pathways in cardiomyocytes.
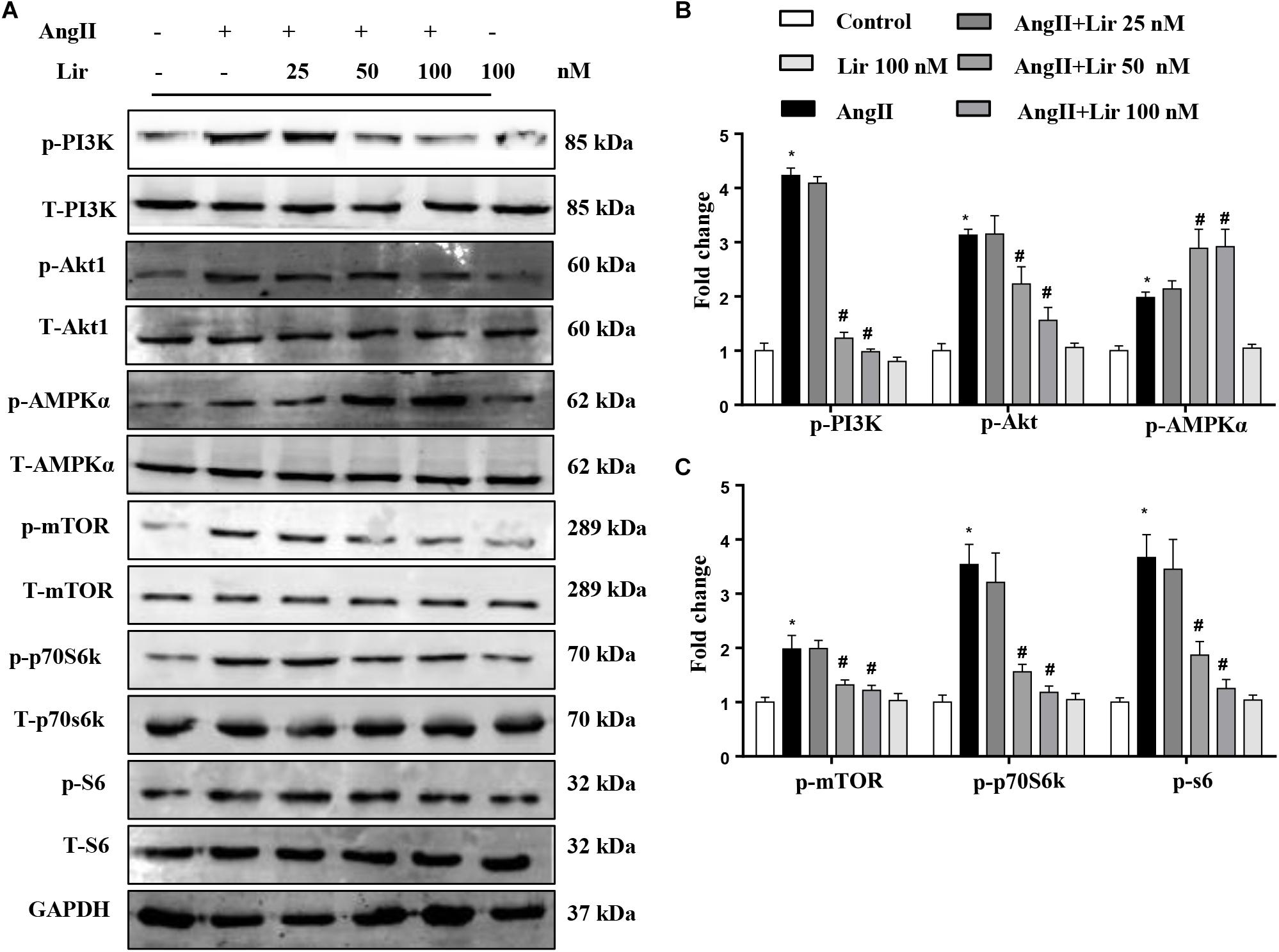
Figure 5. The effect of Lir on AMPKα and Akt/mTOR/70S6K/S6 signaling pathways in response to hypertrophic stimuli in vitro. The protein expression levels of phosphorylated and total PI3K, Akt, AMPKα, mTOR, 70S6K, and S6 in cardiomyocytes treated with AngII (1 μM) and Lir (1, 10, and 30 μM, respectively). Representative WBs (A) and quantitative results (B,C) (n = 6). ∗P < 0.05, compared to the control group; #P < 0.05, compared to the AngII group.
To decipher the effects of Lir on PI3K/Akt1/mTOR and AMPKa signaling, cardiomyocytes were subjected to an mTOR activator (MHY1485, 10 μM, MCE China) or infected with constitutive active Akt1 (Ad.caAkt) to activate Akt1 or Ad-shAMPKa to knockdown AMPKa expression. We found the anti-hypertrophic effect of Lir on cardiomyocytes was abolished by MHY1485, Ad.caAkt, and shAMPKa since the CSA and mRNA expression of hypertrophic markers showed no significant differences among the AngII + Lir + MHY1485/Ad.caAkt/Ad-shAMPKa groups and the AngII group but were increased relative to the AngII + Lir group (Figures 6A–I). These data suggest that both activation of Akt and inhibition of AMPKa could counteract the anti-hypertrophy of Lir in vitro.
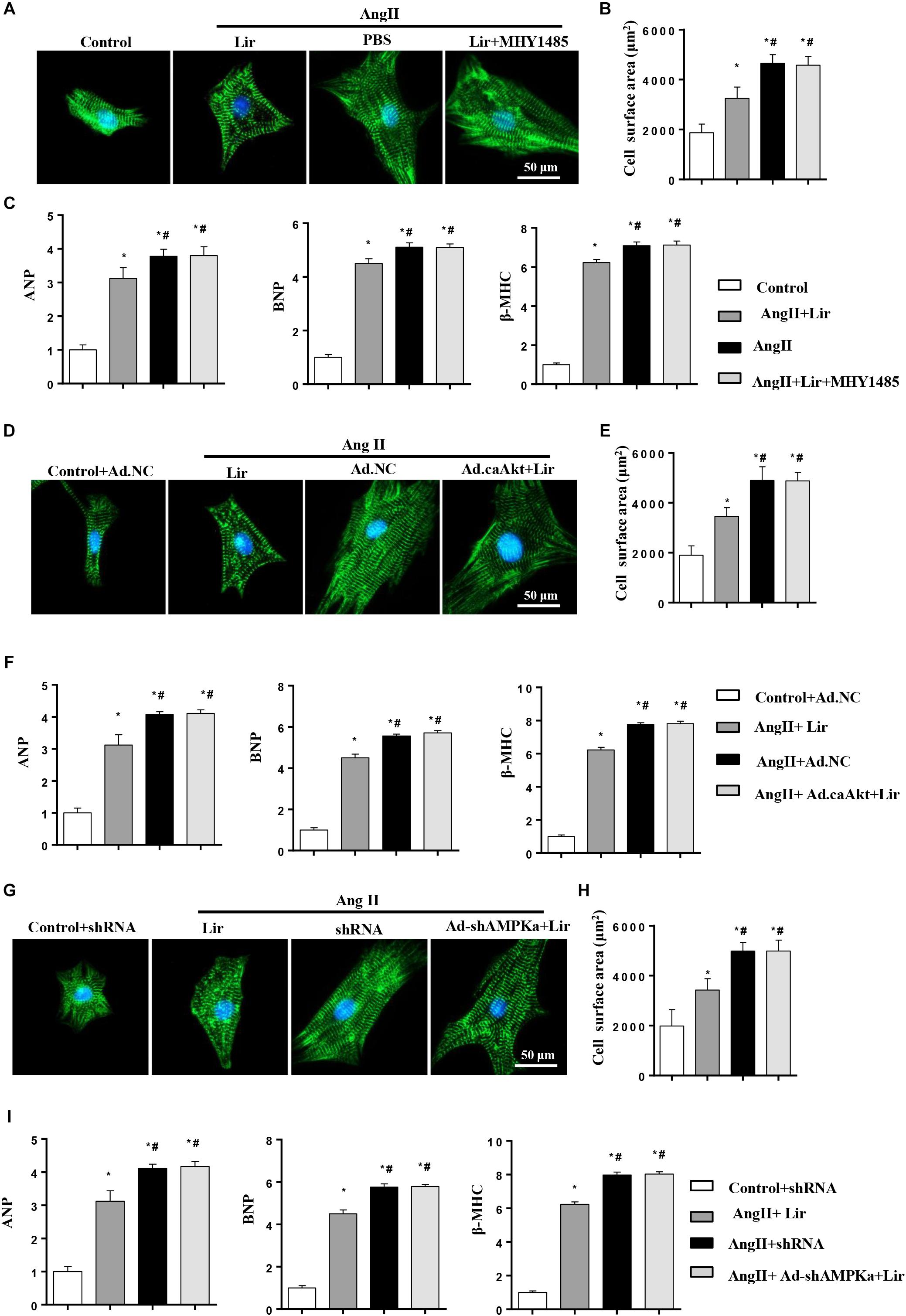
Figure 6. Lir attenuated hypertrophy of myocytes in an AMPKα-dependent manner in vitro. Cardiomyocytes were treated with AngII (1 μM) and Lir (30 μM) with/without MHY1485 (10 μM), Ad.caAkt or Ad-shAMPKa infection. (A,D,G) Representative immunofluorescent staining of α-actin in the indicated group (n = 6). (B,E,H) Quantitative analysis of CSA in the indicated group (n = 50 + cells per group). (C,F,I) RT-PCR analysis of the mRNA levels of ANP, BNP, and β-MHC in the indicated group (n = 6). ∗P < 0.05, compared to the control group (B,C)/Control + Ad.NC (E,F)/Control + shRNA (H,I); #P < 0.05, compared to the AngII group (B,C)/AngII + Ad.NC (E,F)/AngII + shRNA (H,I).
To further investigate whether Lir exerted protective effects through AMPKα in vivo, AMPKa knockout mice were injected with Lir after 2 weeks of AngII infusion. Consistent with the results in vitro, AMPKa knock-out counteracted the protective effects of Lir and was characterized by the same extent of HW/BW, HW/TL, LW to BW ratios, CSA, LV collagen volume, hypertrophic and fibrotic markers transcription levels and the same extent of cardiac dysfunction between Lir treatment in wild type mice and Lir treatment in knockout mice (Figures 7A–K).
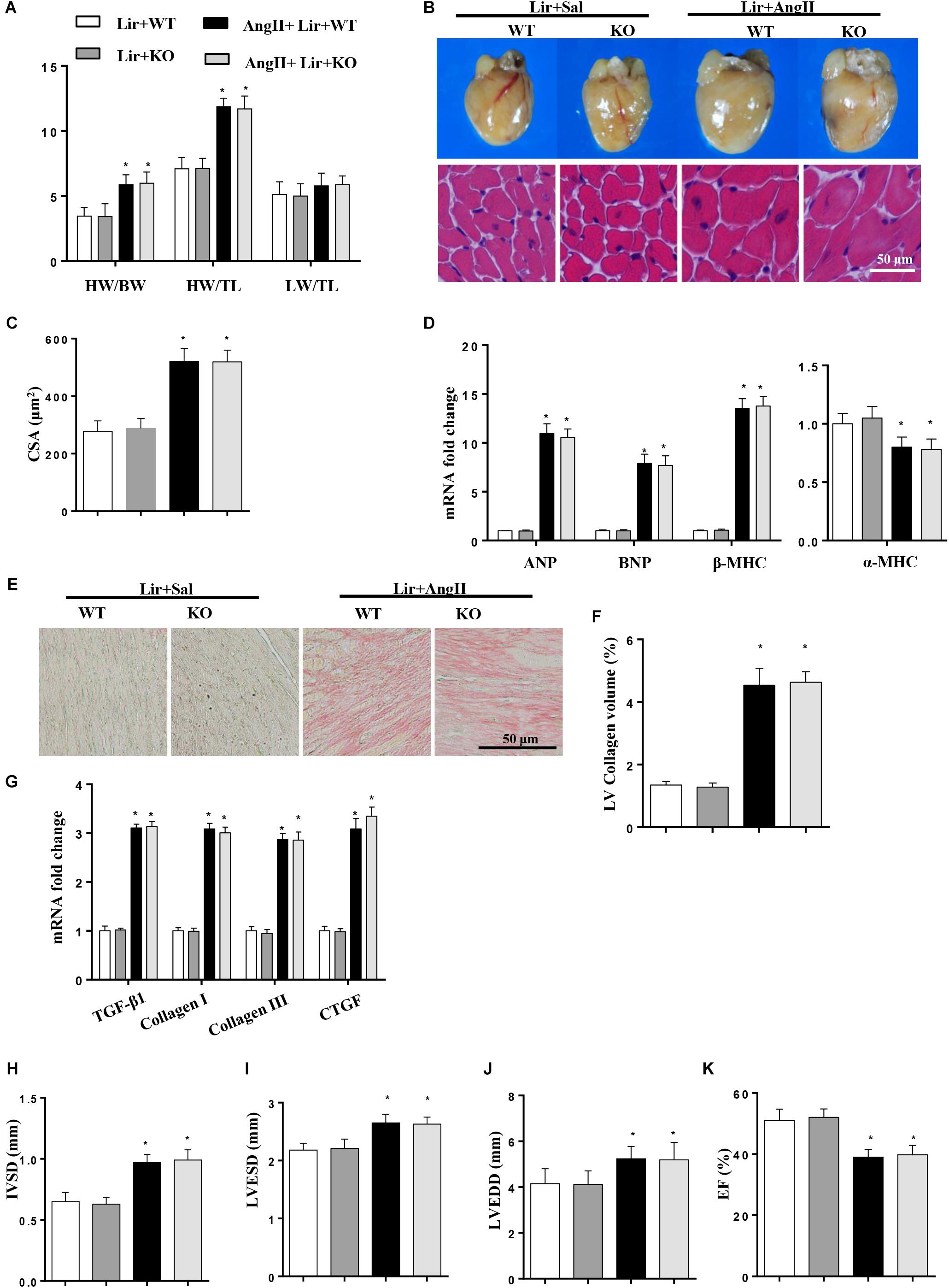
Figure 7. AMPKa deficiency abolished the anti-hypertrophic effects of Lir in vivo. AMPKa knockout mice were injected with Lir 2 weeks after Ang II administration. Statistical results of the HW/BW, HW/TL, and LW/BW ratios (n = 10) at 4 weeks after AngII and Lir treatment (A); the gross heart, HE staining (B) and CSA (n = 100 cells per section) (C) at 4 weeks after AngII infusion; RT-PCR analysis of hypertrophic markers including ANP, BNP, β-MHC, and α-MHC from the heart of different groups (n = 9) (D); Left ventricular interstitial collagen volume fraction in the indicated groups (E,F); RT-PCR analysis of the mRNA expression of TGF-β1, collagen I, collagen III, and CTGF in the myocardium obtained from the indicated groups (n = 9) (G); echocardiography results in the indicated group (H–K, n = 10). ∗P < 0.05, compared to the Lir-WT group; #P < 0.05, compared to the AngII + Lir-WT group.
To confirm the anti-hypertrophy effect of Lir on human cardiomyocytes, AC16 cells were stimulated with Ang II (1 μM) in the presence of Lir (100 nM) and infected with Ad.caAkt to express activated Akt1. Consistent with the results in rat cardiomyocytes, Lir inhibits Ang II induced AC16 cell hypertrophy (a reduced CSA and reduced transcription of ANP and β-MHC) and Ad.caAkt abolished these protective effects of Lir as assessed by the increased CSA and transcription of ANP and β-MHC in the AngII + Ad.caAkt + Lir group compared with that in the AngII group and the AngII + Lir group (Figures 8A–E).
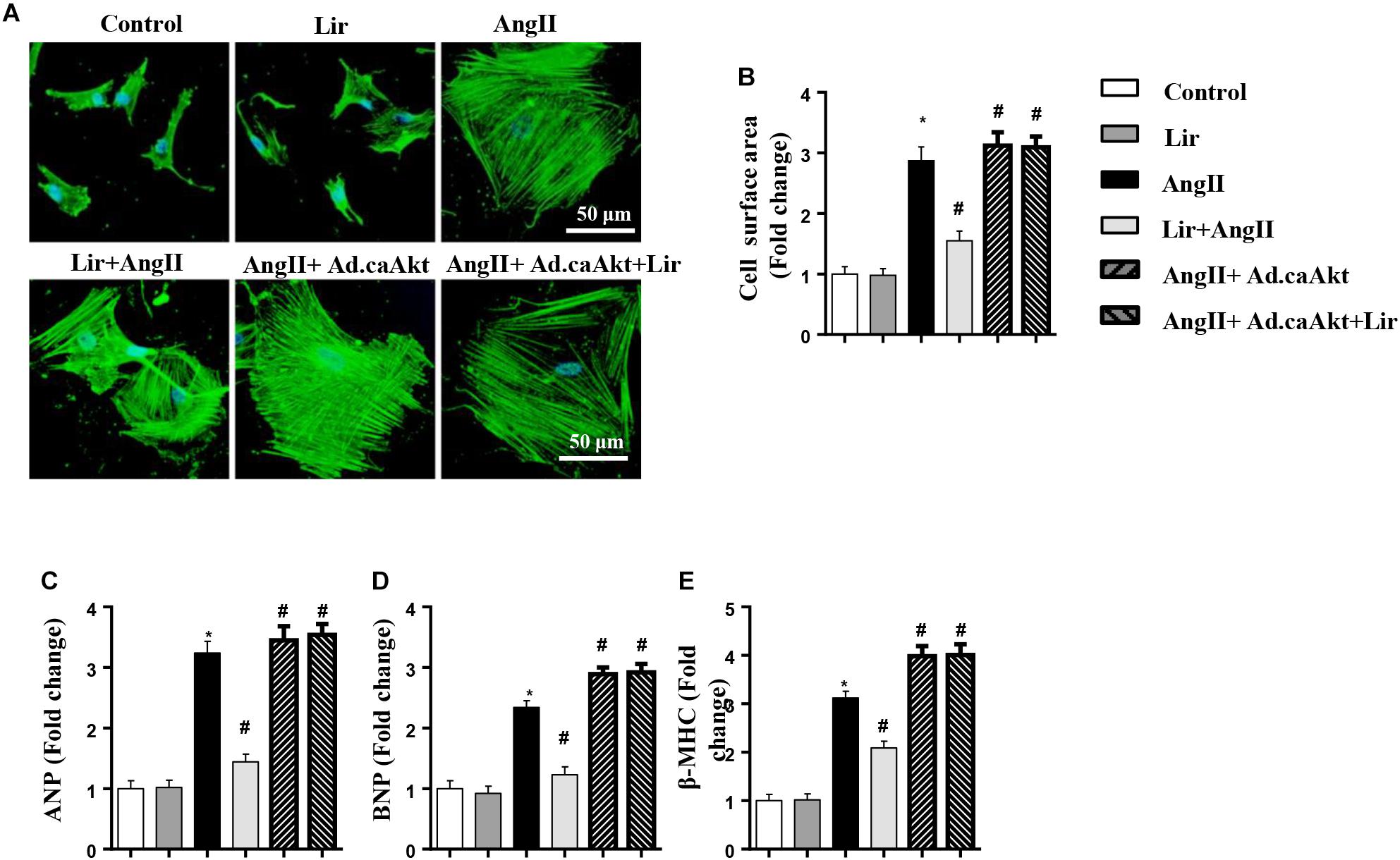
Figure 8. The effect of Lir on human cardiomyocytes. AC16 cells were stimulated with Ang II (1 μM) in the presence of Lir (100 nM) and infected with Ad.caAkt. (A) Representative immunofluorescent staining of α-actin (n = 6). (B) Quantitative analysis of CSA (n > 50 cells per group). (C–E) RT-PCR analysis of the mRNA levels of ANP, BNP, and β-MHC in the indicated groups (n = 6). ∗P < 0.05, compared to the control group; #P < 0.05, compared to the AngII group.
To confirm the effect of Lir on the other cardiac hypertrophy model, mice were subjected to TAC surgery and treated with Lir for 25 days. After 4 weeks of TAC, HW/BW, HW/TL, and LW/TL were remarkably increased in the control-TAC group. The CSA and the transcription levels of the hypertrophic markers were also increased in the control-TAC group (Figures 9A–D). Lir treatment attenuated the extent of the cardiac hypertrophy as assessed by decreased HW/BW, HW/TL, LW/TL, CSA, and reduced transcription levels of these hypertrophic markers (Figures 9A–D). Lir also improved the cardiac dysfunction induced by pressure overload (Figure 9E). The systolic and diastolic blood pressure (SBP/DBP) were measured 4 weeks after TAC surgery. SBP and DBP were found to be elevated in the TAC group and Lir slightly reduced the SBP and DBP (P > 0.05) (Figure 9F).
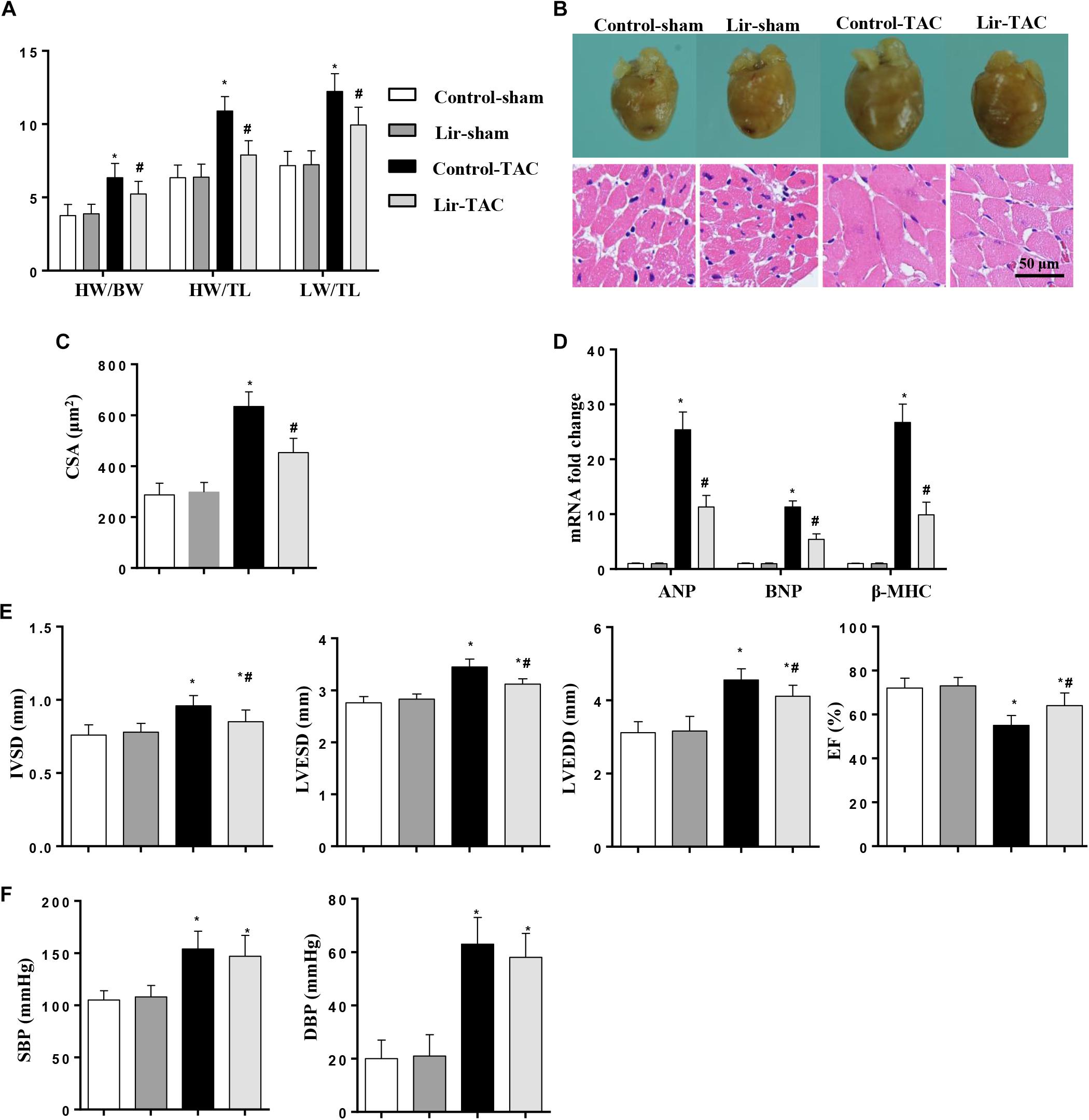
Figure 9. The effect of Lir on pressure overload induced cardiac hypertrophy. Mice were subjected to TAC surgery and treated with Lir for 25 days. Statistical results of the HW/BW, HW/TL, and LW/BW ratios (n = 10) at 4 weeks after TAC (A). The gross heart, HE staining (B) and CSA (n = 100 cells per section) (C) at 4 weeks after TAC. RT-PCR analysis of hypertrophic markers including ANP, BNP, and β-MHC from the heart of the different groups (n = 9) (D). Echocardiography results at 4 weeks after TAC (E, n = 10). Pressure loop measurements of systolic (SBP) and diastolic blood pressure (DBP) in different groups (n = 10 mice/group, F). ∗P < 0.05, compared to the corresponding sham group; #P < 0.05, compared to the control-TAC group.
Cardiac remodeling is an important adaptive response of cardiomyocytes to a variety of mechanical insults such as pressure overload and neurohormonal stimuli such as Ang II, endothelin-1, and adrenaline, which eventually lead to heart failure (Wu J. et al., 2014). It is known that pressure overload can activate the renin-angiotensin system (RAS) and induced the release of AngII, which activates the Galpha(q) protein-coupled receptor (GPCR) signaling pathway (Wu J. et al., 2014; Wu et al., 2015). Thus, we used both Ang II stimuli and a pressure overload model for in vivo study to induce cardiac remodeling in mice. In this study, we demonstrated Lir could attenuate AngII and pressure overload-induced cardiac remodeling, improving the cardiac dysfunction. Lir had a direct effect on the cardiomyocytes that inhibited the AngII-induced cardiomyocytes hypertrophic response. These protective effects were mediated by inhibition of the PI3K/Akt1 pathway and activation of AMPKa. We found that both continuous activation of Akt1 and knock down of AMPKa could abrogate the protective effect of Lir. Our study has revealed a novel approach to the treatment of cardiac remodeling.
The PI3K/Akt pathway is one of the key signaling cascades activated upon IGF-1 receptor (IGF-1R) stimulation. This pathway plays a central role in regulating metabolism, glucose uptake, proliferation and protein synthesis, all of which have the single goal of promoting cell survival (Wu et al., 2017). Mammals express three isoforms of Akt: Akt1, Akt2, and Akt3. Akt1-KO mice show a reduced cardiac muscle mass and display defects in physiological growth (Chen et al., 2001). When subjected to pressure overload, these mice develop an exacerbated form of cardiac hypertrophy (DeBosch et al., 2006). Among the downstream effectors of Akt, mTOR is activated by Akt phosphorylation. mTORC1, a complex form of mTOR, stimulates protein synthesis through the eukaryotic initiation factor 4E-BP1 and p70S6 kinase during protein translation, and the acceleration of protein translation can enhance cell growth and mass (Aoyagi and Matsui, 2011; Wu Q.Q. et al., 2014). A previous study reported that Lir exerted cardioprotection via mTOR/ULK1-dependent autophagy in glucose toxicity-induced cardiac injury (Yu et al., 2018). Lir has also been reported to regulate PI3K/Akt signaling in type 2 diabetic rats (Yang et al., 2018; Yu et al., 2018). In this study, we found that PI3K/Akt1 signaling was activated after 4 weeks of AngII infusion. Lir inhibits PI3K/Akt1 signaling. Activation of mTOR or Akt1 could abolish the protective effects of Lir on cardiomyocyte hypertrophy. These findings suggest that Lir exerted an anti-hypertrophic effect by inhibition of PI3K/Akt signaling.
AMP-activated protein kinase is a sensor of energy status. When the cellular AMP/ATP ratio increases, AMPK is activated by phosphorylation, leading to reduced energy consumption, and increased energy production by the cells (Viollet et al., 2003). AMPK deletion mice display dilated cardiomyopathy and cardiac dysfunction in the absence of pathological stress (Sung et al., 2015), which proves the importance of AMPK in the cardiovascular physiological process. Studies have shown the benefits of many AMPKα activators in protection against LV remodeling (Zong et al., 2013; Xu et al., 2018). Thus, increasing the activation of AMPKa may be a therapeutic target for cardiac hypertrophy. In the heart, insulin stimulates a variety of kinase cascades and controls glucose utilization. Insulin is able to activate Akt and inactivate AMPK in the heart (Kovacic et al., 2003). Studies have reported that Akt and AMPKa are closely associated with each other. Akt activity negatively regulates the phosphorylation of AMPK in cardiomyocytes (Kovacic et al., 2003). Lir was reported to exert neuroprotective effects via activation of AMPKa signaling in a diabetic model (Kong et al., 2018; Yang et al., 2018). In this study, AMPKα activity was increased in AngII stimulated cardiomyocytes. Lir intervention further augmented the activation of AMPKa in cardiomyocytes. AMPKa knockout abolished the protective effects of Lir in vivo. These data indicate that AMPKa signaling regulation also mediated the protective effects of Lir on cardiac hypertrophy. The crosstalk of Akt and AMPKa signaling in cardiomyocytes contributes to the downregulation of the hypertrophic response. Thus, merely knocking out AMPKa or activation of Akt could totally counteract Lir’s cardio-protection effects.
For further confirmation, the effect of Lir on cardiomyocyte hypertrophy was observed in vitro in both neonatal rat cardiomyocytes and human AC16 cardiomyocytes following AngII treatment. The results showed that Lir reduced the AngII-induced hypertrophic response in both neonatal rat cardiomyocytes and human AC16 cardiomyocytes. Furthermore, activation of Akt in both neonatal rat cardiomyocytes and human AC16 cardiomyocytes counteracted Lir’s cardio-protection effects, which is consistent with the in vivo results. These results suggest promising results from application of Lir to treat cardiac hypertrophy patients in clinical practice.
As indicated by a large number of researchers, myocardial fibrosis is associated with cardiac hypertrophy, myocardial infarction, hypertension, and heart failure (Daniel et al., 2016; Shimizu and Minamino, 2016; Wu et al., 2016). Myocardial fibrosis is a common pathological change during the development of various heart diseases. A major presentation of ventricular remodeling, myocardial fibrosis can lead to myocardial stiffness, impaired ventricular diastolic function, decreased coronary flow reserve, and even sudden death (Porter and Turner, 2009; Sirish et al., 2013). Lir was reported to exert anti-renal fibrosis via inhibition of the epithelial-mesenchymal transition (Li et al., 2018). Lir improves myocardial fibrosis after myocardial infarction through inhibition of CTGF by activating cAMP in mice (Huang et al., 2018). Our findings provide valuable experimental data for clinical research on the protective effect of Lir. Thus, it is important to explore the mechanisms that stimulate collagen deposition in the heart and define approaches to limit these processes. As shown here, Lir treatment significantly attenuated cardiac fibrosis after AngII-infusion. These results showed that Lir can inhibit the fibrosis induced by AngII-infusion. The mechanism underlying the anti-fibrosis effect of Lir on cardiac hypertrophy may attribute to the cardio-protection of Lir, which exerts a beneficial communication between cardiomyocytes and fibroblasts. Further study into the anti-fibrosis effect of Lir based on fibroblast experiments is still needed.
As a GLP-1R agonist, whether the antihypertrophic action of Lir is directly mediated by GLP-1R is unclear. Our data suggest that after stimulation with AngII, the expression of GLP-1R was elevated, but Lir did not affect the expression level of GLP-1R. Thus, these data provide indirect evidence that the effects of Lir on AMPKα signaling do not rely on GLP-1R. However, whether GLP-1R mediates the effects needs to be elucidated.
Studies have also reported an anti-hypertension effect of Lir (Rondinelli et al., 2017; Simonds et al., 2019). In our study, we found that Lir treatment could slightly reduce the SBP and DBP induced by both AngII infusion and TAC surgery (without a significant difference). This inconsistency may be attributed to the differences in dosage used and the treatment duration. Overall, in our study the anti-hypertrophy effect of Lir is irrelevant to blood pressure reduction. Since we did not design a group with Ang II-infused/TAC surgery animals sacrificed 2 weeks after the infusion/surgery, a limitation of our study exists in that whether the anti-hypertrophic and anti-fibrotic effects seen with liraglutide treatment were reversing Ang II-induced effects or were only halting the effects of Ang II is unknown. Further studies are needed to clarify this question.
In conclusion, our present study showed for the first time that Lir can ameliorate cardiac hypertrophy by regulating the PI3K/Akt and AMPKa signaling pathways. The advantage of Lir is that Lir is currently in clinical use for treatment of type 2 diabetes. This gives Lir an advantage over many other small molecular peptides, plant extracts, or even gene therapies that are currently undergoing exploration to treat cardiac hypertrophy. Further studies of the comparison of Lir with other clinical anti-hypertrophic agents such as beta receptor blockers, angiotensin converting enzyme inhibitors, and angiotensin II receptor antagonists are needed.
All of the studies were performed in accordance with the guidelines of the NIH (Guide for the Care and Use of Laboratory Animals, 1996) and were approved by the Animal Care and Use Committee of the First Affiliated Hospital of Zhenzhou University.
RL and FW contributed to the conception and designed the experiments. RL, YS, and LG carried out the experiments. XiW and XuW analyzed the experimental results and revised the manuscript. RL and FW wrote and revised the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2019.00537/full#supplementary-material
AMPK, AMP-activated protein kinase; AngII, angiotensin II; ANP, atrial natriuretic peptide; BNP, brain natriuretic peptide; BW, body weight; CSA, cell surface area; CTGF, connective tissue growth factor; EF, ejection fraction; FS, fractional shortening; HE, hematoxylin and eosin; HW, heart weight; IVSd, interventricular septal end diastolic dimension; Lir, lirentin; LV, left ventricular; LVEDd, left ventricular end-diastolic dimension; LVESd, left ventricular end-systolic dimension; LW, lung weight; NC, negative control; PSR, picroSirius red; TAC, thoracic aorta coarctation; TGF-β1, transforming growth factor β1; TL, tibia length; α-MHC, α-myosin heavy chain; β-MHC, β-myosin heavy chain.
Aoyagi, T., and Matsui, T. (2011). Phosphoinositide-3 kinase signaling in cardiac hypertrophy and heart failure. Curr. Pharm. Des. 17, 1818–1824. doi: 10.2174/138161211796390976
Ban, K., Noyan-Ashraf, M., Hoefer, J., Bolz, S. S., Drucker, D. J., and Husain, M. (2008). Cardioprotective and vasodilatory actions of glucagon-like peptide 1 receptor are mediated through both glucagon-like peptide 1 receptor-dependent and -independent pathways. Circulation 117, 2340–2350. doi: 10.1161/CIRCULATIONAHA.107.739938
Chen, W. S., Xu, P. Z., Gottlob, K., Chen, M. L., Sokol, K., Shiyanova, T., et al. (2001). Growth retardation and increased apoptosis in mice with homozygous disruption of the Akt1 gene. Genes Dev. 15, 2203–2208. doi: 10.1101/gad.913901
Dai, Y., Mehta, J., and Chen, M. (2013). Glucagon-like peptide-1 receptor agonist liraglutide inhibits endothelin-1 in endothelial cell by repressing nuclear factor-kappa B activation. Cardiovasc. Drugs Ther. 27, 371–380. doi: 10.1007/s10557-013-6463-z
Daniel, L. L., Scofield, S., Thrasher, P., Dalal, S., Daniels, C. R., Foster, C. R., et al. (2016). Ataxia telangiectasia-mutated kinase deficiency exacerbates left ventricular dysfunction and remodeling late after myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 311, H445–H452. doi: 10.1152/ajpheart.00338.2016
DeBosch, B., Treskov, I., Lupu, T. S., Weinheimer, C., Kovacs, A., Courtois, M., et al. (2006). Akt1 is required for physiological cardiac growth. Circulation 113, 2097–2104. doi: 10.1161/circulationaha.105.595231
Ferrario, C. M. (2016). Cardiac remodelling and RAS inhibition. Ther. Adv. Cardiovasc. Dis. 10, 162–171. doi: 10.1177/1753944716642677
Huang, D. D., Huang, H., Yang, Q., and Chen, X. Q. (2018). Liraglutide improves myocardial fibrosis after myocardial infarction through inhibition of CTGF by activating cAMP in mice. Eur. Rev. Med. Pharmacol. Sci. 22, 4648–4656. doi: 10.26355/eurrev_201807_15524
Kawatani, M., Yamada, Y., and Kawatani, M. (2018). Glucagon-like peptide-1 (GLP-1) action in the mouse area postrema neurons. Peptides 107, 68–74. doi: 10.1016/j.peptides.2018.07.010
Kong, F. J., Wu, J. H., Sun, S. Y., Ma, L. L., and Zhou, J. Q. (2018). Liraglutide ameliorates cognitive decline by promoting autophagy via the AMP-activated protein kinase/mammalian target of rapamycin pathway in a streptozotocin-induced mouse model of diabetes. Neuropharmacology 131, 316–325. doi: 10.1016/j.neuropharm.2018.01.001
Kovacic, S., Soltys, C. L., Barr, A. J., Shiojima, I., Walsh, K., Dyck, J. R., et al. (2003). Akt activity negatively regulates phosphorylation of AMP-activated protein kinase in the heart. J. Biol. Chem. 278, 39422–39427. doi: 10.1074/jbc.m305371200
Kyhl, K., Lønborg, J., Hartmann, B., Kissow, H., Poulsen, S. S., Ali, H. E., et al. (2017). Lack of effect of prolonged treatment with liraglutide on cardiac remodeling in rats after acute myocardial infarction. Peptides 93, 1–12. doi: 10.1016/j.peptides.2017.04.009
Li, Y. K., Ma, D. X., Wang, Z. M., Hu, X. F., Li, S. L., Tian, H. Z., et al. (2018). The glucagon-like peptide-1 (GLP-1) analog liraglutide attenuates renal fibrosis. Pharmacol. Res. 131, 102–111. doi: 10.1016/j.phrs.2018.03.004
Liu, Y., Hu, Z., Liao, H. H., Liu, W., Liu, J., Ma, Z. G., et al. (2015). Toll-like receptor 5 deficiency attenuates interstitial cardiac fibrosis and dysfunction induced by pressure overload by inhibiting inflammation and the endothelial-mesenchymal transition. Biochim. Biophys. Acta 1852, 2456–2466. doi: 10.1016/j.bbadis.2015.08.013
Ma, Z. G., Zhang, X., Yuan, Y. P., Jin, Y. G., Li, N., Kong, C. Y., et al. (2018). A77 1726 (leflunomide) blocks and reverses cardiac hypertrophy and fibrosis in mice. Clin. Sci. 132, 685–699. doi: 10.1042/CS20180160
Mells, J. E., Fu, P., Sharma, S., Olson, D., Cheng, L., Handy, J. A., et al. (2012). Glp-1 analog, liraglutide, ameliorates hepatic steatosis and cardiac hypertrophy in C57BL/6J mice fed a Western diet. Am. J. Physiol. Gastrointest. Liver Physiol. 302, G225–G235. doi: 10.1152/ajpgi.00274.2011
Noyan-Ashraf, M. H., Momen, M., Ban, K., Sadi, A. M., Zhou, Y. Q., Riazi, A. M., et al. (2009). GLP-1R agonist liraglutide activates cytoprotective pathways and improves outcomes after experimental myocardial infarction in mice. Diabetes 58, 975–983. doi: 10.2337/db08-1193
Noyan-Ashraf, M. H., Shikatani, E., Schuiki, I., Mukovozov, I., Wu, J., Li, R. K., et al. (2013). A glucagon-like peptide-1 analog reverses the molecular pathology and cardiac dysfunction of a mouse model of obesity. Circulation 127, 74–85. doi: 10.1161/CIRCULATIONAHA.112.091215
Porter, K. E., and Turner, N. (2009). Cardiac fibroblasts: at the heart of myocardial remodeling. Pharmacol. Ther. 123, 255–278. doi: 10.1016/j.pharmthera.2009.05.002
Rondinelli, M., Rossi, A., Gandolfi, A., Saponaro, F., Bucciarelli, L., Adda, G., et al. (2017). Genovese use of liraglutide in the real world and impact at 36 months on metabolic control, weight, lipid profile, blood pressure, heart rate, and renal function. Clin. Ther. 39, 159–169. doi: 10.1016/j.clinthera.2016.11.001
Shimizu, I., and Minamino, T. (2016). Physiological and pathological cardiac hypertrophy. J. Mol. Cell Cardiol. 97, 245–262. doi: 10.1016/j.yjmcc.2016.06.001
Simonds, S. E., Pryor, J. T., Koegler, F. H., Buch-Rasmussen, A. S., Kelly, L. E., Grove, K. L., et al. (2019). Determining the effects of combined liraglutide and phentermine on metabolic parameters, blood pressure, and heart rate in lean and obese male mice. Diabetes 68, 683–695. doi: 10.2337/db18-1149
Sirish, P., Li, N., Liu, J. Y., Lee, K. S., Hwang, S. H., Qiu, H., et al. (2013). Unique mechanistic insights into the beneficial effects of soluble epoxide hydrolase inhibitors in the prevention of cardiac fibrosis. Proc. Natl. Acad. Sci. U.S.A. 110, 5618–5623. doi: 10.1073/pnas.1221972110
Sung, M. M., Zordoky, B. N., Bujak, A. L., Lally, J. S., Fung, D., Young, M. E., et al. (2015). AMPK deficiency in cardiac muscle results in dilated cardiomyopathy in the absence of changes in energy metabolism. Cardiovasc. Res. 107, 235–245. doi: 10.1093/cvr/cvv166
Timmers, L., Henriques, J., de Kleijn, D. P., Devries, J. H., Kemperman, H., Steendijk, P., et al. (2009). Exenatide reduces infarct size and improves cardiac function in a porcine model of ischemia and reperfusion injury. J. Am. Coll. Cardiol. 53, 501–510. doi: 10.1016/j.jacc.2008.10.033
Trujillo, J. M., and Nuffer, W. (2014). GLP-1 receptor agonists for type 2 diabetes mellitus: recent developments and emerging agents. Pharmacotherapy 34, 1174–1186. doi: 10.1002/phar.1507
Viollet, B., Andreelli, F., Jørgensen, S. B., Perrin, C., Flamez, D., Mu, J., et al. (2003). Physiological role of AMP-activated protein kinase (AMPK): insights from knockout mouse models. Biochem. Soc. Trans. 31(Pt 1), 216–219. doi: 10.1042/bst0310216
Wu, H., Chen, L., Xie, J., Li, R., Li, G. N., Chen, Q. H., et al. (2016). Periostin expression induced by oxidative stress contributes to myocardial fibrosis in a rat model of high salt-induced hypertension. Mol. Med. Rep. 14, 776–782. doi: 10.3892/mmr.2016.5308
Wu, J., You, J., Wang, S., Zhang, L., Gong, H., and Zou, Y. (2014). Insights into the activation and inhibition of angiotensin II type 1 receptor in the mechanically loaded heart. Circ. J. 78, 1283–1289. doi: 10.1253/circj.cj-14-0470
Wu, Q. Q., Xiao, Y., Yuan, Y., Ma, Z. G., Liao, H. H., Liu, C., et al. (2017). Mechanisms contributing to cardiac remodelling. Clin. Sci. 131, 2319–2345. doi: 10.1042/CS20171167
Wu, Q. Q., Xu, M., Yuan, Y., Li, F. F., Yang, Z., Liu, Y., et al. (2015). Cathepsin B deficiency attenuates cardiac remodeling in response to pressure overload via TNF-alpha/ASK1/JNK pathway. Am. J. Physiol. Heart Circ. Physiol. 308, H1143–H1154. doi: 10.1152/ajpheart.00601.2014
Wu, Q. Q., Zong, J., Gao, L., Dai, J., Yang, Z., Xu, M., et al. (2014). Sulforaphane protects H9c2 cardiomyocytes from angiotensin II-induced hypertrophy. Herz 39, 390–396. doi: 10.1007/s00059-013-3849-4
Xiao, Y., Yang, Z., Wu, Q. Q., Jiang, X. H., Yuan, Y., Chang, W., et al. (2017). Cucurbitacin B protects against pressure overload induced cardiac hypertrophy. J. Cell Biochem. 118, 3899–3910. doi: 10.1002/jcb.26041
Xu, M., Xue, R. Q., Lu, Y., Yong, S. Y., Wu, Q., Cui, Y. L., et al. (2018). Choline ameliorates cardiac hypertrophy by regulating metabolic remodelling and UPRmt through SIRT3-AMPK pathway. Cardiovasc. Res. 115, 530–545. doi: 10.1093/cvr/cvy217
Yang, Y., Fang, H., Xu, G., Zhen, Y., Zhang, Y., Tian, J., et al. (2018). Liraglutide improves cognitive impairment via the AMPK and PI3K/Akt signaling pathways in type 2 diabetic rats. Mol. Med. Rep. 18, 2449–2457. doi: 10.3892/mmr.2018.9180
Yu, W., Zha, W., and Ren, J. (2018). Exendin-4 and liraglutide attenuate glucose toxicity-induced cardiac injury through mTOR/ULK1-dependent autophagy. Oxid. Med. Cell Longev. 2018:5396806. doi: 10.1155/2018/5396806
Zhang, L. H., Pang, X. F., Bai, F., Wang, N. P., Shah, A. I., McKallip, R. J., et al. (2015). Preservation of glucagon-like peptide-1 level attenuates angiotensin ii-induced tissue fibrosis by altering AT1/AT 2 receptor expression and angiotensin-converting enzyme 2 activity in rat heart. Cardiovasc. Drugs Ther. 29, 243–255. doi: 10.1007/s10557-015-6592-7
Keywords: liraglutide, cardiac hypertrophy, angiotensin II, AMPK a2, pressure overload
Citation: Li R, Shan Y, Gao L, Wang X, Wang X and Wang F (2019) The Glp-1 Analog Liraglutide Protects Against Angiotensin II and Pressure Overload-Induced Cardiac Hypertrophy via PI3K/Akt1 and AMPKa Signaling. Front. Pharmacol. 10:537. doi: 10.3389/fphar.2019.00537
Received: 06 September 2018; Accepted: 29 April 2019;
Published: 05 June 2019.
Edited by:
Paulo Correia-de-Sá, Universidade do Porto, PortugalReviewed by:
Jwu-Lai Yeh, Kaohsiung Medical University, TaiwanCopyright © 2019 Li, Shan, Gao, Wang, Wang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fang Wang, ZmNjd2FuZ2Y0QHp6dS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.